User login
Agent Orange linked to increased risk of MGUS

Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Despite responses, imetelstat won’t move forward in ET
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Imetelstat in MF: More research needed
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
First biosimilar launched in US
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
Team reports latest results of CTL019 in CLL

Photo from Penn Medicine
The chimeric antigen receptor (CAR) T-cell therapy CTL019 can produce durable responses in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in Science Translational Medicine.
Eight of 14 patients responded to CTL019—4 complete responses (CRs) and 4 partial responses (PRs).
Three of the patients with CRs were still alive and in remission at last follow-up. The longest remission has lasted 53 months.
Only 1 patient with a PR was still alive at last follow-up, and that patient progressed.
Nine patients developed cytokine release syndrome (CRS), some requiring intensive care. And there were 2 cases of tumor lysis syndrome.
These results are the most mature data from this trial. Results from this study were previously presented at ASH 2013 and ASH 2012, and they were published in NEJM and Science Translational Medicine in August 2011.
This study was supported by grants from Novartis, the Leukemia and Lymphoma Society, and the National Institutes of Health. CTL019 was originally developed at the University of Pennsylvania, but the university licensed the technology to Novartis.
Treatment and outcomes
The trial enrolled 23 CLL patients, but only 14 received CTL019. The 14 patients had a median age of 66 (range, 51 to 78), and most (n=14) were male.
They had received a median of 5 prior therapies (range, 1 to 11), and 8 patients had 17p deletion. All patients had active disease at the time of CTL019 infusion.
Patients received CTL019 at doses of 0.14 × 108 to 11 × 108 cells (median, 1.6 × 108 cells). Eight patients responded to the treatment, for an overall response rate of 57%.
Four patients (29%) achieved a CR. One of these patients died while in remission at 21 months due to infectious complications that occurred after removal of a basal cell carcinoma on his leg.
The other 3 CR patients remained alive at the time of analysis, with no evidence of leukemia at 28 months, 52 months, and 53 months after receiving their infusions. They did not receive additional therapy after CTL019.
“The durability of the remissions we have observed in this study are remarkable and have given us great hope that personalized cell therapies are going to be important options for patients whose cancers are no longer treatable with standard approaches,” said study author David L. Porter, MD, of the University of Pennsylvania Perelman School of Medicine in Philadelphia.
Four patients (29%) achieved a PR after receiving CTL019, with responses lasting a median of 7 months. Two of these patients died of disease progression 10 months and 27 months after receiving CTL019.
One PR patient died after suffering a pulmonary embolism 6 months after CTL019 infusion. The last PR patient experienced disease progression at 13 months, but the patient remained alive on other therapies 36 months after receiving CTL019.
Six patients (43%) did not respond to CTL019 and progressed within 1 month to 9 months. Tests revealed that the modified T cells did not expand as robustly in these patients as in those who experienced remissions.
Two of these patients later died from their disease or complications of other therapies, and 4 are receiving other types of treatment.
CRS and other toxicity
The investigators said infusional toxicities were infrequent and mild (less than grade 2). They were primarily low-grade fevers and chills.
The most frequent related adverse events were associated with complications of neutropenia (including fevers) and delayed CRS, which was correlated with in vivo CTL019 expansion. Nine patients (including all 8 responders) developed CRS.
Five patients with CRS required anti-cytokine-directed therapy—tocilizumab (n=4) and/or steroids (n=3). Four patients required intensive care for complications related to CRS, such as hypotension and hypoxia. They remained in the intensive care unit for a median of 6 days (range, 1-9).
Concurrent with CRS were 6 neurologic events in 5 patients—grade 1/2 hallucinations, confusion, or delirium typically associated with high fevers, intensive care, or medication use. There was 1 case of grade 4 confusion that lasted 2 days and was attributed, at least partly, to CTL019.
There were 2 cases of tumor lysis syndrome. And 1 patient died in remission 21 months after CTL019 infusion, having developed overwhelming ecthyma gangrenosum from a pseudomonas wound infection from a skin biopsy site.
CTL019 durability
“Importantly, our tests of patients who experienced complete remissions showed that the modified cells remain in patients’ bodies for years after their infusions, with no sign of cancerous or normal B cells,” said study author Carl H. June, MD, of the University of Pennsylvania Perelman School of Medicine.
“This suggests that at least some of the CTL019 cells retain their ability to hunt for cancerous cells for long periods of time.”
A lab experiment using CAR T cells isolated from one of the first patients to receive CTL019 confirmed the potential for long-term function of these cells. At nearly 3 years after infusion, the patient’s CTL019 cells demonstrated immediate and specific reactivity against cells expressing CD19.
CTL019 development
The investigators did not identify demographic or disease-related factors, such as age or types of prior therapies, that could be used to predict response to CTL019. And there was no association between T-cell dose and patient response.
An ongoing dose-optimization study is exploring this relationship in greater detail. Further future areas of study may include strategies to combine CTL019 with immune checkpoint inhibitors or other therapies to stimulate T-cell recognition of tumor cells.
In addition to CLL, CTL019 is under investigation in patients with acute lymphoblastic leukemia, non-Hodgkin lymphoma, and myeloma. The product has breakthrough designation from the US Food and Drug Administration for acute lymphoblastic leukemia. ![]()
Photo from Penn Medicine
The chimeric antigen receptor (CAR) T-cell therapy CTL019 can produce durable responses in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in Science Translational Medicine.
Eight of 14 patients responded to CTL019—4 complete responses (CRs) and 4 partial responses (PRs).
Three of the patients with CRs were still alive and in remission at last follow-up. The longest remission has lasted 53 months.
Only 1 patient with a PR was still alive at last follow-up, and that patient progressed.
Nine patients developed cytokine release syndrome (CRS), some requiring intensive care. And there were 2 cases of tumor lysis syndrome.
These results are the most mature data from this trial. Results from this study were previously presented at ASH 2013 and ASH 2012, and they were published in NEJM and Science Translational Medicine in August 2011.
This study was supported by grants from Novartis, the Leukemia and Lymphoma Society, and the National Institutes of Health. CTL019 was originally developed at the University of Pennsylvania, but the university licensed the technology to Novartis.
Treatment and outcomes
The trial enrolled 23 CLL patients, but only 14 received CTL019. The 14 patients had a median age of 66 (range, 51 to 78), and most (n=14) were male.
They had received a median of 5 prior therapies (range, 1 to 11), and 8 patients had 17p deletion. All patients had active disease at the time of CTL019 infusion.
Patients received CTL019 at doses of 0.14 × 108 to 11 × 108 cells (median, 1.6 × 108 cells). Eight patients responded to the treatment, for an overall response rate of 57%.
Four patients (29%) achieved a CR. One of these patients died while in remission at 21 months due to infectious complications that occurred after removal of a basal cell carcinoma on his leg.
The other 3 CR patients remained alive at the time of analysis, with no evidence of leukemia at 28 months, 52 months, and 53 months after receiving their infusions. They did not receive additional therapy after CTL019.
“The durability of the remissions we have observed in this study are remarkable and have given us great hope that personalized cell therapies are going to be important options for patients whose cancers are no longer treatable with standard approaches,” said study author David L. Porter, MD, of the University of Pennsylvania Perelman School of Medicine in Philadelphia.
Four patients (29%) achieved a PR after receiving CTL019, with responses lasting a median of 7 months. Two of these patients died of disease progression 10 months and 27 months after receiving CTL019.
One PR patient died after suffering a pulmonary embolism 6 months after CTL019 infusion. The last PR patient experienced disease progression at 13 months, but the patient remained alive on other therapies 36 months after receiving CTL019.
Six patients (43%) did not respond to CTL019 and progressed within 1 month to 9 months. Tests revealed that the modified T cells did not expand as robustly in these patients as in those who experienced remissions.
Two of these patients later died from their disease or complications of other therapies, and 4 are receiving other types of treatment.
CRS and other toxicity
The investigators said infusional toxicities were infrequent and mild (less than grade 2). They were primarily low-grade fevers and chills.
The most frequent related adverse events were associated with complications of neutropenia (including fevers) and delayed CRS, which was correlated with in vivo CTL019 expansion. Nine patients (including all 8 responders) developed CRS.
Five patients with CRS required anti-cytokine-directed therapy—tocilizumab (n=4) and/or steroids (n=3). Four patients required intensive care for complications related to CRS, such as hypotension and hypoxia. They remained in the intensive care unit for a median of 6 days (range, 1-9).
Concurrent with CRS were 6 neurologic events in 5 patients—grade 1/2 hallucinations, confusion, or delirium typically associated with high fevers, intensive care, or medication use. There was 1 case of grade 4 confusion that lasted 2 days and was attributed, at least partly, to CTL019.
