User login
Chronic Urticaria: It’s More Than Just Antihistamines!
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
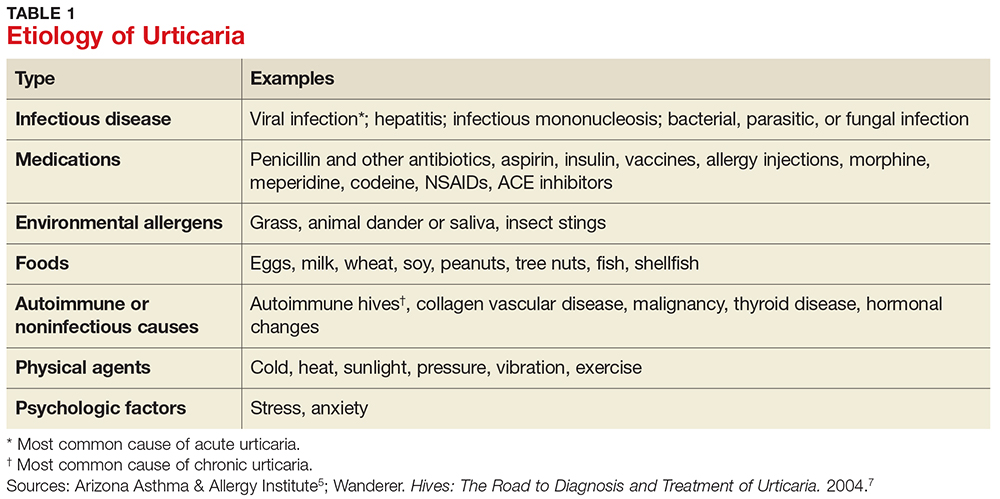
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
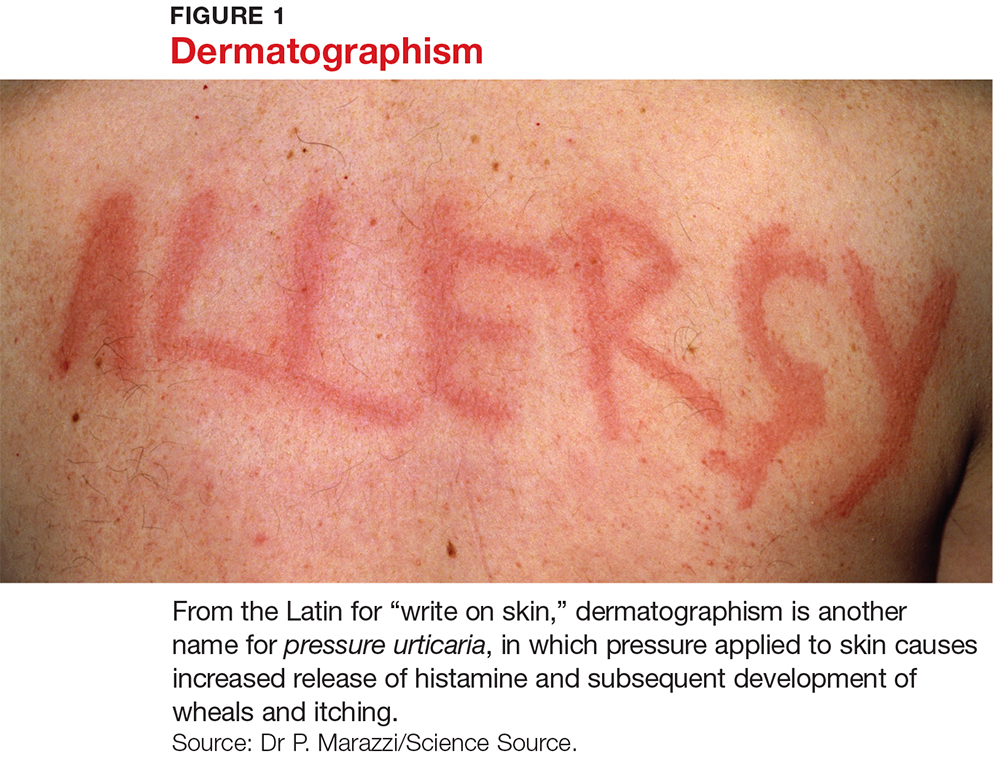
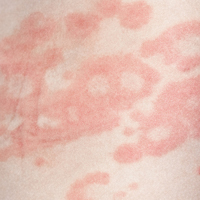
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
CLINICAL FEATURES
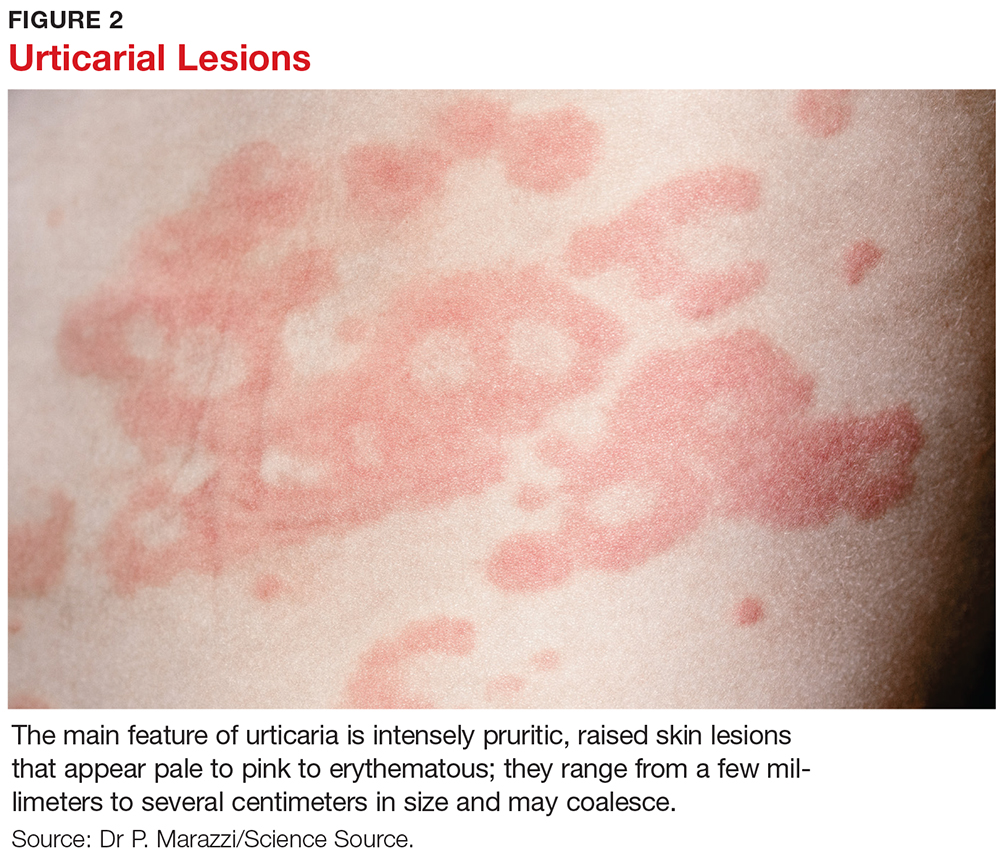
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
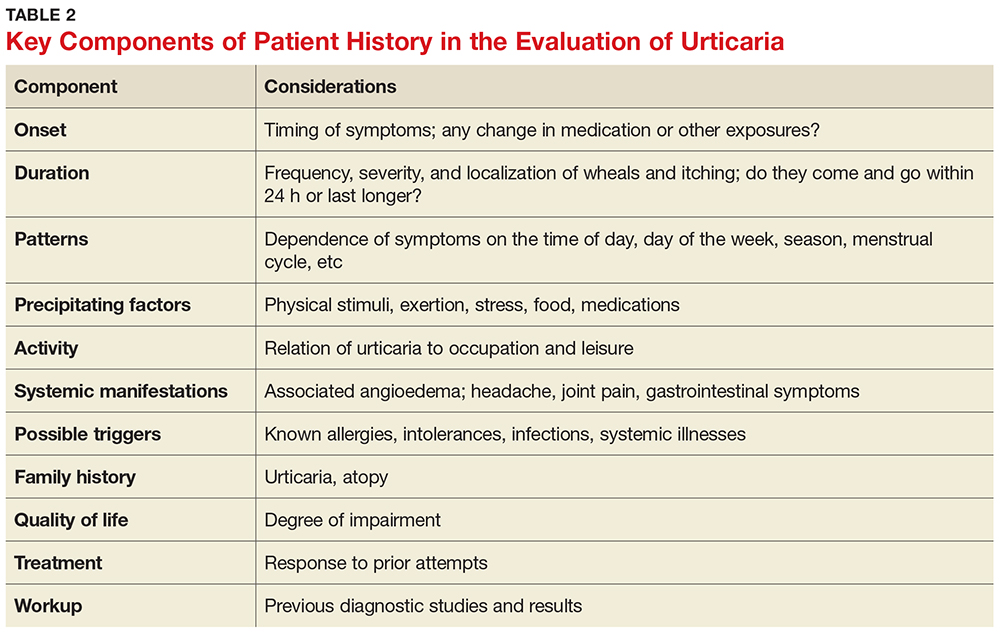
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
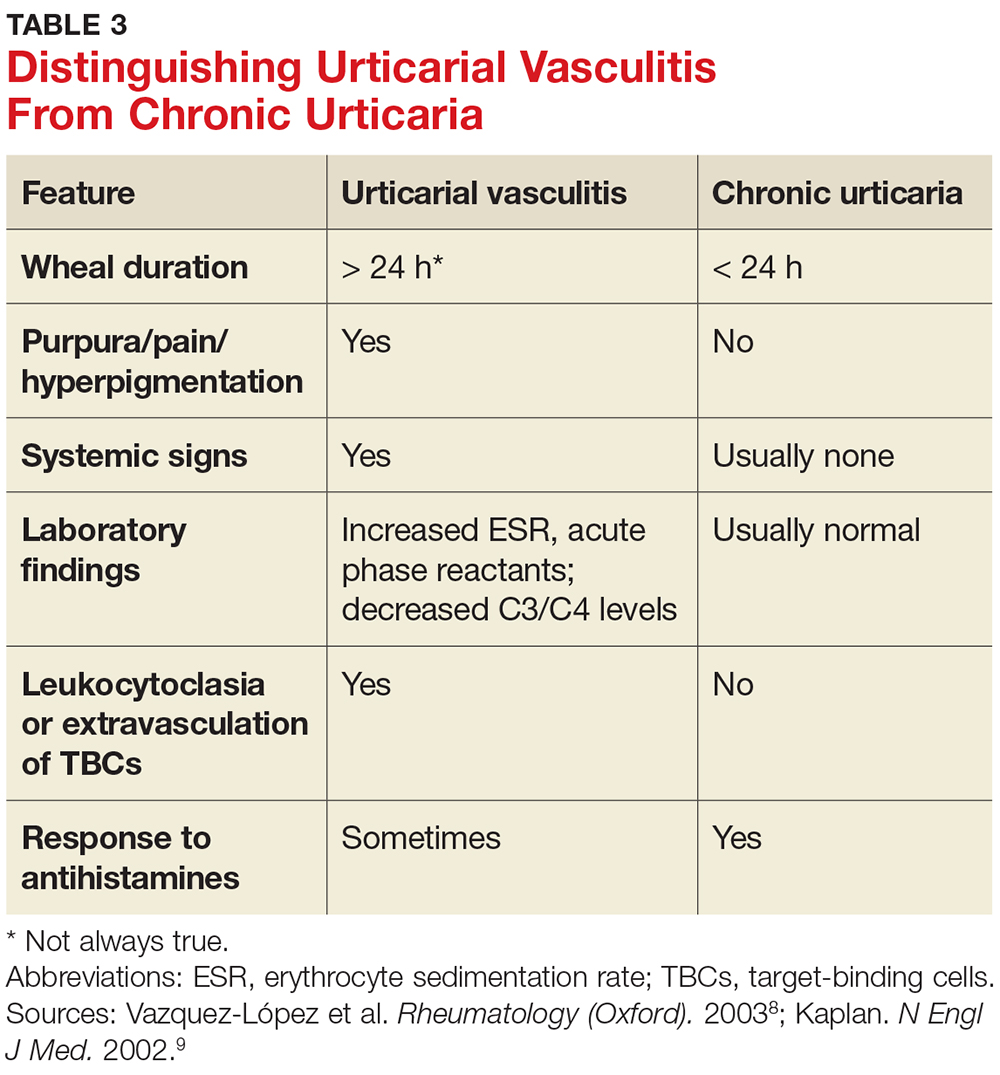
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
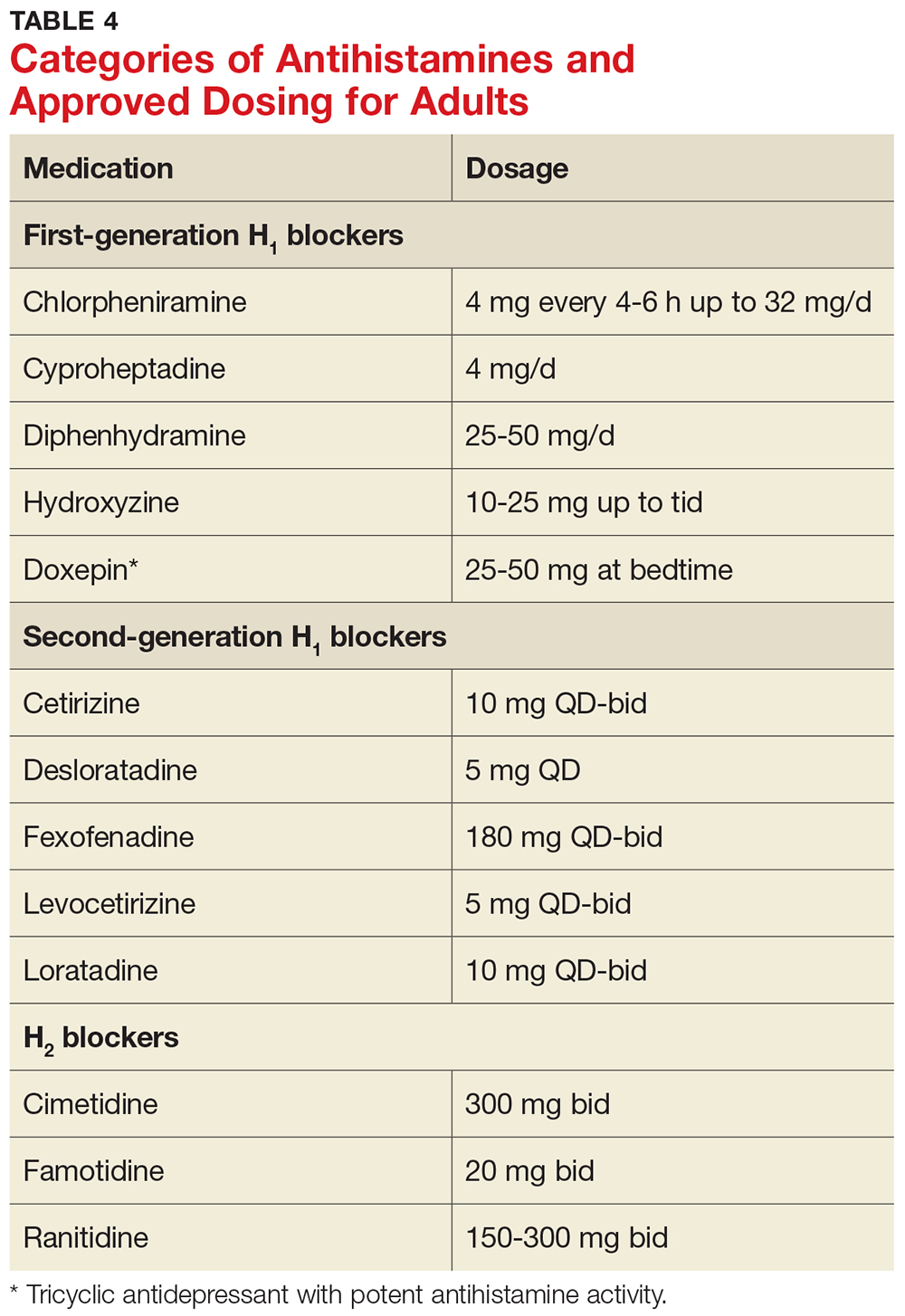
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
CLINICAL FEATURES
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
CE/CME No: CR-1801
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate between acute and chronic urticaria.
• List common history questions required for the diagnosis of chronic urticaria.
• Explain a stepwise plan for treatment of chronic urticaria.
• Describe serologic testing that should be ordered for chronic urticaria.
• Demonstrate knowledge of when to refer patients to a specialist for alternative treatment options.
FACULTY
Randy D. Danielsen is Professor and Dean of the Arizona School of Health Sciences, and Director of the Center for the Future of the Health Professions at A.T. Still University in Mesa, Arizona. Gabriel Ortiz practices at Breathe America El Paso in Texas and is a former AAPA liaison to the American Academy of Allergy, Asthma & Immunology (AAAAI) and National Institutes of Health/National Asthma Education and Prevention Program—Coordinating Committee. Susan Symington has practiced in allergy, asthma, and immunology for more than 10 years. She is the current AAPA liaison to theAAAAI and is President-Elect of the AAPA-Allergy, Asthma, and Immunology subspecialty organization.
The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through December 31, 2018.
Article begins on next page >>
The discomfort caused by an urticarial rash, along with its unpredictable course, can interfere with a patient’s sleep and work/school. Adding to the frustration of patients and providers alike, an underlying cause is seldom identified. But a stepwise treatment approach can bring relief to all.
Urticaria, often referred to as hives, is a common cutaneous disorder with a lifetime incidence between 15% and 25%.1 Urticaria is characterized by recurring pruritic wheals that arise due to allergic and nonallergic reactions to internal and external agents. The name urticaria comes from the Latin word for “nettle,” urtica, derived from the Latin word uro, meaning “to burn.”2
Urticaria can be debilitating for patients, who may complain of a burning sensation. It can last for years in some and reduces quality of life for many. Recently, more successful treatments for urticaria have emerged that can provide tremendous relief.
It is important to understand some of the ways to diagnose and treat patients in a primary care setting and also to know when referral is appropriate. This article will discuss the diagnosis, treatment, and referral process for patients with chronic urticaria.
PATHOPHYSIOLOGY
Hives most commonly arise from an immunologic reaction in the superficial skin layers that results in the release of histamine, which causes swelling, itching, and erythema. The mast cell is the major effector cell in the pathophysiology of urticaria.3 In immunologic urticaria, the antigen binds to immunoglobulin (Ig) E on the mast cell surface, causing degranulation and release of histamine, which accounts for the wheals and itching associated with the condition. Histamine binds to H1 and H2 receptors in the skin to cause arteriolar dilation, venous constriction, and increased capillary permeability, accounting for the accompanying swelling.3 Not all urticaria is mediated by IgE; it can result from systemic disease processes in the body that are immune related but not related to IgE. An example would be autoimmune urticaria.
Urticaria commonly occurs with angioedema, which is marked by a greater degree of swelling and results from mast cell activation in the deeper dermis and subcutaneous tissue. Either condition can occur independently, however. Angioedema typically affects the lips, tongue, face, pharynx, and bilateral extremities; rarely, it affects the gastrointestinal tract. Angioedema may be hereditary, but its nonhereditary causes can be similar to those of urticaria.3 For example, a patient could be severely allergic to cat dander and, when exposed to this allergic trigger, develop swelling of the lips, facial edema, and flushing.
FORMS OF URTICARIA
Urticaria can be broadly divided based on the duration of illness: less than six weeks is termed acute urticaria, and continuous or intermittent presence for six weeks or more, chronic urticaria.4
Acute urticaria may occur in any age group but is most often seen in children.1 Acute urticaria and angioedema frequently resolve within a few days, without an identified cause. An inciting cause can be identified in only about 15% to 20% of cases; the most common cause is viral infection, followed by foods, drugs, insect stings, transfusion reactions, and, rarely, contactants and inhalants (see Table 1).1,5 Acute urticaria that is not associated with angioedema or respiratory distress is usually self-limited. The condition typically resolves before extensive evaluation, including testing for possible allergic triggers, can be done. The associated skin lesions are often self-limited or can be controlled symptomatically with antihistamines and avoidance of known possible triggers.1
Chronic urticaria, sometimes called chronic idiopathic urticaria, is more common in adults, occurs on most days of the week, and, as noted, persists for more than six weeks with no identifiable triggers.6 It affects about 0.5% to 1% of the population (lifetime prevalence).3 Approximately 45% of patients with chronic urticaria have accompanying episodes of angioedema, and 30% to 50% have an autoimmune process involving autoantibodies against the thyroid, IgE, or the high-affinity IgE receptor (FcR1).3 The diagnosis is based primarily on clinical history and presentation; this will guide the determination of what types of diagnostic testing are necessary.
Chronic urticaria requires an extensive, but not indiscriminate, evaluation with history, physical examination, allergy testing, and laboratory testing for immune system, liver, kidney, thyroid, and collagen vascular diseases.3 Unfortunately, an identifiable cause of chronic urticaria is found in only 10% to 20% of patients; most cases are idiopathic.3,7
