User login
Don’t miss neuromuscular complications of cancer immunotherapy
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
EXPERT ANALYSIS FROM AANEM 2019
Armored CAR T cells elicit responses in NHL patients
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
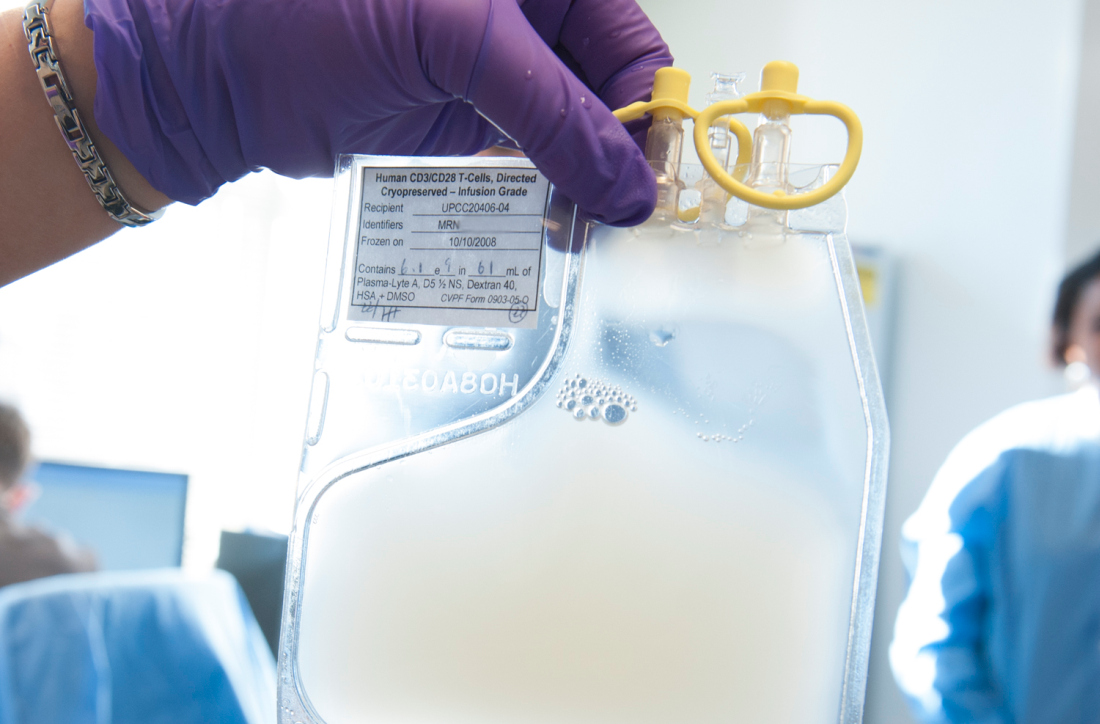
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
REPORTING FROM SITC 2019
FDA approves anemia treatment for transfusion-dependent beta thalassemia patients
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.

Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.
Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.
Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
U.S. deaths from preventable causes occur more often in rural areas
compared with the most urban counties during 2010-2017, according to study published in CDC’s Morbidity and Mortality Weekly Report.
These leading causes of death comprised heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke and accounted for approximately 1.7 million deaths or 61% of all deaths in 2017.
The study presents estimates, percentages, and annual percent changes for potentially excess deaths by urban-rural classification from heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke. Urban-rural categories were identified using the National Center for Health Statistics 2013 urban-rural classification scheme for counties.
The report’s main findings include the following statistics:
- In 2010, 28.7% of deaths from cancer in the most rural counties were potentially preventable, compared with 17.9% in the most urban counties. By 2017, 21.7% of cancer deaths in the most rural counties were potentially preventable, compared with 3.2% in the most urban counties.
- In 2010, 45.1% of deaths from heart disease in the most rural counties were potentially preventable, compared with 33.5% in the most urban counties. By 2017, 44.9% of deaths from heart disease in the most rural counties were potentially preventable, compared with 28.0% in the most urban counties.