There were 2 cases of tumor lysis syndrome. And 1 patient died in remission 21 months after CTL019 infusion, having developed overwhelming ecthyma gangrenosum from a pseudomonas wound infection from a skin biopsy site.
CTL019 durability
“Importantly, our tests of patients who experienced complete remissions showed that the modified cells remain in patients’ bodies for years after their infusions, with no sign of cancerous or normal B cells,” said study author Carl H. June, MD, of the University of Pennsylvania Perelman School of Medicine.
“This suggests that at least some of the CTL019 cells retain their ability to hunt for cancerous cells for long periods of time.”
A lab experiment using CAR T cells isolated from one of the first patients to receive CTL019 confirmed the potential for long-term function of these cells. At nearly 3 years after infusion, the patient’s CTL019 cells demonstrated immediate and specific reactivity against cells expressing CD19.
CTL019 development
The investigators did not identify demographic or disease-related factors, such as age or types of prior therapies, that could be used to predict response to CTL019. And there was no association between T-cell dose and patient response.
An ongoing dose-optimization study is exploring this relationship in greater detail. Further future areas of study may include strategies to combine CTL019 with immune checkpoint inhibitors or other therapies to stimulate T-cell recognition of tumor cells.
In addition to CLL, CTL019 is under investigation in patients with acute lymphoblastic leukemia, non-Hodgkin lymphoma, and myeloma. The product has breakthrough designation from the US Food and Drug Administration for acute lymphoblastic leukemia. ![]()
Photo from Penn Medicine
The chimeric antigen receptor (CAR) T-cell therapy CTL019 can produce durable responses in patients with relapsed/refractory chronic lymphocytic leukemia (CLL), according to research published in Science Translational Medicine.
Eight of 14 patients responded to CTL019—4 complete responses (CRs) and 4 partial responses (PRs).
Three of the patients with CRs were still alive and in remission at last follow-up. The longest remission has lasted 53 months.
Only 1 patient with a PR was still alive at last follow-up, and that patient progressed.
Nine patients developed cytokine release syndrome (CRS), some requiring intensive care. And there were 2 cases of tumor lysis syndrome.
These results are the most mature data from this trial. Results from this study were previously presented at ASH 2013 and ASH 2012, and they were published in NEJM and Science Translational Medicine in August 2011.
This study was supported by grants from Novartis, the Leukemia and Lymphoma Society, and the National Institutes of Health. CTL019 was originally developed at the University of Pennsylvania, but the university licensed the technology to Novartis.
Treatment and outcomes
The trial enrolled 23 CLL patients, but only 14 received CTL019. The 14 patients had a median age of 66 (range, 51 to 78), and most (n=14) were male.
They had received a median of 5 prior therapies (range, 1 to 11), and 8 patients had 17p deletion. All patients had active disease at the time of CTL019 infusion.
Patients received CTL019 at doses of 0.14 × 108 to 11 × 108 cells (median, 1.6 × 108 cells). Eight patients responded to the treatment, for an overall response rate of 57%.
Four patients (29%) achieved a CR. One of these patients died while in remission at 21 months due to infectious complications that occurred after removal of a basal cell carcinoma on his leg.
The other 3 CR patients remained alive at the time of analysis, with no evidence of leukemia at 28 months, 52 months, and 53 months after receiving their infusions. They did not receive additional therapy after CTL019.
“The durability of the remissions we have observed in this study are remarkable and have given us great hope that personalized cell therapies are going to be important options for patients whose cancers are no longer treatable with standard approaches,” said study author David L. Porter, MD, of the University of Pennsylvania Perelman School of Medicine in Philadelphia.
Four patients (29%) achieved a PR after receiving CTL019, with responses lasting a median of 7 months. Two of these patients died of disease progression 10 months and 27 months after receiving CTL019.
One PR patient died after suffering a pulmonary embolism 6 months after CTL019 infusion. The last PR patient experienced disease progression at 13 months, but the patient remained alive on other therapies 36 months after receiving CTL019.
Six patients (43%) did not respond to CTL019 and progressed within 1 month to 9 months. Tests revealed that the modified T cells did not expand as robustly in these patients as in those who experienced remissions.
Two of these patients later died from their disease or complications of other therapies, and 4 are receiving other types of treatment.
CRS and other toxicity
The investigators said infusional toxicities were infrequent and mild (less than grade 2). They were primarily low-grade fevers and chills.
The most frequent related adverse events were associated with complications of neutropenia (including fevers) and delayed CRS, which was correlated with in vivo CTL019 expansion. Nine patients (including all 8 responders) developed CRS.
Five patients with CRS required anti-cytokine-directed therapy—tocilizumab (n=4) and/or steroids (n=3). Four patients required intensive care for complications related to CRS, such as hypotension and hypoxia. They remained in the intensive care unit for a median of 6 days (range, 1-9).
Concurrent with CRS were 6 neurologic events in 5 patients—grade 1/2 hallucinations, confusion, or delirium typically associated with high fevers, intensive care, or medication use. There was 1 case of grade 4 confusion that lasted 2 days and was attributed, at least partly, to CTL019.
There were 2 cases of tumor lysis syndrome. And 1 patient died in remission 21 months after CTL019 infusion, having developed overwhelming ecthyma gangrenosum from a pseudomonas wound infection from a skin biopsy site.
CTL019 durability
“Importantly, our tests of patients who experienced complete remissions showed that the modified cells remain in patients’ bodies for years after their infusions, with no sign of cancerous or normal B cells,” said study author Carl H. June, MD, of the University of Pennsylvania Perelman School of Medicine.
“This suggests that at least some of the CTL019 cells retain their ability to hunt for cancerous cells for long periods of time.”
A lab experiment using CAR T cells isolated from one of the first patients to receive CTL019 confirmed the potential for long-term function of these cells. At nearly 3 years after infusion, the patient’s CTL019 cells demonstrated immediate and specific reactivity against cells expressing CD19.
CTL019 development
The investigators did not identify demographic or disease-related factors, such as age or types of prior therapies, that could be used to predict response to CTL019. And there was no association between T-cell dose and patient response.
An ongoing dose-optimization study is exploring this relationship in greater detail. Further future areas of study may include strategies to combine CTL019 with immune checkpoint inhibitors or other therapies to stimulate T-cell recognition of tumor cells.
In addition to CLL, CTL019 is under investigation in patients with acute lymphoblastic leukemia, non-Hodgkin lymphoma, and myeloma. The product has breakthrough designation from the US Food and Drug Administration for acute lymphoblastic leukemia. ![]()
MATRIX trial update: No clear winner
LONDON—The latest results* of the MATRIX trial haven’t provided any cut-and-dried answers when it comes to anticoagulation in the context of percutaneous coronary intervention (PCI), according to articles published in NEJM and data presented at the ESC Congress 2015.
The trial suggested that, overall, bivalirudin and heparin produce comparable results in patients undergoing PCI.
The drugs produced similar rates of major adverse cardiovascular events (MACE) and net adverse clinical events (NACE).
In addition, among patients in the bivalirudin arm, there was no significant difference in a composite primary outcome between patients who received an additional infusion of bivalirudin after PCI and those who did not.
Results from this trial were published in NEJM alongside a related editorial. At the ESC Congress 2015, the bivalirudin dosing comparison was reported in abstract 6004.
MATRIX was sponsored by the Societa Italiana di Cardiologia Invasiva (GISE), with funding from The Medicines Company and Terumo.
The trial enrolled 7213 patients with acute coronary syndrome who were set to undergo PCI. Patients were randomized to receive bivalirudin (n=3610) or unfractionated heparin (n=3603). Patients in the bivalirudin arm were then randomized to either receive a post-PCI infusion of bivalirudin (n=1799) or not (n=1811).
Primary outcomes for the comparison between bivalirudin and heparin were the occurrence of MACE (a composite of death, myocardial infarction, or stroke) and NACE (a composite of major bleeding or a major adverse cardiovascular event).
The primary outcome for the comparison of post-PCI bivalirudin infusion with no infusion was a composite of urgent target-vessel revascularization, definite stent thrombosis, or NACE.
Heparin vs bivalirudin
There was no significant difference between the bivalirudin and heparin groups with regard to MACE—10.3% and 10.9%, respectively (P=0.44). And the same was true for NACE—11.2% and 12.4%, respectively (P=0.12).
Likewise, there was no significant difference between the bivalirudin arm and the heparin arm with regard to myocardial infarction—8.6% and 8.5%, respectively (P=0.93)—or stroke—0.4% and 0.5%, respectively (P=0.57).
However, bivalirudin was associated with a significantly lower rate of all-cause mortality than heparin—1.7% and 2.3%, respectively (P=0.04)—and a significantly lower rate of cardiac-related death—1.5% and 2.2%, respectively (P=0.03).
