User login
FDA approves IVIG product for kids

Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous human immune globulin (IVIG) product (Gammaplex) for pediatric patients age 2 years and older who have primary humoral immunodeficiencies.
This includes, but is not limited to, the humoral immune defect in common variable immunodeficiency, X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott Aldrich syndrome, and severe combined immunodeficiencies.
The approval was based on data submitted to the FDA as part of a post-marketing commitment following approval of the product for replacement therapy in adults in 2009.
Data supporting the latest approval came from a study of 25 children and adolescents (ages 3 to 16) with primary immunodeficiencies who were treated with IVIG for 12 months.
The study’s primary efficacy endpoint was the incidence of serious, acute bacterial infections (SABIs) as defined by the FDA. Secondary endpoints were safety and tolerability.
Throughout the course of the study, there were 2 SABIs—both pneumonia—resulting in an annual SABI event rate of 0.09, well below the maximum SABI event rate of 0.5 per subject required for approval.
Fourteen subjects (56%) had an adverse event that was possibly related to IVIG. Two patients experienced events that were considered definitely related to the treatment—headache, fatigue, and myalgia.
The most common adverse events, occurring in ≥ 5% of subjects, were dyspnea (2/25, 8%), otitis media acute (2/25, 8%), and tonsillar disorder (2/25, 8%).
Two patients had a serious adverse event of lobar pneumonia. Neither of these was considered related to IVIG, and neither met FDA-defined SABI criteria. None of the subjects withdrew from the study due to adverse events.
IVIG is marketed as Gammaplex by Bio Products Laboratory Limited. For more details on the treatment, see the full prescribing information. ![]()
Drug on fast track to treat aHUS

Image by Kevin MacKenzie
The US Food and Drug Administration (FDA) has granted fast track designation to OMS721 for the treatment of atypical hemolytic uremic syndrome (aHUS).
OMS721 is a monoclonal antibody targeting mannan-binding lectin-associated serine protease-2 (MASP-2), a key regulator of the lectin pathway of the complement system.
The FDA previously granted OMS721 orphan designation for the prevention of thrombotic microangiopathies (TMAs).
Omeros Corporation, the company developing OMS721, has released results from a phase 1 trial of the drug in healthy subjects and an ongoing phase 2 trial in patients with TMAs, including aHUS.
Early positive responses in the phase 2 trial prompted the initiation of a compassionate use program for OMS721 to allow extended treatment of 2 patients who had completed 4 weeks of dosing.
Phase 1 results
In the phase 1 trial of healthy subjects, OMS721 was well tolerated and prompted a high degree of sustained lectin pathway inhibition, according to researchers.
Seven cohorts of subjects received OMS721 or placebo by either subcutaneous injection or intravenous infusion at increasing dose levels. The researchers observed no drug-related adverse events and no clinically significant abnormalities on laboratory tests or electrocardiograms.
At the highest dose evaluated, both routes of administration prompted inhibition of the lectin pathway and achieved the pharmacologic target of sustained inhibition for at least a week.
Phase 2 results and compassionate use
In the ongoing phase 2 study, all patients are receiving OMS721. The researchers said they have observed treatment-related, clinically meaningful improvements in disease markers among the patients treated thus far.
The first cohort in this trial consisted of 3 aHUS patients treated with the lowest dose of OMS721. All 3 patients had improvements in platelet counts after treatment. Serum haptoglobin improved in 2 patients, normalizing in 1.
Serum lactate dehydrogenase levels remained normal in 1 patient, substantially decreased to close to the normal range in another, and remained elevated in the third. Creatinine levels in the 1 patient with independent renal function improved.
One patient was taken off the trial because of a serious adverse event—a localized inflammatory response often related to certain types of infections, one of which the patient previously had for 3 years while on immunosuppressive therapy. All data to date indicate no active infection in this patient.
The patient relapsed after stopping OMS721 treatment. No other significant safety issues were observed in this trial or the phase 1 trial.
The other 2 aHUS patients in this cohort continue to receive OMS721 as part of a compassionate use program. Based on their improvements in disease markers, an investigator requested that Omeros continue to provide OMS721 to these patients.
Following European regulatory approval, Omeros released the shipment of OMS721 so these patients could continue treatment beyond the period that was initially planned for the phase 2 study.
About fast track and orphan designation
The FDA’s fast track program facilitates the development of drugs intended to treat serious or life-threatening conditions and that have the potential to address unmet medical needs. Fast track status affords the company developing a drug greater access to the FDA in order to expedite the drug’s development, review, and potential approval.
Many drugs that receive fast track designation also receive priority review, and their new drug applications may be accepted by the FDA as a rolling submission, in which portions of an application are reviewed before the complete application is submitted. Priority review and rolling submission can each provide further acceleration of the FDA’s approval process.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
Orphan designation provides a drug’s developer with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Image by Kevin MacKenzie
The US Food and Drug Administration (FDA) has granted fast track designation to OMS721 for the treatment of atypical hemolytic uremic syndrome (aHUS).
OMS721 is a monoclonal antibody targeting mannan-binding lectin-associated serine protease-2 (MASP-2), a key regulator of the lectin pathway of the complement system.
The FDA previously granted OMS721 orphan designation for the prevention of thrombotic microangiopathies (TMAs).
Omeros Corporation, the company developing OMS721, has released results from a phase 1 trial of the drug in healthy subjects and an ongoing phase 2 trial in patients with TMAs, including aHUS.
Early positive responses in the phase 2 trial prompted the initiation of a compassionate use program for OMS721 to allow extended treatment of 2 patients who had completed 4 weeks of dosing.
Phase 1 results
In the phase 1 trial of healthy subjects, OMS721 was well tolerated and prompted a high degree of sustained lectin pathway inhibition, according to researchers.
Seven cohorts of subjects received OMS721 or placebo by either subcutaneous injection or intravenous infusion at increasing dose levels. The researchers observed no drug-related adverse events and no clinically significant abnormalities on laboratory tests or electrocardiograms.
At the highest dose evaluated, both routes of administration prompted inhibition of the lectin pathway and achieved the pharmacologic target of sustained inhibition for at least a week.
Phase 2 results and compassionate use
In the ongoing phase 2 study, all patients are receiving OMS721. The researchers said they have observed treatment-related, clinically meaningful improvements in disease markers among the patients treated thus far.
The first cohort in this trial consisted of 3 aHUS patients treated with the lowest dose of OMS721. All 3 patients had improvements in platelet counts after treatment. Serum haptoglobin improved in 2 patients, normalizing in 1.
Serum lactate dehydrogenase levels remained normal in 1 patient, substantially decreased to close to the normal range in another, and remained elevated in the third. Creatinine levels in the 1 patient with independent renal function improved.
One patient was taken off the trial because of a serious adverse event—a localized inflammatory response often related to certain types of infections, one of which the patient previously had for 3 years while on immunosuppressive therapy. All data to date indicate no active infection in this patient.
The patient relapsed after stopping OMS721 treatment. No other significant safety issues were observed in this trial or the phase 1 trial.
The other 2 aHUS patients in this cohort continue to receive OMS721 as part of a compassionate use program. Based on their improvements in disease markers, an investigator requested that Omeros continue to provide OMS721 to these patients.
Following European regulatory approval, Omeros released the shipment of OMS721 so these patients could continue treatment beyond the period that was initially planned for the phase 2 study.
About fast track and orphan designation
The FDA’s fast track program facilitates the development of drugs intended to treat serious or life-threatening conditions and that have the potential to address unmet medical needs. Fast track status affords the company developing a drug greater access to the FDA in order to expedite the drug’s development, review, and potential approval.
Many drugs that receive fast track designation also receive priority review, and their new drug applications may be accepted by the FDA as a rolling submission, in which portions of an application are reviewed before the complete application is submitted. Priority review and rolling submission can each provide further acceleration of the FDA’s approval process.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
Orphan designation provides a drug’s developer with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Image by Kevin MacKenzie
The US Food and Drug Administration (FDA) has granted fast track designation to OMS721 for the treatment of atypical hemolytic uremic syndrome (aHUS).
