User login
Omalizumab for chronic urticaria quells suffocation fears
GENEVA – a widely underappreciated dimension of the impairment caused by this disease, Karsten Weller, MD, said at the annual congress of the European Academy of Dermatology and Venereology.

“Virtually all clinical studies of CSU in recent decades focus on the wheal and pruritus components and not on the angioedema component, even though angioedema is a frequent symptom in the disease. Roughly half of patients with CSU experience wheals and angioedema, and up to 13% experience angioedema only,” said Dr. Weller, a dermatologist at Charité University Hospital in Berlin.
“Angioedema is a major driver of quality-of-life impairment in CSU,” he continued. “We know that these are the patients who particularly suffer from the unpredictability of the disease, from disfigurement, from embarrassment. These are the patients who come to the emergency rooms, who lose working days, and these are the patients who often have the feeling of losing control over their lives.”
X-ACT was a multicenter German study which included 91 patients with moderate to severe CSU marked by at least four angioedema episodes during the 6 months prior to enrollment. Participants also had to be refractory to second-generation H1 antihistamines at two to four times the approved dose. The subjects were randomized to subcutaneous omalizumab at 300 mg every 4 weeks or placebo for 28 weeks; they were then further assessed for changes in quality of life during 8 weeks off omalizumab.
Because assessment of quality of life was such a major part of X-ACT, the investigators pulled out all the stops. Their multimodal evaluation included the Angioedema Quality of Life questionnaire – a patient-reported, 17-item instrument that is the first validated tool for evaluation of angioedema-specific quality of life – as well as the Dermatology Life Quality Index and the weekly Angioedema Activity Score.
Patients were also asked to rate on a 0-4 scale their degree of fearfulness of life-threatening swelling episodes and also their degree of fearfulness of angioedema-related suffocation. “To my knowledge, this is the first time this has been done in a randomized clinical trial,” Dr. Weller noted.
The patient reports were striking: At baseline, 49% indicated that they occasionally, often, or very often were afraid of suffocating caused by swelling episodes; only 4% of patients expressed that fear after 28 weeks on omalizumab, compared with 25% of placebo-treated controls. Similarly, at baseline two-thirds of patients reported occasionally, often, or very often being fearful of life-threatening swelling episodes, a rate that fell to 14% after 28 weeks on omalizumab, compared with 42% for controls.
Scores on the Angioedema Quality of Life Questionnaire improved continuously from a baseline of roughly 60 on a 0-100 scale – indicative of severe impairment – to less than 20 after 28 weeks on omalizumab; these scores steadily worsened again during the 8 weeks following treatment discontinuation. The Dermatology Life Quality Index scores dropped from a mean baseline of 15.6 down to 5 by week 4, remained in the 3-5 range for the remainder of the treatment period, then increased again when treatment was discontinued. The Angioedema Activity Score followed a similar pattern.
One audience member observed that the placebo response was quite strong in the study, with the percentage of patients reporting fear of suffocating caused by angioedema episodes falling from 49% at baseline to 25% after 28 weeks on placebo.
Dr. Weller replied that a potent placebo response is a consistent feature of all clinical trials of CSU therapies. The explanation, he added, is unknown.
He reported receiving research grants from and serving as a consultant to Novartis, which sponsored the X-ACT trial.
SOURCE: Weller K et al. EADV Congress 2017.
GENEVA – a widely underappreciated dimension of the impairment caused by this disease, Karsten Weller, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“Virtually all clinical studies of CSU in recent decades focus on the wheal and pruritus components and not on the angioedema component, even though angioedema is a frequent symptom in the disease. Roughly half of patients with CSU experience wheals and angioedema, and up to 13% experience angioedema only,” said Dr. Weller, a dermatologist at Charité University Hospital in Berlin.
“Angioedema is a major driver of quality-of-life impairment in CSU,” he continued. “We know that these are the patients who particularly suffer from the unpredictability of the disease, from disfigurement, from embarrassment. These are the patients who come to the emergency rooms, who lose working days, and these are the patients who often have the feeling of losing control over their lives.”
X-ACT was a multicenter German study which included 91 patients with moderate to severe CSU marked by at least four angioedema episodes during the 6 months prior to enrollment. Participants also had to be refractory to second-generation H1 antihistamines at two to four times the approved dose. The subjects were randomized to subcutaneous omalizumab at 300 mg every 4 weeks or placebo for 28 weeks; they were then further assessed for changes in quality of life during 8 weeks off omalizumab.
Because assessment of quality of life was such a major part of X-ACT, the investigators pulled out all the stops. Their multimodal evaluation included the Angioedema Quality of Life questionnaire – a patient-reported, 17-item instrument that is the first validated tool for evaluation of angioedema-specific quality of life – as well as the Dermatology Life Quality Index and the weekly Angioedema Activity Score.
Patients were also asked to rate on a 0-4 scale their degree of fearfulness of life-threatening swelling episodes and also their degree of fearfulness of angioedema-related suffocation. “To my knowledge, this is the first time this has been done in a randomized clinical trial,” Dr. Weller noted.
The patient reports were striking: At baseline, 49% indicated that they occasionally, often, or very often were afraid of suffocating caused by swelling episodes; only 4% of patients expressed that fear after 28 weeks on omalizumab, compared with 25% of placebo-treated controls. Similarly, at baseline two-thirds of patients reported occasionally, often, or very often being fearful of life-threatening swelling episodes, a rate that fell to 14% after 28 weeks on omalizumab, compared with 42% for controls.
Scores on the Angioedema Quality of Life Questionnaire improved continuously from a baseline of roughly 60 on a 0-100 scale – indicative of severe impairment – to less than 20 after 28 weeks on omalizumab; these scores steadily worsened again during the 8 weeks following treatment discontinuation. The Dermatology Life Quality Index scores dropped from a mean baseline of 15.6 down to 5 by week 4, remained in the 3-5 range for the remainder of the treatment period, then increased again when treatment was discontinued. The Angioedema Activity Score followed a similar pattern.
One audience member observed that the placebo response was quite strong in the study, with the percentage of patients reporting fear of suffocating caused by angioedema episodes falling from 49% at baseline to 25% after 28 weeks on placebo.
Dr. Weller replied that a potent placebo response is a consistent feature of all clinical trials of CSU therapies. The explanation, he added, is unknown.
He reported receiving research grants from and serving as a consultant to Novartis, which sponsored the X-ACT trial.
SOURCE: Weller K et al. EADV Congress 2017.
GENEVA – a widely underappreciated dimension of the impairment caused by this disease, Karsten Weller, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“Virtually all clinical studies of CSU in recent decades focus on the wheal and pruritus components and not on the angioedema component, even though angioedema is a frequent symptom in the disease. Roughly half of patients with CSU experience wheals and angioedema, and up to 13% experience angioedema only,” said Dr. Weller, a dermatologist at Charité University Hospital in Berlin.
“Angioedema is a major driver of quality-of-life impairment in CSU,” he continued. “We know that these are the patients who particularly suffer from the unpredictability of the disease, from disfigurement, from embarrassment. These are the patients who come to the emergency rooms, who lose working days, and these are the patients who often have the feeling of losing control over their lives.”
X-ACT was a multicenter German study which included 91 patients with moderate to severe CSU marked by at least four angioedema episodes during the 6 months prior to enrollment. Participants also had to be refractory to second-generation H1 antihistamines at two to four times the approved dose. The subjects were randomized to subcutaneous omalizumab at 300 mg every 4 weeks or placebo for 28 weeks; they were then further assessed for changes in quality of life during 8 weeks off omalizumab.
Because assessment of quality of life was such a major part of X-ACT, the investigators pulled out all the stops. Their multimodal evaluation included the Angioedema Quality of Life questionnaire – a patient-reported, 17-item instrument that is the first validated tool for evaluation of angioedema-specific quality of life – as well as the Dermatology Life Quality Index and the weekly Angioedema Activity Score.
Patients were also asked to rate on a 0-4 scale their degree of fearfulness of life-threatening swelling episodes and also their degree of fearfulness of angioedema-related suffocation. “To my knowledge, this is the first time this has been done in a randomized clinical trial,” Dr. Weller noted.
The patient reports were striking: At baseline, 49% indicated that they occasionally, often, or very often were afraid of suffocating caused by swelling episodes; only 4% of patients expressed that fear after 28 weeks on omalizumab, compared with 25% of placebo-treated controls. Similarly, at baseline two-thirds of patients reported occasionally, often, or very often being fearful of life-threatening swelling episodes, a rate that fell to 14% after 28 weeks on omalizumab, compared with 42% for controls.
Scores on the Angioedema Quality of Life Questionnaire improved continuously from a baseline of roughly 60 on a 0-100 scale – indicative of severe impairment – to less than 20 after 28 weeks on omalizumab; these scores steadily worsened again during the 8 weeks following treatment discontinuation. The Dermatology Life Quality Index scores dropped from a mean baseline of 15.6 down to 5 by week 4, remained in the 3-5 range for the remainder of the treatment period, then increased again when treatment was discontinued. The Angioedema Activity Score followed a similar pattern.
One audience member observed that the placebo response was quite strong in the study, with the percentage of patients reporting fear of suffocating caused by angioedema episodes falling from 49% at baseline to 25% after 28 weeks on placebo.
Dr. Weller replied that a potent placebo response is a consistent feature of all clinical trials of CSU therapies. The explanation, he added, is unknown.
He reported receiving research grants from and serving as a consultant to Novartis, which sponsored the X-ACT trial.
SOURCE: Weller K et al. EADV Congress 2017.
REPORTING FROM THE EADV CONGRESS
Key clinical point: Omalizumab relieves the heavy quality-of-life burden associated with CSU.
Major finding: At baseline, 49% of CSU patients indicated they occasionally, often, or very often were afraid of suffocating due to swelling episodes; after 28 weeks on omalizumab, only 4% expressed that fear.
Study details: The X-ACT trial was a phase 3 double-blind, multicenter, placebo-controlled randomized trial including 91 patients with CSU.
Disclosures: The presenter reported receiving research grants from and serving as a consultant to Novartis, which sponsored the X-ACT trial.
Source: Weller K et al. EADV Congress 2017.
Prior biologic exposure doesn’t diminish ixekizumab’s efficacy
REPORTING FROM THE EADV CONGRESS
GENEVA – Psoriasis patients switched to ixekizumab after previous exposure to another interleukin-17 inhibitor respond as well as those who are IL-17 antagonist naive, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
This finding is of importance in real-world clinical practice because it’s not at all uncommon for psoriasis patients on one biologic to have to switch to another because of insufficient efficacy, side effects, or a change in insurance coverage. Physicians would like to know what sort of responses can be expected to whatever agent they prescribe next.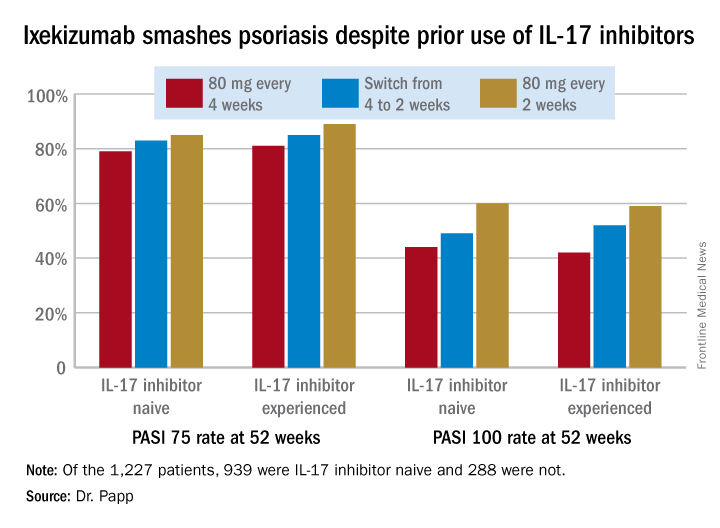
However, that was not a problem in this secondary analysis of a large clinical trial whose primary purpose was to evaluate the relative safety and efficacy of ixekizumab (Taltz) when dosed every 2 weeks versus every 4 weeks.
“I think what we have seen here is a very compelling story: , nor for that matter does it appear to impact safety,” the dermatologist said.
He reported on 1,227 patients with moderate to severe plaque psoriasis who were randomized to ixekizumab at 80 mg every 2 or 4 weeks following an initial 160 mg loading dose. Among those who started out on ixekizumab every 4 weeks, 306 patients got a per-protocol dose adjustment to biweekly therapy because of an insufficient response to monthly dosing as defined by a Physician’s Global Assessment score of 2 or more on two consecutive office visits during study weeks 12-40.
A total of 939 patients were IL-17 inhibitor naive. The other 288 had previously been on the IL-17 antagonists brodalumab (Siliq) or secukinumab (Cosentyx). The two groups had similar baseline demographics with the exception that the experienced cohort had on average a 22.2-year duration of psoriasis, 3.7 years more than IL-17 antagonist-naive patients.
In an intent-to-treat analysis, Psoriasis Area and Severity Index (PASI) 75, 90, and 100 responses at week 52 didn’t differ significantly between the IL-17 inhibitor-naive and -experienced groups. In fact, patients with prior exposure to other IL-17 antagonists showed a consistent trend for slightly higher response rates (see graphic).
It was clear from this analysis that dosing ixekizumab every 2 weeks provides significantly better efficacy than was dosing every 4 weeks, Dr. Papp noted. Yet the approved dosing is 160 mg at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
No new safety issues arose in this study. The only difference between the naive and experienced groups was a lower rate of allergic reactions/hypersensitivity in the experienced group. For example, in patients on ixekizumab every 2 weeks for the entire 52-week study period the incidence of such reactions was 11.5% in the IL-17 antagonist-naive group, compared with 4.1% in the experienced cohort. This isn’t really surprising, according to Dr. Papp.
“Most injection site reactions occur in the newbies,” he said.
The study was sponsored by Eli Lilly. Dr. Papp serves as a consultant and/or adviser to Lilly and numerous other pharmaceutical companies involved in the development of dermatologic therapies.
REPORTING FROM THE EADV CONGRESS
GENEVA – Psoriasis patients switched to ixekizumab after previous exposure to another interleukin-17 inhibitor respond as well as those who are IL-17 antagonist naive, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
This finding is of importance in real-world clinical practice because it’s not at all uncommon for psoriasis patients on one biologic to have to switch to another because of insufficient efficacy, side effects, or a change in insurance coverage. Physicians would like to know what sort of responses can be expected to whatever agent they prescribe next.
However, that was not a problem in this secondary analysis of a large clinical trial whose primary purpose was to evaluate the relative safety and efficacy of ixekizumab (Taltz) when dosed every 2 weeks versus every 4 weeks.
“I think what we have seen here is a very compelling story: , nor for that matter does it appear to impact safety,” the dermatologist said.
He reported on 1,227 patients with moderate to severe plaque psoriasis who were randomized to ixekizumab at 80 mg every 2 or 4 weeks following an initial 160 mg loading dose. Among those who started out on ixekizumab every 4 weeks, 306 patients got a per-protocol dose adjustment to biweekly therapy because of an insufficient response to monthly dosing as defined by a Physician’s Global Assessment score of 2 or more on two consecutive office visits during study weeks 12-40.
A total of 939 patients were IL-17 inhibitor naive. The other 288 had previously been on the IL-17 antagonists brodalumab (Siliq) or secukinumab (Cosentyx). The two groups had similar baseline demographics with the exception that the experienced cohort had on average a 22.2-year duration of psoriasis, 3.7 years more than IL-17 antagonist-naive patients.
In an intent-to-treat analysis, Psoriasis Area and Severity Index (PASI) 75, 90, and 100 responses at week 52 didn’t differ significantly between the IL-17 inhibitor-naive and -experienced groups. In fact, patients with prior exposure to other IL-17 antagonists showed a consistent trend for slightly higher response rates (see graphic).
It was clear from this analysis that dosing ixekizumab every 2 weeks provides significantly better efficacy than was dosing every 4 weeks, Dr. Papp noted. Yet the approved dosing is 160 mg at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
No new safety issues arose in this study. The only difference between the naive and experienced groups was a lower rate of allergic reactions/hypersensitivity in the experienced group. For example, in patients on ixekizumab every 2 weeks for the entire 52-week study period the incidence of such reactions was 11.5% in the IL-17 antagonist-naive group, compared with 4.1% in the experienced cohort. This isn’t really surprising, according to Dr. Papp.
“Most injection site reactions occur in the newbies,” he said.
