User login
Teen vaccines: Where we are now, and how can we go further?
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.
“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.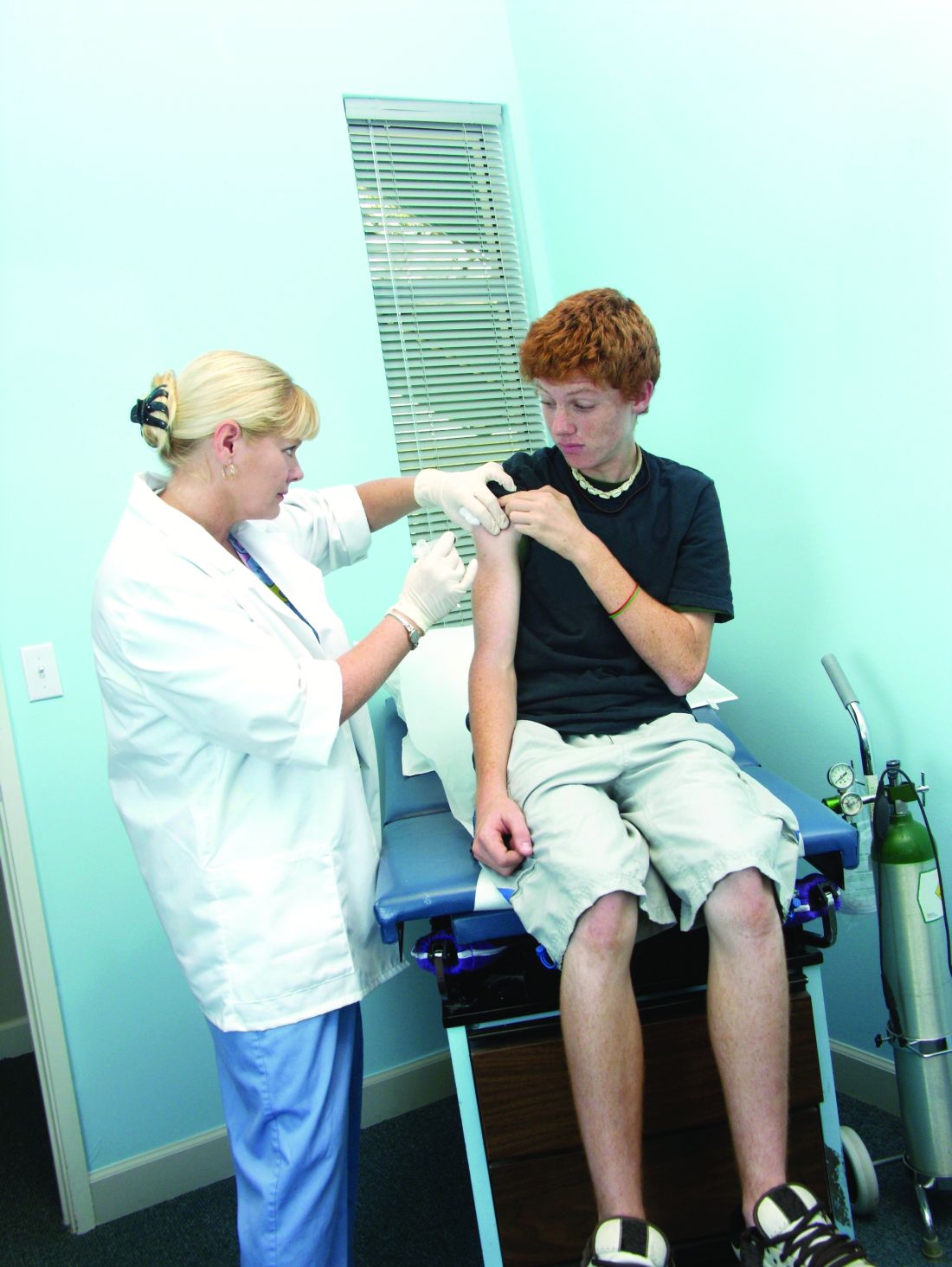
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.
“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.
“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Triclosan sutures halve surgical site infections in children
The use of triclosan-impregnated sutures reduced by half the incidence of surgical site infections in children, a large randomized study has determined.
Overall, the antibiotic-treated sutures cut the number of these infections by 52%, but they were particularly effective in reducing the risk of deep surgical site infections (SSIs), Marjo Renko, MD, wrote (Lancet Infect Dis. 2017;17[1]:50-7).
The study was conducted in clean wounds in healthy children and in a center that already had a very low rate of surgical site infections (just 5%) – showing that improvement is possible even in optimal care settings, wrote Dr. Renko, of the University of Oulu, Finland, and her colleagues.
“This randomized, controlled study shows that even in low-risk settings, where other prophylactic measures are available to use, triclosan-containing sutures effectively prevented the occurrence of SSIs in children,” the team wrote.
The study cohort comprised 1,633 children aged 7-17 who underwent surgery at a single Finnish hospital from 2010-2014. Most were there for planned surgery (87%); the remainder had emergency surgery. The most common surgical site was musculoskeletal (40%), followed by abdominal wall surgery (about 25%), and urogenital surgery (about 13%). The rest were intraabdominal or procedures on the nervous system, chest, and skin or subcutaneous tissue.
The children were randomized to either plain or triclosan-impregnated sutures. The primary outcome was the occurrence of a superficial or deep surgical site infection, based on Centers for Disease Control and Prevention criteria. The procedures were performed by 69 surgeons.
In a modified intent-to-treat analysis, a surgical site infection occurred in 3% of the triclosan-suture group (20 children) and in 5% of the control suture group (42 children). In the control group, these infections were most often of chest incisions (15%), followed by skin incisions (10%) and nervous system, intraabdominal, and musculoskeletal incisions (8% each). In the triclosan group, the most common site of infection was skin (10%), followed by musculoskeletal (4%), nervous system (2%), and urogenital and abdominal wall incisions (1% each).
Compared with control sutures, triclosan sutures reduced the overall risk of a surgical site infection by 52% (relative risk, 0.48; 95% confidence interval, 0.28-0.80). The number needed to treat to avoid one infection was 36.
The sutures were significantly more effective in reducing deep infections than superficial infections. Superficial infections occurred in 2% of the triclosan group (17) and 4% of the control group (28) – a risk reduction of 39% (RR, 0.61; 95% CI, 0.34-1.09) Deep infections occurred in less than 1% of the triclosan group (3) and 2% of the control group (14) – a risk reduction of 79% (RR, 0.21’ CI, 0.07-0.66).
Infections were associated with an increased incidence of wound dehiscence in the control group (6% vs. 4%), the need for additional antimicrobial agents (7% vs. 2%), and wound revisions (2% vs. less than 1%). Children in the control group also had more outpatient visits (8% vs. 4%) and were more often readmitted because of their infection (2% vs. 1%).
The authors noted that triclosan, in the setting of increased household use, “has raised concerns about the toxic effects of the drug on the human body. Observational studies have reported associations between triclosan exposures and altered thyroid hormone levels, body mass index, and waist circumference.”
Two Norwegian studies found that the drug was associated with inhalation allergies and seasonal allergies.
“Because of the agent’s suspected toxicity and to prevent further development of resistant bacteria, use of triclosan should be restricted and reserved only for medical procedures with adequate evidence,” they noted. However, “SSIs cause much morbidity and mortality after surgical procedures, and economic evaluations recommend the use of triclosan-containing material.”
Dr. Renko received grants from the Alma and K.A. Snellman Foundation, the Finnish Medical Foundation, and the Foundation for Pediatric Research.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
The study by Dr. Marjo Renko and her colleagues is impressive in its sheer numbers, if not so much in its findings, Felix J. Hüttner, MD, and Markus K. Diener, MD, wrote in an accompanying editorial (Lancet Infect Dis. 2017;17[1]:3-4).
“We congratulate the authors on successfully doing a pragmatic, large-scale trial in a difficult setting; randomized controlled trials in children are known to pose specific challenges to researchers. However, the monocenter design raises some concerns about the generalizability of the results.”
