User login
For MD-IQ use only
Latest COVID-19 Shot May Cut Severe Outcomes in Veterans
TOPLINE:
Among US veterans, same-day receipt of both the 2024-2025 COVID19 vaccine and the influenza vaccine was associated with lower risks for emergency department visits, hospitalizations, and deaths compared with receipt of the influenza vaccine alone.
METHODOLOGY:
- Researchers conducted an observational study to assess the effectiveness of the 2024-2025 COVID-19 vaccine by comparing veterans who received both the COVID-19 and influenza vaccines on the same day with those who received only the influenza vaccine between September 3 and December 31, 2024.
- Data on participants (mean age, approximately 71.5 years; approximately 92% men) were sourced from electronic health records of the Department of Veterans Affairs and included 164,132 veterans who received both vaccines vs 131,839 who received only the seasonal influenza vaccine, with a follow-up duration of 180 days.
- The vaccines used were mainly the 2024-2025 mRNA COVID19 vaccines: Moderna mRNA1273, Pfizer BNT162b2, and the highdose trivalent 2024-2025 seasonal influenza vaccine.
- Primary outcomes were COVID-19-associated emergency department visits, hospitalizations, and deaths.
TAKEAWAY:
- Receipt of both the COVID-19 and influenza vaccines was associated with a lower risk for COVID-19-associated emergency department visits compared with receipt of the influenza vaccine alone, resulting in a vaccine effectiveness of 29.3% and a risk difference of 18.3 per 10,000 persons (95% CI, 10.8-27.6).
- Similarly, COVID-19 vaccine effectiveness was 39.2% (95% CI, 21.6-54.5) against COVID-19-associated hospitalizations, with a risk difference of 7.5 per 10,000 persons (95% CI, 3.4-13.0).
- For COVID-19-associated deaths, vaccine effectiveness was 64% (95% CI, 23.0-85.8), with a risk difference of 2.2 per 10,000 persons (95% CI, 0.5-6.9).
- Benefits were consistent across age groups (< 65, 65-75, and > 75 years) and among people with various comorbidities, including cardiovascular disease and immunocompromised status.
IN PRACTICE:
“The evidence may help inform ongoing discussions about the value of COVID-19 vaccines in the current epidemiologic landscape,” the authors wrote.
SOURCE:
The study was led by Miao Cai, PhD , Research and Development Service, Veterans Affairs St. Louis Health Care System, and the Veterans Research and Education Foundation of St. Louis, Missouri. It was published online in The New England Journal of Medicine .
LIMITATIONS:
The demographic composition of the cohort — predominantly older, White, male veterans — may limit the generalizability of the study. Although numerous covariates were adjusted for, residual confounding could not be fully ruled out. Safety and variantspecific effectiveness were not assessed.
DISCLOSURES:
The study was supported by a grant from the Department of Veterans Affairs. Two authors disclosed consulting for Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Among US veterans, same-day receipt of both the 2024-2025 COVID19 vaccine and the influenza vaccine was associated with lower risks for emergency department visits, hospitalizations, and deaths compared with receipt of the influenza vaccine alone.
METHODOLOGY:
- Researchers conducted an observational study to assess the effectiveness of the 2024-2025 COVID-19 vaccine by comparing veterans who received both the COVID-19 and influenza vaccines on the same day with those who received only the influenza vaccine between September 3 and December 31, 2024.
- Data on participants (mean age, approximately 71.5 years; approximately 92% men) were sourced from electronic health records of the Department of Veterans Affairs and included 164,132 veterans who received both vaccines vs 131,839 who received only the seasonal influenza vaccine, with a follow-up duration of 180 days.
- The vaccines used were mainly the 2024-2025 mRNA COVID19 vaccines: Moderna mRNA1273, Pfizer BNT162b2, and the highdose trivalent 2024-2025 seasonal influenza vaccine.
- Primary outcomes were COVID-19-associated emergency department visits, hospitalizations, and deaths.
TAKEAWAY:
- Receipt of both the COVID-19 and influenza vaccines was associated with a lower risk for COVID-19-associated emergency department visits compared with receipt of the influenza vaccine alone, resulting in a vaccine effectiveness of 29.3% and a risk difference of 18.3 per 10,000 persons (95% CI, 10.8-27.6).
- Similarly, COVID-19 vaccine effectiveness was 39.2% (95% CI, 21.6-54.5) against COVID-19-associated hospitalizations, with a risk difference of 7.5 per 10,000 persons (95% CI, 3.4-13.0).
- For COVID-19-associated deaths, vaccine effectiveness was 64% (95% CI, 23.0-85.8), with a risk difference of 2.2 per 10,000 persons (95% CI, 0.5-6.9).
- Benefits were consistent across age groups (< 65, 65-75, and > 75 years) and among people with various comorbidities, including cardiovascular disease and immunocompromised status.
IN PRACTICE:
“The evidence may help inform ongoing discussions about the value of COVID-19 vaccines in the current epidemiologic landscape,” the authors wrote.
SOURCE:
The study was led by Miao Cai, PhD , Research and Development Service, Veterans Affairs St. Louis Health Care System, and the Veterans Research and Education Foundation of St. Louis, Missouri. It was published online in The New England Journal of Medicine .
LIMITATIONS:
The demographic composition of the cohort — predominantly older, White, male veterans — may limit the generalizability of the study. Although numerous covariates were adjusted for, residual confounding could not be fully ruled out. Safety and variantspecific effectiveness were not assessed.
DISCLOSURES:
The study was supported by a grant from the Department of Veterans Affairs. Two authors disclosed consulting for Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Among US veterans, same-day receipt of both the 2024-2025 COVID19 vaccine and the influenza vaccine was associated with lower risks for emergency department visits, hospitalizations, and deaths compared with receipt of the influenza vaccine alone.
METHODOLOGY:
- Researchers conducted an observational study to assess the effectiveness of the 2024-2025 COVID-19 vaccine by comparing veterans who received both the COVID-19 and influenza vaccines on the same day with those who received only the influenza vaccine between September 3 and December 31, 2024.
- Data on participants (mean age, approximately 71.5 years; approximately 92% men) were sourced from electronic health records of the Department of Veterans Affairs and included 164,132 veterans who received both vaccines vs 131,839 who received only the seasonal influenza vaccine, with a follow-up duration of 180 days.
- The vaccines used were mainly the 2024-2025 mRNA COVID19 vaccines: Moderna mRNA1273, Pfizer BNT162b2, and the highdose trivalent 2024-2025 seasonal influenza vaccine.
- Primary outcomes were COVID-19-associated emergency department visits, hospitalizations, and deaths.
TAKEAWAY:
- Receipt of both the COVID-19 and influenza vaccines was associated with a lower risk for COVID-19-associated emergency department visits compared with receipt of the influenza vaccine alone, resulting in a vaccine effectiveness of 29.3% and a risk difference of 18.3 per 10,000 persons (95% CI, 10.8-27.6).
- Similarly, COVID-19 vaccine effectiveness was 39.2% (95% CI, 21.6-54.5) against COVID-19-associated hospitalizations, with a risk difference of 7.5 per 10,000 persons (95% CI, 3.4-13.0).
- For COVID-19-associated deaths, vaccine effectiveness was 64% (95% CI, 23.0-85.8), with a risk difference of 2.2 per 10,000 persons (95% CI, 0.5-6.9).
- Benefits were consistent across age groups (< 65, 65-75, and > 75 years) and among people with various comorbidities, including cardiovascular disease and immunocompromised status.
IN PRACTICE:
“The evidence may help inform ongoing discussions about the value of COVID-19 vaccines in the current epidemiologic landscape,” the authors wrote.
SOURCE:
The study was led by Miao Cai, PhD , Research and Development Service, Veterans Affairs St. Louis Health Care System, and the Veterans Research and Education Foundation of St. Louis, Missouri. It was published online in The New England Journal of Medicine .
LIMITATIONS:
The demographic composition of the cohort — predominantly older, White, male veterans — may limit the generalizability of the study. Although numerous covariates were adjusted for, residual confounding could not be fully ruled out. Safety and variantspecific effectiveness were not assessed.
DISCLOSURES:
The study was supported by a grant from the Department of Veterans Affairs. Two authors disclosed consulting for Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Targeted Osteoporosis Program May Benefit At-Risk Older Men
Efforts to identify older men at risk for osteoporosis and treat those who are eligible received a boost from results reported from a Veterans Affairs (VA) study that showed a significant increase in screening, treatment, and medication adherence.
The cluster randomized trial used a centralized nurse-led intervention to assess men for traditional osteoporosis risk factors, offer bone density testing, and recommend treatment for eligible men. Over 2 years, the intervention group had a higher average femoral neck bone density than patients who underwent usual care.
“We designed this study to see if a risk factor-based approach, which is what most of the guidelines use, made sense and was feasible — that men would be accepting of screening and [the approach] would yield a similar proportion of people who need osteoporosis treatment as screening in women, which is widely recommended and implemented. And sure enough, we found that about 85% of the men in the VA primary care practices in our target age range of between 65 and 85 actually met criteria for screening, and over half of them had low bone mass. They were very accepting of screening, very accepting of treatment, and had excellent compliance rates. So, our study, we believe, supports the idea of identifying men with at least one risk factor for fracture and offering them osteoporosis screening starting at age 65, similar to what we do for women,” Cathleen S. Colón-Emeric, MD, MHS, said in an interview. She is the lead author of the study, a physician in the Durham VA Health Care System, and professor of medicine at Duke University School of Medicine, Durham, North Carolina.
