User login
To match bone marrow donors and recipients, ask about grandma

Photo by Chad McNeeley
Asking bone marrow donors about their grandparents’ ancestry may help match donors to the appropriate recipients, according to research published in PLOS ONE.
Investigators found that when donors were self-reporting their race/ethnicity and included information on their grandparents’ ancestry, their responses often matched their genetics better than when they simply selected from standard race/ethnicity categories.
The investigators said these results show that more research is needed to refine methods for collecting and interpreting information on race/ethnicity for medical purposes.
“In medicine, we’ve been under the assumption that it’s just a matter of presenting people with the right check boxes,” said Jill Hollenbach, PhD, of the University of California, San Francisco. “It turns out that it’s much more complex than that.”
She and her colleagues noted that identifying a 10/10 human leukocyte antigen (HLA) match in the National Marrow Donor Program’s Be The Match Registry can be difficult because most of the donors submitted samples at a time when genotyping was much less advanced than it is today.
To find likely matches to pursue, the registry uses bioinformatics to weed out the donors unlikely to have matching HLA genes. Although HLA genes are highly variable, certain variations are found at higher frequencies in some regions of the world than others.
A patient with geographic ancestry in East Asia, for example, is more likely to have the same HLA variation as someone with origins in East Asia, rather than someone with European ancestry.
To obtain such information, most medical facilities use the patients’ race/ethnicity self-identification as a proxy. This can be challenging for people who trace their ancestry to multiple origins.
“The United States census uses a “check all that apply” technique, which is OK, but we want to try to get a little more refined to improve our matching efficiency,” said study author Martin Maiers, director of Bioinformatics Research at the Be The Match Registry.
To assess the extent to which different means of self-identification correspond to genetic ancestry, the investigators recruited 1752 potential donors from the Be The Match Registry.
The team sent participants a questionnaire with multiple measures of self-identification, including race/ethnicity and geographic ancestry, and asked participants to assign geographic ancestry to their grandparents. Participants also submitted cheek swabs as DNA samples.
By analyzing the cheek swabs, the investigators genotyped 93 ancestry informative markers to identify the participants’ genetic ancestry. This revealed a strong correlation between the ancestry markers and HLA genes.
Unfortunately, no measure of self-identification showed complete correspondence with a donor’s genetic ancestry. However, information about the geographic origins of the donors’ grandparents corresponded most closely with genetics, particularly for participants with ancestry from multiple continents.
“The conventional wisdom of the last decade or two has been that race is a social construct,” Dr Hollenbach said. “That’s true, but race is also part of the language of self-identification in [the US]. Let’s understand how these different forms of self-identification and genetic ancestry intersect to improve transplant matching for people of all ancestral backgrounds.” ![]()
Photo by Chad McNeeley
Asking bone marrow donors about their grandparents’ ancestry may help match donors to the appropriate recipients, according to research published in PLOS ONE.
Investigators found that when donors were self-reporting their race/ethnicity and included information on their grandparents’ ancestry, their responses often matched their genetics better than when they simply selected from standard race/ethnicity categories.
The investigators said these results show that more research is needed to refine methods for collecting and interpreting information on race/ethnicity for medical purposes.
“In medicine, we’ve been under the assumption that it’s just a matter of presenting people with the right check boxes,” said Jill Hollenbach, PhD, of the University of California, San Francisco. “It turns out that it’s much more complex than that.”
She and her colleagues noted that identifying a 10/10 human leukocyte antigen (HLA) match in the National Marrow Donor Program’s Be The Match Registry can be difficult because most of the donors submitted samples at a time when genotyping was much less advanced than it is today.
To find likely matches to pursue, the registry uses bioinformatics to weed out the donors unlikely to have matching HLA genes. Although HLA genes are highly variable, certain variations are found at higher frequencies in some regions of the world than others.
A patient with geographic ancestry in East Asia, for example, is more likely to have the same HLA variation as someone with origins in East Asia, rather than someone with European ancestry.
To obtain such information, most medical facilities use the patients’ race/ethnicity self-identification as a proxy. This can be challenging for people who trace their ancestry to multiple origins.
“The United States census uses a “check all that apply” technique, which is OK, but we want to try to get a little more refined to improve our matching efficiency,” said study author Martin Maiers, director of Bioinformatics Research at the Be The Match Registry.
To assess the extent to which different means of self-identification correspond to genetic ancestry, the investigators recruited 1752 potential donors from the Be The Match Registry.
The team sent participants a questionnaire with multiple measures of self-identification, including race/ethnicity and geographic ancestry, and asked participants to assign geographic ancestry to their grandparents. Participants also submitted cheek swabs as DNA samples.
By analyzing the cheek swabs, the investigators genotyped 93 ancestry informative markers to identify the participants’ genetic ancestry. This revealed a strong correlation between the ancestry markers and HLA genes.
Unfortunately, no measure of self-identification showed complete correspondence with a donor’s genetic ancestry. However, information about the geographic origins of the donors’ grandparents corresponded most closely with genetics, particularly for participants with ancestry from multiple continents.
“The conventional wisdom of the last decade or two has been that race is a social construct,” Dr Hollenbach said. “That’s true, but race is also part of the language of self-identification in [the US]. Let’s understand how these different forms of self-identification and genetic ancestry intersect to improve transplant matching for people of all ancestral backgrounds.” ![]()
Photo by Chad McNeeley
Asking bone marrow donors about their grandparents’ ancestry may help match donors to the appropriate recipients, according to research published in PLOS ONE.
Investigators found that when donors were self-reporting their race/ethnicity and included information on their grandparents’ ancestry, their responses often matched their genetics better than when they simply selected from standard race/ethnicity categories.
The investigators said these results show that more research is needed to refine methods for collecting and interpreting information on race/ethnicity for medical purposes.
“In medicine, we’ve been under the assumption that it’s just a matter of presenting people with the right check boxes,” said Jill Hollenbach, PhD, of the University of California, San Francisco. “It turns out that it’s much more complex than that.”
She and her colleagues noted that identifying a 10/10 human leukocyte antigen (HLA) match in the National Marrow Donor Program’s Be The Match Registry can be difficult because most of the donors submitted samples at a time when genotyping was much less advanced than it is today.
To find likely matches to pursue, the registry uses bioinformatics to weed out the donors unlikely to have matching HLA genes. Although HLA genes are highly variable, certain variations are found at higher frequencies in some regions of the world than others.