OMS721 is a monoclonal antibody targeting mannan-binding lectin-associated serine protease-2 (MASP-2), a key regulator of the lectin pathway of the complement system.
The FDA previously granted OMS721 orphan designation for the prevention of thrombotic microangiopathies (TMAs).
Omeros Corporation, the company developing OMS721, has released results from a phase 1 trial of the drug in healthy subjects and an ongoing phase 2 trial in patients with TMAs, including aHUS.
Early positive responses in the phase 2 trial prompted the initiation of a compassionate use program for OMS721 to allow extended treatment of 2 patients who had completed 4 weeks of dosing.
Phase 1 results
In the phase 1 trial of healthy subjects, OMS721 was well tolerated and prompted a high degree of sustained lectin pathway inhibition, according to researchers.
Seven cohorts of subjects received OMS721 or placebo by either subcutaneous injection or intravenous infusion at increasing dose levels. The researchers observed no drug-related adverse events and no clinically significant abnormalities on laboratory tests or electrocardiograms.
At the highest dose evaluated, both routes of administration prompted inhibition of the lectin pathway and achieved the pharmacologic target of sustained inhibition for at least a week.
Phase 2 results and compassionate use
In the ongoing phase 2 study, all patients are receiving OMS721. The researchers said they have observed treatment-related, clinically meaningful improvements in disease markers among the patients treated thus far.
The first cohort in this trial consisted of 3 aHUS patients treated with the lowest dose of OMS721. All 3 patients had improvements in platelet counts after treatment. Serum haptoglobin improved in 2 patients, normalizing in 1.
Serum lactate dehydrogenase levels remained normal in 1 patient, substantially decreased to close to the normal range in another, and remained elevated in the third. Creatinine levels in the 1 patient with independent renal function improved.
One patient was taken off the trial because of a serious adverse event—a localized inflammatory response often related to certain types of infections, one of which the patient previously had for 3 years while on immunosuppressive therapy. All data to date indicate no active infection in this patient.
The patient relapsed after stopping OMS721 treatment. No other significant safety issues were observed in this trial or the phase 1 trial.
The other 2 aHUS patients in this cohort continue to receive OMS721 as part of a compassionate use program. Based on their improvements in disease markers, an investigator requested that Omeros continue to provide OMS721 to these patients.
Following European regulatory approval, Omeros released the shipment of OMS721 so these patients could continue treatment beyond the period that was initially planned for the phase 2 study.
About fast track and orphan designation
The FDA’s fast track program facilitates the development of drugs intended to treat serious or life-threatening conditions and that have the potential to address unmet medical needs. Fast track status affords the company developing a drug greater access to the FDA in order to expedite the drug’s development, review, and potential approval.
Many drugs that receive fast track designation also receive priority review, and their new drug applications may be accepted by the FDA as a rolling submission, in which portions of an application are reviewed before the complete application is submitted. Priority review and rolling submission can each provide further acceleration of the FDA’s approval process.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
Orphan designation provides a drug’s developer with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Method allows for reuse of holdout data sets

Photo by Darren Baker
Researchers say they have devised a method for obtaining statistical validity that allows scientists to reuse their datasets while minimizing the risk of false discoveries.
Historically, to prevent false discoveries, scientists have not been able to reuse data they’ve already tested to test new hypotheses, especially if those new hypotheses were produced after the first round of data analysis.
Such processes may contaminate the data.
This is true even if the data is partitioned into a training set and a holdout set, as is commonly done to help ensure statistical validity.
In this case, Hypotheses generated about correlations between items in the training set can be tested on the holdout set. Real relationships would exist in both sets, while false ones would fail to be replicated.
The problem with using holdouts in that way is that, by nature, they can only be reused if each hypothesis is independent of another. Even a few additional hypotheses chained off one another could quickly lead to false discovery.
So scientists must collect a fresh holdout set each time an analysis depends on the outcomes of previous work.
However, Cynthia Dwork, PhD, of Microsoft Research in Mountain View, California, and her colleagues say they have devised a method that allows scientists to reuse a holdout set many times while still guaranteeing statistical validity.
The researchers described this method in Science.
With the new method, scientists do not test hypotheses on the holdout set directly. Instead, they query the set through a differentially private algorithm.
A differentially private algorithm guarantees that analyses remain functionally identical when applied to two different datasets: one with and one without the data from any single individual.
This means any findings that would rely on idiosyncratic outliers of a given set would disappear when looking at data through a differentially private lens.
To test their algorithm, Dr Dwork and her colleagues performed adaptive analysis on a data set rigged so that it contained nothing but random noise. The set was abstract but could be thought of as one that tested 20,000 patients on 10,000 variables, such as variants in their genomes, for ones that were predictive of lung cancer.
Though, by design, none of the variables in the set were predictive of cancer, reuse of a holdout set in the standard way showed that 500 of the variables had significant predictive power. Performing the same analysis with the researchers’ reusable holdout tool, however, correctly showed the lack of meaningful correlations.
An experiment with a second rigged dataset depicted a more realistic scenario. There, some of the variables did have predictive power, but traditional holdout use created a combination of variables that wildly overestimated this power. The reusable holdout tool correctly identified the 20 that had true statistical significance.
Dr Dwork and her colleagues say their reusable holdout method can prevent accidental overfitting, where predictive trends only apply to a given dataset and can’t be generalized.
And their method can warn users when they are exhausting the validity of a dataset. This is a red flag for what is known as P-hacking, or intentionally gaming data to get a publishable level of significance.
In these ways, the researchers believe that implementing the reusable holdout algorithm will allow scientists to generate stronger, more generalizable findings from smaller amounts of data. ![]()
Photo by Darren Baker
Researchers say they have devised a method for obtaining statistical validity that allows scientists to reuse their datasets while minimizing the risk of false discoveries.
Historically, to prevent false discoveries, scientists have not been able to reuse data they’ve already tested to test new hypotheses, especially if those new hypotheses were produced after the first round of data analysis.
Such processes may contaminate the data.
This is true even if the data is partitioned into a training set and a holdout set, as is commonly done to help ensure statistical validity.
In this case, Hypotheses generated about correlations between items in the training set can be tested on the holdout set. Real relationships would exist in both sets, while false ones would fail to be replicated.
The problem with using holdouts in that way is that, by nature, they can only be reused if each hypothesis is independent of another. Even a few additional hypotheses chained off one another could quickly lead to false discovery.
So scientists must collect a fresh holdout set each time an analysis depends on the outcomes of previous work.
However, Cynthia Dwork, PhD, of Microsoft Research in Mountain View, California, and her colleagues say they have devised a method that allows scientists to reuse a holdout set many times while still guaranteeing statistical validity.
The researchers described this method in Science.
With the new method, scientists do not test hypotheses on the holdout set directly. Instead, they query the set through a differentially private algorithm.
A differentially private algorithm guarantees that analyses remain functionally identical when applied to two different datasets: one with and one without the data from any single individual.
This means any findings that would rely on idiosyncratic outliers of a given set would disappear when looking at data through a differentially private lens.
To test their algorithm, Dr Dwork and her colleagues performed adaptive analysis on a data set rigged so that it contained nothing but random noise. The set was abstract but could be thought of as one that tested 20,000 patients on 10,000 variables, such as variants in their genomes, for ones that were predictive of lung cancer.
Though, by design, none of the variables in the set were predictive of cancer, reuse of a holdout set in the standard way showed that 500 of the variables had significant predictive power. Performing the same analysis with the researchers’ reusable holdout tool, however, correctly showed the lack of meaningful correlations.
An experiment with a second rigged dataset depicted a more realistic scenario. There, some of the variables did have predictive power, but traditional holdout use created a combination of variables that wildly overestimated this power. The reusable holdout tool correctly identified the 20 that had true statistical significance.
Dr Dwork and her colleagues say their reusable holdout method can prevent accidental overfitting, where predictive trends only apply to a given dataset and can’t be generalized.
