User login
FDA Boxed Warning Updates: June 2018
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
VIDEX AND VIDEX EC (DIDANOSINE):
- Edited boxed warning, January 2018
WARNING: PANCREATITIS, LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS
Coadministration of VIDEX or VIDEX EC and stavudine is contraindicated because of increased risk of serious and/or life-threatening events. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occurs.
ZYDELIG (IDELALISIB):
- Edited boxed warning, January 2018
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, and INTESTINAL PERFORATION
Fatal and/or serious hepatotoxicity occurred in 16% to 18% of Zydelig-treated patients…
Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 20% of Zydelig-treated patients…
Fatal and/or serious infections occurred in 21% to 48% of Zydelig-treated patients…
AQUAMEPHYTON (PHYTONADIONE):
- Edited boxed warning, March 2018
WARNING: HYPERSENSITIVITY REACTIONS WITH INTRAVENOUS AND INTRAMUSCULAR USE
Fatal hypersensitivity reactions, including anaphylaxis, have occurred during and immediately after INTRAVENOUS and INTRAMUSCULAR injection of Aqua-MEPHYTON. Reactions have occurred despite dilution to avoid rapid infusion and upon first dose. Avoid the intravenous and intramuscular routes of administration unless the subcutaneous route is not feasible and the serious risk is justified.
FERAHEME (FERUMOXYTOL):
- Edited boxed warning, February 2018
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving feraheme. Initial symptoms may
include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
Only administer feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
METHADONE HYDROCHLORIDE, METHADOSE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, is a risk factor for respiratory depression and death.
Reserve concomitant prescribing of benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting daily methadone dosing.
DOLOPHINE HYDROCHLORIDE (METHADONE HYDROCHLORIDE):
- Edited boxed warning, February 2018
RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
Reserve concomitant prescribing of DOLOPHINE Tablets and benzodiazepines or other CNS depressants for use in patients for whom alternatives to benzodiazepines or other CNS depressants are inadequate.
Limit dosages and durations to the minimum required for patients being treated for pain.
Follow patients for signs and symptoms of respiratory depression and sedation. If the patient is visibly sedated, evaluate the cause of sedation, and consider delaying or omitting the daily methadone dose.
PARNATE (TRANYLCYPROMINE SULFATE):
- Edited boxed warning, January 2018
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS AND HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. PARNATE is not approved for use in pediatric patients.
HYPERTENSIVE CRISIS WITH SIGNIFICANT TYRAMINE USE
Excessive consumption of foods or beverages with significant tyramine content or the use of certain drugs with PARNATE or after PARNATE discontinuation can precipitate hypertensive crisis. Monitor blood pressure and allow for medication-free intervals between administration of PARNATE and interacting drugs. Instruct patients to avoid ingestion of foods and beverages with high tyramine content.
OCALIVA (OBETICHOLIC ACID):
- New boxed warning/Newly added section, February 2018
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended.
The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event.
HIV Update: Which Single-Tablet Regimens, and When (FULL)
CASE: James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.

A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 Years Have Changed Everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the U.S. Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer halflife than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of singletablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of Antiretroviral Therapy
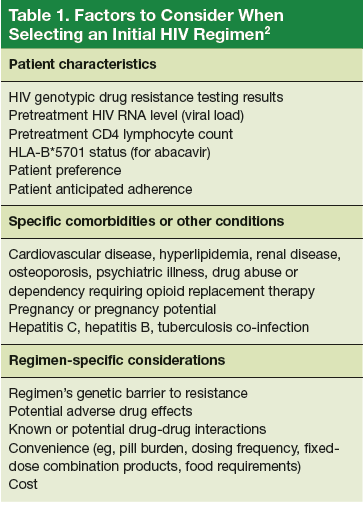
The U.S. Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see Table 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.
The DHHS guidelines divide treatment options into 3 categories2:
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently Recommended First-Line Therapies
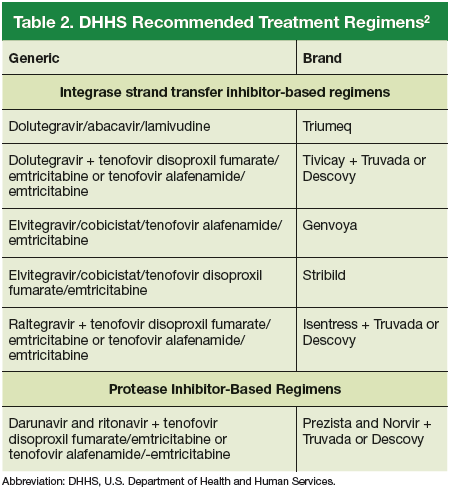
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (Table 2).2
INSTI-Based Regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥ 2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
- Dolutegravir plus tenofovir disoproxil fumarate/ emtricitabine (Tivicay plus Truvada). The combination
of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12 - Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/ cobicistat/tenofovir alafenamide/emtricitabinethat was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
- Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15 Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
- Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/ tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. In May 2017, FDA approved a new 1,200 mg once-daily version of raltegravir as an alternative to the twice daily regimen.20 Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-Based Regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and selflimiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in 2018, it will provide yet another daily STR option.23
The Addition of Fixed-Dose Tenofovir Alafenamide/Emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS "Alternative" And "Other" Regimens
The DHHS guidelines also include “alternative” (Table 3) and “other” regimens (available at http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not.2 These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice. 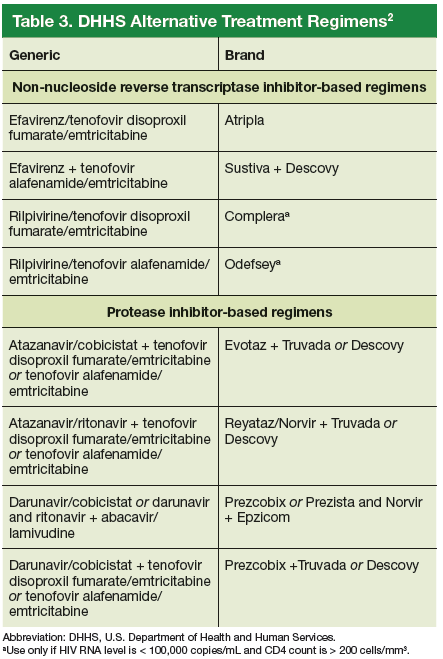
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The Future of Art
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE: At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
Click here to read the digital edition.
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single- tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drug/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatmentnaïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://http://prescribersletter.therapeuticresearch.com/ce/documents/ce_15215-40.pdf. Accessed May 31, 2017.
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
CASE: James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.
A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 Years Have Changed Everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the U.S. Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer halflife than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of singletablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of Antiretroviral Therapy
The U.S. Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see Table 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.
The DHHS guidelines divide treatment options into 3 categories2:
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently Recommended First-Line Therapies
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (Table 2).2
INSTI-Based Regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥ 2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
- Dolutegravir plus tenofovir disoproxil fumarate/ emtricitabine (Tivicay plus Truvada). The combination
of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12 - Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/ cobicistat/tenofovir alafenamide/emtricitabinethat was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
- Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15 Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
- Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/ tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. In May 2017, FDA approved a new 1,200 mg once-daily version of raltegravir as an alternative to the twice daily regimen.20 Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-Based Regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and selflimiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in 2018, it will provide yet another daily STR option.23
The Addition of Fixed-Dose Tenofovir Alafenamide/Emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS "Alternative" And "Other" Regimens
The DHHS guidelines also include “alternative” (Table 3) and “other” regimens (available at http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not.2 These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice.
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The Future of Art
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE: At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
Click here to read the digital edition.
CASE: James G, age 43, recently had blood work performed for a life insurance policy, and his human immunodeficiency virus (HIV) test came back positive. At a follow-up office visit, Mr. G reports having anonymous male sexual partners when traveling to New York on business and rarely using condoms. His last HIV test was “about 4 years ago.” He is otherwise in good health, takes no regular medications, and is not married.
Having recently completed a primary care CME program on HIV disease, you order a CD4/T-cell count, an HIV RNA (viral load) test, and an HIV genotype drug resistance test on Mr. G, along with other baseline lab work, including a complete blood count, chemistry panel, and hepatitis panel. You schedule a follow-up visit with Mr. G in 2 weeks when all of the lab results will be available so that you can discuss his plan of care.
A diagnosis of HIV has moved from being a fatal disease to that of a chronic condition that can be effectively managed with combination antiretroviral therapy (ART) regimens over an almost normal lifespan. As a result, the role of the primary care practitioner in the ongoing care of patients with HIV has grown and will continue to do so, making knowledge of these drug combinations vital.
20 Years Have Changed Everything
Combination ART has existed since 1996 when the first protease inhibitors (PIs) were approved by the U.S. Food and Drug Administration (FDA). Prior to this, treatment was limited to mono or dual therapy with nucleoside reverse transcriptase inhibitors (NRTIs). These agents provided some short-term clinical benefit, but didn’t significantly improve patient survival and ultimately failed due to viral resistance.1
Since the approval of zidovudine (AZT) in 1987, the FDA has approved more than 25 drugs in 6 different classes for the treatment of HIV disease.2 These include the NRTIs, non-nucleoside reverse transcriptase inhibitors (NNRTIs), PIs, a fusion inhibitor (FI), a CCR5 antagonist, and, more recently, integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat and ritonavir, are used solely to improve or “boost” the pharmacokinetic profiles of several antiretroviral drugs.2
Most of these newer agents are more potent, have a higher genetic barrier to resistance, and a longer halflife than their predecessors. Moreover, many are less toxic and thus more tolerable than older drugs. With the progressive development and approval of singletablet regimens (STRs) that contain 3 or 4 drugs, the majority of patients with HIV in the United States now take just one pill per day to treat their infection, facilitating far greater medication adherence.
Initiation of Antiretroviral Therapy
The U.S. Department of Health and Human Services (DHHS) guidelines now recommend that all people infected with HIV, regardless of CD4 cell count, begin ART.2 The evidence for this recommendation comes largely from the START3 and TEMPRANO4 trials, which found that early initiation of ART significantly reduces morbidity and mortality associated with HIV. In addition, the HPTN 052 study concluded that early ART is associated with a 93% lower risk of viral transmission in serodiscordant heterosexual couples.5 The DHHS guidelines do note that when initiating ART, it is important to appropriately educate patients on the benefits of treatment and address strategies to optimize adherence.2 (For more on factors to consider when selecting an initial HIV regimen, see Table 1.2) On a case-by-case basis, ART may be deferred because of clinical and/or psychosocial factors, but it should never be withheld unless the risks clearly outweigh the benefits. Ideally, ART should be initiated as soon as possible after the initial diagnosis of HIV.
The DHHS guidelines divide treatment options into 3 categories2:
- Recommended regimens are backed by randomized controlled trials that show optimal and durable virologic efficacy, they have favorable tolerability and toxicity profiles, and they are easy to use.
- Alternative regimens have less or lower quality supporting data than recommended regimens. Although they are effective and may be optimal for certain individual patients, they have potential disadvantages and/or limitations in certain populations.
- Other regimens have limited supporting data, reduced virologic activity, a higher pill burden, more drug interactions, and greater toxicity.
Currently Recommended First-Line Therapies
An antiretroviral regimen for a treatment-naive patient should consist of 2 NRTIs in combination with a third active antiretroviral drug from one of 3 drug classes. These include: an INSTI, a boosted PI, or, in some situations, an NNRTI. The DHHS guidelines panel currently recommends 6 different ART combinations as first-line treatment in treatment-naive patients (Table 2).2
INSTI-Based Regimens
Dolutegravir/abacavir/lamivudine (Triumeq). Approved by the FDA as a single-tablet regimen in 2014, the combination of dolutegravir/abacavir/lamivudine has proven to be highly effective and well-tolerated in many clinical trials.6-9 However, before this regimen is started, patients must be screened for the HLA-B*5701 allele, which predicts hypersensitivity to abacavir.10 Assessing patients’ risk for cardiovascular disease is also advised because some data suggest that abacavir may increase the risk of cardiovascular events, although this remains controversial.2
Dolutegravir is generally well-tolerated with minimal adverse effects (≥ 2% incidence of headache and insomnia) and toxicity.11 Dolutegravir/abacavir/lamivudine should be taken 2 hours before or 6 hours after taking antacids or laxatives, sucralfate, and oral supplements with iron or calcium. However, it may be taken with calcium or iron supplements if it is also taken with food.11 Dolutegravir increases levels of metformin about 2-fold, so patients should not take more than 1000 mg/d of this oral hypoglycemic agent.11
- Dolutegravir plus tenofovir disoproxil fumarate/ emtricitabine (Tivicay plus Truvada). The combination
of dolutegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine is administered as 2 pills per day. Because tenofovir disoproxil fumarate can cause proximal renal tubular dysfunction, phosphate wasting, and decreased bone mineral density (BMD), avoid prescribing it for patients with underlying renal dysfunction (creatinine clearance [CrCl] <50 mL/min) and prescribe it cautiously for patients with hypertension or diabetes who are at increased risk of renal disease. Emtricitabine is generally safe and well tolerated, but the dose should be reduced in patients with renal insufficiency, which would preclude the use of this fixed-dose combination.12 - Elvitegravir/cobicistat/tenofovir alafenamide/emtricitabine (Genvoya). The newer 4-drug combination of elvitegravir/ cobicistat/tenofovir alafenamide/emtricitabinethat was approved by the FDA in November 2015,13 contains the more recently approved form of tenofovir, which can be used in patients who have a CrCl as low as 30 mL/min. Compared to formulations containing tenofovir disoproxil fumarate, the newer tenofovir alafenamide formulation achieves higher intracellular levels in CD4 lymphocytes (but not in renal tubular cells). This allows for a lower dose of the drug and a smaller tablet size with co-formulation. It does not appear to cause kidney problems or loss of BMD as can be seen with tenofovir disoproxil fumarate.14 This newer single-tablet regimen may be best suited for older patients with HIV or those with comorbidities such as hypertension or diabetes.
- Elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine (Stribild). The FDA approved the combination of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine as a single-tablet regimen in 2012. The integrase inhibitor, elvitegravir, requires boosting with the CYP3A inhibitor, cobicistat, and should be taken with food.15 Two clinical trials demonstrated the superior efficacy of elvitegravir compared to a boosted PI and NNRTI-based regimen.16,17 Elvitegravir is generally well tolerated, but sometimes causes dyspepsia, nausea, or diarrhea.15 Similar to dolutegravir, it should not be taken concurrently with certain supplements—in this case, those containing aluminum, calcium, iron, magnesium, or zinc.15 Because it contains tenofovir disoproxil fumarate as an active agent, it should not be used in patients with a CrCl of <70 mL/min.15 Cobicistat inhibits tubular secretion of creatinine, so it may produce an elevation in serum creatinine without actually affecting glomerular function. Cobicistat may also cause drug-drug interactions with certain antiarrhythmics, sedative-hypnotics, and erectile dysfunction agents, and is contraindicated with some statins, anticonvulsants, and ergot derivatives.18
- Raltegravir plus tenofovir disoproxil fumarate/emtricitabine (Isentress plus Truvada). The combination of the integrase inhibitor raltegravir plus fixed-dose tenofovir disoproxil fumarate and emtricitabine has been recommended by the DHHS as first-line therapy for approximately 5 years. The recommendation is based mainly on data from the STARTMRK trial, a phase III non-inferiority trial that followed more than 500 patients for 5 years and concluded that raltegravir/ tenofovir/emtricitabine has superior efficacy with fewer drug-related adverse effects than efavirenz/tenofovir/emtricitabine.19 The overall pill burden with this regimen is 3 tablets per day. Although highly effective, the main drawbacks of raltegravir are that it must be dosed twice daily (which may be less preferable if adherence is a concern) and the genetic barrier to resistance is lower than that of the other 2 approved integrase inhibitors. In May 2017, FDA approved a new 1,200 mg once-daily version of raltegravir as an alternative to the twice daily regimen.20 Adverse effects and toxicities (except the renal and bone effects due to tenofovir disoproxil fumarate mentioned earlier) and drug interactions with this regimen are infrequent. Raltegravir can be taken with or without food. Concurrent use of antacids that contain aluminum or magnesium may reduce absorption of raltegravir and so should be avoided.21
PI-Based Regimen
Darunavir (Prezista) and ritonavir (Norvir) plus tenofovir disoproxil fumarate/emtricitabine (Truvada). PIs were once the key component of all ART regimens; however, boosted darunavir is now the only PI-based regimen currently recommended as first-line therapy. It is taken as 3 tablets once daily. If the co-formulation with cobicistat is used, just 2 tablets daily are required. One advantage with darunavir with either of the boosting agents is that it does not appear to cause insulin resistance or dyslipidemia as occurs with older PIs, such as indinavir and lopinavir.2 The boosting agents do, however, increase the likelihood of drug-drug interactions. As with all PIs, darunavir has a very high genetic barrier to resistance, which is important in patients for whom adherence is a concern.
Adverse effects of the PIs may include nausea, vomiting, and diarrhea, all of which are typically mild and selflimiting.22 Co-formulation of darunavir with cobicistat, tenofovir alafenamide, and emtricitabine is in phase III studies. Projected to be available in 2018, it will provide yet another daily STR option.23
The Addition of Fixed-Dose Tenofovir Alafenamide/Emtricitabine
In July 2016, the DHHS panel made some additions to their guidelines to reflect the FDA approval of 3 fixed-dose combination products that contain tenofovir alafenamide. Specifically, the combination of tenofovir alafenamide and emtricitabine is recommended for use with the integrase inhibitors—dolutegravir or raltegravir. It is also recommended in combination with ritonavir-boosted darunavir.
DHHS "Alternative" And "Other" Regimens
The DHHS guidelines also include “alternative” (Table 3) and “other” regimens (available at http://aidsinfo.nih.gov/guidelines) that may be used when first-line regimens may not.2 These second-line options are very effective, but have some possible clinical disadvantages or limitations. They are also less well supported by data from clinical trials. However, in certain situations, depending on an individual patient’s comorbidities, inability to tolerate one of the preferred regimens, or personal preferences, an alternative regimen may be the optimal choice.
Under the category of alternative regimens, the panel has included tenofovir alafenamide and emtricitabine in combination with the NNRTI efavirenz or with ritonavir or cobicistat-boosted atazanavir or darunavir.
The third group or “other” regimens have reduced virologic activity, increased toxicity, and even more limited data from clinical trials. Generally, medications from the DHHS “alternative” and “other” categories should be prescribed in consultation with an HIV specialist.
The Future of Art
The currently available drugs are highly effective in fully suppressing HIV and allowing for immune recovery and clinical stability for most patients. Life expectancy for patients living with HIV is estimated to be approaching that of uninfected adults—provided they remain on ART.24 As a way to further simplify ART, current clinical trials are looking at 2-drug regimens including an integrase inhibitor with an NRTI, an INSTI, or an NNRTI, or a PI with one NRTI.25,26 This approach could further reduce pill burden and toxicity and substantially decrease the cost of long-term treatment.27 Also on the horizon are long-acting injectable antiretroviral drugs that will likely be available for clinical use in the next 2 to 3 years.28,29
CASE: At the 2-week follow-up visit, you discuss with Mr. G that his CD4+ count is 390 cells/mm3, his HIV RNA level is 32,450 copies/mL, and his HIV genotype test showed no antiviral drug resistance. Explaining that all patients with HIV should be treated with antiviral therapy regardless of CD4+ count, you recommend that Mr. G begin taking fixed-dose tenofovir disoproxil fumarate/emtricitabine/elvitegravir/cobicistat (Stribild), noting that it is one of the regimens recommended by the DHHS national treatment guidelines. You provide a patient handout that discusses dosing and adverse effects, including nausea and headache. The patient’s pharmacy was contacted and it was determined that Mr. G’s co-pay for the drug would be $50, which he found acceptable.
In addition, you discuss the importance of good adherence to this medication, and instruct Mr. G to contact the office via phone or patient portal for any concerns or questions that arise after starting the medication. Lastly, you advise him to return in 4 weeks for follow-up blood testing, including viral load monitoring, and additional care, if needed, and strongly recommend that he begin using condoms regularly.
Click here to read the digital edition.
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single- tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drug/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatmentnaïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://http://prescribersletter.therapeuticresearch.com/ce/documents/ce_15215-40.pdf. Accessed May 31, 2017.
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
1. Concorde: MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343:871-881.
2. Department of Health and Human Services. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-treatment-guidelines/0. Accessed July 17, 2016.
3. The INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
4. The TEMPRANO ANRS 12136 Study Group. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373:808-822.
5. Cohen MS, Chen YQ, McCauley M, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375:830-839.
6. Molina JM, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomized, open-label, phase 3b study. Lancet HIV. 2015;2:e127-136.
7. Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807-1818.
8. Van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomized, phase 2b trial. Lancet Infect Dis. 2012;12:111-118.
9. Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013; 27:1771-1778.
10. Mallal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568-579.
11. AIDSinfo Drug Database. Dolutegravir. Available at: https://aidsinfo.nih.gov/drugs/509/dolutegravir/0/professional. Accessed July 17, 2016.
12. AIDSinfo Drug Database. Emtricitabine. Available at: https://aidsinfo.nih.gov/drugs/208/emtricitabine/0/patient. Accessed July 17, 2016.
13. AIDSinfo Drug Database. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide fumarate. Available at: https://aidsinfo.nih.gov/drugs/553/genvoya/0/professional. Accessed July 17, 2016.
14. Ray AS, Fordyce MW, Hitchcock, MJM. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016;125:63-70.
15. AIDSinfo Drug Database. Elvitegravir. https://aidsinfo.nih.gov/drugs/421/elvitegravir/0/professional
16. Wohl DA, Cohen C, Gallant JE, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e118-120.
17. Clumeck N, Molina JM, Henry K, et al. A randomized, double-blind comparison of single- tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65:e121-124.
18. AIDSinfo Drug Database. Cobicistat. Available at: https://aidsinfo.nih.gov/drug/537/evotaz/0/patient/. Accessed July 17, 2016.
19. Rockstroh JK, DeJesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatmentnaïve HIV-1 infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63:77-85.
20. Cahn P, Kaplan R, Sax P, et al. Raltegravir (RAL) 1200 mg once daily (QD) is non-inferior to RAL 400 mg twice daily (BID), in combination with tenofovir/emtricitabine, in treatment-naive HIV-1-infected subjects: week 48 results. Abstract FRAB0103LB presented at: 21st International AIDS Conference; July 18-22, 2016; Durban, South Africa.
21. Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48:931-939.
22. Prescriber’s Letter. HIV/AIDS Pharmacotherapy Review. Vol. 2015; Course no. 215. Available at: http://http://prescribersletter.therapeuticresearch.com/ce/documents/ce_15215-40.pdf. Accessed May 31, 2017.
23. AIDSinfo Drug Database. Tenofovir alafenamide. Available at: https://aidsinfo.nih.gov/drugs/514/tenofovir-alafenamide/0/patient. Accessed September 27, 2016.
24. Marcus JL, Chao C, Leyden W, et al. Narrowing the gap in life expectancy for HIV+ compared with HIV- individuals. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016, Boston. Abstract 54.
25. Gubavu C, Prazuck T, Niang M, et al. Dolutegravir-based monotherapy or dual therapy maintains a high proportion of viral suppression even in highly experienced HIV-1-infected patients. J Antimicrob Chemother. 2016;71:1046-1050.
26. Margolis DA, Brinson CC, Smith GHR. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral naïve adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15:1145-1155.
27. Girouard MP, Sax PE, Parker RA, et al. The cost-effectiveness and budget impact of 2-drug dolutegravir-lamivudine regimens for the treatment of HIV infection in the United States. Clin Infect Dis. 2016; 62:784-791.
28. Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, et al. Cabotegravir + rilpivirine as long-acting maintenance therapy: LATTE-2 week 32 results. Abstract number 31 LB. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
29. Murray MI, Markowitz M, Frank I, et al. Tolerability and acceptability of cabotegravir LA injection: results from ECLAIR study. Abstract number 471. Conference on Retroviruses and Opportunistic Infections. February 22-25, 2016; Boston, MA.
Criteria for Use Updates for Enzalutamide, Daratumumab, Elotuzumab, Carfilzomib, and Ixazomib (FULL)
VA Pharmacy Benefit Management Service (PBM) continually issues or revises its guidances for hematology and oncology care providers on a number of cancer care medications. Below are excerpts from recently released Criteria for Use documents. The complete documents, including the inclusion criteria, dosage and administration guidance, monitoring information, and discontinuation criteria should be consulted and can be found at www.pbm.va.gov or vaww.cmopnational.va.gov/cmop/PBM/default.aspx.
ENZALUTAMIDE (XTANDI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive enzalutamide.
- Brain metastases or active epidural disease
- Severe renal impairment (creatinine clearance < 30 mL/min)
- History of seizure (including febrile seizure, loss of consciousness, or transient ischemic attack within the previous 12 months, any condition predisposing to seizure: prior stroke, brain AV malformation, head trauma with loss of consciousness requiring hospitalization)
- ECOG Performance Status > 2
- Inability to swallow capsules
Issues for Consideration
- Enzalutamide is not indicated for use in women. Based on the mechanism of action, can cause fetal harm if used during pregnancy. Pregnancy Category X—use contraindicated during pregnancy. Exclude pregnancy before prescribing enzalutamide, discuss risks if pregnancy occurs, and provide contraceptive counseling.
- Use in patients taking concomitant medications that may lower the seizure threshold was not studied; caution patients about the risk of activities where the sudden loss of consciousness could cause serious harm if concomitant use cannot be avoided.
- Use in patients at risk for or with a strong history of falls: in the phase 3 clinical trial, falls or injuries from falls occurred in 4.6% of enzalutamide patients vs 1.3% of placebo patients.
- Avoid strong inhibitors of CYP2C8 (eg, gemfibrozil); if concomitant use of a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of enzalutamide to 80 mg once daily according to the package insert.
- Co-administration with strong or moderate inducers of CYP3A4 (eg, carbamazepine, phenobarbital, phenytoin, rifampin, bosentan, efavirenz, modafinil, nafcillin, St. John’s Wort) or CYP2C8 (eg, rifampin) should be avoided if possible. If patient must be co-administered a strong CYP3A4 inducer, increase enzalutamide dose from 160 mg to 240 mg once daily.
- Drugs that are substrates of CYP3A4 (eg, alfentanil, cyclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus), CYP2C9 (eg, phenytoin, warfarin), or CYP2C19 (eg, S-mephenytoin) with a narrow therapeutic index should be avoided. If enzalutamide is co-administered with warfarin, additional INR testing should be conducted.
- Use in patients with hepatic impairment: Pharmacokinetics of enzalutamide and its metabolite were examined in volunteers with normal, Child-Pugh Class A, Child-Pugh Class B, and Child-Pugh Class C hepatic impairment. The composite AUC for enzalutamide and its metabolite after a single 160-mg dose was similar across all levels of hepatic impairment compared with normal volunteers.
- There have been postmarketing reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving enzalutamide. PRES is a neurologic disorder presenting with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual/neurological disturbances with or without associated hypertension. Diagnosis of PRES requires brain imaging, preferably by MRI. Enzalutamide should be discontinued in patients developing PRES.
- Sequencing of enzalutamide and abiraterone has been evaluated in several small retrospective analyses; the majority of the analyses are in the post chemotherapy setting. From this limited observational data, it is unclear if there is a preferred sequencing of abiraterone and enzalutamide. There is some evidence for cross-resistance. There are ongoing investigations into mechanisms of resistance to enzalutamide and abiraterone.