A patient with geographic ancestry in East Asia, for example, is more likely to have the same HLA variation as someone with origins in East Asia, rather than someone with European ancestry.
To obtain such information, most medical facilities use the patients’ race/ethnicity self-identification as a proxy. This can be challenging for people who trace their ancestry to multiple origins.
“The United States census uses a “check all that apply” technique, which is OK, but we want to try to get a little more refined to improve our matching efficiency,” said study author Martin Maiers, director of Bioinformatics Research at the Be The Match Registry.
To assess the extent to which different means of self-identification correspond to genetic ancestry, the investigators recruited 1752 potential donors from the Be The Match Registry.
The team sent participants a questionnaire with multiple measures of self-identification, including race/ethnicity and geographic ancestry, and asked participants to assign geographic ancestry to their grandparents. Participants also submitted cheek swabs as DNA samples.
By analyzing the cheek swabs, the investigators genotyped 93 ancestry informative markers to identify the participants’ genetic ancestry. This revealed a strong correlation between the ancestry markers and HLA genes.
Unfortunately, no measure of self-identification showed complete correspondence with a donor’s genetic ancestry. However, information about the geographic origins of the donors’ grandparents corresponded most closely with genetics, particularly for participants with ancestry from multiple continents.
“The conventional wisdom of the last decade or two has been that race is a social construct,” Dr Hollenbach said. “That’s true, but race is also part of the language of self-identification in [the US]. Let’s understand how these different forms of self-identification and genetic ancestry intersect to improve transplant matching for people of all ancestral backgrounds.” ![]()
Blood-cleansing device on the way to the clinic, team says
for Staphylococcus infection
Photo by Bill Branson
Last year, researchers reported promising preclinical results with a device that can treat sepsis by mimicking the human spleen. The device filtered pathogens and toxins from the blood by passing it through a dialysis-like circuit.
Now, the researchers have developed a new, more streamlined device that, they believe, is more likely to translate to the clinic. The new device also synergizes with conventional antibiotic therapies.
The team described this device in Biomaterials.
“The inflammatory cascade that leads to sepsis is triggered by pathogens and, specifically, by the toxins they release,” said study author Donald Ingber, MD, PhD, of the Wyss Institute for Biologically Inspired Engineering at Harvard University in Cambridge, Massachusetts.
“Thus, the most effective strategy is to treat with the best antibiotics you can muster, while also removing the toxins and remaining pathogens from the patient’s blood as quickly as possible.”
How the device works
The researchers say their new blood-cleansing approach can be completed quickly, without even identifying the infectious agent. This is because the device uses a proprietary pathogen-capturing agent, known as FcMBL, that binds all types of live and dead infectious microbes, including bacteria, fungi, viruses, and the toxins they release.
FcMBL is a genetically engineered blood protein inspired by a naturally occurring human molecule called mannose binding lectin (MBL). MBL is found in the innate immune system and binds to toxic invaders, marking them for capture by immune cells in the spleen.
The researchers’ original device concept was similar to how a dialysis machine works. Infected blood in an animal, or potentially one day in a patient, is flowed from one vein through catheters to the device.
There, FcMBL-coated magnetic beads are added to the blood, and the bead-bound pathogens are extracted from the circulating blood by magnets within the device before the cleansed blood is returned to the body through another vein.
The new device removes the complexity, regulatory challenges, and cost associated with the magnetic beads and microfluidic architecture of its predecessor. But it still uses the FcMBL protein to bind to pathogens and toxins.
The new system uses hollow fiber filters similar to those currently used in hemodialysis, but the inner walls of these filters are coated with FcMBL protein to remove pathogens from circulating blood.
Test results
In in vitro tests with human blood, the device reduced the number of Gram-negative bacteria (Escherichia coli), Gram-positive bacteria (Staphylococcus aureus), fungi (Candida albicans), and lipopolysaccharide-endotoxins by 90% to 99%.
In tests with rats, the device reduced levels of circulating pathogens and endotoxins by more than 99%, and it prevented pathogen engraftment and inflammatory cell recruitment in the spleen, lung, liver, and kidney.
The researchers also tested the device in combination with bacteriocidal antibiotics in rats. The antibiotics prompted a “major increase” in the release of microbial fragments, or pathogen-associated molecular patterns (PAMPs).
The device could remove PAMPs from the blood within 2 hours, but high levels of PAMPs remained in rats treated with antibiotics alone.
The researchers said PAMP removal reduced organ pathogen and endotoxin loads, suppressed inflammatory responses, and resulted in more stable vital signs.
“Using the device, alone or alongside antibiotics, we can quickly bring blood back to normal conditions, curtailing an inflammatory response rather than exacerbating it,” said Tohid Fatanat Didar, PhD, of the Wyss Institute.
“If all goes well, physicians will someday be able to use the device in tandem with standard antibiotic treatments to deliver a one-two punch to pathogens, synergistically killing and cleansing all live and dead invaders from the bloodstream.”
As the device has proven effective in small animal experiments, the researchers are planning to move to large animal studies. They must demonstrate proof-of-concept in these models before advancing to clinical trials.
“Our goal is to see this move out of the lab and into hospitals, as well as onto the battlefield,” Dr Ingber said, “where it can save lives within years rather than decades.” ![]()
for Staphylococcus infection
Photo by Bill Branson
Last year, researchers reported promising preclinical results with a device that can treat sepsis by mimicking the human spleen. The device filtered pathogens and toxins from the blood by passing it through a dialysis-like circuit.
Now, the researchers have developed a new, more streamlined device that, they believe, is more likely to translate to the clinic. The new device also synergizes with conventional antibiotic therapies.
The team described this device in Biomaterials.
“The inflammatory cascade that leads to sepsis is triggered by pathogens and, specifically, by the toxins they release,” said study author Donald Ingber, MD, PhD, of the Wyss Institute for Biologically Inspired Engineering at Harvard University in Cambridge, Massachusetts.
“Thus, the most effective strategy is to treat with the best antibiotics you can muster, while also removing the toxins and remaining pathogens from the patient’s blood as quickly as possible.”
How the device works
The researchers say their new blood-cleansing approach can be completed quickly, without even identifying the infectious agent. This is because the device uses a proprietary pathogen-capturing agent, known as FcMBL, that binds all types of live and dead infectious microbes, including bacteria, fungi, viruses, and the toxins they release.