The rate of definite stent thrombosis was significantly higher in the bivalirudin group than the heparin group—1.0% and 0.6%, respectively (P=0.048). But there was no significant difference in the rate of definite or probable stent thrombosis—1.3% and 1.0% (P=0.27).
The rate of any bleeding was significantly lower with bivalirudin than with heparin—11.0% and 13.6%, respectively (P=0.001). And the same was true for major bleeding (BARC 3 or 5)—1.4% and 2.5%, respectively (P<0.001).
Bivalirudin duration
“Post-PCI bivalirudin did not reduce the composite outcome of ischemic and bleeding outcomes, including stent thrombosis risk, as compared to no post-PCI bivalirudin infusion,” said study investigator Marco Valgimigli, MD, PhD, of the Swiss Cardiovascular Center in Bern, Switzerland.
The proportion of patients who met the primary endpoint was 11.0% in the post-PCI infusion group and 11.9% in the no-infusion group (P=0.34).
Similarly, there was no significant difference between the infusion and no-infusion groups in the risk of definite stent thrombosis—1.3% and 0.7%, respectively (P=0.09)—or definite/probable stent thrombosis—1.5% and 1.1%, respectively (P=0.29).
And there was no significant difference in the rate of any bleeding—11.3% and 10.7%, respectively (P=0.62)—but the rate of BARC 3 or 5 bleeding was lower in the group that received the post-PCI infusion—1.0% vs 1.8% (P=0.03).
“Both treatment options are allowed per current European label and observational studies,” Dr Valgimigli said.
“Hence, I believe the option to prolong or stop bivalirudin infusion after PCI remains open for clinicians, who will have to decide based on the ischemic and bleeding risk of individual patients, as well as perhaps based on type of acute coronary syndrome, timing of loading dose, and type of oral P2Y12 inhibitors. This is in keeping with the current labeling of the drug in EU and USA.”
Cost concerns
In the NEJM editorial, Peter Berger, MD, of North Shore–Long Island Jewish Health System in Great Neck, New York, noted that bivalirudin costs more than 400 times the price of heparin.
“Many studies have shown that bivalirudin is safer than heparin—that it causes fewer bleeding complications,” Dr Berger said. “But other studies have shown that heparin is every bit as safe, especially when used in lower doses. And one recent, large study actually suggested that heparin is more effective than bivalirudin.”
Despite these conflicting results, Dr Berger said “nearly everyone” agrees that if the more expensive drug is not superior in some way, the less expensive one should be used.
“Many studies raise more questions than they answer,” he added. “It will be interesting to see whether doctors accept the results of the MATRIX trial or wait for more studies before deciding which blood thinner they prefer.” ![]()
*MATRIX investigators previously compared vascular access sites and found that radial access outperformed femoral access. These results were published earlier this year in The Lancet.
LONDON—The latest results* of the MATRIX trial haven’t provided any cut-and-dried answers when it comes to anticoagulation in the context of percutaneous coronary intervention (PCI), according to articles published in NEJM and data presented at the ESC Congress 2015.
The trial suggested that, overall, bivalirudin and heparin produce comparable results in patients undergoing PCI.
The drugs produced similar rates of major adverse cardiovascular events (MACE) and net adverse clinical events (NACE).
In addition, among patients in the bivalirudin arm, there was no significant difference in a composite primary outcome between patients who received an additional infusion of bivalirudin after PCI and those who did not.
Results from this trial were published in NEJM alongside a related editorial. At the ESC Congress 2015, the bivalirudin dosing comparison was reported in abstract 6004.
MATRIX was sponsored by the Societa Italiana di Cardiologia Invasiva (GISE), with funding from The Medicines Company and Terumo.
The trial enrolled 7213 patients with acute coronary syndrome who were set to undergo PCI. Patients were randomized to receive bivalirudin (n=3610) or unfractionated heparin (n=3603). Patients in the bivalirudin arm were then randomized to either receive a post-PCI infusion of bivalirudin (n=1799) or not (n=1811).
Primary outcomes for the comparison between bivalirudin and heparin were the occurrence of MACE (a composite of death, myocardial infarction, or stroke) and NACE (a composite of major bleeding or a major adverse cardiovascular event).
The primary outcome for the comparison of post-PCI bivalirudin infusion with no infusion was a composite of urgent target-vessel revascularization, definite stent thrombosis, or NACE.
Heparin vs bivalirudin
There was no significant difference between the bivalirudin and heparin groups with regard to MACE—10.3% and 10.9%, respectively (P=0.44). And the same was true for NACE—11.2% and 12.4%, respectively (P=0.12).
Likewise, there was no significant difference between the bivalirudin arm and the heparin arm with regard to myocardial infarction—8.6% and 8.5%, respectively (P=0.93)—or stroke—0.4% and 0.5%, respectively (P=0.57).
However, bivalirudin was associated with a significantly lower rate of all-cause mortality than heparin—1.7% and 2.3%, respectively (P=0.04)—and a significantly lower rate of cardiac-related death—1.5% and 2.2%, respectively (P=0.03).
The rate of definite stent thrombosis was significantly higher in the bivalirudin group than the heparin group—1.0% and 0.6%, respectively (P=0.048). But there was no significant difference in the rate of definite or probable stent thrombosis—1.3% and 1.0% (P=0.27).
The rate of any bleeding was significantly lower with bivalirudin than with heparin—11.0% and 13.6%, respectively (P=0.001). And the same was true for major bleeding (BARC 3 or 5)—1.4% and 2.5%, respectively (P<0.001).
Bivalirudin duration
“Post-PCI bivalirudin did not reduce the composite outcome of ischemic and bleeding outcomes, including stent thrombosis risk, as compared to no post-PCI bivalirudin infusion,” said study investigator Marco Valgimigli, MD, PhD, of the Swiss Cardiovascular Center in Bern, Switzerland.
The proportion of patients who met the primary endpoint was 11.0% in the post-PCI infusion group and 11.9% in the no-infusion group (P=0.34).
Similarly, there was no significant difference between the infusion and no-infusion groups in the risk of definite stent thrombosis—1.3% and 0.7%, respectively (P=0.09)—or definite/probable stent thrombosis—1.5% and 1.1%, respectively (P=0.29).
And there was no significant difference in the rate of any bleeding—11.3% and 10.7%, respectively (P=0.62)—but the rate of BARC 3 or 5 bleeding was lower in the group that received the post-PCI infusion—1.0% vs 1.8% (P=0.03).
“Both treatment options are allowed per current European label and observational studies,” Dr Valgimigli said.
“Hence, I believe the option to prolong or stop bivalirudin infusion after PCI remains open for clinicians, who will have to decide based on the ischemic and bleeding risk of individual patients, as well as perhaps based on type of acute coronary syndrome, timing of loading dose, and type of oral P2Y12 inhibitors. This is in keeping with the current labeling of the drug in EU and USA.”
Cost concerns
In the NEJM editorial, Peter Berger, MD, of North Shore–Long Island Jewish Health System in Great Neck, New York, noted that bivalirudin costs more than 400 times the price of heparin.
“Many studies have shown that bivalirudin is safer than heparin—that it causes fewer bleeding complications,” Dr Berger said. “But other studies have shown that heparin is every bit as safe, especially when used in lower doses. And one recent, large study actually suggested that heparin is more effective than bivalirudin.”
Despite these conflicting results, Dr Berger said “nearly everyone” agrees that if the more expensive drug is not superior in some way, the less expensive one should be used.
“Many studies raise more questions than they answer,” he added. “It will be interesting to see whether doctors accept the results of the MATRIX trial or wait for more studies before deciding which blood thinner they prefer.” ![]()
*MATRIX investigators previously compared vascular access sites and found that radial access outperformed femoral access. These results were published earlier this year in The Lancet.
LONDON—The latest results* of the MATRIX trial haven’t provided any cut-and-dried answers when it comes to anticoagulation in the context of percutaneous coronary intervention (PCI), according to articles published in NEJM and data presented at the ESC Congress 2015.
The trial suggested that, overall, bivalirudin and heparin produce comparable results in patients undergoing PCI.
The drugs produced similar rates of major adverse cardiovascular events (MACE) and net adverse clinical events (NACE).
In addition, among patients in the bivalirudin arm, there was no significant difference in a composite primary outcome between patients who received an additional infusion of bivalirudin after PCI and those who did not.
Results from this trial were published in NEJM alongside a related editorial. At the ESC Congress 2015, the bivalirudin dosing comparison was reported in abstract 6004.
MATRIX was sponsored by the Societa Italiana di Cardiologia Invasiva (GISE), with funding from The Medicines Company and Terumo.