DARATUMUMAB (DARZALEX) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive daratumumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient unable or unwilling to be observed an extended period of time that may be necessary for first infusion (refer to Issues for Consideration)
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 3x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin 2x ULN) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
Issues for Consideration
- Drug infusion time will be dependent upon patient tolerance and exposure to daratumumab. Median duration of the first infusion was ~ 7 hours in the SIRIUS trial, followed by infusion times of 4.2 and 3.4 hours, subsequently.
- Type and screen patients shortly prior to starting treatment. When the sample is provided to the blood bank, inform them that the patient will be receiving daratumumab.
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, C, or HIV were excluded from clinical trials with daratumumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections was slightly higher in the daratumumab-treated arms of the comparative studies. Use of daratumumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
ELOTUZUMAB (EMPLICITI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive elotuzumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient is not a candidate for lenalidomide therapy (ie, is lenalidomide-refractory or possesses contraindications to therapy)
- Patient is not a candidate for high-dose dexamethasone therapy
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 2x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin > 2 mg/dL) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or hepatitis C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
- Patient intends to breastfeed during therapy
Issues for Consideration
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, hepatitis C, or HIV were excluded from clinical trials with elotuzumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections (OI, fungal, viral) was greater in the elotuzumab arm vs control arm of the comparative clinical trial. Use of elotuzumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
- Disappointing response rates as monotherapy in the relapsed/refractory setting suggest that elotuzumab should be given in combination with lenalidomide and dexamethasone.
- Impact of elotuzumab/lenalidomide/dexamethasone on overall survival is not known as these data were not mature at the time ELOQUENT-2 was published.
CARFILZOMIB (KYPROLIS) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive carfilzomib.
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 g/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 1.5 x the upper limit of the normal range or ALT and AST > 3 x ULN
- NYHA Class III and IV heart failure (refer to Issues for Consideration; those at risk for cardiac failure and ischemia were also excluded from clinical trials)
- LVEF < 40%
- Uncontrolled hypertension
- Grade 3 or 4 peripheral neuropathy
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV
Issues for Consideration
- A significant percentage of patients on carfilzomib develop dyspnea. This drug should be used with caution in patients with underlying lung disease. Close monitoring for worsening of dyspnea is advised.
- Risk of cardiac failure increases in those aged > 75 years; those with NYHA Class III and IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications. Refer to Prescribing Information for management recommendations.
- Those aged > 75 years experienced greater toxicity than their younger counterparts.
- Patients on dialysis: administer carfilzomib after the dialysis procedure.
- Phase III evidence in heavily pretreated relapsed/refractory patients (median 5 prior regimens) of carfilzomib vs low-dose steroids ± cyclophosphamide indicates that the median overall survival is not significantly different between these treatment arms.
IXAZOMIB (NILARO) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive ixazomib
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is not a candidate for lenalidomide or dexamethasone therapy
- Patient is refractory to lenalidomide or proteasomeinhibitor therapy (defined as disease progression while on treatment or within 60 days of last dose)
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 75,000/mm3
- ECOG Performance Status > 2
- Patient with CNS involvement
- Patient receiving concurrent therapy with a strong CYP3A inducer (ie, rifampin, phenytoin, carbamazepine) that cannot be discontinued
- Uncontrolled cardiovascular conditions, including uncontrolled hypertension, uncontrolled cardiac arrhythmias, symptomatic congestive heart failure, unstable angina or myocardial infarction within 6 months prior to start
- Ongoing or active systemic infection, including active hepatitis B, hepatitis C, or known HIV
Issues for Consideration
- Indirect comparisons of phase 3 data (KRd vs Rd and IRd vs Rd) show that in similar populations of pretreated relapsed, refractory myeloma patients, those receiving KRd experienced longer PFS (26.3 vs 21 months), greater CR (32% vs 12%), greater ORR (87% vs 78%) and longer duration of response (28.6 vs 20.5 months). Therefore, providers may want to consider using carfilzomib in those meeting its criteria for use.
- Ixazomib is cytotoxic. Capsules should not be opened or crushed. Waste should be considered hazardous.
- Avoid concomitant use of strong CYP3A inducers (rifampin, phenytoin, carbamazepine, and St. John’s Wort).
Click here to read the digital edition.
VA Pharmacy Benefit Management Service (PBM) continually issues or revises its guidances for hematology and oncology care providers on a number of cancer care medications. Below are excerpts from recently released Criteria for Use documents. The complete documents, including the inclusion criteria, dosage and administration guidance, monitoring information, and discontinuation criteria should be consulted and can be found at www.pbm.va.gov or vaww.cmopnational.va.gov/cmop/PBM/default.aspx.
ENZALUTAMIDE (XTANDI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive enzalutamide.
- Brain metastases or active epidural disease
- Severe renal impairment (creatinine clearance < 30 mL/min)
- History of seizure (including febrile seizure, loss of consciousness, or transient ischemic attack within the previous 12 months, any condition predisposing to seizure: prior stroke, brain AV malformation, head trauma with loss of consciousness requiring hospitalization)
- ECOG Performance Status > 2
- Inability to swallow capsules
Issues for Consideration
- Enzalutamide is not indicated for use in women. Based on the mechanism of action, can cause fetal harm if used during pregnancy. Pregnancy Category X—use contraindicated during pregnancy. Exclude pregnancy before prescribing enzalutamide, discuss risks if pregnancy occurs, and provide contraceptive counseling.
- Use in patients taking concomitant medications that may lower the seizure threshold was not studied; caution patients about the risk of activities where the sudden loss of consciousness could cause serious harm if concomitant use cannot be avoided.
- Use in patients at risk for or with a strong history of falls: in the phase 3 clinical trial, falls or injuries from falls occurred in 4.6% of enzalutamide patients vs 1.3% of placebo patients.
- Avoid strong inhibitors of CYP2C8 (eg, gemfibrozil); if concomitant use of a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of enzalutamide to 80 mg once daily according to the package insert.
- Co-administration with strong or moderate inducers of CYP3A4 (eg, carbamazepine, phenobarbital, phenytoin, rifampin, bosentan, efavirenz, modafinil, nafcillin, St. John’s Wort) or CYP2C8 (eg, rifampin) should be avoided if possible. If patient must be co-administered a strong CYP3A4 inducer, increase enzalutamide dose from 160 mg to 240 mg once daily.
- Drugs that are substrates of CYP3A4 (eg, alfentanil, cyclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus), CYP2C9 (eg, phenytoin, warfarin), or CYP2C19 (eg, S-mephenytoin) with a narrow therapeutic index should be avoided. If enzalutamide is co-administered with warfarin, additional INR testing should be conducted.
- Use in patients with hepatic impairment: Pharmacokinetics of enzalutamide and its metabolite were examined in volunteers with normal, Child-Pugh Class A, Child-Pugh Class B, and Child-Pugh Class C hepatic impairment. The composite AUC for enzalutamide and its metabolite after a single 160-mg dose was similar across all levels of hepatic impairment compared with normal volunteers.
- There have been postmarketing reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving enzalutamide. PRES is a neurologic disorder presenting with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual/neurological disturbances with or without associated hypertension. Diagnosis of PRES requires brain imaging, preferably by MRI. Enzalutamide should be discontinued in patients developing PRES.
- Sequencing of enzalutamide and abiraterone has been evaluated in several small retrospective analyses; the majority of the analyses are in the post chemotherapy setting. From this limited observational data, it is unclear if there is a preferred sequencing of abiraterone and enzalutamide. There is some evidence for cross-resistance. There are ongoing investigations into mechanisms of resistance to enzalutamide and abiraterone.
DARATUMUMAB (DARZALEX) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive daratumumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient unable or unwilling to be observed an extended period of time that may be necessary for first infusion (refer to Issues for Consideration)
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 3x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin 2x ULN) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
Issues for Consideration
- Drug infusion time will be dependent upon patient tolerance and exposure to daratumumab. Median duration of the first infusion was ~ 7 hours in the SIRIUS trial, followed by infusion times of 4.2 and 3.4 hours, subsequently.
- Type and screen patients shortly prior to starting treatment. When the sample is provided to the blood bank, inform them that the patient will be receiving daratumumab.
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, C, or HIV were excluded from clinical trials with daratumumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections was slightly higher in the daratumumab-treated arms of the comparative studies. Use of daratumumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
ELOTUZUMAB (EMPLICITI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive elotuzumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient is not a candidate for lenalidomide therapy (ie, is lenalidomide-refractory or possesses contraindications to therapy)
- Patient is not a candidate for high-dose dexamethasone therapy
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 2x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin > 2 mg/dL) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or hepatitis C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
- Patient intends to breastfeed during therapy
Issues for Consideration
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, hepatitis C, or HIV were excluded from clinical trials with elotuzumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections (OI, fungal, viral) was greater in the elotuzumab arm vs control arm of the comparative clinical trial. Use of elotuzumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
- Disappointing response rates as monotherapy in the relapsed/refractory setting suggest that elotuzumab should be given in combination with lenalidomide and dexamethasone.
- Impact of elotuzumab/lenalidomide/dexamethasone on overall survival is not known as these data were not mature at the time ELOQUENT-2 was published.
CARFILZOMIB (KYPROLIS) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive carfilzomib.
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 g/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 1.5 x the upper limit of the normal range or ALT and AST > 3 x ULN
- NYHA Class III and IV heart failure (refer to Issues for Consideration; those at risk for cardiac failure and ischemia were also excluded from clinical trials)
- LVEF < 40%
- Uncontrolled hypertension
- Grade 3 or 4 peripheral neuropathy
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV
Issues for Consideration
- A significant percentage of patients on carfilzomib develop dyspnea. This drug should be used with caution in patients with underlying lung disease. Close monitoring for worsening of dyspnea is advised.
- Risk of cardiac failure increases in those aged > 75 years; those with NYHA Class III and IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications. Refer to Prescribing Information for management recommendations.
- Those aged > 75 years experienced greater toxicity than their younger counterparts.
- Patients on dialysis: administer carfilzomib after the dialysis procedure.
- Phase III evidence in heavily pretreated relapsed/refractory patients (median 5 prior regimens) of carfilzomib vs low-dose steroids ± cyclophosphamide indicates that the median overall survival is not significantly different between these treatment arms.
IXAZOMIB (NILARO) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive ixazomib
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is not a candidate for lenalidomide or dexamethasone therapy
- Patient is refractory to lenalidomide or proteasomeinhibitor therapy (defined as disease progression while on treatment or within 60 days of last dose)
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 75,000/mm3
- ECOG Performance Status > 2
- Patient with CNS involvement
- Patient receiving concurrent therapy with a strong CYP3A inducer (ie, rifampin, phenytoin, carbamazepine) that cannot be discontinued
- Uncontrolled cardiovascular conditions, including uncontrolled hypertension, uncontrolled cardiac arrhythmias, symptomatic congestive heart failure, unstable angina or myocardial infarction within 6 months prior to start
- Ongoing or active systemic infection, including active hepatitis B, hepatitis C, or known HIV
Issues for Consideration
- Indirect comparisons of phase 3 data (KRd vs Rd and IRd vs Rd) show that in similar populations of pretreated relapsed, refractory myeloma patients, those receiving KRd experienced longer PFS (26.3 vs 21 months), greater CR (32% vs 12%), greater ORR (87% vs 78%) and longer duration of response (28.6 vs 20.5 months). Therefore, providers may want to consider using carfilzomib in those meeting its criteria for use.
- Ixazomib is cytotoxic. Capsules should not be opened or crushed. Waste should be considered hazardous.
- Avoid concomitant use of strong CYP3A inducers (rifampin, phenytoin, carbamazepine, and St. John’s Wort).
Click here to read the digital edition.
VA Pharmacy Benefit Management Service (PBM) continually issues or revises its guidances for hematology and oncology care providers on a number of cancer care medications. Below are excerpts from recently released Criteria for Use documents. The complete documents, including the inclusion criteria, dosage and administration guidance, monitoring information, and discontinuation criteria should be consulted and can be found at www.pbm.va.gov or vaww.cmopnational.va.gov/cmop/PBM/default.aspx.
ENZALUTAMIDE (XTANDI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive enzalutamide.
- Brain metastases or active epidural disease
- Severe renal impairment (creatinine clearance < 30 mL/min)
- History of seizure (including febrile seizure, loss of consciousness, or transient ischemic attack within the previous 12 months, any condition predisposing to seizure: prior stroke, brain AV malformation, head trauma with loss of consciousness requiring hospitalization)
- ECOG Performance Status > 2
- Inability to swallow capsules
Issues for Consideration
- Enzalutamide is not indicated for use in women. Based on the mechanism of action, can cause fetal harm if used during pregnancy. Pregnancy Category X—use contraindicated during pregnancy. Exclude pregnancy before prescribing enzalutamide, discuss risks if pregnancy occurs, and provide contraceptive counseling.
- Use in patients taking concomitant medications that may lower the seizure threshold was not studied; caution patients about the risk of activities where the sudden loss of consciousness could cause serious harm if concomitant use cannot be avoided.
- Use in patients at risk for or with a strong history of falls: in the phase 3 clinical trial, falls or injuries from falls occurred in 4.6% of enzalutamide patients vs 1.3% of placebo patients.
- Avoid strong inhibitors of CYP2C8 (eg, gemfibrozil); if concomitant use of a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of enzalutamide to 80 mg once daily according to the package insert.
- Co-administration with strong or moderate inducers of CYP3A4 (eg, carbamazepine, phenobarbital, phenytoin, rifampin, bosentan, efavirenz, modafinil, nafcillin, St. John’s Wort) or CYP2C8 (eg, rifampin) should be avoided if possible. If patient must be co-administered a strong CYP3A4 inducer, increase enzalutamide dose from 160 mg to 240 mg once daily.
- Drugs that are substrates of CYP3A4 (eg, alfentanil, cyclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus), CYP2C9 (eg, phenytoin, warfarin), or CYP2C19 (eg, S-mephenytoin) with a narrow therapeutic index should be avoided. If enzalutamide is co-administered with warfarin, additional INR testing should be conducted.
- Use in patients with hepatic impairment: Pharmacokinetics of enzalutamide and its metabolite were examined in volunteers with normal, Child-Pugh Class A, Child-Pugh Class B, and Child-Pugh Class C hepatic impairment. The composite AUC for enzalutamide and its metabolite after a single 160-mg dose was similar across all levels of hepatic impairment compared with normal volunteers.
- There have been postmarketing reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving enzalutamide. PRES is a neurologic disorder presenting with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual/neurological disturbances with or without associated hypertension. Diagnosis of PRES requires brain imaging, preferably by MRI. Enzalutamide should be discontinued in patients developing PRES.
- Sequencing of enzalutamide and abiraterone has been evaluated in several small retrospective analyses; the majority of the analyses are in the post chemotherapy setting. From this limited observational data, it is unclear if there is a preferred sequencing of abiraterone and enzalutamide. There is some evidence for cross-resistance. There are ongoing investigations into mechanisms of resistance to enzalutamide and abiraterone.
DARATUMUMAB (DARZALEX) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive daratumumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient unable or unwilling to be observed an extended period of time that may be necessary for first infusion (refer to Issues for Consideration)
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 3x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin 2x ULN) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
Issues for Consideration
- Drug infusion time will be dependent upon patient tolerance and exposure to daratumumab. Median duration of the first infusion was ~ 7 hours in the SIRIUS trial, followed by infusion times of 4.2 and 3.4 hours, subsequently.
- Type and screen patients shortly prior to starting treatment. When the sample is provided to the blood bank, inform them that the patient will be receiving daratumumab.
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, C, or HIV were excluded from clinical trials with daratumumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections was slightly higher in the daratumumab-treated arms of the comparative studies. Use of daratumumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
ELOTUZUMAB (EMPLICITI) Criteria for Use, January 2017
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive elotuzumab.
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Patient is not a candidate for lenalidomide therapy (ie, is lenalidomide-refractory or possesses contraindications to therapy)
- Patient is not a candidate for high-dose dexamethasone therapy
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 gm/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 2x the upper limit of the normal range (except for Gilbert syndrome: direct bilirubin > 2 mg/dL) or ALT and AST > 3x ULN
- NYHA Class III or IV heart failure (refer to Issues for Consideration)
- Ongoing or active systemic infection, including active hepatitis B or hepatitis C, or known HIV (refer to Issues for Consideration)
- Positive pregnancy test
- Patient intends to breastfeed during therapy
Issues for Consideration
- Those with NYHA Class III or IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications.
- Patients with active hepatitis B, hepatitis C, or HIV were excluded from clinical trials with elotuzumab, therefore safety and efficacy data are unknown in these patient populations. The risk of infections (OI, fungal, viral) was greater in the elotuzumab arm vs control arm of the comparative clinical trial. Use of elotuzumab should only be considered in those with well-controlled hepatitis B, hepatitis C, or HIV.
- Disappointing response rates as monotherapy in the relapsed/refractory setting suggest that elotuzumab should be given in combination with lenalidomide and dexamethasone.
- Impact of elotuzumab/lenalidomide/dexamethasone on overall survival is not known as these data were not mature at the time ELOQUENT-2 was published.
CARFILZOMIB (KYPROLIS) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive carfilzomib.
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is noncompliant with medication, follow-up, or laboratory appointments
- Hemoglobin < 8 g/dL; Must transfuse to hemoglobin above 8 g/dL prior to therapy initiation
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 50,000/mm3 (< 30,000/mm3 if myeloma involvement in bone marrow > 50%)
- ECOG Performance Status > 2
- Total bilirubin > 1.5 x the upper limit of the normal range or ALT and AST > 3 x ULN
- NYHA Class III and IV heart failure (refer to Issues for Consideration; those at risk for cardiac failure and ischemia were also excluded from clinical trials)
- LVEF < 40%
- Uncontrolled hypertension
- Grade 3 or 4 peripheral neuropathy
- Ongoing or active systemic infection, including active hepatitis B or C, or known HIV
Issues for Consideration
- A significant percentage of patients on carfilzomib develop dyspnea. This drug should be used with caution in patients with underlying lung disease. Close monitoring for worsening of dyspnea is advised.
- Risk of cardiac failure increases in those aged > 75 years; those with NYHA Class III and IV heart failure, recent MI, conduction abnormalities, angina or arrhythmias uncontrolled by medications, were not eligible for clinical trials and may be at greater risk of cardiac complications. Refer to Prescribing Information for management recommendations.
- Those aged > 75 years experienced greater toxicity than their younger counterparts.
- Patients on dialysis: administer carfilzomib after the dialysis procedure.
- Phase III evidence in heavily pretreated relapsed/refractory patients (median 5 prior regimens) of carfilzomib vs low-dose steroids ± cyclophosphamide indicates that the median overall survival is not significantly different between these treatment arms.
IXAZOMIB (NILARO) Criteria for Use, December 2016
Exclusion Criteria: If the answer to ANY item below is met, then the patient should NOT receive ixazomib
- Care for the oncologic disease being treated not provided by a VA or VA purchased care (eg, Choice Program, Fee Basis) oncology provider
- Patient is not a candidate for lenalidomide or dexamethasone therapy
- Patient is refractory to lenalidomide or proteasomeinhibitor therapy (defined as disease progression while on treatment or within 60 days of last dose)
- Absolute neutrophil count (ANC) < 1,000/mm3
- Platelet count < 75,000/mm3
- ECOG Performance Status > 2
- Patient with CNS involvement
- Patient receiving concurrent therapy with a strong CYP3A inducer (ie, rifampin, phenytoin, carbamazepine) that cannot be discontinued
- Uncontrolled cardiovascular conditions, including uncontrolled hypertension, uncontrolled cardiac arrhythmias, symptomatic congestive heart failure, unstable angina or myocardial infarction within 6 months prior to start
- Ongoing or active systemic infection, including active hepatitis B, hepatitis C, or known HIV
Issues for Consideration
- Indirect comparisons of phase 3 data (KRd vs Rd and IRd vs Rd) show that in similar populations of pretreated relapsed, refractory myeloma patients, those receiving KRd experienced longer PFS (26.3 vs 21 months), greater CR (32% vs 12%), greater ORR (87% vs 78%) and longer duration of response (28.6 vs 20.5 months). Therefore, providers may want to consider using carfilzomib in those meeting its criteria for use.
- Ixazomib is cytotoxic. Capsules should not be opened or crushed. Waste should be considered hazardous.
- Avoid concomitant use of strong CYP3A inducers (rifampin, phenytoin, carbamazepine, and St. John’s Wort).
Click here to read the digital edition.
Reducing the Expenditures and Workload Associated With VA Partial-Fill Prescription Processing
According to the US Department of Veterans Affairs (VA) Pharmacy Benefits Management Service, about 80% of all outpatient prescriptions filled by the VA are sent to veterans by mail order, using the Centralized Mail Order Pharmacy (CMOP) network of highly automated pharmacies around the country.1 During fiscal year (FY) 2016, the 7 VA CMOP facilities throughout the US processed 119.7 million outpatient prescriptions. Each day, these CMOPs process nearly 470,000 prescriptions, an evidence of the efficiency provided through this mail-order service.1 The use of CMOP results in lower processing costs and increased convenience for veterans compared with filling prescriptions at pharmacies at individual VA facilities. Notably, VA CMOP has been rated “among the best” mail-order pharmacies in customer satisfaction according to the 2017 J.D. Power US Pharmacy Study.2
Background
Within the Fayetteville VA Medical Center (FVAMC) system in North Carolina, on-site patients receive a new prescription where an on-site pharmacy is available (at health care centers [HCC] and the medical center). For veterans seen at community-based outpatient clinics (CBOCs), emergent new prescriptions are filled through vouchers at contract community pharmacies, and nonemergent new prescriptions are filled by the CMOP (Figure 1). 
The appropriate use of the VA CMOP for refills is intended to allow on-site pharmacy staff to focus on providing customer service for veterans requesting medication counseling from a clinical pharmacist, as well as those with new, changing, or urgent prescription needs. Filling prescriptions through the CMOP also can help control the VA facility pharmacy budget.
Despite the established mail-order process, FVAMC staff noted a high volume of medication refill and partial-fill prescriptions being requested at the on-site pharmacy. When a veteran presented to a pharmacy requesting a medication refill, pharmacy staff members ordered a refill to be filled by the CMOP and provided a limited quantity, otherwise known as a partial fill, to serve as an emergency supply to supplement the veteran until the full quantity of the prescription arrived by mail. Partial fills also were completed for new prescriptions that veterans requested to pick up at the on-site pharmacy. For new 90-day supply prescriptions, pharmacy staff often filled a 30-day supply in addition to submitting the entire 90-day prescription through the CMOP, which led to an unnecessary increase in material expenditures and workload.Preliminary data noted the most frequently used partial-fill days’ supply to be 10 days. Due to the lack of partial-fill criteria, prescriptions of all classes, quantities, and days’ supplies were provided as partial fills. Prior to the implementation of this quality improvement (QI) project, there was no standard approach for how to handle these requests.
Partial fills do not provide copay reimbursement to the facility filling the prescription. In an effort to steward the funds provided to the FVAMC pharmacy department wisely, an evaluation was performed with reference to partial fills. During FY 2016, about $350,000 was spent on medications, materials, and workload associated with the partial filling of FVAMC prescriptions. With respect to unique individuals who were provided care over the course of FY 2015 and 2016, the number of unique patients served by local comparator hospitals increased by only 2%, while the number of unique patients served by FVAMC increased by 12%. This substantial growth in the number of veterans served places further emphasis on the necessity of stewarding the allotted pharmacy budget. Moreover, the excessive number of medication refill and partial-fill prescriptions filled at on-site VA pharmacies can contribute to increased wait times for veterans with urgent prescription needs.
In November 2016, FVAMC implemented an updated partial-fill guidance. Partial-fill process and refill education was provided for VA staff and veterans in an effort to allow all parties involved to use pharmacy services efficiently. This analysis reviewed the reduction in partial-fill expenditures with a secondary focus on workload expenditures following the execution of this education.
Methods
This project was deemed to be a QI project and did not require institutional review board approval. Implementation for this QI project began in November 2016. Baseline raw drug cost, number, and class of prescriptions partialed were retrospectively collected for a 90-day period prior to implementation using all available data. Postintervention data were collected for 90 days following the implementation phase to compare partial-fill expenditures and workload expenditures with baseline data.
Calculations
Materials included in the partial-fill expenditure calculation were prescription vials, prescription vial caps, and prescription labeling. Material cost per partial fill was determined by using the facility’s wholesaler acquisition unit costs to estimate a summed cost for an individual prescription vial and prescription vial cap. The estimated acquisition price of 7 prescription-labeling pages was used in the material calculation, as this was the average number of pages used when performing test partial fills.
The following equation was used to calculate total partial-fill expenditures for any specified time frame:
Total partial-fill expenditure = total raw drug cost + (material cost × number of partial fills)
In addition, the average personnel cost per partial-fill prescription was determined. Average workload expenditure per partial fill was calculated by filling a subset of 10 test prescriptions and multiplying the average time spent by an average of the general station (GS) rate for pharmacists and technicians. The average hourly rate of a GS-12 pharmacist was calculated based on an average of the 10 available pay grades within the GS-12 ranking. The average hourly rate of a GS-6 pharmacy technician was calculated based on an average of the 10 available pay grades within the GS-6 ranking.3 The average workload expenditures were calculated using the following equations:
Average pharmacist workload expenditure per partial fill = time (in hours) × average hourly rate.
Average technician workload expenditure per partial fill = time (in hours) × average hourly rate.
Partial-Fill Guidance
Updated partial-fill guidance was drafted designating acceptable prescriptions to be limited to those responsible for preventing hospitalization and treatment of acute illness. This guidance provided generalized examples of medication classes that could be acceptable for partial filling, though it was not intended to be an all-inclusive list. The guidance also noted examples of classes or groups that should not be partial filled for nonemergent reasons (vitamins, nonprescription items, antilipemics), as well as controlled substances. The refill-process education was reiterated throughout the entirety of the guidance. Specifically, if a pharmacy staff member was to perform a partial fill, a review explaining the appropriate refill process to the veteran also must be provided. If the medication was determined to be of emergent need and not yet transmitted for filling via the CMOP, the directive recommended to fill the entire quantity locally as a onetime fill.
If a onetime on-site fill was determined infeasible, partial-fill quantities were recommended to be limited to only a 7-day supply, and the full quantity filled through the CMOP. Anticipated mail wait time for CMOP prescription delivery was estimated to be less than 7 days based on experience, local pending queues, and guidance from the regional CMOP; however, time could vary among VA and CMOP facilities. Original prescriptions were to be filled for the entire quantity for the first fill at the on-site pharmacy if requested by the veteran. If the pharmacy had an insufficient quantity for an entire initial supply, it could then be partial filled for a 7-day supply and then filled through the CMOP.
The final portion of the partial-fill guidance pertained to the use of partial-fill justification codes. Prior to the execution of the partial-fill guidance, free text was entered into the comments field when processing a partial-fill prescription, as the prescription-processing system used requires a comment to proceed with the partial fill. The use of these codes served to streamline data collection in the postintervention phase and helped identify areas for further education following the close of this project.
Education
Education to the pharmacy staff was disseminated by various modalities, including in-person sessions and written and electronic correspondences. This written guidance was distributed to pharmacy staff by e-mail, the pharmacy newsletter (Rx-tra), signage posted throughout the outpatient pharmacies, and on the facility’s pharmacy Microsoft SharePoint (Redmond, WA) site. In-services provided during pharmacy staff meetings detailed information on the updated partial-fill guidance. A FVAMC Talent Management System (TMS) training module was developed and assigned to all pharmacy service staff to reiterate key points regarding this QI initiative (eAppendix).