The study was sponsored by Eli Lilly. Dr. Papp serves as a consultant and/or adviser to Lilly and numerous other pharmaceutical companies involved in the development of dermatologic therapies.
REPORTING FROM THE EADV CONGRESS
GENEVA – Psoriasis patients switched to ixekizumab after previous exposure to another interleukin-17 inhibitor respond as well as those who are IL-17 antagonist naive, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
This finding is of importance in real-world clinical practice because it’s not at all uncommon for psoriasis patients on one biologic to have to switch to another because of insufficient efficacy, side effects, or a change in insurance coverage. Physicians would like to know what sort of responses can be expected to whatever agent they prescribe next.
However, that was not a problem in this secondary analysis of a large clinical trial whose primary purpose was to evaluate the relative safety and efficacy of ixekizumab (Taltz) when dosed every 2 weeks versus every 4 weeks.
“I think what we have seen here is a very compelling story: , nor for that matter does it appear to impact safety,” the dermatologist said.
He reported on 1,227 patients with moderate to severe plaque psoriasis who were randomized to ixekizumab at 80 mg every 2 or 4 weeks following an initial 160 mg loading dose. Among those who started out on ixekizumab every 4 weeks, 306 patients got a per-protocol dose adjustment to biweekly therapy because of an insufficient response to monthly dosing as defined by a Physician’s Global Assessment score of 2 or more on two consecutive office visits during study weeks 12-40.
A total of 939 patients were IL-17 inhibitor naive. The other 288 had previously been on the IL-17 antagonists brodalumab (Siliq) or secukinumab (Cosentyx). The two groups had similar baseline demographics with the exception that the experienced cohort had on average a 22.2-year duration of psoriasis, 3.7 years more than IL-17 antagonist-naive patients.
In an intent-to-treat analysis, Psoriasis Area and Severity Index (PASI) 75, 90, and 100 responses at week 52 didn’t differ significantly between the IL-17 inhibitor-naive and -experienced groups. In fact, patients with prior exposure to other IL-17 antagonists showed a consistent trend for slightly higher response rates (see graphic).
It was clear from this analysis that dosing ixekizumab every 2 weeks provides significantly better efficacy than was dosing every 4 weeks, Dr. Papp noted. Yet the approved dosing is 160 mg at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
No new safety issues arose in this study. The only difference between the naive and experienced groups was a lower rate of allergic reactions/hypersensitivity in the experienced group. For example, in patients on ixekizumab every 2 weeks for the entire 52-week study period the incidence of such reactions was 11.5% in the IL-17 antagonist-naive group, compared with 4.1% in the experienced cohort. This isn’t really surprising, according to Dr. Papp.
“Most injection site reactions occur in the newbies,” he said.
The study was sponsored by Eli Lilly. Dr. Papp serves as a consultant and/or adviser to Lilly and numerous other pharmaceutical companies involved in the development of dermatologic therapies.
Updosing omalizumab for chronic urticaria pays off
GENEVA – In real-world clinical practice, roughly – and for those who don’t, three-quarters will respond upon updosing to 450 or 600 mg every 4 weeks.
That’s the key message of an open-label study of 286 patients with chronic spontaneous urticaria (CSU) conducted at 15 hospitals by the Catalan and Balearic Chronic Urticaria Network (XUrCB), Jorge Spertino, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

In three published, pivotal, phase 3 randomized trials, the clinical response rate to omalizumab at 300 mg every 4 weeks, as defined by a weekly 7-day Urticaria Activity Score (UAS7) of 6 or less at 12 weeks, was 52% in ASTERIA I, 66% in ASTERIA II, and 52% in GLACIAL. But patients enrolled in formal randomized trials are often quite different from the broader group encountered in daily practice, and the Spanish dermatologists wanted to know if updosing in suboptimal responders was safe and effective. It turns out that it certainly is, according to Dr. Spertino of Teknon Medical Center in Barcelona.
The treatment algorithm followed by the XUrCB investigators was that, if after six doses at the approved dose of 300 mg every 4 weeks a patient didn’t have good control of disease activity, the dose was increased to 450 mg every 4 weeks. If after three doses at that level, there still wasn’t good control of the CSU, the dose was further increased to 600 mg every 4 weeks.
As in the pivotal phase 3 clinical trials, the XUrCB group defined good control of disease activity as a UAS7 score of 6 or less in accord with a study that demonstrated such a score on the 0- to 42-point UAS7 correlates well with minimal or no patient symptoms (Br J Dermatol. 2017 Oct;177[4]:1093-1101).
At baseline, the mean age of the 286 CSU patients was 44.6 years and the mean UAS7 score was 26.5; 74% were women. Forty-seven percent of patients experienced angioedema and 33% had inducible urticaria, most commonly brought forth by pressure or dermographism. One-third of patients had previously been on cyclosporine and half of the patients had a high d-dimer level.
Sixty-five percent of patients achieved good disease control on omalizumab at 300 mg every 4 weeks. Of the 99 patients (35%) who didn’t, 20 patients stopped treatment at their dermatologist’s request because their symptoms remained uncontrolled on the approved dose. But 59 of the 79 who updosed obtained good disease control: 43 on a dose of 450 mg and 16 on a dose of 600 mg.
In multivariate analysis, two predictors of treatment success with updosing were identified: previous treatment with cyclosporine and obesity. Among patients previously on cyclosporine – a marker for more severe disease – only 21% achieved a UAS7 score of 6 or less on the approved dose, while 41% did so upon updosing. And obesity was associated with a 3.7-fold increased likelihood of a favorable response to updosing after lack of treatment success at the approved dose.
Neither a high d-dimer or serum IgE level, baseline UAS7 score, gender, associated angioedema, nor inducible urticaria was significantly associated with an increased treatment success rate upon updosing.
Updosing proved to be safe. All adverse events were mild and infrequent, consisting of headache, local injection site reactions, and arthromyalgia s, each occurring in 1%-2% of patients. Frequencies were similar in updosed patients and those on the approved dosing schedule.
Session cochair Jorgen Serup, MD, DMsc, congratulated Dr. Spertino for supplying physicians with “very-much-needed data.”
“This is very convincing data and highly clinically relevant for those of us who have these patients in our practices,” said Dr. Serup, professor of dermatology at Copenhagen University.
Dr. Spertino reported having no financial conflicts of interest regarding his presentation.
SOURCE: Spertino J et al. EADV Congress
GENEVA – In real-world clinical practice, roughly – and for those who don’t, three-quarters will respond upon updosing to 450 or 600 mg every 4 weeks.
That’s the key message of an open-label study of 286 patients with chronic spontaneous urticaria (CSU) conducted at 15 hospitals by the Catalan and Balearic Chronic Urticaria Network (XUrCB), Jorge Spertino, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
In three published, pivotal, phase 3 randomized trials, the clinical response rate to omalizumab at 300 mg every 4 weeks, as defined by a weekly 7-day Urticaria Activity Score (UAS7) of 6 or less at 12 weeks, was 52% in ASTERIA I, 66% in ASTERIA II, and 52% in GLACIAL. But patients enrolled in formal randomized trials are often quite different from the broader group encountered in daily practice, and the Spanish dermatologists wanted to know if updosing in suboptimal responders was safe and effective. It turns out that it certainly is, according to Dr. Spertino of Teknon Medical Center in Barcelona.
The treatment algorithm followed by the XUrCB investigators was that, if after six doses at the approved dose of 300 mg every 4 weeks a patient didn’t have good control of disease activity, the dose was increased to 450 mg every 4 weeks. If after three doses at that level, there still wasn’t good control of the CSU, the dose was further increased to 600 mg every 4 weeks.
As in the pivotal phase 3 clinical trials, the XUrCB group defined good control of disease activity as a UAS7 score of 6 or less in accord with a study that demonstrated such a score on the 0- to 42-point UAS7 correlates well with minimal or no patient symptoms (Br J Dermatol. 2017 Oct;177[4]:1093-1101).
At baseline, the mean age of the 286 CSU patients was 44.6 years and the mean UAS7 score was 26.5; 74% were women. Forty-seven percent of patients experienced angioedema and 33% had inducible urticaria, most commonly brought forth by pressure or dermographism. One-third of patients had previously been on cyclosporine and half of the patients had a high d-dimer level.
Sixty-five percent of patients achieved good disease control on omalizumab at 300 mg every 4 weeks. Of the 99 patients (35%) who didn’t, 20 patients stopped treatment at their dermatologist’s request because their symptoms remained uncontrolled on the approved dose. But 59 of the 79 who updosed obtained good disease control: 43 on a dose of 450 mg and 16 on a dose of 600 mg.
In multivariate analysis, two predictors of treatment success with updosing were identified: previous treatment with cyclosporine and obesity. Among patients previously on cyclosporine – a marker for more severe disease – only 21% achieved a UAS7 score of 6 or less on the approved dose, while 41% did so upon updosing. And obesity was associated with a 3.7-fold increased likelihood of a favorable response to updosing after lack of treatment success at the approved dose.
Neither a high d-dimer or serum IgE level, baseline UAS7 score, gender, associated angioedema, nor inducible urticaria was significantly associated with an increased treatment success rate upon updosing.
Updosing proved to be safe. All adverse events were mild and infrequent, consisting of headache, local injection site reactions, and arthromyalgia s, each occurring in 1%-2% of patients. Frequencies were similar in updosed patients and those on the approved dosing schedule.
Session cochair Jorgen Serup, MD, DMsc, congratulated Dr. Spertino for supplying physicians with “very-much-needed data.”
“This is very convincing data and highly clinically relevant for those of us who have these patients in our practices,” said Dr. Serup, professor of dermatology at Copenhagen University.
Dr. Spertino reported having no financial conflicts of interest regarding his presentation.
SOURCE: Spertino J et al. EADV Congress
GENEVA – In real-world clinical practice, roughly – and for those who don’t, three-quarters will respond upon updosing to 450 or 600 mg every 4 weeks.
That’s the key message of an open-label study of 286 patients with chronic spontaneous urticaria (CSU) conducted at 15 hospitals by the Catalan and Balearic Chronic Urticaria Network (XUrCB), Jorge Spertino, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
In three published, pivotal, phase 3 randomized trials, the clinical response rate to omalizumab at 300 mg every 4 weeks, as defined by a weekly 7-day Urticaria Activity Score (UAS7) of 6 or less at 12 weeks, was 52% in ASTERIA I, 66% in ASTERIA II, and 52% in GLACIAL. But patients enrolled in formal randomized trials are often quite different from the broader group encountered in daily practice, and the Spanish dermatologists wanted to know if updosing in suboptimal responders was safe and effective. It turns out that it certainly is, according to Dr. Spertino of Teknon Medical Center in Barcelona.
The treatment algorithm followed by the XUrCB investigators was that, if after six doses at the approved dose of 300 mg every 4 weeks a patient didn’t have good control of disease activity, the dose was increased to 450 mg every 4 weeks. If after three doses at that level, there still wasn’t good control of the CSU, the dose was further increased to 600 mg every 4 weeks.
As in the pivotal phase 3 clinical trials, the XUrCB group defined good control of disease activity as a UAS7 score of 6 or less in accord with a study that demonstrated such a score on the 0- to 42-point UAS7 correlates well with minimal or no patient symptoms (Br J Dermatol. 2017 Oct;177[4]:1093-1101).
At baseline, the mean age of the 286 CSU patients was 44.6 years and the mean UAS7 score was 26.5; 74% were women. Forty-seven percent of patients experienced angioedema and 33% had inducible urticaria, most commonly brought forth by pressure or dermographism. One-third of patients had previously been on cyclosporine and half of the patients had a high d-dimer level.
Sixty-five percent of patients achieved good disease control on omalizumab at 300 mg every 4 weeks. Of the 99 patients (35%) who didn’t, 20 patients stopped treatment at their dermatologist’s request because their symptoms remained uncontrolled on the approved dose. But 59 of the 79 who updosed obtained good disease control: 43 on a dose of 450 mg and 16 on a dose of 600 mg.
In multivariate analysis, two predictors of treatment success with updosing were identified: previous treatment with cyclosporine and obesity. Among patients previously on cyclosporine – a marker for more severe disease – only 21% achieved a UAS7 score of 6 or less on the approved dose, while 41% did so upon updosing. And obesity was associated with a 3.7-fold increased likelihood of a favorable response to updosing after lack of treatment success at the approved dose.
Neither a high d-dimer or serum IgE level, baseline UAS7 score, gender, associated angioedema, nor inducible urticaria was significantly associated with an increased treatment success rate upon updosing.
Updosing proved to be safe. All adverse events were mild and infrequent, consisting of headache, local injection site reactions, and arthromyalgia s, each occurring in 1%-2% of patients. Frequencies were similar in updosed patients and those on the approved dosing schedule.
Session cochair Jorgen Serup, MD, DMsc, congratulated Dr. Spertino for supplying physicians with “very-much-needed data.”
“This is very convincing data and highly clinically relevant for those of us who have these patients in our practices,” said Dr. Serup, professor of dermatology at Copenhagen University.
Dr. Spertino reported having no financial conflicts of interest regarding his presentation.
SOURCE: Spertino J et al. EADV Congress
REPORTING FROM THE EADV CONGRESS
Key clinical point: Updosing of omalizumab to a maximum of twice the approved dose is safe and effective in chronic spontaneous urticaria patients unresponsive to the licensed dose.
Major finding: Upon updosing, 75% of nonresponders to the approved dose achieved good disease control with no increase in adverse events.
Study details: This multicenter study of an omalizumab updosing algorithm included 286 patients with chronic spontaneous urticaria.
Disclosures: The study presenter reported having no financial conflicts.
Watchman device PREVAILs for stroke prevention
DENVER – Left atrial appendage closure using the Watchman device is as effective as warfarin in preventing strokes in patients with atrial fibrillation, but the strokes in Watchman recipients are 55% less likely to be disabling, according to a meta-analysis of 5-year outcomes in the PREVAIL and PROTECT AF randomized trials.
The device therapy showed additional advantages over warfarin: significantly reduced risks of mortality, non–procedure-related major bleeding, and hemorrhagic stroke, Saibal Kar, MD, reported in presenting the results of the meta-analysis at the Transcatheter Cardiovascular Therapeutics annual educational meeting.

“We have prevailed,” he declared, referring to device safety concerns that arose early on and have since been laid to rest.
The patient-level meta-analysis of 5-year outcomes included 1,114 patients with atrial fibrillation who were randomized 2:1 to the Watchman device or warfarin, with 4,343 patient-years of follow-up. This was a fairly high–stroke risk population, with CHA2DS2-VASc scores in the 3.6-3.9 range, and 40% of patients aged 75 years or more. At baseline, 23% of subjects had a history of stroke or transient ischemic attack.
At 5 years’ follow-up, the composite endpoint of all stroke or systemic embolism was the same in the two study arms. However, the rate of hemorrhagic stroke was 80% lower in the Watchman group, the risk of disabling or fatal stroke was reduced by 55%, the rate of cardiovascular or unexplained death was 41% lower, all-cause mortality was reduced by 27%, and postprocedure major bleeding was 52% less frequent in the device therapy group. All of these differences achieved statistical significance, the cardiologist reported at the meeting sponsored by the Cardiovascular Research Foundation.
On the downside, the rate of ischemic stroke trended higher in the Watchman group, although the 71% increase in relative risk didn’t achieve statistical significance (P = .08). Dr. Kar asserted that this unhappy trend was a statistical fluke resulting from a low number of events and an implausibly low ischemic stroke rate of 0.73% per year in the warfarin group.
“This is the lowest rate of ischemic stroke in any study of warfarin. In fact, if this was the ischemic stroke rate in any of the NOAC [novel oral anticoagulant] studies, none of those drugs would actually have been approved. Why did we get such an implausibly low ischemic stroke rate? It’s a function of lower numbers and larger confidence intervals,” he said.
Gregg W. Stone, MD, who moderated the discussion panel at the late-breaking clinical trial session, advised Dr. Kar to be less defensive about the ischemic stroke findings.
“I think we have to be a little less apologetic for the great outcomes in the warfarin arm in PREVAIL. We do these randomized trials, and we get what we get,” said Dr. Stone, professor of medicine at Columbia University in New York.
Stephen G. Ellis, MD, said he was particularly impressed with the reduced rate of disabling stroke in the Watchman group.
“Severity of stroke is important. I hadn’t seen that data before,” commented Dr. Ellis, professor of medicine and director of the cardiac catheterization laboratory at the Cleveland Clinic. “The overall message, I think, is that for the patients who would have been candidates to be enrolled in these trials, the device seems to be quite worthwhile. I take note of the overall benefit in terms of cardiovascular death and all-cause death.”
