User login
M. Alexander Otto began his reporting career early in 1999 covering the pharmaceutical industry for a national pharmacists' magazine and freelancing for the Washington Post and other newspapers. He then joined BNA, now part of Bloomberg News, covering health law and the protection of people and animals in medical research. Alex next worked for the McClatchy Company. Based on his work, Alex won a year-long Knight Science Journalism Fellowship to MIT in 2008-2009. He joined the company shortly thereafter. Alex has a newspaper journalism degree from Syracuse (N.Y.) University and a master's degree in medical science -- a physician assistant degree -- from George Washington University. Alex is based in Seattle.
Ivabradine approved to reduce heart failure hospitalizations
The heart rate–lowering agent ivabradine was approved by the Food and Drug Administration on April 15 to reduce hospitalizations in patients with worsening heart failure.
In an April 15 statement, the FDA announced that ivabradine, after undergoing a fast-track evaluation process, is indicated in patients with chronic, stable, symptomatic heart failure and left ventricular ejection fractions at or below 35%; resting heart rates of at least 70 beats per minute; and who are on maximum beta-blockers doses or have beta-blocker contraindications.

Ivabradine “is thought to work by decreasing heart rate and represents the first approved product in [its] drug class,” Dr. Norman Stockbridge, director of the FDA’s division of cardiovascular and renal products, said in a written statement.
The drug was given priority review based on the results of SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which involved 6,505 clinically stable patients, all hospitalized for heart failure in the preceding year and all on standard background therapy, including beta-blockers (89%), ACE inhibitors and/or angiotensin II receptor blockers (91%), diuretics (83%), and antialdosterone agents (60%) (Lancet 2010;376:875-85).
There was a 4.7% absolute risk reduction and a 26% relative risk reduction for hospitalizations as a result of deteriorating heart failure in the 3,241 ivabradine patients, but the drug did not reduce mortality, according to a statement from the drug’s manufacturer, Amgen.
The most common adverse events were bradycardia (10% vs. 2.2% with placebo), hypertension or increased blood pressure (8.9% vs. 7.8% with placebo), atrial fibrillation (8.3% vs. 6.6%), and luminous phenomena or visual brightness (2.8% vs. 0.5%).
Ivabradine is a specific inhibitor of the If (“funny”) current in the sinoatrial node, but not other currents. The drug is contraindicated in patients with acute decompensated heart failure, blood pressure below 90/50 mm Hg, sick sinus syndrome, sinoatrial block, third-degree AV block (unless a functioning demand pacemaker is present), resting heart rate below 60 bpm prior to treatment, severe hepatic impairment, pacemaker dependence, and use of strong cytochrome P450 3A4 inhibitors. Ivabradine increases the risk of atrial fibrillation and can cause fetal toxicity. Bradycardia, sinus arrest, and heart block have been reported with its use, Amgen said in its announcement.
Concurrent use of the calcium channel blockers verapamil or diltiazem increases exposure to the drug and should be avoided. Ivabradine also should be avoided in patients with second-degree AV block unless a functioning demand pacemaker is present.
Ivabradine will be available in 5-mg and 7.5-mg tablets, according to the product’s label. The recommended starting dose is a 5-mg tablet twice daily with meals. After 2 weeks of treatment, the dose should be adjusted depending on heart rate. In patients with a history of conduction defects or others in whom bradycardia could lead to hemodynamic compromise, Amgen said to initiate therapy at 2.5 mg twice daily.
Patients should alert their health care professional if they develop an irregular heartbeat, a pounding or racing heart, chest pressure, worse shortness of breath, dizziness, weakness, or fatigue, Amgen said.
Ivabradine will be available within about a week of the approval under the trade name Corlanor, and will come with a patient medication guide. Wholesale acquisition cost will be $4,500 per year, or $375 per month, and patient costs will vary according to insurance coverage, said Amgen spokesman Cuyler Mayer.
Ivabradine has been available in Europe as Procoralan for several years.
The heart rate–lowering agent ivabradine was approved by the Food and Drug Administration on April 15 to reduce hospitalizations in patients with worsening heart failure.
In an April 15 statement, the FDA announced that ivabradine, after undergoing a fast-track evaluation process, is indicated in patients with chronic, stable, symptomatic heart failure and left ventricular ejection fractions at or below 35%; resting heart rates of at least 70 beats per minute; and who are on maximum beta-blockers doses or have beta-blocker contraindications.
Ivabradine “is thought to work by decreasing heart rate and represents the first approved product in [its] drug class,” Dr. Norman Stockbridge, director of the FDA’s division of cardiovascular and renal products, said in a written statement.
The drug was given priority review based on the results of SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which involved 6,505 clinically stable patients, all hospitalized for heart failure in the preceding year and all on standard background therapy, including beta-blockers (89%), ACE inhibitors and/or angiotensin II receptor blockers (91%), diuretics (83%), and antialdosterone agents (60%) (Lancet 2010;376:875-85).
There was a 4.7% absolute risk reduction and a 26% relative risk reduction for hospitalizations as a result of deteriorating heart failure in the 3,241 ivabradine patients, but the drug did not reduce mortality, according to a statement from the drug’s manufacturer, Amgen.
The most common adverse events were bradycardia (10% vs. 2.2% with placebo), hypertension or increased blood pressure (8.9% vs. 7.8% with placebo), atrial fibrillation (8.3% vs. 6.6%), and luminous phenomena or visual brightness (2.8% vs. 0.5%).
Ivabradine is a specific inhibitor of the If (“funny”) current in the sinoatrial node, but not other currents. The drug is contraindicated in patients with acute decompensated heart failure, blood pressure below 90/50 mm Hg, sick sinus syndrome, sinoatrial block, third-degree AV block (unless a functioning demand pacemaker is present), resting heart rate below 60 bpm prior to treatment, severe hepatic impairment, pacemaker dependence, and use of strong cytochrome P450 3A4 inhibitors. Ivabradine increases the risk of atrial fibrillation and can cause fetal toxicity. Bradycardia, sinus arrest, and heart block have been reported with its use, Amgen said in its announcement.
Concurrent use of the calcium channel blockers verapamil or diltiazem increases exposure to the drug and should be avoided. Ivabradine also should be avoided in patients with second-degree AV block unless a functioning demand pacemaker is present.
Ivabradine will be available in 5-mg and 7.5-mg tablets, according to the product’s label. The recommended starting dose is a 5-mg tablet twice daily with meals. After 2 weeks of treatment, the dose should be adjusted depending on heart rate. In patients with a history of conduction defects or others in whom bradycardia could lead to hemodynamic compromise, Amgen said to initiate therapy at 2.5 mg twice daily.
Patients should alert their health care professional if they develop an irregular heartbeat, a pounding or racing heart, chest pressure, worse shortness of breath, dizziness, weakness, or fatigue, Amgen said.
Ivabradine will be available within about a week of the approval under the trade name Corlanor, and will come with a patient medication guide. Wholesale acquisition cost will be $4,500 per year, or $375 per month, and patient costs will vary according to insurance coverage, said Amgen spokesman Cuyler Mayer.
Ivabradine has been available in Europe as Procoralan for several years.
The heart rate–lowering agent ivabradine was approved by the Food and Drug Administration on April 15 to reduce hospitalizations in patients with worsening heart failure.
In an April 15 statement, the FDA announced that ivabradine, after undergoing a fast-track evaluation process, is indicated in patients with chronic, stable, symptomatic heart failure and left ventricular ejection fractions at or below 35%; resting heart rates of at least 70 beats per minute; and who are on maximum beta-blockers doses or have beta-blocker contraindications.
Ivabradine “is thought to work by decreasing heart rate and represents the first approved product in [its] drug class,” Dr. Norman Stockbridge, director of the FDA’s division of cardiovascular and renal products, said in a written statement.
The drug was given priority review based on the results of SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which involved 6,505 clinically stable patients, all hospitalized for heart failure in the preceding year and all on standard background therapy, including beta-blockers (89%), ACE inhibitors and/or angiotensin II receptor blockers (91%), diuretics (83%), and antialdosterone agents (60%) (Lancet 2010;376:875-85).
There was a 4.7% absolute risk reduction and a 26% relative risk reduction for hospitalizations as a result of deteriorating heart failure in the 3,241 ivabradine patients, but the drug did not reduce mortality, according to a statement from the drug’s manufacturer, Amgen.
The most common adverse events were bradycardia (10% vs. 2.2% with placebo), hypertension or increased blood pressure (8.9% vs. 7.8% with placebo), atrial fibrillation (8.3% vs. 6.6%), and luminous phenomena or visual brightness (2.8% vs. 0.5%).
Ivabradine is a specific inhibitor of the If (“funny”) current in the sinoatrial node, but not other currents. The drug is contraindicated in patients with acute decompensated heart failure, blood pressure below 90/50 mm Hg, sick sinus syndrome, sinoatrial block, third-degree AV block (unless a functioning demand pacemaker is present), resting heart rate below 60 bpm prior to treatment, severe hepatic impairment, pacemaker dependence, and use of strong cytochrome P450 3A4 inhibitors. Ivabradine increases the risk of atrial fibrillation and can cause fetal toxicity. Bradycardia, sinus arrest, and heart block have been reported with its use, Amgen said in its announcement.
Concurrent use of the calcium channel blockers verapamil or diltiazem increases exposure to the drug and should be avoided. Ivabradine also should be avoided in patients with second-degree AV block unless a functioning demand pacemaker is present.
Ivabradine will be available in 5-mg and 7.5-mg tablets, according to the product’s label. The recommended starting dose is a 5-mg tablet twice daily with meals. After 2 weeks of treatment, the dose should be adjusted depending on heart rate. In patients with a history of conduction defects or others in whom bradycardia could lead to hemodynamic compromise, Amgen said to initiate therapy at 2.5 mg twice daily.
Patients should alert their health care professional if they develop an irregular heartbeat, a pounding or racing heart, chest pressure, worse shortness of breath, dizziness, weakness, or fatigue, Amgen said.
