User login
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
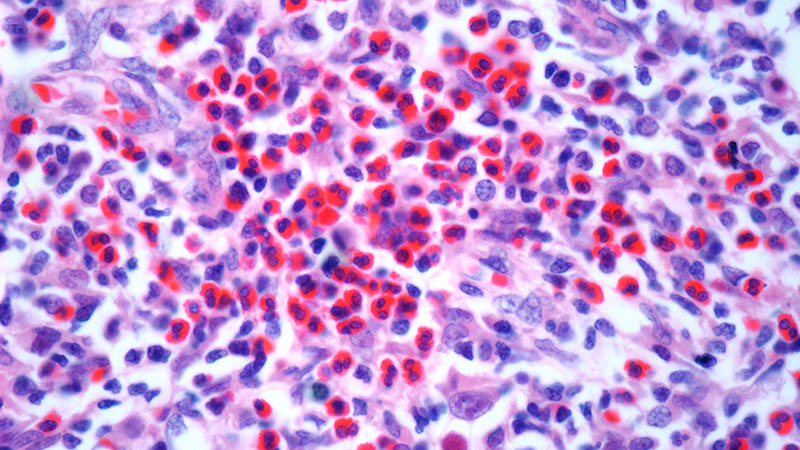
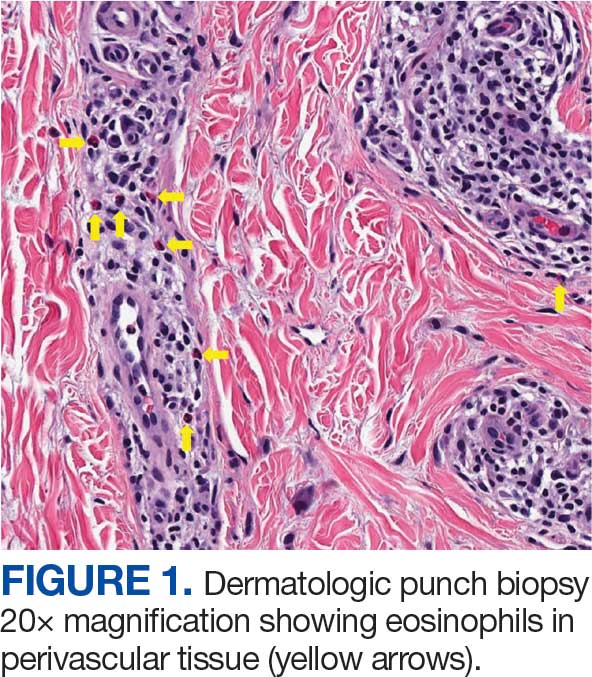
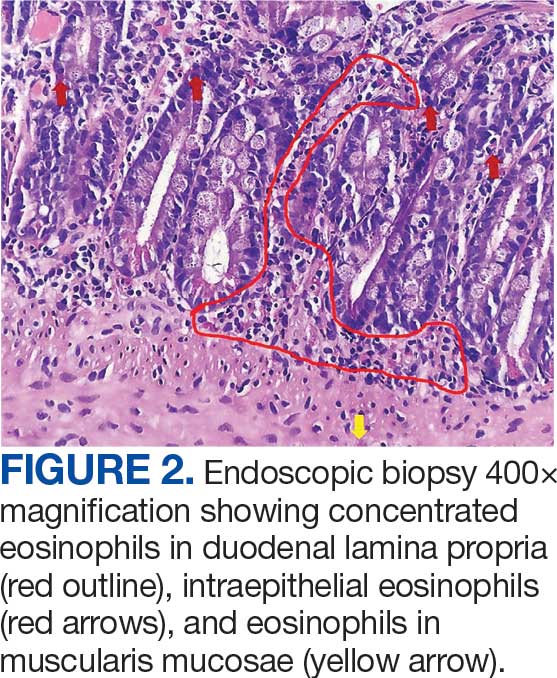
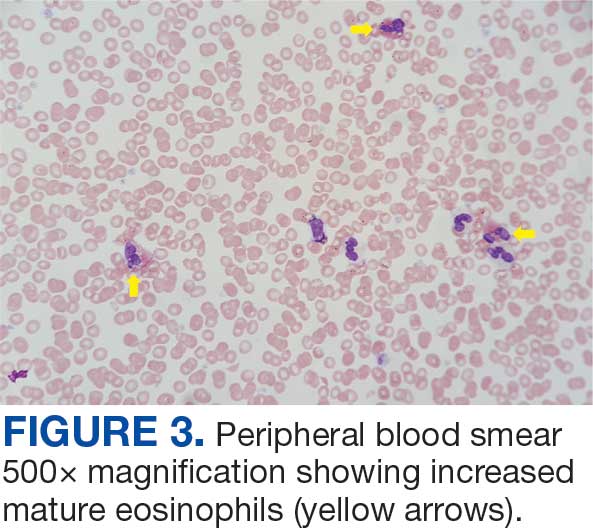
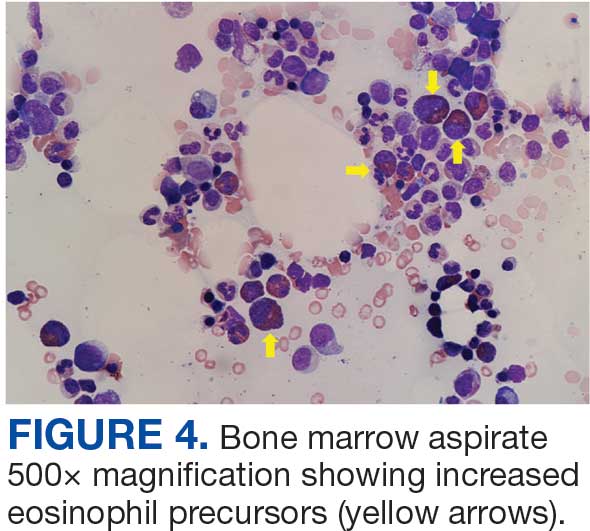
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges