User login
Hairy Cell Leukemia
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14 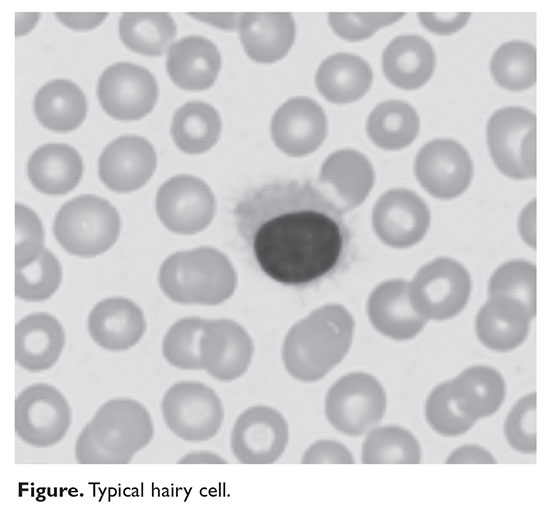
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease. 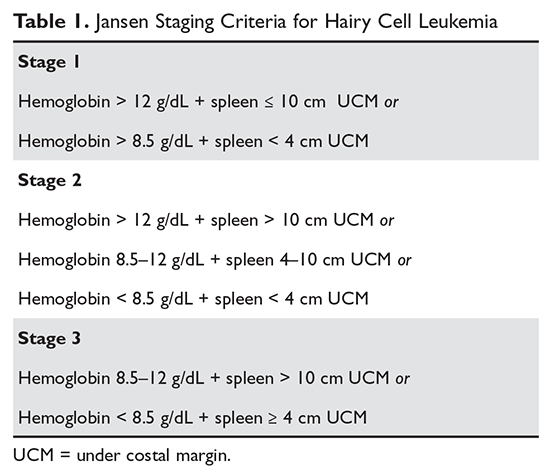
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29 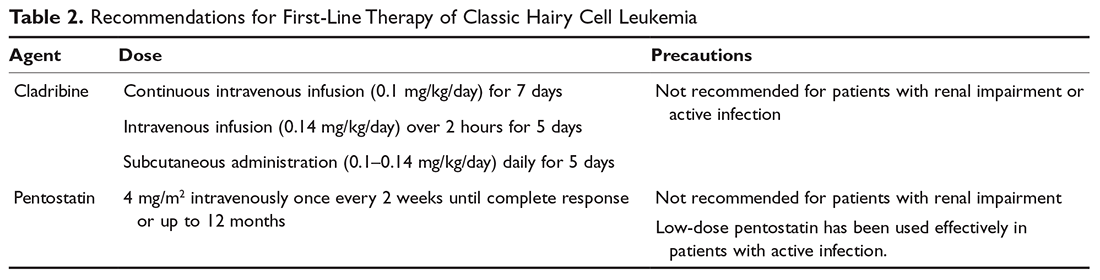
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3. 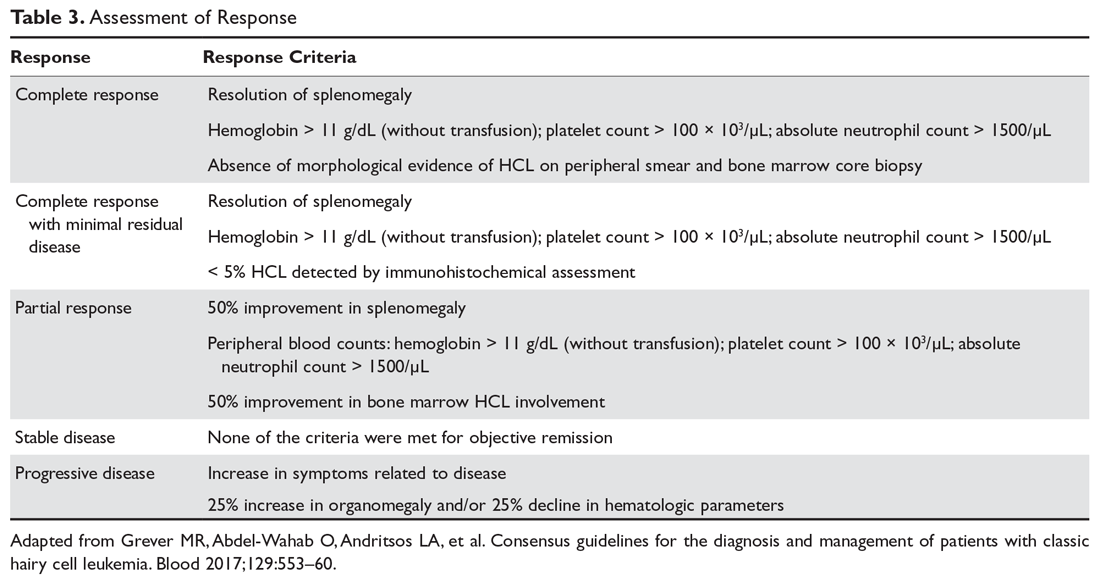
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.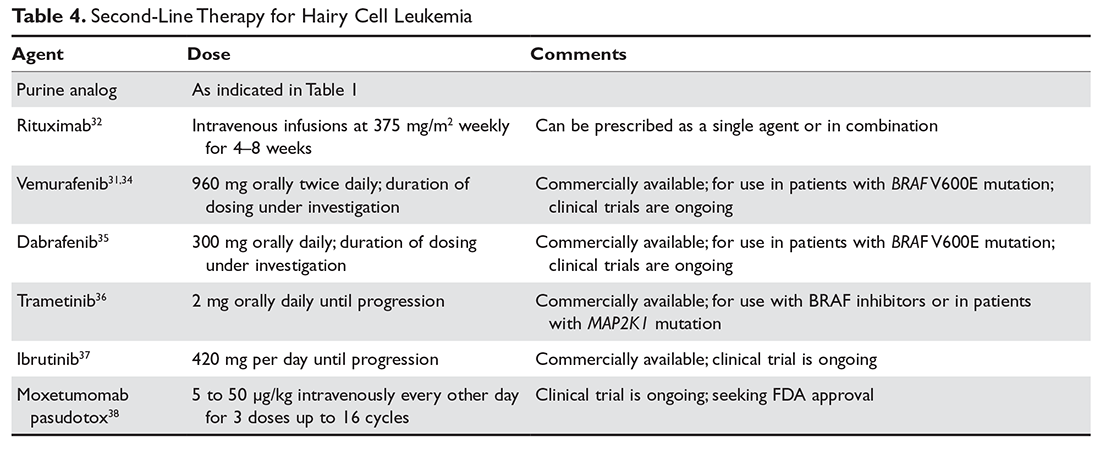
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.
While BRAF V600E is identified in nearly all cases of cHCL, it is rare in vHCL.18 The variant type of HCL was classified as a distinct clinical entity in 2008 and can now often be distinguished from cHCL on the basis of BRAF mutational status, among other differences. Interestingly, in the rare cases of BRAF V600E–negative cHCL, other mutations in BRAF or downstream targets as well as aberrant activation of the RAF-MEK-ERK signaling cascade are observed, indicating that this pathway is critical in HCL and may still represent a viable therapeutic target. Expression of the IGHV4-34 immunoglobulin rearrangement, while more common in vHCL, has also been identified in 10% of cHCL cases and appears to confer poor prognosis.19 Other mutated genes that have been identified in HCL include CDKN1B, TP53, U2AF1, ARID1A, EZH2, and KDM6A.20
Classic HCL is characterized by the immunophenotypic expression of CD11c, CD25, CD103, and CD123, with kappa or lambda light chain restriction indicating clonality; HCL B cells are generally negative for CD5, CD10, CD23, CD27, and CD79b. In contrast, vHCL often lacks expression of CD25 and CD123.18 The B-cell receptor (BCR) is expressed on hairy cells and its activation promotes proliferation and survival in vitro.21 The role of BCR signaling in B-cell malignancies is increasingly recognized, and therapies that target the BCR and associated signaling molecules offer an attractive treatment strategy.22 HCL B cells also typically express CD19, CD20, CD22, CD79a, CD200, CD1d, and annexin A1. Tartrate-resistant acid phosphatase (TRAP) positivity by immunohistochemistry is a hallmark of cHCL. Interestingly, changes to the patient’s original immunophenotype have been observed following treatment and upon disease recurrence, highlighting the importance of tracking immunophenotype throughout the course of disease.