FcMBL is a genetically engineered blood protein inspired by a naturally occurring human molecule called mannose binding lectin (MBL). MBL is found in the innate immune system and binds to toxic invaders, marking them for capture by immune cells in the spleen.
The researchers’ original device concept was similar to how a dialysis machine works. Infected blood in an animal, or potentially one day in a patient, is flowed from one vein through catheters to the device.
There, FcMBL-coated magnetic beads are added to the blood, and the bead-bound pathogens are extracted from the circulating blood by magnets within the device before the cleansed blood is returned to the body through another vein.
The new device removes the complexity, regulatory challenges, and cost associated with the magnetic beads and microfluidic architecture of its predecessor. But it still uses the FcMBL protein to bind to pathogens and toxins.
The new system uses hollow fiber filters similar to those currently used in hemodialysis, but the inner walls of these filters are coated with FcMBL protein to remove pathogens from circulating blood.
Test results
In in vitro tests with human blood, the device reduced the number of Gram-negative bacteria (Escherichia coli), Gram-positive bacteria (Staphylococcus aureus), fungi (Candida albicans), and lipopolysaccharide-endotoxins by 90% to 99%.
In tests with rats, the device reduced levels of circulating pathogens and endotoxins by more than 99%, and it prevented pathogen engraftment and inflammatory cell recruitment in the spleen, lung, liver, and kidney.
The researchers also tested the device in combination with bacteriocidal antibiotics in rats. The antibiotics prompted a “major increase” in the release of microbial fragments, or pathogen-associated molecular patterns (PAMPs).
The device could remove PAMPs from the blood within 2 hours, but high levels of PAMPs remained in rats treated with antibiotics alone.
The researchers said PAMP removal reduced organ pathogen and endotoxin loads, suppressed inflammatory responses, and resulted in more stable vital signs.
“Using the device, alone or alongside antibiotics, we can quickly bring blood back to normal conditions, curtailing an inflammatory response rather than exacerbating it,” said Tohid Fatanat Didar, PhD, of the Wyss Institute.
“If all goes well, physicians will someday be able to use the device in tandem with standard antibiotic treatments to deliver a one-two punch to pathogens, synergistically killing and cleansing all live and dead invaders from the bloodstream.”
As the device has proven effective in small animal experiments, the researchers are planning to move to large animal studies. They must demonstrate proof-of-concept in these models before advancing to clinical trials.
“Our goal is to see this move out of the lab and into hospitals, as well as onto the battlefield,” Dr Ingber said, “where it can save lives within years rather than decades.” ![]()
for Staphylococcus infection
Photo by Bill Branson
Last year, researchers reported promising preclinical results with a device that can treat sepsis by mimicking the human spleen. The device filtered pathogens and toxins from the blood by passing it through a dialysis-like circuit.
Now, the researchers have developed a new, more streamlined device that, they believe, is more likely to translate to the clinic. The new device also synergizes with conventional antibiotic therapies.
The team described this device in Biomaterials.
“The inflammatory cascade that leads to sepsis is triggered by pathogens and, specifically, by the toxins they release,” said study author Donald Ingber, MD, PhD, of the Wyss Institute for Biologically Inspired Engineering at Harvard University in Cambridge, Massachusetts.
“Thus, the most effective strategy is to treat with the best antibiotics you can muster, while also removing the toxins and remaining pathogens from the patient’s blood as quickly as possible.”
How the device works
The researchers say their new blood-cleansing approach can be completed quickly, without even identifying the infectious agent. This is because the device uses a proprietary pathogen-capturing agent, known as FcMBL, that binds all types of live and dead infectious microbes, including bacteria, fungi, viruses, and the toxins they release.
FcMBL is a genetically engineered blood protein inspired by a naturally occurring human molecule called mannose binding lectin (MBL). MBL is found in the innate immune system and binds to toxic invaders, marking them for capture by immune cells in the spleen.
The researchers’ original device concept was similar to how a dialysis machine works. Infected blood in an animal, or potentially one day in a patient, is flowed from one vein through catheters to the device.
There, FcMBL-coated magnetic beads are added to the blood, and the bead-bound pathogens are extracted from the circulating blood by magnets within the device before the cleansed blood is returned to the body through another vein.
The new device removes the complexity, regulatory challenges, and cost associated with the magnetic beads and microfluidic architecture of its predecessor. But it still uses the FcMBL protein to bind to pathogens and toxins.
The new system uses hollow fiber filters similar to those currently used in hemodialysis, but the inner walls of these filters are coated with FcMBL protein to remove pathogens from circulating blood.
Test results
In in vitro tests with human blood, the device reduced the number of Gram-negative bacteria (Escherichia coli), Gram-positive bacteria (Staphylococcus aureus), fungi (Candida albicans), and lipopolysaccharide-endotoxins by 90% to 99%.
In tests with rats, the device reduced levels of circulating pathogens and endotoxins by more than 99%, and it prevented pathogen engraftment and inflammatory cell recruitment in the spleen, lung, liver, and kidney.
The researchers also tested the device in combination with bacteriocidal antibiotics in rats. The antibiotics prompted a “major increase” in the release of microbial fragments, or pathogen-associated molecular patterns (PAMPs).
The device could remove PAMPs from the blood within 2 hours, but high levels of PAMPs remained in rats treated with antibiotics alone.
The researchers said PAMP removal reduced organ pathogen and endotoxin loads, suppressed inflammatory responses, and resulted in more stable vital signs.
“Using the device, alone or alongside antibiotics, we can quickly bring blood back to normal conditions, curtailing an inflammatory response rather than exacerbating it,” said Tohid Fatanat Didar, PhD, of the Wyss Institute.
“If all goes well, physicians will someday be able to use the device in tandem with standard antibiotic treatments to deliver a one-two punch to pathogens, synergistically killing and cleansing all live and dead invaders from the bloodstream.”
As the device has proven effective in small animal experiments, the researchers are planning to move to large animal studies. They must demonstrate proof-of-concept in these models before advancing to clinical trials.
“Our goal is to see this move out of the lab and into hospitals, as well as onto the battlefield,” Dr Ingber said, “where it can save lives within years rather than decades.” ![]()
T-cell finding may have broad implications

resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
NICE drafts guideline for treating myeloma patients

Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has developed a draft guideline detailing “best practices” in caring for patients with myeloma.
It is intended to help ensure “consistently excellent care” for myeloma patients over the age of 16 in England.