Single-center trials can overestimate treatment effects, the colleagues noted. Dr. Renko’s conclusions don’t line up with their own metaanalysis of triclosan-containing sutures for abdominal wall closure. In it, three single-center trials found in favor of the triclosan sutures, but two multicenter trials did not.
The variation in infection rates in each type of surgery is a clue to the difficulty of a one-size-fits-all intervention like the treated sutures. “The differences between the intervention group and the control group vary widely by surgery type – for example, 0% versus 15% for thoracic surgery, compared with 1% versus 1% for surgery of the urinary system and genitals. Thus, triclosan-containing sutures might only be beneficial for specific types of operations and in our opinion, it cannot be concluded that triclosan-containing sutures reduce surgical site infections in all of these indications. Future trials should focus at individual types of pediatric surgery to evaluate a potential beneficial effect.”
Dr. Hüttner and Dr. Diener are surgeons at the University of Heidelberg, Germany. Dr. Hüttner had no financial disclosures. Dr. Diener has received grants from Johnson & Johnson Medical Limited.
The study by Dr. Marjo Renko and her colleagues is impressive in its sheer numbers, if not so much in its findings, Felix J. Hüttner, MD, and Markus K. Diener, MD, wrote in an accompanying editorial (Lancet Infect Dis. 2017;17[1]:3-4).
“We congratulate the authors on successfully doing a pragmatic, large-scale trial in a difficult setting; randomized controlled trials in children are known to pose specific challenges to researchers. However, the monocenter design raises some concerns about the generalizability of the results.”
Single-center trials can overestimate treatment effects, the colleagues noted. Dr. Renko’s conclusions don’t line up with their own metaanalysis of triclosan-containing sutures for abdominal wall closure. In it, three single-center trials found in favor of the triclosan sutures, but two multicenter trials did not.
The variation in infection rates in each type of surgery is a clue to the difficulty of a one-size-fits-all intervention like the treated sutures. “The differences between the intervention group and the control group vary widely by surgery type – for example, 0% versus 15% for thoracic surgery, compared with 1% versus 1% for surgery of the urinary system and genitals. Thus, triclosan-containing sutures might only be beneficial for specific types of operations and in our opinion, it cannot be concluded that triclosan-containing sutures reduce surgical site infections in all of these indications. Future trials should focus at individual types of pediatric surgery to evaluate a potential beneficial effect.”
Dr. Hüttner and Dr. Diener are surgeons at the University of Heidelberg, Germany. Dr. Hüttner had no financial disclosures. Dr. Diener has received grants from Johnson & Johnson Medical Limited.
The study by Dr. Marjo Renko and her colleagues is impressive in its sheer numbers, if not so much in its findings, Felix J. Hüttner, MD, and Markus K. Diener, MD, wrote in an accompanying editorial (Lancet Infect Dis. 2017;17[1]:3-4).
“We congratulate the authors on successfully doing a pragmatic, large-scale trial in a difficult setting; randomized controlled trials in children are known to pose specific challenges to researchers. However, the monocenter design raises some concerns about the generalizability of the results.”
Single-center trials can overestimate treatment effects, the colleagues noted. Dr. Renko’s conclusions don’t line up with their own metaanalysis of triclosan-containing sutures for abdominal wall closure. In it, three single-center trials found in favor of the triclosan sutures, but two multicenter trials did not.
The variation in infection rates in each type of surgery is a clue to the difficulty of a one-size-fits-all intervention like the treated sutures. “The differences between the intervention group and the control group vary widely by surgery type – for example, 0% versus 15% for thoracic surgery, compared with 1% versus 1% for surgery of the urinary system and genitals. Thus, triclosan-containing sutures might only be beneficial for specific types of operations and in our opinion, it cannot be concluded that triclosan-containing sutures reduce surgical site infections in all of these indications. Future trials should focus at individual types of pediatric surgery to evaluate a potential beneficial effect.”
Dr. Hüttner and Dr. Diener are surgeons at the University of Heidelberg, Germany. Dr. Hüttner had no financial disclosures. Dr. Diener has received grants from Johnson & Johnson Medical Limited.
The use of triclosan-impregnated sutures reduced by half the incidence of surgical site infections in children, a large randomized study has determined.
Overall, the antibiotic-treated sutures cut the number of these infections by 52%, but they were particularly effective in reducing the risk of deep surgical site infections (SSIs), Marjo Renko, MD, wrote (Lancet Infect Dis. 2017;17[1]:50-7).
The study was conducted in clean wounds in healthy children and in a center that already had a very low rate of surgical site infections (just 5%) – showing that improvement is possible even in optimal care settings, wrote Dr. Renko, of the University of Oulu, Finland, and her colleagues.
“This randomized, controlled study shows that even in low-risk settings, where other prophylactic measures are available to use, triclosan-containing sutures effectively prevented the occurrence of SSIs in children,” the team wrote.
The study cohort comprised 1,633 children aged 7-17 who underwent surgery at a single Finnish hospital from 2010-2014. Most were there for planned surgery (87%); the remainder had emergency surgery. The most common surgical site was musculoskeletal (40%), followed by abdominal wall surgery (about 25%), and urogenital surgery (about 13%). The rest were intraabdominal or procedures on the nervous system, chest, and skin or subcutaneous tissue.
The children were randomized to either plain or triclosan-impregnated sutures. The primary outcome was the occurrence of a superficial or deep surgical site infection, based on Centers for Disease Control and Prevention criteria. The procedures were performed by 69 surgeons.
In a modified intent-to-treat analysis, a surgical site infection occurred in 3% of the triclosan-suture group (20 children) and in 5% of the control suture group (42 children). In the control group, these infections were most often of chest incisions (15%), followed by skin incisions (10%) and nervous system, intraabdominal, and musculoskeletal incisions (8% each). In the triclosan group, the most common site of infection was skin (10%), followed by musculoskeletal (4%), nervous system (2%), and urogenital and abdominal wall incisions (1% each).
Compared with control sutures, triclosan sutures reduced the overall risk of a surgical site infection by 52% (relative risk, 0.48; 95% confidence interval, 0.28-0.80). The number needed to treat to avoid one infection was 36.
The sutures were significantly more effective in reducing deep infections than superficial infections. Superficial infections occurred in 2% of the triclosan group (17) and 4% of the control group (28) – a risk reduction of 39% (RR, 0.61; 95% CI, 0.34-1.09) Deep infections occurred in less than 1% of the triclosan group (3) and 2% of the control group (14) – a risk reduction of 79% (RR, 0.21’ CI, 0.07-0.66).
Infections were associated with an increased incidence of wound dehiscence in the control group (6% vs. 4%), the need for additional antimicrobial agents (7% vs. 2%), and wound revisions (2% vs. less than 1%). Children in the control group also had more outpatient visits (8% vs. 4%) and were more often readmitted because of their infection (2% vs. 1%).
The authors noted that triclosan, in the setting of increased household use, “has raised concerns about the toxic effects of the drug on the human body. Observational studies have reported associations between triclosan exposures and altered thyroid hormone levels, body mass index, and waist circumference.”
Two Norwegian studies found that the drug was associated with inhalation allergies and seasonal allergies.
“Because of the agent’s suspected toxicity and to prevent further development of resistant bacteria, use of triclosan should be restricted and reserved only for medical procedures with adequate evidence,” they noted. However, “SSIs cause much morbidity and mortality after surgical procedures, and economic evaluations recommend the use of triclosan-containing material.”
Dr. Renko received grants from the Alma and K.A. Snellman Foundation, the Finnish Medical Foundation, and the Foundation for Pediatric Research.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
The use of triclosan-impregnated sutures reduced by half the incidence of surgical site infections in children, a large randomized study has determined.
Overall, the antibiotic-treated sutures cut the number of these infections by 52%, but they were particularly effective in reducing the risk of deep surgical site infections (SSIs), Marjo Renko, MD, wrote (Lancet Infect Dis. 2017;17[1]:50-7).