Robert J. Sommer, MD, said that in patients with high CHA2DS2-VASc scores and previous bleeding on oral anticoagulants, the new data show that the Watchman “is a no-brainer. The patients all want it, the physicians all want it. It’s a very easy decision to make.”
“But you get into other groups who may potentially be interested in the device – particularly the younger patients who are very active and don’t want to be on anticoagulation – I think the ischemic stroke rate in the device arm trending to be higher is a problem for them. And it’s certainly going to be a problem for their physicians. But as we go on, I think with further studies we’ll see expanded indications. Patients with CAD who potentially would need triple therapy – that’s a nice population to study in this area. We’ll also be seeing data on other devices that may have different ischemic stroke rates,” said Dr. Sommer, director of invasive adult congenital heart disease at Columbia University Medical Center.
“I find this data extraordinarily helpful as I think about my conversations with patients about stroke prevention,” said Brian K. Whisenant, MD, medical director of structural heart disease at the Intermountain Medical Center Heart Institute in Salt Lake City.
“I tell them we think of oral anticoagulants as first-line therapy based on a stroke rate of 1% per year or less in most datasets. The data for the Watchman device has been very consistent in that we have a stroke rate that’s a little bit higher, at 1.3%-1.8% per year. And we have extensive data for predicting the stroke rate in the absence of oral anticoagulation: In most of our patients, that rate is in excess of 5% per year. So while the Watchman device may not provide the absolute reduction in ischemic stroke rate that oral anticoagulants do, a stroke rate of less than 2% is a whole lot better than no therapy for many of these patients,” the cardiologist said.
Martin B. Leon, MD, opined that the ischemic stroke data cannot be explained away. But he added that the totality of the meta-analysis data gives him confidence that this is the appropriate treatment in these patients.
“It does leave open the question of whether we can do better with ischemic strokes. Some people are suggesting that maybe adjunctive pharmacotherapy – perhaps a low-dose NOAC – may be a reasonable option in some patients to get even better results. That’s something I believe is open for discussion,” said Dr. Leon, professor of medicine at Columbia University and director of the Center for Interventional Vascular Therapy at New York-Presbyterian/Columbia University Medical Center, New York.
Dr. Stone summed up: “There’s uniformity among the panel that there may be a slightly lower ischemic stroke rate with oral anticoagulation, and the NOACs probably provide some additional benefit, with an additional 50% reduction in hemorrhagic stroke, compared to warfarin. But that being said, I believe that left atrial appendage closure with the Watchman is the viable and now clearly safe approach for patients with any sort of contraindication or strong desire to avoid oral anticoagulation.”
The PREVAIL and PROTECT AF trials and meta-analysis were sponsored by Boston Scientific. Dr. Kar reported receiving research grants from and serving as a consultant to that company as well as Abbott Vascular.
Simultaneously with Dr. Kar’s presentation at TCT 2017, the findings were published online in the Journal of the American College of Cardiology.
SOURCE: Reddy VY et al. TCT 2017; J Am Coll Cardiol. 2017 Nov 4. pii:S0735-1097(17)41187-9. doi: 10.1016/j.jacc.2017.10.021.
DENVER – Left atrial appendage closure using the Watchman device is as effective as warfarin in preventing strokes in patients with atrial fibrillation, but the strokes in Watchman recipients are 55% less likely to be disabling, according to a meta-analysis of 5-year outcomes in the PREVAIL and PROTECT AF randomized trials.
The device therapy showed additional advantages over warfarin: significantly reduced risks of mortality, non–procedure-related major bleeding, and hemorrhagic stroke, Saibal Kar, MD, reported in presenting the results of the meta-analysis at the Transcatheter Cardiovascular Therapeutics annual educational meeting.
“We have prevailed,” he declared, referring to device safety concerns that arose early on and have since been laid to rest.
The patient-level meta-analysis of 5-year outcomes included 1,114 patients with atrial fibrillation who were randomized 2:1 to the Watchman device or warfarin, with 4,343 patient-years of follow-up. This was a fairly high–stroke risk population, with CHA2DS2-VASc scores in the 3.6-3.9 range, and 40% of patients aged 75 years or more. At baseline, 23% of subjects had a history of stroke or transient ischemic attack.
At 5 years’ follow-up, the composite endpoint of all stroke or systemic embolism was the same in the two study arms. However, the rate of hemorrhagic stroke was 80% lower in the Watchman group, the risk of disabling or fatal stroke was reduced by 55%, the rate of cardiovascular or unexplained death was 41% lower, all-cause mortality was reduced by 27%, and postprocedure major bleeding was 52% less frequent in the device therapy group. All of these differences achieved statistical significance, the cardiologist reported at the meeting sponsored by the Cardiovascular Research Foundation.
On the downside, the rate of ischemic stroke trended higher in the Watchman group, although the 71% increase in relative risk didn’t achieve statistical significance (P = .08). Dr. Kar asserted that this unhappy trend was a statistical fluke resulting from a low number of events and an implausibly low ischemic stroke rate of 0.73% per year in the warfarin group.
“This is the lowest rate of ischemic stroke in any study of warfarin. In fact, if this was the ischemic stroke rate in any of the NOAC [novel oral anticoagulant] studies, none of those drugs would actually have been approved. Why did we get such an implausibly low ischemic stroke rate? It’s a function of lower numbers and larger confidence intervals,” he said.
Gregg W. Stone, MD, who moderated the discussion panel at the late-breaking clinical trial session, advised Dr. Kar to be less defensive about the ischemic stroke findings.
“I think we have to be a little less apologetic for the great outcomes in the warfarin arm in PREVAIL. We do these randomized trials, and we get what we get,” said Dr. Stone, professor of medicine at Columbia University in New York.
Stephen G. Ellis, MD, said he was particularly impressed with the reduced rate of disabling stroke in the Watchman group.
“Severity of stroke is important. I hadn’t seen that data before,” commented Dr. Ellis, professor of medicine and director of the cardiac catheterization laboratory at the Cleveland Clinic. “The overall message, I think, is that for the patients who would have been candidates to be enrolled in these trials, the device seems to be quite worthwhile. I take note of the overall benefit in terms of cardiovascular death and all-cause death.”
Robert J. Sommer, MD, said that in patients with high CHA2DS2-VASc scores and previous bleeding on oral anticoagulants, the new data show that the Watchman “is a no-brainer. The patients all want it, the physicians all want it. It’s a very easy decision to make.”
“But you get into other groups who may potentially be interested in the device – particularly the younger patients who are very active and don’t want to be on anticoagulation – I think the ischemic stroke rate in the device arm trending to be higher is a problem for them. And it’s certainly going to be a problem for their physicians. But as we go on, I think with further studies we’ll see expanded indications. Patients with CAD who potentially would need triple therapy – that’s a nice population to study in this area. We’ll also be seeing data on other devices that may have different ischemic stroke rates,” said Dr. Sommer, director of invasive adult congenital heart disease at Columbia University Medical Center.
“I find this data extraordinarily helpful as I think about my conversations with patients about stroke prevention,” said Brian K. Whisenant, MD, medical director of structural heart disease at the Intermountain Medical Center Heart Institute in Salt Lake City.
“I tell them we think of oral anticoagulants as first-line therapy based on a stroke rate of 1% per year or less in most datasets. The data for the Watchman device has been very consistent in that we have a stroke rate that’s a little bit higher, at 1.3%-1.8% per year. And we have extensive data for predicting the stroke rate in the absence of oral anticoagulation: In most of our patients, that rate is in excess of 5% per year. So while the Watchman device may not provide the absolute reduction in ischemic stroke rate that oral anticoagulants do, a stroke rate of less than 2% is a whole lot better than no therapy for many of these patients,” the cardiologist said.
Martin B. Leon, MD, opined that the ischemic stroke data cannot be explained away. But he added that the totality of the meta-analysis data gives him confidence that this is the appropriate treatment in these patients.
“It does leave open the question of whether we can do better with ischemic strokes. Some people are suggesting that maybe adjunctive pharmacotherapy – perhaps a low-dose NOAC – may be a reasonable option in some patients to get even better results. That’s something I believe is open for discussion,” said Dr. Leon, professor of medicine at Columbia University and director of the Center for Interventional Vascular Therapy at New York-Presbyterian/Columbia University Medical Center, New York.
Dr. Stone summed up: “There’s uniformity among the panel that there may be a slightly lower ischemic stroke rate with oral anticoagulation, and the NOACs probably provide some additional benefit, with an additional 50% reduction in hemorrhagic stroke, compared to warfarin. But that being said, I believe that left atrial appendage closure with the Watchman is the viable and now clearly safe approach for patients with any sort of contraindication or strong desire to avoid oral anticoagulation.”
The PREVAIL and PROTECT AF trials and meta-analysis were sponsored by Boston Scientific. Dr. Kar reported receiving research grants from and serving as a consultant to that company as well as Abbott Vascular.
Simultaneously with Dr. Kar’s presentation at TCT 2017, the findings were published online in the Journal of the American College of Cardiology.
SOURCE: Reddy VY et al. TCT 2017; J Am Coll Cardiol. 2017 Nov 4. pii:S0735-1097(17)41187-9. doi: 10.1016/j.jacc.2017.10.021.
DENVER – Left atrial appendage closure using the Watchman device is as effective as warfarin in preventing strokes in patients with atrial fibrillation, but the strokes in Watchman recipients are 55% less likely to be disabling, according to a meta-analysis of 5-year outcomes in the PREVAIL and PROTECT AF randomized trials.
The device therapy showed additional advantages over warfarin: significantly reduced risks of mortality, non–procedure-related major bleeding, and hemorrhagic stroke, Saibal Kar, MD, reported in presenting the results of the meta-analysis at the Transcatheter Cardiovascular Therapeutics annual educational meeting.
“We have prevailed,” he declared, referring to device safety concerns that arose early on and have since been laid to rest.
The patient-level meta-analysis of 5-year outcomes included 1,114 patients with atrial fibrillation who were randomized 2:1 to the Watchman device or warfarin, with 4,343 patient-years of follow-up. This was a fairly high–stroke risk population, with CHA2DS2-VASc scores in the 3.6-3.9 range, and 40% of patients aged 75 years or more. At baseline, 23% of subjects had a history of stroke or transient ischemic attack.
At 5 years’ follow-up, the composite endpoint of all stroke or systemic embolism was the same in the two study arms. However, the rate of hemorrhagic stroke was 80% lower in the Watchman group, the risk of disabling or fatal stroke was reduced by 55%, the rate of cardiovascular or unexplained death was 41% lower, all-cause mortality was reduced by 27%, and postprocedure major bleeding was 52% less frequent in the device therapy group. All of these differences achieved statistical significance, the cardiologist reported at the meeting sponsored by the Cardiovascular Research Foundation.
On the downside, the rate of ischemic stroke trended higher in the Watchman group, although the 71% increase in relative risk didn’t achieve statistical significance (P = .08). Dr. Kar asserted that this unhappy trend was a statistical fluke resulting from a low number of events and an implausibly low ischemic stroke rate of 0.73% per year in the warfarin group.
“This is the lowest rate of ischemic stroke in any study of warfarin. In fact, if this was the ischemic stroke rate in any of the NOAC [novel oral anticoagulant] studies, none of those drugs would actually have been approved. Why did we get such an implausibly low ischemic stroke rate? It’s a function of lower numbers and larger confidence intervals,” he said.
Gregg W. Stone, MD, who moderated the discussion panel at the late-breaking clinical trial session, advised Dr. Kar to be less defensive about the ischemic stroke findings.
“I think we have to be a little less apologetic for the great outcomes in the warfarin arm in PREVAIL. We do these randomized trials, and we get what we get,” said Dr. Stone, professor of medicine at Columbia University in New York.
Stephen G. Ellis, MD, said he was particularly impressed with the reduced rate of disabling stroke in the Watchman group.
“Severity of stroke is important. I hadn’t seen that data before,” commented Dr. Ellis, professor of medicine and director of the cardiac catheterization laboratory at the Cleveland Clinic. “The overall message, I think, is that for the patients who would have been candidates to be enrolled in these trials, the device seems to be quite worthwhile. I take note of the overall benefit in terms of cardiovascular death and all-cause death.”
Robert J. Sommer, MD, said that in patients with high CHA2DS2-VASc scores and previous bleeding on oral anticoagulants, the new data show that the Watchman “is a no-brainer. The patients all want it, the physicians all want it. It’s a very easy decision to make.”
“But you get into other groups who may potentially be interested in the device – particularly the younger patients who are very active and don’t want to be on anticoagulation – I think the ischemic stroke rate in the device arm trending to be higher is a problem for them. And it’s certainly going to be a problem for their physicians. But as we go on, I think with further studies we’ll see expanded indications. Patients with CAD who potentially would need triple therapy – that’s a nice population to study in this area. We’ll also be seeing data on other devices that may have different ischemic stroke rates,” said Dr. Sommer, director of invasive adult congenital heart disease at Columbia University Medical Center.
“I find this data extraordinarily helpful as I think about my conversations with patients about stroke prevention,” said Brian K. Whisenant, MD, medical director of structural heart disease at the Intermountain Medical Center Heart Institute in Salt Lake City.
“I tell them we think of oral anticoagulants as first-line therapy based on a stroke rate of 1% per year or less in most datasets. The data for the Watchman device has been very consistent in that we have a stroke rate that’s a little bit higher, at 1.3%-1.8% per year. And we have extensive data for predicting the stroke rate in the absence of oral anticoagulation: In most of our patients, that rate is in excess of 5% per year. So while the Watchman device may not provide the absolute reduction in ischemic stroke rate that oral anticoagulants do, a stroke rate of less than 2% is a whole lot better than no therapy for many of these patients,” the cardiologist said.
Martin B. Leon, MD, opined that the ischemic stroke data cannot be explained away. But he added that the totality of the meta-analysis data gives him confidence that this is the appropriate treatment in these patients.
“It does leave open the question of whether we can do better with ischemic strokes. Some people are suggesting that maybe adjunctive pharmacotherapy – perhaps a low-dose NOAC – may be a reasonable option in some patients to get even better results. That’s something I believe is open for discussion,” said Dr. Leon, professor of medicine at Columbia University and director of the Center for Interventional Vascular Therapy at New York-Presbyterian/Columbia University Medical Center, New York.
Dr. Stone summed up: “There’s uniformity among the panel that there may be a slightly lower ischemic stroke rate with oral anticoagulation, and the NOACs probably provide some additional benefit, with an additional 50% reduction in hemorrhagic stroke, compared to warfarin. But that being said, I believe that left atrial appendage closure with the Watchman is the viable and now clearly safe approach for patients with any sort of contraindication or strong desire to avoid oral anticoagulation.”
The PREVAIL and PROTECT AF trials and meta-analysis were sponsored by Boston Scientific. Dr. Kar reported receiving research grants from and serving as a consultant to that company as well as Abbott Vascular.
Simultaneously with Dr. Kar’s presentation at TCT 2017, the findings were published online in the Journal of the American College of Cardiology.
SOURCE: Reddy VY et al. TCT 2017; J Am Coll Cardiol. 2017 Nov 4. pii:S0735-1097(17)41187-9. doi: 10.1016/j.jacc.2017.10.021.
REPORTING FROM TCT 2017
Key clinical point:
Major finding: All-cause mortality was reduced by 27% in patients randomized to left atrial appendage closure with the Watchman device, compared with those assigned to warfarin.
Study details: This patient-level meta-analysis included 5-year follow-up data on 1,114 patients with atrial fibrillation at increased stroke risk who were randomized 2:1 to the Watchman device or warfarin.
Disclosures: The study presenter reported receiving research grants from and serving as a consultant to study sponsor Boston Scientific.

MRI-guided neurofeedback improves ADHD long term in adolescent boys
PARIS – Neurofeedback based upon real-time functional magnetic resonance imaging resulted in long-term reduction in attention-deficit/hyperactivity disorder symptoms in adolescents in a randomized controlled proof-of-concept study, Katya Rubia, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
The effect size of the improvement when measured at follow-up 11 months after completing the functional MRI-based neurofeedback (fMRI-NF) training exercises was moderate to large and comparable to that of psychostimulant medication in published placebo-controlled clinical trials. But the effects of the medications last only 24 hours after administration, and the drugs have side effects.