Nonpharmacy staff were educated through staff and in-service meetings and e-mail correspondences. These in-services emphasized how nurses, medical support assistants, and health care providers (HCPs) could assist veterans by knowing the correct refill process, ensuring sufficient refills remained until the next appointment, and providing continual refill-process education.
Following implementation, all veterans receiving prescriptions through the on-site pharmacies in the FVAMC were provided a copy of the refill-process handout with each trip to the pharmacy. Nonpharmacy staff and HCPs also were provided this handout to distribute to patients. The intent of this handout was to clearly detail the various ways in which refills could be ordered and the time frame in which they should be ordered. The pharmacy became involved in new patient orientation classes for all veterans new to FVAMC. Digital signage and messaging was created and circulated throughout several of the FVAMC facilities.
Results
The results of calculations for material cost, personnel time spent, hourly employee rates, and average workload expenditure per partial fill are summarized in Table 1. 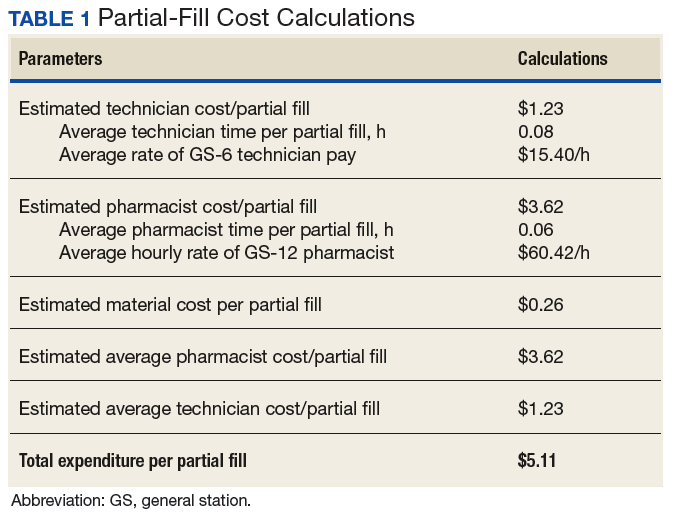
Following the implementation of partial-fill and refill-process education, there was a 54.3% decrease in the total number of partial fills from 5,596 in the 90 days prior to implementation, to 2,555 partials completed over the 90 days postimplementation. Regarding the primary objective, total partial-fill expenditures decreased from $52,015.44 to $44,063.01 (-15.3%). When dissecting the individual components of partial-fill expenditures, material expenditures decreased from $1,454.96 to $664.28 (-54.3%), and raw drug cost expenditures decreased from $50,596.48 to $43,398.73 (-14.3%). Workload expenditures also decreased from $27,140.60 to $12,391.75 (-54.3%).
Several points of descriptive information also were collected. The average days’ supply trended down from a mode of 10 days to 7 days. This reduction in days’ supply likely was seen because staff became more aware of the customary amount required to bridge the veteran until the CMOP supply arrived by mail. Postintervention data showed a 70% utilization of partial-fill reasoning codes. The reasons for partial filling of prescriptions are summarized in Table 2. 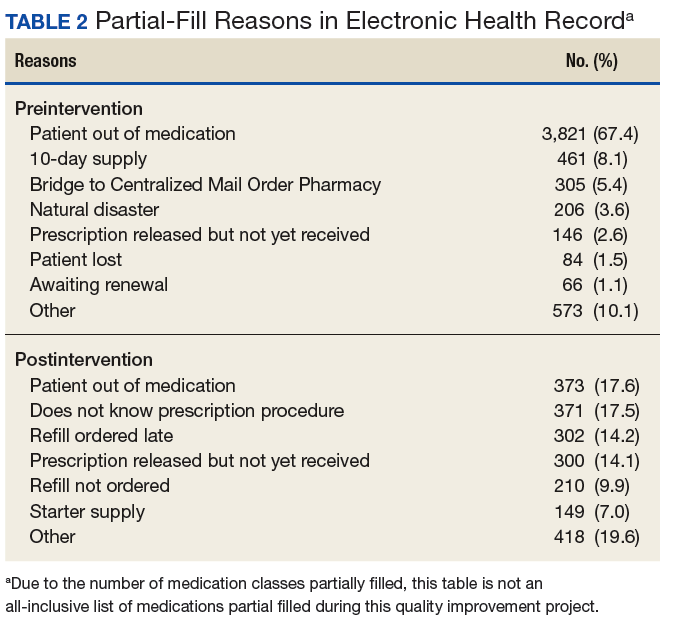
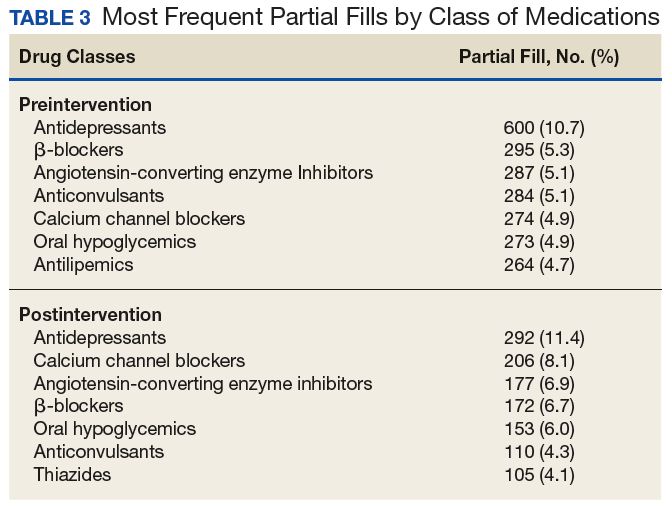
Discussion
Following the implementation of the updated partial-fill guidance and provision of education to the FVAMC veterans and staff, a noteworthy cost savings was observed with respect to both material and workload expenditures. This large reduction in expenditure likely was not related to a reduction in the total prescription volume, as the number of total prescriptions filled by the CMOP and at FVAMC were similar in both the preliminary and postintervention periods. When the results of this 3-month QI project were extrapolated, the annual projected cost avoidance was $91,949.12.
Of note, there was no established process for adjudicating appeals to the partial-fill guidance. Any extenuating circumstances that fell outside the guidance were addressed by the outpatient pharmacy supervisor. There was no formal documentation for these disputed cases. Since there was no prespecified supervisory override code, the most appropriate partial-fill code was entered into the comments field for these scenarios. As such, there is no way to distinguish precisely how many of these partial fills were escalated to a supervisory level for a decision.
The positive fiscal impact noted from the implementation of this project should not be viewed as the only utility for such guidance. Though not directly measured within the confines of this project, a reduction in pharmacy staff time spent on partial-fill prescriptions will likely result in shorter pharmacy wait times, line lengths, and streamlining of pharmacy workflow. When the pharmacy staff is free to work on pressing issues rather than on continually educating veterans on the partial-fill or refill process, many will benefit. Veteran satisfaction was not directly measured during this project but could be an interesting topic to review as a future study.
Each VA facility is unique, with its own challenges for implementation of a project such as this. Nevertheless, the incorporation of a formal guidance and education process, perhaps adapted to the indi vidual facility’s needs may be considered for overall pharmacy operations QI.
Limitations
During the preliminary data collection period, FVAMC and its catchment area were impacted by a natural disaster, Hurricane Matthew. Based on a review of the text entered into the comments field for all partial fills, about 4% of the partial fills completed in the preliminary phase can be attributed to the hurricane. The effects of this hurricane may have potentially increased the number of partial fills completed in the preliminary phase compared with that in the postintervention phase, due to the number of veterans who were temporarily or permanently displaced from their homes. This increase in partial fills and associated expenses preintervention likely caused a slightly higher cost savings to be reflected in the postimplementation phase than what would have traditionally been observed without extenuating factors.
Several other limitations must be considered for this QI project. The implementation phase, during which all education and training was completed, was only 1 month. A longer implementation period and more opportunities to educate veterans and staff might have created a greater impact on the results. Additionally, because there were no data collected on New Patient Orientation attendance for this project, it is unclear exactly how many veterans received refill-process education through this outlet.
Though all staff members were trained on the appropriate process, it was discovered during interim analysis that several pharmacists were not following the partial-fill guidance, potentially negatively impacting the results. It is likely that staff would have benefited from continual reeducation of the process throughout the entirety of the project, as the restriction of partial filling was a novel concept to many. In addition to continual reeducation of current employees, any new hires would likely need this information as part of initial training.
Cost variance in the type of partial fills completed between the preliminary and postintervention phases also may have negatively impacted the results. The postintervention phase contained 2 high-cost classes of drugs (antivirals and immunoglobulins) that received multiple partial fills but were not partialed in the preliminary phase, which increased the raw drug cost in the postintervention phase.
Conclusion
The implementation of partial-fill and process education to FVAMC staff and veterans proved beneficial in reducing the expenditures and workload associated with partial-fill prescription processing. The continued use of the updated partial-fill guidance will provide a standardized approach for pharmacy staff when completing partial-fill prescriptions.
Facilities may consider annual reeducation on their guidance through a local TMS module, as well as occasional process reminders during staff meetings to improve staff adherence to the guidance. Moreover, the sustained incorporation of improved refill process education to new patients and with every prescription pickup could help guide the FVAMC veteran population to use pharmacy services more effectively. The adoption of such procedures may be useful for VA facilities’ health care system looking to maximize the use of funding provided for prescription services as well as improve veterans’ understanding of how to appropriately order prescription refills.
1. US Department of Veterans Affairs. Pharmacy benefits management services. https://www.pbm.va.gov/PBM/CMOP/VA_Mail_Order_Pharmacy.asp. Updated July 14, 2017. Accessed on February 26, 2018.
2. J.D. Power. Decline in pharmacy customer satisfaction driven by prescription drug costs, J. D. Power finds. [press release]. http://www.jdpower.com/press-releases/jd-power-2017-us-pharmacy-study. Published September 5, 2017. Accessed February 26, 2018.
3. US Office of Personnel Management. Pay and leave. https://www.opm.gov/policy-data-oversight/pay-leave/salaries-wages/. Accessed February 26, 2018.
4. Aragon BR, Pierce RA, Jones WN. VA CMOPs: producing a pattern of quality and efficiency in government. J Am Pharm Assoc (2003). 2012;52(6):810-815.
According to the US Department of Veterans Affairs (VA) Pharmacy Benefits Management Service, about 80% of all outpatient prescriptions filled by the VA are sent to veterans by mail order, using the Centralized Mail Order Pharmacy (CMOP) network of highly automated pharmacies around the country.1 During fiscal year (FY) 2016, the 7 VA CMOP facilities throughout the US processed 119.7 million outpatient prescriptions. Each day, these CMOPs process nearly 470,000 prescriptions, an evidence of the efficiency provided through this mail-order service.1 The use of CMOP results in lower processing costs and increased convenience for veterans compared with filling prescriptions at pharmacies at individual VA facilities. Notably, VA CMOP has been rated “among the best” mail-order pharmacies in customer satisfaction according to the 2017 J.D. Power US Pharmacy Study.2
Background
Within the Fayetteville VA Medical Center (FVAMC) system in North Carolina, on-site patients receive a new prescription where an on-site pharmacy is available (at health care centers [HCC] and the medical center). For veterans seen at community-based outpatient clinics (CBOCs), emergent new prescriptions are filled through vouchers at contract community pharmacies, and nonemergent new prescriptions are filled by the CMOP (Figure 1).
The appropriate use of the VA CMOP for refills is intended to allow on-site pharmacy staff to focus on providing customer service for veterans requesting medication counseling from a clinical pharmacist, as well as those with new, changing, or urgent prescription needs. Filling prescriptions through the CMOP also can help control the VA facility pharmacy budget.
Despite the established mail-order process, FVAMC staff noted a high volume of medication refill and partial-fill prescriptions being requested at the on-site pharmacy. When a veteran presented to a pharmacy requesting a medication refill, pharmacy staff members ordered a refill to be filled by the CMOP and provided a limited quantity, otherwise known as a partial fill, to serve as an emergency supply to supplement the veteran until the full quantity of the prescription arrived by mail. Partial fills also were completed for new prescriptions that veterans requested to pick up at the on-site pharmacy. For new 90-day supply prescriptions, pharmacy staff often filled a 30-day supply in addition to submitting the entire 90-day prescription through the CMOP, which led to an unnecessary increase in material expenditures and workload.Preliminary data noted the most frequently used partial-fill days’ supply to be 10 days. Due to the lack of partial-fill criteria, prescriptions of all classes, quantities, and days’ supplies were provided as partial fills. Prior to the implementation of this quality improvement (QI) project, there was no standard approach for how to handle these requests.
Partial fills do not provide copay reimbursement to the facility filling the prescription. In an effort to steward the funds provided to the FVAMC pharmacy department wisely, an evaluation was performed with reference to partial fills. During FY 2016, about $350,000 was spent on medications, materials, and workload associated with the partial filling of FVAMC prescriptions. With respect to unique individuals who were provided care over the course of FY 2015 and 2016, the number of unique patients served by local comparator hospitals increased by only 2%, while the number of unique patients served by FVAMC increased by 12%. This substantial growth in the number of veterans served places further emphasis on the necessity of stewarding the allotted pharmacy budget. Moreover, the excessive number of medication refill and partial-fill prescriptions filled at on-site VA pharmacies can contribute to increased wait times for veterans with urgent prescription needs.
In November 2016, FVAMC implemented an updated partial-fill guidance. Partial-fill process and refill education was provided for VA staff and veterans in an effort to allow all parties involved to use pharmacy services efficiently. This analysis reviewed the reduction in partial-fill expenditures with a secondary focus on workload expenditures following the execution of this education.
Methods
This project was deemed to be a QI project and did not require institutional review board approval. Implementation for this QI project began in November 2016. Baseline raw drug cost, number, and class of prescriptions partialed were retrospectively collected for a 90-day period prior to implementation using all available data. Postintervention data were collected for 90 days following the implementation phase to compare partial-fill expenditures and workload expenditures with baseline data.
Calculations
Materials included in the partial-fill expenditure calculation were prescription vials, prescription vial caps, and prescription labeling. Material cost per partial fill was determined by using the facility’s wholesaler acquisition unit costs to estimate a summed cost for an individual prescription vial and prescription vial cap. The estimated acquisition price of 7 prescription-labeling pages was used in the material calculation, as this was the average number of pages used when performing test partial fills.
The following equation was used to calculate total partial-fill expenditures for any specified time frame:
Total partial-fill expenditure = total raw drug cost + (material cost × number of partial fills)
In addition, the average personnel cost per partial-fill prescription was determined. Average workload expenditure per partial fill was calculated by filling a subset of 10 test prescriptions and multiplying the average time spent by an average of the general station (GS) rate for pharmacists and technicians. The average hourly rate of a GS-12 pharmacist was calculated based on an average of the 10 available pay grades within the GS-12 ranking. The average hourly rate of a GS-6 pharmacy technician was calculated based on an average of the 10 available pay grades within the GS-6 ranking.3 The average workload expenditures were calculated using the following equations:
Average pharmacist workload expenditure per partial fill = time (in hours) × average hourly rate.
Average technician workload expenditure per partial fill = time (in hours) × average hourly rate.
Partial-Fill Guidance
Updated partial-fill guidance was drafted designating acceptable prescriptions to be limited to those responsible for preventing hospitalization and treatment of acute illness. This guidance provided generalized examples of medication classes that could be acceptable for partial filling, though it was not intended to be an all-inclusive list. The guidance also noted examples of classes or groups that should not be partial filled for nonemergent reasons (vitamins, nonprescription items, antilipemics), as well as controlled substances. The refill-process education was reiterated throughout the entirety of the guidance. Specifically, if a pharmacy staff member was to perform a partial fill, a review explaining the appropriate refill process to the veteran also must be provided. If the medication was determined to be of emergent need and not yet transmitted for filling via the CMOP, the directive recommended to fill the entire quantity locally as a onetime fill.
If a onetime on-site fill was determined infeasible, partial-fill quantities were recommended to be limited to only a 7-day supply, and the full quantity filled through the CMOP. Anticipated mail wait time for CMOP prescription delivery was estimated to be less than 7 days based on experience, local pending queues, and guidance from the regional CMOP; however, time could vary among VA and CMOP facilities. Original prescriptions were to be filled for the entire quantity for the first fill at the on-site pharmacy if requested by the veteran. If the pharmacy had an insufficient quantity for an entire initial supply, it could then be partial filled for a 7-day supply and then filled through the CMOP.
The final portion of the partial-fill guidance pertained to the use of partial-fill justification codes. Prior to the execution of the partial-fill guidance, free text was entered into the comments field when processing a partial-fill prescription, as the prescription-processing system used requires a comment to proceed with the partial fill. The use of these codes served to streamline data collection in the postintervention phase and helped identify areas for further education following the close of this project.
Education
Education to the pharmacy staff was disseminated by various modalities, including in-person sessions and written and electronic correspondences. This written guidance was distributed to pharmacy staff by e-mail, the pharmacy newsletter (Rx-tra), signage posted throughout the outpatient pharmacies, and on the facility’s pharmacy Microsoft SharePoint (Redmond, WA) site. In-services provided during pharmacy staff meetings detailed information on the updated partial-fill guidance. A FVAMC Talent Management System (TMS) training module was developed and assigned to all pharmacy service staff to reiterate key points regarding this QI initiative (eAppendix).
Nonpharmacy staff were educated through staff and in-service meetings and e-mail correspondences. These in-services emphasized how nurses, medical support assistants, and health care providers (HCPs) could assist veterans by knowing the correct refill process, ensuring sufficient refills remained until the next appointment, and providing continual refill-process education.
Following implementation, all veterans receiving prescriptions through the on-site pharmacies in the FVAMC were provided a copy of the refill-process handout with each trip to the pharmacy. Nonpharmacy staff and HCPs also were provided this handout to distribute to patients. The intent of this handout was to clearly detail the various ways in which refills could be ordered and the time frame in which they should be ordered. The pharmacy became involved in new patient orientation classes for all veterans new to FVAMC. Digital signage and messaging was created and circulated throughout several of the FVAMC facilities.
Results
The results of calculations for material cost, personnel time spent, hourly employee rates, and average workload expenditure per partial fill are summarized in Table 1.
Following the implementation of partial-fill and refill-process education, there was a 54.3% decrease in the total number of partial fills from 5,596 in the 90 days prior to implementation, to 2,555 partials completed over the 90 days postimplementation. Regarding the primary objective, total partial-fill expenditures decreased from $52,015.44 to $44,063.01 (-15.3%). When dissecting the individual components of partial-fill expenditures, material expenditures decreased from $1,454.96 to $664.28 (-54.3%), and raw drug cost expenditures decreased from $50,596.48 to $43,398.73 (-14.3%). Workload expenditures also decreased from $27,140.60 to $12,391.75 (-54.3%).
Several points of descriptive information also were collected. The average days’ supply trended down from a mode of 10 days to 7 days. This reduction in days’ supply likely was seen because staff became more aware of the customary amount required to bridge the veteran until the CMOP supply arrived by mail. Postintervention data showed a 70% utilization of partial-fill reasoning codes. The reasons for partial filling of prescriptions are summarized in Table 2.
Discussion
Following the implementation of the updated partial-fill guidance and provision of education to the FVAMC veterans and staff, a noteworthy cost savings was observed with respect to both material and workload expenditures. This large reduction in expenditure likely was not related to a reduction in the total prescription volume, as the number of total prescriptions filled by the CMOP and at FVAMC were similar in both the preliminary and postintervention periods. When the results of this 3-month QI project were extrapolated, the annual projected cost avoidance was $91,949.12.
Of note, there was no established process for adjudicating appeals to the partial-fill guidance. Any extenuating circumstances that fell outside the guidance were addressed by the outpatient pharmacy supervisor. There was no formal documentation for these disputed cases. Since there was no prespecified supervisory override code, the most appropriate partial-fill code was entered into the comments field for these scenarios. As such, there is no way to distinguish precisely how many of these partial fills were escalated to a supervisory level for a decision.
The positive fiscal impact noted from the implementation of this project should not be viewed as the only utility for such guidance. Though not directly measured within the confines of this project, a reduction in pharmacy staff time spent on partial-fill prescriptions will likely result in shorter pharmacy wait times, line lengths, and streamlining of pharmacy workflow. When the pharmacy staff is free to work on pressing issues rather than on continually educating veterans on the partial-fill or refill process, many will benefit. Veteran satisfaction was not directly measured during this project but could be an interesting topic to review as a future study.
Each VA facility is unique, with its own challenges for implementation of a project such as this. Nevertheless, the incorporation of a formal guidance and education process, perhaps adapted to the indi vidual facility’s needs may be considered for overall pharmacy operations QI.
Limitations
During the preliminary data collection period, FVAMC and its catchment area were impacted by a natural disaster, Hurricane Matthew. Based on a review of the text entered into the comments field for all partial fills, about 4% of the partial fills completed in the preliminary phase can be attributed to the hurricane. The effects of this hurricane may have potentially increased the number of partial fills completed in the preliminary phase compared with that in the postintervention phase, due to the number of veterans who were temporarily or permanently displaced from their homes. This increase in partial fills and associated expenses preintervention likely caused a slightly higher cost savings to be reflected in the postimplementation phase than what would have traditionally been observed without extenuating factors.
Several other limitations must be considered for this QI project. The implementation phase, during which all education and training was completed, was only 1 month. A longer implementation period and more opportunities to educate veterans and staff might have created a greater impact on the results. Additionally, because there were no data collected on New Patient Orientation attendance for this project, it is unclear exactly how many veterans received refill-process education through this outlet.
Though all staff members were trained on the appropriate process, it was discovered during interim analysis that several pharmacists were not following the partial-fill guidance, potentially negatively impacting the results. It is likely that staff would have benefited from continual reeducation of the process throughout the entirety of the project, as the restriction of partial filling was a novel concept to many. In addition to continual reeducation of current employees, any new hires would likely need this information as part of initial training.
Cost variance in the type of partial fills completed between the preliminary and postintervention phases also may have negatively impacted the results. The postintervention phase contained 2 high-cost classes of drugs (antivirals and immunoglobulins) that received multiple partial fills but were not partialed in the preliminary phase, which increased the raw drug cost in the postintervention phase.
Conclusion
The implementation of partial-fill and process education to FVAMC staff and veterans proved beneficial in reducing the expenditures and workload associated with partial-fill prescription processing. The continued use of the updated partial-fill guidance will provide a standardized approach for pharmacy staff when completing partial-fill prescriptions.
Facilities may consider annual reeducation on their guidance through a local TMS module, as well as occasional process reminders during staff meetings to improve staff adherence to the guidance. Moreover, the sustained incorporation of improved refill process education to new patients and with every prescription pickup could help guide the FVAMC veteran population to use pharmacy services more effectively. The adoption of such procedures may be useful for VA facilities’ health care system looking to maximize the use of funding provided for prescription services as well as improve veterans’ understanding of how to appropriately order prescription refills.
According to the US Department of Veterans Affairs (VA) Pharmacy Benefits Management Service, about 80% of all outpatient prescriptions filled by the VA are sent to veterans by mail order, using the Centralized Mail Order Pharmacy (CMOP) network of highly automated pharmacies around the country.1 During fiscal year (FY) 2016, the 7 VA CMOP facilities throughout the US processed 119.7 million outpatient prescriptions. Each day, these CMOPs process nearly 470,000 prescriptions, an evidence of the efficiency provided through this mail-order service.1 The use of CMOP results in lower processing costs and increased convenience for veterans compared with filling prescriptions at pharmacies at individual VA facilities. Notably, VA CMOP has been rated “among the best” mail-order pharmacies in customer satisfaction according to the 2017 J.D. Power US Pharmacy Study.2
Background
Within the Fayetteville VA Medical Center (FVAMC) system in North Carolina, on-site patients receive a new prescription where an on-site pharmacy is available (at health care centers [HCC] and the medical center). For veterans seen at community-based outpatient clinics (CBOCs), emergent new prescriptions are filled through vouchers at contract community pharmacies, and nonemergent new prescriptions are filled by the CMOP (Figure 1).
The appropriate use of the VA CMOP for refills is intended to allow on-site pharmacy staff to focus on providing customer service for veterans requesting medication counseling from a clinical pharmacist, as well as those with new, changing, or urgent prescription needs. Filling prescriptions through the CMOP also can help control the VA facility pharmacy budget.
Despite the established mail-order process, FVAMC staff noted a high volume of medication refill and partial-fill prescriptions being requested at the on-site pharmacy. When a veteran presented to a pharmacy requesting a medication refill, pharmacy staff members ordered a refill to be filled by the CMOP and provided a limited quantity, otherwise known as a partial fill, to serve as an emergency supply to supplement the veteran until the full quantity of the prescription arrived by mail. Partial fills also were completed for new prescriptions that veterans requested to pick up at the on-site pharmacy. For new 90-day supply prescriptions, pharmacy staff often filled a 30-day supply in addition to submitting the entire 90-day prescription through the CMOP, which led to an unnecessary increase in material expenditures and workload.Preliminary data noted the most frequently used partial-fill days’ supply to be 10 days. Due to the lack of partial-fill criteria, prescriptions of all classes, quantities, and days’ supplies were provided as partial fills. Prior to the implementation of this quality improvement (QI) project, there was no standard approach for how to handle these requests.
Partial fills do not provide copay reimbursement to the facility filling the prescription. In an effort to steward the funds provided to the FVAMC pharmacy department wisely, an evaluation was performed with reference to partial fills. During FY 2016, about $350,000 was spent on medications, materials, and workload associated with the partial filling of FVAMC prescriptions. With respect to unique individuals who were provided care over the course of FY 2015 and 2016, the number of unique patients served by local comparator hospitals increased by only 2%, while the number of unique patients served by FVAMC increased by 12%. This substantial growth in the number of veterans served places further emphasis on the necessity of stewarding the allotted pharmacy budget. Moreover, the excessive number of medication refill and partial-fill prescriptions filled at on-site VA pharmacies can contribute to increased wait times for veterans with urgent prescription needs.
In November 2016, FVAMC implemented an updated partial-fill guidance. Partial-fill process and refill education was provided for VA staff and veterans in an effort to allow all parties involved to use pharmacy services efficiently. This analysis reviewed the reduction in partial-fill expenditures with a secondary focus on workload expenditures following the execution of this education.
Methods
This project was deemed to be a QI project and did not require institutional review board approval. Implementation for this QI project began in November 2016. Baseline raw drug cost, number, and class of prescriptions partialed were retrospectively collected for a 90-day period prior to implementation using all available data. Postintervention data were collected for 90 days following the implementation phase to compare partial-fill expenditures and workload expenditures with baseline data.
Calculations
Materials included in the partial-fill expenditure calculation were prescription vials, prescription vial caps, and prescription labeling. Material cost per partial fill was determined by using the facility’s wholesaler acquisition unit costs to estimate a summed cost for an individual prescription vial and prescription vial cap. The estimated acquisition price of 7 prescription-labeling pages was used in the material calculation, as this was the average number of pages used when performing test partial fills.
The following equation was used to calculate total partial-fill expenditures for any specified time frame:
Total partial-fill expenditure = total raw drug cost + (material cost × number of partial fills)
In addition, the average personnel cost per partial-fill prescription was determined. Average workload expenditure per partial fill was calculated by filling a subset of 10 test prescriptions and multiplying the average time spent by an average of the general station (GS) rate for pharmacists and technicians. The average hourly rate of a GS-12 pharmacist was calculated based on an average of the 10 available pay grades within the GS-12 ranking. The average hourly rate of a GS-6 pharmacy technician was calculated based on an average of the 10 available pay grades within the GS-6 ranking.3 The average workload expenditures were calculated using the following equations:
Average pharmacist workload expenditure per partial fill = time (in hours) × average hourly rate.