The study was conducted in clean wounds in healthy children and in a center that already had a very low rate of surgical site infections (just 5%) – showing that improvement is possible even in optimal care settings, wrote Dr. Renko, of the University of Oulu, Finland, and her colleagues.
“This randomized, controlled study shows that even in low-risk settings, where other prophylactic measures are available to use, triclosan-containing sutures effectively prevented the occurrence of SSIs in children,” the team wrote.
The study cohort comprised 1,633 children aged 7-17 who underwent surgery at a single Finnish hospital from 2010-2014. Most were there for planned surgery (87%); the remainder had emergency surgery. The most common surgical site was musculoskeletal (40%), followed by abdominal wall surgery (about 25%), and urogenital surgery (about 13%). The rest were intraabdominal or procedures on the nervous system, chest, and skin or subcutaneous tissue.
The children were randomized to either plain or triclosan-impregnated sutures. The primary outcome was the occurrence of a superficial or deep surgical site infection, based on Centers for Disease Control and Prevention criteria. The procedures were performed by 69 surgeons.
In a modified intent-to-treat analysis, a surgical site infection occurred in 3% of the triclosan-suture group (20 children) and in 5% of the control suture group (42 children). In the control group, these infections were most often of chest incisions (15%), followed by skin incisions (10%) and nervous system, intraabdominal, and musculoskeletal incisions (8% each). In the triclosan group, the most common site of infection was skin (10%), followed by musculoskeletal (4%), nervous system (2%), and urogenital and abdominal wall incisions (1% each).
Compared with control sutures, triclosan sutures reduced the overall risk of a surgical site infection by 52% (relative risk, 0.48; 95% confidence interval, 0.28-0.80). The number needed to treat to avoid one infection was 36.
The sutures were significantly more effective in reducing deep infections than superficial infections. Superficial infections occurred in 2% of the triclosan group (17) and 4% of the control group (28) – a risk reduction of 39% (RR, 0.61; 95% CI, 0.34-1.09) Deep infections occurred in less than 1% of the triclosan group (3) and 2% of the control group (14) – a risk reduction of 79% (RR, 0.21’ CI, 0.07-0.66).
Infections were associated with an increased incidence of wound dehiscence in the control group (6% vs. 4%), the need for additional antimicrobial agents (7% vs. 2%), and wound revisions (2% vs. less than 1%). Children in the control group also had more outpatient visits (8% vs. 4%) and were more often readmitted because of their infection (2% vs. 1%).
The authors noted that triclosan, in the setting of increased household use, “has raised concerns about the toxic effects of the drug on the human body. Observational studies have reported associations between triclosan exposures and altered thyroid hormone levels, body mass index, and waist circumference.”
Two Norwegian studies found that the drug was associated with inhalation allergies and seasonal allergies.
“Because of the agent’s suspected toxicity and to prevent further development of resistant bacteria, use of triclosan should be restricted and reserved only for medical procedures with adequate evidence,” they noted. However, “SSIs cause much morbidity and mortality after surgical procedures, and economic evaluations recommend the use of triclosan-containing material.”
Dr. Renko received grants from the Alma and K.A. Snellman Foundation, the Finnish Medical Foundation, and the Foundation for Pediatric Research.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
FROM LANCET INFECTIOUS DISEASES
Key clinical point:
Major finding: Overall, the sutures were associated with a 52% decrease in SSIs.
Data source: The study randomized 1,633 children undergoing surgery to the triclosan sutures or to a control suture.
Disclosures: Dr. Renko received grants from the Alma and K.A. Snellman Foundation, the Finnish Medical Foundation, and the Foundation for Pediatric Research.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Incompatible Type A plasma found safe for initial resuscitation of trauma patients
HOLLYWOOD, FLA. – Incompatible Type A plasma appears to be a safe and effective part of an initial resuscitation protocol for trauma patients who need a massive transfusion.
There were no increases in morbidity, mortality, or transfusion-related acute lung injury among 120 patients who received Type A plasma, compared with those who got compatible plasma, Bryan C. Morse, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Type AB blood products are preferred for initial transfusions for trauma patients with unknown blood type. While type AB blood products are universally acceptable to patients, they are also in short supply. In an attempt to mitigate this shortage, some trauma centers are relying on anecdotal data, much drawn from real-life combat experience dating from World War II to present times, suggesting that Type A plasma is safe for initial resuscitation protocols. But the body of data from well-constructed trials is small, said Dr. Morse of Emory University, Atlanta. Thus, EAST sponsored this retrospective registry study, which examined outcomes in 1,536 trauma patients who received plasma transfusions as part of a massive transfusion protocol from 2012 to 2016.
The primary endpoints were overall morbidity, and mortality at four time points: 6 and 24 hours, and 7 and 28 days. Eight trauma centers contributed data to the study.
The group was largely male (75%) with a mean age of 37 years. Patients were seriously injured, with a mean Injury Severity Score (ISS) of 25. About 60% suffered from blunt-force trauma. Among the entire group, 120 (8%) received incompatible type A plasma.
About 28% of patients (434) experienced an adverse event. These were numerically but not significantly more common among the incompatible A plasma group (35% vs. 28%; P = .14). Events included acute respiratory distress syndrome (6% vs. 7.6%), thromboembolism (9% vs. 7%), pneumonia (19% vs. 15%), and acute kidney injury (8% each).
There were two cases of transfusion-related acute lung injury, both of which occurred in the compatible type A group.
Mortality was similar at every time point: 6 hours (16% vs. 15%), 24 hours (25% vs. 22%), 7 days (35% vs. 32%), and 28 days (38% vs. 35%).
A multivariate regression model controlled for treatment center, ISS, units of packed red cells given by 4 hours, mechanism of injury, Type A plasma incompatibility, and age.
In the morbidity analysis, only ISS and units of red blood cells at 4 hours were associated with a significant increase in risk (odd ratio 1.02). Incompatible Type A plasma did not significantly increase the risk of morbidity.
In the mortality analysis, units of red cells, ISS, and age were significantly associated with increased risk. Again, incompatible Type A plasma did not significantly increase the risk of death.
Dr. Morse had no financial declaration.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
HOLLYWOOD, FLA. – Incompatible Type A plasma appears to be a safe and effective part of an initial resuscitation protocol for trauma patients who need a massive transfusion.
There were no increases in morbidity, mortality, or transfusion-related acute lung injury among 120 patients who received Type A plasma, compared with those who got compatible plasma, Bryan C. Morse, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Type AB blood products are preferred for initial transfusions for trauma patients with unknown blood type. While type AB blood products are universally acceptable to patients, they are also in short supply. In an attempt to mitigate this shortage, some trauma centers are relying on anecdotal data, much drawn from real-life combat experience dating from World War II to present times, suggesting that Type A plasma is safe for initial resuscitation protocols. But the body of data from well-constructed trials is small, said Dr. Morse of Emory University, Atlanta. Thus, EAST sponsored this retrospective registry study, which examined outcomes in 1,536 trauma patients who received plasma transfusions as part of a massive transfusion protocol from 2012 to 2016.
The primary endpoints were overall morbidity, and mortality at four time points: 6 and 24 hours, and 7 and 28 days. Eight trauma centers contributed data to the study.
The group was largely male (75%) with a mean age of 37 years. Patients were seriously injured, with a mean Injury Severity Score (ISS) of 25. About 60% suffered from blunt-force trauma. Among the entire group, 120 (8%) received incompatible type A plasma.
About 28% of patients (434) experienced an adverse event. These were numerically but not significantly more common among the incompatible A plasma group (35% vs. 28%; P = .14). Events included acute respiratory distress syndrome (6% vs. 7.6%), thromboembolism (9% vs. 7%), pneumonia (19% vs. 15%), and acute kidney injury (8% each).
There were two cases of transfusion-related acute lung injury, both of which occurred in the compatible type A group.
Mortality was similar at every time point: 6 hours (16% vs. 15%), 24 hours (25% vs. 22%), 7 days (35% vs. 32%), and 28 days (38% vs. 35%).