- In 2010, 60.9% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 25.4% in the most urban counties. By 2017, 64.1% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 47.8% in the most urban counties.
- In 2010, 54.3% of deaths from chronic lower respiratory disease (such as COPD) in the most rural counties were potentially preventable, compared with 23.4% in the most urban counties. By 2017, 57.1% of deaths from chronic lower respiratory disease in the most rural counties were potentially preventable, compared with 13% in the most urban counties.
- In 2010, 41.6% of deaths from stroke in the most rural counties were potentially preventable, compared with 31.7% in most urban areas. By 2017, 37.8% of deaths from stroke in the most rural counties were potentially preventable, compared with 27.4% most urban counties.
“This report demonstrates the value of analyzing potentially excess deaths according to the six 2013 [National Center for Health Statistics] urban-rural county classifications. Reporting trends in potentially excess deaths over an 8-year period highlights differences over time, independent of traditional underlying structural, environmental, and genetic factors,” wrote Macarena C. Garcia, DrPH, and coauthors.
“Because of increasing percentages of potentially excess deaths in recent years for certain causes of death and certain demographic groups, these data can be used, with traditional rate comparisons, by public health practitioners who are involved in planning interventions. Comparing the findings in this report with data from tools such as the CDC Interactive Atlas of Heart Disease and Stroke might help identify the social determinants, health care infrastructures, and public policies that could increase or decrease numbers of deaths in specific nonmetropolitan areas,” they added.
The study authors did not disclose any potential conflicts of interest.
SOURCE: Garcia MC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 8: 68(10);1-11.
compared with the most urban counties during 2010-2017, according to study published in CDC’s Morbidity and Mortality Weekly Report.
These leading causes of death comprised heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke and accounted for approximately 1.7 million deaths or 61% of all deaths in 2017.
The study presents estimates, percentages, and annual percent changes for potentially excess deaths by urban-rural classification from heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke. Urban-rural categories were identified using the National Center for Health Statistics 2013 urban-rural classification scheme for counties.
The report’s main findings include the following statistics:
- In 2010, 28.7% of deaths from cancer in the most rural counties were potentially preventable, compared with 17.9% in the most urban counties. By 2017, 21.7% of cancer deaths in the most rural counties were potentially preventable, compared with 3.2% in the most urban counties.
- In 2010, 45.1% of deaths from heart disease in the most rural counties were potentially preventable, compared with 33.5% in the most urban counties. By 2017, 44.9% of deaths from heart disease in the most rural counties were potentially preventable, compared with 28.0% in the most urban counties.
- In 2010, 60.9% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 25.4% in the most urban counties. By 2017, 64.1% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 47.8% in the most urban counties.
- In 2010, 54.3% of deaths from chronic lower respiratory disease (such as COPD) in the most rural counties were potentially preventable, compared with 23.4% in the most urban counties. By 2017, 57.1% of deaths from chronic lower respiratory disease in the most rural counties were potentially preventable, compared with 13% in the most urban counties.
- In 2010, 41.6% of deaths from stroke in the most rural counties were potentially preventable, compared with 31.7% in most urban areas. By 2017, 37.8% of deaths from stroke in the most rural counties were potentially preventable, compared with 27.4% most urban counties.
“This report demonstrates the value of analyzing potentially excess deaths according to the six 2013 [National Center for Health Statistics] urban-rural county classifications. Reporting trends in potentially excess deaths over an 8-year period highlights differences over time, independent of traditional underlying structural, environmental, and genetic factors,” wrote Macarena C. Garcia, DrPH, and coauthors.
“Because of increasing percentages of potentially excess deaths in recent years for certain causes of death and certain demographic groups, these data can be used, with traditional rate comparisons, by public health practitioners who are involved in planning interventions. Comparing the findings in this report with data from tools such as the CDC Interactive Atlas of Heart Disease and Stroke might help identify the social determinants, health care infrastructures, and public policies that could increase or decrease numbers of deaths in specific nonmetropolitan areas,” they added.