Ivabradine will be available within about a week of the approval under the trade name Corlanor, and will come with a patient medication guide. Wholesale acquisition cost will be $4,500 per year, or $375 per month, and patient costs will vary according to insurance coverage, said Amgen spokesman Cuyler Mayer.
Ivabradine has been available in Europe as Procoralan for several years.

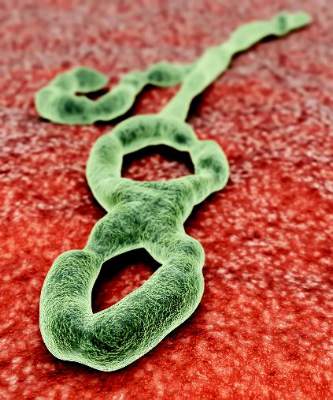
CDC Ebola Vaccine Trial Underway in Sierra Leone
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.

“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.
“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.
“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
FROM A CDC TELECONFERENCE
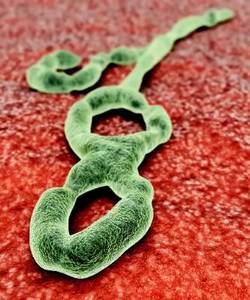
CDC Ebola vaccine trial underway in Sierra Leone
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.
“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.
“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
The Centers for Disease Control and Prevention is looking to enroll 6,000 people in an Ebola vaccine trial now underway in Sierra Leone.
Frontline workers are being randomized to the experimental vaccine rVSV-ZEBOV either up front or 6 months after enrollment. Subjects then will be followed for 6 months to see how well the vaccine protects them from infection. The trial will cost about $25 million, funded mostly by the U.S. government.
“The Ebola epidemic has been devastating for West Africa. I really hope that this is the beginning of a new positive chapter for Sierra Leone and the story of battling this virus,” said Dr. Anne Schuchat, director of CDC’s National Center for Immunization and Respiratory Diseases.
So far, about 200 people have enrolled in the study, and more than 90 have gotten the shot. Foreign aid workers are eligible for the trial, but because subjects need to be in country long enough for follow-up, most will be from Sierra Leone. To cover time and transportation costs, participants are paid $10 for enrollment and $30 after they get the vaccine, Dr. Schuchat said in an April 14 teleconference.
Subjects include doctors, nurses, janitors, lab technicians, security guards, pharmacists, administrators, burial workers, and those who wash the bodies of the newly dead. The study is being conducted in hard-hit areas of Sierra Leone, including the capital of Freetown. Similar studies are underway in Liberia and Guinea.
Ebola cases “are way down” in West Africa lately, “but people are continuing to get” the disease, “and some of the Ebola outbreaks in the past have had long tails,” so health care workers remain at risk, according to Dr. Schuchat.
With infection rates down, it’s unclear whether or not the study will have enough cases to demonstrate efficacy for rVSV-ZEBOV. Even so, “we plan to collect information on safety and immune response that could contribute to licensure pathways even if we are not able to estimate efficacy,” she said.
About 800 people have gotten rVSV-ZEBOV in other trials. Common side effects include fatigue, fever, headaches, and muscle aches. There have also been reports of mild, self-limited joint pain and swelling.
CDC is working with the Sierra Leone College of Medicine and Allied Health Sciences and the Sierra Leone Ministry of Health and Sanitation to conduct the trial, dubbed The Sierra Leone Trial to Introduce a Vaccine against Ebola (STRIVE). About 350 local people have been hired and trained to help with the study.
The vaccine uses a vesicular stomatitis virus carrying a noninfectious Ebola virus gene, and has been shown to produce an immune response against the virus. It was developed by the Public Health Agency of Canada’s National Microbiology Laboratory and is licensed to NewLink Genetics. In 2014, NewLink entered into a licensing and collaboration agreement with Merck to develop rVSV-ZEBOV.
Until it is known whether or not the vaccine works, and how well, CDC cautioned that vaccinated people still need to take full protective measures against Ebola.
FROM A CDC TELECONFERENCE
Estrogen assays need to improve
Direct immunoassays shouldn’t be used to measure estrogens and their metabolites in patients with very low estrogen concentrations, according to a report published online April 7 in the Journal of Clinical Endocrinology and Metabolism.
“The inaccuracy of direct immunoassays is especially problematic in ... men, postmenopausal women, those taking aromatase inhibitors, and prepubertal children. Thus, the use of current direct assays for measuring estradiol concentrations in these groups of patients should be discouraged,” wrote Dr. Laurence Demers of the Pennsylvania State University, Hershey, and his associates.
Mass spectrometry has better sensitivity and specificity, but mass spectrometers are expensive, and assays to quantify estrogen metabolites are demanding. Because of that, many labs continue to use direct immunoassays even when they’re not the best choice.
The report summarized the consensus from a 2014 meeting of experts in Bethesda, MD., on the measurement of the estrogens and their metabolites. In general, participants agreed that measurements need to be standardized and improved (J. Clin. Endocrinol. Metab. 2015 April 7 [doi:10.1210/jc.2015-1040]).
“Assay inaccuracy complicates the comparison of results obtained from different laboratories, from the same laboratory over time, or from different epidemiological and clinical studies and constrains the application of guidelines to ... patients. A set of clinical practice guidelines cannot be implemented unless the assays used are accurate and precise,” according to the investigators.
Even so, “measurement of estradiol is challenging because the physiologically relevant concentration range spans at least four orders of magnitude ... an assay that is suitable for use for diagnosis and management of infertility in adult women may not meet performance specifications for use when evaluating the onset of puberty in a child,” they said.
Current estradiol assays are particularly problematic in postmenopausal women and children, where sensitivity is critical. Reference ranges haven’t been established for either group. The Centers for Disease Control and Prevention are working to address those and other problems under a Hormone Standardization Program.
Meanwhile, “use of currently available direct estradiol immunoassays appears to compromise assay specificity, probably due to antibody cross-reactivity, matrix effects, and suboptimal sensitivity. These direct assays usually yield higher values than [do] those measured by methods with superior specificity, including immunoassays preceded by organic solvent extraction, liquid chromatography, or gas or liquid chromatography mass spectrometry assays,” the authors said.
There’s a need “for a study to evaluate the different approaches for validation of estrogen assay accuracy. It [is also] recommended that journals require a statement regarding accuracy assessment for estrogen and estrogen metabolite assays with a long-term goal of requiring measurements using accurate assays,” they said.
The Bethesda meeting was sponsored primarily by the Endocrine Society, the American Association for Clinical Chemistry, and the Partnership for Accurate Testing of Hormones. One of the nine authors – Dr. Richard J. Santen of the University of Virginia, Charlottesville – has been a Pfizer adviser and is the principal investigator on a grant from the company to his university. The other authors have no disclosures.
Direct immunoassays shouldn’t be used to measure estrogens and their metabolites in patients with very low estrogen concentrations, according to a report published online April 7 in the Journal of Clinical Endocrinology and Metabolism.
“The inaccuracy of direct immunoassays is especially problematic in ... men, postmenopausal women, those taking aromatase inhibitors, and prepubertal children. Thus, the use of current direct assays for measuring estradiol concentrations in these groups of patients should be discouraged,” wrote Dr. Laurence Demers of the Pennsylvania State University, Hershey, and his associates.
Mass spectrometry has better sensitivity and specificity, but mass spectrometers are expensive, and assays to quantify estrogen metabolites are demanding. Because of that, many labs continue to use direct immunoassays even when they’re not the best choice.
The report summarized the consensus from a 2014 meeting of experts in Bethesda, MD., on the measurement of the estrogens and their metabolites. In general, participants agreed that measurements need to be standardized and improved (J. Clin. Endocrinol. Metab. 2015 April 7 [doi:10.1210/jc.2015-1040]).
“Assay inaccuracy complicates the comparison of results obtained from different laboratories, from the same laboratory over time, or from different epidemiological and clinical studies and constrains the application of guidelines to ... patients. A set of clinical practice guidelines cannot be implemented unless the assays used are accurate and precise,” according to the investigators.
Even so, “measurement of estradiol is challenging because the physiologically relevant concentration range spans at least four orders of magnitude ... an assay that is suitable for use for diagnosis and management of infertility in adult women may not meet performance specifications for use when evaluating the onset of puberty in a child,” they said.
Current estradiol assays are particularly problematic in postmenopausal women and children, where sensitivity is critical. Reference ranges haven’t been established for either group. The Centers for Disease Control and Prevention are working to address those and other problems under a Hormone Standardization Program.
Meanwhile, “use of currently available direct estradiol immunoassays appears to compromise assay specificity, probably due to antibody cross-reactivity, matrix effects, and suboptimal sensitivity. These direct assays usually yield higher values than [do] those measured by methods with superior specificity, including immunoassays preceded by organic solvent extraction, liquid chromatography, or gas or liquid chromatography mass spectrometry assays,” the authors said.
There’s a need “for a study to evaluate the different approaches for validation of estrogen assay accuracy. It [is also] recommended that journals require a statement regarding accuracy assessment for estrogen and estrogen metabolite assays with a long-term goal of requiring measurements using accurate assays,” they said.
The Bethesda meeting was sponsored primarily by the Endocrine Society, the American Association for Clinical Chemistry, and the Partnership for Accurate Testing of Hormones. One of the nine authors – Dr. Richard J. Santen of the University of Virginia, Charlottesville – has been a Pfizer adviser and is the principal investigator on a grant from the company to his university. The other authors have no disclosures.
Direct immunoassays shouldn’t be used to measure estrogens and their metabolites in patients with very low estrogen concentrations, according to a report published online April 7 in the Journal of Clinical Endocrinology and Metabolism.
“The inaccuracy of direct immunoassays is especially problematic in ... men, postmenopausal women, those taking aromatase inhibitors, and prepubertal children. Thus, the use of current direct assays for measuring estradiol concentrations in these groups of patients should be discouraged,” wrote Dr. Laurence Demers of the Pennsylvania State University, Hershey, and his associates.
Mass spectrometry has better sensitivity and specificity, but mass spectrometers are expensive, and assays to quantify estrogen metabolites are demanding. Because of that, many labs continue to use direct immunoassays even when they’re not the best choice.
The report summarized the consensus from a 2014 meeting of experts in Bethesda, MD., on the measurement of the estrogens and their metabolites. In general, participants agreed that measurements need to be standardized and improved (J. Clin. Endocrinol. Metab. 2015 April 7 [doi:10.1210/jc.2015-1040]).
“Assay inaccuracy complicates the comparison of results obtained from different laboratories, from the same laboratory over time, or from different epidemiological and clinical studies and constrains the application of guidelines to ... patients. A set of clinical practice guidelines cannot be implemented unless the assays used are accurate and precise,” according to the investigators.
Even so, “measurement of estradiol is challenging because the physiologically relevant concentration range spans at least four orders of magnitude ... an assay that is suitable for use for diagnosis and management of infertility in adult women may not meet performance specifications for use when evaluating the onset of puberty in a child,” they said.
Current estradiol assays are particularly problematic in postmenopausal women and children, where sensitivity is critical. Reference ranges haven’t been established for either group. The Centers for Disease Control and Prevention are working to address those and other problems under a Hormone Standardization Program.
Meanwhile, “use of currently available direct estradiol immunoassays appears to compromise assay specificity, probably due to antibody cross-reactivity, matrix effects, and suboptimal sensitivity. These direct assays usually yield higher values than [do] those measured by methods with superior specificity, including immunoassays preceded by organic solvent extraction, liquid chromatography, or gas or liquid chromatography mass spectrometry assays,” the authors said.
There’s a need “for a study to evaluate the different approaches for validation of estrogen assay accuracy. It [is also] recommended that journals require a statement regarding accuracy assessment for estrogen and estrogen metabolite assays with a long-term goal of requiring measurements using accurate assays,” they said.
The Bethesda meeting was sponsored primarily by the Endocrine Society, the American Association for Clinical Chemistry, and the Partnership for Accurate Testing of Hormones. One of the nine authors – Dr. Richard J. Santen of the University of Virginia, Charlottesville – has been a Pfizer adviser and is the principal investigator on a grant from the company to his university. The other authors have no disclosures.
FROM THE JOURNAL OF CLINICAL ENDOCRINOLOGY AND METABOLISM
No growth hormone/cancer link in kids without malignancy risks
Growth hormone treatment does not increase the likelihood of cancer in children who aren’t at risk for it, but it may increase the risk in children who are predisposed to malignancy, according to a literature review from the Pediatric Endocrine Society published online April 3 in the Journal of Clinical Endocrinology & Metabolism.
CNS tumors, leukemia, and other cancers have been a concern with growth hormone (GH) treatment for years. GH and insulinlike growth factor I (IGF-I) – which GH stimulates – affect tumor growth in vitro, and some studies have found associations with pediatric tumors, while others have not.
To get a sense of the overall direction of the literature, the investigators reviewed decades of studies and reports on PubMed. They found that “in children without prior cancer or known risk factors for developing cancer, the clinical evidence does not affirm an association between GH therapy during childhood and neoplasia.” In those children, “GH therapy can be safely administered without concerns about an increased risk for neoplasia,” said Dr. Sripriya Raman of the University of Kansas Medical Center in Kansas City, Mo., and associates (J. Clin. Endocrinol. Metab. 2015 April 3 [doi:10.1210/jc.2015-1002]).
They cautioned, however, that most of the investigations to date have been “postmarketing surveillance studies lacking rigorous controls.”
A different story emerged in children with genetic or other predispositions to cancer; in them GH might increase the neoplasia risk. When that’s a concern, children “should be critically analyzed on an individual basis, and if [GH is] chosen, appropriate surveillance for malignancies should be undertaken. GH treatment of pediatric cancer survivors does not appear to increase the risk of recurrence, but may increase their risk for subsequent primary neoplasms. Patients and caregivers should be informed that there may be an increase in the risk,” the investigators said.
In children recovering from malignancies, they noted that “no studies specifically address how long one should wait between completion of cancer therapy and initiation of GH treatment. Clinical guidelines from the Pediatric Endocrine Society and expert opinion ... suggest waiting 1 year after cancer treatment is complete to ensure that there is not an early cancer recurrence. ... Other factors to consider when deciding the timing of GH therapy include the chronological and skeletal ages, pubertal status, current height, primary tumor type, overall oncologic prognosis, risk of relapse, and the goals of the patient and caregivers.”
Also, “as most of the cancers associated with acromegaly and/or high GH/IGF-I levels are cancers seen predominantly in adults, long-term studies are needed to determine if treatment with rhGH [recombinant human GH] in childhood is associated with an increased risk for the common adult cancers.”
The team also noted that “there are insufficient data in children to support the practice of titrating rhGH doses to normalize IGF-I levels as a means to modify the risk of malignancy. However, in patients who are treated with rhGH for GH deficiency, it may be reasonable to attempt to maintain age- and Tanner stage–appropriate levels of IGF-I in children undergoing treatment with rhGH; the potential risks conferred by titrating to higher levels are not well defined. This practice has also been proposed for the management of idiopathic short stature and small-for-gestational-age patients, although evidence that this practice improves safety outcomes is not available.”
One of the authors, Dr. Bradley Miller of the University of Minnesota Masonic Children’s Hospital in Minneapolis, is a consultant for Alexion, BioMarin, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Sandoz and has received research support from Abbvie, Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Pfizer, Sandoz, Versartis, and Novo Nordisk. Most of the companies have growth hormone products. Dr. Charles Sklar of Memorial Sloan Kettering Cancer Center in Manhattan, reported honorarium from Sandoz, makers of the GH product Omnitrope. The other authors have no disclosures.
Growth hormone treatment does not increase the likelihood of cancer in children who aren’t at risk for it, but it may increase the risk in children who are predisposed to malignancy, according to a literature review from the Pediatric Endocrine Society published online April 3 in the Journal of Clinical Endocrinology & Metabolism.
CNS tumors, leukemia, and other cancers have been a concern with growth hormone (GH) treatment for years. GH and insulinlike growth factor I (IGF-I) – which GH stimulates – affect tumor growth in vitro, and some studies have found associations with pediatric tumors, while others have not.
To get a sense of the overall direction of the literature, the investigators reviewed decades of studies and reports on PubMed. They found that “in children without prior cancer or known risk factors for developing cancer, the clinical evidence does not affirm an association between GH therapy during childhood and neoplasia.” In those children, “GH therapy can be safely administered without concerns about an increased risk for neoplasia,” said Dr. Sripriya Raman of the University of Kansas Medical Center in Kansas City, Mo., and associates (J. Clin. Endocrinol. Metab. 2015 April 3 [doi:10.1210/jc.2015-1002]).
They cautioned, however, that most of the investigations to date have been “postmarketing surveillance studies lacking rigorous controls.”
A different story emerged in children with genetic or other predispositions to cancer; in them GH might increase the neoplasia risk. When that’s a concern, children “should be critically analyzed on an individual basis, and if [GH is] chosen, appropriate surveillance for malignancies should be undertaken. GH treatment of pediatric cancer survivors does not appear to increase the risk of recurrence, but may increase their risk for subsequent primary neoplasms. Patients and caregivers should be informed that there may be an increase in the risk,” the investigators said.
In children recovering from malignancies, they noted that “no studies specifically address how long one should wait between completion of cancer therapy and initiation of GH treatment. Clinical guidelines from the Pediatric Endocrine Society and expert opinion ... suggest waiting 1 year after cancer treatment is complete to ensure that there is not an early cancer recurrence. ... Other factors to consider when deciding the timing of GH therapy include the chronological and skeletal ages, pubertal status, current height, primary tumor type, overall oncologic prognosis, risk of relapse, and the goals of the patient and caregivers.”
Also, “as most of the cancers associated with acromegaly and/or high GH/IGF-I levels are cancers seen predominantly in adults, long-term studies are needed to determine if treatment with rhGH [recombinant human GH] in childhood is associated with an increased risk for the common adult cancers.”
The team also noted that “there are insufficient data in children to support the practice of titrating rhGH doses to normalize IGF-I levels as a means to modify the risk of malignancy. However, in patients who are treated with rhGH for GH deficiency, it may be reasonable to attempt to maintain age- and Tanner stage–appropriate levels of IGF-I in children undergoing treatment with rhGH; the potential risks conferred by titrating to higher levels are not well defined. This practice has also been proposed for the management of idiopathic short stature and small-for-gestational-age patients, although evidence that this practice improves safety outcomes is not available.”
One of the authors, Dr. Bradley Miller of the University of Minnesota Masonic Children’s Hospital in Minneapolis, is a consultant for Alexion, BioMarin, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Sandoz and has received research support from Abbvie, Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Pfizer, Sandoz, Versartis, and Novo Nordisk. Most of the companies have growth hormone products. Dr. Charles Sklar of Memorial Sloan Kettering Cancer Center in Manhattan, reported honorarium from Sandoz, makers of the GH product Omnitrope. The other authors have no disclosures.
Growth hormone treatment does not increase the likelihood of cancer in children who aren’t at risk for it, but it may increase the risk in children who are predisposed to malignancy, according to a literature review from the Pediatric Endocrine Society published online April 3 in the Journal of Clinical Endocrinology & Metabolism.
CNS tumors, leukemia, and other cancers have been a concern with growth hormone (GH) treatment for years. GH and insulinlike growth factor I (IGF-I) – which GH stimulates – affect tumor growth in vitro, and some studies have found associations with pediatric tumors, while others have not.
To get a sense of the overall direction of the literature, the investigators reviewed decades of studies and reports on PubMed. They found that “in children without prior cancer or known risk factors for developing cancer, the clinical evidence does not affirm an association between GH therapy during childhood and neoplasia.” In those children, “GH therapy can be safely administered without concerns about an increased risk for neoplasia,” said Dr. Sripriya Raman of the University of Kansas Medical Center in Kansas City, Mo., and associates (J. Clin. Endocrinol. Metab. 2015 April 3 [doi:10.1210/jc.2015-1002]).
