User login
Atrophic Areas on the Axillary and Anogenital Anatomy
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
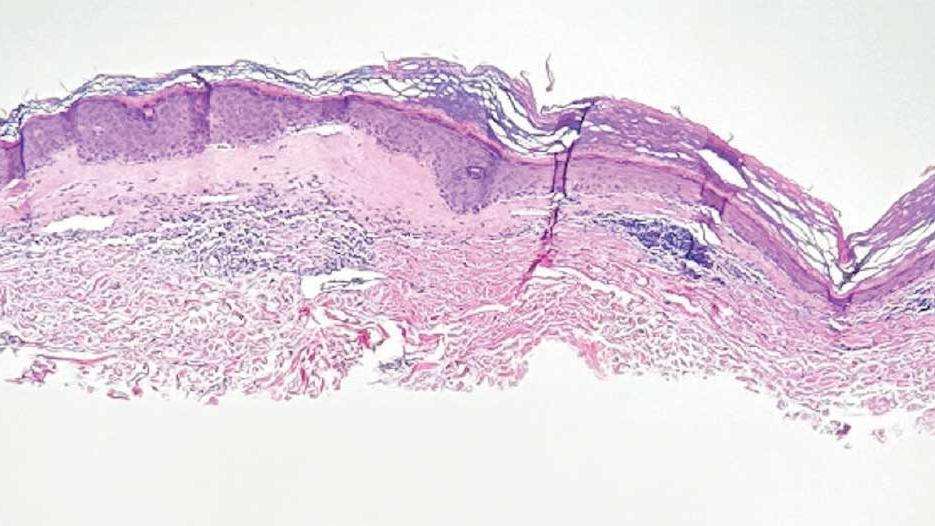
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Atrophic Areas on the Axillary and Anogenital Anatomy
Atrophic Areas on the Axillary and Anogenital Anatomy
A 62-year-old woman presented for a fullbody skin examination and was found to have a rash in her axillae and inframammary regions. The rash was intermittently pruritic, and the patient felt that the inframammary rash had started from contact with brassiere underwires. She had no oral lesions but noted intermittent burning and itching of the vaginal folds and intermittent bleeding near her anus. Physical examination revealed confluent, shiny, white, atrophic, thin papules with surrounding pink and purple patches on bilateral axillae, bilateral inframammary folds, bilateral inner thighs, and on the clitoral hood and labia minora. There was also an hourglass-shaped erythematous patch involving the vagina and anus. A small fissure was noted perianally, and small hemorrhage was noted on the clitoral head, with fusion of the clitoral head and superior labia minora (Figures 1 and 2).
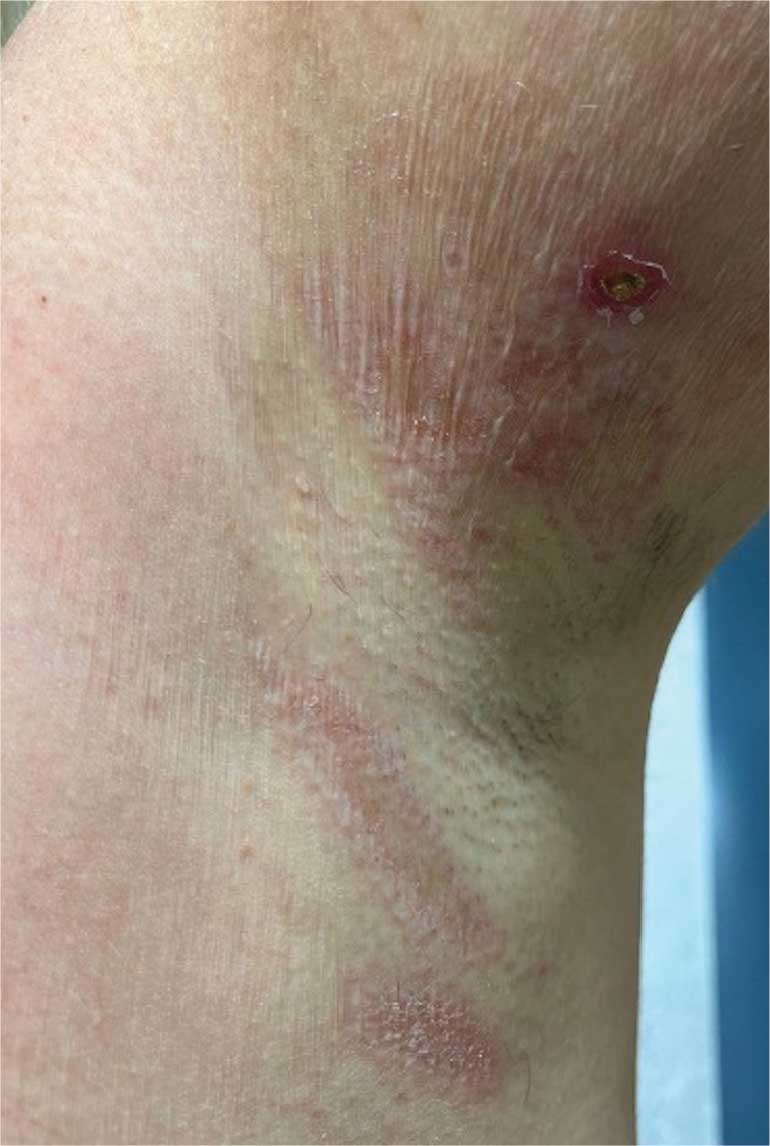
lesion from punch biopsy of the patient’s left axilla.