Several forms of chronic urticaria can be precipitated by physical stimuli, such as exercise, generalized heat, or sweating (cholinergic urticaria); localized heat (localized heat urticaria); low temperatures (cold urticaria); sun exposure (solar urticaria); water (aquagenic urticaria); and vibration.1 In another form (pressure urticaria), pressure on the skin increases histamine release, leading to the development of wheals and itching; this form is also called dermatographism, which means “write on skin” (see Figure 1). These types of urticaria should be evaluated and treated by a board-certified allergist, as there are special evaluations that can confirm the diagnosis.
CLINICAL FEATURES
The main feature of urticaria is raised skin lesions that appear pale to pink to erythematous and most commonly are intensely pruritic (see Figure 2). These lesions range from a few millimeters to several centimeters in size and may coalesce.
Characteristically, evanescent old lesions resolve, and new ones develop over 24 hours, usually without scarring. Scratching generally worsens dermatographism, with new urticaria produced over the scratched area. Any area of the body may be involved.
The lesions of early urticaria may vary in size and blanch when pressure is applied. An individual hive may last minutes or up to 24 hours and may reoccur intermittently on various sites on the body for an unspecified period of time.1,6
DIFFERENTIAL DIAGNOSIS
Other dermatologic conditions may be mistaken for chronic urticaria. Common rashes that may mimic it include anaphylaxis, atopic dermatitis, medication allergy or fixed drug eruption, ACE inhibitor–related angioedema, mastocytosis, contact dermatitis, autoimmune thyroid disease, bullous pemphigoid, and dermatitis herpetiformis.
Patients should be encouraged to bring pictures of the rash to the office visit, since the rash may have waned at the time of the visit and diagnosis based on the patient’s description alone can be challenging. Most rashes in the differential can be identified or eliminated through a careful history and complete physical exam. When necessary, serologic testing and skin punch biopsies can elucidate and confirm the diagnosis.
EVALUATION
History and physical examination
The medical history is the most important part of the evaluation of a patient with urticaria. The information that should be elicited and documented during the history is shown in Table 2.
A general comprehensive physical exam should be undertaken and the findings carefully documented. As noted, it can be helpful for patients to bring in pictures of the rash if the lesions wax and wane. It is also important to assess whether the urticarial lesions blanch when palpated, since this is a characteristic feature of acute and chronic urticarial lesions (but not of those with an autoimmune, cholinergic, or vasculitic cause). Thus, blanching of the wheal is a key finding on physical exam to discriminate between possible causes.8 Lesions pigmented with purpuric areas that scar or last longer than 24 hours suggest urticarial vasculitis; other features that distinguish urticarial vasculitis from chronic urticaria are listed in Table 3.2
Laboratory evaluation
Although there is no consensus regarding appropriate laboratory testing, the following tests should be considered for patients with chronic urticaria after completion of a thorough history and physical exam: complete blood count (CBC) with differential; erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP); chemistry panel and hepatic panel; and thyroid-stimulating hormone, antimicrosomal antibodies, and antithyroglobulin antibodies measurements.7
While the CBC is usually within normal limits, if eosinophilia is present, a workup for an atopic disorder or parasitic infection should be considered. If the ESR/CRP results are positive, consider ordering a larger antinuclear antibody (ANA) panel. Note: The utility of performing these tests routinely for chronic urticaria patients is unclear, as studies have demonstrated that results are usually normal. But it is important to order the appropriate tests to help you rule in or out a likely diagnosis.
Additional testing may be indicated by non-IgE or possible autoimmune findings on the history and/or physical exam. This can include a functional autoantibody assay (for autoantibodies to the high-affinity IgE receptor [FcR1]); complement analysis (eg, C3, C4, CH50), especially when concerned about hereditary angioedema; stool analysis for ova and parasites; Helicobacter pylori workup (there is limited experimental evidence to recommend this, however); hepatitis B and C workup; chest radiograph and/or other imaging studies; ANA panel; rheumatoid factor; cryoglobulin levels; skin biopsy; and urinalysis.7
Local urticaria can occur following contact with allergens via an IgE-mediated mechanism. If an allergen is suspected as a possible trigger, serologic testing to assess allergen-specific IgE levels that may be contributing to the urticaria can be performed in a primary care setting. The specific IgE levels most commonly assessed are for the endemic outdoor aeroallergens (eg, pets [cat, dog], dust mites); measurement of food-specific IgE levels can be ordered if a specific allergy is a concern. Allergy skin prick testing for immediate hypersensitivity and a physical challenge test are usually performed in an allergy office by board-certified allergists.
Skin biopsy should be done on all lesions concerning for urticarial vasculitis (see Table 4).2 Biopsy is also important if the hives are painful rather than pruritic, as this may suggest a different cause. The clinician should consider more detailed lab testing and skin biopsy if urticaria does not respond to therapy as anticipated. Also, specific lab testing may be required screening for certain planned medical therapies (eg, glucose-6-phosphate dehydrogenase enzyme deficiency screening before dapsone or hydroxychloroquine therapy).3
MANAGEMENT
Nonpharmacologic therapy
Treatment of the underlying cause, if identified, may be helpful and should be considered. For example, if a thyroid disorder is found on serologic testing, correcting the disorder may resolve the urticaria.9 Similarly, if a complement deficiency consistent with hereditary angioedema is detected, there are medications to correct it, which can be life-saving.3 Medications for treating hereditary angioedema are best prescribed in an allergy practice.
If triggers are discovered, the patient must be made aware of them and advised to avoid them as much as possible; however, total avoidance can be very difficult. Other common potentiating factors—such as alcohol overuse, excessive tiredness, emotional stress, hyperthermia, and use of aspirin and NSAIDs—should be avoided.10 These factors can worsen what is already triggering the urticaria and make it more difficult to treat; an example would be a patient who develops urticaria from a new household dog and is taking anti-inflammatory drugs for arthritis symptoms.
Topical agents rarely result in any improvement, and their use is therefore discouraged. In fact, high-potency corticosteroids may cause dermal atrophy.11 Also, dietary changes are not indicated for most patients with chronic urticaria, because undiscovered allergy to food or food additives is not likely to be responsible.4