The trial enrolled 7213 patients with acute coronary syndrome who were set to undergo PCI. Patients were randomized to receive bivalirudin (n=3610) or unfractionated heparin (n=3603). Patients in the bivalirudin arm were then randomized to either receive a post-PCI infusion of bivalirudin (n=1799) or not (n=1811).
Primary outcomes for the comparison between bivalirudin and heparin were the occurrence of MACE (a composite of death, myocardial infarction, or stroke) and NACE (a composite of major bleeding or a major adverse cardiovascular event).
The primary outcome for the comparison of post-PCI bivalirudin infusion with no infusion was a composite of urgent target-vessel revascularization, definite stent thrombosis, or NACE.
Heparin vs bivalirudin
There was no significant difference between the bivalirudin and heparin groups with regard to MACE—10.3% and 10.9%, respectively (P=0.44). And the same was true for NACE—11.2% and 12.4%, respectively (P=0.12).
Likewise, there was no significant difference between the bivalirudin arm and the heparin arm with regard to myocardial infarction—8.6% and 8.5%, respectively (P=0.93)—or stroke—0.4% and 0.5%, respectively (P=0.57).
However, bivalirudin was associated with a significantly lower rate of all-cause mortality than heparin—1.7% and 2.3%, respectively (P=0.04)—and a significantly lower rate of cardiac-related death—1.5% and 2.2%, respectively (P=0.03).
The rate of definite stent thrombosis was significantly higher in the bivalirudin group than the heparin group—1.0% and 0.6%, respectively (P=0.048). But there was no significant difference in the rate of definite or probable stent thrombosis—1.3% and 1.0% (P=0.27).
The rate of any bleeding was significantly lower with bivalirudin than with heparin—11.0% and 13.6%, respectively (P=0.001). And the same was true for major bleeding (BARC 3 or 5)—1.4% and 2.5%, respectively (P<0.001).
Bivalirudin duration
“Post-PCI bivalirudin did not reduce the composite outcome of ischemic and bleeding outcomes, including stent thrombosis risk, as compared to no post-PCI bivalirudin infusion,” said study investigator Marco Valgimigli, MD, PhD, of the Swiss Cardiovascular Center in Bern, Switzerland.
The proportion of patients who met the primary endpoint was 11.0% in the post-PCI infusion group and 11.9% in the no-infusion group (P=0.34).
Similarly, there was no significant difference between the infusion and no-infusion groups in the risk of definite stent thrombosis—1.3% and 0.7%, respectively (P=0.09)—or definite/probable stent thrombosis—1.5% and 1.1%, respectively (P=0.29).
And there was no significant difference in the rate of any bleeding—11.3% and 10.7%, respectively (P=0.62)—but the rate of BARC 3 or 5 bleeding was lower in the group that received the post-PCI infusion—1.0% vs 1.8% (P=0.03).
“Both treatment options are allowed per current European label and observational studies,” Dr Valgimigli said.
“Hence, I believe the option to prolong or stop bivalirudin infusion after PCI remains open for clinicians, who will have to decide based on the ischemic and bleeding risk of individual patients, as well as perhaps based on type of acute coronary syndrome, timing of loading dose, and type of oral P2Y12 inhibitors. This is in keeping with the current labeling of the drug in EU and USA.”
Cost concerns
In the NEJM editorial, Peter Berger, MD, of North Shore–Long Island Jewish Health System in Great Neck, New York, noted that bivalirudin costs more than 400 times the price of heparin.
“Many studies have shown that bivalirudin is safer than heparin—that it causes fewer bleeding complications,” Dr Berger said. “But other studies have shown that heparin is every bit as safe, especially when used in lower doses. And one recent, large study actually suggested that heparin is more effective than bivalirudin.”
Despite these conflicting results, Dr Berger said “nearly everyone” agrees that if the more expensive drug is not superior in some way, the less expensive one should be used.
“Many studies raise more questions than they answer,” he added. “It will be interesting to see whether doctors accept the results of the MATRIX trial or wait for more studies before deciding which blood thinner they prefer.” ![]()
*MATRIX investigators previously compared vascular access sites and found that radial access outperformed femoral access. These results were published earlier this year in The Lancet.
FDA approves drug to prevent delayed CINV in adults
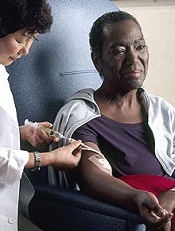
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved rolapitant (Varubi) for use in adult cancer patients receiving initial and repeat courses of emetogenic chemotherapy.
Rolapitant is to be used in combination with other antiemetic agents to prevent delayed chemotherapy-induced nausea and vomiting (CINV).
Tesaro, Inc., the company developing rolapitant, plans to launch the drug in the fourth quarter of this year.
Rolapitant is a selective and competitive antagonist of human substance P/neurokinin 1 (NK-1) receptors, with a plasma half-life of approximately 7 days. Activation of NK-1 receptors plays a central role in CINV, particularly in the delayed phase (the 25- to 120-hour period after chemotherapy administration).
Rolapitant comes in tablet form. The recommended dose is 180 mg, given approximately 1 to 2 hours prior to chemotherapy administration in combination with a 5-HT3 receptor antagonist and dexamethasone. No dosage adjustment is required for dexamethasone when administering rolapitant.
Rolapitant inhibits the CYP2D6 enzyme, so it is contraindicated with the use of thioridazine, a drug metabolized by the CYP2D6 enzyme. Use of these drugs together may increase the amount of thioridazine in the blood and cause an abnormal heart rhythm that can be serious.
Rolapitant clinical trials
Results from three phase 3 trials suggested that rolapitant (at 180 mg) in combination with a 5-HT3 receptor antagonist and dexamethasone was more effective than the 5-HT3 receptor antagonist and dexamethasone on their own (active control).
The 3-drug combination demonstrated a significant reduction in episodes of vomiting or use of rescue medication during the 25- to 120-hour period following administration of highly emetogenic and moderately emetogenic chemotherapy regimens.
In addition, patients who received rolapitant reported experiencing less nausea that interfered with normal daily life and fewer episodes of vomiting or retching over multiple cycles of chemotherapy.
Highly emetogenic chemotherapy
The clinical profile of rolapitant in cisplatin-based, highly emetogenic chemotherapy (HEC) was confirmed in two phase 3 studies: HEC1 and HEC2. Results from these trials were recently published in The Lancet Oncology.
Both trials met their primary endpoint of complete response (CR) and demonstrated statistical superiority of the rolapitant combination compared to active control.
In HEC1, 264 patients received the rolapitant combination, and 262 received active control. The proportion of patients achieving a CR was 72.7% and 58.4%, respectively (P<0.001).
In HEC2, 271 patients received the rolapitant combination, and 273 received active control. The proportion of patients achieving a CR was 70.1% and 61.9%, respectively (P=0.043).
The most common adverse events (≥3%) were neutropenia (9% rolapitant and 8% control), hiccups (5% and 4%), and abdominal pain (3% and 2%).
Moderately emetogenic chemotherapy
Researchers conducted another phase 3 trial to compare the rolapitant combination with active control in 1332 patients receiving moderately emetogenic chemotherapy regimens. Results from this trial were recently published in The Lancet Oncology.
This trial met its primary endpoint of CR and demonstrated statistical superiority of the rolapitant combination compared to active control. The proportion of patients achieving a CR was 71.3% and 61.6%, respectively (P<0.001).
The most common adverse events (≥3%) were decreased appetite (9% rolapitant and 7% control), neutropenia (7% and 6%), dizziness (6% and 4%), dyspepsia (4% and 2%), urinary tract infection (4% and 3%), stomatitis (4% and 2%), and anemia (3% and 2%).
The full prescribing information for rolapitant is available at www.varubirx.com. ![]()
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved rolapitant (Varubi) for use in adult cancer patients receiving initial and repeat courses of emetogenic chemotherapy.
Rolapitant is to be used in combination with other antiemetic agents to prevent delayed chemotherapy-induced nausea and vomiting (CINV).
Tesaro, Inc., the company developing rolapitant, plans to launch the drug in the fourth quarter of this year.
Rolapitant is a selective and competitive antagonist of human substance P/neurokinin 1 (NK-1) receptors, with a plasma half-life of approximately 7 days. Activation of NK-1 receptors plays a central role in CINV, particularly in the delayed phase (the 25- to 120-hour period after chemotherapy administration).
Rolapitant comes in tablet form. The recommended dose is 180 mg, given approximately 1 to 2 hours prior to chemotherapy administration in combination with a 5-HT3 receptor antagonist and dexamethasone. No dosage adjustment is required for dexamethasone when administering rolapitant.