Diagnosis
Prior to the advent of annual screening evaluations with routine examination of complete blood counts (CBC), patients were most often diagnosed with HCL when they presented with symptoms of the disease such as splenomegaly, infections, or complications of anemia or thrombocytopenia.23 In the current era, patients are more likely to be incidentally diagnosed when they are found to have an abnormal value on a CBC. Any blood lineage may be affected and patients may have pancytopenia or isolated cytopenias. Of note, monocytopenia is a common finding in cHCL that is not entirely understood. The cells typical of cHCL do not usually circulate in the peripheral blood, but if present would appear as mature lymphocytes with villous cytoplasmic projections, pale blue cytoplasm, and reniform nuclei with open chromatin (Figure).9 Even if the morphologic examination is highly suggestive of HCL, additional testing is required to differentiate between cHCL, vHCL, and other hematologic malignancies which may also have cytoplasmic projections. A complete assessment of the immunophenotype, molecular profile, and cytogenetic features is required to arrive at this diagnosis.
The international Hairy Cell Leukemia Foundation recently published consensus guidelines for the diagnosis and treatment of HCL.24 These guidelines recommend that patients undergo examination of the peripheral blood for morphology and immunophenotyping and further recommend obtaining bone marrow core and aspirate biopsy samples for immunophenotyping via immunohistochemical staining and flow cytometry. The characteristic immunophenotype of cHCL is a population of monoclonal B lymphocytes which co-express CD19, CD20, CD11c, CD25, CD103, and CD123. Variant HCL is characterized by a very similar immunophenotype but is usually negative for CD25 and CD123. It is notable that CD25 positivity may be lost following treatment, and the absence of this marker should not be used as the sole basis of a cHCL versus vHCL diagnosis. Because marrow fibrosis in HCL may prevent a marrow aspirate from being obtained, many of the key diagnostic studies are performed on the core biopsy, including morphological evaluation and immunohistochemical stains such as CD20 (a pan-B cell antigen), annexin-1 (an anti-inflammatory protein expressed only in cHCL), and VE1 (a BRAF V600E stain).
As noted above, recurrent cytogenetic abnormalities have now been identified that may inform the diagnosis or prognosis of HCL. Next-generation sequencing and other testing of the genetic landscape are taking on a larger role in subtype differentiation, and it is likely that future guidelines will recommend evaluation for significant mutations. Given that BRAF V600E mutation status is a key feature of cHCL and is absent in vHCL, it is important to perform this testing at the time of diagnosis whenever possible. The mutation may be detected via VE1 immunohistochemical staining, allele-specific polymerase chain reaction, or next-generation sequencing. Other less sensitive tests exist but are utilized less frequently.
Minimal Residual Disease
There is currently no accepted standard for minimal residual disease (MRD) monitoring in HCL. While detection of MRD has been clearly associated with increased risk of disease progression, cHCL cells typically do not circulate in the peripheral blood, limiting the use of peripheral blood immunophenotyping for quantitative MRD assessment. For quantitative monitoring of marrow involvement by HCL, immunohistochemical staining of the bone marrow core biopsy is usually required. Staining may be performed for CD20, or, in patients who have received anti-CD20 therapy, DBA.44, VE-1, or CD79a. There is currently not a consensus regarding what level of disease involvement constitutes MRD. One group studied this issue and found that relapse could be predicted by evaluating MRD by percentage of positive cells in the marrow by immunohistochemical staining, with less than 1% involvement having the lowest risk for disease relapse and greater than 5% having the highest risk for disease relapse.25 A recent study evaluated MRD patterns in the peripheral blood of 32 cHCL patients who had completed frontline therapy. This group performed flow cytometry on the peripheral blood of patients at 1, 3, 6, and 12 months following therapy. All patients had achieved a complete response with initial therapy and peripheral blood MRD negativity at the completion of therapy. At a median follow-up of 100 months post therapy, 5 patients converted from peripheral blood–MRD negative to peripheral blood–MRD positive, and 6 patients developed overt disease progression. In all patients who progressed, progression was preceded by an increase in detectable peripheral blood MRD cells.26 Although larger studies are needed, peripheral blood flow cytometric monitoring for MRD may be a useful adjunct to predict ongoing response or impending relapse. In addition, newer, more sensitive methods of disease monitoring may ultimately supplant flow cytometry.
Risk Stratification
Although much progress has been made in the risk stratification profiling of hematologic malignancies in general, HCL has unfortunately lagged behind in this effort. The most recent risk stratification analysis was performed in 1982 by Jansen and colleagues.27 This group of researchers performed a retrospective analysis of 391 HCL patients treated at 22 centers. One of the central questions in their analysis was survival time from diagnosis in patients who had not yet undergone splenectomy (a standard treatment at the time). This group consisted of a total of 154 patients. As this study predated modern pathological and molecular testing, clinical and laboratory features were examined, and these mostly consisted of physical exam findings and analysis of the peripheral blood. This group found that several factors influenced the survival of these patients, including duration of symptoms prior to diagnosis, the degree of splenomegaly, hemoglobin level, and number of hairy cells in the peripheral blood. However, because of interobserver variation for the majority of these variables, only hemoglobin and spleen size were included in the proportional hazard model. Using only these 2 variables, the authors were able to determine 3 clinical stages for HCL (Table 1). The stages were found to correlate with median survival: patients with stage 1 disease had a median survival not reached at 72 months, but patients with stage 2 disease had a median survival of 18 months, which decreased to only 12 months in patients with stage 3 disease.
Because the majority of patients with HCL in the modern era will be diagnosed prior to reaching stage 3, a risk stratification system incorporating clinical features, laboratory parameters, and molecular and genetic testing is of considerable interest and is a subject of ongoing research. Ultimately, the goal will be to identify patients at higher risk of early relapse so that more intensive therapies can be applied to initial treatment that will result in longer treatment-free intervals.
Treatment
Because there is no curative treatment for either cHCL or vHCL outside allogeneic transplantation, and it is not clear that early treatment leads to better outcomes in HCL, patients do not always receive treatment at the time of diagnosis or relapse. The general consensus is that patients should be treated if there is a declining trend in hematologic parameters or they experience symptoms from the disease.24 Current consensus guidelines recommend treatment when any of the following hematologic parameters are met: hemoglobin less than 11 g/dL, platelet count less than 100 × 103/µL, or absolute neutrophil count less than 1000/µL.24 These parameters are surrogate markers that indicate compromised bone marrow function. Cytopenias may also be caused by splenomegaly, and symptomatic splenomegaly with or without cytopenias is an indication for treatment. A small number of patients with HCL (approximately 10%) do not require immediate therapy after diagnosis and are monitored by their provider until treatment is indicated.
First-Line Therapy
Despite advances in targeted therapies for HCL, because no treatment has been shown to extend the treatment-free interval longer than chemotherapy, treatment with a purine nucleoside analog is usually the recommended first-line therapy. This includes either cladribine or pentostatin. Both agents appear to be equally effective, and the choice of therapy is determined by the treating physician based on his or her experience. Cladribine administration has been studied using a number of different schedules and routes: intravenous continuous infusion (0.1 mg/kg) for 7 days, intravenous infusion (0.14 mg/kg/day) over 2 hours on a 5-day regimen, or alternatively subcutaneously (0.1–0.14 mg/kg/day) on a once-per-day or once-per-week regimen (Table 2).28,29
Unlike cHCL, vHCL remains difficult to treat and early disease progression is common. The best outcomes have been seen in patients who have received combination chemo-immunotherapy such as purine nucleoside analog therapy plus rituximab or bendamustine plus rituximab.31 One pilot study of bendamustine plus rituximab in 12 patients found an overall response rate of 100%, with the majority of patients achieving a complete response.31 For patients who achieved a complete response, the median duration of response had not been reached, but patients achieving only a partial response had a median duration of response of only 20 months, indicating there is a subgroup of patients who will require a different treatment approach.32 A randomized phase 2 trial of rituximab with either pentostatin or bendamustine is ongoing.33
Assessment of Response
Response assessment involves physical examination for estimation of spleen size, assessment of hematologic parameters, and a bone marrow biopsy for evaluation of marrow response. It is recommended that the bone marrow biopsy be performed 4 to 6 months following cladribine administration, or after completion of 12 doses of pentostatin. Detailed response assessment criteria are shown in Table 3.