Neurofeedback is an operant conditioning procedure, which, through trial and error, teaches patients to self-regulate specific areas of the brain involved in psychopathology. EEG-based neurofeedback for ADHD has been extensively studied, with generally small to medium effect sizes being reported. Morever, patients need to be very highly motivated in order to succeed at EEG-NF: It takes 30-40 EEG-NF sessions, each an hour long, in order to learn targeted brain self-control in ADHD, whereas in Dr. Rubia’s study, patients learned to self-regulate brain activity in an average of eight fMRI sessions, each lasting 8.5 minutes, over the course of 2 weeks. The far speedier learning curve is probably tied to the superior specificity of spatial localization afforded by fMRI neurofeedback, according to the neuroscientist.
Also, fMRI-NF can reach certain key regions of the brain involved in ADHD that EEG-NF cannot, most notably the inferior frontal cortex (IFC) and basal ganglia, she added.
The target region in the proof-of-concept study was the right IFC, an area important for cognitive control, attention, and timing. Functional neuroimaging studies consistently have shown that the right IFC is underactive in ADHD, and that psychostimulant medications upregulate this area. A dysfunctional right IFC is an ADHD-specific abnormality not present in children with obessive-compulsive disorder (JAMA Psychiatry. 2016 Aug 1;73[8]:815-25), conduct disorder, or autism.
“The IFC seems to be a very good functional biomarker for ADHD,” Dr. Rubia said.
The proof-of-concept study, published in Human Brain Mapping, included 31 boys with a DSM-5 diagnosis of ADHD, aged 12-17, who were randomized to fMRI-NF of the right IFC or, as a control condition, to fMRI-NF targeting the left parahippocampal gyrus. Two patients had the inattentive subtype of ADHD; the rest had the combined hyperactive/inattentive form. Parents and patients were blinded as to their study arm.
So this program uses neuroimaging as a treatment. It is neuroimaging employed as neurotherapy. To make the training experience more attractive to young patients, it was presented as a computer game: By making progress in controlling their brain activity, patients could launch a rocket ship on the screen. With further progress, they could send the rocket through the atmosphere into space and eventually land it on another planet.
The primary study endpoint was change in the ADHD Rating Scale. The group that targeted self-upregulation of right IFC activity showed roughly a 20% improvement in scores, from a baseline mean total score of 36.7 to 30.2 immediately post treatment, further improving to a score of 26.7 at roughly 11 months of follow-up. Mean scores on the inattention subscale improved from 19.8 to 15.9 immediately post treatment and 15.3 at follow-up. Scores on the hyperactivity/impulsivity subscale went from 16.9 before treatment to 14.2 after treatment and 11.5 at follow-up.
There were no side effects of fMRI-NF in either study arm.
However, a degree of uncertainty exists regarding the clinical significance of the results, Dr. Rubia said. That’s because the control group showed a similar degree of improvement in ADHD symptoms immediately after learning to upregulate the left parahippocampal gyrus, although their scores did backslide modestly during 11 months of follow-up, while the IFC group continued to improve.
Dr. Rubia acknowledged that this raises the possibility that the observed improvement in clinical symptoms achieved through fMRI-NF could be attributable to a placebo effect. However, she said she believes this is unlikely for several reasons. For one, brain scans showed that targeting either the right IFC or the left parahippocampal gyrus not only resulted in upregulation of activity in those specific regions, but throughout the broader neural networks of which they are a part. The right IFC upregulators showed activation of a bilateral dorsolateral prefrontal cortex/IFC-insular-striato-cerebellar cognitive control network. In contrast, the boys who targeted the left parahippocampal gyrus experienced activation of associated posterior visual-spatial attention regions, which are relevant to ADHD. This made for a far from ideal control group.
Also, the amount of improvement in ADHD symptoms in the right IFC-targeted group correlated with the degree of activation of that region, indicative of a brain-behavior correlation that speaks against a nonspecific effect.
Because this was a small, unpowered pilot study and interest remains intense in potential nonpharmacologic treatments for ADHD, the U.K. Medical Research Council is funding Dr. Rubia and her colleagues for a new 100-patient study – including a sham fMRI-NF arm – in order to definitively address the possibility of a placebo effect. The study also will attempt to pin down the patient population most likely to benefit from fMRI-NF. “It’s possible that the inattentive subtype of ADHD will respond best. Neurofeedback is, after all, a form of attention training,” she noted.
While real-time fMRI-NF might sound prohibitively expensive for widespread use in clinical practice for a disorder as common as ADHD, which has an estimated prevalence of about 7%, it might actually stack up reasonably well in a cost-benefit analysis, compared with ongoing medication costs and side effects or with a year’s worth of weekly psychotherapy, according to Dr. Rubia.
In parallel with the ongoing sham-controlled fMRI-NF study, Dr. Rubia also is conducting a clinical trial of transcranial direct current stimulation of the right IFC in combination with cognitive training. The idea is to study the clinical impact of directly upregulating activity in this area of the brain, bypassing the added step of training patients to gain self-control over this dysregulated region. The early findings, she said, look promising.
The fMRI-NF study (Hum Brain Mapp. 2017 Jun;38[6]:3190-209) was sponsored by the U.K. National Institute for Health Research and the Maudsley NHS Foundation Trust. Dr. Rubia reported receiving speakers honoraria from Lilly, Shire, and Medice.
Source: Rubia K et al. European College of Neuropsychopharmacology.
PARIS – Neurofeedback based upon real-time functional magnetic resonance imaging resulted in long-term reduction in attention-deficit/hyperactivity disorder symptoms in adolescents in a randomized controlled proof-of-concept study, Katya Rubia, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
The effect size of the improvement when measured at follow-up 11 months after completing the functional MRI-based neurofeedback (fMRI-NF) training exercises was moderate to large and comparable to that of psychostimulant medication in published placebo-controlled clinical trials. But the effects of the medications last only 24 hours after administration, and the drugs have side effects.
Neurofeedback is an operant conditioning procedure, which, through trial and error, teaches patients to self-regulate specific areas of the brain involved in psychopathology. EEG-based neurofeedback for ADHD has been extensively studied, with generally small to medium effect sizes being reported. Morever, patients need to be very highly motivated in order to succeed at EEG-NF: It takes 30-40 EEG-NF sessions, each an hour long, in order to learn targeted brain self-control in ADHD, whereas in Dr. Rubia’s study, patients learned to self-regulate brain activity in an average of eight fMRI sessions, each lasting 8.5 minutes, over the course of 2 weeks. The far speedier learning curve is probably tied to the superior specificity of spatial localization afforded by fMRI neurofeedback, according to the neuroscientist.
Also, fMRI-NF can reach certain key regions of the brain involved in ADHD that EEG-NF cannot, most notably the inferior frontal cortex (IFC) and basal ganglia, she added.
The target region in the proof-of-concept study was the right IFC, an area important for cognitive control, attention, and timing. Functional neuroimaging studies consistently have shown that the right IFC is underactive in ADHD, and that psychostimulant medications upregulate this area. A dysfunctional right IFC is an ADHD-specific abnormality not present in children with obessive-compulsive disorder (JAMA Psychiatry. 2016 Aug 1;73[8]:815-25), conduct disorder, or autism.
“The IFC seems to be a very good functional biomarker for ADHD,” Dr. Rubia said.
The proof-of-concept study, published in Human Brain Mapping, included 31 boys with a DSM-5 diagnosis of ADHD, aged 12-17, who were randomized to fMRI-NF of the right IFC or, as a control condition, to fMRI-NF targeting the left parahippocampal gyrus. Two patients had the inattentive subtype of ADHD; the rest had the combined hyperactive/inattentive form. Parents and patients were blinded as to their study arm.
So this program uses neuroimaging as a treatment. It is neuroimaging employed as neurotherapy. To make the training experience more attractive to young patients, it was presented as a computer game: By making progress in controlling their brain activity, patients could launch a rocket ship on the screen. With further progress, they could send the rocket through the atmosphere into space and eventually land it on another planet.
The primary study endpoint was change in the ADHD Rating Scale. The group that targeted self-upregulation of right IFC activity showed roughly a 20% improvement in scores, from a baseline mean total score of 36.7 to 30.2 immediately post treatment, further improving to a score of 26.7 at roughly 11 months of follow-up. Mean scores on the inattention subscale improved from 19.8 to 15.9 immediately post treatment and 15.3 at follow-up. Scores on the hyperactivity/impulsivity subscale went from 16.9 before treatment to 14.2 after treatment and 11.5 at follow-up.
There were no side effects of fMRI-NF in either study arm.
However, a degree of uncertainty exists regarding the clinical significance of the results, Dr. Rubia said. That’s because the control group showed a similar degree of improvement in ADHD symptoms immediately after learning to upregulate the left parahippocampal gyrus, although their scores did backslide modestly during 11 months of follow-up, while the IFC group continued to improve.
Dr. Rubia acknowledged that this raises the possibility that the observed improvement in clinical symptoms achieved through fMRI-NF could be attributable to a placebo effect. However, she said she believes this is unlikely for several reasons. For one, brain scans showed that targeting either the right IFC or the left parahippocampal gyrus not only resulted in upregulation of activity in those specific regions, but throughout the broader neural networks of which they are a part. The right IFC upregulators showed activation of a bilateral dorsolateral prefrontal cortex/IFC-insular-striato-cerebellar cognitive control network. In contrast, the boys who targeted the left parahippocampal gyrus experienced activation of associated posterior visual-spatial attention regions, which are relevant to ADHD. This made for a far from ideal control group.
Also, the amount of improvement in ADHD symptoms in the right IFC-targeted group correlated with the degree of activation of that region, indicative of a brain-behavior correlation that speaks against a nonspecific effect.
Because this was a small, unpowered pilot study and interest remains intense in potential nonpharmacologic treatments for ADHD, the U.K. Medical Research Council is funding Dr. Rubia and her colleagues for a new 100-patient study – including a sham fMRI-NF arm – in order to definitively address the possibility of a placebo effect. The study also will attempt to pin down the patient population most likely to benefit from fMRI-NF. “It’s possible that the inattentive subtype of ADHD will respond best. Neurofeedback is, after all, a form of attention training,” she noted.
While real-time fMRI-NF might sound prohibitively expensive for widespread use in clinical practice for a disorder as common as ADHD, which has an estimated prevalence of about 7%, it might actually stack up reasonably well in a cost-benefit analysis, compared with ongoing medication costs and side effects or with a year’s worth of weekly psychotherapy, according to Dr. Rubia.
In parallel with the ongoing sham-controlled fMRI-NF study, Dr. Rubia also is conducting a clinical trial of transcranial direct current stimulation of the right IFC in combination with cognitive training. The idea is to study the clinical impact of directly upregulating activity in this area of the brain, bypassing the added step of training patients to gain self-control over this dysregulated region. The early findings, she said, look promising.
The fMRI-NF study (Hum Brain Mapp. 2017 Jun;38[6]:3190-209) was sponsored by the U.K. National Institute for Health Research and the Maudsley NHS Foundation Trust. Dr. Rubia reported receiving speakers honoraria from Lilly, Shire, and Medice.
Source: Rubia K et al. European College of Neuropsychopharmacology.
PARIS – Neurofeedback based upon real-time functional magnetic resonance imaging resulted in long-term reduction in attention-deficit/hyperactivity disorder symptoms in adolescents in a randomized controlled proof-of-concept study, Katya Rubia, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
The effect size of the improvement when measured at follow-up 11 months after completing the functional MRI-based neurofeedback (fMRI-NF) training exercises was moderate to large and comparable to that of psychostimulant medication in published placebo-controlled clinical trials. But the effects of the medications last only 24 hours after administration, and the drugs have side effects.
Neurofeedback is an operant conditioning procedure, which, through trial and error, teaches patients to self-regulate specific areas of the brain involved in psychopathology. EEG-based neurofeedback for ADHD has been extensively studied, with generally small to medium effect sizes being reported. Morever, patients need to be very highly motivated in order to succeed at EEG-NF: It takes 30-40 EEG-NF sessions, each an hour long, in order to learn targeted brain self-control in ADHD, whereas in Dr. Rubia’s study, patients learned to self-regulate brain activity in an average of eight fMRI sessions, each lasting 8.5 minutes, over the course of 2 weeks. The far speedier learning curve is probably tied to the superior specificity of spatial localization afforded by fMRI neurofeedback, according to the neuroscientist.
Also, fMRI-NF can reach certain key regions of the brain involved in ADHD that EEG-NF cannot, most notably the inferior frontal cortex (IFC) and basal ganglia, she added.
The target region in the proof-of-concept study was the right IFC, an area important for cognitive control, attention, and timing. Functional neuroimaging studies consistently have shown that the right IFC is underactive in ADHD, and that psychostimulant medications upregulate this area. A dysfunctional right IFC is an ADHD-specific abnormality not present in children with obessive-compulsive disorder (JAMA Psychiatry. 2016 Aug 1;73[8]:815-25), conduct disorder, or autism.
“The IFC seems to be a very good functional biomarker for ADHD,” Dr. Rubia said.
The proof-of-concept study, published in Human Brain Mapping, included 31 boys with a DSM-5 diagnosis of ADHD, aged 12-17, who were randomized to fMRI-NF of the right IFC or, as a control condition, to fMRI-NF targeting the left parahippocampal gyrus. Two patients had the inattentive subtype of ADHD; the rest had the combined hyperactive/inattentive form. Parents and patients were blinded as to their study arm.
So this program uses neuroimaging as a treatment. It is neuroimaging employed as neurotherapy. To make the training experience more attractive to young patients, it was presented as a computer game: By making progress in controlling their brain activity, patients could launch a rocket ship on the screen. With further progress, they could send the rocket through the atmosphere into space and eventually land it on another planet.
The primary study endpoint was change in the ADHD Rating Scale. The group that targeted self-upregulation of right IFC activity showed roughly a 20% improvement in scores, from a baseline mean total score of 36.7 to 30.2 immediately post treatment, further improving to a score of 26.7 at roughly 11 months of follow-up. Mean scores on the inattention subscale improved from 19.8 to 15.9 immediately post treatment and 15.3 at follow-up. Scores on the hyperactivity/impulsivity subscale went from 16.9 before treatment to 14.2 after treatment and 11.5 at follow-up.
There were no side effects of fMRI-NF in either study arm.
However, a degree of uncertainty exists regarding the clinical significance of the results, Dr. Rubia said. That’s because the control group showed a similar degree of improvement in ADHD symptoms immediately after learning to upregulate the left parahippocampal gyrus, although their scores did backslide modestly during 11 months of follow-up, while the IFC group continued to improve.
Dr. Rubia acknowledged that this raises the possibility that the observed improvement in clinical symptoms achieved through fMRI-NF could be attributable to a placebo effect. However, she said she believes this is unlikely for several reasons. For one, brain scans showed that targeting either the right IFC or the left parahippocampal gyrus not only resulted in upregulation of activity in those specific regions, but throughout the broader neural networks of which they are a part. The right IFC upregulators showed activation of a bilateral dorsolateral prefrontal cortex/IFC-insular-striato-cerebellar cognitive control network. In contrast, the boys who targeted the left parahippocampal gyrus experienced activation of associated posterior visual-spatial attention regions, which are relevant to ADHD. This made for a far from ideal control group.
Also, the amount of improvement in ADHD symptoms in the right IFC-targeted group correlated with the degree of activation of that region, indicative of a brain-behavior correlation that speaks against a nonspecific effect.
Because this was a small, unpowered pilot study and interest remains intense in potential nonpharmacologic treatments for ADHD, the U.K. Medical Research Council is funding Dr. Rubia and her colleagues for a new 100-patient study – including a sham fMRI-NF arm – in order to definitively address the possibility of a placebo effect. The study also will attempt to pin down the patient population most likely to benefit from fMRI-NF. “It’s possible that the inattentive subtype of ADHD will respond best. Neurofeedback is, after all, a form of attention training,” she noted.
While real-time fMRI-NF might sound prohibitively expensive for widespread use in clinical practice for a disorder as common as ADHD, which has an estimated prevalence of about 7%, it might actually stack up reasonably well in a cost-benefit analysis, compared with ongoing medication costs and side effects or with a year’s worth of weekly psychotherapy, according to Dr. Rubia.
In parallel with the ongoing sham-controlled fMRI-NF study, Dr. Rubia also is conducting a clinical trial of transcranial direct current stimulation of the right IFC in combination with cognitive training. The idea is to study the clinical impact of directly upregulating activity in this area of the brain, bypassing the added step of training patients to gain self-control over this dysregulated region. The early findings, she said, look promising.