Average technician workload expenditure per partial fill = time (in hours) × average hourly rate.
Partial-Fill Guidance
Updated partial-fill guidance was drafted designating acceptable prescriptions to be limited to those responsible for preventing hospitalization and treatment of acute illness. This guidance provided generalized examples of medication classes that could be acceptable for partial filling, though it was not intended to be an all-inclusive list. The guidance also noted examples of classes or groups that should not be partial filled for nonemergent reasons (vitamins, nonprescription items, antilipemics), as well as controlled substances. The refill-process education was reiterated throughout the entirety of the guidance. Specifically, if a pharmacy staff member was to perform a partial fill, a review explaining the appropriate refill process to the veteran also must be provided. If the medication was determined to be of emergent need and not yet transmitted for filling via the CMOP, the directive recommended to fill the entire quantity locally as a onetime fill.
If a onetime on-site fill was determined infeasible, partial-fill quantities were recommended to be limited to only a 7-day supply, and the full quantity filled through the CMOP. Anticipated mail wait time for CMOP prescription delivery was estimated to be less than 7 days based on experience, local pending queues, and guidance from the regional CMOP; however, time could vary among VA and CMOP facilities. Original prescriptions were to be filled for the entire quantity for the first fill at the on-site pharmacy if requested by the veteran. If the pharmacy had an insufficient quantity for an entire initial supply, it could then be partial filled for a 7-day supply and then filled through the CMOP.
The final portion of the partial-fill guidance pertained to the use of partial-fill justification codes. Prior to the execution of the partial-fill guidance, free text was entered into the comments field when processing a partial-fill prescription, as the prescription-processing system used requires a comment to proceed with the partial fill. The use of these codes served to streamline data collection in the postintervention phase and helped identify areas for further education following the close of this project.
Education
Education to the pharmacy staff was disseminated by various modalities, including in-person sessions and written and electronic correspondences. This written guidance was distributed to pharmacy staff by e-mail, the pharmacy newsletter (Rx-tra), signage posted throughout the outpatient pharmacies, and on the facility’s pharmacy Microsoft SharePoint (Redmond, WA) site. In-services provided during pharmacy staff meetings detailed information on the updated partial-fill guidance. A FVAMC Talent Management System (TMS) training module was developed and assigned to all pharmacy service staff to reiterate key points regarding this QI initiative (eAppendix).
Nonpharmacy staff were educated through staff and in-service meetings and e-mail correspondences. These in-services emphasized how nurses, medical support assistants, and health care providers (HCPs) could assist veterans by knowing the correct refill process, ensuring sufficient refills remained until the next appointment, and providing continual refill-process education.
Following implementation, all veterans receiving prescriptions through the on-site pharmacies in the FVAMC were provided a copy of the refill-process handout with each trip to the pharmacy. Nonpharmacy staff and HCPs also were provided this handout to distribute to patients. The intent of this handout was to clearly detail the various ways in which refills could be ordered and the time frame in which they should be ordered. The pharmacy became involved in new patient orientation classes for all veterans new to FVAMC. Digital signage and messaging was created and circulated throughout several of the FVAMC facilities.
Results
The results of calculations for material cost, personnel time spent, hourly employee rates, and average workload expenditure per partial fill are summarized in Table 1.
Following the implementation of partial-fill and refill-process education, there was a 54.3% decrease in the total number of partial fills from 5,596 in the 90 days prior to implementation, to 2,555 partials completed over the 90 days postimplementation. Regarding the primary objective, total partial-fill expenditures decreased from $52,015.44 to $44,063.01 (-15.3%). When dissecting the individual components of partial-fill expenditures, material expenditures decreased from $1,454.96 to $664.28 (-54.3%), and raw drug cost expenditures decreased from $50,596.48 to $43,398.73 (-14.3%). Workload expenditures also decreased from $27,140.60 to $12,391.75 (-54.3%).
Several points of descriptive information also were collected. The average days’ supply trended down from a mode of 10 days to 7 days. This reduction in days’ supply likely was seen because staff became more aware of the customary amount required to bridge the veteran until the CMOP supply arrived by mail. Postintervention data showed a 70% utilization of partial-fill reasoning codes. The reasons for partial filling of prescriptions are summarized in Table 2.
Discussion
Following the implementation of the updated partial-fill guidance and provision of education to the FVAMC veterans and staff, a noteworthy cost savings was observed with respect to both material and workload expenditures. This large reduction in expenditure likely was not related to a reduction in the total prescription volume, as the number of total prescriptions filled by the CMOP and at FVAMC were similar in both the preliminary and postintervention periods. When the results of this 3-month QI project were extrapolated, the annual projected cost avoidance was $91,949.12.
Of note, there was no established process for adjudicating appeals to the partial-fill guidance. Any extenuating circumstances that fell outside the guidance were addressed by the outpatient pharmacy supervisor. There was no formal documentation for these disputed cases. Since there was no prespecified supervisory override code, the most appropriate partial-fill code was entered into the comments field for these scenarios. As such, there is no way to distinguish precisely how many of these partial fills were escalated to a supervisory level for a decision.
The positive fiscal impact noted from the implementation of this project should not be viewed as the only utility for such guidance. Though not directly measured within the confines of this project, a reduction in pharmacy staff time spent on partial-fill prescriptions will likely result in shorter pharmacy wait times, line lengths, and streamlining of pharmacy workflow. When the pharmacy staff is free to work on pressing issues rather than on continually educating veterans on the partial-fill or refill process, many will benefit. Veteran satisfaction was not directly measured during this project but could be an interesting topic to review as a future study.
Each VA facility is unique, with its own challenges for implementation of a project such as this. Nevertheless, the incorporation of a formal guidance and education process, perhaps adapted to the indi vidual facility’s needs may be considered for overall pharmacy operations QI.
Limitations
During the preliminary data collection period, FVAMC and its catchment area were impacted by a natural disaster, Hurricane Matthew. Based on a review of the text entered into the comments field for all partial fills, about 4% of the partial fills completed in the preliminary phase can be attributed to the hurricane. The effects of this hurricane may have potentially increased the number of partial fills completed in the preliminary phase compared with that in the postintervention phase, due to the number of veterans who were temporarily or permanently displaced from their homes. This increase in partial fills and associated expenses preintervention likely caused a slightly higher cost savings to be reflected in the postimplementation phase than what would have traditionally been observed without extenuating factors.
Several other limitations must be considered for this QI project. The implementation phase, during which all education and training was completed, was only 1 month. A longer implementation period and more opportunities to educate veterans and staff might have created a greater impact on the results. Additionally, because there were no data collected on New Patient Orientation attendance for this project, it is unclear exactly how many veterans received refill-process education through this outlet.
Though all staff members were trained on the appropriate process, it was discovered during interim analysis that several pharmacists were not following the partial-fill guidance, potentially negatively impacting the results. It is likely that staff would have benefited from continual reeducation of the process throughout the entirety of the project, as the restriction of partial filling was a novel concept to many. In addition to continual reeducation of current employees, any new hires would likely need this information as part of initial training.
Cost variance in the type of partial fills completed between the preliminary and postintervention phases also may have negatively impacted the results. The postintervention phase contained 2 high-cost classes of drugs (antivirals and immunoglobulins) that received multiple partial fills but were not partialed in the preliminary phase, which increased the raw drug cost in the postintervention phase.
Conclusion
The implementation of partial-fill and process education to FVAMC staff and veterans proved beneficial in reducing the expenditures and workload associated with partial-fill prescription processing. The continued use of the updated partial-fill guidance will provide a standardized approach for pharmacy staff when completing partial-fill prescriptions.
Facilities may consider annual reeducation on their guidance through a local TMS module, as well as occasional process reminders during staff meetings to improve staff adherence to the guidance. Moreover, the sustained incorporation of improved refill process education to new patients and with every prescription pickup could help guide the FVAMC veteran population to use pharmacy services more effectively. The adoption of such procedures may be useful for VA facilities’ health care system looking to maximize the use of funding provided for prescription services as well as improve veterans’ understanding of how to appropriately order prescription refills.
1. US Department of Veterans Affairs. Pharmacy benefits management services. https://www.pbm.va.gov/PBM/CMOP/VA_Mail_Order_Pharmacy.asp. Updated July 14, 2017. Accessed on February 26, 2018.
2. J.D. Power. Decline in pharmacy customer satisfaction driven by prescription drug costs, J. D. Power finds. [press release]. http://www.jdpower.com/press-releases/jd-power-2017-us-pharmacy-study. Published September 5, 2017. Accessed February 26, 2018.
3. US Office of Personnel Management. Pay and leave. https://www.opm.gov/policy-data-oversight/pay-leave/salaries-wages/. Accessed February 26, 2018.
4. Aragon BR, Pierce RA, Jones WN. VA CMOPs: producing a pattern of quality and efficiency in government. J Am Pharm Assoc (2003). 2012;52(6):810-815.
1. US Department of Veterans Affairs. Pharmacy benefits management services. https://www.pbm.va.gov/PBM/CMOP/VA_Mail_Order_Pharmacy.asp. Updated July 14, 2017. Accessed on February 26, 2018.
2. J.D. Power. Decline in pharmacy customer satisfaction driven by prescription drug costs, J. D. Power finds. [press release]. http://www.jdpower.com/press-releases/jd-power-2017-us-pharmacy-study. Published September 5, 2017. Accessed February 26, 2018.
3. US Office of Personnel Management. Pay and leave. https://www.opm.gov/policy-data-oversight/pay-leave/salaries-wages/. Accessed February 26, 2018.
4. Aragon BR, Pierce RA, Jones WN. VA CMOPs: producing a pattern of quality and efficiency in government. J Am Pharm Assoc (2003). 2012;52(6):810-815.
FDA Boxed Warning Updates: February 2018
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefit from the drug that it is essential that it be considered in assessing the risks and benefits of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE: EPIVIR-HBV (LAMIVUDINE)
- Added to warning September 2017
WARNING: LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, EXACERBATIONS OF HEPATITIS B, and RISK OF HIV-1 RESISTANCE IF EPIVIR-HBV IS USED IN PATIENTS WITH UNRECOGNIZED OR UNTREATED HIV-1 INFECTION
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues and other antiretrovirals. Discontinue EPIVIR-HBV if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur.
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy (including EPIVIR-HBV). Hepatic function should be monitored closely with both clinical and laboratory followup for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
EPIVIR-HBV is not approved for the treatment of HIV-1 infection because the lamivudine dosage in EPIVIR-HBV is subtherapeutic and monotherapy is inappropriate for the treatment of HIV-1 infection. HIV-1 resistance may emerge in chronic hepatitis B-infected patients with unrecognized or untreated HIV-1 infection. HIV counseling and testing should be offered to all patients before beginning treatment with EPIVIR-HBV and periodically during treatment.
INVOKANA (CANAGLIFLOZIN)
- Added section to warning July 2017
WARNING: LOWER LIMB AMPUTATION
- An approximately 2-fold increased risk of lower limb amputations associated with INVOKANA use was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
Monitor patients receiving INVOKANA for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
INVOKAMET (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Risk of Lower Limb Amputation
- In patients with type 2 diabetes who have established cardiovascular disease (CVD) or at risk for CVD, canagliflozin, a component of INVOKAMET, has been associated with lower limb amputations, most frequently of the toe and midfoot; some also involved the leg.
INVOKAMET XR (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Post-marketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as
malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/ pyruvate ratio; and metformin plasma levels generally >5 mcg/mL. - Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, cationic drugs such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
- Steps to reduce the risk of and manage metforminassociated lactic acidosis in these high risk groups are provided in the full prescribing information.
- If metformin-associated lactic acidosis is suspected, immediately discontinue INVOKAMET and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
Risk of Lower Limb Amputation
- An approximately 2-fold increased risk of lower limb amputations associated with canagliflozin, a component of INVOKAMET, was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
- Monitor patients receiving INVOKAMET for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
JEVTANA KIT (CABAZITAXEL)
- Edited warning September 2017
WARNING: NEUTROPENIA AND HYPERSENSITIVITY
Neutropenia: Neutropenic deaths have been reported. Monitor for neutropenia with frequent blood cell counts. JEVTANA is contraindicated in patients with neutrophil counts of less than or equal to 1,500 cells/mm3. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features.
THYRO-TABS (LEVOTHYROXINE SODIUM)
- Added section to warning August 2017
WARNING: NOT FOR TREATMENT OF OBESITY OR FOR WEIGHT LOSS
Thyroid hormones, including THYRO-TABS, either alone or with other therapeutic agents, should not be used for the treatment of obesity or for weight loss.
In euthyroid patients, doses within the range of daily hormonal requirements are ineffective for weight reduction.
Larger doses may produce serious or even life threatening manifestations of toxicity, particularly when given in association with sympathomimetic amines such as those used for their anorectic effects.
REGLAN (METOCLOPRAMIDE HYDROCHLORIDE)
- Edited warning August 2017
WARNING: TARDIVE DYSKINESIA
- Reglan can cause tardive dyskinesia (TD), a serious movement disorder that is often irreversible. There is no known treatment for TD. The risk of developing TD increases with duration of treatment and total cumulative dosage.
- Discontinue Reglan in patients who develop signs or symptoms of TD. In some patients, symptoms may lessen or resolve after Reglan is stopped.
- Avoid treatment with Reglan for longer than 12 weeks because of the increased risk of developing TD with longer-term use.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefit from the drug that it is essential that it be considered in assessing the risks and benefits of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE: EPIVIR-HBV (LAMIVUDINE)
- Added to warning September 2017
WARNING: LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, EXACERBATIONS OF HEPATITIS B, and RISK OF HIV-1 RESISTANCE IF EPIVIR-HBV IS USED IN PATIENTS WITH UNRECOGNIZED OR UNTREATED HIV-1 INFECTION
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues and other antiretrovirals. Discontinue EPIVIR-HBV if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur.
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy (including EPIVIR-HBV). Hepatic function should be monitored closely with both clinical and laboratory followup for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
EPIVIR-HBV is not approved for the treatment of HIV-1 infection because the lamivudine dosage in EPIVIR-HBV is subtherapeutic and monotherapy is inappropriate for the treatment of HIV-1 infection. HIV-1 resistance may emerge in chronic hepatitis B-infected patients with unrecognized or untreated HIV-1 infection. HIV counseling and testing should be offered to all patients before beginning treatment with EPIVIR-HBV and periodically during treatment.
INVOKANA (CANAGLIFLOZIN)
- Added section to warning July 2017
WARNING: LOWER LIMB AMPUTATION
- An approximately 2-fold increased risk of lower limb amputations associated with INVOKANA use was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
Monitor patients receiving INVOKANA for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
INVOKAMET (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Risk of Lower Limb Amputation
- In patients with type 2 diabetes who have established cardiovascular disease (CVD) or at risk for CVD, canagliflozin, a component of INVOKAMET, has been associated with lower limb amputations, most frequently of the toe and midfoot; some also involved the leg.
INVOKAMET XR (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Post-marketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as
malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/ pyruvate ratio; and metformin plasma levels generally >5 mcg/mL. - Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, cationic drugs such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
- Steps to reduce the risk of and manage metforminassociated lactic acidosis in these high risk groups are provided in the full prescribing information.
- If metformin-associated lactic acidosis is suspected, immediately discontinue INVOKAMET and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
Risk of Lower Limb Amputation
- An approximately 2-fold increased risk of lower limb amputations associated with canagliflozin, a component of INVOKAMET, was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
- Monitor patients receiving INVOKAMET for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
JEVTANA KIT (CABAZITAXEL)
- Edited warning September 2017
WARNING: NEUTROPENIA AND HYPERSENSITIVITY
Neutropenia: Neutropenic deaths have been reported. Monitor for neutropenia with frequent blood cell counts. JEVTANA is contraindicated in patients with neutrophil counts of less than or equal to 1,500 cells/mm3. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features.
THYRO-TABS (LEVOTHYROXINE SODIUM)
- Added section to warning August 2017
WARNING: NOT FOR TREATMENT OF OBESITY OR FOR WEIGHT LOSS
Thyroid hormones, including THYRO-TABS, either alone or with other therapeutic agents, should not be used for the treatment of obesity or for weight loss.
In euthyroid patients, doses within the range of daily hormonal requirements are ineffective for weight reduction.
Larger doses may produce serious or even life threatening manifestations of toxicity, particularly when given in association with sympathomimetic amines such as those used for their anorectic effects.
REGLAN (METOCLOPRAMIDE HYDROCHLORIDE)
- Edited warning August 2017
WARNING: TARDIVE DYSKINESIA
- Reglan can cause tardive dyskinesia (TD), a serious movement disorder that is often irreversible. There is no known treatment for TD. The risk of developing TD increases with duration of treatment and total cumulative dosage.
- Discontinue Reglan in patients who develop signs or symptoms of TD. In some patients, symptoms may lessen or resolve after Reglan is stopped.
- Avoid treatment with Reglan for longer than 12 weeks because of the increased risk of developing TD with longer-term use.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefit from the drug that it is essential that it be considered in assessing the risks and benefits of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE: EPIVIR-HBV (LAMIVUDINE)
- Added to warning September 2017
WARNING: LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, EXACERBATIONS OF HEPATITIS B, and RISK OF HIV-1 RESISTANCE IF EPIVIR-HBV IS USED IN PATIENTS WITH UNRECOGNIZED OR UNTREATED HIV-1 INFECTION
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues and other antiretrovirals. Discontinue EPIVIR-HBV if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur.
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy (including EPIVIR-HBV). Hepatic function should be monitored closely with both clinical and laboratory followup for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
EPIVIR-HBV is not approved for the treatment of HIV-1 infection because the lamivudine dosage in EPIVIR-HBV is subtherapeutic and monotherapy is inappropriate for the treatment of HIV-1 infection. HIV-1 resistance may emerge in chronic hepatitis B-infected patients with unrecognized or untreated HIV-1 infection. HIV counseling and testing should be offered to all patients before beginning treatment with EPIVIR-HBV and periodically during treatment.
INVOKANA (CANAGLIFLOZIN)
- Added section to warning July 2017
WARNING: LOWER LIMB AMPUTATION
- An approximately 2-fold increased risk of lower limb amputations associated with INVOKANA use was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
Monitor patients receiving INVOKANA for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
INVOKAMET (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Risk of Lower Limb Amputation
- In patients with type 2 diabetes who have established cardiovascular disease (CVD) or at risk for CVD, canagliflozin, a component of INVOKAMET, has been associated with lower limb amputations, most frequently of the toe and midfoot; some also involved the leg.
INVOKAMET XR (CANAGLIFLOZIN; METFORMIN HYDROCHLORIDE)
- Edited warning August 2017
WARNING: LACTIC ACIDOSIS and LOWER LIMB AMPUTATION
Lactic Acidosis
- Post-marketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as
malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/ pyruvate ratio; and metformin plasma levels generally >5 mcg/mL. - Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, cationic drugs such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
- Steps to reduce the risk of and manage metforminassociated lactic acidosis in these high risk groups are provided in the full prescribing information.
- If metformin-associated lactic acidosis is suspected, immediately discontinue INVOKAMET and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
Risk of Lower Limb Amputation
- An approximately 2-fold increased risk of lower limb amputations associated with canagliflozin, a component of INVOKAMET, was observed in CANVAS and CANVAS-R, two large, randomized, placebo-controlled trials in patients with type 2 diabetes who had established cardiovascular disease (CVD) or were at risk for CVD.
- Amputations of the toe and midfoot were most frequent; however, amputations involving the leg were also observed. Some patients had multiple amputations, some involving both limbs.
- Before initiating, consider factors that may increase the risk of amputation, such as a history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers.
- Monitor patients receiving INVOKAMET for infection, new pain or tenderness, sores or ulcers involving the lower limbs, and discontinue if these complications occur.
JEVTANA KIT (CABAZITAXEL)
- Edited warning September 2017
WARNING: NEUTROPENIA AND HYPERSENSITIVITY
Neutropenia: Neutropenic deaths have been reported. Monitor for neutropenia with frequent blood cell counts. JEVTANA is contraindicated in patients with neutrophil counts of less than or equal to 1,500 cells/mm3. Primary prophylaxis with G-CSF is recommended in patients with high-risk clinical features.
THYRO-TABS (LEVOTHYROXINE SODIUM)
- Added section to warning August 2017
WARNING: NOT FOR TREATMENT OF OBESITY OR FOR WEIGHT LOSS
Thyroid hormones, including THYRO-TABS, either alone or with other therapeutic agents, should not be used for the treatment of obesity or for weight loss.
In euthyroid patients, doses within the range of daily hormonal requirements are ineffective for weight reduction.
Larger doses may produce serious or even life threatening manifestations of toxicity, particularly when given in association with sympathomimetic amines such as those used for their anorectic effects.
REGLAN (METOCLOPRAMIDE HYDROCHLORIDE)
- Edited warning August 2017
WARNING: TARDIVE DYSKINESIA
- Reglan can cause tardive dyskinesia (TD), a serious movement disorder that is often irreversible. There is no known treatment for TD. The risk of developing TD increases with duration of treatment and total cumulative dosage.
- Discontinue Reglan in patients who develop signs or symptoms of TD. In some patients, symptoms may lessen or resolve after Reglan is stopped.
- Avoid treatment with Reglan for longer than 12 weeks because of the increased risk of developing TD with longer-term use.
Risk Factors Associated With Multidrug-Resistant Pneumonia in Nonhospitalized Patients
Successful treatment of pneumonia depends on timely diagnosis and administration of antibiotics. Multidrug-resistant organisms (MDROs) complicate antibiotic therapies by rendering some antibiotic agents ineffective. Inappropriate initial therapy has been associated with a more than 2-fold increase in the risk of mortality.1 Because culture results are not available immediately, clinicians prescribe antibiotics empirically and must rely on guidelines and knowledge of risk factors associated with MDRO infection to make these selections.
Treatment guidelines exist for hospital-acquired and ventilator-associated pneumonia (HAP/VAP) and community-acquired pneumonia (CAP) to assist with empiric antibiotic selection. For HAP/VAP, 2 to 3 antibiotics with a broad-spectrum of activity are used due to increased prevalence of MDROs in hospitals, whereastreatment of CAP involves more narrow coverage because bacteria that cause this infection typically have fewer antibiotic resistances.2,3 The HAP/VAP guidelines stratify the risk of pneumonia due to the presence of a MDRO acquired during a hospitalization. However, neither the CAP nor HAP/VAP guidelines offer risk-stratification guidance for nonhospitalized patients who develop pneumonia but who may have become colonized with a MDRO during a previous hospitalization or from another exposure to a health care facility.
Health care-associated pneumonia (HCAP) was first described in the 2005 American Thoracic Society and the Infectious Diseases Society of America (ATS/IDSA) nosocomial pneumonia guidelines and was associated with criteria intended to aid clinician identification of nonhospitalized patients at risk for MDRO pneumonia, which warranted empiric broad-spectrum antibiotic therapy.2 According to these guidelines, patients were classified as having HCAP if they had been hospitalized for at least 48 hours in the past 90 days, admitted from a nursing home, received recent intravenous antibiotics, had hemodialysis in the past 30 days, had a history of home infusion therapy or wound care, received intravenous chemotherapy, or had a family member with MDRO colonization.
Since publication of the 2005 guidelines, HCAP has been criticized as being a poor predictor of MDRO infection. A 2014 meta-analysis of 24 studies investigated the discriminating ability of HCAP and reported that the specificity and sensitivity for MDRO infections was 71.2% and 53.7%, respectively.3 In 2016, the ATS/IDSA guidelines were updated to remove HCAP due to the risk of antibiotic overprescribing.4
Literature Review
Although criteria previously defining a patient as having HCAP have been shown to be a poor discriminator of MDRO pneumonia as a whole, MDRO infections still pose a threat to nonhospitalized patients who have exposure to the health care system. A literature review was performed to identify independent HCAP risk factors that may increase the risk of MDRO pneumonia infecting a nonhospitalized patient needing empiric broad-spectrum antibiotic therapy. All included studies were prospective or retrospective observational cohort studies that performed logistic regression analyses to assess the association between MDRO isolation and the previously defined HCAP risk factors (Table 1). 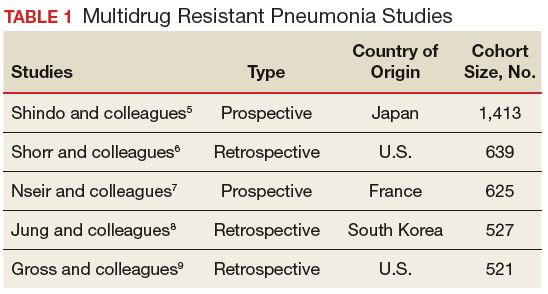
Five studies examined the risk of MDRO infection in patients with a previous hospital admission of 2 days or more in the past 90 days. Shindo and colleagues found a significant increase in MDRO infections by about 2-fold (adjusted odds ratio [AOR], 2.1; 95% confidence interval [CI], 1.2-3.4).5 Shorr and colleagues found a 4-fold increase in likelihood of identifying a MDRO in HCAP (AOR, 4.2; 95% CI, 2.9-6.3).6 Nseir and colleagues and Jung and colleagues found similar results (AOR 3.9, 95% CI 1.7-8.8; AOR 2.7, 95% CI 1.3-5.5, respectively).7,8 Conflicting results were reported by Gross and colleagues who did not find a significant relationship between previous hospitalization and MDRO isolation (AOR 1.2, 95% CI, 0.5-3.2).9
In patients with pneumonia admitted from a nursing home, MDRO infection risk also was evaluated in these 5 studies. Shorr and colleagues, Nseir and colleagues, and Gross and colleagues found significant AORs of 2.7 (95% CI 1.7-4.3), 2.0 (95% CI 1.1-3.7), and 4.2 (95% CI 1.6-11.3), respectively.6,7,9 Shindo and colleagues (AOR 1.1; 95% CI, 0.6-2.0) and Jung and colleagues (AOR 1.9, 95% CI, 0.5-6.9) found this risk factor not significant.5
Receipt of antibiotics within the previous 90 days was assessed in 3 studies. Shindo and colleagues, Nseir and colleagues, and Gross and colleagues all found significant AORs of 2.5 (95% CI 1.2-4.0), 2.3 (95% CI 1.2-4.3), and 2.9 (95% CI 1.1-7.5), respectively.5,7,9 Antibiotic therapy within the previous 90 days is an established risk factor for MDRO pneumonia, and the 2016 ATS/IDSA guidelines consider this a risk factor for HAP and VAP, including pneumonia caused by methicillin resistant Staphylococcus aureus and Pseudomonas aeruginosa.4
The impact of hemodialysis in the previous month on acquisition of MDRO pneumonia was investigated in 4 studies. Shindo and colleagues, Jung and colleagues, and Gross and colleagues concluded that this risk factor was not significantly related to MDRO infection, reporting AORs of 2.2 (95% CI 0.5-9.7), 2.8 (95% CI 0.9-9.2) and 0.7 (95% CI 0.1-5.1), respectively.5,8,9 Shorr and colleagues, however, found a significant AOR of 2.1 (95% CI 1.0-4.3).6
Shindo and colleagues investigated the impact of home infusion therapy on acquisition of pneumonia due to a MDRO and reported a nonsignificant AOR of 0.8 (95% CI 0.4-1.8).5 Gross and colleagues also found a nonsignificant AOR of 0 (P = .1).9 In the Shindo and colleagues study, resistance was found in 107 of 679 patients who did not receive infusion therapy, and 12 of 55 patients who were receiving infusion therapy.5 Gross and colleagues reported that home-infusion therapy was received by 0 of 20 patients with MDRO infection and 4 of the 501 patients without MDRO infection.9
Shindo and colleagues reported that home wound care was not found to be significantly related to MDRO pneumonia as well as did Gross and colleagues: AORs of 3.8 (0.8-18.4) and 1.4 (95% CI 0.5-4.4), respectively.5,9 Jung and colleagues examined IV chemotherapy in the past 30 days, and found this to not significantly impact the odds of MDRO isolation (AOR = 0.62, 95% CI 0.2-1.8).8 No data were available reflecting the risk of a family member with a MDRO.