They cautioned, however, that most of the investigations to date have been “postmarketing surveillance studies lacking rigorous controls.”
A different story emerged in children with genetic or other predispositions to cancer; in them GH might increase the neoplasia risk. When that’s a concern, children “should be critically analyzed on an individual basis, and if [GH is] chosen, appropriate surveillance for malignancies should be undertaken. GH treatment of pediatric cancer survivors does not appear to increase the risk of recurrence, but may increase their risk for subsequent primary neoplasms. Patients and caregivers should be informed that there may be an increase in the risk,” the investigators said.
In children recovering from malignancies, they noted that “no studies specifically address how long one should wait between completion of cancer therapy and initiation of GH treatment. Clinical guidelines from the Pediatric Endocrine Society and expert opinion ... suggest waiting 1 year after cancer treatment is complete to ensure that there is not an early cancer recurrence. ... Other factors to consider when deciding the timing of GH therapy include the chronological and skeletal ages, pubertal status, current height, primary tumor type, overall oncologic prognosis, risk of relapse, and the goals of the patient and caregivers.”
Also, “as most of the cancers associated with acromegaly and/or high GH/IGF-I levels are cancers seen predominantly in adults, long-term studies are needed to determine if treatment with rhGH [recombinant human GH] in childhood is associated with an increased risk for the common adult cancers.”
The team also noted that “there are insufficient data in children to support the practice of titrating rhGH doses to normalize IGF-I levels as a means to modify the risk of malignancy. However, in patients who are treated with rhGH for GH deficiency, it may be reasonable to attempt to maintain age- and Tanner stage–appropriate levels of IGF-I in children undergoing treatment with rhGH; the potential risks conferred by titrating to higher levels are not well defined. This practice has also been proposed for the management of idiopathic short stature and small-for-gestational-age patients, although evidence that this practice improves safety outcomes is not available.”
One of the authors, Dr. Bradley Miller of the University of Minnesota Masonic Children’s Hospital in Minneapolis, is a consultant for Alexion, BioMarin, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Sandoz and has received research support from Abbvie, Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Pfizer, Sandoz, Versartis, and Novo Nordisk. Most of the companies have growth hormone products. Dr. Charles Sklar of Memorial Sloan Kettering Cancer Center in Manhattan, reported honorarium from Sandoz, makers of the GH product Omnitrope. The other authors have no disclosures.
FROM JCEM
Teenage tattoos are most often regretted
SAN FRANCISCO – The younger people are when they get a tattoo, the more likely they are to regret it later, according to a survey of 501 people in the French Quarter of New Orleans.
All the participants were aged 18 years or older and had at least one tattoo. Overall, 16.2% said they regretted at least one tattoo, but that number rose to more than a third (35%) among the 77 people who got their first tattoo when they were 17 years old or younger. Among the 257 people who waited until they were 18-20 years old, 13.6% regretted one or more of their tattoos.
The rest got their first tattoo when they were at least 21 years old; 11.4% had regrets.
About one in five Americans have a tattoo, with the number increasing to about a third or more in people 18-30 years old, according to Walter Liszewski, a medical student at Tulane University in New Orleans.
Previous studies have found a regret rate of 16%-44%. The association with age suggests that it might be wise, when possible, to counsel teenagers to hold off for a while, Mr. Liszewski said. There’s a chance they will listen to that message, too, if it comes from a dermatologist. In the survey, 93% of people thought that tattoo artists were the best source of information on tattoo complications and how to treat them, but dermatologists weren’t far behind, with about 80% of respondents saying that dermatologists also were a trusted source for reliable information about tattoos. “So don’t hesitate to engage with patients about tattoos, complications, and other questions they might have,” Mr. Liszewski said at the annual meeting of the American Academy of Dermatology.
About 70% of the survey participants said they felt comfortable discussing such issues with primary care providers, and 40% said they felt comfortable discussing such issues with pharmacists.
Age requirements for tattoos vary widely from state to state, but some do allow individuals under 18 years of age to get a tattoo, even without parent permission.
In the first few days of 2015, the investigators asked passersby in the French Quarter’s Jackson Square if they had at least one tattoo, then offered the 18-question survey. They ran the project between 11 a.m. and 3 p.m., when people were less likely to be tipsy, Mr. Liszewski said. Respondents came from 38 states, plus the District of Columbia and Puerto Rico; most were from the Southeast, including 39% from Louisiana. Just over half were women, and the majority were white.
“We were also concerned that 21.2% received a tattoo while intoxicated, and 17.6% had at least one tattoo” done at someplace other than a tattoo parlor, he said. That means that if “patients have a lot of tattoos and you are trying to make small talk, feel free to ask them where they’re getting their tattoos done,” he added.
If tattoos were received at a party, or in prison or someplace else, “you may want to consider counseling them on HIV and hepatitis C testing,” Mr. Liszewski noted.
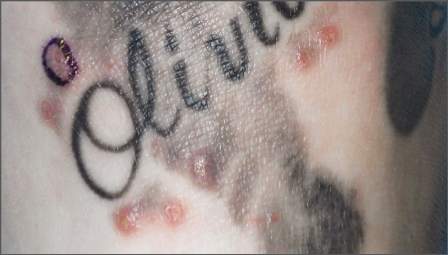
Just over 3% of the participants reported having had an infected tattoo, and while it’s normal for tattoos to be itchy and a little bit painful for the first week or two, 22.6% of the respondents said they had a pruritic tattoo, and 3.8% said they’d had a painful one a month or more after it was done.
“Tattoo complications are not uncommon,” Mr. Liszewski said. Given how much tattooed people seem to trust dermatologists, “there is an opportunity for dermatologists to manage these complications,” he added.
Mr. Liszewski said he had no disclosures.
SAN FRANCISCO – The younger people are when they get a tattoo, the more likely they are to regret it later, according to a survey of 501 people in the French Quarter of New Orleans.
All the participants were aged 18 years or older and had at least one tattoo. Overall, 16.2% said they regretted at least one tattoo, but that number rose to more than a third (35%) among the 77 people who got their first tattoo when they were 17 years old or younger. Among the 257 people who waited until they were 18-20 years old, 13.6% regretted one or more of their tattoos.
The rest got their first tattoo when they were at least 21 years old; 11.4% had regrets.
About one in five Americans have a tattoo, with the number increasing to about a third or more in people 18-30 years old, according to Walter Liszewski, a medical student at Tulane University in New Orleans.
Previous studies have found a regret rate of 16%-44%. The association with age suggests that it might be wise, when possible, to counsel teenagers to hold off for a while, Mr. Liszewski said. There’s a chance they will listen to that message, too, if it comes from a dermatologist. In the survey, 93% of people thought that tattoo artists were the best source of information on tattoo complications and how to treat them, but dermatologists weren’t far behind, with about 80% of respondents saying that dermatologists also were a trusted source for reliable information about tattoos. “So don’t hesitate to engage with patients about tattoos, complications, and other questions they might have,” Mr. Liszewski said at the annual meeting of the American Academy of Dermatology.
About 70% of the survey participants said they felt comfortable discussing such issues with primary care providers, and 40% said they felt comfortable discussing such issues with pharmacists.
Age requirements for tattoos vary widely from state to state, but some do allow individuals under 18 years of age to get a tattoo, even without parent permission.
In the first few days of 2015, the investigators asked passersby in the French Quarter’s Jackson Square if they had at least one tattoo, then offered the 18-question survey. They ran the project between 11 a.m. and 3 p.m., when people were less likely to be tipsy, Mr. Liszewski said. Respondents came from 38 states, plus the District of Columbia and Puerto Rico; most were from the Southeast, including 39% from Louisiana. Just over half were women, and the majority were white.
“We were also concerned that 21.2% received a tattoo while intoxicated, and 17.6% had at least one tattoo” done at someplace other than a tattoo parlor, he said. That means that if “patients have a lot of tattoos and you are trying to make small talk, feel free to ask them where they’re getting their tattoos done,” he added.
If tattoos were received at a party, or in prison or someplace else, “you may want to consider counseling them on HIV and hepatitis C testing,” Mr. Liszewski noted.
Just over 3% of the participants reported having had an infected tattoo, and while it’s normal for tattoos to be itchy and a little bit painful for the first week or two, 22.6% of the respondents said they had a pruritic tattoo, and 3.8% said they’d had a painful one a month or more after it was done.
“Tattoo complications are not uncommon,” Mr. Liszewski said. Given how much tattooed people seem to trust dermatologists, “there is an opportunity for dermatologists to manage these complications,” he added.
Mr. Liszewski said he had no disclosures.
SAN FRANCISCO – The younger people are when they get a tattoo, the more likely they are to regret it later, according to a survey of 501 people in the French Quarter of New Orleans.
All the participants were aged 18 years or older and had at least one tattoo. Overall, 16.2% said they regretted at least one tattoo, but that number rose to more than a third (35%) among the 77 people who got their first tattoo when they were 17 years old or younger. Among the 257 people who waited until they were 18-20 years old, 13.6% regretted one or more of their tattoos.
The rest got their first tattoo when they were at least 21 years old; 11.4% had regrets.
About one in five Americans have a tattoo, with the number increasing to about a third or more in people 18-30 years old, according to Walter Liszewski, a medical student at Tulane University in New Orleans.
Previous studies have found a regret rate of 16%-44%. The association with age suggests that it might be wise, when possible, to counsel teenagers to hold off for a while, Mr. Liszewski said. There’s a chance they will listen to that message, too, if it comes from a dermatologist. In the survey, 93% of people thought that tattoo artists were the best source of information on tattoo complications and how to treat them, but dermatologists weren’t far behind, with about 80% of respondents saying that dermatologists also were a trusted source for reliable information about tattoos. “So don’t hesitate to engage with patients about tattoos, complications, and other questions they might have,” Mr. Liszewski said at the annual meeting of the American Academy of Dermatology.