The fMRI-NF study (Hum Brain Mapp. 2017 Jun;38[6]:3190-209) was sponsored by the U.K. National Institute for Health Research and the Maudsley NHS Foundation Trust. Dr. Rubia reported receiving speakers honoraria from Lilly, Shire, and Medice.
Source: Rubia K et al. European College of Neuropsychopharmacology.
REPORTING FROM THE ECNP CONGRESS
Key clinical point: Neuroimaging can be employed as neurotherapy to improve ADHD nonpharmacologically.
Major finding: Adolescents with ADHD who learned via functional MRI neurofeedback to upregulate activity in their right inferior frontal cortex showed significant improvement in scores on the ADHD Rating Scale, from a baseline mean total score of 36.7 to 30.2 immediately after the training program, further improving to 26.7 at roughly 11 months of follow-up.
Study details: A prospective, randomized, single-blind study of 31 boys aged 12-17 with ADHD.
Disclosures: The study was sponsored by the U.K. National Institute for Health Research and the Maudsley NHS Foundation Trust. The presenter reported receiving speakers honoraria from Lilly, Shire, and Medice.
Source: Rubia K et al. European College of Neuropsychopharmacology.
FFR-guided PCI in stable CAD beats medical management
DENVER – Percutaneous coronary intervention plus optimal medical therapy in stable coronary artery disease (CAD) patients with at least one coronary lesion having an abnormal fractional flow reserve measurement resulted in superior clinical outcomes, better quality of life, and virtually identical cost, compared with optimal medical management alone over 3 years of follow-up in the FAME 2 trial.
“These results reinforce the point that the greater the burden of ischemia, the greater the benefit of revascularization with PCI [percutaneous coronary intervention],” William F. Fearon, MD, said while presenting the FAME 2 findings at the Transcatheter Cardiovascular Therapeutics annual meeting.

FAME 2 was a randomized, multicenter trial designed to help bring clarity regarding the optimal treatment strategy for patients with stable angina and CAD. This is an issue surrounded by considerable controversy. The fog descended a decade ago, when the COURAGE trial created a stir with its conclusion that optimal medical therapy (OMT) alone was as good as PCI plus OMT in terms of clinical outcome and quality of life – and was considerably less expensive as well. And the British ORBITA trial, presented earlier in the same session at TCT 2017 as Dr. Fearon’s report on FAME 2, caused an uproar with its finding that PCI plus OMT was no more effective than sham PCI plus OMT in the setting of stable CAD.
However, FAME 2 (Fractional Flow Reserve versus Angiography for Multivessel Evaluation) differed from those studies in a crucial aspect: randomization in FAME 2 was restricted to patients with physiologically significant cardiac ischemia as evidenced by a fractional flow reserve (FFR) measurement of 0.80 or less.
In contrast, COURAGE and ORBITA randomized patients without FFR guidance. As a result, in those trials PCI was performed in a substantial proportion of patients who actually should not have undergone the intervention because they didn’t have physiologic evidence of clinically important ischemia. The non–physiologically based approach to PCI utilized in COURAGE and ORBITA – disappointingly commonplace in daily clinical practice – diluted any true benefit of the procedure when applied appropriately, explained Dr. Fearon, professor of medicine and director of interventional cardiology at Stanford (Calif.) University.
FAME 2 randomized 888 patients with stable single- or multivessel CAD and an FFR of 0.80 or less to PCI plus OMT or an initial strategy of OMT alone at 28 European and North American sites. The primary outcome was the rate of major adverse cardiac events – death, MI, and urgent revascularization – at 3 years. The rate was 10.1% in the PCI group, compared with 22% in the medically managed cohort, Dr. Fearon reported at the meeting sponsored by the Cardiovascular Research Foundation.
Death or MI occurred in 8.3% of the PCI group versus 10.4% in the OMT group, a trend that didn’t reach significance. Of note, however, fully 44% of patients in the OMT group crossed over to PCI during the 3-year study. In the prespecified intent-to-treat analysis they were counted in the OMT group, whereas an as-treated analysis might well have shown statistically significant reductions in death and MI in the PCI group.
The proportion of patients with class II-IV angina was significantly lower in the PCI plus medical therapy group at every time point, including 5.9% versus 15.2% for OMT alone at 1 year, 5.9% versus 12% at 2 years, and 5.2% versus 9.7% at 3 years. This was the case even though the OMT group received significantly more antianginal therapy in an effort to control symptoms.
FAME 2 featured a first-of-its-kind comprehensive cost-effectiveness analysis of OMT vs. PCI over a 3-year period. It showed that, while mean initial costs were as expected higher in the PCI group ($9,944 versus $4,440), by 3 years the cumulative costs were near identical at $16,792 in the PCI group and $16,737 in the initial OMT group. The incremental cost-effectiveness ratio for PCI, compared with OMT at 3 years was attractive at $1,600 per quality-adjusted life-year gained.
The question on TCT attendees’ minds following presentation of the bombshell ORBITA findings was, what would have happened had FAME 2 featured a sham PCI arm? Could the advantageous outcomes for the initial PCI strategy seen in FAME 2 possibly have been due to a placebo effect?
Extremely unlikely, according to Dr. Fearon. For one thing, when he and his coinvestigators broke down the FFR values in the OMT group into quintiles, they saw a clear dose-response effect: The clinical event rate rose further with worsening quintile of FFR. Also, the study endpoints were death, MI, and urgent revascularization – triggered by ACS in half of cases – which are less susceptible to a placebo effect than, say, treadmill exercise time, the primary endpoint in ORBITA.
Moreover, as noted by Gary S. Mintz, MD, who moderated a press conference highlighting the ORBITA and FAME 2 results, placebo effects don’t last for years.
“Most people would say the placebo effect wanes over time. That’s why these 3-year data, analyzed by intent-to-treat, which allow for crossovers to still be analyzed in the medical therapy arm, are pretty compelling to me,” commented Dr. Mintz, chief medical officer for the Cardiovascular Research Foundation in New York.
FAME 2 was supported by St. Jude Medical. Dr. Fearon reported receiving institutional research support from Medtronic, Abbott Vascular, ACIST Medical, CathWorks, and Edwards LifeSciences.
DENVER – Percutaneous coronary intervention plus optimal medical therapy in stable coronary artery disease (CAD) patients with at least one coronary lesion having an abnormal fractional flow reserve measurement resulted in superior clinical outcomes, better quality of life, and virtually identical cost, compared with optimal medical management alone over 3 years of follow-up in the FAME 2 trial.
“These results reinforce the point that the greater the burden of ischemia, the greater the benefit of revascularization with PCI [percutaneous coronary intervention],” William F. Fearon, MD, said while presenting the FAME 2 findings at the Transcatheter Cardiovascular Therapeutics annual meeting.
FAME 2 was a randomized, multicenter trial designed to help bring clarity regarding the optimal treatment strategy for patients with stable angina and CAD. This is an issue surrounded by considerable controversy. The fog descended a decade ago, when the COURAGE trial created a stir with its conclusion that optimal medical therapy (OMT) alone was as good as PCI plus OMT in terms of clinical outcome and quality of life – and was considerably less expensive as well. And the British ORBITA trial, presented earlier in the same session at TCT 2017 as Dr. Fearon’s report on FAME 2, caused an uproar with its finding that PCI plus OMT was no more effective than sham PCI plus OMT in the setting of stable CAD.
However, FAME 2 (Fractional Flow Reserve versus Angiography for Multivessel Evaluation) differed from those studies in a crucial aspect: randomization in FAME 2 was restricted to patients with physiologically significant cardiac ischemia as evidenced by a fractional flow reserve (FFR) measurement of 0.80 or less.
In contrast, COURAGE and ORBITA randomized patients without FFR guidance. As a result, in those trials PCI was performed in a substantial proportion of patients who actually should not have undergone the intervention because they didn’t have physiologic evidence of clinically important ischemia. The non–physiologically based approach to PCI utilized in COURAGE and ORBITA – disappointingly commonplace in daily clinical practice – diluted any true benefit of the procedure when applied appropriately, explained Dr. Fearon, professor of medicine and director of interventional cardiology at Stanford (Calif.) University.
FAME 2 randomized 888 patients with stable single- or multivessel CAD and an FFR of 0.80 or less to PCI plus OMT or an initial strategy of OMT alone at 28 European and North American sites. The primary outcome was the rate of major adverse cardiac events – death, MI, and urgent revascularization – at 3 years. The rate was 10.1% in the PCI group, compared with 22% in the medically managed cohort, Dr. Fearon reported at the meeting sponsored by the Cardiovascular Research Foundation.
Death or MI occurred in 8.3% of the PCI group versus 10.4% in the OMT group, a trend that didn’t reach significance. Of note, however, fully 44% of patients in the OMT group crossed over to PCI during the 3-year study. In the prespecified intent-to-treat analysis they were counted in the OMT group, whereas an as-treated analysis might well have shown statistically significant reductions in death and MI in the PCI group.
The proportion of patients with class II-IV angina was significantly lower in the PCI plus medical therapy group at every time point, including 5.9% versus 15.2% for OMT alone at 1 year, 5.9% versus 12% at 2 years, and 5.2% versus 9.7% at 3 years. This was the case even though the OMT group received significantly more antianginal therapy in an effort to control symptoms.
FAME 2 featured a first-of-its-kind comprehensive cost-effectiveness analysis of OMT vs. PCI over a 3-year period. It showed that, while mean initial costs were as expected higher in the PCI group ($9,944 versus $4,440), by 3 years the cumulative costs were near identical at $16,792 in the PCI group and $16,737 in the initial OMT group. The incremental cost-effectiveness ratio for PCI, compared with OMT at 3 years was attractive at $1,600 per quality-adjusted life-year gained.
The question on TCT attendees’ minds following presentation of the bombshell ORBITA findings was, what would have happened had FAME 2 featured a sham PCI arm? Could the advantageous outcomes for the initial PCI strategy seen in FAME 2 possibly have been due to a placebo effect?
Extremely unlikely, according to Dr. Fearon. For one thing, when he and his coinvestigators broke down the FFR values in the OMT group into quintiles, they saw a clear dose-response effect: The clinical event rate rose further with worsening quintile of FFR. Also, the study endpoints were death, MI, and urgent revascularization – triggered by ACS in half of cases – which are less susceptible to a placebo effect than, say, treadmill exercise time, the primary endpoint in ORBITA.
Moreover, as noted by Gary S. Mintz, MD, who moderated a press conference highlighting the ORBITA and FAME 2 results, placebo effects don’t last for years.
“Most people would say the placebo effect wanes over time. That’s why these 3-year data, analyzed by intent-to-treat, which allow for crossovers to still be analyzed in the medical therapy arm, are pretty compelling to me,” commented Dr. Mintz, chief medical officer for the Cardiovascular Research Foundation in New York.
FAME 2 was supported by St. Jude Medical. Dr. Fearon reported receiving institutional research support from Medtronic, Abbott Vascular, ACIST Medical, CathWorks, and Edwards LifeSciences.
DENVER – Percutaneous coronary intervention plus optimal medical therapy in stable coronary artery disease (CAD) patients with at least one coronary lesion having an abnormal fractional flow reserve measurement resulted in superior clinical outcomes, better quality of life, and virtually identical cost, compared with optimal medical management alone over 3 years of follow-up in the FAME 2 trial.
“These results reinforce the point that the greater the burden of ischemia, the greater the benefit of revascularization with PCI [percutaneous coronary intervention],” William F. Fearon, MD, said while presenting the FAME 2 findings at the Transcatheter Cardiovascular Therapeutics annual meeting.
FAME 2 was a randomized, multicenter trial designed to help bring clarity regarding the optimal treatment strategy for patients with stable angina and CAD. This is an issue surrounded by considerable controversy. The fog descended a decade ago, when the COURAGE trial created a stir with its conclusion that optimal medical therapy (OMT) alone was as good as PCI plus OMT in terms of clinical outcome and quality of life – and was considerably less expensive as well. And the British ORBITA trial, presented earlier in the same session at TCT 2017 as Dr. Fearon’s report on FAME 2, caused an uproar with its finding that PCI plus OMT was no more effective than sham PCI plus OMT in the setting of stable CAD.
However, FAME 2 (Fractional Flow Reserve versus Angiography for Multivessel Evaluation) differed from those studies in a crucial aspect: randomization in FAME 2 was restricted to patients with physiologically significant cardiac ischemia as evidenced by a fractional flow reserve (FFR) measurement of 0.80 or less.
In contrast, COURAGE and ORBITA randomized patients without FFR guidance. As a result, in those trials PCI was performed in a substantial proportion of patients who actually should not have undergone the intervention because they didn’t have physiologic evidence of clinically important ischemia. The non–physiologically based approach to PCI utilized in COURAGE and ORBITA – disappointingly commonplace in daily clinical practice – diluted any true benefit of the procedure when applied appropriately, explained Dr. Fearon, professor of medicine and director of interventional cardiology at Stanford (Calif.) University.
FAME 2 randomized 888 patients with stable single- or multivessel CAD and an FFR of 0.80 or less to PCI plus OMT or an initial strategy of OMT alone at 28 European and North American sites. The primary outcome was the rate of major adverse cardiac events – death, MI, and urgent revascularization – at 3 years. The rate was 10.1% in the PCI group, compared with 22% in the medically managed cohort, Dr. Fearon reported at the meeting sponsored by the Cardiovascular Research Foundation.
Death or MI occurred in 8.3% of the PCI group versus 10.4% in the OMT group, a trend that didn’t reach significance. Of note, however, fully 44% of patients in the OMT group crossed over to PCI during the 3-year study. In the prespecified intent-to-treat analysis they were counted in the OMT group, whereas an as-treated analysis might well have shown statistically significant reductions in death and MI in the PCI group.
The proportion of patients with class II-IV angina was significantly lower in the PCI plus medical therapy group at every time point, including 5.9% versus 15.2% for OMT alone at 1 year, 5.9% versus 12% at 2 years, and 5.2% versus 9.7% at 3 years. This was the case even though the OMT group received significantly more antianginal therapy in an effort to control symptoms.
FAME 2 featured a first-of-its-kind comprehensive cost-effectiveness analysis of OMT vs. PCI over a 3-year period. It showed that, while mean initial costs were as expected higher in the PCI group ($9,944 versus $4,440), by 3 years the cumulative costs were near identical at $16,792 in the PCI group and $16,737 in the initial OMT group. The incremental cost-effectiveness ratio for PCI, compared with OMT at 3 years was attractive at $1,600 per quality-adjusted life-year gained.
The question on TCT attendees’ minds following presentation of the bombshell ORBITA findings was, what would have happened had FAME 2 featured a sham PCI arm? Could the advantageous outcomes for the initial PCI strategy seen in FAME 2 possibly have been due to a placebo effect?
Extremely unlikely, according to Dr. Fearon. For one thing, when he and his coinvestigators broke down the FFR values in the OMT group into quintiles, they saw a clear dose-response effect: The clinical event rate rose further with worsening quintile of FFR. Also, the study endpoints were death, MI, and urgent revascularization – triggered by ACS in half of cases – which are less susceptible to a placebo effect than, say, treadmill exercise time, the primary endpoint in ORBITA.
Moreover, as noted by Gary S. Mintz, MD, who moderated a press conference highlighting the ORBITA and FAME 2 results, placebo effects don’t last for years.
“Most people would say the placebo effect wanes over time. That’s why these 3-year data, analyzed by intent-to-treat, which allow for crossovers to still be analyzed in the medical therapy arm, are pretty compelling to me,” commented Dr. Mintz, chief medical officer for the Cardiovascular Research Foundation in New York.
FAME 2 was supported by St. Jude Medical. Dr. Fearon reported receiving institutional research support from Medtronic, Abbott Vascular, ACIST Medical, CathWorks, and Edwards LifeSciences.
REPORTING FROM TCT 2017
Key clinical point:
Major finding: The rate of major adverse cardiac events at 3 years was 10.1% in the PCI group and 22% in the medically managed cohort.
Data source: The FAME 2 trial randomized 888 patients with stable single- or multivessel CAD and an FFR of 0.80 or less to PCI plus optimal medical therapy or an initial strategy of optimal medical management alone.
Disclosures: The trial was supported by St. Jude Medical. The presenter reported receiving institutional research support from Medtronic, Abbott Vascular, ACIST Medical, CathWorks, and Edwards LifeSciences.
EVAR, venous CPT coding revamped for 2018
CHICAGO – Current Procedural Terminology coding for endovascular aneurysm repair has been totally overhauled for 2018 with the introduction of a family of 20 new codes and codes for other vascular procedures have also been updated.