“We were able to see a positive effect on bone density in the bone health group, compared with the usual care group, which suggests that if we followed these folks longer and had enough of them, we would be able to show a fracture reduction benefit,” Colón-Emeric said.
There have been few randomized trials of screening interventions in men, leading to inconsistencies in guidelines, according to the authors of the new study, published online in JAMA Internal Medicine . Both the US Preventive Services Task Force and the Veterans Health Administration National Center for Health Promotion and Disease Prevention consider there to be insufficient evidence to recommend for or against screening in men who have not experienced a fracture. Some professional societies recommend such screening, but there are inconsistencies in the recommended criteria, such as age range or risk factors.
Beyond the age of 50 years, one in five men will experience an osteoporosis-related fracture at some point in their life, according to a 2009 study. Treatment is inexpensive and effective in both men and women, and economic models suggest that screening using dual-energy x-ray absorptiometry (DXA) would be cost-effective. Still, screening is rare among men, with fewer than 10% of men getting screened before having an osteoporosis-related fracture.
“It’s important to screen men at risk for osteoporosis due to the dramatically increased mortality men suffer after a fragility fracture compared with women. Within 1 year of a hip fracture, mortality is as high as 36%. Studies have also shown that osteoporosis in men is undertreated, with only 10%-50% being prescribed antifracture treatment within 1 year of a hip fracture. Most individuals do not regain their prior level of function after a hip fracture,” said Joe C. Huang, MD, who was asked for comment. He is a clinical assistant professor of gerontology and geriatric medicine at Harborview Medical Center Senior Care Clinic and Healthy Bones Clinic in Seattle.
Details of the Intervention
The bone health service (BHS) intervention employed an electronic health record case-finding tool and a nurse care manager who undertook screening and treatment monitoring. They identified potential risk factors that included hyperthyroidism, hyperparathyroidism, rheumatoid arthritis, alcohol dependence, chronic lung disease, chronic liver disease, stroke, parkinsonism, prostate cancer, smoking, diabetes, pernicious anemia, gastrectomy, or high-risk medication use in at least 3 months of the prior 2 years. These medications included traditional antiepileptics, glucocorticoids, and androgen deprivation therapy.
The BHS nurse invited eligible men to be screened using an initial letter, followed by up to three phone calls. After DXA screening, the nurse scheduled an electronic consult with an osteoporosis expert, and patients with a T-score between -1 and -2.4 and an elevated 10-year fracture risk as measured by the Fracture Risk Assessment Tool were recommended for osteoporosis medication, vitamin D, and dietary or supplemental calcium. Following the prescription, the nurse provided patient education over the phone and mailed out written instructions. The nurse also made phone calls at 1 month, 6 months, and 12 months to encourage adherence and address common treatment barriers such as forgetting to take medication or dealing with gastrointestinal effects. The researchers recruited 38 primary care physicians from two VA health systems. The study included 3112 male veterans between the ages of 65 and 85 years (40.4% Black and 56% White). Nearly all participants (85.5%) had at least one indication for screening according to VA undersecretary guidelines, and almost a third (32.1%) had been prescribed androgen deprivation therapy, traditional antiepileptic drugs, or glucocorticoids.
Over a mean follow-up of 1.5 years, there was a much higher screening rate in the BHS group (49.2% vs 2.3%; P < .001), with a similar overall yield of DXA results recommending osteoporosis treatment (22.4% vs 27.2%). In the BHS group, 84.4% of patients who had treatment recommended followed through with treatment initiation. The mean persistence over follow-up was 657 days (SD, 366 days), and adherence was high with a mean proportion of days covered of 91.7%.
It was not possible to statistically compare adherence with the usual-care group because there were too few screened patients found to be eligible for treatment in that group, but the historic mean proportion of days covered at the two participating facilities was 52%.
After 2 years, the mean femoral neck T-score tested randomly in a subset of patients was better in the BHS arm, although it did not meet statistical significance according to the Bonferroni corrected criterion of P < .025 (-0.55 vs -0.70; P = .04). Fracture rates were similar between the two groups (1.8% vs 2.0%; P = .69).
Can the Findings Be Translated Across Clinics?
It remains to be seen how well the model could translate to other healthcare settings, according to Kenny Lin, MD, MPH, who was asked for comment on the study. “Outside of the VA health system and perhaps integrated HMOs [health maintenance organizations] such as Kaiser, Geisinger, etc., it seems unlikely that most primary care docs will have access to a centralized bone health service. Who’s going to pay for it? It leaves unanswered the question of whether it’s more efficient to address [osteoporosis] screening on a practice or population level. I suspect the latter is probably superior, but this study doesn’t provide any empiric evidence that this is so,” said Lin, associate director of the Penn Medicine Lancaster General Hospital’s Family Medicine Residency Program, Lancaster, Pennsylvania. The findings could help sway recommendations to screen men for osteoporosis, according to Susan Ott, MD, who was also asked for comment. Guideline committees “have been trying to be very scientific [about it]. I think they overdo it because they only look at one or two kinds of studies, and there are more kinds of science than just a randomized clinical trial. But they’re kind of stuck on that. The fact that this study was a randomized trial maybe they will finally change their recommendation, because there really shouldn’t be any difference in screening for men and for women. The men are actually discriminated against,” said Ott, emeritus professor of medicine at the University of Washington, Seattle.
In fact, she noted that the risks for men are similar to those for women, except that men tend to develop issues 5-10 years later in life. To screen and treat men, healthcare systems can “do the same thing they do with women. Just change the age range,” Ott said.
Lin sounded a different note, suggesting that the focus should remain on improvement of screening and treatment adherence in women. “We know that up to two thirds of women discontinue osteoporosis drugs within a year, and if we can’t figure out how to improve abysmal adherence in women, it’s unlikely we will persuade enough men to take these drugs to make a difference,” he said.
The study was funded by a grant from the VA Health Systems Research. Colón-Emeric, Lin, Ott, and Huang reported having no relevant financial disclosures.
A version of this article first appeared on Medscape.com.
Efforts to identify older men at risk for osteoporosis and treat those who are eligible received a boost from results reported from a Veterans Affairs (VA) study that showed a significant increase in screening, treatment, and medication adherence.
The cluster randomized trial used a centralized nurse-led intervention to assess men for traditional osteoporosis risk factors, offer bone density testing, and recommend treatment for eligible men. Over 2 years, the intervention group had a higher average femoral neck bone density than patients who underwent usual care.
“We designed this study to see if a risk factor-based approach, which is what most of the guidelines use, made sense and was feasible — that men would be accepting of screening and [the approach] would yield a similar proportion of people who need osteoporosis treatment as screening in women, which is widely recommended and implemented. And sure enough, we found that about 85% of the men in the VA primary care practices in our target age range of between 65 and 85 actually met criteria for screening, and over half of them had low bone mass. They were very accepting of screening, very accepting of treatment, and had excellent compliance rates. So, our study, we believe, supports the idea of identifying men with at least one risk factor for fracture and offering them osteoporosis screening starting at age 65, similar to what we do for women,” Cathleen S. Colón-Emeric, MD, MHS, said in an interview. She is the lead author of the study, a physician in the Durham VA Health Care System, and professor of medicine at Duke University School of Medicine, Durham, North Carolina.
“We were able to see a positive effect on bone density in the bone health group, compared with the usual care group, which suggests that if we followed these folks longer and had enough of them, we would be able to show a fracture reduction benefit,” Colón-Emeric said.
There have been few randomized trials of screening interventions in men, leading to inconsistencies in guidelines, according to the authors of the new study, published online in JAMA Internal Medicine . Both the US Preventive Services Task Force and the Veterans Health Administration National Center for Health Promotion and Disease Prevention consider there to be insufficient evidence to recommend for or against screening in men who have not experienced a fracture. Some professional societies recommend such screening, but there are inconsistencies in the recommended criteria, such as age range or risk factors.
Beyond the age of 50 years, one in five men will experience an osteoporosis-related fracture at some point in their life, according to a 2009 study. Treatment is inexpensive and effective in both men and women, and economic models suggest that screening using dual-energy x-ray absorptiometry (DXA) would be cost-effective. Still, screening is rare among men, with fewer than 10% of men getting screened before having an osteoporosis-related fracture.
“It’s important to screen men at risk for osteoporosis due to the dramatically increased mortality men suffer after a fragility fracture compared with women. Within 1 year of a hip fracture, mortality is as high as 36%. Studies have also shown that osteoporosis in men is undertreated, with only 10%-50% being prescribed antifracture treatment within 1 year of a hip fracture. Most individuals do not regain their prior level of function after a hip fracture,” said Joe C. Huang, MD, who was asked for comment. He is a clinical assistant professor of gerontology and geriatric medicine at Harborview Medical Center Senior Care Clinic and Healthy Bones Clinic in Seattle.
Details of the Intervention
The bone health service (BHS) intervention employed an electronic health record case-finding tool and a nurse care manager who undertook screening and treatment monitoring. They identified potential risk factors that included hyperthyroidism, hyperparathyroidism, rheumatoid arthritis, alcohol dependence, chronic lung disease, chronic liver disease, stroke, parkinsonism, prostate cancer, smoking, diabetes, pernicious anemia, gastrectomy, or high-risk medication use in at least 3 months of the prior 2 years. These medications included traditional antiepileptics, glucocorticoids, and androgen deprivation therapy.