And their method can warn users when they are exhausting the validity of a dataset. This is a red flag for what is known as P-hacking, or intentionally gaming data to get a publishable level of significance.
In these ways, the researchers believe that implementing the reusable holdout algorithm will allow scientists to generate stronger, more generalizable findings from smaller amounts of data. ![]()
Photo by Darren Baker
Researchers say they have devised a method for obtaining statistical validity that allows scientists to reuse their datasets while minimizing the risk of false discoveries.
Historically, to prevent false discoveries, scientists have not been able to reuse data they’ve already tested to test new hypotheses, especially if those new hypotheses were produced after the first round of data analysis.
Such processes may contaminate the data.
This is true even if the data is partitioned into a training set and a holdout set, as is commonly done to help ensure statistical validity.
In this case, Hypotheses generated about correlations between items in the training set can be tested on the holdout set. Real relationships would exist in both sets, while false ones would fail to be replicated.
The problem with using holdouts in that way is that, by nature, they can only be reused if each hypothesis is independent of another. Even a few additional hypotheses chained off one another could quickly lead to false discovery.
So scientists must collect a fresh holdout set each time an analysis depends on the outcomes of previous work.
However, Cynthia Dwork, PhD, of Microsoft Research in Mountain View, California, and her colleagues say they have devised a method that allows scientists to reuse a holdout set many times while still guaranteeing statistical validity.
The researchers described this method in Science.
With the new method, scientists do not test hypotheses on the holdout set directly. Instead, they query the set through a differentially private algorithm.
A differentially private algorithm guarantees that analyses remain functionally identical when applied to two different datasets: one with and one without the data from any single individual.
This means any findings that would rely on idiosyncratic outliers of a given set would disappear when looking at data through a differentially private lens.
To test their algorithm, Dr Dwork and her colleagues performed adaptive analysis on a data set rigged so that it contained nothing but random noise. The set was abstract but could be thought of as one that tested 20,000 patients on 10,000 variables, such as variants in their genomes, for ones that were predictive of lung cancer.
Though, by design, none of the variables in the set were predictive of cancer, reuse of a holdout set in the standard way showed that 500 of the variables had significant predictive power. Performing the same analysis with the researchers’ reusable holdout tool, however, correctly showed the lack of meaningful correlations.
An experiment with a second rigged dataset depicted a more realistic scenario. There, some of the variables did have predictive power, but traditional holdout use created a combination of variables that wildly overestimated this power. The reusable holdout tool correctly identified the 20 that had true statistical significance.
Dr Dwork and her colleagues say their reusable holdout method can prevent accidental overfitting, where predictive trends only apply to a given dataset and can’t be generalized.
And their method can warn users when they are exhausting the validity of a dataset. This is a red flag for what is known as P-hacking, or intentionally gaming data to get a publishable level of significance.
In these ways, the researchers believe that implementing the reusable holdout algorithm will allow scientists to generate stronger, more generalizable findings from smaller amounts of data. ![]()
Group proposes revised staging system for MM

Photo by Juan D. Alfonso
Researchers from the International Myeloma Working Group (IMWG) have proposed revising the International Staging System (ISS) used to stratify patients with newly diagnosed multiple myeloma (MM).
The group’s revised ISS (R-ISS) combines the current ISS with tests for chromosomal abnormalities (CAs) and serum lactate dehydrogenase (LDH) in an attempt to refine the system’s prognostic value.
IMWG researchers assessed the R-ISS in more than 3000 newly diagnosed MM patients and found that patients with R-ISS stage I disease had better overall survival (OS) and progression-free survival (PFS) than patients with stage I disease according to the ISS.
And patients with R-ISS stage III disease had worse survival rates than patients with stage III disease according to the ISS. But PFS and OS numbers for stage II disease were the same with both systems.
The researchers reported these results in the Journal of Clinical Oncology.
They noted that the existing ISS relies on tests for serum β2-microglobulin and serum albumin to divide patients into 3 risk-factor stages. But the R-ISS adds interphase fluorescence in situ hybridization to check for CAs, along with separate tests for heightened LDH.
The researchers define the 3 R-ISS groups as follows:
- R-ISS I includes patients with ISS stage I disease (serum β2-microglobulin level < 3.5 mg/L and serum albumin level ≥ 3.5 g/dL), no high-risk CAs (del[17p] and/or t[4;14] and/or t[14;16]), and normal LDH levels (less than the upper limit of normal range).
- R-ISS III includes patients with ISS stage III disease (serum β2-microglobulin level > 5.5 mg/L) and high-risk CAs or high LDH levels.
- R-ISS II includes patients with all other possible combinations.
To evaluate the prognostic value of the R-ISS, the researchers analyzed data from 4445 newly diagnosed MM patients who were enrolled in 11 completed trials. ISS, CA, and LDH data were available for 3060 patients.
At a median follow-up of 46 months, the 5-year OS rate was 82% in the R-ISS I group (n=871), 62% in the R-ISS II group (n=1894), and 40% in the R-ISS III group (n=295). The 5-year PFS rates were 55%, 36%, and 24%, respectively.
In comparison, the 5-year OS rate was 77% for patients with ISS stage I disease (n=1615), 62% for ISS stage II (n=1630), and 47% for ISS stage III (n=987). The 5-year PFS rates were 49%, 36%, and 30%, respectively.
Based on this work, the researchers said the R-ISS is a simple but powerful prognostic staging system, and they recommend its use in future studies to stratify newly diagnosed MM patients effectively.
“The revised staging system can be used by doctors to discuss prognostic results very carefully with individual patients,” added Brian G.M. Durie, MD, chairman of the IMWG.
“It’s helpful to know the expectations and consider how treatments can be modified based on the new ISS system.” ![]()
Photo by Juan D. Alfonso
Researchers from the International Myeloma Working Group (IMWG) have proposed revising the International Staging System (ISS) used to stratify patients with newly diagnosed multiple myeloma (MM).
The group’s revised ISS (R-ISS) combines the current ISS with tests for chromosomal abnormalities (CAs) and serum lactate dehydrogenase (LDH) in an attempt to refine the system’s prognostic value.
IMWG researchers assessed the R-ISS in more than 3000 newly diagnosed MM patients and found that patients with R-ISS stage I disease had better overall survival (OS) and progression-free survival (PFS) than patients with stage I disease according to the ISS.
And patients with R-ISS stage III disease had worse survival rates than patients with stage III disease according to the ISS. But PFS and OS numbers for stage II disease were the same with both systems.
The researchers reported these results in the Journal of Clinical Oncology.
They noted that the existing ISS relies on tests for serum β2-microglobulin and serum albumin to divide patients into 3 risk-factor stages. But the R-ISS adds interphase fluorescence in situ hybridization to check for CAs, along with separate tests for heightened LDH.
The researchers define the 3 R-ISS groups as follows:
- R-ISS I includes patients with ISS stage I disease (serum β2-microglobulin level < 3.5 mg/L and serum albumin level ≥ 3.5 g/dL), no high-risk CAs (del[17p] and/or t[4;14] and/or t[14;16]), and normal LDH levels (less than the upper limit of normal range).
- R-ISS III includes patients with ISS stage III disease (serum β2-microglobulin level > 5.5 mg/L) and high-risk CAs or high LDH levels.
- R-ISS II includes patients with all other possible combinations.
To evaluate the prognostic value of the R-ISS, the researchers analyzed data from 4445 newly diagnosed MM patients who were enrolled in 11 completed trials. ISS, CA, and LDH data were available for 3060 patients.
At a median follow-up of 46 months, the 5-year OS rate was 82% in the R-ISS I group (n=871), 62% in the R-ISS II group (n=1894), and 40% in the R-ISS III group (n=295). The 5-year PFS rates were 55%, 36%, and 24%, respectively.
In comparison, the 5-year OS rate was 77% for patients with ISS stage I disease (n=1615), 62% for ISS stage II (n=1630), and 47% for ISS stage III (n=987). The 5-year PFS rates were 49%, 36%, and 30%, respectively.