About 70% of the survey participants said they felt comfortable discussing such issues with primary care providers, and 40% said they felt comfortable discussing such issues with pharmacists.
Age requirements for tattoos vary widely from state to state, but some do allow individuals under 18 years of age to get a tattoo, even without parent permission.
In the first few days of 2015, the investigators asked passersby in the French Quarter’s Jackson Square if they had at least one tattoo, then offered the 18-question survey. They ran the project between 11 a.m. and 3 p.m., when people were less likely to be tipsy, Mr. Liszewski said. Respondents came from 38 states, plus the District of Columbia and Puerto Rico; most were from the Southeast, including 39% from Louisiana. Just over half were women, and the majority were white.
“We were also concerned that 21.2% received a tattoo while intoxicated, and 17.6% had at least one tattoo” done at someplace other than a tattoo parlor, he said. That means that if “patients have a lot of tattoos and you are trying to make small talk, feel free to ask them where they’re getting their tattoos done,” he added.
If tattoos were received at a party, or in prison or someplace else, “you may want to consider counseling them on HIV and hepatitis C testing,” Mr. Liszewski noted.
Just over 3% of the participants reported having had an infected tattoo, and while it’s normal for tattoos to be itchy and a little bit painful for the first week or two, 22.6% of the respondents said they had a pruritic tattoo, and 3.8% said they’d had a painful one a month or more after it was done.
“Tattoo complications are not uncommon,” Mr. Liszewski said. Given how much tattooed people seem to trust dermatologists, “there is an opportunity for dermatologists to manage these complications,” he added.
Mr. Liszewski said he had no disclosures.
AT THE AAD ANNUAL MEETING
Key clinical point: It might be wise, when possible, to counsel teenagers to postpone tattoos.
Major finding: Overall, 16.2% of tattooed adults said they regretted at least one, but that number rose to 35% among people who got their first tattoo when they were 17 years old or younger.
Data source: An in-person survey of 501 people with at least one tattoo.
Disclosures: The lead investigator had no disclosures.
High testosterone, no growth benefit for boys treated with letrozole
SAN DIEGO – The aromatase inhibitor letrozole didn’t help boys with short stature grow taller, but it did boost their testosterone to concerning levels in a small study at Children’s Hospital Los Angeles.
Estrogens are thought to help close epiphyseal growth plates; the idea of giving aromatase inhibitors (AIs) is to block aromatase from converting androgens to estrogens in boys so that their growth plates stay open a bit longer and they grow taller. In the United States, the drugs are frequently prescribed off label for that purpose in boys with short-stature or rapid-tempo puberty.
It didn’t seem to work, however, in the 16 boys in the study, who were an average of about 12.5 years old when they were started on letrozole.
The seven boys who started out at Tanner stages 1-3 were on the drug for an average of 2.7 years; their Bayley-Pinneau predicted adult height (PAH) fell from 66.9 inches to 66.5 inches. The nine boys who started out at Tanner stages 4-5 went from a PAH of 65.3 inches to 65.4 inches. The changes were not statistically significant.
Meanwhile, testosterone increased from 155 ng/dL to 728 ng/dL in stage 1-3 boys and from 417 ng/dL to 1,192 ng/dL in stage 4-5 boys. Those changes were statistically significant, and testosterone levels were significantly above the upper limit of normal in six (67%) of the later Tanner stage boys. There was a corresponding increase from no or mild acne to moderate acne in later stage boys, and a significant increase in hematocrit, from 43% to 47.5%. Seven (78%) of the later Tanner stage boys had hematocrit levels above the upper limit of normal.

“Short-term AIs were not effective in increasing PAH, regardless of pubertal age. [The] potential consequences of these findings are of concern and require careful long-term study, especially when AIs are started in late puberty,” the team concluded.
The findings “will change my practice with regard to” letrozole, said senior author Dr. Mitchell Geffner, professor of pediatrics at the hospital and president of the Pediatric Endocrine Society. Dr. Geffner treated all but one of the children in the study.
“I might still use” anastrozole, a less-potent AI, “but I have to get a better sense of all the parameters we measured here, a number of which are potentially unhealthy. These drugs are used to treat” boys with short stature and rapid-tempo puberty “very frequently in the United States,” but “I think we’re entering uncharted territory” with them. “We don’t really have a lot of data with use as single agents. We have to be very careful because we are giving a drug that alters, at least in the short term, reproductive hormones. Reproductive health in the future is completely unknown,” he said.
Despite having elevated testosterone levels, all the children had significant change in estradiol levels; in fact, there was a trend toward higher levels in the Tanner stage 4-5 boys, who also had significant increases in follicle-stimulating hormone and luteinizing-hormone levels that were often above the upper limit of normal.
“I was surprised by the results, and how high testosterone went in some of these boys. There’s conflicting data on whether these drugs help gain extra height, but we have to be very careful because we are causing very high testosterone levels,” which – although not seen in the study – could affect bone health and even cause stroke if hematocrit is pushed high enough, said lead investigator Dr. Jessica Ferris of Children’s Hospital Los Angeles.
There was no outside funding for the work. Dr. Ferris had no disclosures. Dr. Geffner is an investigator, consultant, or adviser for several companies, including Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Pfizer.
SAN DIEGO – The aromatase inhibitor letrozole didn’t help boys with short stature grow taller, but it did boost their testosterone to concerning levels in a small study at Children’s Hospital Los Angeles.
Estrogens are thought to help close epiphyseal growth plates; the idea of giving aromatase inhibitors (AIs) is to block aromatase from converting androgens to estrogens in boys so that their growth plates stay open a bit longer and they grow taller. In the United States, the drugs are frequently prescribed off label for that purpose in boys with short-stature or rapid-tempo puberty.
It didn’t seem to work, however, in the 16 boys in the study, who were an average of about 12.5 years old when they were started on letrozole.
The seven boys who started out at Tanner stages 1-3 were on the drug for an average of 2.7 years; their Bayley-Pinneau predicted adult height (PAH) fell from 66.9 inches to 66.5 inches. The nine boys who started out at Tanner stages 4-5 went from a PAH of 65.3 inches to 65.4 inches. The changes were not statistically significant.
Meanwhile, testosterone increased from 155 ng/dL to 728 ng/dL in stage 1-3 boys and from 417 ng/dL to 1,192 ng/dL in stage 4-5 boys. Those changes were statistically significant, and testosterone levels were significantly above the upper limit of normal in six (67%) of the later Tanner stage boys. There was a corresponding increase from no or mild acne to moderate acne in later stage boys, and a significant increase in hematocrit, from 43% to 47.5%. Seven (78%) of the later Tanner stage boys had hematocrit levels above the upper limit of normal.
“Short-term AIs were not effective in increasing PAH, regardless of pubertal age. [The] potential consequences of these findings are of concern and require careful long-term study, especially when AIs are started in late puberty,” the team concluded.
The findings “will change my practice with regard to” letrozole, said senior author Dr. Mitchell Geffner, professor of pediatrics at the hospital and president of the Pediatric Endocrine Society. Dr. Geffner treated all but one of the children in the study.
“I might still use” anastrozole, a less-potent AI, “but I have to get a better sense of all the parameters we measured here, a number of which are potentially unhealthy. These drugs are used to treat” boys with short stature and rapid-tempo puberty “very frequently in the United States,” but “I think we’re entering uncharted territory” with them. “We don’t really have a lot of data with use as single agents. We have to be very careful because we are giving a drug that alters, at least in the short term, reproductive hormones. Reproductive health in the future is completely unknown,” he said.
Despite having elevated testosterone levels, all the children had significant change in estradiol levels; in fact, there was a trend toward higher levels in the Tanner stage 4-5 boys, who also had significant increases in follicle-stimulating hormone and luteinizing-hormone levels that were often above the upper limit of normal.
“I was surprised by the results, and how high testosterone went in some of these boys. There’s conflicting data on whether these drugs help gain extra height, but we have to be very careful because we are causing very high testosterone levels,” which – although not seen in the study – could affect bone health and even cause stroke if hematocrit is pushed high enough, said lead investigator Dr. Jessica Ferris of Children’s Hospital Los Angeles.
There was no outside funding for the work. Dr. Ferris had no disclosures. Dr. Geffner is an investigator, consultant, or adviser for several companies, including Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Pfizer.
SAN DIEGO – The aromatase inhibitor letrozole didn’t help boys with short stature grow taller, but it did boost their testosterone to concerning levels in a small study at Children’s Hospital Los Angeles.
Estrogens are thought to help close epiphyseal growth plates; the idea of giving aromatase inhibitors (AIs) is to block aromatase from converting androgens to estrogens in boys so that their growth plates stay open a bit longer and they grow taller. In the United States, the drugs are frequently prescribed off label for that purpose in boys with short-stature or rapid-tempo puberty.
It didn’t seem to work, however, in the 16 boys in the study, who were an average of about 12.5 years old when they were started on letrozole.
The seven boys who started out at Tanner stages 1-3 were on the drug for an average of 2.7 years; their Bayley-Pinneau predicted adult height (PAH) fell from 66.9 inches to 66.5 inches. The nine boys who started out at Tanner stages 4-5 went from a PAH of 65.3 inches to 65.4 inches. The changes were not statistically significant.
Meanwhile, testosterone increased from 155 ng/dL to 728 ng/dL in stage 1-3 boys and from 417 ng/dL to 1,192 ng/dL in stage 4-5 boys. Those changes were statistically significant, and testosterone levels were significantly above the upper limit of normal in six (67%) of the later Tanner stage boys. There was a corresponding increase from no or mild acne to moderate acne in later stage boys, and a significant increase in hematocrit, from 43% to 47.5%. Seven (78%) of the later Tanner stage boys had hematocrit levels above the upper limit of normal.
“Short-term AIs were not effective in increasing PAH, regardless of pubertal age. [The] potential consequences of these findings are of concern and require careful long-term study, especially when AIs are started in late puberty,” the team concluded.
The findings “will change my practice with regard to” letrozole, said senior author Dr. Mitchell Geffner, professor of pediatrics at the hospital and president of the Pediatric Endocrine Society. Dr. Geffner treated all but one of the children in the study.