Antihistamines
Antihistamines are the most commonly used pharmacologic treatment for chronic urticaria (see Table 4). H2-receptor blockers, taken in combination with first- and second-generation H1-receptor blockers, have been reported to be more efficacious than H1 antihistamines alone for the treatment of chronic urticaria.6 This added efficacy may be related to pharmacologic interactions and increased blood levels achieved with first-generation antihistamines. Increased doses of second-generation antihistamines—as high as four times the standard dose—are advocated by the 2014 Joint Task Force on Practice Parameters (JTFPP) for the diagnosis and management of acute and chronic urticaria.4
A stepwise approach to treatment is imperative. The JTFPP guidelines (available at www.allergyparameters.org) are summarized below.
Step 1: Administer a second-generation antihistamine at the standard therapeutic dose (see Table 4) and avoid triggers, NSAIDs, and other exacerbating factors.
If symptom control is not achieved in one to two weeks, move on to
Step 2: Increase therapy by one or more of the following methods: increase the dose of the second-generation antihistamine used in Step 1 (up to 4x the standard dose); add another second-generation antihistamine to the regimen; add an H2 blocker (ranitidine, famotidine, cimetidine); and/or add a leukotriene-receptor antagonist (montelukast 10 mg/d).
If these measures do not result in adequate symptom control, it’s time for
Step 3: Gradually increase the dose of H1 antihistamine(s) and discontinue any medications added in Step 2 that did not appear beneficial. Add a first-generation antihistamine (hydroxyzine, doxepin, cyproheptadine), which should be taken at bedtime due to risk for sedation.12
If symptoms are not controlled by Step 3 measures, or if the patient is unable to tolerate an increased dose of first-generation antihistamines, the urticaria is considered refractory. At this point, the clinician should consider referral to an allergy specialist for
Step 4: Add an alternative medication, such as cyclosporine (an anti-inflammatory, immunosuppressive agent) or omalizumab (a monoclonal antibody that selectively binds to IgE).
It should be noted that while the recent FDA approval of omalizumab for treatment of chronic urticaria has been life-changing for many patients, the product label does carry a black box warning about anaphylaxis. Because special monitoring is needed (and prior authorization will likely be required by the patient’s insurer), omalizumab is best prescribed in an allergy office.
It is not uncommon for patients with chronic urticaria to require multiple medications to control their symptoms. Once controlled, they will require maintenance and reevaluation on a regular basis.13
When to refer
Clinicians must know when to refer a patient with chronic urticaria to an allergist/immunologist. Referral is indicated when an underlying disorder is suspected, when symptoms are not controlled with Steps 1 to 3 of the management guidelines, or when the patient requires repeated or prolonged treatment with glucocorticoids.
Unfortunately, out of frustration on both the provider and the patient side, glucocorticoids may be started, after determining that that is “all that works” for the patient. There appears to be a limited role for glucocorticoids, so they should be avoided unless absolutely necessary (ie, if there is no response to antihistamines).
If signs and symptoms suggest urticarial vasculitis, it is prudent to consider referral to a specialist in rheumatology. Urticarial vasculitis requires a special skin punch biopsy to confirm the diagnosis.8 The biopsy procedure may be performed by a primary care provider; if the clinician is not comfortable doing so, referral to an appropriate dermatology provider is indicated.
PATIENT EDUCATION/REASSURANCE
Effective patient education is critical, because patients often experience considerable distress as the symptoms of chronic urticaria wax and wane unpredictably. It is not uncommon for patients with this condition to complain of symptoms that interfere with work, school, and sleep. Reassurance can help to alleviate frustration and anxiety. Patients should understand that the symptoms of chronic urticaria can be successfully managed in the majority of patients, and that chronic idiopathic urticaria is rarely permanent, with about 50% of patients experiencing remission within one year.6
CONCLUSION
The diagnosis of chronic urticaria is based primarily on the presentation, clinical history, and laboratory workup. Management of this chronic and uncomfortable condition requires the identification and exclusion of possible triggers, followed by effective patient education/counseling and a personalized management plan. By knowing when to suspect chronic urticaria, being familiar with the approach to evaluation and initial treatment, and knowing when referral to a specialist is indicated, primary care providers can help their patients find a path to relief.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
1. Riedl MA, Ortiz G, Casillas AM. A primary care guide to managing chronic urticaria. JAAPA. 2003;16:WEB.
2. Grieve M. Nettles. http://botanical.com/botanical/mgmh/n/nettle03.html. Accessed December 19, 2017.
3. Powell RJ, Du Toit GL, Siddique N, et al; British Society for Allergy and Clinical Immunology. BSACI guidelines for the management of chronic urticaria and angioedema. Clin Exp Allergy. 2007;37(5):631-650.
4. Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133(5):1270-1277.
5. Arizona Asthma & Allergy Institute. Possible causes of hives. www.azsneeze.com/hives. Accessed December 19, 2017.
6. Kozel MM, Mekkes JR, Bossuyt PM, Bos JD. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45(3):387-391.
7. Wanderer AA. Hives: The Road to Diagnosis and Treatment of Urticaria. Bozeman, MT: Anson Publishing; 2004.
8. Vazquez-López F, Maldonado-Seral C, Soler-Sánchez T, et al. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford). 2003;42(9):1079-1082.
9. Kaplan AP. Chronic urticaria and angioedema. N Engl J Med. 2002;346(3):175-179.
10. Yadav S, Bajaj AK. Management of difficult urticaria. Indian J Dermatol. 2009;54(3):275-279.
11. Ellingsen AR, Thestrup-Pedersen K. Treatment of chronic idiopathic urticaria with topical steroids. An open trial. Acta Derm Venereol. 1996;76(1):43-44.
12. Goldsobel AB, Rohr AS, Siegel SC, et al. Efficacy of doxepin in the treatment of chronic idiopathic urticaria. J Allergy Clin Immunol. 1986;78(5 pt 1):867-873.
13. Ferrer M, Bartra J, Gimenez-Arnau A, et al. Management of urticaria: not too complicated, not too simple. Clin Exp Allergy. 2015;45(4):731-743.
Diagnosing & Treating Neuromyelitis Optica Spectrum Disorder
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.

Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.
Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
Q) How do you know if a neurologic symptom is due to a relapse of neuromyelitis optica spectrum disorder? And how should a confirmed relapse be treated?
Neuromyelitis optica spectrum disorder (NMOSD) is a severe, relapsing autoimmune disease of the central nervous system (CNS) that targets the optic nerves and spinal cord, leading to blindness and paralysis.1,2 Whereas multiple sclerosis (MS) is characterized by demyelination, NMOSD is associated with astrocytic damage and tissue necrosis.3 Because longitudinally extensive inflammatory lesions are typical with NMOSD, permanent CNS damage is common with each relapse.4
Health care providers first need to determine whether a patient with NMOSD who presents with new or worsening symptoms is having a relapse. A relapse is caused by a breach of the blood-brain barrier by the peripheral immune system, which leads to inflammation and damage to the CNS.5 This causes neurologic symptoms that depend on the anatomic location. Once damage has occurred, symptoms may result either from a new relapse in the same location as a previous inflammatory event or from a pseudorelapse.6
Pseudorelapses are triggered by a systemic metabolic imbalance; they exacerbate symptoms from previous CNS damage. Differentiating between a true relapse and a pseudorelapse can be a diagnostic challenge for even the most seasoned of health care providers. Kessler et al retrospectively examined which clinical factors can distinguish relapses from pseudorelapses.6 Their findings suggest that while clinical examination alone may be effective in events involving vision loss, MRI may be necessary when signs and symptoms are attributable to a spinal cord lesion.
In fact, they found that the degree of clinical worsening in patients with spinal cord symptoms caused by a pseudorelapse was similar to that of a true relapse. The most common causes of pseudorelapse included infection, dysautonomia, metabolic abnormalities, and changes to medication regimens. Interestingly, the presence of infection did not rule out a relapse, as patients experiencing relapses were equally likely as those with pseudorelapse to have a urinary tract infection. The authors concluded, based on their data, that an MRI is warranted to verify a relapse in patients who experience worsening of symptoms localized to the spinal cord but is not necessary to rule out a pseudorelapse of optic neu
In contrast to MS, a progressive phase is not believed to be associated with NMOSD.7 Instead, accrual of disability occurs with each relapse. The majority of patients with NMOSD do not return to baseline following an untreated relapse, making it especially important that patients receive adequate acute treatment to mitigate the damage.8
Currently, there are no medications approved by the FDA for the acute or preventive treatment of NMOSD. However, off-label use of immunotherapies, including rituximab, mycophenolate mofetil, azathioprine, prednisone, methotrexate, tocilizumab, and mitoxantrone, have been studied for relapse prevention.2 In addition, there are three ongoing phase III trials investigating eculizumab (C5 complement inhibitor), inebilizumab (CD19 monoclonal antibody), and SA237 (IL6R blocker); results from these studies could potentially widen the landscape of immunotherapy use in NMOSD.2
Less investigation into appropriate acute treatment of new relapses has been conducted, however, leaving clinicians and patients uncertain about how to manage a new inflammatory event. Traditionally, firstline treatment for acute NMOSD relapses has been the same as for MS relapses—high-dose methylprednisolone. However, due to the severity of NMOSD relapses and the relative lack of response to steroids alone, methylprednisolone is commonly followed by plasma exchange (PLEX).2
Most data to guide clinical decision-making suggest that patients with NMOSD relapses recover better when PLEX is added to steroid treatment. Abboud et al found that 65% of patients who received both PLEX and methylprednisolone recovered to their prerelapse baseline, compared to 35% of those who received methylprednisolone alone.9 These findings were supported by a larger retrospective investigation by Kleiter et al, which found improved recovery with treatment escalation in their cohort.8 These data support the recommendation to use PLEX as an adjunct therapy in acute relapses—particularly in relapses with severe presentations.
Because diagnosis and treatment of relapses involve many factors, ranging from accrual of disability, long-term immunotherapy decisions, and medical costs, diligence in provider decision-making is essential when caring for patients with NMOSD. -MAM
Maureen A. Mealy, BSN, MSCN
Neuromyelitis Optica Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, PhD candidate at Johns Hopkins School of Nursing in Baltimore
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
2. Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr Treat Options Neurol. 2016;18(1):2.
3. Popescu BF, Lucchinetti CF. Immunopathology: autoimmune glial diseases and differentiation from multiple sclerosis. Handb Clin Neurol. 2016;133:95-106.
4. Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
5. Orman G, Wang KY, Pekcevik Y, et al. Enhancing brain lesions during acute optic neuritis and/or longitudinally extensive transverse myelitis may portend a higher relapse rate in neuromyelitis optica spectrum disorders. Am J Neuroradiol. 2017;38(5):949-953.
6. Kessler RA, Mealy MA, Levy M. Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e269.
7. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603-605.
8. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79(2):206-216.
9. Abboud H, Petrak A, Mealy M, et al. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. 2016;22(2):185-192.