Based on this work, the researchers said the R-ISS is a simple but powerful prognostic staging system, and they recommend its use in future studies to stratify newly diagnosed MM patients effectively.
“The revised staging system can be used by doctors to discuss prognostic results very carefully with individual patients,” added Brian G.M. Durie, MD, chairman of the IMWG.
“It’s helpful to know the expectations and consider how treatments can be modified based on the new ISS system.” ![]()
Photo by Juan D. Alfonso
Researchers from the International Myeloma Working Group (IMWG) have proposed revising the International Staging System (ISS) used to stratify patients with newly diagnosed multiple myeloma (MM).
The group’s revised ISS (R-ISS) combines the current ISS with tests for chromosomal abnormalities (CAs) and serum lactate dehydrogenase (LDH) in an attempt to refine the system’s prognostic value.
IMWG researchers assessed the R-ISS in more than 3000 newly diagnosed MM patients and found that patients with R-ISS stage I disease had better overall survival (OS) and progression-free survival (PFS) than patients with stage I disease according to the ISS.
And patients with R-ISS stage III disease had worse survival rates than patients with stage III disease according to the ISS. But PFS and OS numbers for stage II disease were the same with both systems.
The researchers reported these results in the Journal of Clinical Oncology.
They noted that the existing ISS relies on tests for serum β2-microglobulin and serum albumin to divide patients into 3 risk-factor stages. But the R-ISS adds interphase fluorescence in situ hybridization to check for CAs, along with separate tests for heightened LDH.
The researchers define the 3 R-ISS groups as follows:
- R-ISS I includes patients with ISS stage I disease (serum β2-microglobulin level < 3.5 mg/L and serum albumin level ≥ 3.5 g/dL), no high-risk CAs (del[17p] and/or t[4;14] and/or t[14;16]), and normal LDH levels (less than the upper limit of normal range).
- R-ISS III includes patients with ISS stage III disease (serum β2-microglobulin level > 5.5 mg/L) and high-risk CAs or high LDH levels.
- R-ISS II includes patients with all other possible combinations.
To evaluate the prognostic value of the R-ISS, the researchers analyzed data from 4445 newly diagnosed MM patients who were enrolled in 11 completed trials. ISS, CA, and LDH data were available for 3060 patients.
At a median follow-up of 46 months, the 5-year OS rate was 82% in the R-ISS I group (n=871), 62% in the R-ISS II group (n=1894), and 40% in the R-ISS III group (n=295). The 5-year PFS rates were 55%, 36%, and 24%, respectively.
In comparison, the 5-year OS rate was 77% for patients with ISS stage I disease (n=1615), 62% for ISS stage II (n=1630), and 47% for ISS stage III (n=987). The 5-year PFS rates were 49%, 36%, and 30%, respectively.
Based on this work, the researchers said the R-ISS is a simple but powerful prognostic staging system, and they recommend its use in future studies to stratify newly diagnosed MM patients effectively.
“The revised staging system can be used by doctors to discuss prognostic results very carefully with individual patients,” added Brian G.M. Durie, MD, chairman of the IMWG.
“It’s helpful to know the expectations and consider how treatments can be modified based on the new ISS system.” ![]()
FDA grants drug orphan designation for ITP

Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan designation to veltuzumab for the treatment of immune thrombocytopenia (ITP).
Veltuzumab is a 2nd-generation, humanized monoclonal antibody targeting CD20. The drug is being developed by Immunomedics as a treatment for ITP, other autoimmune diseases, and non-Hodgkin lymphoma.
Veltuzumab was considered active and well-tolerated in a phase 1 study of adults with ITP. The drug produced responses in about half of patients, with some responses lasting more than 4 years.
The study included 50 patients with primary ITP who had failed 1 or more types of standard therapy, had platelet levels of 30,000/μL or less, and did not have major bleeding. The patients’ median age was 54, and most were female (n=31). Eight patients had undergone splenectomy.
Patients were a median of 2 years from diagnosis. Fourteen had been diagnosed with ITP for a year or less and had received corticosteroids and/or immunoglobulins.
Thirty-six patients had chronic ITP and had received azathioprine or danazol (n=15), thrombopoietin-receptor agonists (n=10), rituximab (n=7), platelets (n=5), and/or chemotherapy (n=4).
The 34 patients assigned to cohort 1 received 2 doses of subcutaneous veltuzumab at 80 mg, 160 mg, or 320 mg, 2 weeks apart (total doses of 160 mg, 320 mg, and 640 mg, respectively). The 18 patients in cohort 2 (which included 2 rollovers) received once-weekly doses at 320 mg for 4 weeks (total dose of 1280 mg).
The researchers said veltuzumab was well tolerated. The only adverse events were grade 1-2, transient injection reactions.
Forty-seven patients were evaluable for response. Forty-seven percent (n=22) had objective responses (ORs), and 28% (n=13) had complete responses (CRs).
Responses did not differ much according to disease duration. Patients with chronic ITP had an OR rate of 42% and a CR rate of 27%. Patients who had ITP for a year or less had an OR rate of 51% and a CR rate of 29%.
The median time to relapse (TTR) did not differ much between patients with CRs and those with partial responses, but there was a sizable difference between patients with chronic ITP and those with newly diagnosed ITP.
The median TTR was 7.9 months for patients with a CR and 7.6 months for patients with a partial response. The median TTR was 6.9 months for patients with chronic ITP and 14.4 months for patients who had ITP for a year or less.
The phase 2 expansion trial of veltuzumab in ITP has completed accrual, and patients are being followed for up to 5 years.
About orphan designation
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US. Orphan designation provides the sponsor of a drug with various development incentives.
The orphan designation for veltuzumab provides Immunomedics with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan designation to veltuzumab for the treatment of immune thrombocytopenia (ITP).
Veltuzumab is a 2nd-generation, humanized monoclonal antibody targeting CD20. The drug is being developed by Immunomedics as a treatment for ITP, other autoimmune diseases, and non-Hodgkin lymphoma.
Veltuzumab was considered active and well-tolerated in a phase 1 study of adults with ITP. The drug produced responses in about half of patients, with some responses lasting more than 4 years.
The study included 50 patients with primary ITP who had failed 1 or more types of standard therapy, had platelet levels of 30,000/μL or less, and did not have major bleeding. The patients’ median age was 54, and most were female (n=31). Eight patients had undergone splenectomy.
Patients were a median of 2 years from diagnosis. Fourteen had been diagnosed with ITP for a year or less and had received corticosteroids and/or immunoglobulins.
Thirty-six patients had chronic ITP and had received azathioprine or danazol (n=15), thrombopoietin-receptor agonists (n=10), rituximab (n=7), platelets (n=5), and/or chemotherapy (n=4).
The 34 patients assigned to cohort 1 received 2 doses of subcutaneous veltuzumab at 80 mg, 160 mg, or 320 mg, 2 weeks apart (total doses of 160 mg, 320 mg, and 640 mg, respectively). The 18 patients in cohort 2 (which included 2 rollovers) received once-weekly doses at 320 mg for 4 weeks (total dose of 1280 mg).
The researchers said veltuzumab was well tolerated. The only adverse events were grade 1-2, transient injection reactions.
Forty-seven patients were evaluable for response. Forty-seven percent (n=22) had objective responses (ORs), and 28% (n=13) had complete responses (CRs).
Responses did not differ much according to disease duration. Patients with chronic ITP had an OR rate of 42% and a CR rate of 27%. Patients who had ITP for a year or less had an OR rate of 51% and a CR rate of 29%.
The median time to relapse (TTR) did not differ much between patients with CRs and those with partial responses, but there was a sizable difference between patients with chronic ITP and those with newly diagnosed ITP.
The median TTR was 7.9 months for patients with a CR and 7.6 months for patients with a partial response. The median TTR was 6.9 months for patients with chronic ITP and 14.4 months for patients who had ITP for a year or less.