The new EVAR CPT codes attempt to capture the work involved in performing the procedures based upon the anatomy of the aneurysm and the treated vessels rather than being device-based, as previously, Matthew J. Sideman, MD, explained in presenting the coding and reimbursement for 2018 at a symposium on vascular surgery sponsored by Northwestern University.

“The new EVAR codes for 2018 have got a lot of gains. There are some losses as well, but overall, I think it’s going to be very positive moving forward,” according to Dr. Sideman, a vascular surgeon at the University of Texas, San Antonio, who serves as chair of the Society for Vascular Surgery Coding and Reimbursement Committee and an adviser to the American Medical Association Relative Value Scale Update Committee (RUC).
“What we gained was a new code for ruptured aneurysm repair, a new code for enhanced fixation, a new code for percutaneous access, new codes for alternative access options, and now all the access codes are add-on codes. But what we traded off was loss due to bundling. So catheterization is now bundled into the main procedure, radiographic supervision and interpretation is now bundled. The big thing that really hurt was we lost all proximal extensions to the renal arteries and all distal extensions to the iliac bifurcations – they’re also bundled into the main procedure,” he said.
Restructuring the EVAR codes was a multiyear collaborative project of the SVS, the American College of Surgeons, the Society of Interventional Radiology, the Society of Thoracic Surgery, the American College of Cardiology, and the Society for Cardiovascular Angiography and Interventions. The impetus was twofold: recognition that the existing codes seriously undervalued the work involved in EVAR because, for example, they didn’t distinguish between ruptured and elective aneurysm repair, nor did they recognize the unique challenges and advantages of percutaneous access.
Also, representatives of the professional societies involved with vascular medicine recognized that they had to develop a detailed proposal for coding restructuring or matters might be taken out of their hands. Bundling of codes has become the prevailing dogma at the RUC and the Centers for Medicare and Medicaid. Their current policy is that when analysis of coding patterns indicates two codes are billed together at least 51% of the time, that’s considered a ‘typical’ situation and a new code must be created combining them. The harsh reality for clinicians is that under what Dr. Sideman called “RUC math,” the new bundled codes invariably pay less than the two old ones.
“There was a little bit of smoke and mirrors – ‘Look at the pretty flashing lights and not what’s going on behind over here’ – as we tried to maintain value as we bundled these EVAR codes,” Dr. Sideman recalled. “I can stand here and tell you I did my very best to push for the best values possible. It can be a painful process, but I thought we came out ok.”
How the new EVAR codes work
Dr. Sideman explained that the impact of the new EVAR codes will depend upon a surgeon’s practice pattern.
He offered as a concrete example a patient undergoing elective EVAR of the aorta and both iliac arteries with percutaneous access and placement of a bifurcated device with one docking limb. In 2017, this might have been handled using CPT codes 34802, 36200-50, and 75952-26, for a total of 31.05 Relative Value Units (RVUs) of work.
In 2018, however, this same surgical strategy would be coded as 34705 (elective endovascular repair of infrarenal aorta and/or iliac artery or arteries) plus 34713 x 2 (percutaneous access and closure), for a total of 34.58 RVUs. Thus, the surgeon would come out 3.53 RVUs ahead in 2018, which at a conversion factor of $35.78/RVU translates to an extra $126.30.
On the other hand, if the surgeon chose to use a bifurcated device with one docking limb, a left iliac bell-bottom extension, a right iliac bell-bottom extension, and percutaneous access, in 2017, this would have been coded as 34802, 34825, 34826, 36200-50, 75952-26, and 75953-26 x 2, for a total of 44.29 RVUs of work. In 2018, this same treatment strategy would be coded as 34705 plus 34713 x 2, for a total of 34.58 RVUs, or a knockdown of 9.71 fewer RVUs compared with the year before, which translates to $347.42 less.
“The more extensions you use, the more you’re going to come out behind going forward,” according to Dr. Sideman.
Other coding changes in 2018
Sclerotherapy of single and multiple veins (codes 36470 and 36471) got down-valued from 1.10 and 2.49 to 0.75 and 1.5 RVUs, respectively.
Angiography of the extremities (75710 and 75716) will be better reimbursed in 2018. In what Dr. Sideman called “a good win,” unilateral angiography will be rated as 1.75 RVUs, up from 1.14 in 2017, while bilateral angiography increased from 1.31 to 1.97 RVUs.
“The other nice thing I can tell you is that through campaigning and lobbying and comments to CMS [Centers for Medicare & Medicaid Services], we got them to reverse their recommendations from 2017 to 2018 on the dialysis family of codes,” the surgeon continued.
Reimbursement for the dialysis codes took a big hit from 2016 to 2017, amounting to several hundred million dollars less in reimbursement, but CMS has reversed its policy on that score. The RVUs for the various dialysis codes have increased from 2017 to 2018 by 5%-21%, with central venous angioplasty (CPT 36907) garnering the biggest increase.
Existing RVUs were retained for 2018 in three of the four selective catheter placement codes. However, reimbursement for 36215 (first order catheterization of the thoracic or brachiocephalic branch) dropped from 4.67 to 4.17 RVUs because physician surveys showed the time involved was less than previously rated. Once the RUC and CMS saw that the time involved in a procedure has decreased, it became impossible to maintain the RVU, Dr. Sideman explained.
And speaking of time involved in procedures, Dr. Sideman offered a final plea to his vascular medicine colleagues:
“When you get surveys from the RUC asking for your input, please, please, please, fill them out because that’s how we get our direct physician input into the valuation of codes.”
He reported having no financial conflicts of interest regarding his presentation.
A detailed listing of many of the codes and changes can be found at the American College of Radiology website, and the Society for Vascular Surgery has coding resources available on their website, as well.
CHICAGO – Current Procedural Terminology coding for endovascular aneurysm repair has been totally overhauled for 2018 with the introduction of a family of 20 new codes and codes for other vascular procedures have also been updated.
The new EVAR CPT codes attempt to capture the work involved in performing the procedures based upon the anatomy of the aneurysm and the treated vessels rather than being device-based, as previously, Matthew J. Sideman, MD, explained in presenting the coding and reimbursement for 2018 at a symposium on vascular surgery sponsored by Northwestern University.
“The new EVAR codes for 2018 have got a lot of gains. There are some losses as well, but overall, I think it’s going to be very positive moving forward,” according to Dr. Sideman, a vascular surgeon at the University of Texas, San Antonio, who serves as chair of the Society for Vascular Surgery Coding and Reimbursement Committee and an adviser to the American Medical Association Relative Value Scale Update Committee (RUC).
“What we gained was a new code for ruptured aneurysm repair, a new code for enhanced fixation, a new code for percutaneous access, new codes for alternative access options, and now all the access codes are add-on codes. But what we traded off was loss due to bundling. So catheterization is now bundled into the main procedure, radiographic supervision and interpretation is now bundled. The big thing that really hurt was we lost all proximal extensions to the renal arteries and all distal extensions to the iliac bifurcations – they’re also bundled into the main procedure,” he said.
Restructuring the EVAR codes was a multiyear collaborative project of the SVS, the American College of Surgeons, the Society of Interventional Radiology, the Society of Thoracic Surgery, the American College of Cardiology, and the Society for Cardiovascular Angiography and Interventions. The impetus was twofold: recognition that the existing codes seriously undervalued the work involved in EVAR because, for example, they didn’t distinguish between ruptured and elective aneurysm repair, nor did they recognize the unique challenges and advantages of percutaneous access.
Also, representatives of the professional societies involved with vascular medicine recognized that they had to develop a detailed proposal for coding restructuring or matters might be taken out of their hands. Bundling of codes has become the prevailing dogma at the RUC and the Centers for Medicare and Medicaid. Their current policy is that when analysis of coding patterns indicates two codes are billed together at least 51% of the time, that’s considered a ‘typical’ situation and a new code must be created combining them. The harsh reality for clinicians is that under what Dr. Sideman called “RUC math,” the new bundled codes invariably pay less than the two old ones.
“There was a little bit of smoke and mirrors – ‘Look at the pretty flashing lights and not what’s going on behind over here’ – as we tried to maintain value as we bundled these EVAR codes,” Dr. Sideman recalled. “I can stand here and tell you I did my very best to push for the best values possible. It can be a painful process, but I thought we came out ok.”
How the new EVAR codes work
Dr. Sideman explained that the impact of the new EVAR codes will depend upon a surgeon’s practice pattern.
He offered as a concrete example a patient undergoing elective EVAR of the aorta and both iliac arteries with percutaneous access and placement of a bifurcated device with one docking limb. In 2017, this might have been handled using CPT codes 34802, 36200-50, and 75952-26, for a total of 31.05 Relative Value Units (RVUs) of work.
In 2018, however, this same surgical strategy would be coded as 34705 (elective endovascular repair of infrarenal aorta and/or iliac artery or arteries) plus 34713 x 2 (percutaneous access and closure), for a total of 34.58 RVUs. Thus, the surgeon would come out 3.53 RVUs ahead in 2018, which at a conversion factor of $35.78/RVU translates to an extra $126.30.
On the other hand, if the surgeon chose to use a bifurcated device with one docking limb, a left iliac bell-bottom extension, a right iliac bell-bottom extension, and percutaneous access, in 2017, this would have been coded as 34802, 34825, 34826, 36200-50, 75952-26, and 75953-26 x 2, for a total of 44.29 RVUs of work. In 2018, this same treatment strategy would be coded as 34705 plus 34713 x 2, for a total of 34.58 RVUs, or a knockdown of 9.71 fewer RVUs compared with the year before, which translates to $347.42 less.
“The more extensions you use, the more you’re going to come out behind going forward,” according to Dr. Sideman.
Other coding changes in 2018
Sclerotherapy of single and multiple veins (codes 36470 and 36471) got down-valued from 1.10 and 2.49 to 0.75 and 1.5 RVUs, respectively.
Angiography of the extremities (75710 and 75716) will be better reimbursed in 2018. In what Dr. Sideman called “a good win,” unilateral angiography will be rated as 1.75 RVUs, up from 1.14 in 2017, while bilateral angiography increased from 1.31 to 1.97 RVUs.
“The other nice thing I can tell you is that through campaigning and lobbying and comments to CMS [Centers for Medicare & Medicaid Services], we got them to reverse their recommendations from 2017 to 2018 on the dialysis family of codes,” the surgeon continued.
Reimbursement for the dialysis codes took a big hit from 2016 to 2017, amounting to several hundred million dollars less in reimbursement, but CMS has reversed its policy on that score. The RVUs for the various dialysis codes have increased from 2017 to 2018 by 5%-21%, with central venous angioplasty (CPT 36907) garnering the biggest increase.
Existing RVUs were retained for 2018 in three of the four selective catheter placement codes. However, reimbursement for 36215 (first order catheterization of the thoracic or brachiocephalic branch) dropped from 4.67 to 4.17 RVUs because physician surveys showed the time involved was less than previously rated. Once the RUC and CMS saw that the time involved in a procedure has decreased, it became impossible to maintain the RVU, Dr. Sideman explained.
And speaking of time involved in procedures, Dr. Sideman offered a final plea to his vascular medicine colleagues:
“When you get surveys from the RUC asking for your input, please, please, please, fill them out because that’s how we get our direct physician input into the valuation of codes.”
He reported having no financial conflicts of interest regarding his presentation.
A detailed listing of many of the codes and changes can be found at the American College of Radiology website, and the Society for Vascular Surgery has coding resources available on their website, as well.
CHICAGO – Current Procedural Terminology coding for endovascular aneurysm repair has been totally overhauled for 2018 with the introduction of a family of 20 new codes and codes for other vascular procedures have also been updated.
The new EVAR CPT codes attempt to capture the work involved in performing the procedures based upon the anatomy of the aneurysm and the treated vessels rather than being device-based, as previously, Matthew J. Sideman, MD, explained in presenting the coding and reimbursement for 2018 at a symposium on vascular surgery sponsored by Northwestern University.
“The new EVAR codes for 2018 have got a lot of gains. There are some losses as well, but overall, I think it’s going to be very positive moving forward,” according to Dr. Sideman, a vascular surgeon at the University of Texas, San Antonio, who serves as chair of the Society for Vascular Surgery Coding and Reimbursement Committee and an adviser to the American Medical Association Relative Value Scale Update Committee (RUC).
“What we gained was a new code for ruptured aneurysm repair, a new code for enhanced fixation, a new code for percutaneous access, new codes for alternative access options, and now all the access codes are add-on codes. But what we traded off was loss due to bundling. So catheterization is now bundled into the main procedure, radiographic supervision and interpretation is now bundled. The big thing that really hurt was we lost all proximal extensions to the renal arteries and all distal extensions to the iliac bifurcations – they’re also bundled into the main procedure,” he said.
Restructuring the EVAR codes was a multiyear collaborative project of the SVS, the American College of Surgeons, the Society of Interventional Radiology, the Society of Thoracic Surgery, the American College of Cardiology, and the Society for Cardiovascular Angiography and Interventions. The impetus was twofold: recognition that the existing codes seriously undervalued the work involved in EVAR because, for example, they didn’t distinguish between ruptured and elective aneurysm repair, nor did they recognize the unique challenges and advantages of percutaneous access.
Also, representatives of the professional societies involved with vascular medicine recognized that they had to develop a detailed proposal for coding restructuring or matters might be taken out of their hands. Bundling of codes has become the prevailing dogma at the RUC and the Centers for Medicare and Medicaid. Their current policy is that when analysis of coding patterns indicates two codes are billed together at least 51% of the time, that’s considered a ‘typical’ situation and a new code must be created combining them. The harsh reality for clinicians is that under what Dr. Sideman called “RUC math,” the new bundled codes invariably pay less than the two old ones.
“There was a little bit of smoke and mirrors – ‘Look at the pretty flashing lights and not what’s going on behind over here’ – as we tried to maintain value as we bundled these EVAR codes,” Dr. Sideman recalled. “I can stand here and tell you I did my very best to push for the best values possible. It can be a painful process, but I thought we came out ok.”
How the new EVAR codes work
Dr. Sideman explained that the impact of the new EVAR codes will depend upon a surgeon’s practice pattern.
He offered as a concrete example a patient undergoing elective EVAR of the aorta and both iliac arteries with percutaneous access and placement of a bifurcated device with one docking limb. In 2017, this might have been handled using CPT codes 34802, 36200-50, and 75952-26, for a total of 31.05 Relative Value Units (RVUs) of work.
In 2018, however, this same surgical strategy would be coded as 34705 (elective endovascular repair of infrarenal aorta and/or iliac artery or arteries) plus 34713 x 2 (percutaneous access and closure), for a total of 34.58 RVUs. Thus, the surgeon would come out 3.53 RVUs ahead in 2018, which at a conversion factor of $35.78/RVU translates to an extra $126.30.
On the other hand, if the surgeon chose to use a bifurcated device with one docking limb, a left iliac bell-bottom extension, a right iliac bell-bottom extension, and percutaneous access, in 2017, this would have been coded as 34802, 34825, 34826, 36200-50, 75952-26, and 75953-26 x 2, for a total of 44.29 RVUs of work. In 2018, this same treatment strategy would be coded as 34705 plus 34713 x 2, for a total of 34.58 RVUs, or a knockdown of 9.71 fewer RVUs compared with the year before, which translates to $347.42 less.
“The more extensions you use, the more you’re going to come out behind going forward,” according to Dr. Sideman.
Other coding changes in 2018
Sclerotherapy of single and multiple veins (codes 36470 and 36471) got down-valued from 1.10 and 2.49 to 0.75 and 1.5 RVUs, respectively.
Angiography of the extremities (75710 and 75716) will be better reimbursed in 2018. In what Dr. Sideman called “a good win,” unilateral angiography will be rated as 1.75 RVUs, up from 1.14 in 2017, while bilateral angiography increased from 1.31 to 1.97 RVUs.
“The other nice thing I can tell you is that through campaigning and lobbying and comments to CMS [Centers for Medicare & Medicaid Services], we got them to reverse their recommendations from 2017 to 2018 on the dialysis family of codes,” the surgeon continued.
Reimbursement for the dialysis codes took a big hit from 2016 to 2017, amounting to several hundred million dollars less in reimbursement, but CMS has reversed its policy on that score. The RVUs for the various dialysis codes have increased from 2017 to 2018 by 5%-21%, with central venous angioplasty (CPT 36907) garnering the biggest increase.