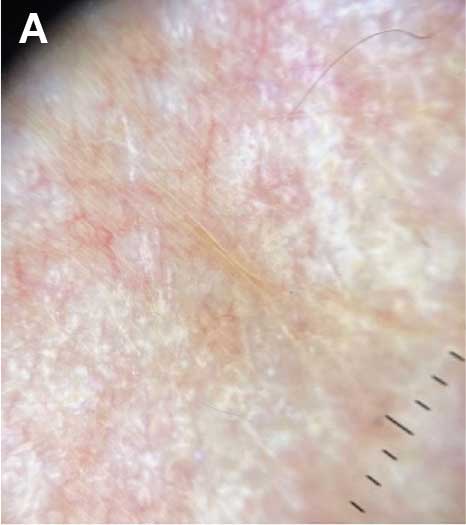
sclerosus plaque showing bright white grouped dots
on a pink background with follicular plugging and linear
branching vessels.
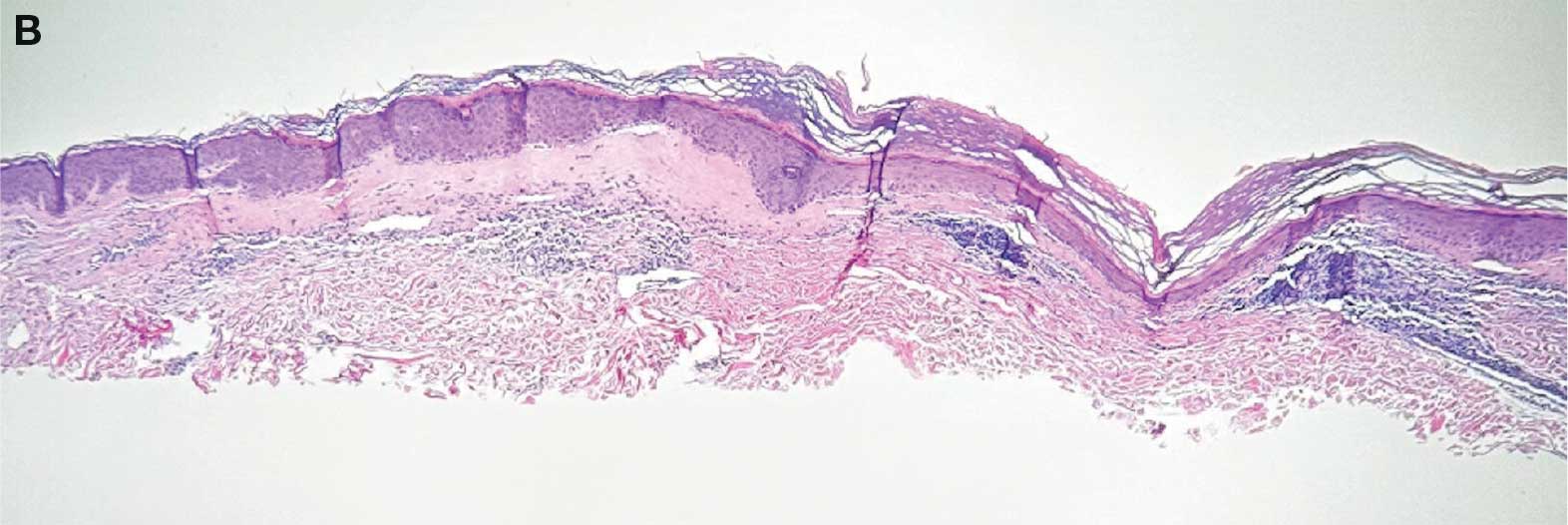
showing a compact corneal layer with a pale papillary
dermis and an underlying lymphocytic infiltrate. These
findings give the “red, white, and blue” appearance.
Low power 20× magnification.
nsbp;