The guideline includes recommendations for diagnosing the disease, managing complications, communicating with patients, and ensuring access to appropriate care.
The draft guideline will remain open for public consultation until October 1, 2015.
“Advances in treatment over the last 15 years have seen more people with myeloma living longer, but there is still no cure,” said Mark Baker, clinical practice director for NICE.
“Our guideline, which is being developed by an independent group of experts, will set out best-practice care to ensure people live as normal a life as possible for as long as possible.”
The guideline’s provisional recommendations are as follows.
Communication and support: Offer prompt psychological assessment and support to myeloma patients at diagnosis and, as appropriate, at the beginning and end of each treatment, when the disease progresses, and when patients start to require end-of-life care.
Laboratory investigations to diagnose myeloma: For patients with suspected myeloma, healthcare professionals should use the same sample for all diagnostic and prognostic tests on bone marrow, so patients only have to have one biopsy.
Scans for patients with suspected myeloma: Offer imaging to all patients with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service delivery: Each hospital treating patients with myeloma who are over the age of 18 should ensure there is regional access to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, particularly early phase trials.
Managing complications: Healthcare professionals should consider extending the pneumococcal vaccination to patients with myeloma who are under 65 in order to prevent infection.
This draft guideline also complements existing NICE guidance on the drug treatment of myeloma. It sets out which treatments—including stem cell transplants—should be used to manage the condition as well as those to prevent and treat bone disease and acute renal disease, which can be caused by myeloma. ![]()
Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has developed a draft guideline detailing “best practices” in caring for patients with myeloma.
It is intended to help ensure “consistently excellent care” for myeloma patients over the age of 16 in England.
The guideline includes recommendations for diagnosing the disease, managing complications, communicating with patients, and ensuring access to appropriate care.
The draft guideline will remain open for public consultation until October 1, 2015.
“Advances in treatment over the last 15 years have seen more people with myeloma living longer, but there is still no cure,” said Mark Baker, clinical practice director for NICE.
“Our guideline, which is being developed by an independent group of experts, will set out best-practice care to ensure people live as normal a life as possible for as long as possible.”
The guideline’s provisional recommendations are as follows.
Communication and support: Offer prompt psychological assessment and support to myeloma patients at diagnosis and, as appropriate, at the beginning and end of each treatment, when the disease progresses, and when patients start to require end-of-life care.
Laboratory investigations to diagnose myeloma: For patients with suspected myeloma, healthcare professionals should use the same sample for all diagnostic and prognostic tests on bone marrow, so patients only have to have one biopsy.
Scans for patients with suspected myeloma: Offer imaging to all patients with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service delivery: Each hospital treating patients with myeloma who are over the age of 18 should ensure there is regional access to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, particularly early phase trials.
Managing complications: Healthcare professionals should consider extending the pneumococcal vaccination to patients with myeloma who are under 65 in order to prevent infection.
This draft guideline also complements existing NICE guidance on the drug treatment of myeloma. It sets out which treatments—including stem cell transplants—should be used to manage the condition as well as those to prevent and treat bone disease and acute renal disease, which can be caused by myeloma. ![]()
Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has developed a draft guideline detailing “best practices” in caring for patients with myeloma.
It is intended to help ensure “consistently excellent care” for myeloma patients over the age of 16 in England.
The guideline includes recommendations for diagnosing the disease, managing complications, communicating with patients, and ensuring access to appropriate care.
The draft guideline will remain open for public consultation until October 1, 2015.
“Advances in treatment over the last 15 years have seen more people with myeloma living longer, but there is still no cure,” said Mark Baker, clinical practice director for NICE.
“Our guideline, which is being developed by an independent group of experts, will set out best-practice care to ensure people live as normal a life as possible for as long as possible.”
The guideline’s provisional recommendations are as follows.
Communication and support: Offer prompt psychological assessment and support to myeloma patients at diagnosis and, as appropriate, at the beginning and end of each treatment, when the disease progresses, and when patients start to require end-of-life care.
Laboratory investigations to diagnose myeloma: For patients with suspected myeloma, healthcare professionals should use the same sample for all diagnostic and prognostic tests on bone marrow, so patients only have to have one biopsy.
Scans for patients with suspected myeloma: Offer imaging to all patients with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service delivery: Each hospital treating patients with myeloma who are over the age of 18 should ensure there is regional access to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, particularly early phase trials.
Managing complications: Healthcare professionals should consider extending the pneumococcal vaccination to patients with myeloma who are under 65 in order to prevent infection.
This draft guideline also complements existing NICE guidance on the drug treatment of myeloma. It sets out which treatments—including stem cell transplants—should be used to manage the condition as well as those to prevent and treat bone disease and acute renal disease, which can be caused by myeloma. ![]()
Team may have found earliest case of leukemia
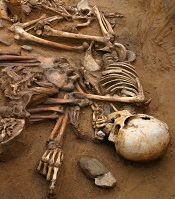
archaeological dig
(male, Late Bronze Age)
Photo by Wessex Archaeology
ZURICH—Researchers may have discovered the earliest known case of leukemia by imaging a 7000-year-old skeleton.
High-resolution CT scans revealed locally defined trabecular bone resorption in the sternum and humerus, which strongly suggests a case of leukemia, according to the researchers.
Heike Scherf, PhD, of Eberhard Karls Universität Tübingen in Tübingen, Germany, and her colleagues presented this discovery at the Evolutionary Medicine Conference: Interdisciplinary Perspectives on Human Health and Disease (abstract P-07).
A poster describing the research is available on ResearchGate.
Dr Scherf and her colleagues described a skeleton recovered from the early Neolithic Linear Pottery Culture site Stuttgart-Muhlhausen in southwest Germany, which dates back to 5700-4900 BP.
The skeleton belonged to a woman who was thought to be 30 to 40 years old at the time of her death. Previous analysis had revealed a severe case of dental caries with alveolar inflammation in this woman.
The high-resolution CT scans revealed loss of trabecular bone in both the humerus and the sternum.
Dr Scherf and her colleagues said the level of trabecular resorption in this skeleton is significantly higher than that observed in specimens of the same age group from the same site, as well as in recent human samples.
The researchers were able to rule out other conditions that might explain the bone resorption, including osteoporosis, hyperparathyroidism, and solid tumor malignancy.
They said the locally restricted resorption at sites of hematopoietic stem cell generation strongly suggests leukemia in its initial stages, although they were unable to identify the subtype.
