User login
U.S. Measles Cases Reported in 2011 Highest Since 1996
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
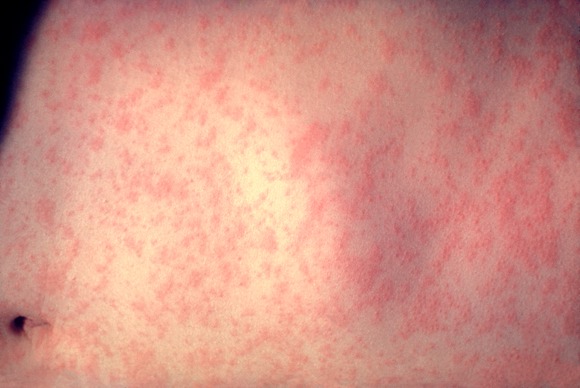
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
FROM THE MMWR
Major Finding: Most of the measles cases – 105 of 118 – were associated with importation from other countries; 105 (89%) had not been vaccinated.
Data Source: Cases reported between Jan. 1, 2011 and May 20, 2011.
Disclosures: The authors had no relevant financial disclosures.
U.S. Measles Cases Reported in 2011 Highest Since 1996
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
FROM THE MMWR
U.S. Measles Cases Reported in 2011 Highest Since 1996
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
As of May 20, 118 cases of measles in people aged 3 months to 68 years had been reported in the United States this year, the highest number reported for this 19-week period in any year since 1996, according to the Centers for Disease Control and Prevention.
Most of the cases (105 or 89%) were associated with importation from other countries, an "unusually large number of importations" that was related to recent increases in measles in countries visited by U.S. travelers, according to the May 24 Morbidity and Mortality Weekly Report (MMWR 60 [Early Release]; 1-4). Most of the cases that were associated with importations from European countries were from France, where about 10,000 cases of measles have been reported from January through April this year. Between 2001 and 2008, the CDC received a median of 56 reports of measles annually.
Fifty-three (45%) of the cases were in people aged 20 years and older; of the rest, 18 (15%) were in children under age 12 months, 24 (20%) were children aged 1-4 years, and 23 (19%) were in individuals aged 5-19 years.
Of the 118 reported cases from 23 states and New York City, 105 (89%) had not been vaccinated. The cases included 45 U.S. residents aged 1-19 years, of whom 39 (87%) had not been vaccinated: In 24 cases, parents had claimed religious or personal exemption and 8 had missed opportunities to be vaccinated.
Of the 42 U.S. residents aged 20 and older who were among the cases, 35 (83%) had not been vaccinated, including 6 who declined vaccination because of "philosophical objections to vaccination." Of the 33 U.S residents who were eligible to receive the vaccine and had traveled abroad, 30 (91%) were not vaccinated, and in one case (3%), the person had received one of the two recommended doses.
The size of the outbreaks ranged from 3 to 21 cases. The 21 cases were in a community in Minnesota where many children were not vaccinated because parents were concerned about the safety of the measles, mumps, and rubella vaccine (MMR). That outbreak resulted in infection of at least seven infants who were too young to receive the MMR vaccine.
Nine outbreaks accounted for 58 (49%) of the 118 cases and, in 6 outbreaks, the index case acquired measles abroad; the source of the other 3 outbreaks could not be determined.
Of the 118 cases reported between Jan. 1, 2011 and May 20, 2011, there were no deaths or cases of encephalitis, but 40% (47) needed to be hospitalized, and there were nine cases of pneumonia.
Because cases of measles continue to be imported into the United States, clinicians should suspect measles in people with a febrile rash illness and "clinically compatible symptoms," such as cough, coryza, and/or conjunctivitis, "who have recently traveled abroad or have had contact with travelers." When measles is suspected, clinicians should isolate the patient, report the case immediately to the local health department, and obtain viral specimens for testing, according to the report.
FROM THE MMWR
Major Finding: Most of the measles cases – 105 of 118 – were associated with importation from other countries; 105 (89%) had not been vaccinated.
Data Source: Cases reported between Jan. 1, 2011 and May 20, 2011.
Disclosures: The authors had no relevant financial disclosures.
FDA Okays Second Hepatitis C Antiviral
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
FROM THE FDA
FDA Okays Second Hepatitis C Antiviral
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
FROM THE FDA
FDA Okays Second Hepatitis C Antiviral
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
The Food and Drug Administration May 23 approved the protease inhibitor telaprevir to treat adults with genotype 1 chronic hepatitis C, in combination with standard therapy – a decision that comes less than 2 weeks after the agency okayed another protease inhibitor, boceprevir, for hepatitis C combination treatment.