Limitations
The variables on which logistic regression were performed differed among the studies. Therefore, results cannot be averaged or compared quantitatively, as AORs varied, depending on the variables included. In addition, data were drawn from multiple geographic locations that may impact MDRO prevalence within each patient population. Finally, this review examines the utility of the risk factors formerly included in HCAP. However, other risk factors for MDRO pneumonia outlined by the ATS/IDSA guidelines still should be considered when evaluating patient risk. The 2016 guidelines recommend local incidence of resistant strains be considered when initiating empiric therapy. Review of medical records for previous positive cultures and duration of current hospitalization also should be considered. Although the 2016 ATS/IDSA HAP guidelines are not intended for immunosuppressed patients, this risk factor also may be taken into account.
Conclusion
Review and synthesis of published literature found previous hospital admission (of ≥ 2 days in the past 90 days), admission from a nursing home, and IV antibiotic therapy in the last 90 days to be independent risk factors for identification of MDRO pneumonia in previously nonhospitalized patients (Table 2). Additionally, although no data were found to support this risk factor, existence of an in-home (close contact) source of MDROs would provide ample opportunity for transmission, so evaluation of known exposure to MDROs from contacts should be considered. When choosing empiric antibiotic therapy for patients admitted to the hospital for treatment of pneumonia, consideration of patient history and risk factors that may contribute to infection with a MDRO are recommended. 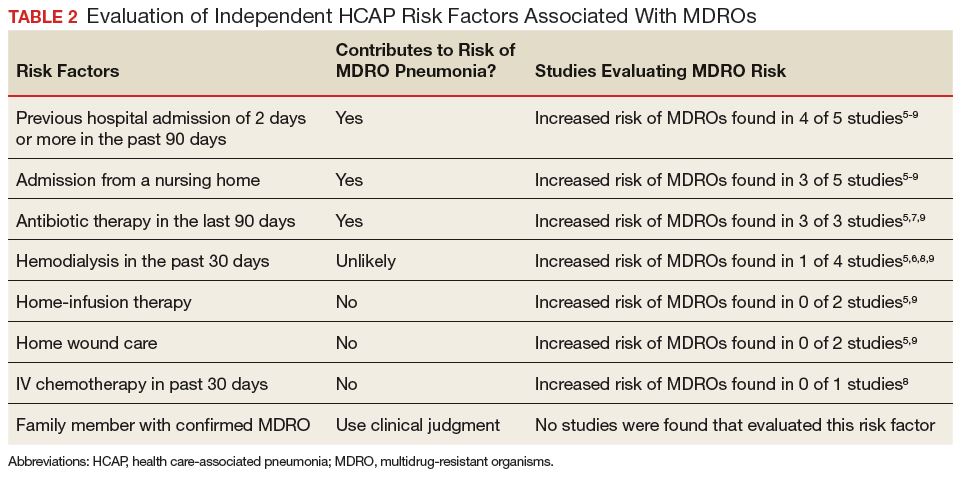
1. Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. 1999;115(2):462-474.
2. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416.
3. Chalmers JD, Rother C, Salih W, Ewig S. Healthcare-associated pneumonia does not accurately identify potentially resistant pathogens: a systematic review and meta-analysis. Clin Infect Dis. 2014;58(3):330-339.
4. Kalil AC, Metersky ML, Klompas M, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61-e111.
5. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995.
6. Shorr AF, Zilberberg MD, Micek ST, Kollef MH. Prediction of infection due to antibiotic-resistant bacteria by select risk factors for health care–associated pneumonia. Arch Intern Med. 2008;168(20):2205-2210.
7. Nseir S, Grailles G, Soury-Lavergne A, Minacori F, Alves I, Durocher A. Accuracy of American Thoracic Society/Infectious Diseases Society of America criteria in predicting infection or colonization with multidrug-resistant bacteria at intensive-care unit admission. Clin Microbiol Infect. 2010;16(7):902-908.
8. Jung JY, Park MS, Kim YS, et al. Healthcare-associated pneumonia among hospitalized patients in a Korean tertiary hospital. BMC Infectious Diseases. 2011;11:61.
9. Gross AE, Van Schooneveld TC, Olsen KM, et al. Epidemiology and predictors of multidrug-resistant community-acquired and health care-associated pneumonia. Antimicrob Agents Chemother. 2014;58(9):5262-5268.
Successful treatment of pneumonia depends on timely diagnosis and administration of antibiotics. Multidrug-resistant organisms (MDROs) complicate antibiotic therapies by rendering some antibiotic agents ineffective. Inappropriate initial therapy has been associated with a more than 2-fold increase in the risk of mortality.1 Because culture results are not available immediately, clinicians prescribe antibiotics empirically and must rely on guidelines and knowledge of risk factors associated with MDRO infection to make these selections.
Treatment guidelines exist for hospital-acquired and ventilator-associated pneumonia (HAP/VAP) and community-acquired pneumonia (CAP) to assist with empiric antibiotic selection. For HAP/VAP, 2 to 3 antibiotics with a broad-spectrum of activity are used due to increased prevalence of MDROs in hospitals, whereastreatment of CAP involves more narrow coverage because bacteria that cause this infection typically have fewer antibiotic resistances.2,3 The HAP/VAP guidelines stratify the risk of pneumonia due to the presence of a MDRO acquired during a hospitalization. However, neither the CAP nor HAP/VAP guidelines offer risk-stratification guidance for nonhospitalized patients who develop pneumonia but who may have become colonized with a MDRO during a previous hospitalization or from another exposure to a health care facility.
Health care-associated pneumonia (HCAP) was first described in the 2005 American Thoracic Society and the Infectious Diseases Society of America (ATS/IDSA) nosocomial pneumonia guidelines and was associated with criteria intended to aid clinician identification of nonhospitalized patients at risk for MDRO pneumonia, which warranted empiric broad-spectrum antibiotic therapy.2 According to these guidelines, patients were classified as having HCAP if they had been hospitalized for at least 48 hours in the past 90 days, admitted from a nursing home, received recent intravenous antibiotics, had hemodialysis in the past 30 days, had a history of home infusion therapy or wound care, received intravenous chemotherapy, or had a family member with MDRO colonization.
Since publication of the 2005 guidelines, HCAP has been criticized as being a poor predictor of MDRO infection. A 2014 meta-analysis of 24 studies investigated the discriminating ability of HCAP and reported that the specificity and sensitivity for MDRO infections was 71.2% and 53.7%, respectively.3 In 2016, the ATS/IDSA guidelines were updated to remove HCAP due to the risk of antibiotic overprescribing.4
Literature Review
Although criteria previously defining a patient as having HCAP have been shown to be a poor discriminator of MDRO pneumonia as a whole, MDRO infections still pose a threat to nonhospitalized patients who have exposure to the health care system. A literature review was performed to identify independent HCAP risk factors that may increase the risk of MDRO pneumonia infecting a nonhospitalized patient needing empiric broad-spectrum antibiotic therapy. All included studies were prospective or retrospective observational cohort studies that performed logistic regression analyses to assess the association between MDRO isolation and the previously defined HCAP risk factors (Table 1).
Five studies examined the risk of MDRO infection in patients with a previous hospital admission of 2 days or more in the past 90 days. Shindo and colleagues found a significant increase in MDRO infections by about 2-fold (adjusted odds ratio [AOR], 2.1; 95% confidence interval [CI], 1.2-3.4).5 Shorr and colleagues found a 4-fold increase in likelihood of identifying a MDRO in HCAP (AOR, 4.2; 95% CI, 2.9-6.3).6 Nseir and colleagues and Jung and colleagues found similar results (AOR 3.9, 95% CI 1.7-8.8; AOR 2.7, 95% CI 1.3-5.5, respectively).7,8 Conflicting results were reported by Gross and colleagues who did not find a significant relationship between previous hospitalization and MDRO isolation (AOR 1.2, 95% CI, 0.5-3.2).9
In patients with pneumonia admitted from a nursing home, MDRO infection risk also was evaluated in these 5 studies. Shorr and colleagues, Nseir and colleagues, and Gross and colleagues found significant AORs of 2.7 (95% CI 1.7-4.3), 2.0 (95% CI 1.1-3.7), and 4.2 (95% CI 1.6-11.3), respectively.6,7,9 Shindo and colleagues (AOR 1.1; 95% CI, 0.6-2.0) and Jung and colleagues (AOR 1.9, 95% CI, 0.5-6.9) found this risk factor not significant.5
Receipt of antibiotics within the previous 90 days was assessed in 3 studies. Shindo and colleagues, Nseir and colleagues, and Gross and colleagues all found significant AORs of 2.5 (95% CI 1.2-4.0), 2.3 (95% CI 1.2-4.3), and 2.9 (95% CI 1.1-7.5), respectively.5,7,9 Antibiotic therapy within the previous 90 days is an established risk factor for MDRO pneumonia, and the 2016 ATS/IDSA guidelines consider this a risk factor for HAP and VAP, including pneumonia caused by methicillin resistant Staphylococcus aureus and Pseudomonas aeruginosa.4
The impact of hemodialysis in the previous month on acquisition of MDRO pneumonia was investigated in 4 studies. Shindo and colleagues, Jung and colleagues, and Gross and colleagues concluded that this risk factor was not significantly related to MDRO infection, reporting AORs of 2.2 (95% CI 0.5-9.7), 2.8 (95% CI 0.9-9.2) and 0.7 (95% CI 0.1-5.1), respectively.5,8,9 Shorr and colleagues, however, found a significant AOR of 2.1 (95% CI 1.0-4.3).6
Shindo and colleagues investigated the impact of home infusion therapy on acquisition of pneumonia due to a MDRO and reported a nonsignificant AOR of 0.8 (95% CI 0.4-1.8).5 Gross and colleagues also found a nonsignificant AOR of 0 (P = .1).9 In the Shindo and colleagues study, resistance was found in 107 of 679 patients who did not receive infusion therapy, and 12 of 55 patients who were receiving infusion therapy.5 Gross and colleagues reported that home-infusion therapy was received by 0 of 20 patients with MDRO infection and 4 of the 501 patients without MDRO infection.9
Shindo and colleagues reported that home wound care was not found to be significantly related to MDRO pneumonia as well as did Gross and colleagues: AORs of 3.8 (0.8-18.4) and 1.4 (95% CI 0.5-4.4), respectively.5,9 Jung and colleagues examined IV chemotherapy in the past 30 days, and found this to not significantly impact the odds of MDRO isolation (AOR = 0.62, 95% CI 0.2-1.8).8 No data were available reflecting the risk of a family member with a MDRO.
Limitations
The variables on which logistic regression were performed differed among the studies. Therefore, results cannot be averaged or compared quantitatively, as AORs varied, depending on the variables included. In addition, data were drawn from multiple geographic locations that may impact MDRO prevalence within each patient population. Finally, this review examines the utility of the risk factors formerly included in HCAP. However, other risk factors for MDRO pneumonia outlined by the ATS/IDSA guidelines still should be considered when evaluating patient risk. The 2016 guidelines recommend local incidence of resistant strains be considered when initiating empiric therapy. Review of medical records for previous positive cultures and duration of current hospitalization also should be considered. Although the 2016 ATS/IDSA HAP guidelines are not intended for immunosuppressed patients, this risk factor also may be taken into account.
Conclusion
Review and synthesis of published literature found previous hospital admission (of ≥ 2 days in the past 90 days), admission from a nursing home, and IV antibiotic therapy in the last 90 days to be independent risk factors for identification of MDRO pneumonia in previously nonhospitalized patients (Table 2). Additionally, although no data were found to support this risk factor, existence of an in-home (close contact) source of MDROs would provide ample opportunity for transmission, so evaluation of known exposure to MDROs from contacts should be considered. When choosing empiric antibiotic therapy for patients admitted to the hospital for treatment of pneumonia, consideration of patient history and risk factors that may contribute to infection with a MDRO are recommended.
Successful treatment of pneumonia depends on timely diagnosis and administration of antibiotics. Multidrug-resistant organisms (MDROs) complicate antibiotic therapies by rendering some antibiotic agents ineffective. Inappropriate initial therapy has been associated with a more than 2-fold increase in the risk of mortality.1 Because culture results are not available immediately, clinicians prescribe antibiotics empirically and must rely on guidelines and knowledge of risk factors associated with MDRO infection to make these selections.
Treatment guidelines exist for hospital-acquired and ventilator-associated pneumonia (HAP/VAP) and community-acquired pneumonia (CAP) to assist with empiric antibiotic selection. For HAP/VAP, 2 to 3 antibiotics with a broad-spectrum of activity are used due to increased prevalence of MDROs in hospitals, whereastreatment of CAP involves more narrow coverage because bacteria that cause this infection typically have fewer antibiotic resistances.2,3 The HAP/VAP guidelines stratify the risk of pneumonia due to the presence of a MDRO acquired during a hospitalization. However, neither the CAP nor HAP/VAP guidelines offer risk-stratification guidance for nonhospitalized patients who develop pneumonia but who may have become colonized with a MDRO during a previous hospitalization or from another exposure to a health care facility.
Health care-associated pneumonia (HCAP) was first described in the 2005 American Thoracic Society and the Infectious Diseases Society of America (ATS/IDSA) nosocomial pneumonia guidelines and was associated with criteria intended to aid clinician identification of nonhospitalized patients at risk for MDRO pneumonia, which warranted empiric broad-spectrum antibiotic therapy.2 According to these guidelines, patients were classified as having HCAP if they had been hospitalized for at least 48 hours in the past 90 days, admitted from a nursing home, received recent intravenous antibiotics, had hemodialysis in the past 30 days, had a history of home infusion therapy or wound care, received intravenous chemotherapy, or had a family member with MDRO colonization.
Since publication of the 2005 guidelines, HCAP has been criticized as being a poor predictor of MDRO infection. A 2014 meta-analysis of 24 studies investigated the discriminating ability of HCAP and reported that the specificity and sensitivity for MDRO infections was 71.2% and 53.7%, respectively.3 In 2016, the ATS/IDSA guidelines were updated to remove HCAP due to the risk of antibiotic overprescribing.4
Literature Review
Although criteria previously defining a patient as having HCAP have been shown to be a poor discriminator of MDRO pneumonia as a whole, MDRO infections still pose a threat to nonhospitalized patients who have exposure to the health care system. A literature review was performed to identify independent HCAP risk factors that may increase the risk of MDRO pneumonia infecting a nonhospitalized patient needing empiric broad-spectrum antibiotic therapy. All included studies were prospective or retrospective observational cohort studies that performed logistic regression analyses to assess the association between MDRO isolation and the previously defined HCAP risk factors (Table 1).
Five studies examined the risk of MDRO infection in patients with a previous hospital admission of 2 days or more in the past 90 days. Shindo and colleagues found a significant increase in MDRO infections by about 2-fold (adjusted odds ratio [AOR], 2.1; 95% confidence interval [CI], 1.2-3.4).5 Shorr and colleagues found a 4-fold increase in likelihood of identifying a MDRO in HCAP (AOR, 4.2; 95% CI, 2.9-6.3).6 Nseir and colleagues and Jung and colleagues found similar results (AOR 3.9, 95% CI 1.7-8.8; AOR 2.7, 95% CI 1.3-5.5, respectively).7,8 Conflicting results were reported by Gross and colleagues who did not find a significant relationship between previous hospitalization and MDRO isolation (AOR 1.2, 95% CI, 0.5-3.2).9
In patients with pneumonia admitted from a nursing home, MDRO infection risk also was evaluated in these 5 studies. Shorr and colleagues, Nseir and colleagues, and Gross and colleagues found significant AORs of 2.7 (95% CI 1.7-4.3), 2.0 (95% CI 1.1-3.7), and 4.2 (95% CI 1.6-11.3), respectively.6,7,9 Shindo and colleagues (AOR 1.1; 95% CI, 0.6-2.0) and Jung and colleagues (AOR 1.9, 95% CI, 0.5-6.9) found this risk factor not significant.5
Receipt of antibiotics within the previous 90 days was assessed in 3 studies. Shindo and colleagues, Nseir and colleagues, and Gross and colleagues all found significant AORs of 2.5 (95% CI 1.2-4.0), 2.3 (95% CI 1.2-4.3), and 2.9 (95% CI 1.1-7.5), respectively.5,7,9 Antibiotic therapy within the previous 90 days is an established risk factor for MDRO pneumonia, and the 2016 ATS/IDSA guidelines consider this a risk factor for HAP and VAP, including pneumonia caused by methicillin resistant Staphylococcus aureus and Pseudomonas aeruginosa.4
The impact of hemodialysis in the previous month on acquisition of MDRO pneumonia was investigated in 4 studies. Shindo and colleagues, Jung and colleagues, and Gross and colleagues concluded that this risk factor was not significantly related to MDRO infection, reporting AORs of 2.2 (95% CI 0.5-9.7), 2.8 (95% CI 0.9-9.2) and 0.7 (95% CI 0.1-5.1), respectively.5,8,9 Shorr and colleagues, however, found a significant AOR of 2.1 (95% CI 1.0-4.3).6
Shindo and colleagues investigated the impact of home infusion therapy on acquisition of pneumonia due to a MDRO and reported a nonsignificant AOR of 0.8 (95% CI 0.4-1.8).5 Gross and colleagues also found a nonsignificant AOR of 0 (P = .1).9 In the Shindo and colleagues study, resistance was found in 107 of 679 patients who did not receive infusion therapy, and 12 of 55 patients who were receiving infusion therapy.5 Gross and colleagues reported that home-infusion therapy was received by 0 of 20 patients with MDRO infection and 4 of the 501 patients without MDRO infection.9
Shindo and colleagues reported that home wound care was not found to be significantly related to MDRO pneumonia as well as did Gross and colleagues: AORs of 3.8 (0.8-18.4) and 1.4 (95% CI 0.5-4.4), respectively.5,9 Jung and colleagues examined IV chemotherapy in the past 30 days, and found this to not significantly impact the odds of MDRO isolation (AOR = 0.62, 95% CI 0.2-1.8).8 No data were available reflecting the risk of a family member with a MDRO.
Limitations
The variables on which logistic regression were performed differed among the studies. Therefore, results cannot be averaged or compared quantitatively, as AORs varied, depending on the variables included. In addition, data were drawn from multiple geographic locations that may impact MDRO prevalence within each patient population. Finally, this review examines the utility of the risk factors formerly included in HCAP. However, other risk factors for MDRO pneumonia outlined by the ATS/IDSA guidelines still should be considered when evaluating patient risk. The 2016 guidelines recommend local incidence of resistant strains be considered when initiating empiric therapy. Review of medical records for previous positive cultures and duration of current hospitalization also should be considered. Although the 2016 ATS/IDSA HAP guidelines are not intended for immunosuppressed patients, this risk factor also may be taken into account.
Conclusion
Review and synthesis of published literature found previous hospital admission (of ≥ 2 days in the past 90 days), admission from a nursing home, and IV antibiotic therapy in the last 90 days to be independent risk factors for identification of MDRO pneumonia in previously nonhospitalized patients (Table 2). Additionally, although no data were found to support this risk factor, existence of an in-home (close contact) source of MDROs would provide ample opportunity for transmission, so evaluation of known exposure to MDROs from contacts should be considered. When choosing empiric antibiotic therapy for patients admitted to the hospital for treatment of pneumonia, consideration of patient history and risk factors that may contribute to infection with a MDRO are recommended.
1. Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. 1999;115(2):462-474.
2. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416.
3. Chalmers JD, Rother C, Salih W, Ewig S. Healthcare-associated pneumonia does not accurately identify potentially resistant pathogens: a systematic review and meta-analysis. Clin Infect Dis. 2014;58(3):330-339.
4. Kalil AC, Metersky ML, Klompas M, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61-e111.
5. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995.
6. Shorr AF, Zilberberg MD, Micek ST, Kollef MH. Prediction of infection due to antibiotic-resistant bacteria by select risk factors for health care–associated pneumonia. Arch Intern Med. 2008;168(20):2205-2210.
7. Nseir S, Grailles G, Soury-Lavergne A, Minacori F, Alves I, Durocher A. Accuracy of American Thoracic Society/Infectious Diseases Society of America criteria in predicting infection or colonization with multidrug-resistant bacteria at intensive-care unit admission. Clin Microbiol Infect. 2010;16(7):902-908.
8. Jung JY, Park MS, Kim YS, et al. Healthcare-associated pneumonia among hospitalized patients in a Korean tertiary hospital. BMC Infectious Diseases. 2011;11:61.
9. Gross AE, Van Schooneveld TC, Olsen KM, et al. Epidemiology and predictors of multidrug-resistant community-acquired and health care-associated pneumonia. Antimicrob Agents Chemother. 2014;58(9):5262-5268.
1. Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. 1999;115(2):462-474.
2. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416.
3. Chalmers JD, Rother C, Salih W, Ewig S. Healthcare-associated pneumonia does not accurately identify potentially resistant pathogens: a systematic review and meta-analysis. Clin Infect Dis. 2014;58(3):330-339.
4. Kalil AC, Metersky ML, Klompas M, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61-e111.
5. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995.
6. Shorr AF, Zilberberg MD, Micek ST, Kollef MH. Prediction of infection due to antibiotic-resistant bacteria by select risk factors for health care–associated pneumonia. Arch Intern Med. 2008;168(20):2205-2210.
7. Nseir S, Grailles G, Soury-Lavergne A, Minacori F, Alves I, Durocher A. Accuracy of American Thoracic Society/Infectious Diseases Society of America criteria in predicting infection or colonization with multidrug-resistant bacteria at intensive-care unit admission. Clin Microbiol Infect. 2010;16(7):902-908.
8. Jung JY, Park MS, Kim YS, et al. Healthcare-associated pneumonia among hospitalized patients in a Korean tertiary hospital. BMC Infectious Diseases. 2011;11:61.
9. Gross AE, Van Schooneveld TC, Olsen KM, et al. Epidemiology and predictors of multidrug-resistant community-acquired and health care-associated pneumonia. Antimicrob Agents Chemother. 2014;58(9):5262-5268.
Impact of Drug Shortages on Patient Safety and Pharmacy Operation Costs
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table). 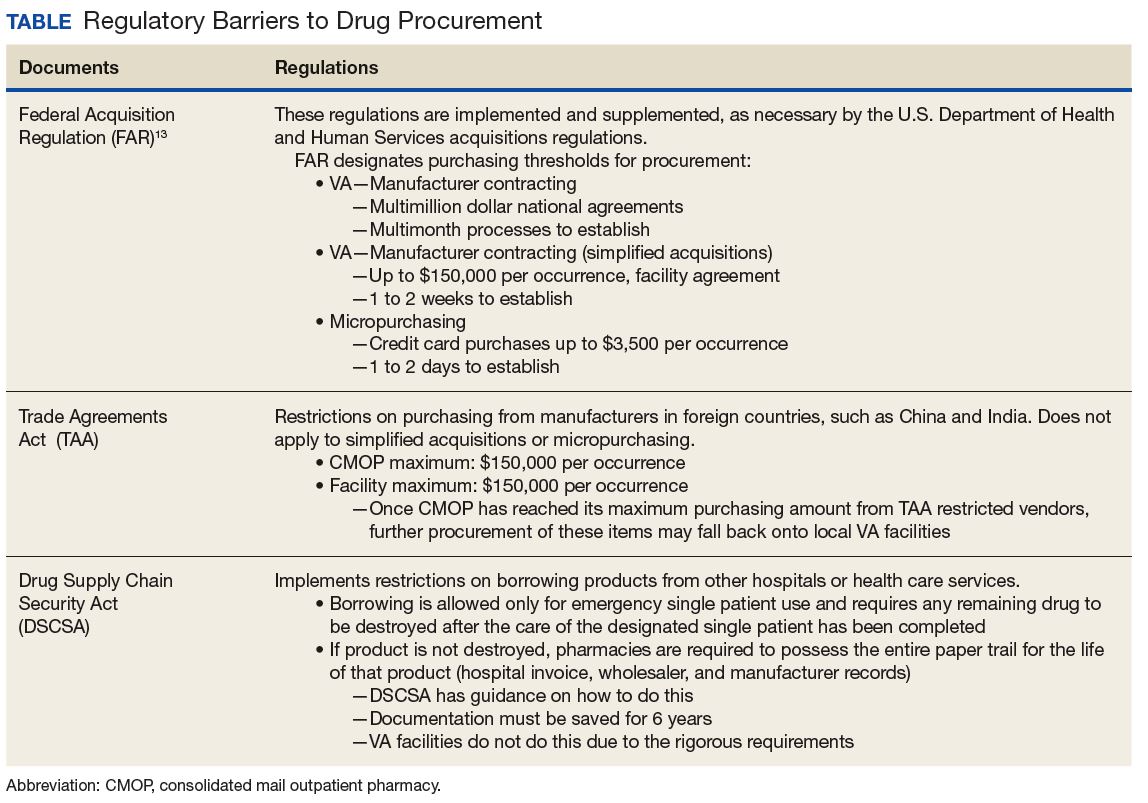
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact. 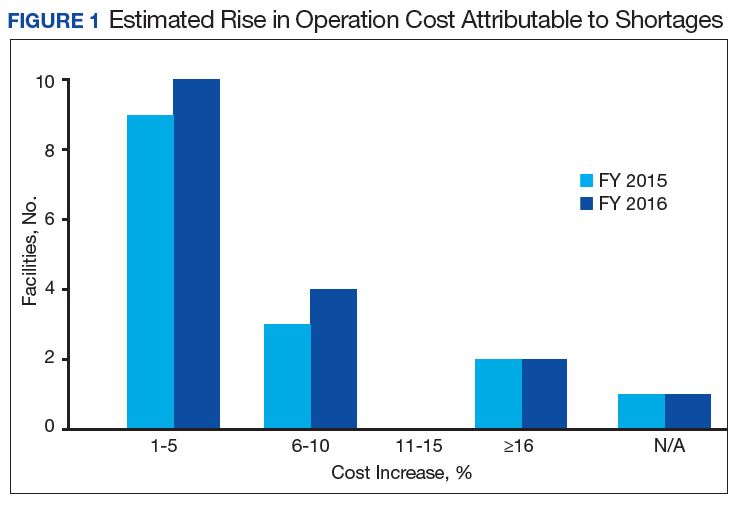
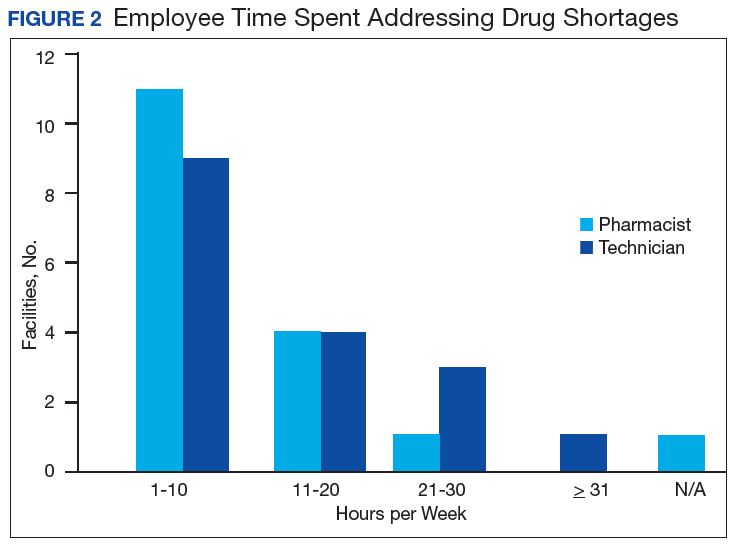
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review. 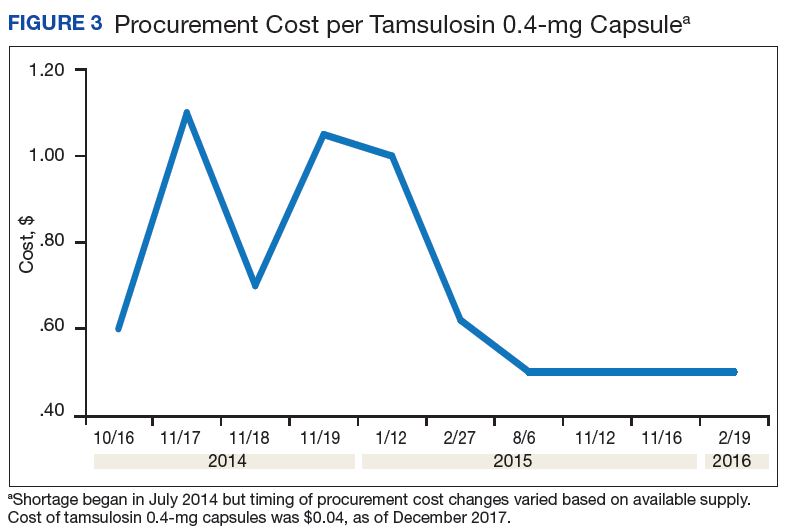
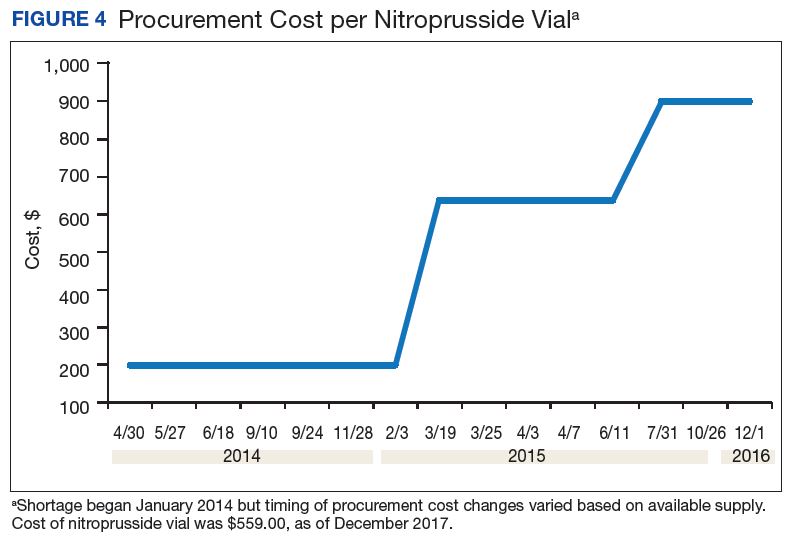
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table).