Still, if the researchers’ interpretation of their findings is correct, this is the earliest case of leukemia reported. ![]()
archaeological dig
(male, Late Bronze Age)
Photo by Wessex Archaeology
ZURICH—Researchers may have discovered the earliest known case of leukemia by imaging a 7000-year-old skeleton.
High-resolution CT scans revealed locally defined trabecular bone resorption in the sternum and humerus, which strongly suggests a case of leukemia, according to the researchers.
Heike Scherf, PhD, of Eberhard Karls Universität Tübingen in Tübingen, Germany, and her colleagues presented this discovery at the Evolutionary Medicine Conference: Interdisciplinary Perspectives on Human Health and Disease (abstract P-07).
A poster describing the research is available on ResearchGate.
Dr Scherf and her colleagues described a skeleton recovered from the early Neolithic Linear Pottery Culture site Stuttgart-Muhlhausen in southwest Germany, which dates back to 5700-4900 BP.
The skeleton belonged to a woman who was thought to be 30 to 40 years old at the time of her death. Previous analysis had revealed a severe case of dental caries with alveolar inflammation in this woman.
The high-resolution CT scans revealed loss of trabecular bone in both the humerus and the sternum.
Dr Scherf and her colleagues said the level of trabecular resorption in this skeleton is significantly higher than that observed in specimens of the same age group from the same site, as well as in recent human samples.
The researchers were able to rule out other conditions that might explain the bone resorption, including osteoporosis, hyperparathyroidism, and solid tumor malignancy.
They said the locally restricted resorption at sites of hematopoietic stem cell generation strongly suggests leukemia in its initial stages, although they were unable to identify the subtype.
Still, if the researchers’ interpretation of their findings is correct, this is the earliest case of leukemia reported. ![]()
archaeological dig
(male, Late Bronze Age)
Photo by Wessex Archaeology
ZURICH—Researchers may have discovered the earliest known case of leukemia by imaging a 7000-year-old skeleton.
High-resolution CT scans revealed locally defined trabecular bone resorption in the sternum and humerus, which strongly suggests a case of leukemia, according to the researchers.
Heike Scherf, PhD, of Eberhard Karls Universität Tübingen in Tübingen, Germany, and her colleagues presented this discovery at the Evolutionary Medicine Conference: Interdisciplinary Perspectives on Human Health and Disease (abstract P-07).
A poster describing the research is available on ResearchGate.
Dr Scherf and her colleagues described a skeleton recovered from the early Neolithic Linear Pottery Culture site Stuttgart-Muhlhausen in southwest Germany, which dates back to 5700-4900 BP.
The skeleton belonged to a woman who was thought to be 30 to 40 years old at the time of her death. Previous analysis had revealed a severe case of dental caries with alveolar inflammation in this woman.
The high-resolution CT scans revealed loss of trabecular bone in both the humerus and the sternum.
Dr Scherf and her colleagues said the level of trabecular resorption in this skeleton is significantly higher than that observed in specimens of the same age group from the same site, as well as in recent human samples.
The researchers were able to rule out other conditions that might explain the bone resorption, including osteoporosis, hyperparathyroidism, and solid tumor malignancy.
They said the locally restricted resorption at sites of hematopoietic stem cell generation strongly suggests leukemia in its initial stages, although they were unable to identify the subtype.
Still, if the researchers’ interpretation of their findings is correct, this is the earliest case of leukemia reported. ![]()
Product can eliminate inhibitors in hemophilia A

A plasma-derived factor VIII/von Willebrand factor product (Octanate) can eliminate inhibitors in patients with hemophilia A, according to research published in Haemophilia.
Octanate eradicated inhibitors in nearly 80% of patients studied, and patients who became inhibitor-free experienced an 86% reduction in monthly bleeding.
Adverse drug reactions occurred in about 42% of patients, and 4 of the 124 adverse events were considered serious.
Wolfhart Kreuz, MD, of HZRM (Haemophilia Centre Rhein Main) in Mörfelden-Walldorf, Germany, and his colleagues conducted this research as part of the ongoing ObsITI study. The trial enrolled 48 patients with hemophilia A and inhibitors.
Most patients–83.3% (n=40)—had 1 or more poor prognostic factors for immune tolerance induction (ITI) success. This includes age of 7 or older, having been diagnosed with inhibitors for 2 years or more, having an inhibitor titer of 10 BU or higher at the start of ITI, and prior ITI failure.
Patients received Octanate as the sole factor VIII product between December 2005 and October 2010. Patients who were “low responders” at the start of ITI (<5 BU) received 50–100 IU factor VIII kg-1 daily or every other day. And “high responders” (≥5 BU) received 100 IU factor VIII kg-1 every 12 hours.
During ITI, 4 patients received prophylaxis with bypassing agents—2 with activated prothrombin complex concentrates and 2 with recombinant factor VIIa.
The researchers assessed ITI efficacy according to 3 criteria. These were: (1) inhibitor titer <0.6 BU, (2) factor VIII recovery ≥80% of the predefined reference value of 1.5% IU-1 kg-1 body weight ≤1 hour post-injection, and (3) factor VIII half-life ≥7 hours.
The researchers defined “complete success” as meeting all 3 efficacy criteria, “partial success” as meeting 2 criteria, and “partial response” as meeting 1 criterion. If none of the criteria were met within the 36-month observation period, this was considered an ITI failure. Withdrawal from the study for administrative reasons was considered an ITI failure as well.
Treatment was deemed a complete success in 70.8% of patients (n=34) and a partial success in 6.3% (n=3). One patient (2.1%) had a partial response. Treatment failed in 20.8% of patients (n=10), all of whom had poor prognostic factors.
Overall, 79.2% of patients (38/48) became negative for inhibitors. These patients saw an 86% reduction in their monthly bleeding rate (P<0.0001), which decreased most dramatically during the initial 4 months of ITI. During that period, the mean monthly bleeding rate fell from 1.05 to 0.26.
The researchers noted that treatment outcome was significantly associated with a patient’s inhibitor titer level at the start of ITI (P=0.0068), the number of poor prognostic factors (P=0.0187), the monthly bleeding rate during ITI (P=0.0005), and peak inhibitor titer during ITI (P=0.0007).
There were 124 adverse drug reactions reported in 20 patients (41.7%). And there were 4 serious adverse drug reactions—intravenous catheter infection, febrile convulsion, gingivitis, and pyrexia. One reaction—allergic dermatitis—was considered possibly or probably related to treatment. ![]()
A plasma-derived factor VIII/von Willebrand factor product (Octanate) can eliminate inhibitors in patients with hemophilia A, according to research published in Haemophilia.