Existing RVUs were retained for 2018 in three of the four selective catheter placement codes. However, reimbursement for 36215 (first order catheterization of the thoracic or brachiocephalic branch) dropped from 4.67 to 4.17 RVUs because physician surveys showed the time involved was less than previously rated. Once the RUC and CMS saw that the time involved in a procedure has decreased, it became impossible to maintain the RVU, Dr. Sideman explained.
And speaking of time involved in procedures, Dr. Sideman offered a final plea to his vascular medicine colleagues:
“When you get surveys from the RUC asking for your input, please, please, please, fill them out because that’s how we get our direct physician input into the valuation of codes.”
He reported having no financial conflicts of interest regarding his presentation.
A detailed listing of many of the codes and changes can be found at the American College of Radiology website, and the Society for Vascular Surgery has coding resources available on their website, as well.
EXPERT ANALYSIS FROM THE NORTHWESTERN VASCULAR SYMPOSIUM
Skin signs spotlight highest-risk SLE patients
GENEVA – Careful warranting prompt initiation of long-term antiplatelet therapy, Dan Lipsker, MD, PhD, said in a plenary lecture at the annual congress of the European Academy of Dermatology and Venereology.
“Those patients are at very high risk. Those are the lupus patients with the poorest prognosis. Those are the lupus patients who still die today,” said Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).

Cutaneous clues suggestive of thrombosis in SLE patients include atrophie blanche, pseudo-Degos lesions, livedo racemosa, acral nonpalpable purpura or reticulate erythema, cutaneous necrosis, splinter hemorrhage, thrombophlebitis, and nailfold telangiectasias. These skin findings can occur simultaneously with or after potentially life-threatening thrombotic events, or in the best of all scenarios, beforehand.
Dr. Lipsker told his audience of dermatologists that, by demonstrating facility in identifying these cutaneous disorders, they can make themselves “indispensable” to the rheumatologists, nephrologists, internists, and/or pediatricians who often provide the bulk of specialized care for SLE patients.
“We know today that, 5 years after initial diagnosis of SLE, the chief causes of morbidity and mortality are thrombotic events. And it can be extremely difficult to distinguish between an acute autoimmune lupus flare and a thrombotic event when, for example, the CNS or eyes are involved. But you will find direct evidence of thrombosis by carefully examining the skin,” the dermatologist maintained.
Some of these cutaneous signs constitute unequivocal evidence of thrombosis in SLE patients. Others are more ambiguous but should raise suspicion of an ongoing systemic thrombotic process unless another explanation is found.
One example of a skin finding that always indicates thrombosis is pseudo-Degos lesions: ivory-colored or white depressed atrophic papules with a raised border composed of telangiectasias. Biopsy shows evidence of a cone-shaped dermal arteriolar infarct.
“We have biopsied dozens of patients with this presentation, and you always find occluded vessels without a single inflammatory cell. These lesions are usually painful, and when you put those patients on low-dose aspirin they do better,” Dr. Lipsker said.
Atrophie blanche in a patient with SLE is also strong evidence of thrombotic vasculopathy. Atrophie blanche is a porcelain-white atrophic white scar with surrounding hyperpigmentation on the lower leg that occurs after skin injury in an area with venous insufficiency.
Livedo racemosa in a patient with SLE is also highly suggestive of a systemic thrombotic process. Characteristic of this dermatologic disorder is an irregular, netlike mottling surrounding pale skin.
“All of these skin signs allow identification of patients who have very high risk of thrombotic events,” Dr. Lipsker stressed.
He reported having no financial conflicts of interest regarding his presentation.
GENEVA – Careful warranting prompt initiation of long-term antiplatelet therapy, Dan Lipsker, MD, PhD, said in a plenary lecture at the annual congress of the European Academy of Dermatology and Venereology.
“Those patients are at very high risk. Those are the lupus patients with the poorest prognosis. Those are the lupus patients who still die today,” said Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
Cutaneous clues suggestive of thrombosis in SLE patients include atrophie blanche, pseudo-Degos lesions, livedo racemosa, acral nonpalpable purpura or reticulate erythema, cutaneous necrosis, splinter hemorrhage, thrombophlebitis, and nailfold telangiectasias. These skin findings can occur simultaneously with or after potentially life-threatening thrombotic events, or in the best of all scenarios, beforehand.
Dr. Lipsker told his audience of dermatologists that, by demonstrating facility in identifying these cutaneous disorders, they can make themselves “indispensable” to the rheumatologists, nephrologists, internists, and/or pediatricians who often provide the bulk of specialized care for SLE patients.
“We know today that, 5 years after initial diagnosis of SLE, the chief causes of morbidity and mortality are thrombotic events. And it can be extremely difficult to distinguish between an acute autoimmune lupus flare and a thrombotic event when, for example, the CNS or eyes are involved. But you will find direct evidence of thrombosis by carefully examining the skin,” the dermatologist maintained.
Some of these cutaneous signs constitute unequivocal evidence of thrombosis in SLE patients. Others are more ambiguous but should raise suspicion of an ongoing systemic thrombotic process unless another explanation is found.
One example of a skin finding that always indicates thrombosis is pseudo-Degos lesions: ivory-colored or white depressed atrophic papules with a raised border composed of telangiectasias. Biopsy shows evidence of a cone-shaped dermal arteriolar infarct.
“We have biopsied dozens of patients with this presentation, and you always find occluded vessels without a single inflammatory cell. These lesions are usually painful, and when you put those patients on low-dose aspirin they do better,” Dr. Lipsker said.
Atrophie blanche in a patient with SLE is also strong evidence of thrombotic vasculopathy. Atrophie blanche is a porcelain-white atrophic white scar with surrounding hyperpigmentation on the lower leg that occurs after skin injury in an area with venous insufficiency.
Livedo racemosa in a patient with SLE is also highly suggestive of a systemic thrombotic process. Characteristic of this dermatologic disorder is an irregular, netlike mottling surrounding pale skin.
“All of these skin signs allow identification of patients who have very high risk of thrombotic events,” Dr. Lipsker stressed.
He reported having no financial conflicts of interest regarding his presentation.
GENEVA – Careful warranting prompt initiation of long-term antiplatelet therapy, Dan Lipsker, MD, PhD, said in a plenary lecture at the annual congress of the European Academy of Dermatology and Venereology.
“Those patients are at very high risk. Those are the lupus patients with the poorest prognosis. Those are the lupus patients who still die today,” said Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
Cutaneous clues suggestive of thrombosis in SLE patients include atrophie blanche, pseudo-Degos lesions, livedo racemosa, acral nonpalpable purpura or reticulate erythema, cutaneous necrosis, splinter hemorrhage, thrombophlebitis, and nailfold telangiectasias. These skin findings can occur simultaneously with or after potentially life-threatening thrombotic events, or in the best of all scenarios, beforehand.
Dr. Lipsker told his audience of dermatologists that, by demonstrating facility in identifying these cutaneous disorders, they can make themselves “indispensable” to the rheumatologists, nephrologists, internists, and/or pediatricians who often provide the bulk of specialized care for SLE patients.
“We know today that, 5 years after initial diagnosis of SLE, the chief causes of morbidity and mortality are thrombotic events. And it can be extremely difficult to distinguish between an acute autoimmune lupus flare and a thrombotic event when, for example, the CNS or eyes are involved. But you will find direct evidence of thrombosis by carefully examining the skin,” the dermatologist maintained.
Some of these cutaneous signs constitute unequivocal evidence of thrombosis in SLE patients. Others are more ambiguous but should raise suspicion of an ongoing systemic thrombotic process unless another explanation is found.
One example of a skin finding that always indicates thrombosis is pseudo-Degos lesions: ivory-colored or white depressed atrophic papules with a raised border composed of telangiectasias. Biopsy shows evidence of a cone-shaped dermal arteriolar infarct.
“We have biopsied dozens of patients with this presentation, and you always find occluded vessels without a single inflammatory cell. These lesions are usually painful, and when you put those patients on low-dose aspirin they do better,” Dr. Lipsker said.
Atrophie blanche in a patient with SLE is also strong evidence of thrombotic vasculopathy. Atrophie blanche is a porcelain-white atrophic white scar with surrounding hyperpigmentation on the lower leg that occurs after skin injury in an area with venous insufficiency.
Livedo racemosa in a patient with SLE is also highly suggestive of a systemic thrombotic process. Characteristic of this dermatologic disorder is an irregular, netlike mottling surrounding pale skin.
“All of these skin signs allow identification of patients who have very high risk of thrombotic events,” Dr. Lipsker stressed.
He reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Guselkumab crushes skin disease in psoriatic arthritis patients
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.

The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.
The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
GENEVA – The interleukin-23 inhibitor guselkumab generates the same impressive improvement in skin disease in psoriatic arthritis patients as has been seen in psoriasis without joint disease, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
However, psoriatic arthritis patients’ improvement in Dermatology Life Quality Index (DLQI) scores is less robust than in patients with psoriasis only, added Dr. Kimball, professor of dermatology at Harvard Medical School, Boston, and CEO of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center.
The psoriatic arthritis group as a whole had more severe psoriasis, with a baseline mean PASI score of 24.3 and involvement of 32.7% of their body surface area as compared with a PASI score of 21.2 and 27.2% BSA in psoriasis patients without arthritis. A total of 28% of the psoriatic arthritis patients had previously been on other biologics and 77% had been on nonbiologic systemic agents, compared with 19% and 60% of the psoriasis patients, respectively. The psoriatic arthritis group had a mean 19.2-year history of psoriasis, 1.9 years longer than the psoriasis-only group.
Participants were randomized to 100 mg of guselkumab administered subcutaneously at weeks 0, 4, 12, and 20; placebo through week 12, followed by a switch to adalimumab (Humira); or adalimumab at 80 mg at week 0, then 40 mg at week 2 and 40 mg again every 2 weeks until week 23.
The key findings:
The PASI 90 response rate – that is, at least a 90% improvement in Psoriasis Area and Severity Index – in guselkumab-treated patients at week 16 was 72% in patients with psoriatic arthritis and 71% in those without. At week 24, the PASI 90 rate was 74% in guselkumab-treated patients with psoriatic arthritis and similar at 78% in those without. In contrast, the PASI 90 rate at week 24 in patients on adalimumab was significantly lower: 48% in the psoriatic arthritis group and 55% in those with psoriasis only. The PASI 90 rate in placebo-treated controls was single digit.
At week 24, 82% of psoriatic arthritis patients on guselkumab had clear or almost clear skin as reflected in an Investigator’s Global Assessment score of 0 or 1, as did 84% of psoriasis-only patients.
A DLQI score of 0 or 1, meaning the dermatologic disease had no impact on patient quality of life, was documented at week 16 in 46% of psoriatic arthritis patients and 55% of psoriasis-only patients, a trend that didn’t achieve statistical significance. However, by week 24 the difference became significant, with a DLQI of 0 or 1 in 48% of the psoriatic arthritis patients, compared with 62% of psoriasis-only patients.
VOYAGE 1 and 2 were dermatologic studies that didn’t measure changes in joint symptom scores or other psoriatic arthritis outcomes. Guselkumab as a potential treatment for psoriatic arthritis is under investigation in other studies.
The VOYAGE trials and this analysis were sponsored by Janssen. Dr. Kimball reported receiving research funding from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
SOURCE: Kimball A et al. https://eadvgeneva2017.org/
REPORTING FROM THE EADV CONGRESS
Key clinical point:
Major finding: After 16 weeks on guselkumab, 72% of psoriatic arthritis patients and 71% with psoriasis-only had a PASI 90 response.
Study details: This was a comparison of skin and DLQI outcomes in 335 patients with psoriatic arthritis and 1,494 with psoriasis only who participated in two randomized, double-blind, phase 3 clinical trials.
Disclosures: Janssen sponsored the study. The presenter reported receiving research grants from and serving as a consultant to Janssen and numerous other pharmaceutical companies.
Source: Kimball A et al. https://eadvgeneva2017.org/.
JAK inhibitors for atopic dermatitis might hit JAK-pot
GENEVA – at the annual congress of the European Academy of Dermatology and Venereology, with three positive phase 2 randomized trials featuring one topical and two oral agents presented to enthusiastic audiences.
The way has already been paved for dermatologic researchers by veterinarians, who developed oclacitinib (Apoquel), a relatively selective Janus kinase 1 (JAK1) inhibitor, for canine AD. The medication was approved by the Food and Drug Administration in 2013 for treating AD and for controlling pruritus associated with allergic dermatitis in dogs.

PF-04965842
“Get out your pencils, everyone. This is why you’re all here at 8 o’clock on a Sunday morning,” Melinda Gooderham, MD, said, standing before a packed house at the main arena of the Geneva Convention Center, as she launched into the results of a phase II randomized, double-blind, placebo-controlled, 12-week trial of a JAK inhibitor known for now as PF-04965842. This is a JAK1-selective agent with a good effect on interleukin-4 and -13, key mediators of the Th2 cytokines implicated in the pathogenesis of AD.
The dose-ranging study included 250 adults with AD and an inadequate response to or intolerance of topical therapy. Their mean baseline Eczema Area and Severity Index (EASI) score was 25 with a 60/40 ratio of moderate to severe AD. The five-arm trial randomized patients to PF-04965842 at 10 mg, 30 mg, 100 mg, or 200 mg once daily or placebo.
The primary endpoint was the proportion of patients achieving an Investigator Global Assessment (IGA) score of 0 or 1 – clear or almost clear – along with at least a 2-grade improvement from baseline at week 12. A clear dose-response effect was evident, with the 100- and 200-mg doses achieving response rates of 28% and 45%, respectively, compared with 6% in placebo-treated controls, reported Dr. Gooderham, medical director of the Skin Center for Dermatology in Peterborough, Ont., and a dermatologist at Queen’s University in Kingston, Ont.
Onset of action was speedy: patients in the 200-mg group reached their full improvement in IGA score by week 4 and maintained that response through week 12. Maximum improvement in EASI score – a mean 80% reduction – was achieved by week 6 and sustained thereafter. The proportion of patients in the 200-mg group achieving at least a 4-point improvement on the Pruritus Numeric Rating Scale significantly exceeded that in the placebo group as early as day 2 of the trial. At week 12, 64% of patients in the 200-mg group had achieved this level of improvement in itch, compared with 26% of controls.
A dose-dependent drop in platelet count occurred in the study, reaching a 30% decline at the 4-week nadir in the 200-mg group, followed by gradual on-treatment recovery. Both LDL and HDL cholesterol rose on active therapy – a class effect of JAK inhibitors – but the ratio between the two lipid levels remained unchanged. The two serious adverse events deemed treatment related were a case of eczema herpeticum in a patient on the 100-mg dose and pneumonia in a patient on the 200-mg dose.
Baricitinib
This once-daily oral JAK1/2 inhibitor is approved for treatment of rheumatoid arthritis in Europe and Japan. Emma Guttman-Yassky, MD, PhD, presented a phase 2 study of baricitinib in 124 adults with moderate to severe AD. Notably, prior to enrollment, all participants had to have failed to respond to a 4-week run-in period of supervised treatment with 0.1% triamcinolone cream, a midpotency topical steroid. They were then randomized to 2 mg or 4 mg of once-daily baricitinib or placebo, in all cases supplemented as needed with the topical steroid. Their median baseline EASI score was 21.
The primary endpoint was the proportion of patients achieving at least a 50% improvement in EASI score, or EASI 50 response, by week 16 from baseline in a nonresponder imputation analysis. This was achieved in 65% of patients on the 4-mg dose of baricitinib, 64% on the 2-mg dose, and 46% of controls on placebo plus the topical steroid. A statistically significant difference in EASI 50 response between the baricitinib groups and controls was seen at 1 week, with nearly the maximum effect achieved at week 4. Patients with a baseline EASI score above the median had a much more impressive treatment response because the placebo effect was smaller in participants with more severe AD.
“I think this drug can be an exciting new addition to the field,” declared Dr. Guttman-Yassky, professor and vice chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
From a baseline total SCORAD (Scoring Atopic Dermatitis) score of 55, major improvements were seen in the JAK inhibitor–treated patients by week 4. At week 16, the average reduction from baseline was 47% in the 4-mg group, 41% in the 2-mg group, and 21% in the placebo group. Both the SCORAD pruritus and sleep loss subscores showed significantly more robust improvement in the baricitinib groups than in controls. Indeed, a significant drop in pruritus scores was noted within the first week.
The 4-mg dose was associated with greater improvement and faster onset of action than the 2-mg dose on some but not all disease measures.