The phase 2 expansion trial of veltuzumab in ITP has completed accrual, and patients are being followed for up to 5 years.
About orphan designation
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US. Orphan designation provides the sponsor of a drug with various development incentives.
The orphan designation for veltuzumab provides Immunomedics with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted orphan designation to veltuzumab for the treatment of immune thrombocytopenia (ITP).
Veltuzumab is a 2nd-generation, humanized monoclonal antibody targeting CD20. The drug is being developed by Immunomedics as a treatment for ITP, other autoimmune diseases, and non-Hodgkin lymphoma.
Veltuzumab was considered active and well-tolerated in a phase 1 study of adults with ITP. The drug produced responses in about half of patients, with some responses lasting more than 4 years.
The study included 50 patients with primary ITP who had failed 1 or more types of standard therapy, had platelet levels of 30,000/μL or less, and did not have major bleeding. The patients’ median age was 54, and most were female (n=31). Eight patients had undergone splenectomy.
Patients were a median of 2 years from diagnosis. Fourteen had been diagnosed with ITP for a year or less and had received corticosteroids and/or immunoglobulins.
Thirty-six patients had chronic ITP and had received azathioprine or danazol (n=15), thrombopoietin-receptor agonists (n=10), rituximab (n=7), platelets (n=5), and/or chemotherapy (n=4).
The 34 patients assigned to cohort 1 received 2 doses of subcutaneous veltuzumab at 80 mg, 160 mg, or 320 mg, 2 weeks apart (total doses of 160 mg, 320 mg, and 640 mg, respectively). The 18 patients in cohort 2 (which included 2 rollovers) received once-weekly doses at 320 mg for 4 weeks (total dose of 1280 mg).
The researchers said veltuzumab was well tolerated. The only adverse events were grade 1-2, transient injection reactions.
Forty-seven patients were evaluable for response. Forty-seven percent (n=22) had objective responses (ORs), and 28% (n=13) had complete responses (CRs).
Responses did not differ much according to disease duration. Patients with chronic ITP had an OR rate of 42% and a CR rate of 27%. Patients who had ITP for a year or less had an OR rate of 51% and a CR rate of 29%.
The median time to relapse (TTR) did not differ much between patients with CRs and those with partial responses, but there was a sizable difference between patients with chronic ITP and those with newly diagnosed ITP.
The median TTR was 7.9 months for patients with a CR and 7.6 months for patients with a partial response. The median TTR was 6.9 months for patients with chronic ITP and 14.4 months for patients who had ITP for a year or less.
The phase 2 expansion trial of veltuzumab in ITP has completed accrual, and patients are being followed for up to 5 years.
About orphan designation
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US. Orphan designation provides the sponsor of a drug with various development incentives.
The orphan designation for veltuzumab provides Immunomedics with opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, 7 years of US marketing exclusivity if the drug is approved, and other benefits. ![]()
Reporting requirements may affect trial outcomes

Photo by Esther Dyson
The reporting requirements developed to increase transparency in US medical research may lead to fewer positive trial outcomes, according to a study published in PLOS ONE.
Researchers analyzed data from large-budget trials funded by the National Heart, Lung and Blood Institute (NHLBI).
And they found evidence suggesting the reporting requirements may have contributed to a significant reduction in studies with positive findings.
The reporting standards were phased in around 2000. They require researchers conducting drug or dietary supplement trials using human subjects to identify
projected outcomes and register their trials on ClinicalTrials.gov before they begin to collect data.
When entering their trial into the database, researchers are required to state the specific outcome on which they will focus. In the past, a researcher might have published an aspect of a study that was successful, even if the study overall did not produce the expected results.
But the new requirements mean researchers are less likely to change their analysis plan to consider another outcome that may have shown a positive result, said Veronica L. Irvin, PhD, of Oregon State University in Corvallis.
Dr Irvin began working on this project with the study’s lead author, Robert M. Kaplan, PhD, of the Agency for Healthcare Research and Quality in Rockville,
Maryland, while the two worked together in the National Institutes of Health’s Office of Behavior and Social Science Research.
The pair reviewed all large-budget clinical trials evaluating drugs or dietary supplements for the treatment or prevention of cardiovascular disease that had received funding from the NHLBI between 1970 and 2012.
They chose large-budget, NHLBI-funded trials in part because outcomes from the trials were more likely to be published, even if they did not produce the expected result.
Fifty-five studies were included in the research. Thirty were published prior to the reporting changes in 2000 (1970 to 1999), and 25 were published after the changes (2000 to 2012).
Of the studies published after 2000, only 2 (8%) showed positive outcomes, while 17 (57%) of the studies published before 2000 showed positive results.
Drs Kaplan and Irvin acknowledged that factors other than the reporting requirements may be contributing to the decline in positive outcomes, but they were unable to identify other compelling explanations.
For example, one suggestion was that older trials were more likely to compare new treatments to placebos, while newer trials were more likely to compare new
treatments to established treatments.
But when Drs Kaplan and Irvin examined the data, they found that 60% of trials published before 2000 used placebo comparators and nearly the same amount, 64%, of trials published after 2000 used placebos.
The researchers noted that although this work focused on clinical trials related to cardiovascular health, it would be reasonable to see similar changes in results
across other disease types.
“We don’t know if this decrease in positive outcomes also affects drug trials for prevention and treatment of cancer, diabetes, or other diseases,” Dr Irvin said. “But it would not be surprising because they have the same reporting requirements.” ![]()
Photo by Esther Dyson
The reporting requirements developed to increase transparency in US medical research may lead to fewer positive trial outcomes, according to a study published in PLOS ONE.
Researchers analyzed data from large-budget trials funded by the National Heart, Lung and Blood Institute (NHLBI).
And they found evidence suggesting the reporting requirements may have contributed to a significant reduction in studies with positive findings.
The reporting standards were phased in around 2000. They require researchers conducting drug or dietary supplement trials using human subjects to identify
projected outcomes and register their trials on ClinicalTrials.gov before they begin to collect data.
When entering their trial into the database, researchers are required to state the specific outcome on which they will focus. In the past, a researcher might have published an aspect of a study that was successful, even if the study overall did not produce the expected results.
But the new requirements mean researchers are less likely to change their analysis plan to consider another outcome that may have shown a positive result, said Veronica L. Irvin, PhD, of Oregon State University in Corvallis.
Dr Irvin began working on this project with the study’s lead author, Robert M. Kaplan, PhD, of the Agency for Healthcare Research and Quality in Rockville,
Maryland, while the two worked together in the National Institutes of Health’s Office of Behavior and Social Science Research.
The pair reviewed all large-budget clinical trials evaluating drugs or dietary supplements for the treatment or prevention of cardiovascular disease that had received funding from the NHLBI between 1970 and 2012.
They chose large-budget, NHLBI-funded trials in part because outcomes from the trials were more likely to be published, even if they did not produce the expected result.
Fifty-five studies were included in the research. Thirty were published prior to the reporting changes in 2000 (1970 to 1999), and 25 were published after the changes (2000 to 2012).
Of the studies published after 2000, only 2 (8%) showed positive outcomes, while 17 (57%) of the studies published before 2000 showed positive results.
Drs Kaplan and Irvin acknowledged that factors other than the reporting requirements may be contributing to the decline in positive outcomes, but they were unable to identify other compelling explanations.
For example, one suggestion was that older trials were more likely to compare new treatments to placebos, while newer trials were more likely to compare new
treatments to established treatments.
But when Drs Kaplan and Irvin examined the data, they found that 60% of trials published before 2000 used placebo comparators and nearly the same amount, 64%, of trials published after 2000 used placebos.
The researchers noted that although this work focused on clinical trials related to cardiovascular health, it would be reasonable to see similar changes in results
across other disease types.
“We don’t know if this decrease in positive outcomes also affects drug trials for prevention and treatment of cancer, diabetes, or other diseases,” Dr Irvin said. “But it would not be surprising because they have the same reporting requirements.” ![]()
Photo by Esther Dyson
The reporting requirements developed to increase transparency in US medical research may lead to fewer positive trial outcomes, according to a study published in PLOS ONE.