The BHS nurse invited eligible men to be screened using an initial letter, followed by up to three phone calls. After DXA screening, the nurse scheduled an electronic consult with an osteoporosis expert, and patients with a T-score between -1 and -2.4 and an elevated 10-year fracture risk as measured by the Fracture Risk Assessment Tool were recommended for osteoporosis medication, vitamin D, and dietary or supplemental calcium. Following the prescription, the nurse provided patient education over the phone and mailed out written instructions. The nurse also made phone calls at 1 month, 6 months, and 12 months to encourage adherence and address common treatment barriers such as forgetting to take medication or dealing with gastrointestinal effects. The researchers recruited 38 primary care physicians from two VA health systems. The study included 3112 male veterans between the ages of 65 and 85 years (40.4% Black and 56% White). Nearly all participants (85.5%) had at least one indication for screening according to VA undersecretary guidelines, and almost a third (32.1%) had been prescribed androgen deprivation therapy, traditional antiepileptic drugs, or glucocorticoids.
Over a mean follow-up of 1.5 years, there was a much higher screening rate in the BHS group (49.2% vs 2.3%; P < .001), with a similar overall yield of DXA results recommending osteoporosis treatment (22.4% vs 27.2%). In the BHS group, 84.4% of patients who had treatment recommended followed through with treatment initiation. The mean persistence over follow-up was 657 days (SD, 366 days), and adherence was high with a mean proportion of days covered of 91.7%.
It was not possible to statistically compare adherence with the usual-care group because there were too few screened patients found to be eligible for treatment in that group, but the historic mean proportion of days covered at the two participating facilities was 52%.
After 2 years, the mean femoral neck T-score tested randomly in a subset of patients was better in the BHS arm, although it did not meet statistical significance according to the Bonferroni corrected criterion of P < .025 (-0.55 vs -0.70; P = .04). Fracture rates were similar between the two groups (1.8% vs 2.0%; P = .69).
Can the Findings Be Translated Across Clinics?
It remains to be seen how well the model could translate to other healthcare settings, according to Kenny Lin, MD, MPH, who was asked for comment on the study. “Outside of the VA health system and perhaps integrated HMOs [health maintenance organizations] such as Kaiser, Geisinger, etc., it seems unlikely that most primary care docs will have access to a centralized bone health service. Who’s going to pay for it? It leaves unanswered the question of whether it’s more efficient to address [osteoporosis] screening on a practice or population level. I suspect the latter is probably superior, but this study doesn’t provide any empiric evidence that this is so,” said Lin, associate director of the Penn Medicine Lancaster General Hospital’s Family Medicine Residency Program, Lancaster, Pennsylvania. The findings could help sway recommendations to screen men for osteoporosis, according to Susan Ott, MD, who was also asked for comment. Guideline committees “have been trying to be very scientific [about it]. I think they overdo it because they only look at one or two kinds of studies, and there are more kinds of science than just a randomized clinical trial. But they’re kind of stuck on that. The fact that this study was a randomized trial maybe they will finally change their recommendation, because there really shouldn’t be any difference in screening for men and for women. The men are actually discriminated against,” said Ott, emeritus professor of medicine at the University of Washington, Seattle.
In fact, she noted that the risks for men are similar to those for women, except that men tend to develop issues 5-10 years later in life. To screen and treat men, healthcare systems can “do the same thing they do with women. Just change the age range,” Ott said.
Lin sounded a different note, suggesting that the focus should remain on improvement of screening and treatment adherence in women. “We know that up to two thirds of women discontinue osteoporosis drugs within a year, and if we can’t figure out how to improve abysmal adherence in women, it’s unlikely we will persuade enough men to take these drugs to make a difference,” he said.
The study was funded by a grant from the VA Health Systems Research. Colón-Emeric, Lin, Ott, and Huang reported having no relevant financial disclosures.
A version of this article first appeared on Medscape.com.
Efforts to identify older men at risk for osteoporosis and treat those who are eligible received a boost from results reported from a Veterans Affairs (VA) study that showed a significant increase in screening, treatment, and medication adherence.
The cluster randomized trial used a centralized nurse-led intervention to assess men for traditional osteoporosis risk factors, offer bone density testing, and recommend treatment for eligible men. Over 2 years, the intervention group had a higher average femoral neck bone density than patients who underwent usual care.
“We designed this study to see if a risk factor-based approach, which is what most of the guidelines use, made sense and was feasible — that men would be accepting of screening and [the approach] would yield a similar proportion of people who need osteoporosis treatment as screening in women, which is widely recommended and implemented. And sure enough, we found that about 85% of the men in the VA primary care practices in our target age range of between 65 and 85 actually met criteria for screening, and over half of them had low bone mass. They were very accepting of screening, very accepting of treatment, and had excellent compliance rates. So, our study, we believe, supports the idea of identifying men with at least one risk factor for fracture and offering them osteoporosis screening starting at age 65, similar to what we do for women,” Cathleen S. Colón-Emeric, MD, MHS, said in an interview. She is the lead author of the study, a physician in the Durham VA Health Care System, and professor of medicine at Duke University School of Medicine, Durham, North Carolina.
“We were able to see a positive effect on bone density in the bone health group, compared with the usual care group, which suggests that if we followed these folks longer and had enough of them, we would be able to show a fracture reduction benefit,” Colón-Emeric said.
There have been few randomized trials of screening interventions in men, leading to inconsistencies in guidelines, according to the authors of the new study, published online in JAMA Internal Medicine . Both the US Preventive Services Task Force and the Veterans Health Administration National Center for Health Promotion and Disease Prevention consider there to be insufficient evidence to recommend for or against screening in men who have not experienced a fracture. Some professional societies recommend such screening, but there are inconsistencies in the recommended criteria, such as age range or risk factors.
Beyond the age of 50 years, one in five men will experience an osteoporosis-related fracture at some point in their life, according to a 2009 study. Treatment is inexpensive and effective in both men and women, and economic models suggest that screening using dual-energy x-ray absorptiometry (DXA) would be cost-effective. Still, screening is rare among men, with fewer than 10% of men getting screened before having an osteoporosis-related fracture.
“It’s important to screen men at risk for osteoporosis due to the dramatically increased mortality men suffer after a fragility fracture compared with women. Within 1 year of a hip fracture, mortality is as high as 36%. Studies have also shown that osteoporosis in men is undertreated, with only 10%-50% being prescribed antifracture treatment within 1 year of a hip fracture. Most individuals do not regain their prior level of function after a hip fracture,” said Joe C. Huang, MD, who was asked for comment. He is a clinical assistant professor of gerontology and geriatric medicine at Harborview Medical Center Senior Care Clinic and Healthy Bones Clinic in Seattle.
Details of the Intervention
The bone health service (BHS) intervention employed an electronic health record case-finding tool and a nurse care manager who undertook screening and treatment monitoring. They identified potential risk factors that included hyperthyroidism, hyperparathyroidism, rheumatoid arthritis, alcohol dependence, chronic lung disease, chronic liver disease, stroke, parkinsonism, prostate cancer, smoking, diabetes, pernicious anemia, gastrectomy, or high-risk medication use in at least 3 months of the prior 2 years. These medications included traditional antiepileptics, glucocorticoids, and androgen deprivation therapy.
The BHS nurse invited eligible men to be screened using an initial letter, followed by up to three phone calls. After DXA screening, the nurse scheduled an electronic consult with an osteoporosis expert, and patients with a T-score between -1 and -2.4 and an elevated 10-year fracture risk as measured by the Fracture Risk Assessment Tool were recommended for osteoporosis medication, vitamin D, and dietary or supplemental calcium. Following the prescription, the nurse provided patient education over the phone and mailed out written instructions. The nurse also made phone calls at 1 month, 6 months, and 12 months to encourage adherence and address common treatment barriers such as forgetting to take medication or dealing with gastrointestinal effects. The researchers recruited 38 primary care physicians from two VA health systems. The study included 3112 male veterans between the ages of 65 and 85 years (40.4% Black and 56% White). Nearly all participants (85.5%) had at least one indication for screening according to VA undersecretary guidelines, and almost a third (32.1%) had been prescribed androgen deprivation therapy, traditional antiepileptic drugs, or glucocorticoids.
Over a mean follow-up of 1.5 years, there was a much higher screening rate in the BHS group (49.2% vs 2.3%; P < .001), with a similar overall yield of DXA results recommending osteoporosis treatment (22.4% vs 27.2%). In the BHS group, 84.4% of patients who had treatment recommended followed through with treatment initiation. The mean persistence over follow-up was 657 days (SD, 366 days), and adherence was high with a mean proportion of days covered of 91.7%.
It was not possible to statistically compare adherence with the usual-care group because there were too few screened patients found to be eligible for treatment in that group, but the historic mean proportion of days covered at the two participating facilities was 52%.
After 2 years, the mean femoral neck T-score tested randomly in a subset of patients was better in the BHS arm, although it did not meet statistical significance according to the Bonferroni corrected criterion of P < .025 (-0.55 vs -0.70; P = .04). Fracture rates were similar between the two groups (1.8% vs 2.0%; P = .69).
Can the Findings Be Translated Across Clinics?