The study authors did not disclose any potential conflicts of interest.
SOURCE: Garcia MC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 8: 68(10);1-11.
compared with the most urban counties during 2010-2017, according to study published in CDC’s Morbidity and Mortality Weekly Report.
These leading causes of death comprised heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke and accounted for approximately 1.7 million deaths or 61% of all deaths in 2017.
The study presents estimates, percentages, and annual percent changes for potentially excess deaths by urban-rural classification from heart disease, cancer, unintentional injury, chronic lower respiratory disease, and stroke. Urban-rural categories were identified using the National Center for Health Statistics 2013 urban-rural classification scheme for counties.
The report’s main findings include the following statistics:
- In 2010, 28.7% of deaths from cancer in the most rural counties were potentially preventable, compared with 17.9% in the most urban counties. By 2017, 21.7% of cancer deaths in the most rural counties were potentially preventable, compared with 3.2% in the most urban counties.
- In 2010, 45.1% of deaths from heart disease in the most rural counties were potentially preventable, compared with 33.5% in the most urban counties. By 2017, 44.9% of deaths from heart disease in the most rural counties were potentially preventable, compared with 28.0% in the most urban counties.
- In 2010, 60.9% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 25.4% in the most urban counties. By 2017, 64.1% of deaths from unintentional injury in the most rural counties were potentially preventable, compared with 47.8% in the most urban counties.
- In 2010, 54.3% of deaths from chronic lower respiratory disease (such as COPD) in the most rural counties were potentially preventable, compared with 23.4% in the most urban counties. By 2017, 57.1% of deaths from chronic lower respiratory disease in the most rural counties were potentially preventable, compared with 13% in the most urban counties.
- In 2010, 41.6% of deaths from stroke in the most rural counties were potentially preventable, compared with 31.7% in most urban areas. By 2017, 37.8% of deaths from stroke in the most rural counties were potentially preventable, compared with 27.4% most urban counties.
“This report demonstrates the value of analyzing potentially excess deaths according to the six 2013 [National Center for Health Statistics] urban-rural county classifications. Reporting trends in potentially excess deaths over an 8-year period highlights differences over time, independent of traditional underlying structural, environmental, and genetic factors,” wrote Macarena C. Garcia, DrPH, and coauthors.
“Because of increasing percentages of potentially excess deaths in recent years for certain causes of death and certain demographic groups, these data can be used, with traditional rate comparisons, by public health practitioners who are involved in planning interventions. Comparing the findings in this report with data from tools such as the CDC Interactive Atlas of Heart Disease and Stroke might help identify the social determinants, health care infrastructures, and public policies that could increase or decrease numbers of deaths in specific nonmetropolitan areas,” they added.
The study authors did not disclose any potential conflicts of interest.
SOURCE: Garcia MC et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 8: 68(10);1-11.
Best practice alerts really can work
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
REPORTING FROM AABB 2019
Fresh RBCs offer no benefit over older cells in pediatric ICU
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.

Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
REPORTING FROM AABB 2019
Levofloxacin prophylaxis improves survival in newly diagnosed myeloma
Adding levofloxacin to antimyeloma therapy improved survival and reduced infections in patients with newly diagnosed myeloma, findings from a phase 3 trial suggest.
The advantages of levofloxacin prophylaxis appear to offset the potential risks in patients with newly diagnosed disease, explained Mark T. Drayson, MBChB, PhD, of the University of Birmingham (England) and colleagues. The study was published in the Lancet Oncology.
The randomized, placebo-controlled, phase 3 TEAMM study enrolled 977 patients with newly diagnosed myeloma. The effects of antimicrobial prophylaxis on infection risk and infection-related mortality were evaluated across 93 hospitals throughout the United Kingdom.