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact.
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review.
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
Drug product shortages threaten health care quality and public health by creating barriers to optimal care. The frequency of drug shortages has risen dramatically since 2005 and now influences broad areas of health care practice. More than 400 generic drug products have been affected, forcing institutions to purchase costly brand-name products, substitute alternative therapies, or procure from gray market vendors at increased institutional costs.1 Scarcity and cost have potential to negatively impact patient outcomes and the ability of health care organizations to respond to the needs of their patients.
Background
Although constantly fluctuating, the number of active shortages reached a height of 320 products at the end the third quarter of 2014.2 A 2011 analysis from Premier Healthcare Alliance estimated the added cost of purchasing brand, generic, or alternative drugs due to shortage may have inflated hospital costs by $200 million annually.1 In 2016, the number of active shortages dropped to 176, suggesting a downward trend. However, the drug supply chain remains a concern for pharmacies in the U.S.
Despite creative approaches to shortage management, the variable characteristics of shortages make planning difficult. For example, the drug product in short supply may or may not have an alternative for use in similar clinical scenarios. The impact of shortages of medications lacking an equivalent alternative product has been documented, such as the past shortage of succinylcholine for anesthesia, resulting in surgery cancellations when an alternative paralytic agent was not appropriate.3 In 2016, the Cleveland Clinic reported undertaking “military-style triage” in determining patients who required use of aminocaproic acid during open heart surgery due to its limited supply.4 Decisions to reserve drug supply for emergency use and prefilling syringes under pharmacy supervision to extend stability and shelf life are short-term solutions to larger, systemic issues. Unfortunately, these scenarios have the potential to disrupt patient care and diminish health outcomes.
Shortages of products that have an available therapeutic substitution may seem easily manageable, but additional considerations may be present. Bacillus Calmette-Guérin (BCG) is considered the drug of choice for bladder cancer. In 2011, there was a shortage of the BCG vaccine after mold was discovered in the formulation.5 Providers were forced to choose between reducing or reallocating the dose of BCG, turning away patient, or substituting mitomycin C, which is less effective and costlier. When tamsulosin capsules became difficult to obtain in 2014, some institutions began switching patients to alfuzosin.6 Although alfuzosin is similar in mechanism to tamsulosin, it may prolong the QTc interval. Not only did this substitution present a contraindication for patients with elevated QTc intervals or who were already receiving concomitant medications that prolonged the QTc interval, but also it required additional cost and resources needed to update electrocardiograms.
VA Consolidated Mail Outpatient Pharmacies
The VHA serves nearly 9 million patients at more than 1,200 facilities across the U.S.7 This large patient population results in an estimated 149 million outpatient prescriptions annually.8 About 80% of these are distributed by mail through 7 VA consolidated mail outpatient pharmacies (CMOPs). When drug scarcity impedes the ability of the CMOP to respond to medication demand, the local facility must fill these prescriptions. These rejections sent back to the facility impact workload, patient wait times, and access to medication therapy. Barriers to medication procurement in the VA also stem from regulations based on legislation, including the Trade Agreements Act, Drug Supply Chain Security Act, and the Federal Acquisition Regulation (FAR) (Table).
The impact of drug shortages has been described previously in the private sector, particularly for emergency medicine and chemotherapy.9,10 However, the impact of drug shortages on health care provision to veteran populations within the VA has not previously been analyzed. Due to the unique procurement regulations that influence the VA and the importance of continuing to provide optimal health care services to veterans, assessing the impact of drug shortages on patient safety and health care costs is necessary in informing policy decisions and guiding recommendations for mitigation strategies. The purpose of this study was to assess the influence of drug shortages on institutional costs and patient care within VA facilities and formulate recommendations for enhanced mitigation of this issue.
Methods
The primary outcome of this study was to characterize the impact of drug shortages on institutional cost and patient safety events among VHA facilities. Secondary outcomes included subgroup evaluation in reported drug shortage impact among 1a, 1b, and 1c complexity VA facility survey respondents and assessment of drug shortage impact on CMOP prescription order fulfillment and operation cost.
Definitions
The complexity ranking system is a facility grouping method used within the VA to characterize the level of service provision, teaching and research opportunities, patient volume, intensive care unit level, and other factors offered by a VA site. Rankings start from 1 (highest level of services offered) to 3 (lowest level of services offered), with level 1 facilities further divided into a, b, and c subdivisions. A level 1a facility will be larger with more services offered than a 1b, which is larger and offers more services than a 1c facility. The VA facilities are further characterized by regional distribution. Sites are grouped under VISNs of which there are currently 21.
The CMOP program was responsible for dispensing about 119 million outpatient prescriptions in 2016 and includes designated sites for the dispensing of controlled substances and supply items. The VA Pharmacy Benefits Management Service (PBM) oversees formulary management, plans national drug policy, promotes safe and appropriate drug therapy, and delivers high-quality and sustainable pharmacy benefits for veterans.
Study Design
A descriptive study was initiated to characterize the impact of drug shortages among VA facilities. An analysis of administrative medication safety event reporting and institutional costs data at the Denver VAMC in Colorado was done, focusing on predetermined drug products involved in a recent shortage. The analysis was accomplished through a review of the VA adverse drug events reporting system (VA ADERS) reports and a local medication errors quality improvement database and paper procurement records, respectively. Concurrently, a survey was disseminated among qualifying VA facilities across the country that sought to characterize the impact of drug shortages nationally.
Sample Selection
Denver VAMC. The Denver VAMC, where the authors were located, was selected as the local sample site. The intention was to compare the strategies used locally with strategies used among similar (level 1a, 1b, and 1c) facilities. Preselected “cost-impacting” drug products were identified through a review of historic shortages with a significant local impact. These drugs were defined as low cost/high utilization (eg, tamsulosin 0.4-mg capsules and ketorolac solution), medium cost/utilization (eg, piperacillin/tazobactam IV solutions and aminocaproic acid solution), and high cost/low utilization (eg, nitroprusside IV solution and BCG vaccine solution). Additionally, patient safety event data reported internally for quality improvement and locally via VA ADERS were reviewed for preselected “safety impact” drug products and included BCG vaccine, tamsulosin capsules, IV fluid products, calcium gluconate and chloride injections, and aminocaproic acid injection.
National Survey. The authors identified 84 level 1 complexity facilities and used the PBM pharmacy directory to contact the administrative personnel representing each facility. These representatives identified a point of contact to aid in survey completion. A separate survey also was sent to the CMOP facilities (survey outlines available at www.fedprac.com).
Data Collection
Denver VAMC. Financial data were sampled through a manual review of paper procurement records stored by date in the inpatient pharmacy of the Denver VAMC. Variables included units of product used over the period of drug shortage, cost per unit during shortage, and cost per unit before shortage. This information also was supplemented with data from the prescription processing software’s drug file. Patient safety data were gathered through query of the identified event reporting databases for the prespecified drug on shortage. These variables included the type of error and the effect the error had on the patient.
National Survey. Data collection focused on notable drug shortages and patient safety reporting between January 1, 2013 and December 31, 2016. The survey was maintained in a facility-specific spreadsheet. Editing capabilities were disabled for all actions other than responding to questions. Recipients were followed up with a courtesy e-mail after 2 weeks and another 2 times unless a survey was received. Data were de-identified and aggregated for analyses.
Statistical Analyses
Excel 2010 (Microsoft, Redmond, WA) descriptive statistics were used to relay information from this assessment. Extrapolations from procurement cost data and drug product utilization were used to estimate the enhanced direct cost associated with identified drug shortages. Similar extrapolations were used to estimate the cost associated with shortages leading to CMOP rejection and local fill.
Results
Survey completion totaled 20% of invited facilities (n = 17). Good geographic and VISN distribution was noted with representatives from VISNs 2, 4, 8, 9, 10, 12, 15, 16, 21, and 22. VISNs 10 and 12 provided the most representation with 3 participants, each. Level 1a facilities participated most (n = 9), followed by 1b (n = 6) and 1c (n = 2). Participating facilities reported a mean (SD) of 54 (21.5) pharmacists and 34 (15.3) pharmacy technician staff members employed. The most common reason for not participating was lack of personnel resources and competing demands. The CMOP participation was 100% (n = 7) and completed through a coordinated response.
Results of the budgetary increase and staff member time allocation survey assessments are provided (Figures 1 and 2). Five facilities provided an annual estimate of increased cost due to acquisition of drugs on shortage through open market purchases that ranged from about $150,000 to $750,000. Nearly half of the surveyed facilities endorsed having a drug shortage task force (n = 8) to respond to drug shortages and mitigate their impact.
Regarding drug product allocation, only 2 facilities did not have current restrictions for use due to a shortage. Many had between 1 and 10 of these restrictions implemented to conserve supply (n = 11, 64%), 2 facilities reported 11 to 20 restrictions, and 2 facilities noted more than 30 restrictions. Similarly, 3 facilities had not needed to revise any current treatment protocols due to drug shortages. The majority of facilities had revised 1 to 5 current protocols (n = 12, 70%), 1 revised 6 to 10 protocols, and 1 facility revised more than 10 protocols.
In assessing patient safety concerns, 1 facility identified a history of transferring patients to alternative medical sites for the patients to obtain necessary medication impacted by a local shortage. Additionally, during the BCG vaccine shortage, 6 facilities (35.3%) substituted mitomycin C for the treatment of urinary bladder cancer.
Most participants either agreed (n = 8, 47.0%) or strongly agreed (n = 4, 23.5%) that modifications to FAR to increase purchasing opportunities from foreign distributors during drug shortage would help mitigate the impact of such shortages. Similarly, most participants agreed (n = 10, 58.8%) or strongly agreed (n = 3, 17.6%) that PBM guidance on drug shortage management would help efficiently and effectively respond to issues that might arise. The consensus of participants also agreed (n = 13, 76.5%) that organized collaborations or working groups within each VISN might help assist in drug shortage management.
The CMOP facility data revealed that 2 sites did not require dedicated staffing to respond to shortages, and 3 sites had not experienced cost increases because of shortages. Pharmacist use varied between sites, with 2 facilities using 1 to 10 pharmacist h/wk, and 1 facility using 11 to 20 pharmacist h/wk, and 1 facility using 21 to 30 pharmacist h/wk. Technician utilization was more pronounced, with 2 facilities using more than 30 technician h/wk, and 2 facilities using 1 to 10 technician h/wk. Workload and costs may have been influenced in other ways as 3 sites endorsed using overtime pay, shifting product responsibility between CMOPs, prolonging patient wait times, and close monitoring for each. In fiscal year 2015, some sites experienced a 1% to 5% (n = 2) and 6% to 10% (n = 1) increase in operation cost attributable to shortage. Results from fiscal year 2016 showed that some sites continued to see a 1% to 5% (n = 1) and 6% to 10% (n = 2) increase in operation cost attributable to shortage.
Through aggregation of CMOP responses on the number of prescriptions sent back to local facility for fill due to back order, a downward trend in the total number of rejections was seen over the 2.5 fiscal years assessed. This amounted to more than 1 million rejections in fiscal year 2015, about 788,000 rejections in 2016, and about 318,000 rejections through the first 2 quarters of 2017.
A consistent rise in the medication procurement budget requirement was characterized within the single VA facility review. The quarterly median increase was 2.7% over 2.5 years (min: -1.4%; max: 6.6%) for total outpatient medication costs, excluding hepatitis C antiviral therapies. Procurement cost records were insufficient to characterize historic expenditures for 4 of the prespecified drug products. The data collected on tamsulosin capsule and nitroprusside vial procurement during shortage is provided (Figures 3 and 4). Over the time frame of procurement records found on review, the added costs of nitroprusside vials and tamsulosin capsules were $22,766.09 (+167.9% of base cost) and $17,433.70 (+657.3% of base cost), respectively. No patient safety data were found on review.
Discussion
Drug product shortages represent a barrier to quality and efficiency across health care institutions. A survey of health system pharmacies in the southeastern U.S. found that the majority of respondents tracking shortage data reported a 300% to 500% markup by alternative or gray market suppliers for hard-to-find medications.11 These reports are similar to the authors’ analyses of the trends in increased procurement expenditures documented during the tamsulosin capsule and nitroprusside vial shortages and indirectly correlate with the survey results indicating that most facilities endorsed a trend in operation cost increase attributable to drugs product shortage. The estimated annual costs for open market purchases further informs the financial burden aggregated by this issue.
Indirect costs from drug shortage further complicated quantifying the impact of shortages. Many facilities acknowledged the indirect influence drug shortages have on staffing and workload due to the implementation of mitigation strategies. Most participants found it necessary to establish restrictions for use in addition to altering protocols. These required the time investment of essential personnel from development through execution and education. Situations also can arise for mass therapeutic substitution. In this example, pharmacy staff may be required to oversee medication transition from the product on shortage to an appropriate alternative. When substitution involves hundreds or thousands of outpatient prescriptions, such as the tamsulosin shortage, the process may be tedious and time consuming, depending on the level of clinical decision making needed to determine patient candidacy for transitioning products.
Improving institutional cost efficiency becomes a significant challenge with persistent drug shortages. Professional advocacy groups, such as the American Society of Health-System Pharmacists (ASHP), help provide guidance to organizations constrained by specific drug shortages.12 Staff knowledgeable in allocation, supply considerations, and product repackaging and stability data also are essential. Other mitigation strategies include automatic substitutions, restrictions for use or inventory control strategies, and open market procurement, or borrowing from other institutions.
Data gathered from the survey of CMOP facilities also helped elucidate strategies used to mitigate drug shortage impacts for those respondents impacted by shortage. Likely, the 2 CMOP facilities without dedicated staff focused on shortages are those whose outpatient prescription fulfillment responsibility were focused on supply items or controlled substances. The impacted CMOP respondents cited overtime pay, shifting product responsibility, and prolonging patient wait times as the most frequently employed mitigation strategies. When these and other strategies fail to manage a shortage, prescriptions are often sent back to the local facility to be filled. Unfortunately for these facilities, the same mitigation strategies used by CMOP are not always feasible. Overtime pay may not be possible given staffing and budgetary resources, sending prescriptions back to facilities in itself prolongs patient wait times, and local medical centers do not have the option of shifting product responsibility between sites or sending the prescription to another facility. Herein lies 1 rationale for the CMOP effort to reduce the volume of prescriptions sent back to local medical centers.
Multiple offices within the FDA have roles in the mitigation of national drug shortages within their regulatory purview. Much of the recent focus stems from provisions enacted under Title X of the FDA Safety and Innovation Act of 2012, which addresses problems in the drug-supply chain.12 Rectifying a shortage involves short- and long-term strategic planning to address supply, distribution, and market reaction to need. Collaboration between the FDA and manufacturers is one method by which demand can be satisfied through the coordination of resources, expedition of inspections, and root cause analysis of the shortage.
Similar collaborations within the VA were viewed favorably by respondents and might yield productive relationships if regional or VISN working groups were to be established. Alternative long-term strategies are executed through regulation, particularly concerning the importation of foreign manufactured drugs and regulatory discretion on supplier vetting. Despite a strong respondent consensus that regulatory modifications of foreign product importation in the setting of a drug shortage may be beneficial, such a change would require a congressional action and is not likely to be timely. Unfortunately, gray market pharmaceutical distribution, driven by wholesaler stockpiling to raise prices, is separate from manufacturer driven shortages and falls outside the FDA’s regulatory purview and institutional mitigation strategies.
Although based on this limited survey, general agreement existed on the importance of greater national collaboration and communication regarding drug shortage management strategies. This could include PBM guidance on specific shortage management opportunities or establishing collaborations by region or VISN. These possibilities may be more realistically attainable in comparison to modifying federal regulations on drug product procurement during active shortages, which requires an act of Congress. Many of the survey participants endorsed a drug shortage task force within their facility. Coordinating interaction between preexisting or newly established task forces or working groups on a monthly or quarterly basis may provide fruitful interactions and the exchange of strategies to reduce shortage impact on institutional cost, efficiency, and patient care.
Limitations
Quantifying the extent of drug shortage impact on patient safety and institutional costs is a difficult task. The procurement records data used for the analysis of a single VAMC were gathered through manual review of stored paper invoices, opening the possibility for missing data. It is also difficult to extrapolate the sum of indirect costs such as process changes, alternative product utilization, and pharmacy staffing resources as additional financial burdens to the affected institution. Any quantifiable cost assessment also is biased by contract terms between the VA and wholesalers in which unavailable products that must be purchased off-contract are subsequently reimbursed through credit or alternative means.
Patient safety events are frequently underreported, leading to underestimation of true safety event incidence. Given that these events are documented by multiple disciplines and that many of these documenters may not be aware consistently of the drug products and volume impacted by shortage, elucidating safety events unfolding in relation to shortage also is difficult to quantify.
The response rate for the survey was low but near the expected rate for this methodology. Feedback from several facilities was received, citing competing demands and workforce shortage as barriers to participation. The survey also was limited by reporting bias and recall bias. As assessment of prespecified past drug shortages may require intimate knowledge of pharmacy department processes and mitigation strategies, the accuracy of question answering may have been limited to the length of time the points of contact had been in their current position.
Conclusion
Drug shortages are a pervasive barrier to patient care within larger facilities of the VA health care system, similar to what has been characterized in the private sector. As a result of these shortages and the mitigation strategies to reduce their burden, many facilities endorsed trends in increasing workload for staff, institutional operation costs, and risk for patient safety and care quality concerns. Due to the demands of shortages, some facilities have implemented drug shortage task forces or equivalent groups to specifically manage these issues. Moving forward, the VA health care system may benefit from similar task forces or working groups at the VISN level, to aid in collaborative efforts to respond to shortage. Support for revising federal regulations on procurement in times of shortage and enhanced PBM drug shortage management guidance also was endorsed.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.
1. Cherici C, Frazier J, Feldman M, et al. Navigating drug shortages in American healthcare: a premier healthcare alliance analysis. https://www.heartland.org/_template-assets/documents/publications/30103.pdf. Published March 2011. Accessed December 5, 2017.
2. American Society of Health-System Pharmacists. ASHP drug shortage statistics. https://www.ashp.org/Drug-Shortages/Shortage-Resources/Drug-Shortages-Statistics. Updated 2017. Accessed December 5, 2017.
3. Dooren JC. Most hospitals face drug shortages. The Wall Street Journal. http://www.wsj.com/articles/SB10001424052702304584404576442211187884744. Published July 13, 2011. Accessed December 5, 2017.
4. Fink S. Drug shortages forcing hard decisions on rationing treatment. The New York Times. http://www.nytimes.com/2016/01/29/us/drug-shortages-forcing-hard-decisions-on-rationing-treatments.html. Published January 29, 2016. Accessed December 5, 2017.
5. Loftus P. Drug shortages frustrate doctors, patients. The Wall Street Journal. http://www.wsj.com/articles/u-s-drug-shortages-frustrate-doctors-patients-1433125793. Published May, 31, 2015. Accessed December 5, 2017.
6. U.S. Food and Drug Administration. Strategic plan for preventing and mitigating drug shortages. http://www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf. Published October 2013. Accessed August 22, 2016.
7. U.S. Department of Veteran Affairs, National Center for Veterans Analysis and Statistics. Quick facts. https://www.va.gov/vetdata/Quick_Facts.asp. Updated November 20, 2017. Accessed December 5, 2017.
8. U.S. Department of Veterans Affairs, Office of the Inspector General. Audit of Consolidated Mail Outpatient Pharmacy Program. https://www.va.gov/oig/pubs/VAOIG-15-05255-422.pdf. Accessed December 11, 2017.
9. Mazer-Amirshahi M, Pourmand A, Singer S, Pines JM, van den Anker J. Critical drug shortages: implications for emergency medicine. Acad Emerg Med. 2014;21(6):704-711.
10. McBride A, Holle LM, Westendorf C, et al. National survey on the effect of oncology drug shortages on cancer care. Am J Health Syst Pharm. 2013;70(7):609-617.
11. Caulder CR, Mehta B, Bookstaver PB, Sims LD, Stevenson B; South Carolina Society of Health-System Pharmacists. Impact of drug shortages on health system pharmacies in the southeastern United States. Hosp Pharm. 2015;50(4):279-286.
12. Florida Society of Health-System Pharmacists. Conservation strategies for IV fluids. http://www.fshp.org/news/165998/Conservation-Strategies-for-IV-Fluid.htm. Accessed December 11, 2017.
13. Federal Acquisition Regulation Site. FAR—Part 13 Simplified Acquisition Procedures, 13 CFR §§ 201-302. http://farsite.hill.af.mil/reghtml/regs/far2afmcfars/fardfars/far/13.htm. Updated January 13, 2017. Accessed December 5, 2017.

FDA Boxed Warning Updates: December 2017
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE
- Edited and updated warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRARAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Codeine sulfate tablets are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of codeine sulfate tablets in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a CYP2D6 polymorphism. Tuxarin ER is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuxarin ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Concomitant Use with Benzodiazepines, CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound
sedation, respiratory depression, coma, and death. Avoid use of opioid cough medications in patients taking benzodiazepines, other CNS depressants, or alcohol.
TUZISTRA XR (CHLORPHENIRAMINE POLISTIREX; CODEINE POLISTIREX):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a
CYP2D6 polymorphism. Tuzistra XR is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuzistra XR in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORICET W/CODEINE (ACETAMINOPHEN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and HEPATOTOXICITY
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Butalbital, acetaminophen, caffeine, and codeine phosphate capsules are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of butalbital, acetaminophen, caffeine, and codeine phosphate capsules in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORINAL W/CODEINE (ASPIRIN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; and INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES.
Risks From Concomitant Use With Benzodiazepines or Other CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
- Reserve concomitant prescribing of Fiorinal with codeine and benzodiazepines or other CNS depressants for use in patients for whom alternative treatment options are inadequate.
- Limit dosages and durations to the minimum required.
- Follow patients for signs and symptoms of respiratory depression and sedation.
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. Fiorinal with codeine is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Fiorinal with codeine in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
PHENERGAN VC W/CODEINE (CODEINE PHOSPHATE; PHENYLEPHRINE HYDROCHLORIDE; PROMETHAZINE HYDROCHLORIDE): PHENERGAN W/CODEINE (CODEINE PHOSPHATE; PROMETHAZINE HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Promethazine HCl and codeine phosphate oral solution is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of promethazine HCl and codeine phosphate oral solution in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Promethazine and Respiratory Depression in Children
Postmarketing cases of respiratory depression, including fatalities have been reported with use of promethazine in pediatric patients. Children may be particularly sensitive to the additive respiratory depressant effects when promethazine is combined with other respiratory depressants, including codeine.
SYNALGOS-DC (ASPIRIN; CAFFEINE; DIHYDROCODEINE BITARTRATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF DIHYDROCODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Synalgos-DC is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Synalgos-DC in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of dihydrocodeine.
CONZIP (TRAMADOL HYDROCHLORIDE): ULTRAM (TRAMADOL HYDROCHLORIDE): ULTRACET (ACETAMINOPHEN; TRAMADOL HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFETHREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF TRAMADOL AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received tramadol. Some of the reported cases followed tonsillectomy and/or adenoidectomy; and at least one case, the child had evidence of being an ultra-rapid metabolizer of tramadol due to a CYP2D6 polymorphism. Ultram is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Ultram in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of tramadol.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE
- Edited and updated warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRARAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Codeine sulfate tablets are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of codeine sulfate tablets in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a CYP2D6 polymorphism. Tuxarin ER is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuxarin ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Concomitant Use with Benzodiazepines, CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound
sedation, respiratory depression, coma, and death. Avoid use of opioid cough medications in patients taking benzodiazepines, other CNS depressants, or alcohol.
TUZISTRA XR (CHLORPHENIRAMINE POLISTIREX; CODEINE POLISTIREX):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a
CYP2D6 polymorphism. Tuzistra XR is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuzistra XR in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORICET W/CODEINE (ACETAMINOPHEN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and HEPATOTOXICITY
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Butalbital, acetaminophen, caffeine, and codeine phosphate capsules are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of butalbital, acetaminophen, caffeine, and codeine phosphate capsules in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORINAL W/CODEINE (ASPIRIN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; and INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES.
Risks From Concomitant Use With Benzodiazepines or Other CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
- Reserve concomitant prescribing of Fiorinal with codeine and benzodiazepines or other CNS depressants for use in patients for whom alternative treatment options are inadequate.
- Limit dosages and durations to the minimum required.
- Follow patients for signs and symptoms of respiratory depression and sedation.