It remains to be seen how well the model could translate to other healthcare settings, according to Kenny Lin, MD, MPH, who was asked for comment on the study. “Outside of the VA health system and perhaps integrated HMOs [health maintenance organizations] such as Kaiser, Geisinger, etc., it seems unlikely that most primary care docs will have access to a centralized bone health service. Who’s going to pay for it? It leaves unanswered the question of whether it’s more efficient to address [osteoporosis] screening on a practice or population level. I suspect the latter is probably superior, but this study doesn’t provide any empiric evidence that this is so,” said Lin, associate director of the Penn Medicine Lancaster General Hospital’s Family Medicine Residency Program, Lancaster, Pennsylvania. The findings could help sway recommendations to screen men for osteoporosis, according to Susan Ott, MD, who was also asked for comment. Guideline committees “have been trying to be very scientific [about it]. I think they overdo it because they only look at one or two kinds of studies, and there are more kinds of science than just a randomized clinical trial. But they’re kind of stuck on that. The fact that this study was a randomized trial maybe they will finally change their recommendation, because there really shouldn’t be any difference in screening for men and for women. The men are actually discriminated against,” said Ott, emeritus professor of medicine at the University of Washington, Seattle.
In fact, she noted that the risks for men are similar to those for women, except that men tend to develop issues 5-10 years later in life. To screen and treat men, healthcare systems can “do the same thing they do with women. Just change the age range,” Ott said.
Lin sounded a different note, suggesting that the focus should remain on improvement of screening and treatment adherence in women. “We know that up to two thirds of women discontinue osteoporosis drugs within a year, and if we can’t figure out how to improve abysmal adherence in women, it’s unlikely we will persuade enough men to take these drugs to make a difference,” he said.
The study was funded by a grant from the VA Health Systems Research. Colón-Emeric, Lin, Ott, and Huang reported having no relevant financial disclosures.
A version of this article first appeared on Medscape.com.

U.S. Health Chief Kennedy Targets Vaccine Injury Compensation Program
WASHINGTON (Reuters) - U.S. Health Secretary Robert F. Kennedy Jr. said on July 28 that he will work to “fix” the program that compensates victims of vaccine injuries, the National Vaccine Injury Compensation Program.
Kennedy, a long-time vaccine skeptic and former vaccine injury plaintiff lawyer, accused the program and its so-called “Vaccine Court” of corruption and inefficiency in a post on X. He has long been an outspoken critic of the program.
“I will not allow the VICP to continue to ignore its mandate and fail its mission of quickly and fairly compensating vaccine-injured individuals,” he wrote, adding he was working with Attorney General Pam Bondi. “Together, we will steer the Vaccine Court back to its original congressional intent.”
He said the structure disadvantaged claimants because the Department of Health & Human Services – which he now leads – is the defendant, as opposed to vaccine makers.
Changing the VICP would be the latest in a series of far-reaching actions by Kennedy to reshape U.S. regulation of vaccines, food and medicine.
In June, he fired all 17 members of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices, a panel of vaccine experts, replacing them with 7 handpicked members, including known vaccine skeptics.
One of them earned thousands of dollars as an expert witness in litigation against Merck’s, Gardasil vaccine, court records show. Kennedy himself played an instrumental role in organizing mass litigation over the vaccine.
He also is planning to remove all the members of another advisory panel that determines what preventive health measures insurers must cover, the Wall Street Journal reported on July 25. An HHS spokesperson said Kennedy had not yet made a decision regarding the 16-member U.S. Preventive Services Task Force.
Kennedy has for years sown doubt about the safety and efficacy of vaccines. He has a history of clashing with the medical establishment and spreading misinformation about vaccines, including promoting a debunked link between vaccines and autism despite scientific evidence to the contrary.
He has also said the measles vaccine contains cells from aborted fetuses and that the mumps vaccination does not work, comments he made as the U.S. battles one of its worst outbreaks of measles in 25 years.
Kennedy made millions over the years from advocating against vaccines through case referrals, book sales, and consulting fees paid by a nonprofit he founded, according to ethics disclosures.
(Reporting by Ahmed Aboulenein; Additional reporting by Ryan Patrick Jones in Toronto; Editing by Doina Chiacu and Nia Williams)
A version of this article appeared on Medscape.com.
WASHINGTON (Reuters) - U.S. Health Secretary Robert F. Kennedy Jr. said on July 28 that he will work to “fix” the program that compensates victims of vaccine injuries, the National Vaccine Injury Compensation Program.
Kennedy, a long-time vaccine skeptic and former vaccine injury plaintiff lawyer, accused the program and its so-called “Vaccine Court” of corruption and inefficiency in a post on X. He has long been an outspoken critic of the program.
“I will not allow the VICP to continue to ignore its mandate and fail its mission of quickly and fairly compensating vaccine-injured individuals,” he wrote, adding he was working with Attorney General Pam Bondi. “Together, we will steer the Vaccine Court back to its original congressional intent.”
He said the structure disadvantaged claimants because the Department of Health & Human Services – which he now leads – is the defendant, as opposed to vaccine makers.
Changing the VICP would be the latest in a series of far-reaching actions by Kennedy to reshape U.S. regulation of vaccines, food and medicine.
In June, he fired all 17 members of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices, a panel of vaccine experts, replacing them with 7 handpicked members, including known vaccine skeptics.
One of them earned thousands of dollars as an expert witness in litigation against Merck’s, Gardasil vaccine, court records show. Kennedy himself played an instrumental role in organizing mass litigation over the vaccine.
He also is planning to remove all the members of another advisory panel that determines what preventive health measures insurers must cover, the Wall Street Journal reported on July 25. An HHS spokesperson said Kennedy had not yet made a decision regarding the 16-member U.S. Preventive Services Task Force.
Kennedy has for years sown doubt about the safety and efficacy of vaccines. He has a history of clashing with the medical establishment and spreading misinformation about vaccines, including promoting a debunked link between vaccines and autism despite scientific evidence to the contrary.
He has also said the measles vaccine contains cells from aborted fetuses and that the mumps vaccination does not work, comments he made as the U.S. battles one of its worst outbreaks of measles in 25 years.
Kennedy made millions over the years from advocating against vaccines through case referrals, book sales, and consulting fees paid by a nonprofit he founded, according to ethics disclosures.
(Reporting by Ahmed Aboulenein; Additional reporting by Ryan Patrick Jones in Toronto; Editing by Doina Chiacu and Nia Williams)
A version of this article appeared on Medscape.com.
WASHINGTON (Reuters) - U.S. Health Secretary Robert F. Kennedy Jr. said on July 28 that he will work to “fix” the program that compensates victims of vaccine injuries, the National Vaccine Injury Compensation Program.
Kennedy, a long-time vaccine skeptic and former vaccine injury plaintiff lawyer, accused the program and its so-called “Vaccine Court” of corruption and inefficiency in a post on X. He has long been an outspoken critic of the program.
“I will not allow the VICP to continue to ignore its mandate and fail its mission of quickly and fairly compensating vaccine-injured individuals,” he wrote, adding he was working with Attorney General Pam Bondi. “Together, we will steer the Vaccine Court back to its original congressional intent.”
He said the structure disadvantaged claimants because the Department of Health & Human Services – which he now leads – is the defendant, as opposed to vaccine makers.
Changing the VICP would be the latest in a series of far-reaching actions by Kennedy to reshape U.S. regulation of vaccines, food and medicine.
In June, he fired all 17 members of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices, a panel of vaccine experts, replacing them with 7 handpicked members, including known vaccine skeptics.
One of them earned thousands of dollars as an expert witness in litigation against Merck’s, Gardasil vaccine, court records show. Kennedy himself played an instrumental role in organizing mass litigation over the vaccine.
He also is planning to remove all the members of another advisory panel that determines what preventive health measures insurers must cover, the Wall Street Journal reported on July 25. An HHS spokesperson said Kennedy had not yet made a decision regarding the 16-member U.S. Preventive Services Task Force.
Kennedy has for years sown doubt about the safety and efficacy of vaccines. He has a history of clashing with the medical establishment and spreading misinformation about vaccines, including promoting a debunked link between vaccines and autism despite scientific evidence to the contrary.
He has also said the measles vaccine contains cells from aborted fetuses and that the mumps vaccination does not work, comments he made as the U.S. battles one of its worst outbreaks of measles in 25 years.
Kennedy made millions over the years from advocating against vaccines through case referrals, book sales, and consulting fees paid by a nonprofit he founded, according to ethics disclosures.
(Reporting by Ahmed Aboulenein; Additional reporting by Ryan Patrick Jones in Toronto; Editing by Doina Chiacu and Nia Williams)
A version of this article appeared on Medscape.com.
Rurality and Age May Shape Phone-Only Mental Health Care Access Among Veterans
TOPLINE:
Patients living in rural areas and those aged ≥ 65 y had increased odds of receiving mental health care exclusively by phone.
METHODOLOGY:
- Researchers explored factors linked to receiving phone-only mental health care among patients within the Department of Veterans Affairs.
- They included data for 1,156,146 veteran patients with at least one mental health-specific outpatient encounter between October 2021 and September 2022 and at least one between October 2022 and September 2023.
- Patients were categorized as those who received care through phone only (n = 49,125) and those who received care through other methods (n = 1,107,021. Care was received exclusively through video (6.39%), in-person (6.63%), or a combination of in-person, video, and/or phone (86.98%).
- Demographic and clinical predictors, including rurality, age, sex, race, ethnicity, and the number of mental health diagnoses (< 3 vs ≥ 3), were evaluated.
TAKEAWAY:
- The phone-only group had a mean of 6.27 phone visits, whereas those who received care through other methods had a mean of 4.79 phone visits.