Researchers analyzed data from large-budget trials funded by the National Heart, Lung and Blood Institute (NHLBI).
And they found evidence suggesting the reporting requirements may have contributed to a significant reduction in studies with positive findings.
The reporting standards were phased in around 2000. They require researchers conducting drug or dietary supplement trials using human subjects to identify
projected outcomes and register their trials on ClinicalTrials.gov before they begin to collect data.
When entering their trial into the database, researchers are required to state the specific outcome on which they will focus. In the past, a researcher might have published an aspect of a study that was successful, even if the study overall did not produce the expected results.
But the new requirements mean researchers are less likely to change their analysis plan to consider another outcome that may have shown a positive result, said Veronica L. Irvin, PhD, of Oregon State University in Corvallis.
Dr Irvin began working on this project with the study’s lead author, Robert M. Kaplan, PhD, of the Agency for Healthcare Research and Quality in Rockville,
Maryland, while the two worked together in the National Institutes of Health’s Office of Behavior and Social Science Research.
The pair reviewed all large-budget clinical trials evaluating drugs or dietary supplements for the treatment or prevention of cardiovascular disease that had received funding from the NHLBI between 1970 and 2012.
They chose large-budget, NHLBI-funded trials in part because outcomes from the trials were more likely to be published, even if they did not produce the expected result.
Fifty-five studies were included in the research. Thirty were published prior to the reporting changes in 2000 (1970 to 1999), and 25 were published after the changes (2000 to 2012).
Of the studies published after 2000, only 2 (8%) showed positive outcomes, while 17 (57%) of the studies published before 2000 showed positive results.
Drs Kaplan and Irvin acknowledged that factors other than the reporting requirements may be contributing to the decline in positive outcomes, but they were unable to identify other compelling explanations.
For example, one suggestion was that older trials were more likely to compare new treatments to placebos, while newer trials were more likely to compare new
treatments to established treatments.
But when Drs Kaplan and Irvin examined the data, they found that 60% of trials published before 2000 used placebo comparators and nearly the same amount, 64%, of trials published after 2000 used placebos.
The researchers noted that although this work focused on clinical trials related to cardiovascular health, it would be reasonable to see similar changes in results
across other disease types.
“We don’t know if this decrease in positive outcomes also affects drug trials for prevention and treatment of cancer, diabetes, or other diseases,” Dr Irvin said. “But it would not be surprising because they have the same reporting requirements.” ![]()
Lenalidomide can treat pulmonary sarcoidosis in MDS

Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide. ![]()
Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide. ![]()
Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide.
Nanocapsules exploit biology to destroy blood clots

Image by Andre E.X. Brown
Scientists say they have created drug-loaded nanocapsules that can target and destroy blood clots by exploiting the intrinsic properties of thrombosis.
These polymer capsules are “inherently responsive” to thrombus microenvironments.
They home to activated platelets, where exposure to thrombin prompts the capsules to degrade and release a thrombolytic drug—urokinase plasminogen activator—at the site of thrombosis.
Christoph Hagemeyer, PhD, of Baker IDI Heart and Diabetes Institute in Melbourne, Victoria, Australia, and his colleagues described the creation and testing of these capsules in Advanced Materials.
“We’ve created a nanocapsule that contains a clot-busting drug,” Dr Hagemeyer explained. “The drug-loaded nanocapsule is coated with an antibody that specifically targets activated platelets . . . .”
“Once located at the site of the blood clot, thrombin (a molecule at the center of the clotting process) breaks open the outer layer of the nanocapsule, releasing the clot-busting drug. We are effectively hijacking the blood-clotting system to initiate the removal of the blockage in the blood vessel.”
Specifically, Dr Hagemeyer and his colleagues created the capsules via layer-by-layer assembly of brushlike poly(2-ethyl-2-oxazoline) with alkyne functional groups on mesoporous silica particle templates.
The team loaded the capsules with the thrombolytic agent urokinase plasminogen activator and incorporated a thrombin-sensitive cross-linker to activate capsule degradation and drug release at the site of thrombosis.
So the capsules would target activated platelets, the researchers attached an antibody to the capsules’ surface. The team used a phage-display-derived single-chain antibody that is specific for the fibrinogen receptor GPIIb/IIIa in its activated form.
Flow chamber experiments showed that the capsules do target surface-bound GPIIb/IIIa receptors expressed on activated platelets.
And exposing the capsules to thrombin revealed concentration-dependent degradation and drug release. The researchers exposed loaded capsules to simulated thrombotic conditions with a range of thrombin concentrations.
In the presence of 1 unit mL−1, capsules degraded/released their cargo after 4 hours. With higher thrombin concentrations—5 or 10 units mL−1—capsules released their cargo within 15 minutes.
Image by Andre E.X. Brown
Scientists say they have created drug-loaded nanocapsules that can target and destroy blood clots by exploiting the intrinsic properties of thrombosis.
These polymer capsules are “inherently responsive” to thrombus microenvironments.
They home to activated platelets, where exposure to thrombin prompts the capsules to degrade and release a thrombolytic drug—urokinase plasminogen activator—at the site of thrombosis.
Christoph Hagemeyer, PhD, of Baker IDI Heart and Diabetes Institute in Melbourne, Victoria, Australia, and his colleagues described the creation and testing of these capsules in Advanced Materials.
“We’ve created a nanocapsule that contains a clot-busting drug,” Dr Hagemeyer explained. “The drug-loaded nanocapsule is coated with an antibody that specifically targets activated platelets . . . .”
“Once located at the site of the blood clot, thrombin (a molecule at the center of the clotting process) breaks open the outer layer of the nanocapsule, releasing the clot-busting drug. We are effectively hijacking the blood-clotting system to initiate the removal of the blockage in the blood vessel.”
Specifically, Dr Hagemeyer and his colleagues created the capsules via layer-by-layer assembly of brushlike poly(2-ethyl-2-oxazoline) with alkyne functional groups on mesoporous silica particle templates.
The team loaded the capsules with the thrombolytic agent urokinase plasminogen activator and incorporated a thrombin-sensitive cross-linker to activate capsule degradation and drug release at the site of thrombosis.
So the capsules would target activated platelets, the researchers attached an antibody to the capsules’ surface. The team used a phage-display-derived single-chain antibody that is specific for the fibrinogen receptor GPIIb/IIIa in its activated form.
Flow chamber experiments showed that the capsules do target surface-bound GPIIb/IIIa receptors expressed on activated platelets.
And exposing the capsules to thrombin revealed concentration-dependent degradation and drug release. The researchers exposed loaded capsules to simulated thrombotic conditions with a range of thrombin concentrations.
In the presence of 1 unit mL−1, capsules degraded/released their cargo after 4 hours. With higher thrombin concentrations—5 or 10 units mL−1—capsules released their cargo within 15 minutes.
Image by Andre E.X. Brown
Scientists say they have created drug-loaded nanocapsules that can target and destroy blood clots by exploiting the intrinsic properties of thrombosis.
These polymer capsules are “inherently responsive” to thrombus microenvironments.
They home to activated platelets, where exposure to thrombin prompts the capsules to degrade and release a thrombolytic drug—urokinase plasminogen activator—at the site of thrombosis.
Christoph Hagemeyer, PhD, of Baker IDI Heart and Diabetes Institute in Melbourne, Victoria, Australia, and his colleagues described the creation and testing of these capsules in Advanced Materials.
“We’ve created a nanocapsule that contains a clot-busting drug,” Dr Hagemeyer explained. “The drug-loaded nanocapsule is coated with an antibody that specifically targets activated platelets . . . .”
“Once located at the site of the blood clot, thrombin (a molecule at the center of the clotting process) breaks open the outer layer of the nanocapsule, releasing the clot-busting drug. We are effectively hijacking the blood-clotting system to initiate the removal of the blockage in the blood vessel.”