Rolapitant inhibits the CYP2D6 enzyme, so it is contraindicated with the use of thioridazine, a drug metabolized by the CYP2D6 enzyme. Use of these drugs together may increase the amount of thioridazine in the blood and cause an abnormal heart rhythm that can be serious.
Rolapitant clinical trials
Results from three phase 3 trials suggested that rolapitant (at 180 mg) in combination with a 5-HT3 receptor antagonist and dexamethasone was more effective than the 5-HT3 receptor antagonist and dexamethasone on their own (active control).
The 3-drug combination demonstrated a significant reduction in episodes of vomiting or use of rescue medication during the 25- to 120-hour period following administration of highly emetogenic and moderately emetogenic chemotherapy regimens.
In addition, patients who received rolapitant reported experiencing less nausea that interfered with normal daily life and fewer episodes of vomiting or retching over multiple cycles of chemotherapy.
Highly emetogenic chemotherapy
The clinical profile of rolapitant in cisplatin-based, highly emetogenic chemotherapy (HEC) was confirmed in two phase 3 studies: HEC1 and HEC2. Results from these trials were recently published in The Lancet Oncology.
Both trials met their primary endpoint of complete response (CR) and demonstrated statistical superiority of the rolapitant combination compared to active control.
In HEC1, 264 patients received the rolapitant combination, and 262 received active control. The proportion of patients achieving a CR was 72.7% and 58.4%, respectively (P<0.001).
In HEC2, 271 patients received the rolapitant combination, and 273 received active control. The proportion of patients achieving a CR was 70.1% and 61.9%, respectively (P=0.043).
The most common adverse events (≥3%) were neutropenia (9% rolapitant and 8% control), hiccups (5% and 4%), and abdominal pain (3% and 2%).
Moderately emetogenic chemotherapy
Researchers conducted another phase 3 trial to compare the rolapitant combination with active control in 1332 patients receiving moderately emetogenic chemotherapy regimens. Results from this trial were recently published in The Lancet Oncology.
This trial met its primary endpoint of CR and demonstrated statistical superiority of the rolapitant combination compared to active control. The proportion of patients achieving a CR was 71.3% and 61.6%, respectively (P<0.001).
The most common adverse events (≥3%) were decreased appetite (9% rolapitant and 7% control), neutropenia (7% and 6%), dizziness (6% and 4%), dyspepsia (4% and 2%), urinary tract infection (4% and 3%), stomatitis (4% and 2%), and anemia (3% and 2%).
The full prescribing information for rolapitant is available at www.varubirx.com. ![]()
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved rolapitant (Varubi) for use in adult cancer patients receiving initial and repeat courses of emetogenic chemotherapy.
Rolapitant is to be used in combination with other antiemetic agents to prevent delayed chemotherapy-induced nausea and vomiting (CINV).
Tesaro, Inc., the company developing rolapitant, plans to launch the drug in the fourth quarter of this year.
Rolapitant is a selective and competitive antagonist of human substance P/neurokinin 1 (NK-1) receptors, with a plasma half-life of approximately 7 days. Activation of NK-1 receptors plays a central role in CINV, particularly in the delayed phase (the 25- to 120-hour period after chemotherapy administration).
Rolapitant comes in tablet form. The recommended dose is 180 mg, given approximately 1 to 2 hours prior to chemotherapy administration in combination with a 5-HT3 receptor antagonist and dexamethasone. No dosage adjustment is required for dexamethasone when administering rolapitant.
Rolapitant inhibits the CYP2D6 enzyme, so it is contraindicated with the use of thioridazine, a drug metabolized by the CYP2D6 enzyme. Use of these drugs together may increase the amount of thioridazine in the blood and cause an abnormal heart rhythm that can be serious.
Rolapitant clinical trials
Results from three phase 3 trials suggested that rolapitant (at 180 mg) in combination with a 5-HT3 receptor antagonist and dexamethasone was more effective than the 5-HT3 receptor antagonist and dexamethasone on their own (active control).
The 3-drug combination demonstrated a significant reduction in episodes of vomiting or use of rescue medication during the 25- to 120-hour period following administration of highly emetogenic and moderately emetogenic chemotherapy regimens.
In addition, patients who received rolapitant reported experiencing less nausea that interfered with normal daily life and fewer episodes of vomiting or retching over multiple cycles of chemotherapy.
Highly emetogenic chemotherapy
The clinical profile of rolapitant in cisplatin-based, highly emetogenic chemotherapy (HEC) was confirmed in two phase 3 studies: HEC1 and HEC2. Results from these trials were recently published in The Lancet Oncology.
Both trials met their primary endpoint of complete response (CR) and demonstrated statistical superiority of the rolapitant combination compared to active control.
In HEC1, 264 patients received the rolapitant combination, and 262 received active control. The proportion of patients achieving a CR was 72.7% and 58.4%, respectively (P<0.001).
In HEC2, 271 patients received the rolapitant combination, and 273 received active control. The proportion of patients achieving a CR was 70.1% and 61.9%, respectively (P=0.043).
The most common adverse events (≥3%) were neutropenia (9% rolapitant and 8% control), hiccups (5% and 4%), and abdominal pain (3% and 2%).
Moderately emetogenic chemotherapy
Researchers conducted another phase 3 trial to compare the rolapitant combination with active control in 1332 patients receiving moderately emetogenic chemotherapy regimens. Results from this trial were recently published in The Lancet Oncology.
This trial met its primary endpoint of CR and demonstrated statistical superiority of the rolapitant combination compared to active control. The proportion of patients achieving a CR was 71.3% and 61.6%, respectively (P<0.001).
The most common adverse events (≥3%) were decreased appetite (9% rolapitant and 7% control), neutropenia (7% and 6%), dizziness (6% and 4%), dyspepsia (4% and 2%), urinary tract infection (4% and 3%), stomatitis (4% and 2%), and anemia (3% and 2%).
The full prescribing information for rolapitant is available at www.varubirx.com.
Eltrombopag approved to treat SAA in EU

The European Commission has approved eltrombopag (Revolade) for the treatment of adults with severe aplastic anemia (SAA) who were either refractory to prior immunosuppressive therapy or heavily pretreated and are unsuitable for hematopoietic stem cell transplant.
Eltrombopag is the first therapy approved in the European Union (EU) for this patient population.
The approval applies to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
Trials of eltrombopag in SAA
The European Commission’s approval is based primarily on results of a phase 2 pilot study (NCT00922883) conducted by the National Heart, Lung and Blood Institute at the National Institutes of Health.
Results from the ongoing study were published in NEJM in 2012 and Blood in 2013. The trial has enrolled 43 patients with SAA who had an insufficient response to at least 1 prior immunosuppressive therapy and who had a platelet count of 30 x 109/L or less.
At baseline, the median platelet count was 20 x 109/L, hemoglobin was 8.4 g/dL, the absolute neutrophil count was 0.58 x 109/L, and absolute reticulocyte count was 24.3 x 109/L.
Patients had a median age of 45 (range, 17 to 77), and 56% were male. The majority of patients (84%) had received at least 2 prior immunosuppressive therapies.
Patients received eltrombopag at an initial dose of 50 mg once daily for 2 weeks. The dose increased over 2-week periods to a maximum of 150 mg once daily.
The study’s primary endpoint was hematologic response, which was initially assessed after 12 weeks of treatment. Treatment was discontinued after 16 weeks in patients who did not exhibit a hematologic response.
Forty percent of patients (n=17) experienced a hematologic response in at least 1 lineage—platelets, red blood cells (RBCs), or white blood cells—after week 12.
In the extension phase of the study, 8 patients achieved a multilineage response. Four of these patients subsequently tapered off treatment and maintained the response. The median follow-up was 8.1 months (range, 7.2 to 10.6 months).
Ninety-one percent of patients were platelet-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require platelet transfusions for a median of 200 days (range, 8 to 1096 days).
Eighty-six percent of patients were RBC-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require RBC transfusions for a median of 208 days (range, 15 to 1082 days).
The most common adverse events (≥20%) associated with eltrombopag were nausea (33%), fatigue (28%), cough (23%), diarrhea (21%), and headache (21%).
Patients were also evaluated for cytogenetic abnormalities. Eight patients had a new cytogenetic abnormality after treatment, including 5 patients who had complex changes in chromosome 7.
Patients who develop new cytogenetic abnormalities while on eltrombopag may need to be taken off treatment.
About eltrombopag
Eltrombopag is already approved to treat SAA in the US and Canada. The drug recently gained approval in the US to treat children age 1 and older who have chronic immune thrombocytopenia and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is approved in more than 100 countries to treat adults with chronic immune thrombocytopenia who have had an inadequate response to or are intolerant of other treatments.
The drug is approved in more than 45 countries for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow them to initiate and maintain interferon-based therapy.