Bladder Complications in MS
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month's responses were authored by Denise R. Bruen, MSN, APRN-BC, MSCN, who is with the University of Virginia in Charlottesville, and Maureen A. Mealy, BSN, MSCN, who is Neuromyelitis Optica Clinical Research Program Manager, Senior Research Nurse of the Transverse Myelitis & Multiple Sclerosis Centers, and PhD candidate at Johns Hopkins School of Nursing in Baltimore.
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
Q) My patient has multiple sclerosis and complains of feeling weaker, but denies urinary symptoms. Why have I been told to check for urinary tract infection and not just administer steroids?
Bladder complications are extremely common in patients living with multiple sclerosis (MS), occurring in around 80% of this population.1 These complications—which include urinary urgency, failure to fully empty the bladder, incontinence, and difficulty getting to a toilet in time—can increase risk for urinary tract infection (UTI). And because many patients with MS also have sensory problems (eg, neurogenic bladder), they do not always present with the hallmark UTI symptoms of burning or pain with urination.
Often, presenting symptoms include generalized weakness, increased spasticity, or intensified neurologic issues. These can lead patients to believe they are having a relapse, when in fact, a UTI is causing a pseudoexacerbation of their baseline neurologic issues. In addition, frequent nocturia can disrupt sleep and further contribute to MS-related fatigue. Patients may self-induce dehydration by limiting their daytime fluid intake in an effort to avoid bathroom visits.1
In partnership with urology colleagues, you can help mitigate bladder complications in patients with MS; this can entail use of medication or interventions such as in-and-out or straight catheterization, timed voids, Botox, or pelvic floor physical therapy. Behavior modifications—ie, minimizing caffeine intake, limiting alcohol consumption, and stopping fluids early in the evening—can also be beneficial.1,2
Before initiating bladder medication, it is important to review potential adverse effects with the patient. It’s also crucial to ensure that patients are fully emptying their bladders before starting anticholinergic medications, as these can worsen retention.
Which treatment should you choose? Insurance companies tend to prefer generic, older-generation anticholinergics, but bear in mind that these can cause or contribute to cognitive issues (which many patients with MS already have).3 Another medication, such as mirabegron, may be preferable; it’s less likely than anticholinergics to cause dry mouth, which may help with compliance. Also, be aware that anticholinergics can cause blurred vision, which might lead patients to believe they are having optic neuritis or another MS-related visual change.4
That said, it is possible for patients to have a relapse and a UTI simultaneously. Due to potential adverse effects, it is essential to balance the risks and benefits of steroid therapy. Steroids could worsen an untreated infection and may not be appropriate for the patient’s symptoms or chief complaint.
Addressing bladder symptoms can not only help prevent UTIs but can also improve skin integrity, sleep quality, independence, and overall quality of life. A thorough exam and history-taking can alleviate secondary and tertiary urinary complications, as well as avoid unnecessary use of corticosteroids. -DRB
Denise R. Bruen, MSN, APRN-BC, MSCN
University of Virgina, Charlottesville
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.
1. Sheehan J. Coping with MS bladder dysfunction. www.everydayhealth.com/multiple-sclerosis/symptoms/coping-with-bladder-dysfunction/. Accessed November 18, 2017.
2. Mayo Clinic. Bladder control: medications for urinary problems. www.mayoclinic.org/diseases-conditions/urinary-incontinence/in-depth/bladder-control-problems/art-20044220. Accessed November 18, 2017.
3. Staskin DR, Zoltan E. Anticholinergics and central nervous system effects: are we confused? Rev Urol. 2007;9(4):191-196.
4. Geller EJ, Crane AK, Wells EC, et al. Effect of anticholinergic use for the treatment of overactive bladder on cognitive function in post-menopausal women. Clin Drug Investig. 2012;32(10):697-705.
To have not and then to have: A challenging immune paradox