Second-Line Therapy
Although the majority of patients treated with purine analogs will achieve durable remissions, approximately 40% of patients will eventually require second-line therapy. Criteria for treatment at relapse are the same as the criteria for initial therapy, including symptomatic disease or progressive anemia, thrombocytopenia, or neutropenia. The choice of treatment is based on clinical parameters and the duration of the previous remission. If the initial remission was longer than 65 months and the patient is eligible to receive chemotherapy, re-treatment with initial therapy is recommended. For a remission between 24 and 65 months, re-treatment with a purine analog combined with an anti-CD20 monoclonal antibody may be considered.34 If the first remission is shorter than 24 months, confirmation of the original diagnosis as well as consideration for testing for additional mutations with therapeutic targets (BRAF V600E, MAP2K1) should be considered before a treatment decision is made. For these patients, alternative therapies, including investigational agents, should be considered.24
Monoclonal antibody therapy has been studied in both the up-front setting and in relapsed or refractory HCL.35 An initial study of 15 patients with relapsed HCL found an overall response rate of 80%, with 8 patients achieving a complete response. A subsequent study of 26 patients who relapsed after cladribine therapy found an overall response rate of 80%, with a complete response rate of 32%. Median relapse-free survival was 27 months.36 Ravandi and others studied rituximab in the up-front setting in combination with cladribine, and found an overall response rate of 100%, including in patients with vHCL. At the time of publication of the study results, the median survival had not been reached.37 As has been seen with other lymphoid malignancies, concurrent therapy with rituximab appears to enhance the activity of the agent with which it is combined. While its use in the up-front setting remains an area of active investigation, there is a clear role for chemo-immunotherapy in the relapsed setting.
In patients with cHCL, excellent results including complete remissions have been reported with the use of BRAF inhibitors, both as a single agent and when combined with anti-CD20 therapy. The 2 commercially available BRAF inhibitors are vemurafenib and dabrafenib, and both have been tested in relapsed cHCL.38,39 The first study of vemurafenib was reported by Tiacci and colleagues, who found an overall response rate of 96% after a median of 8 weeks and a 100% response rate after a median of 12 weeks, with complete response rates up to 42%.38 The median relapse-free survival was 23 months (decreasing to only 6 months in patients who achieved only a partial remission), indicating that these agents will likely need to be administered in combination with other effective therapies with non-overlapping toxicities. Vemurafenib has been administered concurrently with rituximab, and preliminary results of this combination therapy showed early rates of complete responses.40 Dabrafenib has been reported for use as a single agent in cHCL and clinical trials are underway evaluating its efficacy when administered with trametinib, a MEK inhibitor.39,41 Of note, patients receiving BRAF inhibitors frequently develop cutaneous complications of RAF inhibition including cutaneous squamous cell carcinomas and keratoacanthomas, and close dermatologic surveillance is required.
Variant HCL does not harbor the BRAF V600E mutation, but up to half of patients have been found to have mutations of MAP2K1, which upregulates MEK1 expression.42 Trametinib is approved by the US Food and Drug Administration for the treatment of patients with melanoma at a dose of 2 mg orally daily, and has been successfully used to treat 1 patient with vHCL.43 Further evaluation of this targeted therapy is underway.
Ibrutinib, a Bruton tyrosine kinase inhibitor, and moxetumomab pasudotox, an immunotoxin conjugate, are currently being studied in National Institutes of Health–sponsored multi-institutional trials for patients with HCL. Ibrutinib is administered orally at 420 mg per day until relapse.44 Moxetumomab pasudotox was tested at different doses between 5 and 50 μg/kg intravenously every other day for 3 doses for up to 16 cycles unless they experienced disease progression or developed neutralizing antibodies.45 Both agents have been shown to have significant activity in cHCL and vHCL and will likely be included in the treatment armamentarium once trials are completed. Second-line therapy options are summarized in Table 4.
Complications and Supportive Care
The complications of HCL may be separated into the pre-, intra-, and post-treatment periods. At the time of diagnosis and prior to the initiation of therapy, marrow infiltration by HCL frequently leads to cytopenias which cause symptomatic anemia, infection, and/or bleeding complications. Many patients develop splenomegaly, which may further lower the blood counts and which is experienced as abdominal fullness or distention, with early satiety leading to weight loss. Patients may also experience constitutional symptoms with fatigue, fevers in the absence of infection, and unintentional weight loss even without splenomegaly.
For patients who initiate therapy with purine nucleoside analogs, the early part of treatment is associated with the greatest risk of morbidity and mortality. Chemotherapy leads to both immunosuppression (altered cellular immunity) as well as myelosuppression. Thus, patients who are already in need of treatment because of disease-related cytopenias will experience an abrupt and sometimes significant decline in the peripheral blood counts. The treatment period prior to recovery of neutrophils requires the greatest vigilance. Because patients are profoundly immunocompromised, febrile neutropenia is a common complication leading to hospital admission and the cause is often difficult to identify. Treatment with broad-spectrum antibiotics, investigation for opportunistic and viral infections, and considerations for antifungal prophylaxis or therapy are required in this setting. It is recommended that all patients treated with purine nucleoside analogs receive prophylactic antimicrobials for herpes simplex virus and varicella zoster virus, as well as prophylaxis against Pneumocystis jirovecii. Unfortunately, growth factor support has not proven successful in this patient population but is not contraindicated.46
Following successful completion of therapy, patients may remain functionally immunocompromised for a significant period of time even with a normal neutrophil count. Monitoring of the CD4 count may help to determine when prophylactic antimicrobials may be discontinued. A CD4 count greater than 200 cells/µL is generally considered to be adequate for prevention of opportunistic infections. Although immunizations have not been well studied in HCL, it is recommended that patients receive annual influenza immunizations as well as age-appropriate immunizations against Streptococcus pneumoniae and other infectious illnesses as indicated. Live viral vaccines such as the currently available herpes zoster vaccine can lead to infections in this patient population and are not recommended.
Like many hematologic malignancies, HCL may be associated with comorbid conditions related to immune dysfunction. There is a known association with an increased risk of second primary malignancies, which may predate the diagnosis of HCL.47 Therefore, it is recommended that patients continue annual cancer screenings as well as undergo prompt evaluation for potential symptoms of second malignancies. In addition, it is thought that there may be an increased risk for autoimmune disorders such as inflammatory arthritis or immune-mediated cytopenias. One case-control study found a possible association between autoimmune diseases and HCL, noting that at times these diseases are diagnosed concurrently.48 However, because of the rarity of the disease it has been difficult to quantify these associated conditions in a systematic way. There is currently an international patient data registry under development for the systematic study of HCL and its complications which may answer many of these questions.
Survivorship and quality of life are important considerations in chronic diseases. It is not uncommon for patients to develop anxiety related to the trauma of diagnosis and treatment, especially when intensive care has been required. Patients may have lingering fears regarding concerns of developing infections due to exposure to ill persons or fears regarding risk of relapse and need for re-treatment. A proactive approach with partnership with psychosocial oncology may be of benefit, especially when symptoms of post-traumatic stress disorder are evident.
Conclusion
HCL is a rare, chronic lymphoid malignancy that is now subclassified into classic and variant HCL. Further investigations into the disease subtypes will allow more precise disease definitions, and these studies are underway. Renewed efforts toward updated risk stratification and clinical staging systems will be important aspects of these investigations. Refinements in treatment and supportive care have resulted in greatly improved overall survival, which has translated into larger numbers of people living with HCL. However, new treatment paradigms for vHCL are needed as the progression-free survival in this disease remains significantly lower than that of cHCL. Future efforts toward understanding survivorship issues and management of long-term treatment and disease-related complications will be critical for ensuring good quality of life for patients living with HCL.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
1. Teras LR, Desantis DE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC; 2008.
3. Yetgin S, Olcay L, Yenicesu I, et al. Relapse in hairy cell leukemia due to isolated nodular skin infiltration. Pediatr Hematol Oncol 2001;18:415–7.
4. Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:175–9.
5. Veterans and agent orange: update 2014. Mil Med 2017;182:1619–20.
6. Villemagne B, Bay JO, Tournilhac O, et al. Two new cases of familial hairy cell leukemia associated with HLA haplotypes A2, B7, Bw4, Bw6. Leuk Lymphoma 2005;46:243–5.
7. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol 2013;163:407–9.
8. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood 1958;13:609–30.
9. Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood 1966;27:199–211.
10. Polliack A, Tadmor T. Surface topography of hairy cell leukemia cells compared to other leukemias as seen by scanning electron microscopy. Leuk Lymphoma 2011;52 Suppl 2:14–7.
11. Miranda RN, Cousar JB, Hammer RD, et al. Somatic mutation analysis of IgH variable regions reveals that tumor cells of most parafollicular (monocytoid) B-cell lymphoma, splenic marginal zone B-cell lymphoma, and some hairy cell leukemia are composed of memory B lymphocytes. Hum Pathol 1999;30:306–12.
12. Vanhentenrijk V, Tierens A, Wlodarska I, et al. V(H) gene analysis of hairy cell leukemia reveals a homogeneous mutation status and suggests its marginal zone B-cell origin. Leukemia 2004;18:1729–32.