- Highly rural patients had 1.50 times higher odds of receiving phone-only mental health care than their urban counterparts (adjusted odds ratio [aOR], 1.50; P < .0001).
- Patients aged 65 years or older were more than twice as likely to receive phone-only care than those younger than 30 years (aOR, ≥ 2.17; P < .0001).
- Having fewer than three mental health diagnoses and more than 50% of mental health visits conducted by medical providers was associated with higher odds of receiving mental health care exclusively by phone (aORs, 2.03 and 1.87, respectively; P < .0001).
IN PRACTICE:
“The results of this work help to characterize the phone-only patient population and can serve to inform future implementation efforts to ensure that patients are receiving care via the modality that best meets their needs,” the authors wrote.
SOURCE:
This study was led by Samantha L. Connolly, PhD, at the VA Boston Healthcare System in Boston. It was published online in The Journal of Rural Health.
LIMITATIONS:
This study focused on a veteran population which may limit the generalizability of the findings to other groups. Additionally, its cross-sectional design restricted the ability to determine cause-and-effect relationships between factors and phone-only care.
DISCLOSURES:
This study was supported by the US Department of Veterans Affairs. The authors declared having no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Patients living in rural areas and those aged ≥ 65 y had increased odds of receiving mental health care exclusively by phone.
METHODOLOGY:
- Researchers explored factors linked to receiving phone-only mental health care among patients within the Department of Veterans Affairs.
- They included data for 1,156,146 veteran patients with at least one mental health-specific outpatient encounter between October 2021 and September 2022 and at least one between October 2022 and September 2023.
- Patients were categorized as those who received care through phone only (n = 49,125) and those who received care through other methods (n = 1,107,021. Care was received exclusively through video (6.39%), in-person (6.63%), or a combination of in-person, video, and/or phone (86.98%).
- Demographic and clinical predictors, including rurality, age, sex, race, ethnicity, and the number of mental health diagnoses (< 3 vs ≥ 3), were evaluated.
TAKEAWAY:
- The phone-only group had a mean of 6.27 phone visits, whereas those who received care through other methods had a mean of 4.79 phone visits.
- Highly rural patients had 1.50 times higher odds of receiving phone-only mental health care than their urban counterparts (adjusted odds ratio [aOR], 1.50; P < .0001).
- Patients aged 65 years or older were more than twice as likely to receive phone-only care than those younger than 30 years (aOR, ≥ 2.17; P < .0001).
- Having fewer than three mental health diagnoses and more than 50% of mental health visits conducted by medical providers was associated with higher odds of receiving mental health care exclusively by phone (aORs, 2.03 and 1.87, respectively; P < .0001).
IN PRACTICE:
“The results of this work help to characterize the phone-only patient population and can serve to inform future implementation efforts to ensure that patients are receiving care via the modality that best meets their needs,” the authors wrote.
SOURCE:
This study was led by Samantha L. Connolly, PhD, at the VA Boston Healthcare System in Boston. It was published online in The Journal of Rural Health.
LIMITATIONS:
This study focused on a veteran population which may limit the generalizability of the findings to other groups. Additionally, its cross-sectional design restricted the ability to determine cause-and-effect relationships between factors and phone-only care.
DISCLOSURES:
This study was supported by the US Department of Veterans Affairs. The authors declared having no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Patients living in rural areas and those aged ≥ 65 y had increased odds of receiving mental health care exclusively by phone.
METHODOLOGY:
- Researchers explored factors linked to receiving phone-only mental health care among patients within the Department of Veterans Affairs.
- They included data for 1,156,146 veteran patients with at least one mental health-specific outpatient encounter between October 2021 and September 2022 and at least one between October 2022 and September 2023.
- Patients were categorized as those who received care through phone only (n = 49,125) and those who received care through other methods (n = 1,107,021. Care was received exclusively through video (6.39%), in-person (6.63%), or a combination of in-person, video, and/or phone (86.98%).
- Demographic and clinical predictors, including rurality, age, sex, race, ethnicity, and the number of mental health diagnoses (< 3 vs ≥ 3), were evaluated.
TAKEAWAY:
- The phone-only group had a mean of 6.27 phone visits, whereas those who received care through other methods had a mean of 4.79 phone visits.
- Highly rural patients had 1.50 times higher odds of receiving phone-only mental health care than their urban counterparts (adjusted odds ratio [aOR], 1.50; P < .0001).
- Patients aged 65 years or older were more than twice as likely to receive phone-only care than those younger than 30 years (aOR, ≥ 2.17; P < .0001).
- Having fewer than three mental health diagnoses and more than 50% of mental health visits conducted by medical providers was associated with higher odds of receiving mental health care exclusively by phone (aORs, 2.03 and 1.87, respectively; P < .0001).
IN PRACTICE:
“The results of this work help to characterize the phone-only patient population and can serve to inform future implementation efforts to ensure that patients are receiving care via the modality that best meets their needs,” the authors wrote.
SOURCE:
This study was led by Samantha L. Connolly, PhD, at the VA Boston Healthcare System in Boston. It was published online in The Journal of Rural Health.
LIMITATIONS:
This study focused on a veteran population which may limit the generalizability of the findings to other groups. Additionally, its cross-sectional design restricted the ability to determine cause-and-effect relationships between factors and phone-only care.
DISCLOSURES:
This study was supported by the US Department of Veterans Affairs. The authors declared having no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.

Searching for the Optimal CRC Surveillance Test
About a third of the US population are eligible for colorectal cancer screening but aren’t up to date on screening.
Many patients are reluctant to test for colon cancer for a variety of reasons, said Jeffrey K. Lee, MD, MPH, a research scientist at the Kaiser Permanente Northern California Division of Research and an attending gastroenterologist at Kaiser Permanente San Francisco Medical Center.
“As a gastroenterologist, I strongly believe we should emphasize the importance of colorectal cancer screening. And there’s many tests available, not just a colonoscopy, to help reduce your chances of developing colorectal cancer and even dying from colorectal cancer,” said Dr. Lee.
Many patients prefer a test that’s more convenient, that doesn’t require them to take time out of their busy schedules. “We must educate our patients that there are some noninvasive screening options that are helpful, and to be able to share with them some of the benefits, but also some of the drawbacks compared to colonoscopy and allow them to have a choice,” he advised.

He is a recipient of the AGA Research Scholar Award, and has in turn supported other researchers by contributing to the AGA Research Foundation. In 2012, Dr. Lee received a grant from the Sylvia Allison Kaplan Clinical Research Fund to fund a study on long-term colorectal cancer risk in patients with normal colonoscopy results.
The findings, published in JAMA Internal Medicine, determined that 10 years after a negative colonoscopy, Kaiser Permanente members had a 46% lower risk of being diagnosed with CRC and were 88% less likely to die from disease compared with patients who didn’t undergo screening.
“Furthermore, the reduced risk of developing colorectal cancer, even dying from it, persisted for more than 12 years after the examination compared with an unscreened population,” said Dr. Lee. “I firmly believe our study really supports the ten-year screening interval after a normal colonoscopy, as currently recommended by our guidelines.”
In an interview, he discussed his research efforts to find the best detection regimens for CRC, and the mentors who guided his career path as a GI scientist.
Q: Why did you choose GI?
During medical school I was fortunate to work in the lab of Dr. John M. Carethers at UC San Diego. He introduced me to GI and inspired me to choose GI as a career. His mentorship was invaluable because he not only solidified my interest in GI, but also inspired me to become a physician scientist, focusing on colorectal cancer prevention and control. His amazing mentorship drew me to this field.
Q: One of your clinical focus areas is hereditary gastrointestinal cancer syndromes. How did you become interested in this area of GI medicine?
My interest in hereditary GI cancer syndromes stemmed from my work as a medical student in Dr. Carethers’ lab. One of my research projects was looking at certain gene mutations among patients with hereditary GI cancer syndromes, specifically, familial hamartomatous polyposis syndrome. It was through these research projects and seeing how these genetic mutations impacted their risk of developing colorectal cancer, inspired me to care for patients with hereditary GI cancer syndromes.
Q: Have you been doing any research on the reasons why more young people are getting colon cancer?
We recently published work looking at the potential factors that may be driving the rising rates of early onset colorectal cancer. One hypothesis that’s been floating around is antibiotic exposure in early adulthood or childhood because of its effect on the microbiome. Using our large database at Kaiser Permanente Northern California, we did not find an association between oral antibiotic use during early adulthood and the risk of early-onset colorectal cancer.
You have the usual suspects like obesity and diabetes, but it’s not explaining all that risk. While familial colorectal cancer syndromes contribute to a small proportion of early-onset colorectal, these syndromes are not increasing across generations. I really do feel it’s something in the diet or how foods are processed and environmental factors that’s driving some of the risk of early onset colorectal cancer and this should be explored further.
Q: In 2018, you issued a landmark study which found an association between a 10-year follow-up after negative colonoscopy and reduced risk of disease and mortality. Has there been any updates to these findings over the last 6 years?
We recently saw a study in JAMA Oncology of a Swedish cohort that showed a negative colonoscopy result was associated with a reduced risk of developing and even dying from colorectal cancer 15 years from that examination, compared to the general population of Sweden. I think there’s some things that we need to be cautious about regarding that study. We have to think about the comparison group that they used and the lack of information regarding the indication of the colonoscopy and the quality of the examination. So, it remains uncertain whether future guidelines are going to stretch out that 10-year interval to 15 years.