“I might still use” anastrozole, a less-potent AI, “but I have to get a better sense of all the parameters we measured here, a number of which are potentially unhealthy. These drugs are used to treat” boys with short stature and rapid-tempo puberty “very frequently in the United States,” but “I think we’re entering uncharted territory” with them. “We don’t really have a lot of data with use as single agents. We have to be very careful because we are giving a drug that alters, at least in the short term, reproductive hormones. Reproductive health in the future is completely unknown,” he said.
Despite having elevated testosterone levels, all the children had significant change in estradiol levels; in fact, there was a trend toward higher levels in the Tanner stage 4-5 boys, who also had significant increases in follicle-stimulating hormone and luteinizing-hormone levels that were often above the upper limit of normal.
“I was surprised by the results, and how high testosterone went in some of these boys. There’s conflicting data on whether these drugs help gain extra height, but we have to be very careful because we are causing very high testosterone levels,” which – although not seen in the study – could affect bone health and even cause stroke if hematocrit is pushed high enough, said lead investigator Dr. Jessica Ferris of Children’s Hospital Los Angeles.
There was no outside funding for the work. Dr. Ferris had no disclosures. Dr. Geffner is an investigator, consultant, or adviser for several companies, including Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Pfizer.
AT ENDO 2015
Key clinical point: Aromatase inhibitors don’t help shorter boys grow taller.
Major finding: The seven boys who started out at Tanner stages 1-3 were on letrozole for an average of 2.7 years; their Bayley-Pinneau predicted adult height fell from 66.9 inches to 66.5 inches. The nine boys who started out at Tanner stages 4-5 went from a PAH of 65.3 inches to 65.4 inches.
Data source: A review of outcomes for 16 boys treated at Children’s Hospital Los Angeles.
Disclosures: There was no outside funding for the work. The senior investigator is a consultant, adviser, or researcher for several companies, including Eli Lilly, Endo Pharmaceuticals, Genentech, Ipsen, Novo Nordisk, and Pfizer.

Worse melanoma outcomes found in pregnant women
SAN FRANCISCO – Pregnancy increases the risk of poor outcomes in melanoma, according to a review of melanoma cases at the Cleveland Clinic.
The effect of pregnancy on melanoma has been debated for more than a decade. Some studies have found evidence of worse outcomes, but others have not. That prompted Dr. Natasha Mesinkovska, a dermatologist at the clinic, and her colleagues to review their own melanoma outcomes over the past 20 years. They compared 49 women who were pregnant or within a year of pregnancy at diagnosis, with 418 women of childbearing age who were not pregnant. All the patients had at least 2 years follow-up.
Mortality (20% vs. 10.3%; P = .06); recurrence (12.5% vs. 1.4%; P < .001); metastasis (25% vs. 12.7%; P = .03); and the use of radiation and chemotherapy were all more common in the pregnancy group. On logistic regression, women who were pregnant or recently pregnant at the time of melanoma diagnosis were 5.1 times more likely to die of the disease than those who weren’t (P = .03), Dr. Mesinkovska reported at the annual meeting of the American Academy of Dermatology.
The findings prompted the investigators to compare histologic specimens from 17 pregnant and 14 nonpregnant women to find an explanation.
Pregnancy melanomas had reduced PD-1 [programmed cell death] expression (18.3 vs. 45 cells per high-power field [hpf]); decreased CD-3 [cluster of differentiation 3] (191.7 vs. 265.7 cells/hpf); and increased CD-3/PD-1 ratios (57.4 vs. 8.3).
The findings were statistically significant and may have treatment implications for the use in recently pregnant women of antibodies against PD-1 cell-surface receptors, a class of biologics that include nivolumab and pembrolizumab, both approved in 2014 for advanced melanoma. With reduced expression of PD-1, tumors arising around pregnancy might not be as sensitive to such agents.
Labeling for both biologics, however, note the risk of fetal harm, and advise the use of effective contraception while on the agents and for several months after their discontinuation. Both agents should also be discontinued during breastfeeding.
“Immune ignorance may predominate in melanoma specimens in pregnancy. Most novel melanoma therapies target immune modulation. There’s a need in some pregnant women for neoadjuvant treatment prior to immunotherapy to convert them to an inflamed phenotype,” Dr. Mesinkovska said.
Dr. Mesinkovska said she has no relevant financial disclosures.
SAN FRANCISCO – Pregnancy increases the risk of poor outcomes in melanoma, according to a review of melanoma cases at the Cleveland Clinic.
The effect of pregnancy on melanoma has been debated for more than a decade. Some studies have found evidence of worse outcomes, but others have not. That prompted Dr. Natasha Mesinkovska, a dermatologist at the clinic, and her colleagues to review their own melanoma outcomes over the past 20 years. They compared 49 women who were pregnant or within a year of pregnancy at diagnosis, with 418 women of childbearing age who were not pregnant. All the patients had at least 2 years follow-up.
Mortality (20% vs. 10.3%; P = .06); recurrence (12.5% vs. 1.4%; P < .001); metastasis (25% vs. 12.7%; P = .03); and the use of radiation and chemotherapy were all more common in the pregnancy group. On logistic regression, women who were pregnant or recently pregnant at the time of melanoma diagnosis were 5.1 times more likely to die of the disease than those who weren’t (P = .03), Dr. Mesinkovska reported at the annual meeting of the American Academy of Dermatology.
The findings prompted the investigators to compare histologic specimens from 17 pregnant and 14 nonpregnant women to find an explanation.
Pregnancy melanomas had reduced PD-1 [programmed cell death] expression (18.3 vs. 45 cells per high-power field [hpf]); decreased CD-3 [cluster of differentiation 3] (191.7 vs. 265.7 cells/hpf); and increased CD-3/PD-1 ratios (57.4 vs. 8.3).
The findings were statistically significant and may have treatment implications for the use in recently pregnant women of antibodies against PD-1 cell-surface receptors, a class of biologics that include nivolumab and pembrolizumab, both approved in 2014 for advanced melanoma. With reduced expression of PD-1, tumors arising around pregnancy might not be as sensitive to such agents.
Labeling for both biologics, however, note the risk of fetal harm, and advise the use of effective contraception while on the agents and for several months after their discontinuation. Both agents should also be discontinued during breastfeeding.
“Immune ignorance may predominate in melanoma specimens in pregnancy. Most novel melanoma therapies target immune modulation. There’s a need in some pregnant women for neoadjuvant treatment prior to immunotherapy to convert them to an inflamed phenotype,” Dr. Mesinkovska said.
Dr. Mesinkovska said she has no relevant financial disclosures.
SAN FRANCISCO – Pregnancy increases the risk of poor outcomes in melanoma, according to a review of melanoma cases at the Cleveland Clinic.
The effect of pregnancy on melanoma has been debated for more than a decade. Some studies have found evidence of worse outcomes, but others have not. That prompted Dr. Natasha Mesinkovska, a dermatologist at the clinic, and her colleagues to review their own melanoma outcomes over the past 20 years. They compared 49 women who were pregnant or within a year of pregnancy at diagnosis, with 418 women of childbearing age who were not pregnant. All the patients had at least 2 years follow-up.
Mortality (20% vs. 10.3%; P = .06); recurrence (12.5% vs. 1.4%; P < .001); metastasis (25% vs. 12.7%; P = .03); and the use of radiation and chemotherapy were all more common in the pregnancy group. On logistic regression, women who were pregnant or recently pregnant at the time of melanoma diagnosis were 5.1 times more likely to die of the disease than those who weren’t (P = .03), Dr. Mesinkovska reported at the annual meeting of the American Academy of Dermatology.
The findings prompted the investigators to compare histologic specimens from 17 pregnant and 14 nonpregnant women to find an explanation.
Pregnancy melanomas had reduced PD-1 [programmed cell death] expression (18.3 vs. 45 cells per high-power field [hpf]); decreased CD-3 [cluster of differentiation 3] (191.7 vs. 265.7 cells/hpf); and increased CD-3/PD-1 ratios (57.4 vs. 8.3).
The findings were statistically significant and may have treatment implications for the use in recently pregnant women of antibodies against PD-1 cell-surface receptors, a class of biologics that include nivolumab and pembrolizumab, both approved in 2014 for advanced melanoma. With reduced expression of PD-1, tumors arising around pregnancy might not be as sensitive to such agents.
Labeling for both biologics, however, note the risk of fetal harm, and advise the use of effective contraception while on the agents and for several months after their discontinuation. Both agents should also be discontinued during breastfeeding.
“Immune ignorance may predominate in melanoma specimens in pregnancy. Most novel melanoma therapies target immune modulation. There’s a need in some pregnant women for neoadjuvant treatment prior to immunotherapy to convert them to an inflamed phenotype,” Dr. Mesinkovska said.
Dr. Mesinkovska said she has no relevant financial disclosures.
AT AAD 2015
Key clinical point: Heighten melanoma surveillance in and around pregnancy, and treat lesions aggressively.
Major finding: Melanoma mortality (20% vs. 10.3%; P = .06); recurrence (12.5% vs. 1.4%; P < .001); and metastasis (25% vs. 12.7%; P = .03) were higher in 49 women diagnosed with melanoma while pregnant or within a year of pregnancy, compared with 418 women of childbearing age who were not pregnant.
Data source: Review of 467 melanoma cases at the Cleveland Clinic.
Disclosures: The lead investigator has no relevant financial disclosures.
Avoid voriconazole in transplant patients at risk for skin cancer
SAN FRANCISCO – Voriconazole increased the risk of squamous cell carcinoma by 73% in a review of 455 lung transplant patients at the University of California, San Francisco.
The increase was for any exposure to the drug after transplant (adjusted hazard ratio, 1.73; P = .03). The investigators also found that each additional 30-day exposure at 200 mg of voriconazole twice daily increased the risk of squamous cell carcinoma (SCC) by 3.0% (HR, 1.03; P < .001). The results were adjusted for age at transplant, sex, and race. Overall, SCC risk was highest among white men aged 50 years or older at the time of transplant.