A multivariate regression model controlled for treatment center, ISS, units of packed red cells given by 4 hours, mechanism of injury, Type A plasma incompatibility, and age.
In the morbidity analysis, only ISS and units of red blood cells at 4 hours were associated with a significant increase in risk (odd ratio 1.02). Incompatible Type A plasma did not significantly increase the risk of morbidity.
In the mortality analysis, units of red cells, ISS, and age were significantly associated with increased risk. Again, incompatible Type A plasma did not significantly increase the risk of death.
Dr. Morse had no financial declaration.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
HOLLYWOOD, FLA. – Incompatible Type A plasma appears to be a safe and effective part of an initial resuscitation protocol for trauma patients who need a massive transfusion.
There were no increases in morbidity, mortality, or transfusion-related acute lung injury among 120 patients who received Type A plasma, compared with those who got compatible plasma, Bryan C. Morse, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Type AB blood products are preferred for initial transfusions for trauma patients with unknown blood type. While type AB blood products are universally acceptable to patients, they are also in short supply. In an attempt to mitigate this shortage, some trauma centers are relying on anecdotal data, much drawn from real-life combat experience dating from World War II to present times, suggesting that Type A plasma is safe for initial resuscitation protocols. But the body of data from well-constructed trials is small, said Dr. Morse of Emory University, Atlanta. Thus, EAST sponsored this retrospective registry study, which examined outcomes in 1,536 trauma patients who received plasma transfusions as part of a massive transfusion protocol from 2012 to 2016.
The primary endpoints were overall morbidity, and mortality at four time points: 6 and 24 hours, and 7 and 28 days. Eight trauma centers contributed data to the study.
The group was largely male (75%) with a mean age of 37 years. Patients were seriously injured, with a mean Injury Severity Score (ISS) of 25. About 60% suffered from blunt-force trauma. Among the entire group, 120 (8%) received incompatible type A plasma.
About 28% of patients (434) experienced an adverse event. These were numerically but not significantly more common among the incompatible A plasma group (35% vs. 28%; P = .14). Events included acute respiratory distress syndrome (6% vs. 7.6%), thromboembolism (9% vs. 7%), pneumonia (19% vs. 15%), and acute kidney injury (8% each).
There were two cases of transfusion-related acute lung injury, both of which occurred in the compatible type A group.
Mortality was similar at every time point: 6 hours (16% vs. 15%), 24 hours (25% vs. 22%), 7 days (35% vs. 32%), and 28 days (38% vs. 35%).
A multivariate regression model controlled for treatment center, ISS, units of packed red cells given by 4 hours, mechanism of injury, Type A plasma incompatibility, and age.
In the morbidity analysis, only ISS and units of red blood cells at 4 hours were associated with a significant increase in risk (odd ratio 1.02). Incompatible Type A plasma did not significantly increase the risk of morbidity.
In the mortality analysis, units of red cells, ISS, and age were significantly associated with increased risk. Again, incompatible Type A plasma did not significantly increase the risk of death.
Dr. Morse had no financial declaration.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
AT THE EAST SCIENTIFIC ASSEMBLY
Key clinical point:
Major finding: Adverse events were not significantly more common among the incompatible A plasma group (35% vs. 28%; P = .14).
Data source: The retrospective study comprised 1,536 patients.
Disclosures: Dr. Morse had no financial disclosures.
Interval cholecystectomy may be a risky business
HOLLYWOOD, FLA – Interval cholecystectomy remains a challenging procedure, with longer operative times and ICU stays, greater blood loss, more biliary and bowel injuries, and even hints of increased mortality, compared with immediate cholecystectomy, according to the findings from a retrospective study of 404 patients.
The staged procedure, completed after antibiotic therapy and percutaneous cholecystostomy, has been increasing in frequency over the past 10 years, but has not been rigorously studied, James Ackerman, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“Looking at Medicare data over the past decade, we see a 50% increase in this procedure, which is marked by some striking regional variation,” from 5% of acute cholecystitis cases in the Northeast to less than 1% in some other regions. “This shows that as a group, we really don’t know what to do with this procedure.”
The revised Tokyo Guidelines for the management of acute cholangitis and cholecystitis aren’t hugely helpful either, noted Dr. Ackerman of the University of Pittsburgh Medical Center. While the guidelines are fairly straightforward for patients with grade 1 and grade 3 disease, “there’s a lot of gray area in grade 2.”
Treatment for these patients should include biliary drainage with antibiotics, but, he said, the recommendations for surgery, and whether it should be elective, immediate, or delayed, can be confusing for this group.
Dr. Ackerman’s retrospective analysis comprised 177 patients with acute cholecystitis who underwent an interval cholecystectomy (IC) after percutaneous cholecystostomy, and 227 controls who underwent an immediate cholecystectomy. The analysis spanned 2008-2013 and used data from seven hospitals in one health care system.
Patients who had the IC were older (70 vs. 55 years), had a worse American Society of Anesthesiologists class (3 vs. 2.5), and a worse Tokyo Grade (2 vs. 1).
Most of the IC procedures (119) were laparoscopic. There were 43 conversions to open and 15 were planned open surgeries. Among the immediate cholecystectomies, most (192) were laparoscopic. There were 28 conversions to open and six planned open surgeries.
The conversion rate was significantly higher among the IC group (28% vs.13%). The most common reasons for conversion were hostile abdomen (48% vs. 16%) and hostile right upper quadrant (34% vs. 58%).
Operating time was significantly longer in the IC group (121 vs. 90 minutes). Estimated blood loss was also significantly higher (30 vs. 15 cc). Total hospital stay was significantly longer (7 vs. 5 days), as was ICU stay (1 vs. 0.1 day).
There were no biliary tract injuries in the cholecystectomy group, while 5.7% of IC patients sustained such an injury. Bowel injuries, most often serosal, were also more common in the IC group (6% vs. 0.4%). The IC group had more surgical site infections as well (12% vs. 0.44%).
There was no significant difference in 30-day mortality, but at 1 year, IC patients were significantly more likely to have died (15% vs. 0.44%).
The ongoing CHOCOLATE trial (Acute cholecystitis in high risk surgical patients: percutaneous cholecystostomy versus laparoscopic cholecystectomy) may help clarify the issue further, Dr. Ackerman said. The study being conducted in the Netherlands is randomizing high-risk cholecystitis patients to either laparoscopic cholecystectomy or percutaneous drainage.
Dr. Ackerman had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
HOLLYWOOD, FLA – Interval cholecystectomy remains a challenging procedure, with longer operative times and ICU stays, greater blood loss, more biliary and bowel injuries, and even hints of increased mortality, compared with immediate cholecystectomy, according to the findings from a retrospective study of 404 patients.
The staged procedure, completed after antibiotic therapy and percutaneous cholecystostomy, has been increasing in frequency over the past 10 years, but has not been rigorously studied, James Ackerman, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“Looking at Medicare data over the past decade, we see a 50% increase in this procedure, which is marked by some striking regional variation,” from 5% of acute cholecystitis cases in the Northeast to less than 1% in some other regions. “This shows that as a group, we really don’t know what to do with this procedure.”
The revised Tokyo Guidelines for the management of acute cholangitis and cholecystitis aren’t hugely helpful either, noted Dr. Ackerman of the University of Pittsburgh Medical Center. While the guidelines are fairly straightforward for patients with grade 1 and grade 3 disease, “there’s a lot of gray area in grade 2.”
Treatment for these patients should include biliary drainage with antibiotics, but, he said, the recommendations for surgery, and whether it should be elective, immediate, or delayed, can be confusing for this group.
Dr. Ackerman’s retrospective analysis comprised 177 patients with acute cholecystitis who underwent an interval cholecystectomy (IC) after percutaneous cholecystostomy, and 227 controls who underwent an immediate cholecystectomy. The analysis spanned 2008-2013 and used data from seven hospitals in one health care system.
Patients who had the IC were older (70 vs. 55 years), had a worse American Society of Anesthesiologists class (3 vs. 2.5), and a worse Tokyo Grade (2 vs. 1).