13. Basso K, Liso A, Tiacci E, et al. Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59–68.
14. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med 2014;6:238ra71.
15. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305–15.
16. Kamiguti AS, Harris RJ, Slupsky JR, et al. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003;22:2272–84.
17. Rahman MA, Salajegheh A, Smith RA, Lam AK. BRAF inhibitors: From the laboratory to clinical trials. Crit Rev Oncol Hematol 2014;90:220–32.
18. Shao H, Calvo KR, Gronborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res 2013;37:401–9.
19. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330–2.
20. Jain P, Pemmaraju N, Ravandi F. Update on the biology and treatment options for hairy cell leukemia. Curr Treat Options Oncol 2014;15:187–209.
21. Sivina M, Kreitman RJ, Arons E, et al. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol 2014;166:177–88.
22. Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood 2015;126:842–50.
23. Andritsos LA, Grever MR. Historical overview of hairy cell leukemia. Best Pract Res Clin Haematol 2015;28:166–74.
24. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood 2017;129:553–60.
25. Mhawech-Fauceglia P, Oberholzer M, Aschenafi S, et al. Potential predictive patterns of minimal residual disease detected by immunohistochemistry on bone marrow biopsy specimens during a long-term follow-up in patients treated with cladribine for hairy cell leukemia. Arch Pathol Lab Med 2006;130:374–7.
26. Ortiz-Maldonado V, Villamor N, Baumann T, et al., Is there a role for minimal residual disease monitoring in the management of patients with hairy-cell leukaemia? Br J Haematol 2017 Aug 18.
27. Jansen J, Hermans J. Clinical staging system for hairy-cell leukemia. Blood 1982;60:571–7.
28. Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583–90.
29. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
30. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974–82.
31. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res 2013;19:6873–81.
32. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res 2013;19:6313–21.
33. Randomized phase II trial of rituximab with either pentostatin or bendamustine for multiply relapsed or refractory hairy cell leukemia. 2017 [cited 2017 Oct 26]; NCT01059786. https://clinicaltrials.gov/ct2/show/NCT01059786.
34. Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52 Suppl 2:75–8.
35. Thomas DA, O’Brien S, Bueso-Ramos C, et al. Rituximab in relapsed or refractory hairy cell leukemia. Blood 2003;102:3906–11.
36. Zenhäusern R, Simcock M, Gratwohl A, et al. Rituximab in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine (SAKK 31/98). Haematologica 2008;93(9):1426–8.
37. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818–23.
38. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 2015;373:1733–47.
39. Blachly JS, Lozanski G, Lucas DM, et al. Cotreatment of hairy cell leukemia and melanoma with the BRAF inhibitor dabrafenib. J Natl Compr Canc Netw 2015;13:9–13.
40. Tiacci E, De Carolis L, Zaja F, et al. Vemurafenib plus rituximab in hairy cell leukemia: a promisingchemotherapy-free regimen for relapsed or refractory patients. Blood 2016;128:1.
41. A phase II, open-label study in subjects with BRAF V600E-mutated rare cancers with several histologies to investigate the clinical efficacy and safety of the combination therapy of dabrafenib and trametinib. 2017 [cited 2017 Oct 26]; NCT02034110. https://clinicaltrials.gov/ct2/show/NCT02034110.
42. Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014;46:8–10.
43. Andritsos LA, Grieselhuber NR, Anghelina M, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma 2017 Sep 18:1–4.
44. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–506.
45. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30:1822–8.
46. Saven A, Burian C, Adusumalli J, Koziol JA. Filgrastim for cladribine-induced neutropenic fever in patients with hairy cell leukemia. Blood 1999;93:2471–7.
47. Cornet E, Tomowiak C, Tanguy-Schmidt A, et al. Long-term follow-up and second malignancies in 487 patients with hairy cell leukaemia. Br J Haematol 2014;166:390–400.
48. Anderson LA, Engels EA. Autoimmune conditions and hairy cell leukemia: an exploratory case-control study. J Hematol Oncol 2010;3:35.
Researchers identify potential gene target for AML drug development
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
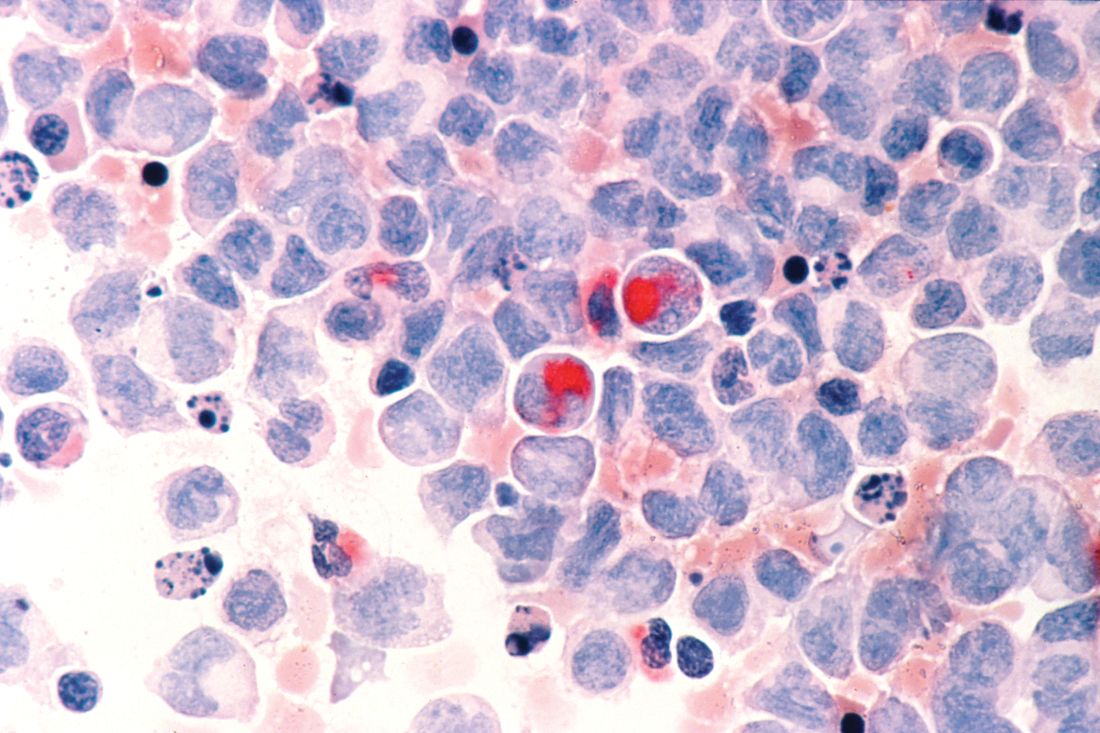
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
FROM NATURE
Study reveals predictor of early mortality in AML

New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
How CLL patients weigh treatment efficacy, safety, and cost

New research suggests patients with chronic lymphocytic leukemia (CLL) are willing to trade treatment efficacy for a reduced risk of side effects, but the cost of treatment may trump other factors.
The patients studied placed the highest value on treatments that deliver the longest progression-free survival (PFS), but the patients were also willing to swap some efficacy for a reduced risk of serious adverse events (AEs).
Study results also indicated that factoring out-of-pocket costs into the decision-making process can significantly influence a patient’s choice of treatment.
Carol Mansfield, PhD, of RTI Health Solutions in Research Triangle Park, North Carolina, and her colleagues conducted this study and reported the results in Blood Advances. The study was supported by funding from Genentech, Inc., to RTI Health Solutions.
The researchers surveyed 384 patients with CLL. Patients were asked to choose between hypothetical treatment options, each of which was defined by 5 variable attributes—PFS, mode of administration, typical severity of diarrhea, chance of serious infection, and chance of organ damage.
The attribute patients ranked highest was a change in PFS from 10 months to 60 months. This was followed by a change in infection risk from 30% to 0%, a change in the risk of organ damage from 8% to 0%, a change in diarrhea from severe to none, and a change in the mode of administration from intravenous to oral.
On average, a gain in PFS of 35.9 months was needed for patients to accept a 30% risk of serious infection. A gain in PFS of 26.3 months was needed for patients to accept an 8% risk of organ damage.
A gain in PFS of 21.6 months was needed for patients to accept severe diarrhea. And a gain in PFS of 3.5 months was needed for patients to accept the change from a daily pill to intravenous administration for 6 months.
There were no significant differences in preferences among treatment-naïve patients, first-line patients, and relapsed/refractory patients.
Impact of cost
When the researchers conducted a supplemental cost analysis, they found that out-of-pocket cost had a substantial impact on treatment choice.