Although voriconazole did protect against posttransplant Aspergillus colonization (aHR, 0.50; P < .001), it did not reduce the risk of invasive aspergillosis. The drug reduced all-cause mortality only among colonized subjects (aHR, 0.34; P = .03), and offered no mortality benefit among those who were not colonized.
There was no difference in all-cause mortality between patients who had any exposure to voriconazole and those who did not, “but we actually found a 2% increased risk of death for each 1 month on the medication. Patients who weren’t colonized were the ones contributing to this increased risk of death,” said lead investigator Matthew Mansh, now a medical student at Stanford (Calif.) University.
There was no increased risk of SCC with alternative antifungals, including inhaled amphotericin and posaconazole. These alternatives should be considered instead of voriconazole in people at higher risk for skin cancer after lung transplants, according to the study, Mr. Mansh noted.
Voriconazole, which is widely used for antifungal prophylaxis after solid organ transplants, has been linked to skin cancer. The reason for the carcinogenic effect is not known; researchers are working to unravel the molecular mechanisms.
“Physicians should be cautious when using voriconazole in the care of transplant recipients. If you see a patient who is developing phototoxicity” with voriconazole, “and if they don’t have evidence of Aspergillus colonization, you may want to limit exposure to high doses of this drug or suggest an alternative,” Mr. Mansh said.
“We have now demonstrated that the alternatives “don’t carry this increased risk of cutaneous SCC,” Mr. Mansh said at the American Academy of Dermatology annual meeting.
The mean age of the study patients at transplant was 52 years, and the majority of patients were white; slightly more than half were men. Most had bilateral lung transplants, with pulmonary fibrosis at the leading indication.
Voriconazole was used in 85% of the patients for an average of 10 months. A quarter of voriconazole patients developed SCC within 5 years of transplant, and 43% within 10 years. Among patients who did not receive the drug, 15% developed SCC within 5 years of transplant, and 28% developed SCC within 10 years of transplant.
“The benefit of voriconazole in terms of death was limited to patients with evidence of Aspergillus colonization, and it wasn’t dose dependent. Patients who had a higher cumulative exposure did not get more benefit,” Mr. Mansh said.
Mr. Mansh had no relevant disclosures.
 |
Dr. Paul T. Nghiem |
This is a carefully done study with a practical message: voriconazole patients are at a prolonged increased risk for squamous cell carcinoma. If patients develop phototoxicity or are fair-skinned, have sun damage, a history of squamous cell carcinoma or other risk factors, I think it’s highly appropriate to suggest an alternative. The alternatives are not at all associated with phototoxicity or squamous cell carcinoma.
Dr. Paul T. Nghiem moderated the late-breaker presentation in which the study was presented and is a professor of dermatology at the University of Washington, Seattle. Dr. Nghiem had no disclosures related to the study.
|
Dr. Paul T. Nghiem |
This is a carefully done study with a practical message: voriconazole patients are at a prolonged increased risk for squamous cell carcinoma. If patients develop phototoxicity or are fair-skinned, have sun damage, a history of squamous cell carcinoma or other risk factors, I think it’s highly appropriate to suggest an alternative. The alternatives are not at all associated with phototoxicity or squamous cell carcinoma.
Dr. Paul T. Nghiem moderated the late-breaker presentation in which the study was presented and is a professor of dermatology at the University of Washington, Seattle. Dr. Nghiem had no disclosures related to the study.
|
Dr. Paul T. Nghiem |
This is a carefully done study with a practical message: voriconazole patients are at a prolonged increased risk for squamous cell carcinoma. If patients develop phototoxicity or are fair-skinned, have sun damage, a history of squamous cell carcinoma or other risk factors, I think it’s highly appropriate to suggest an alternative. The alternatives are not at all associated with phototoxicity or squamous cell carcinoma.
Dr. Paul T. Nghiem moderated the late-breaker presentation in which the study was presented and is a professor of dermatology at the University of Washington, Seattle. Dr. Nghiem had no disclosures related to the study.
SAN FRANCISCO – Voriconazole increased the risk of squamous cell carcinoma by 73% in a review of 455 lung transplant patients at the University of California, San Francisco.
The increase was for any exposure to the drug after transplant (adjusted hazard ratio, 1.73; P = .03). The investigators also found that each additional 30-day exposure at 200 mg of voriconazole twice daily increased the risk of squamous cell carcinoma (SCC) by 3.0% (HR, 1.03; P < .001). The results were adjusted for age at transplant, sex, and race. Overall, SCC risk was highest among white men aged 50 years or older at the time of transplant.
Although voriconazole did protect against posttransplant Aspergillus colonization (aHR, 0.50; P < .001), it did not reduce the risk of invasive aspergillosis. The drug reduced all-cause mortality only among colonized subjects (aHR, 0.34; P = .03), and offered no mortality benefit among those who were not colonized.
There was no difference in all-cause mortality between patients who had any exposure to voriconazole and those who did not, “but we actually found a 2% increased risk of death for each 1 month on the medication. Patients who weren’t colonized were the ones contributing to this increased risk of death,” said lead investigator Matthew Mansh, now a medical student at Stanford (Calif.) University.
There was no increased risk of SCC with alternative antifungals, including inhaled amphotericin and posaconazole. These alternatives should be considered instead of voriconazole in people at higher risk for skin cancer after lung transplants, according to the study, Mr. Mansh noted.
Voriconazole, which is widely used for antifungal prophylaxis after solid organ transplants, has been linked to skin cancer. The reason for the carcinogenic effect is not known; researchers are working to unravel the molecular mechanisms.
“Physicians should be cautious when using voriconazole in the care of transplant recipients. If you see a patient who is developing phototoxicity” with voriconazole, “and if they don’t have evidence of Aspergillus colonization, you may want to limit exposure to high doses of this drug or suggest an alternative,” Mr. Mansh said.
“We have now demonstrated that the alternatives “don’t carry this increased risk of cutaneous SCC,” Mr. Mansh said at the American Academy of Dermatology annual meeting.
The mean age of the study patients at transplant was 52 years, and the majority of patients were white; slightly more than half were men. Most had bilateral lung transplants, with pulmonary fibrosis at the leading indication.
Voriconazole was used in 85% of the patients for an average of 10 months. A quarter of voriconazole patients developed SCC within 5 years of transplant, and 43% within 10 years. Among patients who did not receive the drug, 15% developed SCC within 5 years of transplant, and 28% developed SCC within 10 years of transplant.
“The benefit of voriconazole in terms of death was limited to patients with evidence of Aspergillus colonization, and it wasn’t dose dependent. Patients who had a higher cumulative exposure did not get more benefit,” Mr. Mansh said.
Mr. Mansh had no relevant disclosures.
SAN FRANCISCO – Voriconazole increased the risk of squamous cell carcinoma by 73% in a review of 455 lung transplant patients at the University of California, San Francisco.
The increase was for any exposure to the drug after transplant (adjusted hazard ratio, 1.73; P = .03). The investigators also found that each additional 30-day exposure at 200 mg of voriconazole twice daily increased the risk of squamous cell carcinoma (SCC) by 3.0% (HR, 1.03; P < .001). The results were adjusted for age at transplant, sex, and race. Overall, SCC risk was highest among white men aged 50 years or older at the time of transplant.
Although voriconazole did protect against posttransplant Aspergillus colonization (aHR, 0.50; P < .001), it did not reduce the risk of invasive aspergillosis. The drug reduced all-cause mortality only among colonized subjects (aHR, 0.34; P = .03), and offered no mortality benefit among those who were not colonized.
There was no difference in all-cause mortality between patients who had any exposure to voriconazole and those who did not, “but we actually found a 2% increased risk of death for each 1 month on the medication. Patients who weren’t colonized were the ones contributing to this increased risk of death,” said lead investigator Matthew Mansh, now a medical student at Stanford (Calif.) University.
There was no increased risk of SCC with alternative antifungals, including inhaled amphotericin and posaconazole. These alternatives should be considered instead of voriconazole in people at higher risk for skin cancer after lung transplants, according to the study, Mr. Mansh noted.
Voriconazole, which is widely used for antifungal prophylaxis after solid organ transplants, has been linked to skin cancer. The reason for the carcinogenic effect is not known; researchers are working to unravel the molecular mechanisms.
“Physicians should be cautious when using voriconazole in the care of transplant recipients. If you see a patient who is developing phototoxicity” with voriconazole, “and if they don’t have evidence of Aspergillus colonization, you may want to limit exposure to high doses of this drug or suggest an alternative,” Mr. Mansh said.
“We have now demonstrated that the alternatives “don’t carry this increased risk of cutaneous SCC,” Mr. Mansh said at the American Academy of Dermatology annual meeting.
The mean age of the study patients at transplant was 52 years, and the majority of patients were white; slightly more than half were men. Most had bilateral lung transplants, with pulmonary fibrosis at the leading indication.
Voriconazole was used in 85% of the patients for an average of 10 months. A quarter of voriconazole patients developed SCC within 5 years of transplant, and 43% within 10 years. Among patients who did not receive the drug, 15% developed SCC within 5 years of transplant, and 28% developed SCC within 10 years of transplant.
“The benefit of voriconazole in terms of death was limited to patients with evidence of Aspergillus colonization, and it wasn’t dose dependent. Patients who had a higher cumulative exposure did not get more benefit,” Mr. Mansh said.
Mr. Mansh had no relevant disclosures.
AT AAD 2015
Key clinical point: Use an alternative antifungal after lung transplant in white men aged 50 years and older.
Major finding: Exposure to voriconazole increased the risk of squamous cell carcinoma by 73% after lung transplant (aHR, 1.73; P = .03); each additional 30‐day exposure at 200 mg twice daily increased the risk by 3.0% (HR 1.03; P < .001).
Data source: Retrospective cohort study of 455 lung transplant patients
Disclosures: The lead investigator had no relevant disclosures.