Most of the IC procedures (119) were laparoscopic. There were 43 conversions to open and 15 were planned open surgeries. Among the immediate cholecystectomies, most (192) were laparoscopic. There were 28 conversions to open and six planned open surgeries.
The conversion rate was significantly higher among the IC group (28% vs.13%). The most common reasons for conversion were hostile abdomen (48% vs. 16%) and hostile right upper quadrant (34% vs. 58%).
Operating time was significantly longer in the IC group (121 vs. 90 minutes). Estimated blood loss was also significantly higher (30 vs. 15 cc). Total hospital stay was significantly longer (7 vs. 5 days), as was ICU stay (1 vs. 0.1 day).
There were no biliary tract injuries in the cholecystectomy group, while 5.7% of IC patients sustained such an injury. Bowel injuries, most often serosal, were also more common in the IC group (6% vs. 0.4%). The IC group had more surgical site infections as well (12% vs. 0.44%).
There was no significant difference in 30-day mortality, but at 1 year, IC patients were significantly more likely to have died (15% vs. 0.44%).
The ongoing CHOCOLATE trial (Acute cholecystitis in high risk surgical patients: percutaneous cholecystostomy versus laparoscopic cholecystectomy) may help clarify the issue further, Dr. Ackerman said. The study being conducted in the Netherlands is randomizing high-risk cholecystitis patients to either laparoscopic cholecystectomy or percutaneous drainage.
Dr. Ackerman had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
HOLLYWOOD, FLA – Interval cholecystectomy remains a challenging procedure, with longer operative times and ICU stays, greater blood loss, more biliary and bowel injuries, and even hints of increased mortality, compared with immediate cholecystectomy, according to the findings from a retrospective study of 404 patients.
The staged procedure, completed after antibiotic therapy and percutaneous cholecystostomy, has been increasing in frequency over the past 10 years, but has not been rigorously studied, James Ackerman, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“Looking at Medicare data over the past decade, we see a 50% increase in this procedure, which is marked by some striking regional variation,” from 5% of acute cholecystitis cases in the Northeast to less than 1% in some other regions. “This shows that as a group, we really don’t know what to do with this procedure.”
The revised Tokyo Guidelines for the management of acute cholangitis and cholecystitis aren’t hugely helpful either, noted Dr. Ackerman of the University of Pittsburgh Medical Center. While the guidelines are fairly straightforward for patients with grade 1 and grade 3 disease, “there’s a lot of gray area in grade 2.”
Treatment for these patients should include biliary drainage with antibiotics, but, he said, the recommendations for surgery, and whether it should be elective, immediate, or delayed, can be confusing for this group.
Dr. Ackerman’s retrospective analysis comprised 177 patients with acute cholecystitis who underwent an interval cholecystectomy (IC) after percutaneous cholecystostomy, and 227 controls who underwent an immediate cholecystectomy. The analysis spanned 2008-2013 and used data from seven hospitals in one health care system.
Patients who had the IC were older (70 vs. 55 years), had a worse American Society of Anesthesiologists class (3 vs. 2.5), and a worse Tokyo Grade (2 vs. 1).
Most of the IC procedures (119) were laparoscopic. There were 43 conversions to open and 15 were planned open surgeries. Among the immediate cholecystectomies, most (192) were laparoscopic. There were 28 conversions to open and six planned open surgeries.
The conversion rate was significantly higher among the IC group (28% vs.13%). The most common reasons for conversion were hostile abdomen (48% vs. 16%) and hostile right upper quadrant (34% vs. 58%).
Operating time was significantly longer in the IC group (121 vs. 90 minutes). Estimated blood loss was also significantly higher (30 vs. 15 cc). Total hospital stay was significantly longer (7 vs. 5 days), as was ICU stay (1 vs. 0.1 day).
There were no biliary tract injuries in the cholecystectomy group, while 5.7% of IC patients sustained such an injury. Bowel injuries, most often serosal, were also more common in the IC group (6% vs. 0.4%). The IC group had more surgical site infections as well (12% vs. 0.44%).
There was no significant difference in 30-day mortality, but at 1 year, IC patients were significantly more likely to have died (15% vs. 0.44%).
The ongoing CHOCOLATE trial (Acute cholecystitis in high risk surgical patients: percutaneous cholecystostomy versus laparoscopic cholecystectomy) may help clarify the issue further, Dr. Ackerman said. The study being conducted in the Netherlands is randomizing high-risk cholecystitis patients to either laparoscopic cholecystectomy or percutaneous drainage.
Dr. Ackerman had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
AT THE EAST ANNUAL SCIENTIFIC ASSEMBLY
Key clinical point:
Major finding: Interval cholecystectomy was associated with greater blood loss, more conversions to open surgery, bowel and biliary injuries, and even higher 1-year mortality (15% vs. 0.44%).
Data source: A retrospective review comparing 177 patients with interval surgery to 227 who had immediate surgery.
Disclosures: Dr. Ackerman had no financial disclosures.
Tertiary center repeat CT scans find additional trauma injuries
HOLLYWOOD, FLA. – Imaging obtained at nontertiary trauma centers probably doesn’t tell the whole story of a trauma patent’s injuries, according to a new retrospective study.
Repeat scans done at a Level 1 trauma center identified new injuries in 76% of patients who were transferred, Morgan Bonds, MD, reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma. About half of these previously unobserved injuries were considered clinically significant, said Dr. Bonds, a surgical resident at the University of Oklahoma, Oklahoma City.
Her study examined imaging and clinical assessment of 203 trauma patients who were initially worked up at a nontertiary trauma center (NTC), and then transferred to the Level 1 University of Oklahoma tertiary trauma Center (TTC). The facility’s primary radiologist reviewed all of the initial CT scans while blinded to the NTC interpretation. The initial scans and interpretations were then compared with those done at the TTC.
The team split imaging and interpretation disconnects into four categories:
• Type A errors: A missed injury on the NTC scan. “This represents the expertise and experience of our primary radiologist,” Dr. Bonds said.
• Type B errors: Missed injuries on scans where NTC radiologists saw other injuries that the TTC radiologist did not confirm. “This represents the experience of our radiologist and also the inexperience and overreaction of the NTC radiologists.
• Type C errors: New injuries seen on additional TTC imaging of the same body area. “This represents the quality of the image.”
• Type D errors: New injuries found upon any new imaging, whether of a previously scanned or newly scanned body area. “This represents quality of work-up – the decision of the trauma team to more fully investigate the patient’s injuries, as well as the quality of the CT tech performing the scan.”
During the study period, 203 patients presented at the TTC with prior scans conducted at an NTC.
The mean age of the patients was 43 years; most (67%) were men. The mean Injury Severity Score was 16; 97% had experienced blunt trauma. Shock was present in 3% and a traumatic brain injury in 8%. Repeat scans were most common for neck and cervical spine injuries (54%) and thoracic/lumbar spine injuries (53%) and least common for chest injuries (32%).
An inadequate NTC work-up as judged by the TTC attending was the most common reason for getting new images (76%). Poor image quality was the next most common reason (31%).
Among the 203 patients, 99 (49%) had a type A error. Of these injuries missed on the initial scan, 90% were considered to be clinically significant.
Type B errors occurred in 15% of patients.
Type C errors (new injuries in different body area) occurred in 54% of patients and, of these, 76% were considered clinically significant. Type D errors (new injuries seen in any imaging of any area) occurred in 73% of patients.
“This study confirms that images are often repeated or completed after having images done at nontertiary trauma centers,” Dr. Bonds said. “Relying on NTC image interpretation can lead to undertreating our patients. One potential solution to this issue could be image sharing between NTCs and TTCs. This might reduce both the rate of missed injuries and the need for repeat scans.”
Dr. Bonds had no financial disclosures.
On Twitter @Alz_Gal
HOLLYWOOD, FLA. – Imaging obtained at nontertiary trauma centers probably doesn’t tell the whole story of a trauma patent’s injuries, according to a new retrospective study.