The cost analysis included 2 treatments—medicines A and B. Based on the prior analysis, the researchers predicted that 91% of patients would choose medicine B if cost were not a concern because B offered longer PFS than A.
“We used the results from the discrete-choice experiment to forecast the probability that a respondent would pick each hypothetical drug without any mention of cost and then compared that to the choices people made when out-of-pocket costs for these medicines were included,” Dr Mansfield explained.
Patients were asked to choose between medicines A and B under 2 circumstances in which B cost more than A.
When medicine B had a monthly out-of-pocket cost that was $75 more than medicine A, 50% of patients chose medicine A.
When medicine B had a monthly out-of-pocket cost that was $400 more than medicine A, 74% of patients chose medicine A.
“Cost is clearly something that has an impact,” Dr Mansfield said. “When patients get prescribed something they can’t afford, they have to make very difficult choices.”
Dr Mansfield and her colleagues believe their findings will help doctors and patients focus on treatments that account for a patient’s unique circumstances and goals.
“Patients don’t always know that they could be making these tradeoffs,” Dr Mansfield said. “We hope that our findings can help doctors to have frank discussions with their patients about the differences between treatments and how these might affect their lives.” ![]()
New research suggests patients with chronic lymphocytic leukemia (CLL) are willing to trade treatment efficacy for a reduced risk of side effects, but the cost of treatment may trump other factors.
The patients studied placed the highest value on treatments that deliver the longest progression-free survival (PFS), but the patients were also willing to swap some efficacy for a reduced risk of serious adverse events (AEs).
Study results also indicated that factoring out-of-pocket costs into the decision-making process can significantly influence a patient’s choice of treatment.
Carol Mansfield, PhD, of RTI Health Solutions in Research Triangle Park, North Carolina, and her colleagues conducted this study and reported the results in Blood Advances. The study was supported by funding from Genentech, Inc., to RTI Health Solutions.
The researchers surveyed 384 patients with CLL. Patients were asked to choose between hypothetical treatment options, each of which was defined by 5 variable attributes—PFS, mode of administration, typical severity of diarrhea, chance of serious infection, and chance of organ damage.
The attribute patients ranked highest was a change in PFS from 10 months to 60 months. This was followed by a change in infection risk from 30% to 0%, a change in the risk of organ damage from 8% to 0%, a change in diarrhea from severe to none, and a change in the mode of administration from intravenous to oral.
On average, a gain in PFS of 35.9 months was needed for patients to accept a 30% risk of serious infection. A gain in PFS of 26.3 months was needed for patients to accept an 8% risk of organ damage.
A gain in PFS of 21.6 months was needed for patients to accept severe diarrhea. And a gain in PFS of 3.5 months was needed for patients to accept the change from a daily pill to intravenous administration for 6 months.
There were no significant differences in preferences among treatment-naïve patients, first-line patients, and relapsed/refractory patients.
Impact of cost
When the researchers conducted a supplemental cost analysis, they found that out-of-pocket cost had a substantial impact on treatment choice.
The cost analysis included 2 treatments—medicines A and B. Based on the prior analysis, the researchers predicted that 91% of patients would choose medicine B if cost were not a concern because B offered longer PFS than A.
“We used the results from the discrete-choice experiment to forecast the probability that a respondent would pick each hypothetical drug without any mention of cost and then compared that to the choices people made when out-of-pocket costs for these medicines were included,” Dr Mansfield explained.
Patients were asked to choose between medicines A and B under 2 circumstances in which B cost more than A.
When medicine B had a monthly out-of-pocket cost that was $75 more than medicine A, 50% of patients chose medicine A.
When medicine B had a monthly out-of-pocket cost that was $400 more than medicine A, 74% of patients chose medicine A.
“Cost is clearly something that has an impact,” Dr Mansfield said. “When patients get prescribed something they can’t afford, they have to make very difficult choices.”
Dr Mansfield and her colleagues believe their findings will help doctors and patients focus on treatments that account for a patient’s unique circumstances and goals.
“Patients don’t always know that they could be making these tradeoffs,” Dr Mansfield said. “We hope that our findings can help doctors to have frank discussions with their patients about the differences between treatments and how these might affect their lives.” ![]()
New research suggests patients with chronic lymphocytic leukemia (CLL) are willing to trade treatment efficacy for a reduced risk of side effects, but the cost of treatment may trump other factors.
The patients studied placed the highest value on treatments that deliver the longest progression-free survival (PFS), but the patients were also willing to swap some efficacy for a reduced risk of serious adverse events (AEs).
Study results also indicated that factoring out-of-pocket costs into the decision-making process can significantly influence a patient’s choice of treatment.
Carol Mansfield, PhD, of RTI Health Solutions in Research Triangle Park, North Carolina, and her colleagues conducted this study and reported the results in Blood Advances. The study was supported by funding from Genentech, Inc., to RTI Health Solutions.
The researchers surveyed 384 patients with CLL. Patients were asked to choose between hypothetical treatment options, each of which was defined by 5 variable attributes—PFS, mode of administration, typical severity of diarrhea, chance of serious infection, and chance of organ damage.
The attribute patients ranked highest was a change in PFS from 10 months to 60 months. This was followed by a change in infection risk from 30% to 0%, a change in the risk of organ damage from 8% to 0%, a change in diarrhea from severe to none, and a change in the mode of administration from intravenous to oral.
On average, a gain in PFS of 35.9 months was needed for patients to accept a 30% risk of serious infection. A gain in PFS of 26.3 months was needed for patients to accept an 8% risk of organ damage.
A gain in PFS of 21.6 months was needed for patients to accept severe diarrhea. And a gain in PFS of 3.5 months was needed for patients to accept the change from a daily pill to intravenous administration for 6 months.
There were no significant differences in preferences among treatment-naïve patients, first-line patients, and relapsed/refractory patients.
Impact of cost
When the researchers conducted a supplemental cost analysis, they found that out-of-pocket cost had a substantial impact on treatment choice.
The cost analysis included 2 treatments—medicines A and B. Based on the prior analysis, the researchers predicted that 91% of patients would choose medicine B if cost were not a concern because B offered longer PFS than A.
“We used the results from the discrete-choice experiment to forecast the probability that a respondent would pick each hypothetical drug without any mention of cost and then compared that to the choices people made when out-of-pocket costs for these medicines were included,” Dr Mansfield explained.
Patients were asked to choose between medicines A and B under 2 circumstances in which B cost more than A.
When medicine B had a monthly out-of-pocket cost that was $75 more than medicine A, 50% of patients chose medicine A.
When medicine B had a monthly out-of-pocket cost that was $400 more than medicine A, 74% of patients chose medicine A.
“Cost is clearly something that has an impact,” Dr Mansfield said. “When patients get prescribed something they can’t afford, they have to make very difficult choices.”
Dr Mansfield and her colleagues believe their findings will help doctors and patients focus on treatments that account for a patient’s unique circumstances and goals.
“Patients don’t always know that they could be making these tradeoffs,” Dr Mansfield said. “We hope that our findings can help doctors to have frank discussions with their patients about the differences between treatments and how these might affect their lives.” ![]()
CCSs have increased risk of hypertension
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
Late-breaking abstracts highlight treatment advances in CLL, myeloma, and more
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
.
In a preplanned interim analysis of data from 389 patients in the randomized phase III Murano trial, venetoclax and rituximab therapy proved “superior to the standard of care and well tolerated, and a major advance in the management of [relapsed/refractory] CLL,” ASH President Kenneth C. Anderson, MD said during a premeeting preview session for the media.
In Murano, venetoclax plus rituximab bettered bendamustine plus rituximab in progression-free survival, overall survival, overall and complete response rates, and number of patients achieving minimal residual disease (MRD) negativity, said Dr. Anderson, who is also director of the Lebow Institute for Myeloma Therapeutics and Jerome Lipper Myeloma Center at Dana-Farber Cancer Institute, Boston.
The results were consistent in all risk subsets, including patients who had high-risk disease by virtue of chromosome 17p deletion, according Dr. Anderson.
In another late-breaking randomized phase III study, known as ALCYONE, adding the CD38-targeting monoclonal antibody daratumumab to standard therapy with bortezomib, melphalan, and prednisone (VMP) resulted in a “doubling” of progression-free survival in patients who had newly diagnosed multiple myeloma and were ineligible for transplantation, he reported.
In the trial of more than 700 patients, daratumumab plus VMP as initial treatment for nontransplant patients was well tolerated and improved outcomes, including overall response rate and the percent of patients who achieved MRD negative status.
“As we saw in CLL, so it’s true in this abstract in myeloma: this is a very major advance,” Dr. Anderson said.