"The availability of new therapies that significantly increase responses while potentially decreasing the overall duration of treatment is a major step forward in the battle against chronic hepatitis C infection," said Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, in a statement.
Telaprevir’s approved indication is for treatment (in combination with peginterferon alfa and ribavirin) of genotype 1 chronic hepatitis C in adults "with compensated liver disease, including cirrhosis, who are [treatment naive] or who have been previously treated with interferon-based treatment, including prior null responders, partial responders, and relapsers."
At a meeting in April, the FDA’s antiviral drugs advisory panel unanimously supported the approval of telaprevir.
The FDA based its approval decision on the results of three phase III studies comparing treatment with peginterferon alfa and ribavirin (standard treatment) alone or in combination with telaprevir in about 2,250 treatment-naive and treatment-experienced adults.
The sustained virologic response (SVR) rate was 20%-45% higher among patients on telaprevir plus standard treatment, compared with those on standard treatment, according to the FDA statement announcing the approval.
Fewer than half of the patients responded to standard treatment with peginterferon alfa and ribavirin, which is administered for 48 weeks. However, the telaprevir studies indicated that treatment with telaprevir can be shortened to 24 weeks, "in most patients," according to the FDA, which noted that 60% of previously untreated patients achieved an early response after 24 weeks of treatment, with an SVR of 90%.
The recommended dosing is 750 mg of telaprevir taken three times a day, with food. It is administered with peginterferon alfa and ribavirin for 12 weeks, "followed by a response-guided regimen of either 12 or 36 additional weeks of peginterferon alfa and ribavirin depending on viral response and prior response status," according to the label.
The most commonly reported side effects associated with telaprevir in clinical trials included rash (which can be severe and treatment limiting), anemia, nausea, fatigue, headache, diarrhea, pruritus, and anal or rectal irritation and pain. Rash can be serious and may require stopping treatment with telaprevir or all three drugs, the FDA statement said.
Vertex Pharmaceuticals will market telaprevir as Incivek. Merck will market boceprevir as Victrelis, which the FDA approved May 13.
FROM THE FDA
FDA Panel Divided on Trilipix Labeling Changes
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY PANEL
FDA Panel Divided on Trilipix Labeling Changes
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY PANEL
FDA Panel Divided on Trilipix Labeling Changes
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
SILVER SPRING, MD. — Members of a Food and Drug Administration advisory panel were divided on whether the approved indication for fenofibric acid, when co-administered with a statin, should be revised based on trial results showing no benefit of the drug in reducing cardiovascular disease risk when added to a statin in patients with type 2 diabetes.
At a meeting May 19, 6 of the 13 members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the marketing of fenofibric acid should be allowed to continue, with the addition to the label of the main findings of the study – the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Lipid trial. Four panelists, however, voted to recommend that the co-administration indication be withdrawn, citing the lack of solid evidence to support the indication. The remaining three panelists voted to allow continued marketing with no changes to the label.
Fenofibric acid, marketed by Abbott Laboratories as Trilipix, was approved by the FDA in December 2008. The drug’s indication includes its use with a statin as an adjunct to diet to reduce serum triglyceride (TG) levels and increase serum HDL cholesterol levels in patients with mixed dyslipidemia and coronary heart disease (CHD) or a CHD risk equivalent who are on "optimal statin therapy" to achieve their LDL cholesterol goal. Fenofibric acid is the active ingredient of fenofibrate, a fibrate approved in 1993 that is now available in generic formulations.
The FDA convened the panel to review the co-administration indication of fenofibric acid in the context of the ACCORD Lipid study results. That study found no benefit of the combination treatment on major cardiovascular events, compared with treatment with simvastatin alone, over a mean of almost 5 years of follow-up. The study enrolled more than 5,500 patients with type 2 diabetes at high risk of cardiovascular disease, with a range of TG and HDL cholesterol levels. Among women in the study, the rate of major adverse cardiovascular events was higher than that among those on the statin alone. But in another subgroup of patients, those with elevated TG levels (204 mg/dL or higher) and reduced HDL cholesterol levels (34 mg/dL or lower), there was a suggestion of benefit with combination therapy (N. Engl. J. Med. 2010;362:1563-74).
Panelists at the meeting said they were not comfortable drawing any conclusions from analyses of patient subgroups in a negative trial. They voted 13-0 that the FDA should require Abbott to conduct a study to test the hypothesis that add-on therapy with fenofibric acid, compared with placebo, significantly lowers the risk of major adverse cardiovascular events in high-risk patients who have reached their LDL cholesterol goal with a statin but have residual high serum TG and low serum HDL cholesterol levels.
The ACCORD Lipid study was sponsored by the National Heart, Lung and Blood Institute.