Repeat scans done at a Level 1 trauma center identified new injuries in 76% of patients who were transferred, Morgan Bonds, MD, reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma. About half of these previously unobserved injuries were considered clinically significant, said Dr. Bonds, a surgical resident at the University of Oklahoma, Oklahoma City.
Her study examined imaging and clinical assessment of 203 trauma patients who were initially worked up at a nontertiary trauma center (NTC), and then transferred to the Level 1 University of Oklahoma tertiary trauma Center (TTC). The facility’s primary radiologist reviewed all of the initial CT scans while blinded to the NTC interpretation. The initial scans and interpretations were then compared with those done at the TTC.
The team split imaging and interpretation disconnects into four categories:
• Type A errors: A missed injury on the NTC scan. “This represents the expertise and experience of our primary radiologist,” Dr. Bonds said.
• Type B errors: Missed injuries on scans where NTC radiologists saw other injuries that the TTC radiologist did not confirm. “This represents the experience of our radiologist and also the inexperience and overreaction of the NTC radiologists.
• Type C errors: New injuries seen on additional TTC imaging of the same body area. “This represents the quality of the image.”
• Type D errors: New injuries found upon any new imaging, whether of a previously scanned or newly scanned body area. “This represents quality of work-up – the decision of the trauma team to more fully investigate the patient’s injuries, as well as the quality of the CT tech performing the scan.”
During the study period, 203 patients presented at the TTC with prior scans conducted at an NTC.
The mean age of the patients was 43 years; most (67%) were men. The mean Injury Severity Score was 16; 97% had experienced blunt trauma. Shock was present in 3% and a traumatic brain injury in 8%. Repeat scans were most common for neck and cervical spine injuries (54%) and thoracic/lumbar spine injuries (53%) and least common for chest injuries (32%).
An inadequate NTC work-up as judged by the TTC attending was the most common reason for getting new images (76%). Poor image quality was the next most common reason (31%).
Among the 203 patients, 99 (49%) had a type A error. Of these injuries missed on the initial scan, 90% were considered to be clinically significant.
Type B errors occurred in 15% of patients.
Type C errors (new injuries in different body area) occurred in 54% of patients and, of these, 76% were considered clinically significant. Type D errors (new injuries seen in any imaging of any area) occurred in 73% of patients.
“This study confirms that images are often repeated or completed after having images done at nontertiary trauma centers,” Dr. Bonds said. “Relying on NTC image interpretation can lead to undertreating our patients. One potential solution to this issue could be image sharing between NTCs and TTCs. This might reduce both the rate of missed injuries and the need for repeat scans.”
Dr. Bonds had no financial disclosures.
On Twitter @Alz_Gal
HOLLYWOOD, FLA. – Imaging obtained at nontertiary trauma centers probably doesn’t tell the whole story of a trauma patent’s injuries, according to a new retrospective study.
Repeat scans done at a Level 1 trauma center identified new injuries in 76% of patients who were transferred, Morgan Bonds, MD, reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma. About half of these previously unobserved injuries were considered clinically significant, said Dr. Bonds, a surgical resident at the University of Oklahoma, Oklahoma City.
Her study examined imaging and clinical assessment of 203 trauma patients who were initially worked up at a nontertiary trauma center (NTC), and then transferred to the Level 1 University of Oklahoma tertiary trauma Center (TTC). The facility’s primary radiologist reviewed all of the initial CT scans while blinded to the NTC interpretation. The initial scans and interpretations were then compared with those done at the TTC.
The team split imaging and interpretation disconnects into four categories:
• Type A errors: A missed injury on the NTC scan. “This represents the expertise and experience of our primary radiologist,” Dr. Bonds said.
• Type B errors: Missed injuries on scans where NTC radiologists saw other injuries that the TTC radiologist did not confirm. “This represents the experience of our radiologist and also the inexperience and overreaction of the NTC radiologists.
• Type C errors: New injuries seen on additional TTC imaging of the same body area. “This represents the quality of the image.”
• Type D errors: New injuries found upon any new imaging, whether of a previously scanned or newly scanned body area. “This represents quality of work-up – the decision of the trauma team to more fully investigate the patient’s injuries, as well as the quality of the CT tech performing the scan.”
During the study period, 203 patients presented at the TTC with prior scans conducted at an NTC.
The mean age of the patients was 43 years; most (67%) were men. The mean Injury Severity Score was 16; 97% had experienced blunt trauma. Shock was present in 3% and a traumatic brain injury in 8%. Repeat scans were most common for neck and cervical spine injuries (54%) and thoracic/lumbar spine injuries (53%) and least common for chest injuries (32%).
An inadequate NTC work-up as judged by the TTC attending was the most common reason for getting new images (76%). Poor image quality was the next most common reason (31%).
Among the 203 patients, 99 (49%) had a type A error. Of these injuries missed on the initial scan, 90% were considered to be clinically significant.
Type B errors occurred in 15% of patients.
Type C errors (new injuries in different body area) occurred in 54% of patients and, of these, 76% were considered clinically significant. Type D errors (new injuries seen in any imaging of any area) occurred in 73% of patients.
“This study confirms that images are often repeated or completed after having images done at nontertiary trauma centers,” Dr. Bonds said. “Relying on NTC image interpretation can lead to undertreating our patients. One potential solution to this issue could be image sharing between NTCs and TTCs. This might reduce both the rate of missed injuries and the need for repeat scans.”
Dr. Bonds had no financial disclosures.
On Twitter @Alz_Gal
Key clinical point:
Major finding: Overall, 76% of patients had missed injuries on their initial CT scans.
Data source: A study of 203 trauma patients.
Disclosures: Dr. Bonds had no financial disclosures.
Gastrografin IDs, treats suspected small bowel obstruction
HOLLYWOOD, FLA – The radiopaque contrast agent Gastrografin accurately diagnosed the majority of small bowel obstructions, allowing surgeons to identify which patients needed emergent surgery and which could be managed conservatively.
When instilled via nasogastric tube, the diatrizoate solution had a 92% positive predictive value for adhesive small bowel obstruction, Martin D. Zielinski, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Dr. Zielinski of the Mayo Clinic, Rochester, Minn., examined the diagnostic accuracy of the Gastrografin challenge, a small bowel obstruction diagnosis and treatment protocol he developed at the center. The challenge begins with 2 hours of nasogastric suctioning. Patients then receive 100 mL Gastrografin mixed with 50 mL water via the nasogastric tube. The tube is clamped for 8 hours, and then patients have an abdominal x-ray. If the contrast material appears in the colon, or if the patient has a bowel movement in the interim, then the challenge is passed, the tube can be removed, and diet advanced.
If there is no contrast in the colon, or if the patient has no bowel movement, then the surgeon assumes the obstruction remains, and exploratory surgery proceeds.
Dr. Zielinski’s study comprised 316 patients with a suspected adhesive small bowel obstruction. Of these, 173 were managed with the Gastrografin challenge; they were compared to 143 patients who were managed without the contrast agent.
Patients were a mean of 58 years. There were no significant differences in the rate of prior abdominal operations; duration of obstipation; or small bowel feces sign.
The comparator group was managed by a clinical algorithm in which any patient with initial signs of ischemia underwent exploratory surgery, and those without signs of ischemia were managed symptomatically. Patients in the Gastrografin arm who passed the trial were similarly managed, while those who failed it underwent exploratory surgery.
Among those who had the challenge, 130 (75%) passed. Gastrografin had a high diagnostic accuracy for small bowel obstruction, with 87% sensitivity, 71% specificity; and 92% positive predictive value. The negative predictive value was not as good, at 59%.
The Gastrografin protocol was associated with significantly fewer exploratory surgeries (21% vs. 44%), and significantly fewer small bowel resections (7% vs. 21%). That advantage was maintained even among patients in both groups who underwent exploratory surgery, with an ultimate resection rate of 34% vs. 49%. The length of stay was also significantly less in the Gastrografin group, 4 vs. 5 days).
There was no difference in the overall complication rate (12.5% vs. 18%). Complications included acute kidney injury (6% vs. 9%); pneumonia (4% vs. 5%), organ space infection (1% vs. 4%), surgical site infection (3.5% vs. 5%), and anastomotic leak (2% each group).