Octanate eradicated inhibitors in nearly 80% of patients studied, and patients who became inhibitor-free experienced an 86% reduction in monthly bleeding.
Adverse drug reactions occurred in about 42% of patients, and 4 of the 124 adverse events were considered serious.
Wolfhart Kreuz, MD, of HZRM (Haemophilia Centre Rhein Main) in Mörfelden-Walldorf, Germany, and his colleagues conducted this research as part of the ongoing ObsITI study. The trial enrolled 48 patients with hemophilia A and inhibitors.
Most patients–83.3% (n=40)—had 1 or more poor prognostic factors for immune tolerance induction (ITI) success. This includes age of 7 or older, having been diagnosed with inhibitors for 2 years or more, having an inhibitor titer of 10 BU or higher at the start of ITI, and prior ITI failure.
Patients received Octanate as the sole factor VIII product between December 2005 and October 2010. Patients who were “low responders” at the start of ITI (<5 BU) received 50–100 IU factor VIII kg-1 daily or every other day. And “high responders” (≥5 BU) received 100 IU factor VIII kg-1 every 12 hours.
During ITI, 4 patients received prophylaxis with bypassing agents—2 with activated prothrombin complex concentrates and 2 with recombinant factor VIIa.
The researchers assessed ITI efficacy according to 3 criteria. These were: (1) inhibitor titer <0.6 BU, (2) factor VIII recovery ≥80% of the predefined reference value of 1.5% IU-1 kg-1 body weight ≤1 hour post-injection, and (3) factor VIII half-life ≥7 hours.
The researchers defined “complete success” as meeting all 3 efficacy criteria, “partial success” as meeting 2 criteria, and “partial response” as meeting 1 criterion. If none of the criteria were met within the 36-month observation period, this was considered an ITI failure. Withdrawal from the study for administrative reasons was considered an ITI failure as well.
Treatment was deemed a complete success in 70.8% of patients (n=34) and a partial success in 6.3% (n=3). One patient (2.1%) had a partial response. Treatment failed in 20.8% of patients (n=10), all of whom had poor prognostic factors.
Overall, 79.2% of patients (38/48) became negative for inhibitors. These patients saw an 86% reduction in their monthly bleeding rate (P<0.0001), which decreased most dramatically during the initial 4 months of ITI. During that period, the mean monthly bleeding rate fell from 1.05 to 0.26.
The researchers noted that treatment outcome was significantly associated with a patient’s inhibitor titer level at the start of ITI (P=0.0068), the number of poor prognostic factors (P=0.0187), the monthly bleeding rate during ITI (P=0.0005), and peak inhibitor titer during ITI (P=0.0007).
There were 124 adverse drug reactions reported in 20 patients (41.7%). And there were 4 serious adverse drug reactions—intravenous catheter infection, febrile convulsion, gingivitis, and pyrexia. One reaction—allergic dermatitis—was considered possibly or probably related to treatment. ![]()
A plasma-derived factor VIII/von Willebrand factor product (Octanate) can eliminate inhibitors in patients with hemophilia A, according to research published in Haemophilia.
Octanate eradicated inhibitors in nearly 80% of patients studied, and patients who became inhibitor-free experienced an 86% reduction in monthly bleeding.
Adverse drug reactions occurred in about 42% of patients, and 4 of the 124 adverse events were considered serious.
Wolfhart Kreuz, MD, of HZRM (Haemophilia Centre Rhein Main) in Mörfelden-Walldorf, Germany, and his colleagues conducted this research as part of the ongoing ObsITI study. The trial enrolled 48 patients with hemophilia A and inhibitors.
Most patients–83.3% (n=40)—had 1 or more poor prognostic factors for immune tolerance induction (ITI) success. This includes age of 7 or older, having been diagnosed with inhibitors for 2 years or more, having an inhibitor titer of 10 BU or higher at the start of ITI, and prior ITI failure.
Patients received Octanate as the sole factor VIII product between December 2005 and October 2010. Patients who were “low responders” at the start of ITI (<5 BU) received 50–100 IU factor VIII kg-1 daily or every other day. And “high responders” (≥5 BU) received 100 IU factor VIII kg-1 every 12 hours.
During ITI, 4 patients received prophylaxis with bypassing agents—2 with activated prothrombin complex concentrates and 2 with recombinant factor VIIa.
The researchers assessed ITI efficacy according to 3 criteria. These were: (1) inhibitor titer <0.6 BU, (2) factor VIII recovery ≥80% of the predefined reference value of 1.5% IU-1 kg-1 body weight ≤1 hour post-injection, and (3) factor VIII half-life ≥7 hours.
The researchers defined “complete success” as meeting all 3 efficacy criteria, “partial success” as meeting 2 criteria, and “partial response” as meeting 1 criterion. If none of the criteria were met within the 36-month observation period, this was considered an ITI failure. Withdrawal from the study for administrative reasons was considered an ITI failure as well.
Treatment was deemed a complete success in 70.8% of patients (n=34) and a partial success in 6.3% (n=3). One patient (2.1%) had a partial response. Treatment failed in 20.8% of patients (n=10), all of whom had poor prognostic factors.
Overall, 79.2% of patients (38/48) became negative for inhibitors. These patients saw an 86% reduction in their monthly bleeding rate (P<0.0001), which decreased most dramatically during the initial 4 months of ITI. During that period, the mean monthly bleeding rate fell from 1.05 to 0.26.
The researchers noted that treatment outcome was significantly associated with a patient’s inhibitor titer level at the start of ITI (P=0.0068), the number of poor prognostic factors (P=0.0187), the monthly bleeding rate during ITI (P=0.0005), and peak inhibitor titer during ITI (P=0.0007).
There were 124 adverse drug reactions reported in 20 patients (41.7%). And there were 4 serious adverse drug reactions—intravenous catheter infection, febrile convulsion, gingivitis, and pyrexia. One reaction—allergic dermatitis—was considered possibly or probably related to treatment. ![]()
FDA clears automated system for testing blood

Photo courtesy of
Ortho Clinical Diagnostics
The US Food and Drug Administration (FDA) has granted 510(k) clearance to the ORTHO VISIONTM Analyzer, a system that automates in vitro testing of human blood.