The FDA usually follows the recommendations of its advisory panels. Members of FDA advisory panels have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, they may be given a waiver, but not at this meeting.
FROM A MEETING OF THE FDA'S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY PANEL
Antiretroviral Therapy Cuts Risk of Sexually Transmitted HIV
The risk of sexually transmitted HIV was reduced by 96% when the infected partner received a combination of three or more antiretroviral drugs, according to the findings of an international study involving 1,763 serodiscordant couples.
The results "are the first from a major randomized trial to indicate that treating an HIV-infected individual can reduce the risk of sexual transmission of HIV to an uninfected partner," according to a statement issued by the National Institute of Allergy and Infectious Diseases (NIAID), which sponsored the study.
Previous studies on the ability of antiretrovirals to prevent sexual transmission of HIV were either observational and epidemiologic.
Results from the current randomized study were so striking that the trial, which began in April 2005 and was slated to continue into 2015, was halted early. The study has not yet been published; the results were reported in the NIAID statement.
"This new finding convincingly demonstrates that treating the infected individual – and doing so sooner rather than later – can have a major impact on reducing HIV transmission," NIAID director Dr. Anthony Fauci said in the statement.
The study enrolled 1,763 mostly heterosexual serodiscordant couples aged 18 years and older in several African countries, Brazil, India, Thailand, and the United States. The HIV-infected partners were randomly assigned to either immediately start triple or quadruple antiretroviral therapy, or to a deferred-treatment group that started therapy only after their CD4 counts dropped below 250 cells/mm3, or they developed pneumocystis pneumonia, or another AIDS-related event. All couples received information and counseling about safe sex and were treated for any HIV-related complications.
There were 39 previously uninfected partners who became infected with HIV; genetic analyses of the viruses in both partners determined that 28 of these individuals were infected from their HIV-infected partners, 7 were infected by another sexual partner, and the results of the remaining 4 have not yet been determined.
Of the 28 cases that were determined to be from the sexual partner in the study, 27 were among the 877 serodiscordant couples in the deferred-treatment group; only one case occurred among those in the immediate-treatment group. This highly significant difference corresponded to a 96% risk reduction attributed to immediate therapy, and spurred the independent data and safety monitoring board’s recommendation that the deferred-treatment arm be stopped early.
In addition to evaluating the impact of antiretroviral therapy on sexual transmission of HIV, the investigators looked at whether immediate treatment altered the course of disease in the HIV-infected individuals. Among the HIV-infected partners in the deferred-treatment group, there were 17 cases of extrapulmonary tuberculosis, compared with 3 cases among the infected partners in the immediate-treatment group, a significant difference. The number of deaths, however, was not significantly different: Ten deaths occurred in the immediate-treatment group versus 13 in the deferred-treatment group.
The study was conducted by the HIV Prevention Trials Network, largely funded by NIAID, and also received funding from the National Institute on Drug Abuse and the National Institute of Mental Health; the AIDS Clinical Trials Group, funded by NIAID, also provided support for the study. The 11 antiretroviral drugs used in various combinations in the in the study were provided by the manufacturers, Abbott Laboratories; Boehringer Ingelheim Pharmaceuticals; Bristol-Myers Squibb; Gilead Sciences; GlaxoSmithKline/ViiV Healthcare; and Merck.
The risk of sexually transmitted HIV was reduced by 96% when the infected partner received a combination of three or more antiretroviral drugs, according to the findings of an international study involving 1,763 serodiscordant couples.
The results "are the first from a major randomized trial to indicate that treating an HIV-infected individual can reduce the risk of sexual transmission of HIV to an uninfected partner," according to a statement issued by the National Institute of Allergy and Infectious Diseases (NIAID), which sponsored the study.
Previous studies on the ability of antiretrovirals to prevent sexual transmission of HIV were either observational and epidemiologic.
Results from the current randomized study were so striking that the trial, which began in April 2005 and was slated to continue into 2015, was halted early. The study has not yet been published; the results were reported in the NIAID statement.
"This new finding convincingly demonstrates that treating the infected individual – and doing so sooner rather than later – can have a major impact on reducing HIV transmission," NIAID director Dr. Anthony Fauci said in the statement.
The study enrolled 1,763 mostly heterosexual serodiscordant couples aged 18 years and older in several African countries, Brazil, India, Thailand, and the United States. The HIV-infected partners were randomly assigned to either immediately start triple or quadruple antiretroviral therapy, or to a deferred-treatment group that started therapy only after their CD4 counts dropped below 250 cells/mm3, or they developed pneumocystis pneumonia, or another AIDS-related event. All couples received information and counseling about safe sex and were treated for any HIV-related complications.