Study patients were randomly assigned to receive 500 mg of oral levofloxacin once daily or placebo for a total of 12 weeks. If applicable, dose adjustments were made based on estimated glomerular filtration rate.
At baseline, the team collected stool samples and nasal swabs, and follow-up assessment occurred every 4 weeks for up to 1 year. The primary endpoint was time to death (all causes) or first febrile event from the start of prophylactic therapy to 12 weeks.
After a median follow-up of 12 months, first febrile episodes or deaths were significantly lower for patients in the levofloxacin arm (19%), compared with the placebo arm (27%) for a hazard ratio for time to first event of 0.66 (95% confidence interval, 0.51-0.86; P = .0018).
With respect to safety, the rates of serious adverse events were similar between the study arms, with the exception of tendinitis in the levofloxacin group (1%). Among all patients, a total of 597 serious toxicities were observed from baseline to 16 weeks (52% in the levofloxacin arm vs. 48% in the placebo arm).
“To our knowledge, this is the first time that the use of prophylactic antibiotics has shown a survival benefit in patients with newly diagnosed myeloma,” the researchers reported.
One key limitation of the study was the younger patient population relative to the general population. As a result, differences in survival estimates could exist between the trial and real-world populations, they noted.
“Patients with newly diagnosed myeloma could benefit from levofloxacin prophylaxis, although local antibiotic resistance proportions must be considered,” the researchers cautioned.
The study was funded by the National Institute for Health Research in the United Kingdom. The authors reported financial affiliations with Actelion, Astellas, Celgene, Gilead, Janssen, Pfizer, Takeda, and other companies.
SOURCE: Drayson MT et al. Lancet Oncol. 2019 Oct 23. doi: 10.1016/S1470-2045(19)30506-6.
Adding levofloxacin to antimyeloma therapy improved survival and reduced infections in patients with newly diagnosed myeloma, findings from a phase 3 trial suggest.
The advantages of levofloxacin prophylaxis appear to offset the potential risks in patients with newly diagnosed disease, explained Mark T. Drayson, MBChB, PhD, of the University of Birmingham (England) and colleagues. The study was published in the Lancet Oncology.
The randomized, placebo-controlled, phase 3 TEAMM study enrolled 977 patients with newly diagnosed myeloma. The effects of antimicrobial prophylaxis on infection risk and infection-related mortality were evaluated across 93 hospitals throughout the United Kingdom.
Study patients were randomly assigned to receive 500 mg of oral levofloxacin once daily or placebo for a total of 12 weeks. If applicable, dose adjustments were made based on estimated glomerular filtration rate.
At baseline, the team collected stool samples and nasal swabs, and follow-up assessment occurred every 4 weeks for up to 1 year. The primary endpoint was time to death (all causes) or first febrile event from the start of prophylactic therapy to 12 weeks.
After a median follow-up of 12 months, first febrile episodes or deaths were significantly lower for patients in the levofloxacin arm (19%), compared with the placebo arm (27%) for a hazard ratio for time to first event of 0.66 (95% confidence interval, 0.51-0.86; P = .0018).
With respect to safety, the rates of serious adverse events were similar between the study arms, with the exception of tendinitis in the levofloxacin group (1%). Among all patients, a total of 597 serious toxicities were observed from baseline to 16 weeks (52% in the levofloxacin arm vs. 48% in the placebo arm).
“To our knowledge, this is the first time that the use of prophylactic antibiotics has shown a survival benefit in patients with newly diagnosed myeloma,” the researchers reported.
One key limitation of the study was the younger patient population relative to the general population. As a result, differences in survival estimates could exist between the trial and real-world populations, they noted.
“Patients with newly diagnosed myeloma could benefit from levofloxacin prophylaxis, although local antibiotic resistance proportions must be considered,” the researchers cautioned.
The study was funded by the National Institute for Health Research in the United Kingdom. The authors reported financial affiliations with Actelion, Astellas, Celgene, Gilead, Janssen, Pfizer, Takeda, and other companies.