The system is now commercially available in the US and Puerto Rico.
The ORTHO VISION Analyzer automates test processing functions, including liquid pipetting, reagent handling, incubation, centrifugation, reaction grading, and interpretation and data management requirements using ID-MTS Gel Cards and digital image processing.
Tests that can be performed with the system include:
- ABO/Rh/grouping
- ABO/Rh confirmation
- Antibody screen
- Antibody identification
- Selected cell panel
- Rh phenotype (C,c,E,e)
- Donor confirmation
- Crossmatch (AHG)
- Antigen typing
- Serial dilutions for titration studies
- DAT (polyspecific)
- DAT (IGG)
- Cord blood testing
The system can be used as a standalone instrument or interfaced to a laboratory information system.
The ORTHO VISION Analyzer was designed with secure monitoring technologies for safety checks and balances, and it allows transfusion medicine professionals to track steps in the immunohematology testing process.
Through Ortho Clinical Diagnostics’ proprietary Intellicheck Technology, the ORTHO VISION Analyzer verifies and documents diagnostic checks throughout the testing process, while e-Connectivity Technology provides 24/7 remote data tracking that monitors instrument performance while maximizing uptime. Laboratory personnel can log on anytime, anywhere to collaborate on interpreting results in real time.
“With the launch of the ORTHO VISION Analyzer, our goal is to help improve the safety of blood transfusions by reducing the lab’s reliance on manual methods,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics, the company developing the ORTHO VISION Analyzer.
A version of the ORTHO VISION Analyzer is already commercially available in Europe, Japan, Latin America, Canada, and Australia.
For more information on the system, visit the Ortho Clinical Diagnostics website. ![]()
Photo courtesy of
Ortho Clinical Diagnostics
The US Food and Drug Administration (FDA) has granted 510(k) clearance to the ORTHO VISIONTM Analyzer, a system that automates in vitro testing of human blood.
The system is now commercially available in the US and Puerto Rico.
The ORTHO VISION Analyzer automates test processing functions, including liquid pipetting, reagent handling, incubation, centrifugation, reaction grading, and interpretation and data management requirements using ID-MTS Gel Cards and digital image processing.
Tests that can be performed with the system include:
- ABO/Rh/grouping
- ABO/Rh confirmation
- Antibody screen
- Antibody identification
- Selected cell panel
- Rh phenotype (C,c,E,e)
- Donor confirmation
- Crossmatch (AHG)
- Antigen typing
- Serial dilutions for titration studies
- DAT (polyspecific)
- DAT (IGG)
- Cord blood testing
The system can be used as a standalone instrument or interfaced to a laboratory information system.
The ORTHO VISION Analyzer was designed with secure monitoring technologies for safety checks and balances, and it allows transfusion medicine professionals to track steps in the immunohematology testing process.
Through Ortho Clinical Diagnostics’ proprietary Intellicheck Technology, the ORTHO VISION Analyzer verifies and documents diagnostic checks throughout the testing process, while e-Connectivity Technology provides 24/7 remote data tracking that monitors instrument performance while maximizing uptime. Laboratory personnel can log on anytime, anywhere to collaborate on interpreting results in real time.
“With the launch of the ORTHO VISION Analyzer, our goal is to help improve the safety of blood transfusions by reducing the lab’s reliance on manual methods,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics, the company developing the ORTHO VISION Analyzer.
A version of the ORTHO VISION Analyzer is already commercially available in Europe, Japan, Latin America, Canada, and Australia.
For more information on the system, visit the Ortho Clinical Diagnostics website. ![]()
Photo courtesy of
Ortho Clinical Diagnostics
The US Food and Drug Administration (FDA) has granted 510(k) clearance to the ORTHO VISIONTM Analyzer, a system that automates in vitro testing of human blood.
The system is now commercially available in the US and Puerto Rico.
The ORTHO VISION Analyzer automates test processing functions, including liquid pipetting, reagent handling, incubation, centrifugation, reaction grading, and interpretation and data management requirements using ID-MTS Gel Cards and digital image processing.
Tests that can be performed with the system include:
- ABO/Rh/grouping
- ABO/Rh confirmation
- Antibody screen
- Antibody identification
- Selected cell panel
- Rh phenotype (C,c,E,e)
- Donor confirmation
- Crossmatch (AHG)
- Antigen typing
- Serial dilutions for titration studies
- DAT (polyspecific)
- DAT (IGG)
- Cord blood testing
The system can be used as a standalone instrument or interfaced to a laboratory information system.
The ORTHO VISION Analyzer was designed with secure monitoring technologies for safety checks and balances, and it allows transfusion medicine professionals to track steps in the immunohematology testing process.
Through Ortho Clinical Diagnostics’ proprietary Intellicheck Technology, the ORTHO VISION Analyzer verifies and documents diagnostic checks throughout the testing process, while e-Connectivity Technology provides 24/7 remote data tracking that monitors instrument performance while maximizing uptime. Laboratory personnel can log on anytime, anywhere to collaborate on interpreting results in real time.
“With the launch of the ORTHO VISION Analyzer, our goal is to help improve the safety of blood transfusions by reducing the lab’s reliance on manual methods,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics, the company developing the ORTHO VISION Analyzer.
A version of the ORTHO VISION Analyzer is already commercially available in Europe, Japan, Latin America, Canada, and Australia.
For more information on the system, visit the Ortho Clinical Diagnostics website.
New HMA shows early promise for MDS/AML

Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy.
Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy.
Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy.
Toward a single, whole-body scan to detect blood clots

Image by Andre E.X. Brown
BOSTON—Researchers have devised a method that may someday allow healthcare providers to quickly scan the entire body for a blood clot.
The team developed a fibrin-specific probe, known as 64Cu-FBP8, by attaching a radionuclide to a fibrin-binding peptide.
They administered the probe to rats with thrombosis and found they could locate clots via PET imaging.
Peter Caravan, PhD, of the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital in Boston, presented details on this work at the 250th National Meeting & Exposition of the American Chemical Society.
He and his colleagues also reported results with this method in Arteriosclerosis, Thrombosis, and Vascular Biology.
Dr Caravan noted that, at present, a physician may need to use 3 different methods to locate a blood clot: ultrasound to check the carotid arteries or legs, MRI to scan the heart, and CT to view the lungs.
“It’s a shot in the dark,” he said. “Patients could end up being scanned multiple times by multiple techniques in order to locate a clot. We sought a method that could detect blood clots anywhere in the body with a single, whole-body scan.”