The rate of missed small bowel strangulation was significantly lower among the Gastrografin group as well (0.6% vs. 7.7%). There were no cases of Gastrografin pneumonitis.
Dr. Zielinski had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
HOLLYWOOD, FLA – The radiopaque contrast agent Gastrografin accurately diagnosed the majority of small bowel obstructions, allowing surgeons to identify which patients needed emergent surgery and which could be managed conservatively.
When instilled via nasogastric tube, the diatrizoate solution had a 92% positive predictive value for adhesive small bowel obstruction, Martin D. Zielinski, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Dr. Zielinski of the Mayo Clinic, Rochester, Minn., examined the diagnostic accuracy of the Gastrografin challenge, a small bowel obstruction diagnosis and treatment protocol he developed at the center. The challenge begins with 2 hours of nasogastric suctioning. Patients then receive 100 mL Gastrografin mixed with 50 mL water via the nasogastric tube. The tube is clamped for 8 hours, and then patients have an abdominal x-ray. If the contrast material appears in the colon, or if the patient has a bowel movement in the interim, then the challenge is passed, the tube can be removed, and diet advanced.
If there is no contrast in the colon, or if the patient has no bowel movement, then the surgeon assumes the obstruction remains, and exploratory surgery proceeds.
Dr. Zielinski’s study comprised 316 patients with a suspected adhesive small bowel obstruction. Of these, 173 were managed with the Gastrografin challenge; they were compared to 143 patients who were managed without the contrast agent.
Patients were a mean of 58 years. There were no significant differences in the rate of prior abdominal operations; duration of obstipation; or small bowel feces sign.
The comparator group was managed by a clinical algorithm in which any patient with initial signs of ischemia underwent exploratory surgery, and those without signs of ischemia were managed symptomatically. Patients in the Gastrografin arm who passed the trial were similarly managed, while those who failed it underwent exploratory surgery.
Among those who had the challenge, 130 (75%) passed. Gastrografin had a high diagnostic accuracy for small bowel obstruction, with 87% sensitivity, 71% specificity; and 92% positive predictive value. The negative predictive value was not as good, at 59%.
The Gastrografin protocol was associated with significantly fewer exploratory surgeries (21% vs. 44%), and significantly fewer small bowel resections (7% vs. 21%). That advantage was maintained even among patients in both groups who underwent exploratory surgery, with an ultimate resection rate of 34% vs. 49%. The length of stay was also significantly less in the Gastrografin group, 4 vs. 5 days).
There was no difference in the overall complication rate (12.5% vs. 18%). Complications included acute kidney injury (6% vs. 9%); pneumonia (4% vs. 5%), organ space infection (1% vs. 4%), surgical site infection (3.5% vs. 5%), and anastomotic leak (2% each group).
The rate of missed small bowel strangulation was significantly lower among the Gastrografin group as well (0.6% vs. 7.7%). There were no cases of Gastrografin pneumonitis.
Dr. Zielinski had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
HOLLYWOOD, FLA – The radiopaque contrast agent Gastrografin accurately diagnosed the majority of small bowel obstructions, allowing surgeons to identify which patients needed emergent surgery and which could be managed conservatively.
When instilled via nasogastric tube, the diatrizoate solution had a 92% positive predictive value for adhesive small bowel obstruction, Martin D. Zielinski, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Dr. Zielinski of the Mayo Clinic, Rochester, Minn., examined the diagnostic accuracy of the Gastrografin challenge, a small bowel obstruction diagnosis and treatment protocol he developed at the center. The challenge begins with 2 hours of nasogastric suctioning. Patients then receive 100 mL Gastrografin mixed with 50 mL water via the nasogastric tube. The tube is clamped for 8 hours, and then patients have an abdominal x-ray. If the contrast material appears in the colon, or if the patient has a bowel movement in the interim, then the challenge is passed, the tube can be removed, and diet advanced.
If there is no contrast in the colon, or if the patient has no bowel movement, then the surgeon assumes the obstruction remains, and exploratory surgery proceeds.
Dr. Zielinski’s study comprised 316 patients with a suspected adhesive small bowel obstruction. Of these, 173 were managed with the Gastrografin challenge; they were compared to 143 patients who were managed without the contrast agent.
Patients were a mean of 58 years. There were no significant differences in the rate of prior abdominal operations; duration of obstipation; or small bowel feces sign.
The comparator group was managed by a clinical algorithm in which any patient with initial signs of ischemia underwent exploratory surgery, and those without signs of ischemia were managed symptomatically. Patients in the Gastrografin arm who passed the trial were similarly managed, while those who failed it underwent exploratory surgery.
Among those who had the challenge, 130 (75%) passed. Gastrografin had a high diagnostic accuracy for small bowel obstruction, with 87% sensitivity, 71% specificity; and 92% positive predictive value. The negative predictive value was not as good, at 59%.
The Gastrografin protocol was associated with significantly fewer exploratory surgeries (21% vs. 44%), and significantly fewer small bowel resections (7% vs. 21%). That advantage was maintained even among patients in both groups who underwent exploratory surgery, with an ultimate resection rate of 34% vs. 49%. The length of stay was also significantly less in the Gastrografin group, 4 vs. 5 days).
There was no difference in the overall complication rate (12.5% vs. 18%). Complications included acute kidney injury (6% vs. 9%); pneumonia (4% vs. 5%), organ space infection (1% vs. 4%), surgical site infection (3.5% vs. 5%), and anastomotic leak (2% each group).
The rate of missed small bowel strangulation was significantly lower among the Gastrografin group as well (0.6% vs. 7.7%). There were no cases of Gastrografin pneumonitis.
Dr. Zielinski had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT THE EAST ANNUAL SCIENTIFIC ASSEMBLY
Key clinical point: The bowel-imaging agent Gastrografin can both diagnose and treat small bowel obstruction.
Major finding: The agent had a 92% positive predictive value; it was associated with fewer bowel resections (7% vs. 21%) and a day shorter length of stay, compared with those who didn’t receive it.
Data source: The prospective study comprised 316 patients, 173 of whom underwent the Gastrografin challenge.
Disclosures: Dr. Zielinski had no financial disclosures.

LMWH trumps unfractionated heparin in reducing posttrauma thrombotic events
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT THE EAST ANNUAL SCIENTIFIC ASSEMBLY
Key clinical point:
Major finding: Overall mortality was reduced by 37% with LMWH, compared with unfractionated heparin.
Data source: The review comprised 37,868 patients included in the Michigan Trauma Quality Improvement Program.
Disclosures: Dr. Jacobs had no financial disclosures.
Older adults can sustain asymptomatic cervical fractures
HOLLYWOOD, FLA. –
The 4-year review found that 21% of older patients with a confirmed C-spine fracture reported no pain on history or physical exam, and that 76% of these fractures needed treatment – twin findings suggesting that asymptomatic neck fractures may be undiagnosed and untreated in this population.
The 183-patient study also found no significant pain differences between age groups: The silent injuries were just as common among 55-year-olds as among 65-year-olds, Christopher Healey, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“I guess we can say, ‘55 is the new 65,’ when it comes to this injury,” said Dr. Healey of the Iowa Methodist Medical Center, Des Moines. “In our study the rate of a pain-free neck fracture in 55-year-olds was equivalent to that in their older counterpoints, so they also represent a group at increased risk.”
Dr. Healey and his colleagues conducted a 4-year review of trauma patients aged 55 years and older who were treated for a C-spine fracture. All of the patients had a Glasgow Coma 
There were no differences in the mechanism of injury between the pain-free and pain-positive groups. The most common source of injury was a fall from the person’s own height (39% vs. 49%), followed by a high fall (21% vs. 19%). Motor vehicle crashes came in third (37% vs. 30%), with other methods of injury accounting for about 3% of each group.
The level of fracture was widely dispersed among both groups and not associated with pain. Asymptomatic fractures occurred at C1 and C2 vertabrae (about 13%); C3 (26%); C4 (10%); C5 (12%); and C6 and C7 (30%).