Eltrombopag is marketed under the brand name Promacta in the US and Revolade in most other countries. For more details on the drug, see the European Medicines Agency’s Summary of Product Characteristics.
The European Commission has approved eltrombopag (Revolade) for the treatment of adults with severe aplastic anemia (SAA) who were either refractory to prior immunosuppressive therapy or heavily pretreated and are unsuitable for hematopoietic stem cell transplant.
Eltrombopag is the first therapy approved in the European Union (EU) for this patient population.
The approval applies to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
Trials of eltrombopag in SAA
The European Commission’s approval is based primarily on results of a phase 2 pilot study (NCT00922883) conducted by the National Heart, Lung and Blood Institute at the National Institutes of Health.
Results from the ongoing study were published in NEJM in 2012 and Blood in 2013. The trial has enrolled 43 patients with SAA who had an insufficient response to at least 1 prior immunosuppressive therapy and who had a platelet count of 30 x 109/L or less.
At baseline, the median platelet count was 20 x 109/L, hemoglobin was 8.4 g/dL, the absolute neutrophil count was 0.58 x 109/L, and absolute reticulocyte count was 24.3 x 109/L.
Patients had a median age of 45 (range, 17 to 77), and 56% were male. The majority of patients (84%) had received at least 2 prior immunosuppressive therapies.
Patients received eltrombopag at an initial dose of 50 mg once daily for 2 weeks. The dose increased over 2-week periods to a maximum of 150 mg once daily.
The study’s primary endpoint was hematologic response, which was initially assessed after 12 weeks of treatment. Treatment was discontinued after 16 weeks in patients who did not exhibit a hematologic response.
Forty percent of patients (n=17) experienced a hematologic response in at least 1 lineage—platelets, red blood cells (RBCs), or white blood cells—after week 12.
In the extension phase of the study, 8 patients achieved a multilineage response. Four of these patients subsequently tapered off treatment and maintained the response. The median follow-up was 8.1 months (range, 7.2 to 10.6 months).
Ninety-one percent of patients were platelet-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require platelet transfusions for a median of 200 days (range, 8 to 1096 days).
Eighty-six percent of patients were RBC-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require RBC transfusions for a median of 208 days (range, 15 to 1082 days).
The most common adverse events (≥20%) associated with eltrombopag were nausea (33%), fatigue (28%), cough (23%), diarrhea (21%), and headache (21%).
Patients were also evaluated for cytogenetic abnormalities. Eight patients had a new cytogenetic abnormality after treatment, including 5 patients who had complex changes in chromosome 7.
Patients who develop new cytogenetic abnormalities while on eltrombopag may need to be taken off treatment.
About eltrombopag
Eltrombopag is already approved to treat SAA in the US and Canada. The drug recently gained approval in the US to treat children age 1 and older who have chronic immune thrombocytopenia and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is approved in more than 100 countries to treat adults with chronic immune thrombocytopenia who have had an inadequate response to or are intolerant of other treatments.
The drug is approved in more than 45 countries for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow them to initiate and maintain interferon-based therapy.
Eltrombopag is marketed under the brand name Promacta in the US and Revolade in most other countries. For more details on the drug, see the European Medicines Agency’s Summary of Product Characteristics.
The European Commission has approved eltrombopag (Revolade) for the treatment of adults with severe aplastic anemia (SAA) who were either refractory to prior immunosuppressive therapy or heavily pretreated and are unsuitable for hematopoietic stem cell transplant.
Eltrombopag is the first therapy approved in the European Union (EU) for this patient population.
The approval applies to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
Trials of eltrombopag in SAA
The European Commission’s approval is based primarily on results of a phase 2 pilot study (NCT00922883) conducted by the National Heart, Lung and Blood Institute at the National Institutes of Health.
Results from the ongoing study were published in NEJM in 2012 and Blood in 2013. The trial has enrolled 43 patients with SAA who had an insufficient response to at least 1 prior immunosuppressive therapy and who had a platelet count of 30 x 109/L or less.
At baseline, the median platelet count was 20 x 109/L, hemoglobin was 8.4 g/dL, the absolute neutrophil count was 0.58 x 109/L, and absolute reticulocyte count was 24.3 x 109/L.
Patients had a median age of 45 (range, 17 to 77), and 56% were male. The majority of patients (84%) had received at least 2 prior immunosuppressive therapies.
Patients received eltrombopag at an initial dose of 50 mg once daily for 2 weeks. The dose increased over 2-week periods to a maximum of 150 mg once daily.
The study’s primary endpoint was hematologic response, which was initially assessed after 12 weeks of treatment. Treatment was discontinued after 16 weeks in patients who did not exhibit a hematologic response.
Forty percent of patients (n=17) experienced a hematologic response in at least 1 lineage—platelets, red blood cells (RBCs), or white blood cells—after week 12.
In the extension phase of the study, 8 patients achieved a multilineage response. Four of these patients subsequently tapered off treatment and maintained the response. The median follow-up was 8.1 months (range, 7.2 to 10.6 months).
Ninety-one percent of patients were platelet-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require platelet transfusions for a median of 200 days (range, 8 to 1096 days).
Eighty-six percent of patients were RBC-transfusion-dependent at baseline. Patients who responded to eltrombopag did not require RBC transfusions for a median of 208 days (range, 15 to 1082 days).
The most common adverse events (≥20%) associated with eltrombopag were nausea (33%), fatigue (28%), cough (23%), diarrhea (21%), and headache (21%).
Patients were also evaluated for cytogenetic abnormalities. Eight patients had a new cytogenetic abnormality after treatment, including 5 patients who had complex changes in chromosome 7.
Patients who develop new cytogenetic abnormalities while on eltrombopag may need to be taken off treatment.
About eltrombopag
Eltrombopag is already approved to treat SAA in the US and Canada. The drug recently gained approval in the US to treat children age 1 and older who have chronic immune thrombocytopenia and have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is approved in more than 100 countries to treat adults with chronic immune thrombocytopenia who have had an inadequate response to or are intolerant of other treatments.
The drug is approved in more than 45 countries for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow them to initiate and maintain interferon-based therapy.
Eltrombopag is marketed under the brand name Promacta in the US and Revolade in most other countries. For more details on the drug, see the European Medicines Agency’s Summary of Product Characteristics.
Rivaroxaban appears safe, effective in the real world

Photo courtesy of the CDC
LONDON—Patients with atrial fibrillation (AF) who receive the anticoagulant rivaroxaban as stroke prophylaxis have low rates of major bleeding and stroke, according to real-world data from the XANTUS trial.
Investigators said this finding is similar to clinical trial results with rivaroxaban and suggest the drug is safe and effective for stroke prevention in patients with AF who have a high or low risk of thromboembolic events.
The team reported results of the XANTUS trial in the European Heart Journal and at the ESC Congress 2015 (abstract 5072). The study was sponsored by Bayer, the company developing rivaroxaban in cooperation with Janssen Pharmaceuticals, Inc.
“The findings reaffirm the positive benefit-risk profile of rivaroxaban established in the phase 3 clinical trial ROCKET AF, in which rivaroxaban was shown to provide effective stroke prevention with a similar overall bleeding profile and significantly lower rates of the most feared intracranial and fatal bleeds compared with vitamin K antagonists,” said John A. Camm, MD, of St. George’s University of London in the UK.
“The patients included in ROCKET AF were at moderate to high risk of stroke, with a mean CHADS2 score of 3.5, and the incidence of major bleeding in those taking rivaroxaban was 3.6 per 100 person-years. In XANTUS, patients seen in daily clinical practice had a lower risk of stroke, with a mean CHADS2 score of 2.0, and the incidence rate of major bleeding was lower, at 2.1 per 100 person-years.”
Patients and treatment
For the XANTUS trial, Dr Camm and his colleagues evaluated the outcomes of rivaroxaban use in 6784 patients with non-valvular AF who were treated at 311 centers across Europe and Canada in routine clinical practice.
Patients had newly diagnosed AF (18.5%), paroxysmal AF (40.6%), persistent AF (13.6%), and permanent AF (27%). For 0.2% of patients, data on their exact diagnosis was missing.
Comorbidities included hypertension (74.7%), diabetes mellitus (19.6%), prior stroke/non-central nervous system systemic embolism/transient ischemic attack (19%), congestive heart failure (18.6%), and prior myocardial infarction (10.1%).
The patients’ mean age was 71.5, and 37.2% were older than 75. Their mean CHADS2 score was 2, and their mean CHA2DS2VASc score was 3.4. A little more than half (54.5%) of patients were vitamin K agonist-naïve.
All treatment and dosing decisions for rivaroxaban were at the discretion of the treating physicians. Patients received the drug at 15 mg (n=1410), 20 mg (n=5336), or other doses (n=35).