SOURCE: Drayson MT et al. Lancet Oncol. 2019 Oct 23. doi: 10.1016/S1470-2045(19)30506-6.
Adding levofloxacin to antimyeloma therapy improved survival and reduced infections in patients with newly diagnosed myeloma, findings from a phase 3 trial suggest.
The advantages of levofloxacin prophylaxis appear to offset the potential risks in patients with newly diagnosed disease, explained Mark T. Drayson, MBChB, PhD, of the University of Birmingham (England) and colleagues. The study was published in the Lancet Oncology.
The randomized, placebo-controlled, phase 3 TEAMM study enrolled 977 patients with newly diagnosed myeloma. The effects of antimicrobial prophylaxis on infection risk and infection-related mortality were evaluated across 93 hospitals throughout the United Kingdom.
Study patients were randomly assigned to receive 500 mg of oral levofloxacin once daily or placebo for a total of 12 weeks. If applicable, dose adjustments were made based on estimated glomerular filtration rate.
At baseline, the team collected stool samples and nasal swabs, and follow-up assessment occurred every 4 weeks for up to 1 year. The primary endpoint was time to death (all causes) or first febrile event from the start of prophylactic therapy to 12 weeks.
After a median follow-up of 12 months, first febrile episodes or deaths were significantly lower for patients in the levofloxacin arm (19%), compared with the placebo arm (27%) for a hazard ratio for time to first event of 0.66 (95% confidence interval, 0.51-0.86; P = .0018).
With respect to safety, the rates of serious adverse events were similar between the study arms, with the exception of tendinitis in the levofloxacin group (1%). Among all patients, a total of 597 serious toxicities were observed from baseline to 16 weeks (52% in the levofloxacin arm vs. 48% in the placebo arm).
“To our knowledge, this is the first time that the use of prophylactic antibiotics has shown a survival benefit in patients with newly diagnosed myeloma,” the researchers reported.
One key limitation of the study was the younger patient population relative to the general population. As a result, differences in survival estimates could exist between the trial and real-world populations, they noted.
“Patients with newly diagnosed myeloma could benefit from levofloxacin prophylaxis, although local antibiotic resistance proportions must be considered,” the researchers cautioned.
The study was funded by the National Institute for Health Research in the United Kingdom. The authors reported financial affiliations with Actelion, Astellas, Celgene, Gilead, Janssen, Pfizer, Takeda, and other companies.
SOURCE: Drayson MT et al. Lancet Oncol. 2019 Oct 23. doi: 10.1016/S1470-2045(19)30506-6.
FROM LANCET ONCOLOGY
Atraumatic splenic rupture in acute myeloid leukemia
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
 and transverse (right) views on initial computed tomography of the abdomen without contrast showed massive splenomegaly (white arrow).")
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
 and transverse (right) views showed irregular splenic margins (red arrows), intraparenchymal hemorrhages (black arrows), and hemoperitoneum (white arrows).")
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
A 50-year-old man with acute myeloid leukemia (AML) with a complex karyotype was admitted to the hospital with several days of dull, left-sided abdominal pain. His most recent bone marrow biopsy showed 30% blasts, and immunophenotyping was suggestive of persistent AML (CD13+, CD34+, CD117+, CD33+, CD7+, MPO–). He was on treatment with venetoclax and cytarabine after induction therapy had failed.
On admission, his heart rate was 101 beats per minute and his blood pressure was 122/85 mm Hg. Abdominal examination revealed mild distention, hepatomegaly, and previously known massive splenomegaly, with the splenic tip extending to the umbilicus, and mild tenderness.
Results of laboratory testing revealed persistent pancytopenia:
- Hemoglobin level 6.8 g/dL (reference range 13.0–17.0)
- Total white blood cell count 0.8 × 109/L (4.5–11.0)
- Platelet count 8 × 109/L (150–400).