Add baseline DHEAS when screening adrenal incidentalomas for subclinical hypercortisolism
SAN DIEGO – A single baseline measurement of dehydroepiandrosterone sulfate (DHEAS) outperforms overnight dexamethasone suppression and other standard tests for the detection of subclinical hypercortisolism in patients with adrenal incidentalomas, according to a prospective, blinded study of 185 consecutive patients at Cambridge University in England.
It’s an important finding because 1-mg overnight dexamethasone suppression testing is the current screening standard for detection of the condition, said Dr. Michael Dennedy, a former endocrinology fellow at Cambridge and now a senior lecturer at the National University of Ireland (NUI), Galway.

“On the basis of these data, we advocate the use of a single measurement of DHEAS as a convenient, reliable, and robust test for screening of SH [subclinical hypercortisolism] in the context of adrenal incidentalomas,” Dr. Dennedy said at the meeting of the Endocrine Society.
DHEAS avoids the sampling problems and high false positives of older tests and “reliably differentiates between SH and non-SH etiologies of adrenal nodules. We use it now as a standard test for patients who have adrenal nodules” at Cambridge and NUI Galway, he said.
The theory for checking DHEAS is straightforward. Like DHEA, it’s regulated by pituitary ACTH; sustained suppression of central ACTH leads to reductions in both. However, DHEA does not work for screening because it has a short half-life – about 25 minutes – and has circadian secretion patterns similar to ACTH. The half-life of DHEAS, on the other hand, is 10-16 hours and levels stay relatively stable throughout the day, which makes it a more attractive marker for detecting chronically suppressed ACTH.
At baseline, the team measured DHEAS by immunoassay and then put their 185 subjects through standard workups for newly diagnosed adrenal incidentalomas, including plasma metanephrines, 1-mg overnight dexamethasone suppression, 24-hour urinary free cortisol, and paired renin and aldosterone measurements. They then unblinded their DHEAS measurements to see how they fared against the standard approaches.
Because DHEAS levels are determined by a person’s age and gender, the investigators used a ratio of the actual baseline measurement divided by the lower limit of the appropriate reference range; for instance, 1.2 mmol/L in younger patients and 0.4 mmol/L in older patients.
A baseline DHEAS ratio at or below 1.12 was 100% sensitive and 92% specific for the diagnosis of SH in patients with adrenal incidentalomas. In contrast, a cortisol cutoff of 1.9 mcg/dL following 1-mg dexamethasone suppression was 100% sensitive but 82.9% specific and 24-hour urinary free cortisol was 69% sensitive and 68% specific.
The patients were aged 25 years to over 85, with an average age of about 65 years; 29 (16%) were diagnosed with SH. Most had nonfunctional adenomas, and a few had adrenal cortical carcinomas.
“Once upon a time, if there was a positive overnight dexamethasone or a positive urinary free cortisol, or both, we would send” patients on for a full inpatient Cushing’s workup. “Now we have a third criterion,” he said: suppressed DHEAS. Also, “in the presence of a high DHEAS with a failed dexamethasone suppression test, you would worry about adrenal corticocarcinoma, pituitary Cushing’s, or adrenal metastasis,” Dr. Dennedy said.
The investigators had no relevant disclosures, and there was no outside funding for the research.
SAN DIEGO – A single baseline measurement of dehydroepiandrosterone sulfate (DHEAS) outperforms overnight dexamethasone suppression and other standard tests for the detection of subclinical hypercortisolism in patients with adrenal incidentalomas, according to a prospective, blinded study of 185 consecutive patients at Cambridge University in England.
It’s an important finding because 1-mg overnight dexamethasone suppression testing is the current screening standard for detection of the condition, said Dr. Michael Dennedy, a former endocrinology fellow at Cambridge and now a senior lecturer at the National University of Ireland (NUI), Galway.
“On the basis of these data, we advocate the use of a single measurement of DHEAS as a convenient, reliable, and robust test for screening of SH [subclinical hypercortisolism] in the context of adrenal incidentalomas,” Dr. Dennedy said at the meeting of the Endocrine Society.
DHEAS avoids the sampling problems and high false positives of older tests and “reliably differentiates between SH and non-SH etiologies of adrenal nodules. We use it now as a standard test for patients who have adrenal nodules” at Cambridge and NUI Galway, he said.
The theory for checking DHEAS is straightforward. Like DHEA, it’s regulated by pituitary ACTH; sustained suppression of central ACTH leads to reductions in both. However, DHEA does not work for screening because it has a short half-life – about 25 minutes – and has circadian secretion patterns similar to ACTH. The half-life of DHEAS, on the other hand, is 10-16 hours and levels stay relatively stable throughout the day, which makes it a more attractive marker for detecting chronically suppressed ACTH.
At baseline, the team measured DHEAS by immunoassay and then put their 185 subjects through standard workups for newly diagnosed adrenal incidentalomas, including plasma metanephrines, 1-mg overnight dexamethasone suppression, 24-hour urinary free cortisol, and paired renin and aldosterone measurements. They then unblinded their DHEAS measurements to see how they fared against the standard approaches.
Because DHEAS levels are determined by a person’s age and gender, the investigators used a ratio of the actual baseline measurement divided by the lower limit of the appropriate reference range; for instance, 1.2 mmol/L in younger patients and 0.4 mmol/L in older patients.
A baseline DHEAS ratio at or below 1.12 was 100% sensitive and 92% specific for the diagnosis of SH in patients with adrenal incidentalomas. In contrast, a cortisol cutoff of 1.9 mcg/dL following 1-mg dexamethasone suppression was 100% sensitive but 82.9% specific and 24-hour urinary free cortisol was 69% sensitive and 68% specific.
The patients were aged 25 years to over 85, with an average age of about 65 years; 29 (16%) were diagnosed with SH. Most had nonfunctional adenomas, and a few had adrenal cortical carcinomas.
“Once upon a time, if there was a positive overnight dexamethasone or a positive urinary free cortisol, or both, we would send” patients on for a full inpatient Cushing’s workup. “Now we have a third criterion,” he said: suppressed DHEAS. Also, “in the presence of a high DHEAS with a failed dexamethasone suppression test, you would worry about adrenal corticocarcinoma, pituitary Cushing’s, or adrenal metastasis,” Dr. Dennedy said.
The investigators had no relevant disclosures, and there was no outside funding for the research.
SAN DIEGO – A single baseline measurement of dehydroepiandrosterone sulfate (DHEAS) outperforms overnight dexamethasone suppression and other standard tests for the detection of subclinical hypercortisolism in patients with adrenal incidentalomas, according to a prospective, blinded study of 185 consecutive patients at Cambridge University in England.
It’s an important finding because 1-mg overnight dexamethasone suppression testing is the current screening standard for detection of the condition, said Dr. Michael Dennedy, a former endocrinology fellow at Cambridge and now a senior lecturer at the National University of Ireland (NUI), Galway.
“On the basis of these data, we advocate the use of a single measurement of DHEAS as a convenient, reliable, and robust test for screening of SH [subclinical hypercortisolism] in the context of adrenal incidentalomas,” Dr. Dennedy said at the meeting of the Endocrine Society.
DHEAS avoids the sampling problems and high false positives of older tests and “reliably differentiates between SH and non-SH etiologies of adrenal nodules. We use it now as a standard test for patients who have adrenal nodules” at Cambridge and NUI Galway, he said.
The theory for checking DHEAS is straightforward. Like DHEA, it’s regulated by pituitary ACTH; sustained suppression of central ACTH leads to reductions in both. However, DHEA does not work for screening because it has a short half-life – about 25 minutes – and has circadian secretion patterns similar to ACTH. The half-life of DHEAS, on the other hand, is 10-16 hours and levels stay relatively stable throughout the day, which makes it a more attractive marker for detecting chronically suppressed ACTH.
At baseline, the team measured DHEAS by immunoassay and then put their 185 subjects through standard workups for newly diagnosed adrenal incidentalomas, including plasma metanephrines, 1-mg overnight dexamethasone suppression, 24-hour urinary free cortisol, and paired renin and aldosterone measurements. They then unblinded their DHEAS measurements to see how they fared against the standard approaches.
Because DHEAS levels are determined by a person’s age and gender, the investigators used a ratio of the actual baseline measurement divided by the lower limit of the appropriate reference range; for instance, 1.2 mmol/L in younger patients and 0.4 mmol/L in older patients.
A baseline DHEAS ratio at or below 1.12 was 100% sensitive and 92% specific for the diagnosis of SH in patients with adrenal incidentalomas. In contrast, a cortisol cutoff of 1.9 mcg/dL following 1-mg dexamethasone suppression was 100% sensitive but 82.9% specific and 24-hour urinary free cortisol was 69% sensitive and 68% specific.
The patients were aged 25 years to over 85, with an average age of about 65 years; 29 (16%) were diagnosed with SH. Most had nonfunctional adenomas, and a few had adrenal cortical carcinomas.
“Once upon a time, if there was a positive overnight dexamethasone or a positive urinary free cortisol, or both, we would send” patients on for a full inpatient Cushing’s workup. “Now we have a third criterion,” he said: suppressed DHEAS. Also, “in the presence of a high DHEAS with a failed dexamethasone suppression test, you would worry about adrenal corticocarcinoma, pituitary Cushing’s, or adrenal metastasis,” Dr. Dennedy said.
The investigators had no relevant disclosures, and there was no outside funding for the research.
AT ENDO 2015
Key clinical point: A single baseline measurement of dehydroepiandrosterone sulfate (DHEAS) beats 1-mg overnight dexamethasone suppression testing for detecting subclinical hypercortisolism.
Major finding: A baseline DHEAS ratio at or below 1.12 was 100% sensitive and 92% specific for the diagnosis of SH in patients with adrenal incidentalomas. In contrast, 1-mg dexamethasone suppression testing was 100% sensitive but 82.9% specific and 24-hour urinary free cortisol was 69% sensitive and 68% specific.
Data source: Prospective, blinded study of 185 patients worked up for adrenal incidentalomas.
Disclosures: The investigators had no relevant disclosures, and there was no outside funding for the work.