Specifically, Dr Hagemeyer and his colleagues created the capsules via layer-by-layer assembly of brushlike poly(2-ethyl-2-oxazoline) with alkyne functional groups on mesoporous silica particle templates.
The team loaded the capsules with the thrombolytic agent urokinase plasminogen activator and incorporated a thrombin-sensitive cross-linker to activate capsule degradation and drug release at the site of thrombosis.
So the capsules would target activated platelets, the researchers attached an antibody to the capsules’ surface. The team used a phage-display-derived single-chain antibody that is specific for the fibrinogen receptor GPIIb/IIIa in its activated form.
Flow chamber experiments showed that the capsules do target surface-bound GPIIb/IIIa receptors expressed on activated platelets.
And exposing the capsules to thrombin revealed concentration-dependent degradation and drug release. The researchers exposed loaded capsules to simulated thrombotic conditions with a range of thrombin concentrations.
In the presence of 1 unit mL−1, capsules degraded/released their cargo after 4 hours. With higher thrombin concentrations—5 or 10 units mL−1—capsules released their cargo within 15 minutes.
Drug gets orphan designation for CTCL
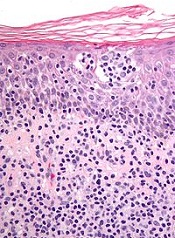
The European Commission has granted orphan drug designation to synthetic hypericin, the active pharmaceutical ingredient in SGX301, for the treatment of cutaneous T-cell lymphoma (CTCL).
SGX301 is a first-in-class, photodynamic therapy utilizing safe, visible light for activation. Synthetic hypericin is a potent photosensitizer that is topically applied to skin lesions and activated by visible fluorescent light 16 to 24 hours later.
This treatment approach is intended to prevent the secondary malignancies that may occur following chemotherapy or photodynamic therapies that are dependent on ultraviolet exposure.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients.
Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
Soligenix, Inc., the company developing SGX301, is currently working with CTCL centers, the National Organization for Rare Disorders, and the Cutaneous Lymphoma Foundation to begin a 120-subject phase 3 trial of SGX301.
About orphan designation
The European Commission grants orphan designation to medicines designed to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons in the European Union and has no satisfactory treatment available.
In addition to a 10-year period of marketing exclusivity after product approval, orphan drug designation provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase, as well as direct access to the centralized authorization procedure.
SGX301 has both orphan designation and fast track designation from the US Food and Drug Administration for the first-line treatment of CTCL.
The European Commission has granted orphan drug designation to synthetic hypericin, the active pharmaceutical ingredient in SGX301, for the treatment of cutaneous T-cell lymphoma (CTCL).
SGX301 is a first-in-class, photodynamic therapy utilizing safe, visible light for activation. Synthetic hypericin is a potent photosensitizer that is topically applied to skin lesions and activated by visible fluorescent light 16 to 24 hours later.
This treatment approach is intended to prevent the secondary malignancies that may occur following chemotherapy or photodynamic therapies that are dependent on ultraviolet exposure.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients.
Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
Soligenix, Inc., the company developing SGX301, is currently working with CTCL centers, the National Organization for Rare Disorders, and the Cutaneous Lymphoma Foundation to begin a 120-subject phase 3 trial of SGX301.
About orphan designation
The European Commission grants orphan designation to medicines designed to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons in the European Union and has no satisfactory treatment available.
In addition to a 10-year period of marketing exclusivity after product approval, orphan drug designation provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase, as well as direct access to the centralized authorization procedure.
SGX301 has both orphan designation and fast track designation from the US Food and Drug Administration for the first-line treatment of CTCL.
The European Commission has granted orphan drug designation to synthetic hypericin, the active pharmaceutical ingredient in SGX301, for the treatment of cutaneous T-cell lymphoma (CTCL).
SGX301 is a first-in-class, photodynamic therapy utilizing safe, visible light for activation. Synthetic hypericin is a potent photosensitizer that is topically applied to skin lesions and activated by visible fluorescent light 16 to 24 hours later.
This treatment approach is intended to prevent the secondary malignancies that may occur following chemotherapy or photodynamic therapies that are dependent on ultraviolet exposure.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients.
Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
Soligenix, Inc., the company developing SGX301, is currently working with CTCL centers, the National Organization for Rare Disorders, and the Cutaneous Lymphoma Foundation to begin a 120-subject phase 3 trial of SGX301.
About orphan designation
The European Commission grants orphan designation to medicines designed to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons in the European Union and has no satisfactory treatment available.
In addition to a 10-year period of marketing exclusivity after product approval, orphan drug designation provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase, as well as direct access to the centralized authorization procedure.
SGX301 has both orphan designation and fast track designation from the US Food and Drug Administration for the first-line treatment of CTCL.
DAPT may be better for older patients after PCI

Photo courtesy of the CDC
A new study suggests less is more when it comes to antithrombotic therapy for higher-risk older patients with atrial fibrillation who have a heart attack and undergo percutaneous coronary intervention (PCI).
At 2 years of follow-up, patients who had received triple therapy—warfarin, aspirin, and P2Y12 inhibitor—after PCI had similar rates of major adverse cardiac events (MACE) as patients who received dual antiplatelet therapy (DAPT)—aspirin and P2Y12 inhibitor.
But patients on triple therapy had a higher incidence of intracranial hemorrhage and bleeding that required hospitalization.
These results appear in the Journal of the American College of Cardiology alongside a related editorial.
Researchers examined data from the National Cardiovascular Data Registry ACTION Registry-GWTG linked with Centers for Medicare and Medicaid Services data, looking at records from January 2007 through December 2010.
They identified 4959 patients aged 65 and older with a history of atrial fibrillation who presented with acute myocardial infarction (MI) and underwent PCI. Most patients (72.4%, n=3589) were discharged on DAPT, but 27.6% (n=1370) were discharged on triple therapy.
In the DAPT arm, 97.2% of patients (n=3490) received clopidogrel, 2.5% (n=89) received prasugrel, and 0.3% (n=10) received ticlopidine. In the triple therapy arm, 98.2% of patients (n=1346) received clopidogrel, 1.4% (n=19) received prasugrel, and 0.4% (n=5) received ticlopidine.
Patients receiving triple therapy were more likely to be male, have a history of either angioplasty or coronary artery bypass surgery, and have a history of stroke. These patients were frequently already on warfarin before they were admitted to the hospital.
Patients who were released on DAPT were more likely to have had an in-hospital major bleeding event.
Incidence of MACE
Two years after discharge, the risk of MACE—death, hospital readmission for MI, or stroke readmission—was similar between the DAPT and triple therapy arms. The unadjusted cumulative incidence rate of MACE was 32.6% in the triple therapy arm and 32.7% in the DAPT arm (P=0.99).
The unadjusted cumulative incidence rates of the individual MACE components were also similar between the triple therapy and DAPT arms. All-cause mortality occurred in 23.8% and 24.8%, respectively (P=0.70), MI readmission occurred in 8.5% and 8.1%, respectively (P=0.54), and stroke readmission occurred in 4.7% and 5.3%, respectively (P=0.23).
After the researchers adjusted for patient, treatment, and hospital characteristics, there was still no significant difference between the arms with regard to the incidence of MACE or MACE components.
The adjusted hazard ratio (HR) was 0.99 for MACE (P=0.94), 0.98 for all-cause mortality (P=0.82), 1.03 for MI readmission (P=0.83), and 0.85 for stroke readmission (P=0.38).
Bleeding incidence
The cumulative incidence of bleeding requiring hospitalization within 2 years of discharge after PCI was significantly higher for the triple therapy arm than the DAPT arm—17.6% and 11.0%, respectively (P<0.0001).
The difference remained significant after the researchers adjusted for patient, treatment, and hospital characteristics. The adjusted HR was 1.61 (P<0.0001).
Similarly, the unadjusted cumulative incidence of intracranial hemorrhage was significantly higher for the triple therapy arm than the DAPT arm—3.4% and 1.5%, respectively (P<0.001).
This difference remained significant after adjustment. The adjusted HR was 2.04 (P<0.01).