The next day, he developed severe, acute-onset left-sided abdominal pain. A check of vital signs showed worsening sinus tachycardia at 132 beats per minute and a drop in blood pressure to 90/56 mm Hg. He had worsening diffuse abdominal tenderness with sluggish bowel sounds. His hemoglobin concentration was 6.4 g/dL and platelet count 12 × 109/L.
He received supportive transfusions of blood products. Surgical exploration was deemed risky, given his overall condition and severe thrombocytopenia. Splenic angiography showed no evidence of pseudoaneurysm or focal contrast extravasation. He underwent empiric embolization of the midsplenic artery, after which his hemodynamic status stabilized. He died 4 weeks later of acute respiratory failure from pneumonia.
SPLENIC RUPTURE IN AML
Atraumatic splenic rupture is rare but potentially life-threatening, especially if the diagnosis is delayed. Conditions that can cause splenomegaly and predispose to rupture include infection (infectious mononucleosis, malaria), malignant hematologic disorders (leukemia, lymphoma), other neoplasms, and amyloidosis.1
The literature includes a few reports of splenic rupture in patients with AML.2–4 The proposed mechanisms include bleeding from infarction sites or tumor foci, dysregulated hemostasis, and leukostasis.
The classic presentation of splenic rupture is acute-onset left-sided abdominal pain associated with hypotension and decreasing hemoglobin levels. CT of the abdomen is confirmatory, and resuscitation with crystalloids and blood products is a vital initial step in management. Choice of treatment depends on the patient’s surgical risk and hemodynamic status; options include conservative medical management, splenic artery embolization, and exploratory laparotomy.
In patients with AML and splenomegaly presenting with acute abdominal pain, clinicians need to be aware of this potential hematologic emergency.
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
- Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg 2009; 96(10):1114–1121. doi:10.1002/bjs.6737
- Gardner JA, Bao L, Ornstein DL. Spontaneous splenic rupture in acute myeloid leukemia with mixed-lineage leukemia gene rearrangement. Med Rep Case Stud 2016; 1:119. doi:10.4172/2572-5130.1000119