The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.

Drug reaction or metastatic lung cancer?
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
. The left lung appeared normal (B).")

Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
Anaphylaxis Controversy and Consensus

Abdominal pain and bloody diarrhea in a 32-year-old woman
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.

Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.

No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.

Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.

Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.
Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.
No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.
Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.
Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
A 32-year-old woman presented to our emergency department with chest pain and painful ulcerations on her arms, abdomen, back, groin, axillae, and in her mouth. She first noticed the ulcers 7 days earlier.
She also reported bloody diarrhea, which had started 2 years earlier, with 10 or more bowel movements daily. She described her stools as semiformed and associated with urgency and painful abdominal cramps.
Medical history
Her medical history included obstructive sleep apnea and morbid obesity. She had first presented 2 years earlier to another hospital with diarrhea, abdominal pain, and rectal bleeding. At that time, results of esophagogastroduodenoscopy and colonoscopy were reported as normal. Later, she became pregnant, and her symptoms went away. She had a normal pregnancy and delivery.
About 1 year postpartum, her abdominal pain and bloody diarrhea recurred. Colonoscopy showed severe sigmoid inflammation with small, shallow ulcerations and friable mucosa interrupted by areas of normal mucosa. Histopathologic study of the colonic mucosa indicated mild to moderate chronic active colitis consisting of focal areas of cryptitis with occasional crypt abscess formation. She was diagnosed with Crohn colitis based on the endoscopic appearance, histopathology, and clinical presentation. The endoscope, however, could not be advanced beyond the sigmoid colon, which suggested stenosis. She was started on 5-aminosalicylic acid (5-ASA) but developed visual hallucinations, and the medication was stopped.
Her symptoms continued, and she developed worsening rectal bleeding and anemia that required hospitalization and blood transfusions. Another colonoscopy performed 1 month before this emergency department visit had shown multiple mucosal ulcerations, but again, the colonoscope could not be advanced beyond the sigmoid colon. She was started on oral corticosteroids, which provided only minimal clinical improvement.
Her current medications included atenolol (for sinus tachycardia), prednisone (initial dose 60 mg/day tapered to 20 mg/day at presentation), and ciprofloxacin.
Her family history was unknown because she had been adopted.
About 1 week before presentation, she had noticed ulcers developing on her arms, abdomen, back, groin, oral mucosa, and axillae. The ulcers were large and painful, with occasional spontaneous bleeding. She also reported pustules and ulcerations at sites of previous skin punctures, consistent with pathergy.
Findings on presentation
- Temperature 99.5°F (37.5°C)
- Heart rate 124 beats per minute
- Respiratory rate 22 breaths per minute
- Oxygen saturation 100% on room air
- Blood pressure 128/81 mm Hg
- Body mass index 67 kg/m2 (morbidly obese).
She had multiple greyish-white patches and erosions over the soft palate, tongue, and upper and lower lip mucosa, erythematous pustules in the axillae bilaterally, and large erythematous, sharply demarcated ulcerations with a fibrinous base bilaterally covering her arms, thighs, groin, and abdomen.
Blood testing showed multiple abnormal results (Table 1). Urinalysis revealed a urine protein concentration of 100 mg/dL (reference range 0), more than 25 white blood cells per high-power field (reference range < 5), 6 to 10 red blood cells per high-power field (0–3), and more than 10 casts per low-power field (0), which suggested a urinary tract infection with hematuria.
Computed tomography (CT) of the abdomen and pelvis with intravenous and oral contrast showed diffuse fatty infiltration of the liver and wall thickening of the rectum and sigmoid colon.
She was admitted to the medical intensive care unit for potential septic shock. Intravenous vancomycin and ciprofloxacin were started (the latter owing to penicillin allergy).
CAUSES OF DIARRHEA AND SKIN CHANGES
1. What is the most likely diagnosis in our patient?
- Ulcerative colitis
- Crohn disease
- Behçet disease
- Intestinal tuberculosis
- Herpes simplex virus infection
- Cytomegalovirus infection
All of the above can cause diarrhea in combination with mucocutaneous lesions and other manifestations.
Ulcerative colitis and Crohn disease: Mucocutaneous findings
Extraintestinal manifestations of inflammatory bowel diseases (Crohn disease, ulcerative colitis, and Behçet disease) include arthritis, ocular involvement, mucocutaneous manifestations, and liver involvement in the form of primary sclerosing cholangitis. Less common extraintestinal manifestations include vascular, renal, pulmonary, cardiac, and neurologic involvement.
Mucocutaneous findings are observed in 5% to 10% of patients with ulcerative colitis and 20% to 75% of patients with Crohn disease.1–3 The most common are erythema nodosum and pyoderma gangrenosum.4
Yüksel et al5 reported that of 352 patients with inflammatory bowel disease, 7.4% had erythema nodosum and 2.3% had pyoderma gangrenosum. Erythema nodosum was significantly more common in patients with Crohn disease than in those with ulcerative colitis, and its severity was linked with higher disease activity. Lesions frequently resolved when bowel disease subsided.
Lebwohl and Lebwohl6 reported that pyoderma gangrenosum occurred in up to 20% of patients with Crohn disease and up to 10% of those with ulcerative colitis. It is not known whether pyoderma gangrenosum correlates with intestinal disease severity.
Other mucocutaneous manifestations of inflammatory bowel disease include oral aphthous ulcers, acute febrile neutrophilic dermatosis (Sweet syndrome), and metastatic Crohn disease. Aphthous ulcers in the oral cavity, often observed in both Crohn disease and ulcerative colitis, cannot be differentiated on clinical examination from herpes simplex virus (HSV) type 1-induced or idiopathic mucous membrane ulcers. The most common ulcer locations are the lips and buccal mucosa. If biopsied (seldom required), noncaseating granulomas can be identified that are comparable with intestinal mucosal granulomas found in Crohn disease.7
Behçet disease has similar signs
Oral aphthous ulcers are also the most frequent symptom in Behçet disease, occurring in 97% to 100% of cases.8 They most commonly affect the tongue, lips, buccal mucosa, and gingiva.
Cutaneous manifestations include erythema nodosum-like lesions, which present as erythematous painful nodules over pretibial surfaces of the lower limbs but can also affect the arms and thighs; they can also present as papulopustular rosacea eruptions composed of papules, pustules, and noninflammatory comedones, most commonly on the chest, back, and shoulders.8,9
Pathergy, ie, skin hyperresponse to minor trauma such as a bump or bruise, is a typical trait of Behçet disease. A positive pathergy test (ie, skin hyperreactivity to a needlestick or intracutaneous injection) has a specificity of 98.4% in patients with Behçet disease.10
Interestingly, there appears to be a regional difference in the susceptibility to pathergy. While a pathergy response in patients with Behçet disease is rare in the United States and the United Kingdom, it is very common in Japan, Turkey, and Israel.11
Patient demographics also distinguish Behçet disease from Crohn disease. The prevalence of Behçet disease is highest along the Silk Road from the Mediterranean Basin to East Asia and lowest in North America and Northern Europe.12 The mean age at onset is around the third and fourth decades. In males, the prevalence is highest in Mediterranean, Middle Eastern, and Asian countries. In females, the prevalence is highest in the United States, Northern Europe, and East Asia.10
Tuberculosis
Tubercular skin lesions can present in different forms.13 Lupus vulgaris, the most common, occurs after primary infection and presents as translucent brown nodules, mainly over the face and neck. So-called scrofuloderma is common at the site of a lymph node. It appears as a gradually enlarging subcutaneous nodule followed by skin breaks and ulcerations. Tuberculosis verrucosa cutis, also known as warty tuberculosis, is common in developing countries and presents as warty plaque over the hands, knees, and buttocks.14 Tuberculids are skin reactions to systemic tuberculosis infection.
Herpes simplex virus
Mucocutaneous manifestations of herpes simplex virus affect the oral cavity (gingivostomatitis, pharyngitis, and lip border lesions), the entire integumentary system, the eyes (HSV-1), and the genital region (HSV-2). The classic presentation is systemic symptoms (fever and malaise) associated with multiple vesicles on an erythematous base in a distinct region of skin. The virus can remain latent with reactivation occurring because of illness, immunosuppression, or stress. Pruritus and pain precede the appearance of these lesions.
Cytomegalovirus
Primary cytomegalovirus infection is subclinical in almost all cases unless the patient is immunocompromised, and it presents similarly to mononucleosis induced by Epstein-Barr virus. The skin manifestations are nonspecific and can include macular, maculopapular, morbilliform, and urticarial rashes, but usually not ulcerations.15
OUR PATIENT: BEHÇET DISEASE OR CROHN DISEASE?
In our patient, oral mucosal aphthous ulcers and the location of pustular skin lesions, in addition to pathergy, were highly suggestive of Behçet disease. However, Crohn disease with mucocutaneous manifestations remained in the differential diagnosis.
Because there is significant overlap between these diseases, it is important to know the key distinguishing features. Oral aphthous ulcers, pathergy, uveitis, skin and genital lesions, and neurologic involvement are much more common in Behçet disease than in Crohn disease.16,17 Demographic information was not helpful in this case, given that the patient was adopted.
FURTHER WORKUP
2. What should be the next step in the work-up?
- CT enterography
- Skin biopsy
- Colonoscopy with biopsy
- C-reactive protein, erythrocyte sedimentation rate, and fecal calprotecting testing
The endoscopic appearance and histopathology of the affected tissues are crucial for the diagnosis. Differentiating between Crohn disease and Behçet disease can be particularly challenging because of significant overlap between the intestinal and extraintestinal manifestations of the two diseases, especially the oral lesions and arthralgias. Thus, both colonoscopy with biopsy of the intestinal lesions and biopsy of a cutaneous ulceration should be pursued.
No single test or feature is pathognomonic for Behçet disease. Although many diagnostic criteria have been established, those of the International Study Group (Table 2) are the most widely used.18 Their sensitivity for Behçet disease has been found to be 92%, and their specificity 97%.19
Both CT enterography and inflammatory markers would depict inflammation, but since this is present in both Crohn disease and Behçet disease, these tests would not be helpful in this situation.
Endoscopic appearance of Crohn disease and Behçet disease
Intestinal Behçet disease, like Crohn disease, is an inflammatory bowel disease occurring throughout the gastrointestinal tract (small and large bowel). Both are chronic diseases with a waxing and waning course and have similar extraintestinal manifestations. Typical endoscopic lesions are deep, sharply demarcated (“punched-out”), round ulcers. The intestinal Behçet disease and Crohn disease ulcer phenotype and distribution can look the same, and in both entities, rectal sparing and “skip lesions” have been described.20–22
Nevertheless, findings on endoscopy have been analyzed to try to differentiate between Crohn disease and Behçet disease.
In 2009, Lee et al23 published a simple and accurate strategy for distinguishing the two diseases endoscopically. The authors reviewed 250 patients (115 with Behçet disease, 135 with Crohn disease) with ulcers on colonoscopy and identified 5 endoscopic findings indicative of intestinal Behçet disease:
- Round ulcers
- Focal single or focal multiple distribution of ulcers
- Fewer than 6 ulcers
- Absence of a “cobblestone” appearance
- Absence of aphthous lesions.
The two most accurate factors were absence of a cobblestone appearance (sensitivity 100%) and round ulcer shape (specificity 97.5 %). When more than one factor was present, specificity increased but sensitivity decreased.
Using a classification and regression tree analysis, the investigators created an algorithm that endoscopically differentiates between Crohn disease and Behçet disease (Figure 1) with an accuracy of 92 %.23
Histopathologic analysis of both colonic and skin lesions can provide additional clues to the correct diagnosis. Vasculitis suggests Behçet disease, whereas granulomas suggest Crohn disease.
CASE CONTINUED: SKIN BIOPSY AND COLONOSCOPY
Punch biopsy of the skin was performed on the right anterior thigh. Histopathologic analysis revealed acanthotic epidermis, a discrete full-thickness necrotic ulcer with a neutrophilic base, granulation tissue, and vasculitic changes. There were no vasculitic changes or granulomas outside the ulcer base. Cytomegalovirus staining was negative. An interferon-gamma release assay for tuberculosis was negative. Eye examination results were normal.
Colonoscopy showed multiple deep, round, and confluent ulcers with a punched-out appearance and fissures with normal intervening mucosa in the entire examined colon (Figure 2). The terminal ileal mucosa was normal. Colonic biopsies were consistent with cryptitis and rare crypt abscesses. Vasculitis was not identified.
Although the histologic changes were nonspecific, at this point we considered Behçet disease to be more likely than Crohn disease, given the typical endoscopic appearance and skin changes.
TREATING INTESTINAL BEHÇET DISEASE
3. Which is not considered a standard treatment for intestinal Behçet disease?
- Mesalamine (5-ASA)
- Corticosteroids
- Immunosuppressants
- Mycophenolate mofetil
- Surgery
Overall, data on the management of intestinal Behçet disease are limited. The data that do exist have shown that 5-ASA, corticosteroids, immunosuppressants, and surgery are options, but not mycophenolate mofetil.
Consensus recommendations from the Japanese IBD Research Group,24 published in 2007, included 5-ASA, corticosteroids, immunosuppressants, enteral and total parenteral nutrition, and surgical resection. In 2014, the group published a second consensus statement, adding anti-tumor necrosis factor (TNF) agents as standard therapy for this disease.22
Mycophenolate mofetil has not been shown to be effective in the treatment of mucocutaneous Behçet disease,25 although it may be effective in the treatment of its neurologic manifestations.26 Data regarding its efficacy in intestinal Behçet disease are sparse.
Differences in treatment for Crohn and Behçet disease
Although the treatment options are comparable for Behçet disease and Crohn disease, certain features differ.
Doses of 5-ASA and immunnosuppressive agents are typically higher in Crohn disease. For example, the optimal dose of 5-ASA is up to 3 g/day for Behçet disease but up to 4.8 g/day for Crohn disease.
Standard dosing for azathioprine is 50 to 100 mg/day for Behçet disease but 2 to 2.5 mg/kg/day (eg, 168 to 210 mg/day for a 185-lb patient) for Crohn disease.
In addition, evidence supporting the use of biologic agents such as anti-TNF agents or vedolizumab is more abundant in Crohn disease.
Finally, data on monitoring drug levels of immunomodulators or biologics are available only for patients with Crohn disease, not Behçet disease. Thus, an accurate diagnosis is important.
CASE CONTINUED: EMERGENCY LAPAROTOMY
Our patient continued to experience abdominal pain and bloody diarrhea despite receiving corticosteroids intravenously in high doses. We were also considering anti-TNF therapy.
At this point, CT of her abdomen and pelvis was repeated and showed free intraperitoneal air consistent with a perforation of the transverse colon.
She underwent emergency exploratory laparotomy. Intraoperative findings included pneumoperitoneum but no gross peritoneal contamination, extensive colitis with a contained splenic flexure perforation, and normal small-bowel features without evidence of enteritis. Subtotal colectomy, implantation of the rectal stump into the subcutaneous tissue, and end-ileostomy were performed.
After 23 days of recovery in the hospital, she was discharged on oral antibiotics and 4 weeks of steroid taper.
PROGNOSIS OF INTESTINAL BEHÇET DISEASE
4. What can the patient expect from her intestinal Behçet disease in the future?
- The disease is cured after resection of the diseased segments
- Behçet disease is a progressive lifelong disorder that can recur after surgery
Like Crohn disease, Behçet disease should be considered a lifelong progressive disorder, even after surgical resection of diseased segments.
It is unclear which patients will have a complicated disease course and need treatment with stronger immunosuppression. In patients with intestinal Behçet disease whose disease is in remission on thiopurine therapy, the 1-year relapse rate has been reported as 5.8%, and the 5-year relapse rate 51.7%.27,28 After surgical resection, the 5-year recurrence rate was 47.2%, and 30.6% of patients needed repeat surgery.29 Predictors of poor prognosis were younger age, higher erythrocyte sedimentation rate, higher C-reactive protein level, low albumin level at diagnosis, and a high disease-activity index for intestinal Behçet disease.30
The Korean IBD Study Group has developed and validated a disease activity index for intestinal Behçet disease.28 The index has a list of weighted scores for 8 symptoms, which provides for a more objective assessment of disease activity for determining the best treatment approach.
CASE CONTINUED
The patient has continued with her follow-up care and appointments in gastroenterology, rheumatology, and dermatology clinics. She still complains of intermittent abdominal pain, occasional bleeding at the rectal stump, intermittent skin lesions mainly in the form of pustular lesions, and intermittent joint pain. If symptoms persist, anti-TNF therapy is an option.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol 1981; 5:689–695.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol 2008; 22:1033–1043.
- Timani S, Mutasim DF. Skin manifestations of inflammatory bowel disease. Clin Dermatol 2008; 26:265–273.
- Tavarela Veloso F. Skin complications associated with inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20(suppl 4):50–53.
- Yüksel I, Basar O, Ataseven H, et al. Mucocutaneous manifestations in inflammatory bowel disease. Inflamm Bowel Dis 2009; 15:546–550.
- Lebwohl M, Lebwohl O. Cutaneous manifestations of inflammatory bowel disease. Inflamm Bowel Dis 1998; 4:142–148.
- Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (NY) 2011; 7:235–241.
- Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y. Behçet’s syndrome: facts and controversies. Clin Dermatol 2013; 31:352–361.
- Lee ES, Bangz D, Lee S. Dermatologic manifestation of Behçet’s disease. Yonsei Med J 1997; 38:380–389.
- Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behçet’s disease according to the change of incidence over the time. Clin Rheumatol 2011; 30:1151–1155.
- Friedman-Birnbaum R, Bergman R, Aizen E. Sensitivity and specificity of pathergy test results in Israeli patients with Behçet’s disease. Cutis 1990; 45:261–264.
- Mahr A, Maldini C. Epidemiology of Behçet’s disease. Rev Med Interne 2014; 35:81–89. French.
- Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous tuberculosis. Am J Clin Dermatol 2002; 3:319–328.
- Padmavathy L, Lakshmana Rao L, Ethirajan N, Ramakrishna Rao M, Subrahmanyan EN, Manohar U. Tuberculosis verrucosa cutis (TBVC)—foot with miliary tuberculosis. Indian J Tuberc 2007; 54:145–148.
- Drago F, Aragone MG, Lugani C, Rebora A. Cytomegalovirus infection in normal and immunocompromised humans. A review. Dermatology 2000; 200:189–195.
- Yazısız V. Similarities and differences between Behçet’s disease and Crohn’s disease. World J Gastrointest Pathophysiol 2014; 5:228–238.
- Chin AB, Kumar AS. Behçet colitis. Clin Colon Rectal Surg 2015; 28:99–102.
- International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335:1078–1080.
- Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int 2012; 2012:607921.
- Chang DK, Kim JJ, Choi H, et al. Double balloon endoscopy in small intestinal Crohn’s disease and other inflammatory diseases such as cryptogenic multifocal ulcerous stenosing enteritis (CMUSE). Gastrointest Endosc 2007; 66(suppl):S96–S98.
- Hamdulay SS, Cheent K, Ghosh C, Stocks J, Ghosh S, Haskard DO. Wireless capsule endoscopy in the investigation of intestinal Behçet’s syndrome. Rheumatology (Oxford) 2008; 47:1231–1234.
- Hisamatsu T, Ueno F, Matsumoto T, et al. The 2nd edition of consensus statements for the diagnosis and management of intestinal Behçet’s disease: indication of anti-TNFa monoclonal antibodies. J Gastroenterol 2014; 49:156–162.
- Lee SK, Kim BK, Kim TI, Kim WH. Differential diagnosis of intestinal Behçet’s disease and Crohn’s disease by colonoscopic findings. Endoscopy 2009; 41:9–16.
- Kobayashi K, Ueno F, Bito S, et al. Development of consensus statements for the diagnosis and management of intestinal Behçet’s disease using a modified Delphi approach. J Gastroenterol 2007; 42:737–745.
- Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous Adamantiades-Behçet’s disease. Dermatology 2001; 203:322–324.
- Shugaiv E, Tüzün E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behçet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol 2011; 29(suppl 67):S64–S67.
- Jung YS, Cheon JH, Hong SP, Kim TI, Kim WH. Clinical outcomes and prognostic factors for thiopurine maintenance therapy in patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2012; 18:750–757.
- Cheon JH, Han DS, Park JY, et al; Korean IBD Study Group. Development, validation, and responsiveness of a novel disease activity index for intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:605–613.
- Jung YS, Yoon JY, Lee JH, et al. Prognostic factors and long-term clinical outcomes for surgical patients with intestinal Behçet’s disease. Inflamm Bowel Dis 2011; 17:1594–1602.
- Jung YS, Cheon JH, Park SJ, Hong SP, Kim TI, Kim WH. Clinical course of intestinal Behçet’s disease during the first five years. Dig Dis Sci 2013; 58:496–503.
Tdap during pregnancy, or before, offers infants pertussis protection
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.

Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.
Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
during the early months of life, according to Tami H. Skoff of the Centers for Disease Control and Prevention, Atlanta, and her associates.
In an analysis of 240 infants younger than 2 months with pertussis cough onset between 2011 and 2015 and 535 control infants, 57% of case mothers and 67% of control mothers had at least one valid Tdap dose; 13% of vaccinated case mothers and 14% of vaccinated control mothers had more than one valid dose of Tdap reported.
Of Tdap doses received during pregnancy in 22 cases and 117 controls, 77% were received during the third trimester, most during the Advisory Committee on Immunization Practices’ recommended 27-36 weeks of gestation. Of the Tdap doses received before pregnancy in mothers of 24 cases and 67 controls, 25% of the case mothers and 67% of the control mothers received Tdap 2 or fewer years before pregnancy.
The effectiveness of Tdap vaccination during the third trimester of pregnancy was 78%, and effectiveness during the first or second trimester was 64%. Effectiveness of Tdap given 2 or fewer years before pregnancy was 83%. This study was not powered to determine a difference if the vaccine was administered in the ACIP-recommended time period during the third trimester.
A reported 49% of U.S. pregnant women received Tdap during the 2015-2016 flu season, an increase of 22% from the 2013-2014 season, according to a CDC Internet panel survey.
“While maternal immunization during pregnancy will help bridge the gap until next-generation pertussis vaccines are licensed and available for use, this highly effective strategy will likely remain an integral component of pertussis prevention and control, even in the setting of new vaccines,” the investigators said.
Read more in Clinical Infectious Diseases (2017 Sep 28. doi: 10.1093/cid/cix724).
FROM CLINICAL INFECTIOUS DISEASES
Acute monocular vision loss: Don’t lose sight of the differential
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.

Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history

A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.

An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations

A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).

For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
Immunologic testing is key to diagnosing autoimmune blistering diseases
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
SAN FRANCISCO –
“You have to have some kind of immunological test,” according to Peter Marinkovich, MD. “Pathologists will try to give you as much information as they can on the routine histology, but don’t use that as a diagnostic.”
If not properly identified, autoimmune blistering diseases can lead to chronic overexposure to steroids and resultant side effects without addressing the underlying problem, said Dr. Marinkovich of the department of dermatology at Stanford (Calif.) University.
Dr. Marinkovich gave one example of a patient who had been diagnosed with bullous pemphigoid several years before, and who was becoming Cushingoid as a result of steroids. But the diagnosis was made on the basis of histopathology and clinical appearance alone.
“Nobody had done the immunofluorescence test,” he explained at the annual meeting of the Pacific Dermatologic Association. “I did it, and it turned out she had linear IgA disease. The patient went through 2 years of toxicity just because nobody had done the immunofluorescence test.” Instead, the patient improved when placed on dapsone, which is much less toxic than prednisone.
Direct/indirect immunofluorescence is the highest-yield test for patients with blistering disease. “It’s the best way, I believe, to make the diagnosis,” Dr. Marinkovich said. If that test isn’t available, serum taken during an active phase can also be used. But serum samples can turn up false negatives, so dermatologists should consider collecting and testing serum samples several times.
Another useful tool is salt-split skin analysis, which will demarcate antigens to the roof or floor of the blister. Specifically, it helps distinguish bullous pemphigoid and epidermolysis bullosa acquisita.
In the future, Dr. Marinkovich said, ELISA (enzyme-linked immunosorbent assay) testing will have greater importance for diagnosis and disease monitoring, not just for pemphigus but for subepidermal bullous disorders as well.
Autoimmune blistering diseases do respond to prednisone treatment, although not as well as some other conditions. However, symptom improvement can mask the true cause of the disease.
“It’s easy for physicians to give steroids, and the patients will be happy for the time being; but that doesn’t solve the problem in the long term,” Dr. Marinkovich cautioned. “These are chronic conditions, and the patient will continue to require prednisone, and they’ll get more and more side effects, which could have been avoided if someone had done a more thorough investigation.”
Topical agents such as tetracycline, niacinamide, and topical steroids are more useful in pemphigoid than for pemphigus, because pemphigoid involves local immune factors that the agents can target, while pemphigus can be driven by antibodies alone, which are not as responsive to these treatments.
When systemic therapies are necessary, prednisone is a useful tool, but aim for the lowest possible dose, he said. Reducing prednisone dose is challenging in and of itself. Dropping the dose too quickly can lead to more long-term exposure, because a steep drop can lead to a rebound in the disease, which leads to a higher dose.
“The patient is on this roller coaster ride, up and down, up and down, and that alone can ramp up disease activity,” said Dr. Marinkovich. “Lowering steroid more steadily is a better way to go. This calms the disease down by itself.”
When steroids can’t be completely tapered, which is almost always the case in pemphigus and common in pemphigoid, add steroid-sparing agents such as mycophenolate and azathioprine.
If the steroid-sparing agents don’t get patients down to 10 mg/day prednisone or below, then consider using rituximab and intravenous IgG.
In Europe, physicians are using rituximab earlier in the course of disease, a strategy that appeared effective in a study published in the Lancet (2017 May 20;389[10083]:2031-40). “The evidence suggests to me that earlier use of rituximab tends to reduce the total amount of steroids that the patients are using and has the potential to reduce the duration of the disease,” Dr. Marinkovich said. “That’s a trend that will be going on in the next couple of years here in the United States.”
Dr. Marinkovich is an investigator on a clinical trial funded by Syntimmune.
AT PDA 2017