Perhaps surprisingly, patients who didn’t report pain had significantly higher Injury Severity Scores (15 vs. 10). They were also significantly more likely to have injuries to other body regions (71% vs. 47%), including the head (22% vs. 16%), thorax/abdomen (39% vs. 20%), and extremities (33% vs. 20%).
A third of those with a pain-free fracture had breaks at multiple levels. Their hospital stays were significantly longer than were those of patients with painful fractures (7 vs. 5 days).
These findings of more severe injuries in the asymptomatic group led Dr. Healey to suggest that distracting pain might be playing a part in the phenomenon.
When the group was split into 10-year age increments, asymptomatic fractures occurred in about 20% of each group from 55-64 years up to 85 years and older.
The majority of both groups required treatment (91% with pain; 76% asymptomatic), Dr. Healey said. The most common treatment in each group was a cervical collar (61% with pain vs. 46% asymptomatic ). A cervical-thoracic-lumbar-sacral orthosis brace was used in 8% of those with pain and 11% of the asymptomatic patients.
Invasive procedures were performed in just as many of the asymptomatic patients as in those who had pain. These included vertebral fusion (11% asymptomatic vs. 9% with pain) and cervical halo (8% vs. 13%).
The lesson here, Dr. Healey concluded, is that pain is not always a reliable indicator of neck injury in older patients. “Older adults can break their neck and have no pain at all. This is concerning, because the presence or absence of neck pain is a major component in many clearance protocols for C-spine trauma. This begs the question whether we should be treating our older patients by general adult guidelines. I would advocate the development of trauma guidelines that are specific for the older or geriatric patient.”
Dr. Healey had no financial disclosures.
HOLLYWOOD, FLA. –
The 4-year review found that 21% of older patients with a confirmed C-spine fracture reported no pain on history or physical exam, and that 76% of these fractures needed treatment – twin findings suggesting that asymptomatic neck fractures may be undiagnosed and untreated in this population.
The 183-patient study also found no significant pain differences between age groups: The silent injuries were just as common among 55-year-olds as among 65-year-olds, Christopher Healey, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“I guess we can say, ‘55 is the new 65,’ when it comes to this injury,” said Dr. Healey of the Iowa Methodist Medical Center, Des Moines. “In our study the rate of a pain-free neck fracture in 55-year-olds was equivalent to that in their older counterpoints, so they also represent a group at increased risk.”
Dr. Healey and his colleagues conducted a 4-year review of trauma patients aged 55 years and older who were treated for a C-spine fracture. All of the patients had a Glasgow Coma
There were no differences in the mechanism of injury between the pain-free and pain-positive groups. The most common source of injury was a fall from the person’s own height (39% vs. 49%), followed by a high fall (21% vs. 19%). Motor vehicle crashes came in third (37% vs. 30%), with other methods of injury accounting for about 3% of each group.
The level of fracture was widely dispersed among both groups and not associated with pain. Asymptomatic fractures occurred at C1 and C2 vertabrae (about 13%); C3 (26%); C4 (10%); C5 (12%); and C6 and C7 (30%).
Perhaps surprisingly, patients who didn’t report pain had significantly higher Injury Severity Scores (15 vs. 10). They were also significantly more likely to have injuries to other body regions (71% vs. 47%), including the head (22% vs. 16%), thorax/abdomen (39% vs. 20%), and extremities (33% vs. 20%).
A third of those with a pain-free fracture had breaks at multiple levels. Their hospital stays were significantly longer than were those of patients with painful fractures (7 vs. 5 days).
These findings of more severe injuries in the asymptomatic group led Dr. Healey to suggest that distracting pain might be playing a part in the phenomenon.
When the group was split into 10-year age increments, asymptomatic fractures occurred in about 20% of each group from 55-64 years up to 85 years and older.
The majority of both groups required treatment (91% with pain; 76% asymptomatic), Dr. Healey said. The most common treatment in each group was a cervical collar (61% with pain vs. 46% asymptomatic ). A cervical-thoracic-lumbar-sacral orthosis brace was used in 8% of those with pain and 11% of the asymptomatic patients.
Invasive procedures were performed in just as many of the asymptomatic patients as in those who had pain. These included vertebral fusion (11% asymptomatic vs. 9% with pain) and cervical halo (8% vs. 13%).
The lesson here, Dr. Healey concluded, is that pain is not always a reliable indicator of neck injury in older patients. “Older adults can break their neck and have no pain at all. This is concerning, because the presence or absence of neck pain is a major component in many clearance protocols for C-spine trauma. This begs the question whether we should be treating our older patients by general adult guidelines. I would advocate the development of trauma guidelines that are specific for the older or geriatric patient.”
Dr. Healey had no financial disclosures.
HOLLYWOOD, FLA. –
The 4-year review found that 21% of older patients with a confirmed C-spine fracture reported no pain on history or physical exam, and that 76% of these fractures needed treatment – twin findings suggesting that asymptomatic neck fractures may be undiagnosed and untreated in this population.
The 183-patient study also found no significant pain differences between age groups: The silent injuries were just as common among 55-year-olds as among 65-year-olds, Christopher Healey, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
“I guess we can say, ‘55 is the new 65,’ when it comes to this injury,” said Dr. Healey of the Iowa Methodist Medical Center, Des Moines. “In our study the rate of a pain-free neck fracture in 55-year-olds was equivalent to that in their older counterpoints, so they also represent a group at increased risk.”
Dr. Healey and his colleagues conducted a 4-year review of trauma patients aged 55 years and older who were treated for a C-spine fracture. All of the patients had a Glasgow Coma
There were no differences in the mechanism of injury between the pain-free and pain-positive groups. The most common source of injury was a fall from the person’s own height (39% vs. 49%), followed by a high fall (21% vs. 19%). Motor vehicle crashes came in third (37% vs. 30%), with other methods of injury accounting for about 3% of each group.
The level of fracture was widely dispersed among both groups and not associated with pain. Asymptomatic fractures occurred at C1 and C2 vertabrae (about 13%); C3 (26%); C4 (10%); C5 (12%); and C6 and C7 (30%).
Perhaps surprisingly, patients who didn’t report pain had significantly higher Injury Severity Scores (15 vs. 10). They were also significantly more likely to have injuries to other body regions (71% vs. 47%), including the head (22% vs. 16%), thorax/abdomen (39% vs. 20%), and extremities (33% vs. 20%).
A third of those with a pain-free fracture had breaks at multiple levels. Their hospital stays were significantly longer than were those of patients with painful fractures (7 vs. 5 days).
These findings of more severe injuries in the asymptomatic group led Dr. Healey to suggest that distracting pain might be playing a part in the phenomenon.
When the group was split into 10-year age increments, asymptomatic fractures occurred in about 20% of each group from 55-64 years up to 85 years and older.
The majority of both groups required treatment (91% with pain; 76% asymptomatic), Dr. Healey said. The most common treatment in each group was a cervical collar (61% with pain vs. 46% asymptomatic ). A cervical-thoracic-lumbar-sacral orthosis brace was used in 8% of those with pain and 11% of the asymptomatic patients.
Invasive procedures were performed in just as many of the asymptomatic patients as in those who had pain. These included vertebral fusion (11% asymptomatic vs. 9% with pain) and cervical halo (8% vs. 13%).
The lesson here, Dr. Healey concluded, is that pain is not always a reliable indicator of neck injury in older patients. “Older adults can break their neck and have no pain at all. This is concerning, because the presence or absence of neck pain is a major component in many clearance protocols for C-spine trauma. This begs the question whether we should be treating our older patients by general adult guidelines. I would advocate the development of trauma guidelines that are specific for the older or geriatric patient.”
Dr. Healey had no financial disclosures.
Key clinical point: Older trauma patients can present with pain-free cervical fractures. Major finding: Cervical fractures were asymptomatic in 21% of the study group; of these, 76% required treatment.
Data source: The 4-year retrospective study comprised 183 older patients with confirmed cervical fractures.
Disclosures: Dr. Healey had no financial disclosures.