Q: What other CRC studies are you working on now?
We have several studies that we are working on right now. One is called the PREVENT CRC study, which is looking at whether a polygenic risk score can improve risk stratification following adenoma removal for colorectal cancer prevention and tailoring post-polypectomy surveillance. This is a large observational cohort study that we have teamed up with the Fred Hutchinson Cancer Center, Erasmus University, and Kaiser Permanente Northwest to answer this important question that may have implications for personalized medicine.
Then there’s the COOP study, funded by the Patient-Centered Outcomes Research Institute. This is looking at the best surveillance test to use among older adults 65 years and older with a history of polyps. The trial is randomizing them to either getting a colonoscopy for surveillance or annual fecal immunochemical test (FIT) for surveillance. This is to see which test is best for detecting colorectal cancer among older adults with a history of polyps.
Q: Do you think FIT tests could eventually replace colonoscopy, given that it’s less invasive?
Although FIT and other stool-based tests are less invasive and have been shown to have high accuracy for detecting colorectal cancer, I personally do not think they are going to replace colonoscopy as the most popular screening modality in the United States. Colonoscopy remains the gold standard for detecting and removing precancerous polyps and has the highest accuracy for detecting colorectal cancer.
Q: Besides Dr. Carethers, what teacher or mentor had the greatest impact on you?
Clinically it’s been Dr. Jonathan Terdiman from UCSF, who taught me everything I know about clinical GI, and the art of colonoscopy. In addition, Douglas A. Corley, MD, PhD, the Permanente Medical Group’s chief research officer, has made the greatest impact on my research career. He’s really taught me how to rigorously design a research study to answer important clinically relevant questions, and has given me the skill set to write NIH grants. I would not be here without these mentors who are truly giants in the field of GI.
Q: When you’re not being a GI, how do you spend your free weekend afternoons? Are you still a “Cal Bears” fan at your alma mater, UC Berkeley?
I spend a lot of time taking my kids to their activities on the weekends. I just took my son to a Cal Bears Game Day, which was hosted by ESPN at Berkeley.

It was an incredible experience hearing sports analyst Pat McAfee lead all the Cal chants, seeing Nick Saban from the University of Alabama take off his red tie and replace it with a Cal Bears tie, and watching a Cal student win a hundred thousand dollars by kicking a football through the goal posts wearing checkered vans.
Lightning Round
Texting or talking?
Text
Favorite breakfast?
Taiwanese breakfast
Place you most want to travel to?
Japan
Favorite junk food?
Trader Joe’s chili lime chips
Favorite season?
Springtime, baseball season
Favorite ice cream flavor?
Mint chocolate chip
How many cups of coffee do you drink per day?
2-3
Last movie you watched?
Oppenheimer
Best place you ever went on vacation?
Hawaii
If you weren’t a gastroenterologist, what would you be?
Barber
Best Halloween costume you ever wore?
SpongeBob SquarePants
Favorite sport?
Tennis
What song do you have to sing along with when you hear it?
Any classic 80s song
Introvert or extrovert?
Introvert
About a third of the US population are eligible for colorectal cancer screening but aren’t up to date on screening.
Many patients are reluctant to test for colon cancer for a variety of reasons, said Jeffrey K. Lee, MD, MPH, a research scientist at the Kaiser Permanente Northern California Division of Research and an attending gastroenterologist at Kaiser Permanente San Francisco Medical Center.
“As a gastroenterologist, I strongly believe we should emphasize the importance of colorectal cancer screening. And there’s many tests available, not just a colonoscopy, to help reduce your chances of developing colorectal cancer and even dying from colorectal cancer,” said Dr. Lee.
Many patients prefer a test that’s more convenient, that doesn’t require them to take time out of their busy schedules. “We must educate our patients that there are some noninvasive screening options that are helpful, and to be able to share with them some of the benefits, but also some of the drawbacks compared to colonoscopy and allow them to have a choice,” he advised.
He is a recipient of the AGA Research Scholar Award, and has in turn supported other researchers by contributing to the AGA Research Foundation. In 2012, Dr. Lee received a grant from the Sylvia Allison Kaplan Clinical Research Fund to fund a study on long-term colorectal cancer risk in patients with normal colonoscopy results.
The findings, published in JAMA Internal Medicine, determined that 10 years after a negative colonoscopy, Kaiser Permanente members had a 46% lower risk of being diagnosed with CRC and were 88% less likely to die from disease compared with patients who didn’t undergo screening.
“Furthermore, the reduced risk of developing colorectal cancer, even dying from it, persisted for more than 12 years after the examination compared with an unscreened population,” said Dr. Lee. “I firmly believe our study really supports the ten-year screening interval after a normal colonoscopy, as currently recommended by our guidelines.”
In an interview, he discussed his research efforts to find the best detection regimens for CRC, and the mentors who guided his career path as a GI scientist.
Q: Why did you choose GI?
During medical school I was fortunate to work in the lab of Dr. John M. Carethers at UC San Diego. He introduced me to GI and inspired me to choose GI as a career. His mentorship was invaluable because he not only solidified my interest in GI, but also inspired me to become a physician scientist, focusing on colorectal cancer prevention and control. His amazing mentorship drew me to this field.
Q: One of your clinical focus areas is hereditary gastrointestinal cancer syndromes. How did you become interested in this area of GI medicine?
My interest in hereditary GI cancer syndromes stemmed from my work as a medical student in Dr. Carethers’ lab. One of my research projects was looking at certain gene mutations among patients with hereditary GI cancer syndromes, specifically, familial hamartomatous polyposis syndrome. It was through these research projects and seeing how these genetic mutations impacted their risk of developing colorectal cancer, inspired me to care for patients with hereditary GI cancer syndromes.
Q: Have you been doing any research on the reasons why more young people are getting colon cancer?
We recently published work looking at the potential factors that may be driving the rising rates of early onset colorectal cancer. One hypothesis that’s been floating around is antibiotic exposure in early adulthood or childhood because of its effect on the microbiome. Using our large database at Kaiser Permanente Northern California, we did not find an association between oral antibiotic use during early adulthood and the risk of early-onset colorectal cancer.
You have the usual suspects like obesity and diabetes, but it’s not explaining all that risk. While familial colorectal cancer syndromes contribute to a small proportion of early-onset colorectal, these syndromes are not increasing across generations. I really do feel it’s something in the diet or how foods are processed and environmental factors that’s driving some of the risk of early onset colorectal cancer and this should be explored further.
Q: In 2018, you issued a landmark study which found an association between a 10-year follow-up after negative colonoscopy and reduced risk of disease and mortality. Has there been any updates to these findings over the last 6 years?
We recently saw a study in JAMA Oncology of a Swedish cohort that showed a negative colonoscopy result was associated with a reduced risk of developing and even dying from colorectal cancer 15 years from that examination, compared to the general population of Sweden. I think there’s some things that we need to be cautious about regarding that study. We have to think about the comparison group that they used and the lack of information regarding the indication of the colonoscopy and the quality of the examination. So, it remains uncertain whether future guidelines are going to stretch out that 10-year interval to 15 years.
Q: What other CRC studies are you working on now?
We have several studies that we are working on right now. One is called the PREVENT CRC study, which is looking at whether a polygenic risk score can improve risk stratification following adenoma removal for colorectal cancer prevention and tailoring post-polypectomy surveillance. This is a large observational cohort study that we have teamed up with the Fred Hutchinson Cancer Center, Erasmus University, and Kaiser Permanente Northwest to answer this important question that may have implications for personalized medicine.
Then there’s the COOP study, funded by the Patient-Centered Outcomes Research Institute. This is looking at the best surveillance test to use among older adults 65 years and older with a history of polyps. The trial is randomizing them to either getting a colonoscopy for surveillance or annual fecal immunochemical test (FIT) for surveillance. This is to see which test is best for detecting colorectal cancer among older adults with a history of polyps.
Q: Do you think FIT tests could eventually replace colonoscopy, given that it’s less invasive?
Although FIT and other stool-based tests are less invasive and have been shown to have high accuracy for detecting colorectal cancer, I personally do not think they are going to replace colonoscopy as the most popular screening modality in the United States. Colonoscopy remains the gold standard for detecting and removing precancerous polyps and has the highest accuracy for detecting colorectal cancer.
Q: Besides Dr. Carethers, what teacher or mentor had the greatest impact on you?
Clinically it’s been Dr. Jonathan Terdiman from UCSF, who taught me everything I know about clinical GI, and the art of colonoscopy. In addition, Douglas A. Corley, MD, PhD, the Permanente Medical Group’s chief research officer, has made the greatest impact on my research career. He’s really taught me how to rigorously design a research study to answer important clinically relevant questions, and has given me the skill set to write NIH grants. I would not be here without these mentors who are truly giants in the field of GI.
Q: When you’re not being a GI, how do you spend your free weekend afternoons? Are you still a “Cal Bears” fan at your alma mater, UC Berkeley?
I spend a lot of time taking my kids to their activities on the weekends. I just took my son to a Cal Bears Game Day, which was hosted by ESPN at Berkeley.
It was an incredible experience hearing sports analyst Pat McAfee lead all the Cal chants, seeing Nick Saban from the University of Alabama take off his red tie and replace it with a Cal Bears tie, and watching a Cal student win a hundred thousand dollars by kicking a football through the goal posts wearing checkered vans.
Lightning Round
Texting or talking?
Text
Favorite breakfast?
Taiwanese breakfast
Place you most want to travel to?
Japan
Favorite junk food?
Trader Joe’s chili lime chips
Favorite season?
Springtime, baseball season
Favorite ice cream flavor?
Mint chocolate chip
How many cups of coffee do you drink per day?
2-3
Last movie you watched?
Oppenheimer
Best place you ever went on vacation?
Hawaii
If you weren’t a gastroenterologist, what would you be?
Barber
Best Halloween costume you ever wore?
SpongeBob SquarePants
Favorite sport?
Tennis
What song do you have to sing along with when you hear it?
Any classic 80s song
Introvert or extrovert?
Introvert
About a third of the US population are eligible for colorectal cancer screening but aren’t up to date on screening.
Many patients are reluctant to test for colon cancer for a variety of reasons, said Jeffrey K. Lee, MD, MPH, a research scientist at the Kaiser Permanente Northern California Division of Research and an attending gastroenterologist at Kaiser Permanente San Francisco Medical Center.
“As a gastroenterologist, I strongly believe we should emphasize the importance of colorectal cancer screening. And there’s many tests available, not just a colonoscopy, to help reduce your chances of developing colorectal cancer and even dying from colorectal cancer,” said Dr. Lee.
Many patients prefer a test that’s more convenient, that doesn’t require them to take time out of their busy schedules. “We must educate our patients that there are some noninvasive screening options that are helpful, and to be able to share with them some of the benefits, but also some of the drawbacks compared to colonoscopy and allow them to have a choice,” he advised.
He is a recipient of the AGA Research Scholar Award, and has in turn supported other researchers by contributing to the AGA Research Foundation. In 2012, Dr. Lee received a grant from the Sylvia Allison Kaplan Clinical Research Fund to fund a study on long-term colorectal cancer risk in patients with normal colonoscopy results.
The findings, published in JAMA Internal Medicine, determined that 10 years after a negative colonoscopy, Kaiser Permanente members had a 46% lower risk of being diagnosed with CRC and were 88% less likely to die from disease compared with patients who didn’t undergo screening.
“Furthermore, the reduced risk of developing colorectal cancer, even dying from it, persisted for more than 12 years after the examination compared with an unscreened population,” said Dr. Lee. “I firmly believe our study really supports the ten-year screening interval after a normal colonoscopy, as currently recommended by our guidelines.”
In an interview, he discussed his research efforts to find the best detection regimens for CRC, and the mentors who guided his career path as a GI scientist.
Q: Why did you choose GI?
During medical school I was fortunate to work in the lab of Dr. John M. Carethers at UC San Diego. He introduced me to GI and inspired me to choose GI as a career. His mentorship was invaluable because he not only solidified my interest in GI, but also inspired me to become a physician scientist, focusing on colorectal cancer prevention and control. His amazing mentorship drew me to this field.
Q: One of your clinical focus areas is hereditary gastrointestinal cancer syndromes. How did you become interested in this area of GI medicine?
My interest in hereditary GI cancer syndromes stemmed from my work as a medical student in Dr. Carethers’ lab. One of my research projects was looking at certain gene mutations among patients with hereditary GI cancer syndromes, specifically, familial hamartomatous polyposis syndrome. It was through these research projects and seeing how these genetic mutations impacted their risk of developing colorectal cancer, inspired me to care for patients with hereditary GI cancer syndromes.
Q: Have you been doing any research on the reasons why more young people are getting colon cancer?
We recently published work looking at the potential factors that may be driving the rising rates of early onset colorectal cancer. One hypothesis that’s been floating around is antibiotic exposure in early adulthood or childhood because of its effect on the microbiome. Using our large database at Kaiser Permanente Northern California, we did not find an association between oral antibiotic use during early adulthood and the risk of early-onset colorectal cancer.
You have the usual suspects like obesity and diabetes, but it’s not explaining all that risk. While familial colorectal cancer syndromes contribute to a small proportion of early-onset colorectal, these syndromes are not increasing across generations. I really do feel it’s something in the diet or how foods are processed and environmental factors that’s driving some of the risk of early onset colorectal cancer and this should be explored further.
Q: In 2018, you issued a landmark study which found an association between a 10-year follow-up after negative colonoscopy and reduced risk of disease and mortality. Has there been any updates to these findings over the last 6 years?
We recently saw a study in JAMA Oncology of a Swedish cohort that showed a negative colonoscopy result was associated with a reduced risk of developing and even dying from colorectal cancer 15 years from that examination, compared to the general population of Sweden. I think there’s some things that we need to be cautious about regarding that study. We have to think about the comparison group that they used and the lack of information regarding the indication of the colonoscopy and the quality of the examination. So, it remains uncertain whether future guidelines are going to stretch out that 10-year interval to 15 years.
Q: What other CRC studies are you working on now?
We have several studies that we are working on right now. One is called the PREVENT CRC study, which is looking at whether a polygenic risk score can improve risk stratification following adenoma removal for colorectal cancer prevention and tailoring post-polypectomy surveillance. This is a large observational cohort study that we have teamed up with the Fred Hutchinson Cancer Center, Erasmus University, and Kaiser Permanente Northwest to answer this important question that may have implications for personalized medicine.
Then there’s the COOP study, funded by the Patient-Centered Outcomes Research Institute. This is looking at the best surveillance test to use among older adults 65 years and older with a history of polyps. The trial is randomizing them to either getting a colonoscopy for surveillance or annual fecal immunochemical test (FIT) for surveillance. This is to see which test is best for detecting colorectal cancer among older adults with a history of polyps.
Q: Do you think FIT tests could eventually replace colonoscopy, given that it’s less invasive?
Although FIT and other stool-based tests are less invasive and have been shown to have high accuracy for detecting colorectal cancer, I personally do not think they are going to replace colonoscopy as the most popular screening modality in the United States. Colonoscopy remains the gold standard for detecting and removing precancerous polyps and has the highest accuracy for detecting colorectal cancer.
Q: Besides Dr. Carethers, what teacher or mentor had the greatest impact on you?
Clinically it’s been Dr. Jonathan Terdiman from UCSF, who taught me everything I know about clinical GI, and the art of colonoscopy. In addition, Douglas A. Corley, MD, PhD, the Permanente Medical Group’s chief research officer, has made the greatest impact on my research career. He’s really taught me how to rigorously design a research study to answer important clinically relevant questions, and has given me the skill set to write NIH grants. I would not be here without these mentors who are truly giants in the field of GI.
Q: When you’re not being a GI, how do you spend your free weekend afternoons? Are you still a “Cal Bears” fan at your alma mater, UC Berkeley?
I spend a lot of time taking my kids to their activities on the weekends. I just took my son to a Cal Bears Game Day, which was hosted by ESPN at Berkeley.
It was an incredible experience hearing sports analyst Pat McAfee lead all the Cal chants, seeing Nick Saban from the University of Alabama take off his red tie and replace it with a Cal Bears tie, and watching a Cal student win a hundred thousand dollars by kicking a football through the goal posts wearing checkered vans.
Lightning Round
Texting or talking?
Text
Favorite breakfast?
Taiwanese breakfast
Place you most want to travel to?
Japan
Favorite junk food?
Trader Joe’s chili lime chips
Favorite season?
Springtime, baseball season
Favorite ice cream flavor?
Mint chocolate chip
How many cups of coffee do you drink per day?
2-3
Last movie you watched?
Oppenheimer
Best place you ever went on vacation?
Hawaii
If you weren’t a gastroenterologist, what would you be?
Barber
Best Halloween costume you ever wore?
SpongeBob SquarePants
Favorite sport?
Tennis
What song do you have to sing along with when you hear it?
Any classic 80s song
Introvert or extrovert?
Introvert

US Military Requires Flu Vaccine for Some After Outbreak in Texas Training Center
June 24 (Reuters) - The US military has resumed requiring flu vaccines for some service members in an exception to Defense Secretary Pete Hegseth’s guidance declaring the shots voluntary 2 months ago.
The decision follows a flu outbreak among recruits at Joint Base San Antonio-Lackland in Texas and sharp criticism of the policy from public health experts. More than 220 recruits have been diagnosed with influenza and 4 hospitalized in the outbreak, according to media reports.
Hegseth said in April that the annual flu vaccine would become optional for all US military personnel under the Pentagon’s new vaccine policy. It had previously been mandated and considered critical to troop preparedness.
The Under Secretary for War Personnel and Readiness approved exception requests for the Army, Navy, Air Force, National Security Agency, and Defense Health Agency, according to a statement from chief Pentagon spokesperson Sean Parnell on Wednesday.
“The decisions were based upon thorough risk assessments and are designed to maximize operational readiness, lethality, and force generation, while safeguarding at-risk populations,” Parnell said.
Each department is responsible for implementation, the spokesperson added.
The World Health Organization recommends the flu shot for those aged ≥ 6 months.
Trump administration Health Secretary Robert F. Kennedy Jr., a longtime antivaccine activist, has enacted policies that have decreased the use of inoculations in the US, including dropping its 2025 flu vaccine campaign. Kennedy’s Make America Healthy Again movement has sought to weaken school enrollment mandates around the country.
Flu vaccines from Sanofi, CSL Seqirus, GSK and AstraZeneca are approved in the United States.
(Reporting by Idrees Ali, Mariam Sunny and Mrinalika Roy; Editing by Caroline Humer and Bill Berkrot)
A version of this article first appeared on Medscape.com.