Dr. Guttman-Yassky described baricitinib as having “an overall acceptable safety profile,” with no serious treatment-related adverse events noted. Headache, nasopharyngitis, and asymptomatic increases in serum creatinine phosphokinase were more common in baricitinib-treated patients than with placebo.
JTE-052
Hidemi Nakagawa, MD, presented a phase 2 study of topical JTE-052 ointment in 327 Japanese adults with moderate to severe AD. The drug inhibits JAK1/2/3 as well as the tyrosine kinase pathway. It also promotes keratinocyte production of filaggrin in the skin barrier. Participants were randomized to twice-daily application of JTE-052 ointment (at 0.25%, 0.5%, 1%, or 3%), vehicle ointment, or 0.1% tacrolimus ointment twice a day for 4 weeks. The primary outcome was the change from baseline in modified EASI score in the active treatment groups compared with placebo. All doses of JTE-052 proved significantly more effective than vehicle. A dose-response effect was noted, with a 42% reduction from baseline in modified EASI score in the 0.25% JTE-052 group, a 57% reduction with 0.5%, a 55% reduction with 1% ointment, and a 73% reduction with 3%, compared with a 12% reduction decrease in patients who received vehicle. The topical tacrolimus group showed a 62% reduction from baseline, reported Dr. Nakagawa, professor and head of the division of dermatology at Jikei University, Tokyo.
All doses of JTE-052 were also significantly more effective than placebo on all secondary endpoints, which included IGA, percent body surface area affected, and Pruritus Numeric Rating Scale score.
At all but the weakest concentration, JTE-052 resulted in significant reduction in pruritus starting with the second dose on day 1 of the trial, he added.
Mild nasopharyngitis occurred in 3.4% of JTE-052–treated patients. There were no serious adverse events and no changes in laboratory parameters in the study. One patient discontinued JTE-052 because of application-site contact dermatitis, another because of application-site irritation. The results of this study were recently published in the British Journal of Dermatology (Br J Dermatol. 2017 Sep 28. doi: 10.1111/bjd.16014).
Dr. Nakagawa reported receiving research grants from and serving as a consultant to Japan Tobacco, which is developing JTE-052. Dr. Guttman-Yassky reported having financial relationships with Eli Lilly and Incyte, which sponsored the baricitinib study, as well as most other pharmaceutical companies developing therapies for AD. Dr. Gooderham reported receiving research funding from and serving as a consultant to Pfizer, which sponsored the PF-04965842 study, as well as numerous other pharmaceutical companies.
GENEVA – at the annual congress of the European Academy of Dermatology and Venereology, with three positive phase 2 randomized trials featuring one topical and two oral agents presented to enthusiastic audiences.
The way has already been paved for dermatologic researchers by veterinarians, who developed oclacitinib (Apoquel), a relatively selective Janus kinase 1 (JAK1) inhibitor, for canine AD. The medication was approved by the Food and Drug Administration in 2013 for treating AD and for controlling pruritus associated with allergic dermatitis in dogs.
PF-04965842
“Get out your pencils, everyone. This is why you’re all here at 8 o’clock on a Sunday morning,” Melinda Gooderham, MD, said, standing before a packed house at the main arena of the Geneva Convention Center, as she launched into the results of a phase II randomized, double-blind, placebo-controlled, 12-week trial of a JAK inhibitor known for now as PF-04965842. This is a JAK1-selective agent with a good effect on interleukin-4 and -13, key mediators of the Th2 cytokines implicated in the pathogenesis of AD.
The dose-ranging study included 250 adults with AD and an inadequate response to or intolerance of topical therapy. Their mean baseline Eczema Area and Severity Index (EASI) score was 25 with a 60/40 ratio of moderate to severe AD. The five-arm trial randomized patients to PF-04965842 at 10 mg, 30 mg, 100 mg, or 200 mg once daily or placebo.
The primary endpoint was the proportion of patients achieving an Investigator Global Assessment (IGA) score of 0 or 1 – clear or almost clear – along with at least a 2-grade improvement from baseline at week 12. A clear dose-response effect was evident, with the 100- and 200-mg doses achieving response rates of 28% and 45%, respectively, compared with 6% in placebo-treated controls, reported Dr. Gooderham, medical director of the Skin Center for Dermatology in Peterborough, Ont., and a dermatologist at Queen’s University in Kingston, Ont.
Onset of action was speedy: patients in the 200-mg group reached their full improvement in IGA score by week 4 and maintained that response through week 12. Maximum improvement in EASI score – a mean 80% reduction – was achieved by week 6 and sustained thereafter. The proportion of patients in the 200-mg group achieving at least a 4-point improvement on the Pruritus Numeric Rating Scale significantly exceeded that in the placebo group as early as day 2 of the trial. At week 12, 64% of patients in the 200-mg group had achieved this level of improvement in itch, compared with 26% of controls.
A dose-dependent drop in platelet count occurred in the study, reaching a 30% decline at the 4-week nadir in the 200-mg group, followed by gradual on-treatment recovery. Both LDL and HDL cholesterol rose on active therapy – a class effect of JAK inhibitors – but the ratio between the two lipid levels remained unchanged. The two serious adverse events deemed treatment related were a case of eczema herpeticum in a patient on the 100-mg dose and pneumonia in a patient on the 200-mg dose.
Baricitinib
This once-daily oral JAK1/2 inhibitor is approved for treatment of rheumatoid arthritis in Europe and Japan. Emma Guttman-Yassky, MD, PhD, presented a phase 2 study of baricitinib in 124 adults with moderate to severe AD. Notably, prior to enrollment, all participants had to have failed to respond to a 4-week run-in period of supervised treatment with 0.1% triamcinolone cream, a midpotency topical steroid. They were then randomized to 2 mg or 4 mg of once-daily baricitinib or placebo, in all cases supplemented as needed with the topical steroid. Their median baseline EASI score was 21.
The primary endpoint was the proportion of patients achieving at least a 50% improvement in EASI score, or EASI 50 response, by week 16 from baseline in a nonresponder imputation analysis. This was achieved in 65% of patients on the 4-mg dose of baricitinib, 64% on the 2-mg dose, and 46% of controls on placebo plus the topical steroid. A statistically significant difference in EASI 50 response between the baricitinib groups and controls was seen at 1 week, with nearly the maximum effect achieved at week 4. Patients with a baseline EASI score above the median had a much more impressive treatment response because the placebo effect was smaller in participants with more severe AD.
“I think this drug can be an exciting new addition to the field,” declared Dr. Guttman-Yassky, professor and vice chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
From a baseline total SCORAD (Scoring Atopic Dermatitis) score of 55, major improvements were seen in the JAK inhibitor–treated patients by week 4. At week 16, the average reduction from baseline was 47% in the 4-mg group, 41% in the 2-mg group, and 21% in the placebo group. Both the SCORAD pruritus and sleep loss subscores showed significantly more robust improvement in the baricitinib groups than in controls. Indeed, a significant drop in pruritus scores was noted within the first week.
The 4-mg dose was associated with greater improvement and faster onset of action than the 2-mg dose on some but not all disease measures.
Dr. Guttman-Yassky described baricitinib as having “an overall acceptable safety profile,” with no serious treatment-related adverse events noted. Headache, nasopharyngitis, and asymptomatic increases in serum creatinine phosphokinase were more common in baricitinib-treated patients than with placebo.
JTE-052
Hidemi Nakagawa, MD, presented a phase 2 study of topical JTE-052 ointment in 327 Japanese adults with moderate to severe AD. The drug inhibits JAK1/2/3 as well as the tyrosine kinase pathway. It also promotes keratinocyte production of filaggrin in the skin barrier. Participants were randomized to twice-daily application of JTE-052 ointment (at 0.25%, 0.5%, 1%, or 3%), vehicle ointment, or 0.1% tacrolimus ointment twice a day for 4 weeks. The primary outcome was the change from baseline in modified EASI score in the active treatment groups compared with placebo. All doses of JTE-052 proved significantly more effective than vehicle. A dose-response effect was noted, with a 42% reduction from baseline in modified EASI score in the 0.25% JTE-052 group, a 57% reduction with 0.5%, a 55% reduction with 1% ointment, and a 73% reduction with 3%, compared with a 12% reduction decrease in patients who received vehicle. The topical tacrolimus group showed a 62% reduction from baseline, reported Dr. Nakagawa, professor and head of the division of dermatology at Jikei University, Tokyo.
All doses of JTE-052 were also significantly more effective than placebo on all secondary endpoints, which included IGA, percent body surface area affected, and Pruritus Numeric Rating Scale score.
At all but the weakest concentration, JTE-052 resulted in significant reduction in pruritus starting with the second dose on day 1 of the trial, he added.
Mild nasopharyngitis occurred in 3.4% of JTE-052–treated patients. There were no serious adverse events and no changes in laboratory parameters in the study. One patient discontinued JTE-052 because of application-site contact dermatitis, another because of application-site irritation. The results of this study were recently published in the British Journal of Dermatology (Br J Dermatol. 2017 Sep 28. doi: 10.1111/bjd.16014).
Dr. Nakagawa reported receiving research grants from and serving as a consultant to Japan Tobacco, which is developing JTE-052. Dr. Guttman-Yassky reported having financial relationships with Eli Lilly and Incyte, which sponsored the baricitinib study, as well as most other pharmaceutical companies developing therapies for AD. Dr. Gooderham reported receiving research funding from and serving as a consultant to Pfizer, which sponsored the PF-04965842 study, as well as numerous other pharmaceutical companies.
GENEVA – at the annual congress of the European Academy of Dermatology and Venereology, with three positive phase 2 randomized trials featuring one topical and two oral agents presented to enthusiastic audiences.
The way has already been paved for dermatologic researchers by veterinarians, who developed oclacitinib (Apoquel), a relatively selective Janus kinase 1 (JAK1) inhibitor, for canine AD. The medication was approved by the Food and Drug Administration in 2013 for treating AD and for controlling pruritus associated with allergic dermatitis in dogs.
PF-04965842
“Get out your pencils, everyone. This is why you’re all here at 8 o’clock on a Sunday morning,” Melinda Gooderham, MD, said, standing before a packed house at the main arena of the Geneva Convention Center, as she launched into the results of a phase II randomized, double-blind, placebo-controlled, 12-week trial of a JAK inhibitor known for now as PF-04965842. This is a JAK1-selective agent with a good effect on interleukin-4 and -13, key mediators of the Th2 cytokines implicated in the pathogenesis of AD.
The dose-ranging study included 250 adults with AD and an inadequate response to or intolerance of topical therapy. Their mean baseline Eczema Area and Severity Index (EASI) score was 25 with a 60/40 ratio of moderate to severe AD. The five-arm trial randomized patients to PF-04965842 at 10 mg, 30 mg, 100 mg, or 200 mg once daily or placebo.
The primary endpoint was the proportion of patients achieving an Investigator Global Assessment (IGA) score of 0 or 1 – clear or almost clear – along with at least a 2-grade improvement from baseline at week 12. A clear dose-response effect was evident, with the 100- and 200-mg doses achieving response rates of 28% and 45%, respectively, compared with 6% in placebo-treated controls, reported Dr. Gooderham, medical director of the Skin Center for Dermatology in Peterborough, Ont., and a dermatologist at Queen’s University in Kingston, Ont.
Onset of action was speedy: patients in the 200-mg group reached their full improvement in IGA score by week 4 and maintained that response through week 12. Maximum improvement in EASI score – a mean 80% reduction – was achieved by week 6 and sustained thereafter. The proportion of patients in the 200-mg group achieving at least a 4-point improvement on the Pruritus Numeric Rating Scale significantly exceeded that in the placebo group as early as day 2 of the trial. At week 12, 64% of patients in the 200-mg group had achieved this level of improvement in itch, compared with 26% of controls.
A dose-dependent drop in platelet count occurred in the study, reaching a 30% decline at the 4-week nadir in the 200-mg group, followed by gradual on-treatment recovery. Both LDL and HDL cholesterol rose on active therapy – a class effect of JAK inhibitors – but the ratio between the two lipid levels remained unchanged. The two serious adverse events deemed treatment related were a case of eczema herpeticum in a patient on the 100-mg dose and pneumonia in a patient on the 200-mg dose.
Baricitinib
This once-daily oral JAK1/2 inhibitor is approved for treatment of rheumatoid arthritis in Europe and Japan. Emma Guttman-Yassky, MD, PhD, presented a phase 2 study of baricitinib in 124 adults with moderate to severe AD. Notably, prior to enrollment, all participants had to have failed to respond to a 4-week run-in period of supervised treatment with 0.1% triamcinolone cream, a midpotency topical steroid. They were then randomized to 2 mg or 4 mg of once-daily baricitinib or placebo, in all cases supplemented as needed with the topical steroid. Their median baseline EASI score was 21.
The primary endpoint was the proportion of patients achieving at least a 50% improvement in EASI score, or EASI 50 response, by week 16 from baseline in a nonresponder imputation analysis. This was achieved in 65% of patients on the 4-mg dose of baricitinib, 64% on the 2-mg dose, and 46% of controls on placebo plus the topical steroid. A statistically significant difference in EASI 50 response between the baricitinib groups and controls was seen at 1 week, with nearly the maximum effect achieved at week 4. Patients with a baseline EASI score above the median had a much more impressive treatment response because the placebo effect was smaller in participants with more severe AD.
“I think this drug can be an exciting new addition to the field,” declared Dr. Guttman-Yassky, professor and vice chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
From a baseline total SCORAD (Scoring Atopic Dermatitis) score of 55, major improvements were seen in the JAK inhibitor–treated patients by week 4. At week 16, the average reduction from baseline was 47% in the 4-mg group, 41% in the 2-mg group, and 21% in the placebo group. Both the SCORAD pruritus and sleep loss subscores showed significantly more robust improvement in the baricitinib groups than in controls. Indeed, a significant drop in pruritus scores was noted within the first week.
The 4-mg dose was associated with greater improvement and faster onset of action than the 2-mg dose on some but not all disease measures.
Dr. Guttman-Yassky described baricitinib as having “an overall acceptable safety profile,” with no serious treatment-related adverse events noted. Headache, nasopharyngitis, and asymptomatic increases in serum creatinine phosphokinase were more common in baricitinib-treated patients than with placebo.
JTE-052
Hidemi Nakagawa, MD, presented a phase 2 study of topical JTE-052 ointment in 327 Japanese adults with moderate to severe AD. The drug inhibits JAK1/2/3 as well as the tyrosine kinase pathway. It also promotes keratinocyte production of filaggrin in the skin barrier. Participants were randomized to twice-daily application of JTE-052 ointment (at 0.25%, 0.5%, 1%, or 3%), vehicle ointment, or 0.1% tacrolimus ointment twice a day for 4 weeks. The primary outcome was the change from baseline in modified EASI score in the active treatment groups compared with placebo. All doses of JTE-052 proved significantly more effective than vehicle. A dose-response effect was noted, with a 42% reduction from baseline in modified EASI score in the 0.25% JTE-052 group, a 57% reduction with 0.5%, a 55% reduction with 1% ointment, and a 73% reduction with 3%, compared with a 12% reduction decrease in patients who received vehicle. The topical tacrolimus group showed a 62% reduction from baseline, reported Dr. Nakagawa, professor and head of the division of dermatology at Jikei University, Tokyo.
All doses of JTE-052 were also significantly more effective than placebo on all secondary endpoints, which included IGA, percent body surface area affected, and Pruritus Numeric Rating Scale score.
At all but the weakest concentration, JTE-052 resulted in significant reduction in pruritus starting with the second dose on day 1 of the trial, he added.
Mild nasopharyngitis occurred in 3.4% of JTE-052–treated patients. There were no serious adverse events and no changes in laboratory parameters in the study. One patient discontinued JTE-052 because of application-site contact dermatitis, another because of application-site irritation. The results of this study were recently published in the British Journal of Dermatology (Br J Dermatol. 2017 Sep 28. doi: 10.1111/bjd.16014).
Dr. Nakagawa reported receiving research grants from and serving as a consultant to Japan Tobacco, which is developing JTE-052. Dr. Guttman-Yassky reported having financial relationships with Eli Lilly and Incyte, which sponsored the baricitinib study, as well as most other pharmaceutical companies developing therapies for AD. Dr. Gooderham reported receiving research funding from and serving as a consultant to Pfizer, which sponsored the PF-04965842 study, as well as numerous other pharmaceutical companies.
EXPERT ANALYSIS FROM THE EADV CONGRESS