There were 39 previously uninfected partners who became infected with HIV; genetic analyses of the viruses in both partners determined that 28 of these individuals were infected from their HIV-infected partners, 7 were infected by another sexual partner, and the results of the remaining 4 have not yet been determined.
Of the 28 cases that were determined to be from the sexual partner in the study, 27 were among the 877 serodiscordant couples in the deferred-treatment group; only one case occurred among those in the immediate-treatment group. This highly significant difference corresponded to a 96% risk reduction attributed to immediate therapy, and spurred the independent data and safety monitoring board’s recommendation that the deferred-treatment arm be stopped early.
In addition to evaluating the impact of antiretroviral therapy on sexual transmission of HIV, the investigators looked at whether immediate treatment altered the course of disease in the HIV-infected individuals. Among the HIV-infected partners in the deferred-treatment group, there were 17 cases of extrapulmonary tuberculosis, compared with 3 cases among the infected partners in the immediate-treatment group, a significant difference. The number of deaths, however, was not significantly different: Ten deaths occurred in the immediate-treatment group versus 13 in the deferred-treatment group.
The study was conducted by the HIV Prevention Trials Network, largely funded by NIAID, and also received funding from the National Institute on Drug Abuse and the National Institute of Mental Health; the AIDS Clinical Trials Group, funded by NIAID, also provided support for the study. The 11 antiretroviral drugs used in various combinations in the in the study were provided by the manufacturers, Abbott Laboratories; Boehringer Ingelheim Pharmaceuticals; Bristol-Myers Squibb; Gilead Sciences; GlaxoSmithKline/ViiV Healthcare; and Merck.
The risk of sexually transmitted HIV was reduced by 96% when the infected partner received a combination of three or more antiretroviral drugs, according to the findings of an international study involving 1,763 serodiscordant couples.
The results "are the first from a major randomized trial to indicate that treating an HIV-infected individual can reduce the risk of sexual transmission of HIV to an uninfected partner," according to a statement issued by the National Institute of Allergy and Infectious Diseases (NIAID), which sponsored the study.
Previous studies on the ability of antiretrovirals to prevent sexual transmission of HIV were either observational and epidemiologic.
Results from the current randomized study were so striking that the trial, which began in April 2005 and was slated to continue into 2015, was halted early. The study has not yet been published; the results were reported in the NIAID statement.
"This new finding convincingly demonstrates that treating the infected individual – and doing so sooner rather than later – can have a major impact on reducing HIV transmission," NIAID director Dr. Anthony Fauci said in the statement.
The study enrolled 1,763 mostly heterosexual serodiscordant couples aged 18 years and older in several African countries, Brazil, India, Thailand, and the United States. The HIV-infected partners were randomly assigned to either immediately start triple or quadruple antiretroviral therapy, or to a deferred-treatment group that started therapy only after their CD4 counts dropped below 250 cells/mm3, or they developed pneumocystis pneumonia, or another AIDS-related event. All couples received information and counseling about safe sex and were treated for any HIV-related complications.
There were 39 previously uninfected partners who became infected with HIV; genetic analyses of the viruses in both partners determined that 28 of these individuals were infected from their HIV-infected partners, 7 were infected by another sexual partner, and the results of the remaining 4 have not yet been determined.
Of the 28 cases that were determined to be from the sexual partner in the study, 27 were among the 877 serodiscordant couples in the deferred-treatment group; only one case occurred among those in the immediate-treatment group. This highly significant difference corresponded to a 96% risk reduction attributed to immediate therapy, and spurred the independent data and safety monitoring board’s recommendation that the deferred-treatment arm be stopped early.
In addition to evaluating the impact of antiretroviral therapy on sexual transmission of HIV, the investigators looked at whether immediate treatment altered the course of disease in the HIV-infected individuals. Among the HIV-infected partners in the deferred-treatment group, there were 17 cases of extrapulmonary tuberculosis, compared with 3 cases among the infected partners in the immediate-treatment group, a significant difference. The number of deaths, however, was not significantly different: Ten deaths occurred in the immediate-treatment group versus 13 in the deferred-treatment group.
The study was conducted by the HIV Prevention Trials Network, largely funded by NIAID, and also received funding from the National Institute on Drug Abuse and the National Institute of Mental Health; the AIDS Clinical Trials Group, funded by NIAID, also provided support for the study. The 11 antiretroviral drugs used in various combinations in the in the study were provided by the manufacturers, Abbott Laboratories; Boehringer Ingelheim Pharmaceuticals; Bristol-Myers Squibb; Gilead Sciences; GlaxoSmithKline/ViiV Healthcare; and Merck.
FROM THE NATIONAL INSTITUTE OF ALLERGY AND INFECTIOUS DISEASES