Also during the preview session, ASH Secretary Robert A. Brodsky, MD, discussed the randomized, phase III HERCULES study results, which showed that patients with acquired thrombotic thrombocytopenic purpura (TTP) may benefit when caplacizumab is added to standard therapy. Caplacizumab targets the A1 domain of von Willebrand factor, which inhibits interaction between ultra-large von Willebrand factor and platelets.
In the trial, 145 patients were randomized to receive either plasma exchange alone or plasma exchange and caplacizumab.
Preliminary results suggest “this was a very positive trial” with a primary endpoint of time to platelet response that “greatly favored the caplacizumab arm,” said Dr. Brodsky, professor of medicine and oncology and director of the division of hematology at Johns Hopkins University, Baltimore. “Even the secondary composite endpoint of death, recurrence, and/or major thromboembolic events was much improved with caplacizumab, so this is a very positive trial and potentially a game-changing drug for the management of TTP, which can be very challenging.”
Dr. Brodsky also discussed the Hokusai VTE-Cancer Study, a randomized, open-label, blinded outcome assessment trial that showed the oral factor Xa inhibitor edoxaban was noninferior to subcutaneous dalteparin for the prevention of cancer-associated venous thromboembolism.
With more than 1,000 patients enrolled in 114 centers, the Hokusai VTE-Cancer Study had a primary outcome of the composite of the first recurrent VTE or major bleeding event during follow-up. The primary outcome occurred in 12.8% of patients in the edoxaban group, compared with 13.5% of patients in the dalteparin group (P = .0056 for noninferiority), according to the preliminary published results.
The key question addressed by the trial is whether a newer oral anticoagulant, edoxaban, can substitute for the older, subcutaneously administered low-molecular-weight heparin, dalteparin. The results “confirmed that a newer oral anticoagulant is at least as good and as safe as the low molecular weight heparin,” allowing patients the convenience of an oral therapy, Dr. Brodsky noted.
This year’s late-breaking abstracts at ASH are:
LBA-1 Results of the Randomized, Double-Blind, Placebo-Controlled, Phase III Hercules Study of Caplacizumab in Patients with Acquired Thrombotic Thrombocytopenic Purpura.
LBA-2 Venetoclax Plus Rituximab Is Superior to Bendamustine Plus Rituximab in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia - Results from Pre-Planned Interim Analysis of the Randomized Phase III Murano Study.
LBA-3 Mutations in SRP54 Gene Cause Severe Primary Neutropenia As Well As Shwachman-Diamond-like Syndrome.
LBA-4 Phase III Randomized Study of Daratumumab Plus Bortezomib, Melphalan, and Prednisone (D-VMP) Versus Bortezomib, Melphalan, and Prednisone (VMP) in Newly Diagnosed Multiple Myeloma (NDMM) Patients (Pts) Ineligible for Transplant (ALCYONE).
LBA-5 Prospective Molecular MRD Detection By NGS: A Powerful Independent Predictor for Relapse and Survival in Adults with Newly Diagnosed AML.
LBA-6 A Randomized, Open-Label, Blinded Outcome Assessment Trial Evaluating the Efficacy and Safety of LMWH/Edoxaban Versus Dalteparin for Venous Thromboembolism Associated with Cancer: Hokusai VTE-Cancer Study
FROM ASH 2017
Method identifies effective treatments for leukemias, lymphomas

An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
PTSD can persist in cancer survivors

Cancer patients may experience lasting post-traumatic stress disorder (PTSD), according to a study published in the journal Cancer.
Approximately one-fifth of patients involved in the study experienced PTSD several months after their cancer diagnosis, and roughly a third of these patients continued to live with PTSD 4 years later.
Researchers say these findings highlight the need for early identification, careful monitoring, and treatment of PTSD in cancer survivors.
Caryn Mei Hsien Chan, PhD, of the National University of Malaysia in Kuala Lumpur, and her colleagues conducted this research.
The study included 469 adults with various cancers who were within 1 month of cancer diagnosis at enrollment.
Patients who had significant psychological distress (defined as a Hospital Anxiety and Depression Scale total cutoff score of 16 or higher) underwent
testing for PTSD at 6 months of follow-up. All patients were tested for PTSD at 4 years of follow-up (regardless of their Hospital Anxiety and Depression Scale score).
The incidence of PTSD was 21.7% at 6 months and 6.1% at 4 years. Although overall rates of PTSD decreased with time, roughly one-third of patients initially diagnosed with PTSD were found to have persistent or worsening symptoms 4 years later.
“Many cancer patients believe they need to adopt a ‘warrior mentality’ and remain positive and optimistic from diagnosis through treatment to stand a better chance of beating their cancer,” Dr Chan said.
“To these patients, seeking help for the emotional issues they face is akin to admitting weakness. There needs to be greater awareness that there is nothing wrong with getting help to manage the emotional upheaval—particularly depression, anxiety, and PTSD—post-cancer.”
Dr Chan also stressed that many patients live in fear that their cancer may come back, and they may think the cancer has returned with every lump or bump, pain or ache, fatigue or fever.
In addition, cancer survivors might skip visits to their oncologists or other physicians to avoid triggering memories of their past cancer experience. This can lead to delays in seeking help for new symptoms or even refusal of treatment for unrelated conditions.
“We need psychological evaluation and support services for patients with cancer at an initial stage and at continued follows-up because psychological well-being and mental health—and by extension, quality of life—are just as important as physical health,” Dr Chan noted. ![]()
Cancer patients may experience lasting post-traumatic stress disorder (PTSD), according to a study published in the journal Cancer.
Approximately one-fifth of patients involved in the study experienced PTSD several months after their cancer diagnosis, and roughly a third of these patients continued to live with PTSD 4 years later.
Researchers say these findings highlight the need for early identification, careful monitoring, and treatment of PTSD in cancer survivors.
Caryn Mei Hsien Chan, PhD, of the National University of Malaysia in Kuala Lumpur, and her colleagues conducted this research.
The study included 469 adults with various cancers who were within 1 month of cancer diagnosis at enrollment.
Patients who had significant psychological distress (defined as a Hospital Anxiety and Depression Scale total cutoff score of 16 or higher) underwent
testing for PTSD at 6 months of follow-up. All patients were tested for PTSD at 4 years of follow-up (regardless of their Hospital Anxiety and Depression Scale score).
The incidence of PTSD was 21.7% at 6 months and 6.1% at 4 years. Although overall rates of PTSD decreased with time, roughly one-third of patients initially diagnosed with PTSD were found to have persistent or worsening symptoms 4 years later.
“Many cancer patients believe they need to adopt a ‘warrior mentality’ and remain positive and optimistic from diagnosis through treatment to stand a better chance of beating their cancer,” Dr Chan said.
“To these patients, seeking help for the emotional issues they face is akin to admitting weakness. There needs to be greater awareness that there is nothing wrong with getting help to manage the emotional upheaval—particularly depression, anxiety, and PTSD—post-cancer.”
Dr Chan also stressed that many patients live in fear that their cancer may come back, and they may think the cancer has returned with every lump or bump, pain or ache, fatigue or fever.
In addition, cancer survivors might skip visits to their oncologists or other physicians to avoid triggering memories of their past cancer experience. This can lead to delays in seeking help for new symptoms or even refusal of treatment for unrelated conditions.
“We need psychological evaluation and support services for patients with cancer at an initial stage and at continued follows-up because psychological well-being and mental health—and by extension, quality of life—are just as important as physical health,” Dr Chan noted. ![]()
Cancer patients may experience lasting post-traumatic stress disorder (PTSD), according to a study published in the journal Cancer.
Approximately one-fifth of patients involved in the study experienced PTSD several months after their cancer diagnosis, and roughly a third of these patients continued to live with PTSD 4 years later.
Researchers say these findings highlight the need for early identification, careful monitoring, and treatment of PTSD in cancer survivors.
Caryn Mei Hsien Chan, PhD, of the National University of Malaysia in Kuala Lumpur, and her colleagues conducted this research.
The study included 469 adults with various cancers who were within 1 month of cancer diagnosis at enrollment.
Patients who had significant psychological distress (defined as a Hospital Anxiety and Depression Scale total cutoff score of 16 or higher) underwent
testing for PTSD at 6 months of follow-up. All patients were tested for PTSD at 4 years of follow-up (regardless of their Hospital Anxiety and Depression Scale score).
The incidence of PTSD was 21.7% at 6 months and 6.1% at 4 years. Although overall rates of PTSD decreased with time, roughly one-third of patients initially diagnosed with PTSD were found to have persistent or worsening symptoms 4 years later.