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. Fiorinal with codeine is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Fiorinal with codeine in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
PHENERGAN VC W/CODEINE (CODEINE PHOSPHATE; PHENYLEPHRINE HYDROCHLORIDE; PROMETHAZINE HYDROCHLORIDE): PHENERGAN W/CODEINE (CODEINE PHOSPHATE; PROMETHAZINE HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Promethazine HCl and codeine phosphate oral solution is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of promethazine HCl and codeine phosphate oral solution in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Promethazine and Respiratory Depression in Children
Postmarketing cases of respiratory depression, including fatalities have been reported with use of promethazine in pediatric patients. Children may be particularly sensitive to the additive respiratory depressant effects when promethazine is combined with other respiratory depressants, including codeine.
SYNALGOS-DC (ASPIRIN; CAFFEINE; DIHYDROCODEINE BITARTRATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF DIHYDROCODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Synalgos-DC is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Synalgos-DC in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of dihydrocodeine.
CONZIP (TRAMADOL HYDROCHLORIDE): ULTRAM (TRAMADOL HYDROCHLORIDE): ULTRACET (ACETAMINOPHEN; TRAMADOL HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFETHREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF TRAMADOL AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received tramadol. Some of the reported cases followed tonsillectomy and/or adenoidectomy; and at least one case, the child had evidence of being an ultra-rapid metabolizer of tramadol due to a CYP2D6 polymorphism. Ultram is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Ultram in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of tramadol.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
CODEINE SULFATE
- Edited and updated warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRARAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Codeine sulfate tablets are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of codeine sulfate tablets in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN AND RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a CYP2D6 polymorphism. Tuxarin ER is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuxarin ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Concomitant Use with Benzodiazepines, CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound
sedation, respiratory depression, coma, and death. Avoid use of opioid cough medications in patients taking benzodiazepines, other CNS depressants, or alcohol.
TUZISTRA XR (CHLORPHENIRAMINE POLISTIREX; CODEINE POLISTIREX):
- Edited warning August 2017
ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultrarapid metabolizer of codeine due to a
CYP2D6 polymorphism. Tuzistra XR is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Tuzistra XR in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORICET W/CODEINE (ACETAMINOPHEN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and HEPATOTOXICITY
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Butalbital, acetaminophen, caffeine, and codeine phosphate capsules are contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of butalbital, acetaminophen, caffeine, and codeine phosphate capsules in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
FIORINAL W/CODEINE (ASPIRIN; BUTALBITAL; CAFFEINE; CODEINE PHOSPHATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; and INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES.
Risks From Concomitant Use With Benzodiazepines or Other CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death.
- Reserve concomitant prescribing of Fiorinal with codeine and benzodiazepines or other CNS depressants for use in patients for whom alternative treatment options are inadequate.
- Limit dosages and durations to the minimum required.
- Follow patients for signs and symptoms of respiratory depression and sedation.
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. Fiorinal with codeine is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Fiorinal with codeine in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
PHENERGAN VC W/CODEINE (CODEINE PHOSPHATE; PHENYLEPHRINE HYDROCHLORIDE; PROMETHAZINE HYDROCHLORIDE): PHENERGAN W/CODEINE (CODEINE PHOSPHATE; PROMETHAZINE HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Promethazine HCl and codeine phosphate oral solution is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of promethazine HCl and codeine phosphate oral solution in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine.
Promethazine and Respiratory Depression in Children
Postmarketing cases of respiratory depression, including fatalities have been reported with use of promethazine in pediatric patients. Children may be particularly sensitive to the additive respiratory depressant effects when promethazine is combined with other respiratory depressants, including codeine.
SYNALGOS-DC (ASPIRIN; CAFFEINE; DIHYDROCODEINE BITARTRATE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF DIHYDROCODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Life-threatening respiratory depression and death have occurred in children who received codeine; most cases followed tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a CYP2D6 polymorphism. Synalgos-DC is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Synalgos-DC in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of dihydrocodeine.
CONZIP (TRAMADOL HYDROCHLORIDE): ULTRAM (TRAMADOL HYDROCHLORIDE): ULTRACET (ACETAMINOPHEN; TRAMADOL HYDROCHLORIDE):
- Edited warning August 2017
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFETHREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF TRAMADOL AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; NEONATAL OPIOID WITHDRAWAL SYNDROME; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children
Life-threatening respiratory depression and death have occurred in children who received tramadol. Some of the reported cases followed tonsillectomy and/or adenoidectomy; and at least one case, the child had evidence of being an ultra-rapid metabolizer of tramadol due to a CYP2D6 polymorphism. Ultram is contraindicated in children younger than 12 years of age and in children younger than 18 years of age following tonsillectomy and/or adenoidectomy. Avoid the use of Ultram in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of tramadol.
FDA Boxed Warning Updates
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
The Role of Methicillin-Resistant Staphylococcus aureus Polymerase Chain Reaction Nasal Swabs in Clinical Decision Making
Methicillin-resistant Staphylococcus aureus (MRSA) is a Gram positive, round bacterium. The bacteria has evolved to withstand attacks from antibiotics and has made MRSA resistant to traditional antibiotics, such as β-lactams, resulting in difficult-to-treat infections. The presence of a genetic mutation within the mecA gene, which codes for the penicillin-binding protein 2a (PBP2a), differentiates MRSA from methicillin-susceptible Staphylococcus aureus (MSSA). Presence of the PBP2a protein allows Staphylococcus aureus (S aureus)to overcome β-lactam antibiotics’ method of killing by allowing the bacteria to continue to divide and grow.
β-lactam antibiotics cause cell death in susceptible isolates by binding to penicillin-binding proteins, which inhibits transpeptidation within the cell wall via inactivation of the penicillin-binding protein. By inhibiting cell wall synthesis, the cell loses its integrity and leaks its contents, causing cell death. Penicillin-binding protein 2a is a modified protein that has a low affinity for β-lactam antibiotics, allowing MRSA to survive and making it dangerous and difficult to eradicate.
First described in 1961, MRSA’s prevalence steadily increased in the following decades. It is the most common cause of skin and soft tissue infections presenting to emergency departments in the U.S.1 About 20% of bloodstream infections are caused by S aureus, and in 2003, nearly two-thirds of hospital-onset S aureus infections were methicillin-resistant in U.S. intensive-care units (ICUs).2 It has been shown that patients with MRSA bacteremia have worse overall outcomes, including increased mortality, greater lengths of stay, and increased costs, compared with those with MSSA infections.2,3 In 2011, MRSA infections caused an estimated 11,000 deaths, making fast and accurate detection of MRSA a crucial step in appropriate antimicrobial therapy selection.4
Currently, the Clinical and Laboratory Standards Institute (CLSI) recommends testing for MRSA by using phenotypic or genotypic methods. Phenotypic methods test for the observable characteristics of an organism, whereas a genotypic method identifies the specific gene that the organism carries. Recommended phenotypic methods include the latex agglutination test for PBP2a, the cefoxitin disk screen test, and a plate containing 6 μg/mL of oxacillin in Mueller-Hinton agar supplemented with sodium chloride.5 These methods have varying sensitivity and specificity and take between 48 to 72 hours to provide a result.
Within the past 15 years, a newer, genotypic, method of MRSA detection was approved by the FDA with high sensitivity and specificity. This method uses polymerase chain reaction (PCR) to identify the mecA gene. Polymerase chain reaction is a technique used to copy and amplify a specific segment of DNA, making thousands to millions of copies. If present, the MRSA PCR amplifies the mecA gene that makes S aureus resistant to methicillin and other β-lactams, which confirms that the specimen contains MRSA. The FDA has approved the use of MRSA PCR nasal swabs to detect MRSA in patients at risk of nasal colonization. While previously discussed methods may take between 2 and 3 days to confirm presence of MRSA, PCR can identify MRSA in about 1 hour.6
If a S aureus infection is suspected, empiric therapy often includes coverage of both MSSA and MRSA, due to the high morbidity and mortality associated with these infections. However, continuing an unneeded or unduly broad antibiotic, such as those that cover MRSA, can cause unintended consequences, such as toxicities, emerging resistance, or selection for pathogenic organisms.7 Therefore, empiric broad antibiotic therapy should be de-escalated as soon as possible, which further emphasizes the need for quick and accurate detection of the infecting organism. De-escalation of therapy can lead to a shorter length of stay and decreased mortality.8,9 Conversely, quick identification of infections caused by MRSA would allow therapy to be broadened to cover MRSA in infected patients, which could potentially decrease patient morbidity and mortality.
Nasal MRSA PCR Colonization
Rapid identification of a causative organism is crucial to determine appropriate antibiotic therapy. Fortunately, PCR is a very rapid method of detecting MRSA, and the use of MRSA PCR nasal swabs may be an effective way to predict whether MRSA is the organism causing an infection at various anatomical sites. If a patient has a suspected infection on admission, a MRSA PCR nasal swab often is completed to determine whether a patient’s nares are colonized with MRSA. However, there is no clear consensus in the literature regarding the correlation between MRSA nasal colonization and an infection caused by MRSA, making it difficult for clinicians to confidently de-escalate therapy on a negative MRSA PCR or broaden therapy on a positive result. The purpose of this literature review was to determine whether a MRSA PCR nasal swab can be used as a surrogate marker for MRSA infections at various sites.
Pneumonia has many potential causative organisms, many of which are covered empirically with guideline-directed therapy. The predictive power of MRSA PCR nasal swabs may allow clinicians to prescribe earlier directed therapy. A retrospective cohort study performed at a tertiary care center looked at the clinical usefulness of a MRSA PCR nasal swab in the treatment of pneumonia.10 Patients were included in the trial if they had a MRSA PCR nasal swab within 1 month of their blood or sputum culture as well as confirmed pneumonia. After analysis of 435 patients, the MRSA PCR nasal swab showed the following performance characteristics for detecting culture-proven MRSA: 88.0% sensitivity, 90.1% specificity, 35.4% positive predictive value (PPV), and 99.2% negative predictive value (NPV). Due to the high negative predictive value, the results indicated that discontinuation of MRSA antibiotic coverage would be appropriate for noncritically ill patients with pneumonia who had a negative MRSA PCR nasal swab.
Another retrospective study was performed by Johnson and colleagues to determine the association between MRSA PCR nasal swabs and the causative organism in pneumonia.11 Patients were included in the trial if they had a MRSA PCR nasal swab and a lower respiratory culture yielding S aureus within 48 hours of hospital admission. After analysis of 72 patients, MRSA PCR nasal swabs demonstrated the following diagnostic characteristics for detecting culture-proven MRSA: 93.3% sensitivity, 95.2% specificity, 93.3%PPV, and 95.2% NPV. These results suggest that early nasal swab MRSA PCR tests can predict the absence of MRSA reliably and may help guide the discontinuation of MRSA-directed empiric antibiotic therapy.
In addition, Giancola retrospectively studied the relationship between MRSA PCR nasal swabs and the likelihood of pneumonia caused by MRSA in intensive and intermediate care units.12 An analysis of 200 patients revealed high concordance between respiratory cultures and MRSA PCR nasal swab results with the following characteristics: 90.5% sensitivity, 79.9% specificity, 34.5% PPV, and 98.6% NPV. These test characteristics suggested that MRSA PCR nasal swabs might be a useful stewardship tool to allow for discontinuation of anti-MRSA therapy in critically ill patients with confirmed pneumonia.
Another retrospective analysis conducted by Baby and colleagues took a different approach to determine the clinical usefulness of MRSA PCR nasal swabs in the treatment of pneumonia.13 The primary outcome, mean duration of MRSA-targeted therapy, was reduced by 46.6 hours in the group who received a pharmacist-ordered MRSA PCR nasal swab compared with the group that did not receive a MRSA PCR nasal swab (P < .01) Per protocol, pharmacists were authorized to order a MRSA PCR nasal swab for patients who were prescribed vancomycin or linezolid for pneumonia. On receipt of the MRSA PCR nasal swab results, pharmacists were instructed to recommend discontinuation of anti-MRSA therapy if the PCR was negative for MRSA.
Results of this study indicated there were no significant differences in time to clinical improvement between preprotocol and postprotocol implementation (1.8 days vs 2.3 days, respectively; P = .54), length of stay (11.0 days vs 8.2 days, respectively; P = .22), or mortality (14.8% vs 6.7%, respectively; P = .41). The MRSA PCR nasal swabs allowed for a reduction in duration of anti-MRSA therapy without adverse effects on outcomes and provided a statistically significant reduction in the incidence of acute kidney injury during therapy in the postprotocol implementation group (26% vs 3.3%; P = .02), likely due to decreased exposure to vancomycin. Collectively, these studies indicate that MRSA PCR nasal swabs can be clinically useful in making decisions regarding discontinuation of MRSA-targeted therapy in pneumonia when MRSA PCR nasal swabs are negative.
A wider variety of infection sites were studied in a 2008 retrospective review of nearly 5,800 MRSA PCR nasal swabs taken within 24 hours (before or after) of a clinical culture that resulted growth of any organism.14 The goal of this study was to determine whether MRSA nasal colonization could predict MRSA involvement at various suspected infection sites. Overall, 217 patients (67.2%) with positive MRSA clinical cultures had a positive MRSA PCR nasal swab. The concordance between MRSA PCR nasal swabs and infection sites was highest with positive urine cultures (77%) and lowest in “other” infection sites (60%, primarily abdomen, buttock, and breast). Respiratory infections showed a 75% concordance between MRSA PCR nasal swabs and infection sites, as well as the following characteristics: 75% sensitivity, 90% specificity, 30% PPV, and 98% NPV. Additionally, infection site concordance was higher when clinical cultures grew clindamycin-resistant MRSA (71.3%) vs clindamycin-susceptible MRSA (59.3%; P = .04).
Overall, a positive MRSA PCR nasal swab increased the likelihood of MRSA at the primary infection site but was not clinically significant or consistent across infection sites. As seen in other studies, a negative MRSA PCR nasal swab could be useful for lowering concern for MRSA involvement in the primary infection, as evidenced by the following characteristics for all infection sites: 67% sensitivity, 90% specificity, 27% PPV, and 98% NPV.
Sarkionda and colleagues evaluated the clinical usefulness of MRSA PCR nasal swabs in the ICU setting in patients with a lower respiratory tract infection (RTI) or bloodstream infection.15 A total of 749 patients received a MRSA PCR nasal swab before admission to the ICU and were included in this study. The concordance between MRSA PCR nasal swabs and the causative organism was analyzed in patients who developed a MRSA lower respiratory infection (N = 120) and a MRSA bloodstream infection (N = 78) and demonstrated the following characteristics: 24.2% sensitivity, 78.5% specificity, 17.7% PPV, and 84.4% NPV; and 23.1% sensitivity, 78.2% specificity, 11.0% PPV, and 89.7% NPV, respectively. The authors concluded that the MRSA nasal swab results are not useful for making decisions regarding the need of empiric antimicrobial therapy targeting MRSA infections in lower respiratory infections and bloodstream infections. However, due to the high NPV in this study, one might conclude that negative MRSA PCR nasal swabs could still be used to de-escalate therapy, which is in agreement with the results from Dangerfield and Johnson.10,11
Similarly, results from a retrospective chart review demonstrated a lack of predictive value by the MRSA PCR nasal swab.16 Of 1,203 adult patients admitted to an ICU at a single center, 57 positive MRSA colonized and 122 negative MRSA colonized patients’ charts were randomly selected. The presence of MRSA lower RTI or bloodstream infections was found to be 3.51% vs 2.46% in the colonized and noncolonized groups, respectively (P = .46). These results led to the conclusion that a positive MRSA PCR nasal swab alone should not be used to make decisions regarding empiric MRSA antibiotic coverage.
An alternative approach to MRSA surveillance was taken by Harris in a prospective cohort of 12,080 adults with a suspected infection on admission to a non-ICU.17 Patients were screened with a 2-question tool to determine whether they were high risk for a MRSA infection. The 2 questions were “Have you been admitted to any health care facility in the last 12 months?” and “Do you have a skin infection (eg, boil, abscess, spider bite, or cellulitis) at this time?” If patients answered yes to either question, they were considered high risk, and a MRSA PCR nasal swab was ordered.
Patients who answered no to both questions were considered low risk and did not receive a MRSA PCR nasal swab. In total, 623 of 5,609 patients (11.1%) identified as high risk had a positive MRSA PCR nasal swab, and 148 of these 623 patients (23.8%) developed a MRSA-positive clinical culture. Only 121 of 4,986 patients (2.4%) who were high risk and had a negative MRSA PCR nasal swab went on to develop a MRSA-positive clinical culture (98% NPV). Additionally, 104 of 6,741 patients (1.6%) who answered no to both screening questions developed a MRSA-positive clinical culture (98% NPV). Results indicated that a high percentage of patients who were at high risk for MRSA (yes response to either question) and had a positive MRSA PCR nasal swab also had a positive clinical culture for MRSA. Conversely, a very small percentage of high-risk patients with a negative MRSA PCR nasal swab developed a positive clinical culture for MRSA.
The screening tool proved very effective as the low-risk group had the lowest number of patients (1.6%) develop a positive clinical culture for MRSA. It may be deduced that combination use of MRSA colonization testing via PCR nasal swabs in conjunction with a screening tool may be an effective method to identify patients in whom anti-MRSA therapy can be safely discontinued.
Conclusion
Based on the results of previously described studies, sufficient data may exist to support the discontinuation of MRSA-targeted therapy in noncritically ill patients with confirmed or suspected pneumonia and a negative MRSA PCR nasal swab. Insufficient evidence exists, however, to support a broadening of antimicrobial therapy to include anti-MRSA coverage in individuals with a positive MRSA PCR nasal swab, regardless of the infection site.
Clinical judgment should be used when determining empiric antimicrobial therapy and for appropriateness of de-escalation of therapy in critically ill patients. Once a patient stabilizes, a negative MRSA PCR nasal swab could be considered as supporting evidence to discontinue anti-MRSA therapy, especially in patients with lower respiratory infections, such as pneumonia.
1. Moran GJ, Krishnadasan A, Gorwitz RJ, et al; EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
2. Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763-1771.
3. Cosgrove SE, Fowler VG Jr. Management of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(suppl 5):S386-S393.
4. Dantes R, Mu Y, Belflower R, et al; Emerging Infections Program-Active Bacterial Core Surveillance MRSA Surveillance Investigators. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013;173(21):1970-1978.
5. Pillai MM, Latha R, Sarkar G. Detection of methicillin resistance in Staphylococcus aureus by polymerase chain reaction and conventional methods: a comparative study. J Lab Physicians. 2012;4(2):83-88.
6. Peterson LR, Liesenfeld O, Woods CW, et al. Multicenter evaluation of the LightCycler methicillin-resistant Staphylococcus aureus (MRSA) advanced test as a rapid method for detection of MRSA in nasal surveillance swabs. J Clin Microbiol. 2010;48(5):1661-1666.
7. File TM Jr, Srinivasan A, Bartlett JG. Antimicrobial stewardship: important for patient and public health. Clin Infect Dis. 2014;59(suppl 3):S93-S96.
8. Viasus D, Simonetti AF, Garcia-Vidal C, Niubó J, Dorca J, Carratalà J. Impact of antibiotic de-escalation on clinical outcomes in community-acquired pneumococcal pneumonia. J Antimicrob Chemother. 2017;72(2):547-5553.
9. Paul M, Dickstein Y, Raz-Pasteur A. Antibiotic de-escalation for bloodstream infections and pneumonia: a systematic review and meta-analysis. Clin Microbiol Infect. 2016;22(12):960-967.
10. Dangerfield B, Chung A, Webb B, Seville MT. Predictive value of methicillin-resistant Staphylococcus aureus (MRSA) nasal swab PCR assay for MRSA pneumonia. Antimicrob Agents Chemother. 2014;58(2):859-864.
11. Johnson JA, Wright ME, Sheperd LA, Musher DM, Dang BN. Nasal methicillin-resistant Staphylococcus aureus polymerase chain reaction a potential use in guiding antibiotic therapy for pneumonia. Perm J. 2015;19(1):34-36.
12. Giancola SE, Nguyen AT, Le B, et al. Clinical utility of a nasal swab methicillin-resistant Staphylococcus aureus polymerase chain reaction test in intensive and intermediate care unit patients with pneumonia. Diagn Microbiol Infect Dis. 2016;86(3):307-310.
13. Baby N, Faust AC, Smith T, Sheperd LA, Knoll L, Goodman EL. Nasal methicillin-resistant Staphylococcus aureus (MRSA) PCR testing reduces the duration of MRSA-targeted therapy in patients with suspected MRSA pneumonia. Antimicrob Agents Chemother. 2017;61(4):e02432-16.
14. Robicsek A, Suseno M, Beaumont JL, Thomson RB Jr, Peterson LR. Prediction of methicillin-resistant Staphylococcus aureus involvement in disease sites by concomitant nasal sampling. J Clin Microbiol. 2008;46(2):588-592.
15. Sarkionda KV, Micek ST, Dohery JA, Reichley RM, Warren D, Kollef MH. Methicillin-resistant Staphylococcus aureus nasal colonization is a poor predictor of intensive care unit-acquired methicillin-resistant Staphylococcus aureus infections requiring antibiotic treatment. Crit Care Med. 2010;38(10):1991-1995.
16. Ghidey F, Igbinosa O, Igbinosa E. Nasal colonization of methicillin resistant Staphylococcus aureus (MRSA) does not predict subsequent infection in the intensive care unit. Beni-Seuf University J Basic Appl Sci
17. Harris, AD, Furuno JP, Roghmann MC, et al. Targeted surveillance of methicillin-resistant Staphylococcus aureus and its potential use to guide empiric antibiotic therapy. Antimicrob Agents Chemother. 2010;54(8):3143-3148.
Methicillin-resistant Staphylococcus aureus (MRSA) is a Gram positive, round bacterium. The bacteria has evolved to withstand attacks from antibiotics and has made MRSA resistant to traditional antibiotics, such as β-lactams, resulting in difficult-to-treat infections. The presence of a genetic mutation within the mecA gene, which codes for the penicillin-binding protein 2a (PBP2a), differentiates MRSA from methicillin-susceptible Staphylococcus aureus (MSSA). Presence of the PBP2a protein allows Staphylococcus aureus (S aureus)to overcome β-lactam antibiotics’ method of killing by allowing the bacteria to continue to divide and grow.
β-lactam antibiotics cause cell death in susceptible isolates by binding to penicillin-binding proteins, which inhibits transpeptidation within the cell wall via inactivation of the penicillin-binding protein. By inhibiting cell wall synthesis, the cell loses its integrity and leaks its contents, causing cell death. Penicillin-binding protein 2a is a modified protein that has a low affinity for β-lactam antibiotics, allowing MRSA to survive and making it dangerous and difficult to eradicate.
First described in 1961, MRSA’s prevalence steadily increased in the following decades. It is the most common cause of skin and soft tissue infections presenting to emergency departments in the U.S.1 About 20% of bloodstream infections are caused by S aureus, and in 2003, nearly two-thirds of hospital-onset S aureus infections were methicillin-resistant in U.S. intensive-care units (ICUs).2 It has been shown that patients with MRSA bacteremia have worse overall outcomes, including increased mortality, greater lengths of stay, and increased costs, compared with those with MSSA infections.2,3 In 2011, MRSA infections caused an estimated 11,000 deaths, making fast and accurate detection of MRSA a crucial step in appropriate antimicrobial therapy selection.4
Currently, the Clinical and Laboratory Standards Institute (CLSI) recommends testing for MRSA by using phenotypic or genotypic methods. Phenotypic methods test for the observable characteristics of an organism, whereas a genotypic method identifies the specific gene that the organism carries. Recommended phenotypic methods include the latex agglutination test for PBP2a, the cefoxitin disk screen test, and a plate containing 6 μg/mL of oxacillin in Mueller-Hinton agar supplemented with sodium chloride.5 These methods have varying sensitivity and specificity and take between 48 to 72 hours to provide a result.
Within the past 15 years, a newer, genotypic, method of MRSA detection was approved by the FDA with high sensitivity and specificity. This method uses polymerase chain reaction (PCR) to identify the mecA gene. Polymerase chain reaction is a technique used to copy and amplify a specific segment of DNA, making thousands to millions of copies. If present, the MRSA PCR amplifies the mecA gene that makes S aureus resistant to methicillin and other β-lactams, which confirms that the specimen contains MRSA. The FDA has approved the use of MRSA PCR nasal swabs to detect MRSA in patients at risk of nasal colonization. While previously discussed methods may take between 2 and 3 days to confirm presence of MRSA, PCR can identify MRSA in about 1 hour.6
If a S aureus infection is suspected, empiric therapy often includes coverage of both MSSA and MRSA, due to the high morbidity and mortality associated with these infections. However, continuing an unneeded or unduly broad antibiotic, such as those that cover MRSA, can cause unintended consequences, such as toxicities, emerging resistance, or selection for pathogenic organisms.7 Therefore, empiric broad antibiotic therapy should be de-escalated as soon as possible, which further emphasizes the need for quick and accurate detection of the infecting organism. De-escalation of therapy can lead to a shorter length of stay and decreased mortality.8,9 Conversely, quick identification of infections caused by MRSA would allow therapy to be broadened to cover MRSA in infected patients, which could potentially decrease patient morbidity and mortality.
Nasal MRSA PCR Colonization
Rapid identification of a causative organism is crucial to determine appropriate antibiotic therapy. Fortunately, PCR is a very rapid method of detecting MRSA, and the use of MRSA PCR nasal swabs may be an effective way to predict whether MRSA is the organism causing an infection at various anatomical sites. If a patient has a suspected infection on admission, a MRSA PCR nasal swab often is completed to determine whether a patient’s nares are colonized with MRSA. However, there is no clear consensus in the literature regarding the correlation between MRSA nasal colonization and an infection caused by MRSA, making it difficult for clinicians to confidently de-escalate therapy on a negative MRSA PCR or broaden therapy on a positive result. The purpose of this literature review was to determine whether a MRSA PCR nasal swab can be used as a surrogate marker for MRSA infections at various sites.
Pneumonia has many potential causative organisms, many of which are covered empirically with guideline-directed therapy. The predictive power of MRSA PCR nasal swabs may allow clinicians to prescribe earlier directed therapy. A retrospective cohort study performed at a tertiary care center looked at the clinical usefulness of a MRSA PCR nasal swab in the treatment of pneumonia.10 Patients were included in the trial if they had a MRSA PCR nasal swab within 1 month of their blood or sputum culture as well as confirmed pneumonia. After analysis of 435 patients, the MRSA PCR nasal swab showed the following performance characteristics for detecting culture-proven MRSA: 88.0% sensitivity, 90.1% specificity, 35.4% positive predictive value (PPV), and 99.2% negative predictive value (NPV). Due to the high negative predictive value, the results indicated that discontinuation of MRSA antibiotic coverage would be appropriate for noncritically ill patients with pneumonia who had a negative MRSA PCR nasal swab.
Another retrospective study was performed by Johnson and colleagues to determine the association between MRSA PCR nasal swabs and the causative organism in pneumonia.11 Patients were included in the trial if they had a MRSA PCR nasal swab and a lower respiratory culture yielding S aureus within 48 hours of hospital admission. After analysis of 72 patients, MRSA PCR nasal swabs demonstrated the following diagnostic characteristics for detecting culture-proven MRSA: 93.3% sensitivity, 95.2% specificity, 93.3%PPV, and 95.2% NPV. These results suggest that early nasal swab MRSA PCR tests can predict the absence of MRSA reliably and may help guide the discontinuation of MRSA-directed empiric antibiotic therapy.