They were followed for 1 year or until 30 days after premature treatment discontinuation. Bleeding events and major thromboembolic events were centrally adjudicated by an independent committee.
Results
By the end of the observation period, most patients (96.1%) had not experienced treatment-emergent major bleeding, all-cause death, or stroke/systemic embolism. And the majority of patients (80%) continued taking rivaroxaban throughout the 1-year study period.
The rate of stroke was 0.7% per year, and the annual rate of stroke/systemic embolism was 0.8%.
Overall, 2.1% of patients per year experienced treatment-emergent major bleeding. Non-major bleeding events occurred in 15.4% of patients per year.
The yearly rate of fatal bleeding was 0.2%, critical organ bleeding was 0.7%, intracranial hemorrhage was 0.4%, mucosal bleeding was 1.0%, and gastrointestinal bleeding was 0.9%. In addition, 0.9% of patients required transfusions of 2 or more units of packed red blood cells or whole blood per year.
The rate of on-treatment, all-cause mortality was 1.9% per year. There were 118 deaths in all, and some patients had multiple reasons reported as the cause of death. Causes of death were cardiovascular events (n=49), cancer (n=23), bleeding (n=12), infectious disease (n=10), other (n=16), and unexplained (n=9).
“These real-world insights from XANTUS complement and expand on what we already know from clinical trials and provide physicians with reassurance to prescribe rivaroxaban as an effective and well-tolerated treatment option for the broad range of patients with AF seen in their everyday clinical practice,” Dr Camm concluded.
Photo courtesy of the CDC
LONDON—Patients with atrial fibrillation (AF) who receive the anticoagulant rivaroxaban as stroke prophylaxis have low rates of major bleeding and stroke, according to real-world data from the XANTUS trial.
Investigators said this finding is similar to clinical trial results with rivaroxaban and suggest the drug is safe and effective for stroke prevention in patients with AF who have a high or low risk of thromboembolic events.
The team reported results of the XANTUS trial in the European Heart Journal and at the ESC Congress 2015 (abstract 5072). The study was sponsored by Bayer, the company developing rivaroxaban in cooperation with Janssen Pharmaceuticals, Inc.
“The findings reaffirm the positive benefit-risk profile of rivaroxaban established in the phase 3 clinical trial ROCKET AF, in which rivaroxaban was shown to provide effective stroke prevention with a similar overall bleeding profile and significantly lower rates of the most feared intracranial and fatal bleeds compared with vitamin K antagonists,” said John A. Camm, MD, of St. George’s University of London in the UK.
“The patients included in ROCKET AF were at moderate to high risk of stroke, with a mean CHADS2 score of 3.5, and the incidence of major bleeding in those taking rivaroxaban was 3.6 per 100 person-years. In XANTUS, patients seen in daily clinical practice had a lower risk of stroke, with a mean CHADS2 score of 2.0, and the incidence rate of major bleeding was lower, at 2.1 per 100 person-years.”
Patients and treatment
For the XANTUS trial, Dr Camm and his colleagues evaluated the outcomes of rivaroxaban use in 6784 patients with non-valvular AF who were treated at 311 centers across Europe and Canada in routine clinical practice.
Patients had newly diagnosed AF (18.5%), paroxysmal AF (40.6%), persistent AF (13.6%), and permanent AF (27%). For 0.2% of patients, data on their exact diagnosis was missing.
Comorbidities included hypertension (74.7%), diabetes mellitus (19.6%), prior stroke/non-central nervous system systemic embolism/transient ischemic attack (19%), congestive heart failure (18.6%), and prior myocardial infarction (10.1%).
The patients’ mean age was 71.5, and 37.2% were older than 75. Their mean CHADS2 score was 2, and their mean CHA2DS2VASc score was 3.4. A little more than half (54.5%) of patients were vitamin K agonist-naïve.
All treatment and dosing decisions for rivaroxaban were at the discretion of the treating physicians. Patients received the drug at 15 mg (n=1410), 20 mg (n=5336), or other doses (n=35).
They were followed for 1 year or until 30 days after premature treatment discontinuation. Bleeding events and major thromboembolic events were centrally adjudicated by an independent committee.
Results
By the end of the observation period, most patients (96.1%) had not experienced treatment-emergent major bleeding, all-cause death, or stroke/systemic embolism. And the majority of patients (80%) continued taking rivaroxaban throughout the 1-year study period.
The rate of stroke was 0.7% per year, and the annual rate of stroke/systemic embolism was 0.8%.
Overall, 2.1% of patients per year experienced treatment-emergent major bleeding. Non-major bleeding events occurred in 15.4% of patients per year.
The yearly rate of fatal bleeding was 0.2%, critical organ bleeding was 0.7%, intracranial hemorrhage was 0.4%, mucosal bleeding was 1.0%, and gastrointestinal bleeding was 0.9%. In addition, 0.9% of patients required transfusions of 2 or more units of packed red blood cells or whole blood per year.
The rate of on-treatment, all-cause mortality was 1.9% per year. There were 118 deaths in all, and some patients had multiple reasons reported as the cause of death. Causes of death were cardiovascular events (n=49), cancer (n=23), bleeding (n=12), infectious disease (n=10), other (n=16), and unexplained (n=9).
“These real-world insights from XANTUS complement and expand on what we already know from clinical trials and provide physicians with reassurance to prescribe rivaroxaban as an effective and well-tolerated treatment option for the broad range of patients with AF seen in their everyday clinical practice,” Dr Camm concluded.
Photo courtesy of the CDC
LONDON—Patients with atrial fibrillation (AF) who receive the anticoagulant rivaroxaban as stroke prophylaxis have low rates of major bleeding and stroke, according to real-world data from the XANTUS trial.
Investigators said this finding is similar to clinical trial results with rivaroxaban and suggest the drug is safe and effective for stroke prevention in patients with AF who have a high or low risk of thromboembolic events.
The team reported results of the XANTUS trial in the European Heart Journal and at the ESC Congress 2015 (abstract 5072). The study was sponsored by Bayer, the company developing rivaroxaban in cooperation with Janssen Pharmaceuticals, Inc.
“The findings reaffirm the positive benefit-risk profile of rivaroxaban established in the phase 3 clinical trial ROCKET AF, in which rivaroxaban was shown to provide effective stroke prevention with a similar overall bleeding profile and significantly lower rates of the most feared intracranial and fatal bleeds compared with vitamin K antagonists,” said John A. Camm, MD, of St. George’s University of London in the UK.
“The patients included in ROCKET AF were at moderate to high risk of stroke, with a mean CHADS2 score of 3.5, and the incidence of major bleeding in those taking rivaroxaban was 3.6 per 100 person-years. In XANTUS, patients seen in daily clinical practice had a lower risk of stroke, with a mean CHADS2 score of 2.0, and the incidence rate of major bleeding was lower, at 2.1 per 100 person-years.”
Patients and treatment
For the XANTUS trial, Dr Camm and his colleagues evaluated the outcomes of rivaroxaban use in 6784 patients with non-valvular AF who were treated at 311 centers across Europe and Canada in routine clinical practice.
Patients had newly diagnosed AF (18.5%), paroxysmal AF (40.6%), persistent AF (13.6%), and permanent AF (27%). For 0.2% of patients, data on their exact diagnosis was missing.
Comorbidities included hypertension (74.7%), diabetes mellitus (19.6%), prior stroke/non-central nervous system systemic embolism/transient ischemic attack (19%), congestive heart failure (18.6%), and prior myocardial infarction (10.1%).
The patients’ mean age was 71.5, and 37.2% were older than 75. Their mean CHADS2 score was 2, and their mean CHA2DS2VASc score was 3.4. A little more than half (54.5%) of patients were vitamin K agonist-naïve.
All treatment and dosing decisions for rivaroxaban were at the discretion of the treating physicians. Patients received the drug at 15 mg (n=1410), 20 mg (n=5336), or other doses (n=35).
They were followed for 1 year or until 30 days after premature treatment discontinuation. Bleeding events and major thromboembolic events were centrally adjudicated by an independent committee.
Results
By the end of the observation period, most patients (96.1%) had not experienced treatment-emergent major bleeding, all-cause death, or stroke/systemic embolism. And the majority of patients (80%) continued taking rivaroxaban throughout the 1-year study period.
The rate of stroke was 0.7% per year, and the annual rate of stroke/systemic embolism was 0.8%.
Overall, 2.1% of patients per year experienced treatment-emergent major bleeding. Non-major bleeding events occurred in 15.4% of patients per year.
The yearly rate of fatal bleeding was 0.2%, critical organ bleeding was 0.7%, intracranial hemorrhage was 0.4%, mucosal bleeding was 1.0%, and gastrointestinal bleeding was 0.9%. In addition, 0.9% of patients required transfusions of 2 or more units of packed red blood cells or whole blood per year.