“Many cancer patients believe they need to adopt a ‘warrior mentality’ and remain positive and optimistic from diagnosis through treatment to stand a better chance of beating their cancer,” Dr Chan said.
“To these patients, seeking help for the emotional issues they face is akin to admitting weakness. There needs to be greater awareness that there is nothing wrong with getting help to manage the emotional upheaval—particularly depression, anxiety, and PTSD—post-cancer.”
Dr Chan also stressed that many patients live in fear that their cancer may come back, and they may think the cancer has returned with every lump or bump, pain or ache, fatigue or fever.
In addition, cancer survivors might skip visits to their oncologists or other physicians to avoid triggering memories of their past cancer experience. This can lead to delays in seeking help for new symptoms or even refusal of treatment for unrelated conditions.
“We need psychological evaluation and support services for patients with cancer at an initial stage and at continued follows-up because psychological well-being and mental health—and by extension, quality of life—are just as important as physical health,” Dr Chan noted. ![]()
Nilotinib approved to treat kids with CML in EU
The European Commission has approved nilotinib (Tasigna®) for the treatment of pediatric patients.
The drug is now approved to treat children age 2 and older with newly diagnosed, Philadelphia chromosome-positive (Ph+), chronic phase (CP) chronic myeloid leukemia (CML) or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
Nilotinib is the only second-generation tyrosine kinase inhibitor currently approved in the European Union (EU) for the treatment of Ph+ CP-CML in children. The approval applies to all EU member states.
According to Novartis, the expanded indication for nilotinib is based on 2 prospective studies of the drug in children with Ph+ CP-CML, which were part of a formal “pediatric investigation plan” agreed upon with the European Medicines Agency.
The company said 69 patients received nilotinib in these studies. The patients ranged in age from 2 to 18. They had either newly diagnosed Ph+ CP-CML or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% (95% CI: 38.7, 78.9) at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% (95% CI: 26.3, 56.8) at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation. ![]()
The European Commission has approved nilotinib (Tasigna®) for the treatment of pediatric patients.
The drug is now approved to treat children age 2 and older with newly diagnosed, Philadelphia chromosome-positive (Ph+), chronic phase (CP) chronic myeloid leukemia (CML) or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
Nilotinib is the only second-generation tyrosine kinase inhibitor currently approved in the European Union (EU) for the treatment of Ph+ CP-CML in children. The approval applies to all EU member states.
According to Novartis, the expanded indication for nilotinib is based on 2 prospective studies of the drug in children with Ph+ CP-CML, which were part of a formal “pediatric investigation plan” agreed upon with the European Medicines Agency.
The company said 69 patients received nilotinib in these studies. The patients ranged in age from 2 to 18. They had either newly diagnosed Ph+ CP-CML or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% (95% CI: 38.7, 78.9) at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% (95% CI: 26.3, 56.8) at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation. ![]()
The European Commission has approved nilotinib (Tasigna®) for the treatment of pediatric patients.
The drug is now approved to treat children age 2 and older with newly diagnosed, Philadelphia chromosome-positive (Ph+), chronic phase (CP) chronic myeloid leukemia (CML) or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
Nilotinib is the only second-generation tyrosine kinase inhibitor currently approved in the European Union (EU) for the treatment of Ph+ CP-CML in children. The approval applies to all EU member states.
According to Novartis, the expanded indication for nilotinib is based on 2 prospective studies of the drug in children with Ph+ CP-CML, which were part of a formal “pediatric investigation plan” agreed upon with the European Medicines Agency.
The company said 69 patients received nilotinib in these studies. The patients ranged in age from 2 to 18. They had either newly diagnosed Ph+ CP-CML or Ph+ CP-CML with resistance or intolerance to prior therapy, including imatinib.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% (95% CI: 38.7, 78.9) at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% (95% CI: 26.3, 56.8) at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation. ![]()
CD22-CAR therapy shows activity in rel/ref B-ALL

Researchers say they have reported the first results demonstrating clinical activity of a CD22-directed chimeric antigen receptor (CAR) T-cell therapy in B-cell acute lymphoblastic leukemia (B-ALL).
The team conducted a phase 1 study of the therapy in 21 children and adults with relapsed/refractory B-ALL.
Twelve patients achieved a complete response (CR) to the treatment, with 3 patients still in CR at last follow-up.
Sixteen patients developed cytokine release syndrome (CRS), all grade 1 or 2.
Crystal Mackall, MD, of Stanford University in California, and her colleagues reported these results in Nature Medicine.*
“This is the first time that we’ve seen response rates anything like we achieved when we were first testing the CD19 CAR T therapy,” Dr Mackall said.
“We were all a little worried that we wouldn’t find anything comparable, but this study gives hope to the idea that there may be another similar, very potent treatment.”
Patients
Dr Mackall and her colleagues studied the CD22-CAR T-cell therapy in 21 patients with relapsed/refractory B-ALL. They had a median age of 19 (range, 7 to 30).
All of the patients had received a hematopoietic stem cell transplant at least once, and 2 patients had 2 prior transplants each. Seventeen patients had received prior CD19-directed immunotherapy. Fifteen had received CD19-directed CAR T-cell therapy, and 2 had received blinatumomab.
Lymphoblasts were CD19− or CD19dim in 10 patients (9 who had received a CD19-CAR and 1 treated with blinatumomab).
The median CD22 site density was 2839 molecules per cell (range, 613 to 13,452).
Dosing and DLTs
Patients received the CD22-CAR T-cell therapy at 1 of 3 dose levels:
- 0.3 × 106 CD22-CAR T cells per kg body weight (n=6)
- 1 × 106 cells per kg (n=13)
- 3 × 106 cells per kg (n=2).
There was 1 dose-limiting toxicity (DLT) at the first dose level. It was grade 3, self-limited, noninfectious diarrhea that occurred during CRS and resolved with supportive care.
The other DLT occurred in a patient who received treatment at the third dose level. This patient had grade 4 hypoxia that was associated with rapid disease progression. The patient required brief intubation, and the hypoxia was resolved within 24 hours of starting steroid treatment.
Based on these results, the second dose level became the recommended phase 2 dose.
Other adverse events
The researchers said the primary toxicity was CRS, which occurred in 16 patients. Nine patients had grade 1 CRS, and 7 had grade 2.
There were no cases of irreversible neurotoxicity or seizure reported. Among the first 16 patients with complete assessments, there were cases of transient visual hallucinations (n=2), mild unresponsiveness (n=1), mild disorientation (n=1), and mild to moderate pain (n=2). However, these incidents resolved by day 28.
One patient died from gram-negative rod sepsis that developed after the resolution of CRS and neutrophil count recovery to >1000 cells/μL blood. The patient had a history of multi-organ failure due to sepsis.
Response
Twelve patients (57%) had a CR, and 9 of them were minimal residual disease negative.
One CR occurred at the lowest dose of therapy, 1 occurred at the highest dose, and the remaining 10 CRs occurred in patients who received dose level 2.
The researchers said there was no evidence to suggest that previous CD19-directed immunotherapy or diminished surface expression of CD19 impacted response to the CD22-CAR T-cell therapy.
Of the 9 patients who did not respond, 4 progressed and 5 had stable disease.
The researchers said 4 non-responders had “very high disease burden with rapid disease progression.” And 2 non-responders expressed diminished or partial CD22 on leukemic blasts at the time of enrollment.
The median duration of response was 6 months (range, 1.5 to 21+ months). Three patients are still in CR at 6, 9, and 21 months of follow-up.
“The take-home message is that we’ve found another CAR T-cell therapy that displays high-level activity in this phase 1 trial,” Dr Mackall said. “But the relapse rate was also high. So this forces the field to get even more sophisticated. How much of a target is needed for successful, long-lasting treatment? What happens if we target both CD19 and CD22 simultaneously?”
The researchers are already tackling the last question by testing a CAR T-cell therapy that recognizes both CD19 and CD22. They’ve confirmed this therapy can kill cancer cells in vitro and in vivo. Now, they’re testing it in a clinical trial that has opened at Stanford University and will open soon at the National Cancer Institute. ![]()
*This research was supported, in part, by the Intramural Research Program, National Cancer Institute and NIH Clinical Center, National Institutes of Health; by a Stand Up to Cancer–St. Baldrick’s Pediatric Dream Team translational research grant; and by a St. Baldrick’s Foundation Scholar Award.
Researchers say they have reported the first results demonstrating clinical activity of a CD22-directed chimeric antigen receptor (CAR) T-cell therapy in B-cell acute lymphoblastic leukemia (B-ALL).
The team conducted a phase 1 study of the therapy in 21 children and adults with relapsed/refractory B-ALL.
Twelve patients achieved a complete response (CR) to the treatment, with 3 patients still in CR at last follow-up.