“The increased risk of bleeding without apparent benefit of triple therapy observed in this study suggests that clinicians should carefully consider the risk-to-benefit ratio of triple therapy use in older atrial fibrillation patients who have had a heart attack treated with angioplasty,” said Connie N. Hess, MD, of the Duke University School of Medicine in Durham, North Carolina.
“Further prospective studies of different combinations of anticlotting agents are needed to define the optimal treatment regimen for this population.”
Photo courtesy of the CDC
A new study suggests less is more when it comes to antithrombotic therapy for higher-risk older patients with atrial fibrillation who have a heart attack and undergo percutaneous coronary intervention (PCI).
At 2 years of follow-up, patients who had received triple therapy—warfarin, aspirin, and P2Y12 inhibitor—after PCI had similar rates of major adverse cardiac events (MACE) as patients who received dual antiplatelet therapy (DAPT)—aspirin and P2Y12 inhibitor.
But patients on triple therapy had a higher incidence of intracranial hemorrhage and bleeding that required hospitalization.
These results appear in the Journal of the American College of Cardiology alongside a related editorial.
Researchers examined data from the National Cardiovascular Data Registry ACTION Registry-GWTG linked with Centers for Medicare and Medicaid Services data, looking at records from January 2007 through December 2010.
They identified 4959 patients aged 65 and older with a history of atrial fibrillation who presented with acute myocardial infarction (MI) and underwent PCI. Most patients (72.4%, n=3589) were discharged on DAPT, but 27.6% (n=1370) were discharged on triple therapy.
In the DAPT arm, 97.2% of patients (n=3490) received clopidogrel, 2.5% (n=89) received prasugrel, and 0.3% (n=10) received ticlopidine. In the triple therapy arm, 98.2% of patients (n=1346) received clopidogrel, 1.4% (n=19) received prasugrel, and 0.4% (n=5) received ticlopidine.
Patients receiving triple therapy were more likely to be male, have a history of either angioplasty or coronary artery bypass surgery, and have a history of stroke. These patients were frequently already on warfarin before they were admitted to the hospital.
Patients who were released on DAPT were more likely to have had an in-hospital major bleeding event.
Incidence of MACE
Two years after discharge, the risk of MACE—death, hospital readmission for MI, or stroke readmission—was similar between the DAPT and triple therapy arms. The unadjusted cumulative incidence rate of MACE was 32.6% in the triple therapy arm and 32.7% in the DAPT arm (P=0.99).
The unadjusted cumulative incidence rates of the individual MACE components were also similar between the triple therapy and DAPT arms. All-cause mortality occurred in 23.8% and 24.8%, respectively (P=0.70), MI readmission occurred in 8.5% and 8.1%, respectively (P=0.54), and stroke readmission occurred in 4.7% and 5.3%, respectively (P=0.23).
After the researchers adjusted for patient, treatment, and hospital characteristics, there was still no significant difference between the arms with regard to the incidence of MACE or MACE components.
The adjusted hazard ratio (HR) was 0.99 for MACE (P=0.94), 0.98 for all-cause mortality (P=0.82), 1.03 for MI readmission (P=0.83), and 0.85 for stroke readmission (P=0.38).
Bleeding incidence
The cumulative incidence of bleeding requiring hospitalization within 2 years of discharge after PCI was significantly higher for the triple therapy arm than the DAPT arm—17.6% and 11.0%, respectively (P<0.0001).
The difference remained significant after the researchers adjusted for patient, treatment, and hospital characteristics. The adjusted HR was 1.61 (P<0.0001).
Similarly, the unadjusted cumulative incidence of intracranial hemorrhage was significantly higher for the triple therapy arm than the DAPT arm—3.4% and 1.5%, respectively (P<0.001).
This difference remained significant after adjustment. The adjusted HR was 2.04 (P<0.01).
“The increased risk of bleeding without apparent benefit of triple therapy observed in this study suggests that clinicians should carefully consider the risk-to-benefit ratio of triple therapy use in older atrial fibrillation patients who have had a heart attack treated with angioplasty,” said Connie N. Hess, MD, of the Duke University School of Medicine in Durham, North Carolina.
“Further prospective studies of different combinations of anticlotting agents are needed to define the optimal treatment regimen for this population.”
Photo courtesy of the CDC
A new study suggests less is more when it comes to antithrombotic therapy for higher-risk older patients with atrial fibrillation who have a heart attack and undergo percutaneous coronary intervention (PCI).
At 2 years of follow-up, patients who had received triple therapy—warfarin, aspirin, and P2Y12 inhibitor—after PCI had similar rates of major adverse cardiac events (MACE) as patients who received dual antiplatelet therapy (DAPT)—aspirin and P2Y12 inhibitor.
But patients on triple therapy had a higher incidence of intracranial hemorrhage and bleeding that required hospitalization.
These results appear in the Journal of the American College of Cardiology alongside a related editorial.
Researchers examined data from the National Cardiovascular Data Registry ACTION Registry-GWTG linked with Centers for Medicare and Medicaid Services data, looking at records from January 2007 through December 2010.
They identified 4959 patients aged 65 and older with a history of atrial fibrillation who presented with acute myocardial infarction (MI) and underwent PCI. Most patients (72.4%, n=3589) were discharged on DAPT, but 27.6% (n=1370) were discharged on triple therapy.
In the DAPT arm, 97.2% of patients (n=3490) received clopidogrel, 2.5% (n=89) received prasugrel, and 0.3% (n=10) received ticlopidine. In the triple therapy arm, 98.2% of patients (n=1346) received clopidogrel, 1.4% (n=19) received prasugrel, and 0.4% (n=5) received ticlopidine.
Patients receiving triple therapy were more likely to be male, have a history of either angioplasty or coronary artery bypass surgery, and have a history of stroke. These patients were frequently already on warfarin before they were admitted to the hospital.
Patients who were released on DAPT were more likely to have had an in-hospital major bleeding event.
Incidence of MACE
Two years after discharge, the risk of MACE—death, hospital readmission for MI, or stroke readmission—was similar between the DAPT and triple therapy arms. The unadjusted cumulative incidence rate of MACE was 32.6% in the triple therapy arm and 32.7% in the DAPT arm (P=0.99).
The unadjusted cumulative incidence rates of the individual MACE components were also similar between the triple therapy and DAPT arms. All-cause mortality occurred in 23.8% and 24.8%, respectively (P=0.70), MI readmission occurred in 8.5% and 8.1%, respectively (P=0.54), and stroke readmission occurred in 4.7% and 5.3%, respectively (P=0.23).
After the researchers adjusted for patient, treatment, and hospital characteristics, there was still no significant difference between the arms with regard to the incidence of MACE or MACE components.
The adjusted hazard ratio (HR) was 0.99 for MACE (P=0.94), 0.98 for all-cause mortality (P=0.82), 1.03 for MI readmission (P=0.83), and 0.85 for stroke readmission (P=0.38).
Bleeding incidence
The cumulative incidence of bleeding requiring hospitalization within 2 years of discharge after PCI was significantly higher for the triple therapy arm than the DAPT arm—17.6% and 11.0%, respectively (P<0.0001).
The difference remained significant after the researchers adjusted for patient, treatment, and hospital characteristics. The adjusted HR was 1.61 (P<0.0001).
Similarly, the unadjusted cumulative incidence of intracranial hemorrhage was significantly higher for the triple therapy arm than the DAPT arm—3.4% and 1.5%, respectively (P<0.001).
This difference remained significant after adjustment. The adjusted HR was 2.04 (P<0.01).
“The increased risk of bleeding without apparent benefit of triple therapy observed in this study suggests that clinicians should carefully consider the risk-to-benefit ratio of triple therapy use in older atrial fibrillation patients who have had a heart attack treated with angioplasty,” said Connie N. Hess, MD, of the Duke University School of Medicine in Durham, North Carolina.
“Further prospective studies of different combinations of anticlotting agents are needed to define the optimal treatment regimen for this population.”