He and his colleagues reported results with an early prototype of their fibrin-specific probe in 2011. With the current work, they believe they have optimized the probe, and they are now ready to proceed to clinical studies.
The researchers used different radionuclides and peptides, as well as different chemical groups for linking the radionuclide to the peptide, to identify which combination would provide the brightest PET signal in clots. They ultimately constructed and tested 15 candidate probes.
The team first analyzed how well each probe bound to fibrin in vitro. Then, they studied how well the probe detected thrombi in rats.
“The probes all had a similar affinity to fibrin in vitro, but, in rats, their performances were quite different,” Dr Caravan said.
He attributed these differences to metabolism. Some probes were broken down quickly in the body and could no longer bind to a thrombus, but others were resistant to the metabolism.
Dr Caravan said the best probe was the one that was most stable. And that proved to be 64Cu-FBP8 (copper-64—fibrin binding probe #8).
To test 64Cu-FBP8 in rats, the researchers induced thrombosis via ferric chloride application on both the carotid artery and femoral vein. They then performed 64Cu-FBP8-PET/CT imaging 1, 3, or 7 days after thrombosis induction.
The team found they could locate arterial and venous thrombi on fused PET/CT images with 97.6% accuracy. A single, whole-body PET/MRI could reveal the location of both arterial and venous thrombi as well.
Dr Caravan said the question now is how well this method will work in humans. To answer that, his group is hoping to start testing the probe in clinical trials in the fall.
Image by Andre E.X. Brown
BOSTON—Researchers have devised a method that may someday allow healthcare providers to quickly scan the entire body for a blood clot.
The team developed a fibrin-specific probe, known as 64Cu-FBP8, by attaching a radionuclide to a fibrin-binding peptide.
They administered the probe to rats with thrombosis and found they could locate clots via PET imaging.
Peter Caravan, PhD, of the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital in Boston, presented details on this work at the 250th National Meeting & Exposition of the American Chemical Society.
He and his colleagues also reported results with this method in Arteriosclerosis, Thrombosis, and Vascular Biology.
Dr Caravan noted that, at present, a physician may need to use 3 different methods to locate a blood clot: ultrasound to check the carotid arteries or legs, MRI to scan the heart, and CT to view the lungs.
“It’s a shot in the dark,” he said. “Patients could end up being scanned multiple times by multiple techniques in order to locate a clot. We sought a method that could detect blood clots anywhere in the body with a single, whole-body scan.”
He and his colleagues reported results with an early prototype of their fibrin-specific probe in 2011. With the current work, they believe they have optimized the probe, and they are now ready to proceed to clinical studies.
The researchers used different radionuclides and peptides, as well as different chemical groups for linking the radionuclide to the peptide, to identify which combination would provide the brightest PET signal in clots. They ultimately constructed and tested 15 candidate probes.
The team first analyzed how well each probe bound to fibrin in vitro. Then, they studied how well the probe detected thrombi in rats.
“The probes all had a similar affinity to fibrin in vitro, but, in rats, their performances were quite different,” Dr Caravan said.
He attributed these differences to metabolism. Some probes were broken down quickly in the body and could no longer bind to a thrombus, but others were resistant to the metabolism.
Dr Caravan said the best probe was the one that was most stable. And that proved to be 64Cu-FBP8 (copper-64—fibrin binding probe #8).
To test 64Cu-FBP8 in rats, the researchers induced thrombosis via ferric chloride application on both the carotid artery and femoral vein. They then performed 64Cu-FBP8-PET/CT imaging 1, 3, or 7 days after thrombosis induction.
The team found they could locate arterial and venous thrombi on fused PET/CT images with 97.6% accuracy. A single, whole-body PET/MRI could reveal the location of both arterial and venous thrombi as well.
Dr Caravan said the question now is how well this method will work in humans. To answer that, his group is hoping to start testing the probe in clinical trials in the fall.
Image by Andre E.X. Brown
BOSTON—Researchers have devised a method that may someday allow healthcare providers to quickly scan the entire body for a blood clot.
The team developed a fibrin-specific probe, known as 64Cu-FBP8, by attaching a radionuclide to a fibrin-binding peptide.
They administered the probe to rats with thrombosis and found they could locate clots via PET imaging.
Peter Caravan, PhD, of the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital in Boston, presented details on this work at the 250th National Meeting & Exposition of the American Chemical Society.
He and his colleagues also reported results with this method in Arteriosclerosis, Thrombosis, and Vascular Biology.
Dr Caravan noted that, at present, a physician may need to use 3 different methods to locate a blood clot: ultrasound to check the carotid arteries or legs, MRI to scan the heart, and CT to view the lungs.
“It’s a shot in the dark,” he said. “Patients could end up being scanned multiple times by multiple techniques in order to locate a clot. We sought a method that could detect blood clots anywhere in the body with a single, whole-body scan.”
He and his colleagues reported results with an early prototype of their fibrin-specific probe in 2011. With the current work, they believe they have optimized the probe, and they are now ready to proceed to clinical studies.
The researchers used different radionuclides and peptides, as well as different chemical groups for linking the radionuclide to the peptide, to identify which combination would provide the brightest PET signal in clots. They ultimately constructed and tested 15 candidate probes.
The team first analyzed how well each probe bound to fibrin in vitro. Then, they studied how well the probe detected thrombi in rats.
“The probes all had a similar affinity to fibrin in vitro, but, in rats, their performances were quite different,” Dr Caravan said.
He attributed these differences to metabolism. Some probes were broken down quickly in the body and could no longer bind to a thrombus, but others were resistant to the metabolism.
Dr Caravan said the best probe was the one that was most stable. And that proved to be 64Cu-FBP8 (copper-64—fibrin binding probe #8).
To test 64Cu-FBP8 in rats, the researchers induced thrombosis via ferric chloride application on both the carotid artery and femoral vein. They then performed 64Cu-FBP8-PET/CT imaging 1, 3, or 7 days after thrombosis induction.
The team found they could locate arterial and venous thrombi on fused PET/CT images with 97.6% accuracy. A single, whole-body PET/MRI could reveal the location of both arterial and venous thrombi as well.
Dr Caravan said the question now is how well this method will work in humans. To answer that, his group is hoping to start testing the probe in clinical trials in the fall.
FDA approves brentuximab vedotin as consolidation

Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).
Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).
Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).