- Zeidan AM, Mitchell M, Khatri R, et al. Spontaneous splenic rupture during induction chemotherapy for acute myeloid leukemia. Leuk Lymphoma 2014; 55(1):209–212. doi:10.3109/10428194.2013.796060
- Fahmi Y, Elabbasi T, Khaiz D, et al. Splenic spontaneous rupture associated with acute myeloïd leukemia: report of a case and literature review. Surgery Curr Res 2014; 4:170. doi:10.4172/2161-1076.1000170
2019 at a glance: Hem-onc U.S. drug approvals
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.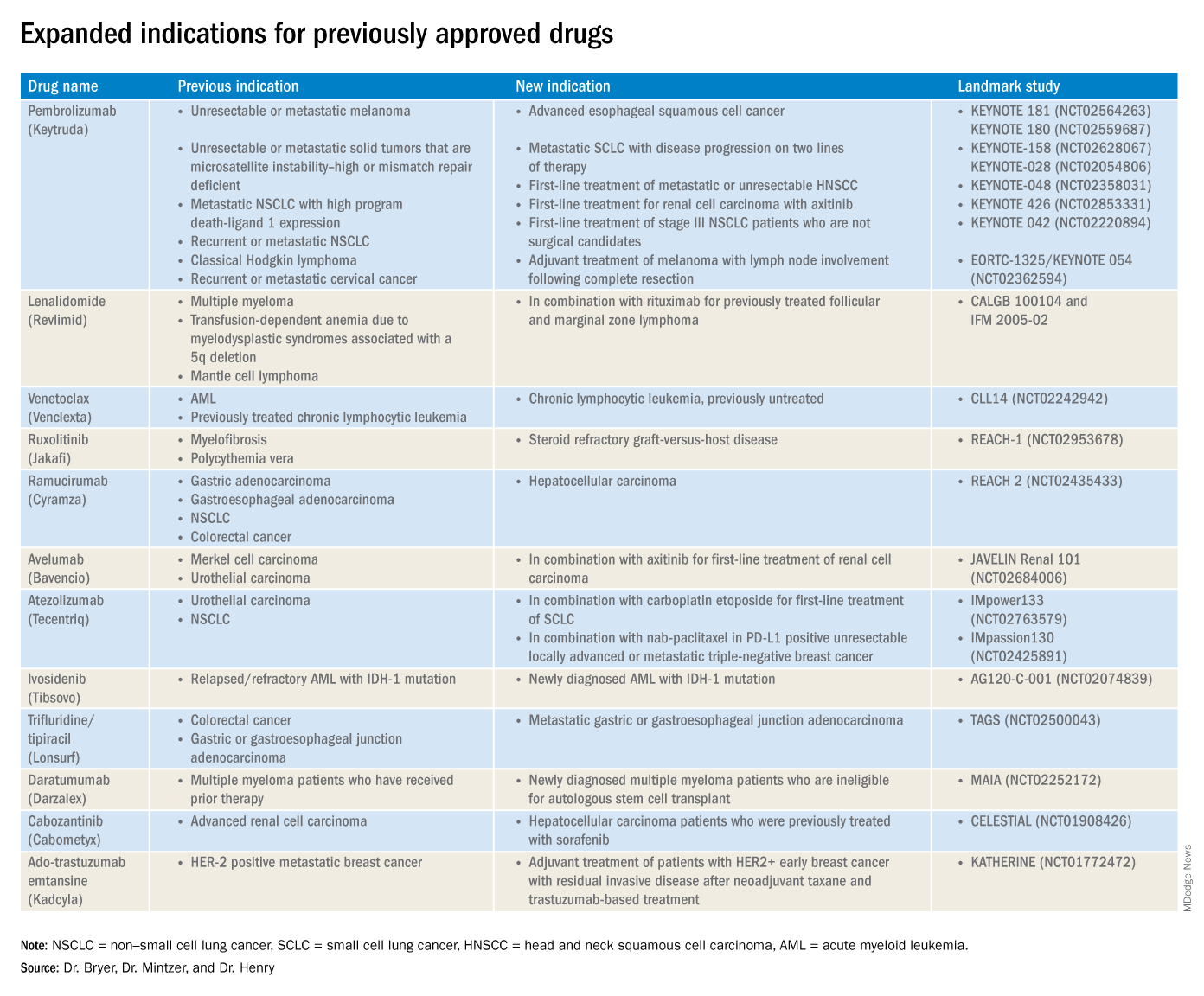
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
Dose-reduced NOACs may be safer than warfarin in some AFib patients
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.

Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.
Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.
Background: Prior studies have suggested that NOACs have a favorable risk-benefit profile when compared with warfarin, but it is unclear if this advantage also is present for those high-risk patients for whom NOAC dose reduction is recommended.
Study design: A meta-analysis.
Setting: Three phase 3 randomized, control trials.
Synopsis: From the three randomized, control trials, the authors identified 7,351 of the 46,426 patients as being eligible for dose-reduced NOACs. Of these patients, 3,702 were randomized to take a NOAC and 3,649 were randomized to take warfarin. For the primary outcomes of stroke or systemic embolism, there was no significant difference between patients randomized to receive dose-reduced NOAC versus warfarin. For outcomes of major bleeding, hemorrhagic stroke, intracranial hemorrhage, and fatal bleeding, dose-reduced NOACs had a significantly lower risk, compared with warfarin.
Bottom line: In patients eligible for dose-reduced NOACs, the use of dose-reduced NOACs may have a better safety profile without significant difference in the rate of ischemic stroke or systemic embolism.
Citation: Wang KL et al. Efficacy and safety of reduced-dose non–vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: A meta-analysis of randomized controlled trials. Eur Heart J. 2018 Dec 22. doi: 10.1093/eurheartj/ehy802.
Dr. Biddick is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine Harvard Medical School.