In addition, Giancola retrospectively studied the relationship between MRSA PCR nasal swabs and the likelihood of pneumonia caused by MRSA in intensive and intermediate care units.12 An analysis of 200 patients revealed high concordance between respiratory cultures and MRSA PCR nasal swab results with the following characteristics: 90.5% sensitivity, 79.9% specificity, 34.5% PPV, and 98.6% NPV. These test characteristics suggested that MRSA PCR nasal swabs might be a useful stewardship tool to allow for discontinuation of anti-MRSA therapy in critically ill patients with confirmed pneumonia.
Another retrospective analysis conducted by Baby and colleagues took a different approach to determine the clinical usefulness of MRSA PCR nasal swabs in the treatment of pneumonia.13 The primary outcome, mean duration of MRSA-targeted therapy, was reduced by 46.6 hours in the group who received a pharmacist-ordered MRSA PCR nasal swab compared with the group that did not receive a MRSA PCR nasal swab (P < .01) Per protocol, pharmacists were authorized to order a MRSA PCR nasal swab for patients who were prescribed vancomycin or linezolid for pneumonia. On receipt of the MRSA PCR nasal swab results, pharmacists were instructed to recommend discontinuation of anti-MRSA therapy if the PCR was negative for MRSA.
Results of this study indicated there were no significant differences in time to clinical improvement between preprotocol and postprotocol implementation (1.8 days vs 2.3 days, respectively; P = .54), length of stay (11.0 days vs 8.2 days, respectively; P = .22), or mortality (14.8% vs 6.7%, respectively; P = .41). The MRSA PCR nasal swabs allowed for a reduction in duration of anti-MRSA therapy without adverse effects on outcomes and provided a statistically significant reduction in the incidence of acute kidney injury during therapy in the postprotocol implementation group (26% vs 3.3%; P = .02), likely due to decreased exposure to vancomycin. Collectively, these studies indicate that MRSA PCR nasal swabs can be clinically useful in making decisions regarding discontinuation of MRSA-targeted therapy in pneumonia when MRSA PCR nasal swabs are negative.
A wider variety of infection sites were studied in a 2008 retrospective review of nearly 5,800 MRSA PCR nasal swabs taken within 24 hours (before or after) of a clinical culture that resulted growth of any organism.14 The goal of this study was to determine whether MRSA nasal colonization could predict MRSA involvement at various suspected infection sites. Overall, 217 patients (67.2%) with positive MRSA clinical cultures had a positive MRSA PCR nasal swab. The concordance between MRSA PCR nasal swabs and infection sites was highest with positive urine cultures (77%) and lowest in “other” infection sites (60%, primarily abdomen, buttock, and breast). Respiratory infections showed a 75% concordance between MRSA PCR nasal swabs and infection sites, as well as the following characteristics: 75% sensitivity, 90% specificity, 30% PPV, and 98% NPV. Additionally, infection site concordance was higher when clinical cultures grew clindamycin-resistant MRSA (71.3%) vs clindamycin-susceptible MRSA (59.3%; P = .04).
Overall, a positive MRSA PCR nasal swab increased the likelihood of MRSA at the primary infection site but was not clinically significant or consistent across infection sites. As seen in other studies, a negative MRSA PCR nasal swab could be useful for lowering concern for MRSA involvement in the primary infection, as evidenced by the following characteristics for all infection sites: 67% sensitivity, 90% specificity, 27% PPV, and 98% NPV.
Sarkionda and colleagues evaluated the clinical usefulness of MRSA PCR nasal swabs in the ICU setting in patients with a lower respiratory tract infection (RTI) or bloodstream infection.15 A total of 749 patients received a MRSA PCR nasal swab before admission to the ICU and were included in this study. The concordance between MRSA PCR nasal swabs and the causative organism was analyzed in patients who developed a MRSA lower respiratory infection (N = 120) and a MRSA bloodstream infection (N = 78) and demonstrated the following characteristics: 24.2% sensitivity, 78.5% specificity, 17.7% PPV, and 84.4% NPV; and 23.1% sensitivity, 78.2% specificity, 11.0% PPV, and 89.7% NPV, respectively. The authors concluded that the MRSA nasal swab results are not useful for making decisions regarding the need of empiric antimicrobial therapy targeting MRSA infections in lower respiratory infections and bloodstream infections. However, due to the high NPV in this study, one might conclude that negative MRSA PCR nasal swabs could still be used to de-escalate therapy, which is in agreement with the results from Dangerfield and Johnson.10,11
Similarly, results from a retrospective chart review demonstrated a lack of predictive value by the MRSA PCR nasal swab.16 Of 1,203 adult patients admitted to an ICU at a single center, 57 positive MRSA colonized and 122 negative MRSA colonized patients’ charts were randomly selected. The presence of MRSA lower RTI or bloodstream infections was found to be 3.51% vs 2.46% in the colonized and noncolonized groups, respectively (P = .46). These results led to the conclusion that a positive MRSA PCR nasal swab alone should not be used to make decisions regarding empiric MRSA antibiotic coverage.
An alternative approach to MRSA surveillance was taken by Harris in a prospective cohort of 12,080 adults with a suspected infection on admission to a non-ICU.17 Patients were screened with a 2-question tool to determine whether they were high risk for a MRSA infection. The 2 questions were “Have you been admitted to any health care facility in the last 12 months?” and “Do you have a skin infection (eg, boil, abscess, spider bite, or cellulitis) at this time?” If patients answered yes to either question, they were considered high risk, and a MRSA PCR nasal swab was ordered.
Patients who answered no to both questions were considered low risk and did not receive a MRSA PCR nasal swab. In total, 623 of 5,609 patients (11.1%) identified as high risk had a positive MRSA PCR nasal swab, and 148 of these 623 patients (23.8%) developed a MRSA-positive clinical culture. Only 121 of 4,986 patients (2.4%) who were high risk and had a negative MRSA PCR nasal swab went on to develop a MRSA-positive clinical culture (98% NPV). Additionally, 104 of 6,741 patients (1.6%) who answered no to both screening questions developed a MRSA-positive clinical culture (98% NPV). Results indicated that a high percentage of patients who were at high risk for MRSA (yes response to either question) and had a positive MRSA PCR nasal swab also had a positive clinical culture for MRSA. Conversely, a very small percentage of high-risk patients with a negative MRSA PCR nasal swab developed a positive clinical culture for MRSA.
The screening tool proved very effective as the low-risk group had the lowest number of patients (1.6%) develop a positive clinical culture for MRSA. It may be deduced that combination use of MRSA colonization testing via PCR nasal swabs in conjunction with a screening tool may be an effective method to identify patients in whom anti-MRSA therapy can be safely discontinued.
Conclusion
Based on the results of previously described studies, sufficient data may exist to support the discontinuation of MRSA-targeted therapy in noncritically ill patients with confirmed or suspected pneumonia and a negative MRSA PCR nasal swab. Insufficient evidence exists, however, to support a broadening of antimicrobial therapy to include anti-MRSA coverage in individuals with a positive MRSA PCR nasal swab, regardless of the infection site.
Clinical judgment should be used when determining empiric antimicrobial therapy and for appropriateness of de-escalation of therapy in critically ill patients. Once a patient stabilizes, a negative MRSA PCR nasal swab could be considered as supporting evidence to discontinue anti-MRSA therapy, especially in patients with lower respiratory infections, such as pneumonia.
Methicillin-resistant Staphylococcus aureus (MRSA) is a Gram positive, round bacterium. The bacteria has evolved to withstand attacks from antibiotics and has made MRSA resistant to traditional antibiotics, such as β-lactams, resulting in difficult-to-treat infections. The presence of a genetic mutation within the mecA gene, which codes for the penicillin-binding protein 2a (PBP2a), differentiates MRSA from methicillin-susceptible Staphylococcus aureus (MSSA). Presence of the PBP2a protein allows Staphylococcus aureus (S aureus)to overcome β-lactam antibiotics’ method of killing by allowing the bacteria to continue to divide and grow.
β-lactam antibiotics cause cell death in susceptible isolates by binding to penicillin-binding proteins, which inhibits transpeptidation within the cell wall via inactivation of the penicillin-binding protein. By inhibiting cell wall synthesis, the cell loses its integrity and leaks its contents, causing cell death. Penicillin-binding protein 2a is a modified protein that has a low affinity for β-lactam antibiotics, allowing MRSA to survive and making it dangerous and difficult to eradicate.
First described in 1961, MRSA’s prevalence steadily increased in the following decades. It is the most common cause of skin and soft tissue infections presenting to emergency departments in the U.S.1 About 20% of bloodstream infections are caused by S aureus, and in 2003, nearly two-thirds of hospital-onset S aureus infections were methicillin-resistant in U.S. intensive-care units (ICUs).2 It has been shown that patients with MRSA bacteremia have worse overall outcomes, including increased mortality, greater lengths of stay, and increased costs, compared with those with MSSA infections.2,3 In 2011, MRSA infections caused an estimated 11,000 deaths, making fast and accurate detection of MRSA a crucial step in appropriate antimicrobial therapy selection.4
Currently, the Clinical and Laboratory Standards Institute (CLSI) recommends testing for MRSA by using phenotypic or genotypic methods. Phenotypic methods test for the observable characteristics of an organism, whereas a genotypic method identifies the specific gene that the organism carries. Recommended phenotypic methods include the latex agglutination test for PBP2a, the cefoxitin disk screen test, and a plate containing 6 μg/mL of oxacillin in Mueller-Hinton agar supplemented with sodium chloride.5 These methods have varying sensitivity and specificity and take between 48 to 72 hours to provide a result.
Within the past 15 years, a newer, genotypic, method of MRSA detection was approved by the FDA with high sensitivity and specificity. This method uses polymerase chain reaction (PCR) to identify the mecA gene. Polymerase chain reaction is a technique used to copy and amplify a specific segment of DNA, making thousands to millions of copies. If present, the MRSA PCR amplifies the mecA gene that makes S aureus resistant to methicillin and other β-lactams, which confirms that the specimen contains MRSA. The FDA has approved the use of MRSA PCR nasal swabs to detect MRSA in patients at risk of nasal colonization. While previously discussed methods may take between 2 and 3 days to confirm presence of MRSA, PCR can identify MRSA in about 1 hour.6
If a S aureus infection is suspected, empiric therapy often includes coverage of both MSSA and MRSA, due to the high morbidity and mortality associated with these infections. However, continuing an unneeded or unduly broad antibiotic, such as those that cover MRSA, can cause unintended consequences, such as toxicities, emerging resistance, or selection for pathogenic organisms.7 Therefore, empiric broad antibiotic therapy should be de-escalated as soon as possible, which further emphasizes the need for quick and accurate detection of the infecting organism. De-escalation of therapy can lead to a shorter length of stay and decreased mortality.8,9 Conversely, quick identification of infections caused by MRSA would allow therapy to be broadened to cover MRSA in infected patients, which could potentially decrease patient morbidity and mortality.
Nasal MRSA PCR Colonization
Rapid identification of a causative organism is crucial to determine appropriate antibiotic therapy. Fortunately, PCR is a very rapid method of detecting MRSA, and the use of MRSA PCR nasal swabs may be an effective way to predict whether MRSA is the organism causing an infection at various anatomical sites. If a patient has a suspected infection on admission, a MRSA PCR nasal swab often is completed to determine whether a patient’s nares are colonized with MRSA. However, there is no clear consensus in the literature regarding the correlation between MRSA nasal colonization and an infection caused by MRSA, making it difficult for clinicians to confidently de-escalate therapy on a negative MRSA PCR or broaden therapy on a positive result. The purpose of this literature review was to determine whether a MRSA PCR nasal swab can be used as a surrogate marker for MRSA infections at various sites.
Pneumonia has many potential causative organisms, many of which are covered empirically with guideline-directed therapy. The predictive power of MRSA PCR nasal swabs may allow clinicians to prescribe earlier directed therapy. A retrospective cohort study performed at a tertiary care center looked at the clinical usefulness of a MRSA PCR nasal swab in the treatment of pneumonia.10 Patients were included in the trial if they had a MRSA PCR nasal swab within 1 month of their blood or sputum culture as well as confirmed pneumonia. After analysis of 435 patients, the MRSA PCR nasal swab showed the following performance characteristics for detecting culture-proven MRSA: 88.0% sensitivity, 90.1% specificity, 35.4% positive predictive value (PPV), and 99.2% negative predictive value (NPV). Due to the high negative predictive value, the results indicated that discontinuation of MRSA antibiotic coverage would be appropriate for noncritically ill patients with pneumonia who had a negative MRSA PCR nasal swab.
Another retrospective study was performed by Johnson and colleagues to determine the association between MRSA PCR nasal swabs and the causative organism in pneumonia.11 Patients were included in the trial if they had a MRSA PCR nasal swab and a lower respiratory culture yielding S aureus within 48 hours of hospital admission. After analysis of 72 patients, MRSA PCR nasal swabs demonstrated the following diagnostic characteristics for detecting culture-proven MRSA: 93.3% sensitivity, 95.2% specificity, 93.3%PPV, and 95.2% NPV. These results suggest that early nasal swab MRSA PCR tests can predict the absence of MRSA reliably and may help guide the discontinuation of MRSA-directed empiric antibiotic therapy.
In addition, Giancola retrospectively studied the relationship between MRSA PCR nasal swabs and the likelihood of pneumonia caused by MRSA in intensive and intermediate care units.12 An analysis of 200 patients revealed high concordance between respiratory cultures and MRSA PCR nasal swab results with the following characteristics: 90.5% sensitivity, 79.9% specificity, 34.5% PPV, and 98.6% NPV. These test characteristics suggested that MRSA PCR nasal swabs might be a useful stewardship tool to allow for discontinuation of anti-MRSA therapy in critically ill patients with confirmed pneumonia.
Another retrospective analysis conducted by Baby and colleagues took a different approach to determine the clinical usefulness of MRSA PCR nasal swabs in the treatment of pneumonia.13 The primary outcome, mean duration of MRSA-targeted therapy, was reduced by 46.6 hours in the group who received a pharmacist-ordered MRSA PCR nasal swab compared with the group that did not receive a MRSA PCR nasal swab (P < .01) Per protocol, pharmacists were authorized to order a MRSA PCR nasal swab for patients who were prescribed vancomycin or linezolid for pneumonia. On receipt of the MRSA PCR nasal swab results, pharmacists were instructed to recommend discontinuation of anti-MRSA therapy if the PCR was negative for MRSA.
Results of this study indicated there were no significant differences in time to clinical improvement between preprotocol and postprotocol implementation (1.8 days vs 2.3 days, respectively; P = .54), length of stay (11.0 days vs 8.2 days, respectively; P = .22), or mortality (14.8% vs 6.7%, respectively; P = .41). The MRSA PCR nasal swabs allowed for a reduction in duration of anti-MRSA therapy without adverse effects on outcomes and provided a statistically significant reduction in the incidence of acute kidney injury during therapy in the postprotocol implementation group (26% vs 3.3%; P = .02), likely due to decreased exposure to vancomycin. Collectively, these studies indicate that MRSA PCR nasal swabs can be clinically useful in making decisions regarding discontinuation of MRSA-targeted therapy in pneumonia when MRSA PCR nasal swabs are negative.
A wider variety of infection sites were studied in a 2008 retrospective review of nearly 5,800 MRSA PCR nasal swabs taken within 24 hours (before or after) of a clinical culture that resulted growth of any organism.14 The goal of this study was to determine whether MRSA nasal colonization could predict MRSA involvement at various suspected infection sites. Overall, 217 patients (67.2%) with positive MRSA clinical cultures had a positive MRSA PCR nasal swab. The concordance between MRSA PCR nasal swabs and infection sites was highest with positive urine cultures (77%) and lowest in “other” infection sites (60%, primarily abdomen, buttock, and breast). Respiratory infections showed a 75% concordance between MRSA PCR nasal swabs and infection sites, as well as the following characteristics: 75% sensitivity, 90% specificity, 30% PPV, and 98% NPV. Additionally, infection site concordance was higher when clinical cultures grew clindamycin-resistant MRSA (71.3%) vs clindamycin-susceptible MRSA (59.3%; P = .04).
Overall, a positive MRSA PCR nasal swab increased the likelihood of MRSA at the primary infection site but was not clinically significant or consistent across infection sites. As seen in other studies, a negative MRSA PCR nasal swab could be useful for lowering concern for MRSA involvement in the primary infection, as evidenced by the following characteristics for all infection sites: 67% sensitivity, 90% specificity, 27% PPV, and 98% NPV.
Sarkionda and colleagues evaluated the clinical usefulness of MRSA PCR nasal swabs in the ICU setting in patients with a lower respiratory tract infection (RTI) or bloodstream infection.15 A total of 749 patients received a MRSA PCR nasal swab before admission to the ICU and were included in this study. The concordance between MRSA PCR nasal swabs and the causative organism was analyzed in patients who developed a MRSA lower respiratory infection (N = 120) and a MRSA bloodstream infection (N = 78) and demonstrated the following characteristics: 24.2% sensitivity, 78.5% specificity, 17.7% PPV, and 84.4% NPV; and 23.1% sensitivity, 78.2% specificity, 11.0% PPV, and 89.7% NPV, respectively. The authors concluded that the MRSA nasal swab results are not useful for making decisions regarding the need of empiric antimicrobial therapy targeting MRSA infections in lower respiratory infections and bloodstream infections. However, due to the high NPV in this study, one might conclude that negative MRSA PCR nasal swabs could still be used to de-escalate therapy, which is in agreement with the results from Dangerfield and Johnson.10,11
Similarly, results from a retrospective chart review demonstrated a lack of predictive value by the MRSA PCR nasal swab.16 Of 1,203 adult patients admitted to an ICU at a single center, 57 positive MRSA colonized and 122 negative MRSA colonized patients’ charts were randomly selected. The presence of MRSA lower RTI or bloodstream infections was found to be 3.51% vs 2.46% in the colonized and noncolonized groups, respectively (P = .46). These results led to the conclusion that a positive MRSA PCR nasal swab alone should not be used to make decisions regarding empiric MRSA antibiotic coverage.
An alternative approach to MRSA surveillance was taken by Harris in a prospective cohort of 12,080 adults with a suspected infection on admission to a non-ICU.17 Patients were screened with a 2-question tool to determine whether they were high risk for a MRSA infection. The 2 questions were “Have you been admitted to any health care facility in the last 12 months?” and “Do you have a skin infection (eg, boil, abscess, spider bite, or cellulitis) at this time?” If patients answered yes to either question, they were considered high risk, and a MRSA PCR nasal swab was ordered.
Patients who answered no to both questions were considered low risk and did not receive a MRSA PCR nasal swab. In total, 623 of 5,609 patients (11.1%) identified as high risk had a positive MRSA PCR nasal swab, and 148 of these 623 patients (23.8%) developed a MRSA-positive clinical culture. Only 121 of 4,986 patients (2.4%) who were high risk and had a negative MRSA PCR nasal swab went on to develop a MRSA-positive clinical culture (98% NPV). Additionally, 104 of 6,741 patients (1.6%) who answered no to both screening questions developed a MRSA-positive clinical culture (98% NPV). Results indicated that a high percentage of patients who were at high risk for MRSA (yes response to either question) and had a positive MRSA PCR nasal swab also had a positive clinical culture for MRSA. Conversely, a very small percentage of high-risk patients with a negative MRSA PCR nasal swab developed a positive clinical culture for MRSA.
The screening tool proved very effective as the low-risk group had the lowest number of patients (1.6%) develop a positive clinical culture for MRSA. It may be deduced that combination use of MRSA colonization testing via PCR nasal swabs in conjunction with a screening tool may be an effective method to identify patients in whom anti-MRSA therapy can be safely discontinued.
Conclusion
Based on the results of previously described studies, sufficient data may exist to support the discontinuation of MRSA-targeted therapy in noncritically ill patients with confirmed or suspected pneumonia and a negative MRSA PCR nasal swab. Insufficient evidence exists, however, to support a broadening of antimicrobial therapy to include anti-MRSA coverage in individuals with a positive MRSA PCR nasal swab, regardless of the infection site.
Clinical judgment should be used when determining empiric antimicrobial therapy and for appropriateness of de-escalation of therapy in critically ill patients. Once a patient stabilizes, a negative MRSA PCR nasal swab could be considered as supporting evidence to discontinue anti-MRSA therapy, especially in patients with lower respiratory infections, such as pneumonia.
1. Moran GJ, Krishnadasan A, Gorwitz RJ, et al; EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
2. Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763-1771.
3. Cosgrove SE, Fowler VG Jr. Management of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(suppl 5):S386-S393.
4. Dantes R, Mu Y, Belflower R, et al; Emerging Infections Program-Active Bacterial Core Surveillance MRSA Surveillance Investigators. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013;173(21):1970-1978.
5. Pillai MM, Latha R, Sarkar G. Detection of methicillin resistance in Staphylococcus aureus by polymerase chain reaction and conventional methods: a comparative study. J Lab Physicians. 2012;4(2):83-88.
6. Peterson LR, Liesenfeld O, Woods CW, et al. Multicenter evaluation of the LightCycler methicillin-resistant Staphylococcus aureus (MRSA) advanced test as a rapid method for detection of MRSA in nasal surveillance swabs. J Clin Microbiol. 2010;48(5):1661-1666.
7. File TM Jr, Srinivasan A, Bartlett JG. Antimicrobial stewardship: important for patient and public health. Clin Infect Dis. 2014;59(suppl 3):S93-S96.
8. Viasus D, Simonetti AF, Garcia-Vidal C, Niubó J, Dorca J, Carratalà J. Impact of antibiotic de-escalation on clinical outcomes in community-acquired pneumococcal pneumonia. J Antimicrob Chemother. 2017;72(2):547-5553.
9. Paul M, Dickstein Y, Raz-Pasteur A. Antibiotic de-escalation for bloodstream infections and pneumonia: a systematic review and meta-analysis. Clin Microbiol Infect. 2016;22(12):960-967.
10. Dangerfield B, Chung A, Webb B, Seville MT. Predictive value of methicillin-resistant Staphylococcus aureus (MRSA) nasal swab PCR assay for MRSA pneumonia. Antimicrob Agents Chemother. 2014;58(2):859-864.
11. Johnson JA, Wright ME, Sheperd LA, Musher DM, Dang BN. Nasal methicillin-resistant Staphylococcus aureus polymerase chain reaction a potential use in guiding antibiotic therapy for pneumonia. Perm J. 2015;19(1):34-36.
12. Giancola SE, Nguyen AT, Le B, et al. Clinical utility of a nasal swab methicillin-resistant Staphylococcus aureus polymerase chain reaction test in intensive and intermediate care unit patients with pneumonia. Diagn Microbiol Infect Dis. 2016;86(3):307-310.
13. Baby N, Faust AC, Smith T, Sheperd LA, Knoll L, Goodman EL. Nasal methicillin-resistant Staphylococcus aureus (MRSA) PCR testing reduces the duration of MRSA-targeted therapy in patients with suspected MRSA pneumonia. Antimicrob Agents Chemother. 2017;61(4):e02432-16.
14. Robicsek A, Suseno M, Beaumont JL, Thomson RB Jr, Peterson LR. Prediction of methicillin-resistant Staphylococcus aureus involvement in disease sites by concomitant nasal sampling. J Clin Microbiol. 2008;46(2):588-592.
15. Sarkionda KV, Micek ST, Dohery JA, Reichley RM, Warren D, Kollef MH. Methicillin-resistant Staphylococcus aureus nasal colonization is a poor predictor of intensive care unit-acquired methicillin-resistant Staphylococcus aureus infections requiring antibiotic treatment. Crit Care Med. 2010;38(10):1991-1995.
16. Ghidey F, Igbinosa O, Igbinosa E. Nasal colonization of methicillin resistant Staphylococcus aureus (MRSA) does not predict subsequent infection in the intensive care unit. Beni-Seuf University J Basic Appl Sci
17. Harris, AD, Furuno JP, Roghmann MC, et al. Targeted surveillance of methicillin-resistant Staphylococcus aureus and its potential use to guide empiric antibiotic therapy. Antimicrob Agents Chemother. 2010;54(8):3143-3148.
1. Moran GJ, Krishnadasan A, Gorwitz RJ, et al; EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666-674.
2. Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763-1771.
3. Cosgrove SE, Fowler VG Jr. Management of methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(suppl 5):S386-S393.
4. Dantes R, Mu Y, Belflower R, et al; Emerging Infections Program-Active Bacterial Core Surveillance MRSA Surveillance Investigators. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med. 2013;173(21):1970-1978.
5. Pillai MM, Latha R, Sarkar G. Detection of methicillin resistance in Staphylococcus aureus by polymerase chain reaction and conventional methods: a comparative study. J Lab Physicians. 2012;4(2):83-88.
6. Peterson LR, Liesenfeld O, Woods CW, et al. Multicenter evaluation of the LightCycler methicillin-resistant Staphylococcus aureus (MRSA) advanced test as a rapid method for detection of MRSA in nasal surveillance swabs. J Clin Microbiol. 2010;48(5):1661-1666.
7. File TM Jr, Srinivasan A, Bartlett JG. Antimicrobial stewardship: important for patient and public health. Clin Infect Dis. 2014;59(suppl 3):S93-S96.
8. Viasus D, Simonetti AF, Garcia-Vidal C, Niubó J, Dorca J, Carratalà J. Impact of antibiotic de-escalation on clinical outcomes in community-acquired pneumococcal pneumonia. J Antimicrob Chemother. 2017;72(2):547-5553.
9. Paul M, Dickstein Y, Raz-Pasteur A. Antibiotic de-escalation for bloodstream infections and pneumonia: a systematic review and meta-analysis. Clin Microbiol Infect. 2016;22(12):960-967.
10. Dangerfield B, Chung A, Webb B, Seville MT. Predictive value of methicillin-resistant Staphylococcus aureus (MRSA) nasal swab PCR assay for MRSA pneumonia. Antimicrob Agents Chemother. 2014;58(2):859-864.
11. Johnson JA, Wright ME, Sheperd LA, Musher DM, Dang BN. Nasal methicillin-resistant Staphylococcus aureus polymerase chain reaction a potential use in guiding antibiotic therapy for pneumonia. Perm J. 2015;19(1):34-36.
12. Giancola SE, Nguyen AT, Le B, et al. Clinical utility of a nasal swab methicillin-resistant Staphylococcus aureus polymerase chain reaction test in intensive and intermediate care unit patients with pneumonia. Diagn Microbiol Infect Dis. 2016;86(3):307-310.
13. Baby N, Faust AC, Smith T, Sheperd LA, Knoll L, Goodman EL. Nasal methicillin-resistant Staphylococcus aureus (MRSA) PCR testing reduces the duration of MRSA-targeted therapy in patients with suspected MRSA pneumonia. Antimicrob Agents Chemother. 2017;61(4):e02432-16.
14. Robicsek A, Suseno M, Beaumont JL, Thomson RB Jr, Peterson LR. Prediction of methicillin-resistant Staphylococcus aureus involvement in disease sites by concomitant nasal sampling. J Clin Microbiol. 2008;46(2):588-592.
15. Sarkionda KV, Micek ST, Dohery JA, Reichley RM, Warren D, Kollef MH. Methicillin-resistant Staphylococcus aureus nasal colonization is a poor predictor of intensive care unit-acquired methicillin-resistant Staphylococcus aureus infections requiring antibiotic treatment. Crit Care Med. 2010;38(10):1991-1995.
16. Ghidey F, Igbinosa O, Igbinosa E. Nasal colonization of methicillin resistant Staphylococcus aureus (MRSA) does not predict subsequent infection in the intensive care unit. Beni-Seuf University J Basic Appl Sci
17. Harris, AD, Furuno JP, Roghmann MC, et al. Targeted surveillance of methicillin-resistant Staphylococcus aureus and its potential use to guide empiric antibiotic therapy. Antimicrob Agents Chemother. 2010;54(8):3143-3148.