The rate of on-treatment, all-cause mortality was 1.9% per year. There were 118 deaths in all, and some patients had multiple reasons reported as the cause of death. Causes of death were cardiovascular events (n=49), cancer (n=23), bleeding (n=12), infectious disease (n=10), other (n=16), and unexplained (n=9).
“These real-world insights from XANTUS complement and expand on what we already know from clinical trials and provide physicians with reassurance to prescribe rivaroxaban as an effective and well-tolerated treatment option for the broad range of patients with AF seen in their everyday clinical practice,” Dr Camm concluded.
Extending DAPT may provide benefit for some, speaker says
LONDON—Extending dual antiplatelet therapy (DAPT) beyond the recommended 12 months after coronary stenting “should be considered” in patients with a drug-eluting stent (DES) who are at low risk for bleeding, according to investigators from the OPTIDUAL trial.
The study showed that receiving DAPT—aspirin and clopidogrel—for an additional 36 months did not decrease the rate of major adverse cardiovascular and cerebrovascular events (MACCE).
However, there was a suggestion that it might reduce ischemic outcomes without excess bleeding.
“Given the lack of harm and the signal for benefit of prolonged DAPT in the OPTIDUAL trial, and the results from prior randomized trials testing long durations of DAPT, prolongation of DAPT beyond 12 months should be considered in patients without high-risk bleeding who have received a drug-eluting coronary stent and are event-free at 12 months,” said study investigator Gérard Helft, MD, PhD, of Hôpital Universitaire Pitié-Salpétrière in Paris, France.
He presented results of the trial at the ESC Congress 2015 (abstract 3159).
The study included 1385 patients from 58 French sites who had undergone percutaneous coronary intervention with placement of at least 1 DES for either stable coronary artery disease or acute coronary syndrome.
All patients had been on DAPT for 1 year and were randomly assigned to continue or to remain on aspirin alone for an additional 36 months.
There was no significant difference between the groups with regard to MACCE, a composite of all-cause death, myocardial infarction, stroke, and major bleeding. These events occurred in 5.8% of patients in the extended-DAPT group and 7.5% in the aspirin-only group (hazard ratio (HR=0.75, P=0.17).
Likewise, there was no significant difference between the treatment groups for any of the components of MACCE—stroke (HR=0.69, P=0.53), myocardial infarction (HR=0.67, P=0.31), major bleeding (HR=0.98, P=0.95), or death (HR=0.65, P=0.18).
There was a non-significant (but borderline) reduction in ischemic outcomes—a post-hoc outcome composite rate of death, myocardial infarction, or stroke—with extended DAPT. These events occurred in 4.2% of patients in the extended-DAPT group and 6.4% in the aspirin-only group (HR=0.64, P=0.06).
“The results are consistent with the recent findings on ischemic outcomes from the DAPT trial regarding the value of prolonging DAPT after DES placement,” Dr Helft said.
“There was no apparent harm, and the post-hoc efficacy signal on MACCE is consistent with the benefit seen in the DAPT trial. Thus, OPTIDUAL adds to the evidence suggesting benefit to extended DAPT after DES in patients who are event-free at 12 months.”
This study was funded by Assistance Publique - Hôpitaux de Paris and the Fédération Française de Cardiologie, with unrestricted grants from Cordis, Boston, Medtronic, Terumo, and Biotronik.
LONDON—Extending dual antiplatelet therapy (DAPT) beyond the recommended 12 months after coronary stenting “should be considered” in patients with a drug-eluting stent (DES) who are at low risk for bleeding, according to investigators from the OPTIDUAL trial.
The study showed that receiving DAPT—aspirin and clopidogrel—for an additional 36 months did not decrease the rate of major adverse cardiovascular and cerebrovascular events (MACCE).
However, there was a suggestion that it might reduce ischemic outcomes without excess bleeding.
“Given the lack of harm and the signal for benefit of prolonged DAPT in the OPTIDUAL trial, and the results from prior randomized trials testing long durations of DAPT, prolongation of DAPT beyond 12 months should be considered in patients without high-risk bleeding who have received a drug-eluting coronary stent and are event-free at 12 months,” said study investigator Gérard Helft, MD, PhD, of Hôpital Universitaire Pitié-Salpétrière in Paris, France.
He presented results of the trial at the ESC Congress 2015 (abstract 3159).
The study included 1385 patients from 58 French sites who had undergone percutaneous coronary intervention with placement of at least 1 DES for either stable coronary artery disease or acute coronary syndrome.
All patients had been on DAPT for 1 year and were randomly assigned to continue or to remain on aspirin alone for an additional 36 months.
There was no significant difference between the groups with regard to MACCE, a composite of all-cause death, myocardial infarction, stroke, and major bleeding. These events occurred in 5.8% of patients in the extended-DAPT group and 7.5% in the aspirin-only group (hazard ratio (HR=0.75, P=0.17).
Likewise, there was no significant difference between the treatment groups for any of the components of MACCE—stroke (HR=0.69, P=0.53), myocardial infarction (HR=0.67, P=0.31), major bleeding (HR=0.98, P=0.95), or death (HR=0.65, P=0.18).
There was a non-significant (but borderline) reduction in ischemic outcomes—a post-hoc outcome composite rate of death, myocardial infarction, or stroke—with extended DAPT. These events occurred in 4.2% of patients in the extended-DAPT group and 6.4% in the aspirin-only group (HR=0.64, P=0.06).
“The results are consistent with the recent findings on ischemic outcomes from the DAPT trial regarding the value of prolonging DAPT after DES placement,” Dr Helft said.
“There was no apparent harm, and the post-hoc efficacy signal on MACCE is consistent with the benefit seen in the DAPT trial. Thus, OPTIDUAL adds to the evidence suggesting benefit to extended DAPT after DES in patients who are event-free at 12 months.”
This study was funded by Assistance Publique - Hôpitaux de Paris and the Fédération Française de Cardiologie, with unrestricted grants from Cordis, Boston, Medtronic, Terumo, and Biotronik.
LONDON—Extending dual antiplatelet therapy (DAPT) beyond the recommended 12 months after coronary stenting “should be considered” in patients with a drug-eluting stent (DES) who are at low risk for bleeding, according to investigators from the OPTIDUAL trial.
The study showed that receiving DAPT—aspirin and clopidogrel—for an additional 36 months did not decrease the rate of major adverse cardiovascular and cerebrovascular events (MACCE).
However, there was a suggestion that it might reduce ischemic outcomes without excess bleeding.
“Given the lack of harm and the signal for benefit of prolonged DAPT in the OPTIDUAL trial, and the results from prior randomized trials testing long durations of DAPT, prolongation of DAPT beyond 12 months should be considered in patients without high-risk bleeding who have received a drug-eluting coronary stent and are event-free at 12 months,” said study investigator Gérard Helft, MD, PhD, of Hôpital Universitaire Pitié-Salpétrière in Paris, France.
He presented results of the trial at the ESC Congress 2015 (abstract 3159).
The study included 1385 patients from 58 French sites who had undergone percutaneous coronary intervention with placement of at least 1 DES for either stable coronary artery disease or acute coronary syndrome.
All patients had been on DAPT for 1 year and were randomly assigned to continue or to remain on aspirin alone for an additional 36 months.
There was no significant difference between the groups with regard to MACCE, a composite of all-cause death, myocardial infarction, stroke, and major bleeding. These events occurred in 5.8% of patients in the extended-DAPT group and 7.5% in the aspirin-only group (hazard ratio (HR=0.75, P=0.17).
Likewise, there was no significant difference between the treatment groups for any of the components of MACCE—stroke (HR=0.69, P=0.53), myocardial infarction (HR=0.67, P=0.31), major bleeding (HR=0.98, P=0.95), or death (HR=0.65, P=0.18).
There was a non-significant (but borderline) reduction in ischemic outcomes—a post-hoc outcome composite rate of death, myocardial infarction, or stroke—with extended DAPT. These events occurred in 4.2% of patients in the extended-DAPT group and 6.4% in the aspirin-only group (HR=0.64, P=0.06).
“The results are consistent with the recent findings on ischemic outcomes from the DAPT trial regarding the value of prolonging DAPT after DES placement,” Dr Helft said.
“There was no apparent harm, and the post-hoc efficacy signal on MACCE is consistent with the benefit seen in the DAPT trial. Thus, OPTIDUAL adds to the evidence suggesting benefit to extended DAPT after DES in patients who are event-free at 12 months.”
This study was funded by Assistance Publique - Hôpitaux de Paris and the Fédération Française de Cardiologie, with unrestricted grants from Cordis, Boston, Medtronic, Terumo, and Biotronik.