Sixteen patients developed cytokine release syndrome (CRS), all grade 1 or 2.
Crystal Mackall, MD, of Stanford University in California, and her colleagues reported these results in Nature Medicine.*
“This is the first time that we’ve seen response rates anything like we achieved when we were first testing the CD19 CAR T therapy,” Dr Mackall said.
“We were all a little worried that we wouldn’t find anything comparable, but this study gives hope to the idea that there may be another similar, very potent treatment.”
Patients
Dr Mackall and her colleagues studied the CD22-CAR T-cell therapy in 21 patients with relapsed/refractory B-ALL. They had a median age of 19 (range, 7 to 30).
All of the patients had received a hematopoietic stem cell transplant at least once, and 2 patients had 2 prior transplants each. Seventeen patients had received prior CD19-directed immunotherapy. Fifteen had received CD19-directed CAR T-cell therapy, and 2 had received blinatumomab.
Lymphoblasts were CD19− or CD19dim in 10 patients (9 who had received a CD19-CAR and 1 treated with blinatumomab).
The median CD22 site density was 2839 molecules per cell (range, 613 to 13,452).
Dosing and DLTs
Patients received the CD22-CAR T-cell therapy at 1 of 3 dose levels:
- 0.3 × 106 CD22-CAR T cells per kg body weight (n=6)
- 1 × 106 cells per kg (n=13)
- 3 × 106 cells per kg (n=2).
There was 1 dose-limiting toxicity (DLT) at the first dose level. It was grade 3, self-limited, noninfectious diarrhea that occurred during CRS and resolved with supportive care.
The other DLT occurred in a patient who received treatment at the third dose level. This patient had grade 4 hypoxia that was associated with rapid disease progression. The patient required brief intubation, and the hypoxia was resolved within 24 hours of starting steroid treatment.
Based on these results, the second dose level became the recommended phase 2 dose.
Other adverse events
The researchers said the primary toxicity was CRS, which occurred in 16 patients. Nine patients had grade 1 CRS, and 7 had grade 2.
There were no cases of irreversible neurotoxicity or seizure reported. Among the first 16 patients with complete assessments, there were cases of transient visual hallucinations (n=2), mild unresponsiveness (n=1), mild disorientation (n=1), and mild to moderate pain (n=2). However, these incidents resolved by day 28.
One patient died from gram-negative rod sepsis that developed after the resolution of CRS and neutrophil count recovery to >1000 cells/μL blood. The patient had a history of multi-organ failure due to sepsis.
Response
Twelve patients (57%) had a CR, and 9 of them were minimal residual disease negative.
One CR occurred at the lowest dose of therapy, 1 occurred at the highest dose, and the remaining 10 CRs occurred in patients who received dose level 2.
The researchers said there was no evidence to suggest that previous CD19-directed immunotherapy or diminished surface expression of CD19 impacted response to the CD22-CAR T-cell therapy.
Of the 9 patients who did not respond, 4 progressed and 5 had stable disease.
The researchers said 4 non-responders had “very high disease burden with rapid disease progression.” And 2 non-responders expressed diminished or partial CD22 on leukemic blasts at the time of enrollment.
The median duration of response was 6 months (range, 1.5 to 21+ months). Three patients are still in CR at 6, 9, and 21 months of follow-up.
“The take-home message is that we’ve found another CAR T-cell therapy that displays high-level activity in this phase 1 trial,” Dr Mackall said. “But the relapse rate was also high. So this forces the field to get even more sophisticated. How much of a target is needed for successful, long-lasting treatment? What happens if we target both CD19 and CD22 simultaneously?”
The researchers are already tackling the last question by testing a CAR T-cell therapy that recognizes both CD19 and CD22. They’ve confirmed this therapy can kill cancer cells in vitro and in vivo. Now, they’re testing it in a clinical trial that has opened at Stanford University and will open soon at the National Cancer Institute. ![]()
*This research was supported, in part, by the Intramural Research Program, National Cancer Institute and NIH Clinical Center, National Institutes of Health; by a Stand Up to Cancer–St. Baldrick’s Pediatric Dream Team translational research grant; and by a St. Baldrick’s Foundation Scholar Award.
Researchers say they have reported the first results demonstrating clinical activity of a CD22-directed chimeric antigen receptor (CAR) T-cell therapy in B-cell acute lymphoblastic leukemia (B-ALL).
The team conducted a phase 1 study of the therapy in 21 children and adults with relapsed/refractory B-ALL.
Twelve patients achieved a complete response (CR) to the treatment, with 3 patients still in CR at last follow-up.
Sixteen patients developed cytokine release syndrome (CRS), all grade 1 or 2.
Crystal Mackall, MD, of Stanford University in California, and her colleagues reported these results in Nature Medicine.*
“This is the first time that we’ve seen response rates anything like we achieved when we were first testing the CD19 CAR T therapy,” Dr Mackall said.
“We were all a little worried that we wouldn’t find anything comparable, but this study gives hope to the idea that there may be another similar, very potent treatment.”
Patients
Dr Mackall and her colleagues studied the CD22-CAR T-cell therapy in 21 patients with relapsed/refractory B-ALL. They had a median age of 19 (range, 7 to 30).
All of the patients had received a hematopoietic stem cell transplant at least once, and 2 patients had 2 prior transplants each. Seventeen patients had received prior CD19-directed immunotherapy. Fifteen had received CD19-directed CAR T-cell therapy, and 2 had received blinatumomab.
Lymphoblasts were CD19− or CD19dim in 10 patients (9 who had received a CD19-CAR and 1 treated with blinatumomab).
The median CD22 site density was 2839 molecules per cell (range, 613 to 13,452).
Dosing and DLTs
Patients received the CD22-CAR T-cell therapy at 1 of 3 dose levels:
- 0.3 × 106 CD22-CAR T cells per kg body weight (n=6)
- 1 × 106 cells per kg (n=13)
- 3 × 106 cells per kg (n=2).
There was 1 dose-limiting toxicity (DLT) at the first dose level. It was grade 3, self-limited, noninfectious diarrhea that occurred during CRS and resolved with supportive care.
The other DLT occurred in a patient who received treatment at the third dose level. This patient had grade 4 hypoxia that was associated with rapid disease progression. The patient required brief intubation, and the hypoxia was resolved within 24 hours of starting steroid treatment.
Based on these results, the second dose level became the recommended phase 2 dose.
Other adverse events
The researchers said the primary toxicity was CRS, which occurred in 16 patients. Nine patients had grade 1 CRS, and 7 had grade 2.
There were no cases of irreversible neurotoxicity or seizure reported. Among the first 16 patients with complete assessments, there were cases of transient visual hallucinations (n=2), mild unresponsiveness (n=1), mild disorientation (n=1), and mild to moderate pain (n=2). However, these incidents resolved by day 28.
One patient died from gram-negative rod sepsis that developed after the resolution of CRS and neutrophil count recovery to >1000 cells/μL blood. The patient had a history of multi-organ failure due to sepsis.
Response
Twelve patients (57%) had a CR, and 9 of them were minimal residual disease negative.
One CR occurred at the lowest dose of therapy, 1 occurred at the highest dose, and the remaining 10 CRs occurred in patients who received dose level 2.
The researchers said there was no evidence to suggest that previous CD19-directed immunotherapy or diminished surface expression of CD19 impacted response to the CD22-CAR T-cell therapy.
Of the 9 patients who did not respond, 4 progressed and 5 had stable disease.
The researchers said 4 non-responders had “very high disease burden with rapid disease progression.” And 2 non-responders expressed diminished or partial CD22 on leukemic blasts at the time of enrollment.
The median duration of response was 6 months (range, 1.5 to 21+ months). Three patients are still in CR at 6, 9, and 21 months of follow-up.
“The take-home message is that we’ve found another CAR T-cell therapy that displays high-level activity in this phase 1 trial,” Dr Mackall said. “But the relapse rate was also high. So this forces the field to get even more sophisticated. How much of a target is needed for successful, long-lasting treatment? What happens if we target both CD19 and CD22 simultaneously?”
The researchers are already tackling the last question by testing a CAR T-cell therapy that recognizes both CD19 and CD22. They’ve confirmed this therapy can kill cancer cells in vitro and in vivo. Now, they’re testing it in a clinical trial that has opened at Stanford University and will open soon at the National Cancer Institute.
*This research was supported, in part, by the Intramural Research Program, National Cancer Institute and NIH Clinical Center, National Institutes of Health; by a Stand Up to Cancer–St. Baldrick’s Pediatric Dream Team translational research grant; and by a St. Baldrick’s Foundation Scholar Award.