June 24 (Reuters) - The US military has resumed requiring flu vaccines for some service members in an exception to Defense Secretary Pete Hegseth’s guidance declaring the shots voluntary 2 months ago.
The decision follows a flu outbreak among recruits at Joint Base San Antonio-Lackland in Texas and sharp criticism of the policy from public health experts. More than 220 recruits have been diagnosed with influenza and 4 hospitalized in the outbreak, according to media reports.
Hegseth said in April that the annual flu vaccine would become optional for all US military personnel under the Pentagon’s new vaccine policy. It had previously been mandated and considered critical to troop preparedness.
The Under Secretary for War Personnel and Readiness approved exception requests for the Army, Navy, Air Force, National Security Agency, and Defense Health Agency, according to a statement from chief Pentagon spokesperson Sean Parnell on Wednesday.
“The decisions were based upon thorough risk assessments and are designed to maximize operational readiness, lethality, and force generation, while safeguarding at-risk populations,” Parnell said.
Each department is responsible for implementation, the spokesperson added.
The World Health Organization recommends the flu shot for those aged ≥ 6 months.
Trump administration Health Secretary Robert F. Kennedy Jr., a longtime antivaccine activist, has enacted policies that have decreased the use of inoculations in the US, including dropping its 2025 flu vaccine campaign. Kennedy’s Make America Healthy Again movement has sought to weaken school enrollment mandates around the country.
Flu vaccines from Sanofi, CSL Seqirus, GSK and AstraZeneca are approved in the United States.
(Reporting by Idrees Ali, Mariam Sunny and Mrinalika Roy; Editing by Caroline Humer and Bill Berkrot)
A version of this article first appeared on Medscape.com.
June 24 (Reuters) - The US military has resumed requiring flu vaccines for some service members in an exception to Defense Secretary Pete Hegseth’s guidance declaring the shots voluntary 2 months ago.
The decision follows a flu outbreak among recruits at Joint Base San Antonio-Lackland in Texas and sharp criticism of the policy from public health experts. More than 220 recruits have been diagnosed with influenza and 4 hospitalized in the outbreak, according to media reports.
Hegseth said in April that the annual flu vaccine would become optional for all US military personnel under the Pentagon’s new vaccine policy. It had previously been mandated and considered critical to troop preparedness.
The Under Secretary for War Personnel and Readiness approved exception requests for the Army, Navy, Air Force, National Security Agency, and Defense Health Agency, according to a statement from chief Pentagon spokesperson Sean Parnell on Wednesday.
“The decisions were based upon thorough risk assessments and are designed to maximize operational readiness, lethality, and force generation, while safeguarding at-risk populations,” Parnell said.
Each department is responsible for implementation, the spokesperson added.
The World Health Organization recommends the flu shot for those aged ≥ 6 months.
Trump administration Health Secretary Robert F. Kennedy Jr., a longtime antivaccine activist, has enacted policies that have decreased the use of inoculations in the US, including dropping its 2025 flu vaccine campaign. Kennedy’s Make America Healthy Again movement has sought to weaken school enrollment mandates around the country.
Flu vaccines from Sanofi, CSL Seqirus, GSK and AstraZeneca are approved in the United States.
(Reporting by Idrees Ali, Mariam Sunny and Mrinalika Roy; Editing by Caroline Humer and Bill Berkrot)
A version of this article first appeared on Medscape.com.
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
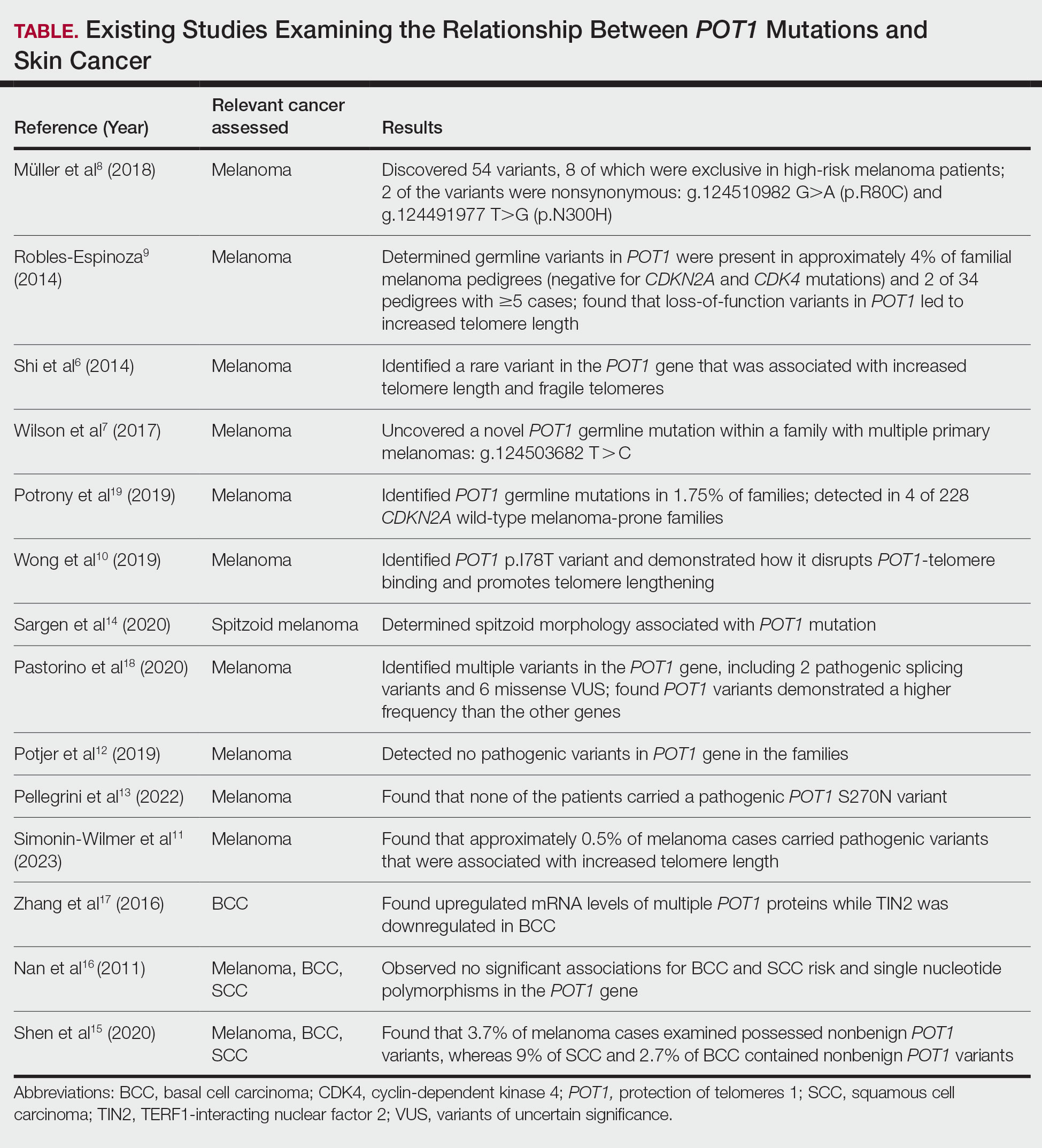
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
PRACTICE POINTS
- Dermatologists should consider referring patients with both a history of skin cancer and a strong family history of internal malignancy for genetic testing for POT1 (protection of telomeres 1) mutations.
- Although melanoma, chronic lymphocytic leukemia, angiosarcoma, and gliomas are most commonly associated with POT1 mutations, this case suggests a broader and more heterogeneous malignancy spectrum than previously recognized.

Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
A 78-year-old man was referred to our dermatology clinic for evaluation of nontender erythematous plaques and nodules with central ulceration on the right leg of 5 months’ duration. The patient’s medical history was remarkable for hyperlipidemia, gastroesophageal reflux disease, prostate cancer, and colon cancer status post resection. He denied any relevant travel history but noted that he was an avid hiker and suspected he may have obtained a puncture wound from a bush or a mosquito bite prior to the appearance of the lesions. Previous therapies prescribed by outside physicians and our practice included trimethoprim/sulfamethoxazole, ceftriaxone, levofloxacin, mupirocin, and topical corticosteroids, all with minimal benefit. Clinical examination on initial presentation revealed multiple ulcerations of the lower extremities present for more than 2 months. Punch biopsy of a sample lesion at the current presentation revealed granulomatous change, focal necrosis, and a mixed inflammatory cell infiltrate. Grocott-Gomori methenamine silver and periodic acid–Schiff stains were negative for fungal organisms. The initial acid-fast bacilli stain was negative for mycobacteria, and tissue culture showed no growth.

Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
A 35-year-old woman presented with an insidious onset of multiple grouped erythematous to violaceous papules over the left preauricular area of 3 months’ duration (top quiz image). The lesions were soft, itchy, nontender, and friable and were associated with bleeding on excoriation and preauricular lymphadenopathy. Serology for HIV was nonreactive, and Gram staining revealed no bacilli. Laboratory assessment including a complete blood count, urinalysis, and liver and renal function tests was normal.
On dermoscopy (middle quiz image), multiple linear and dotted vessels (circle), reddish lacunae (clods), hemorrhagic crusting (blue arrow), white scaling (black arrow), a brown pigment network (square), white structureless areas (yellow arrow), and white lines were seen over a pale-pink background (green arrow). Scaling and crusting over some lesions, along with a peripheral rim of scaling and brownish pigmentation, also was appreciated. Histopathology revealed a proliferation of vascular channels admixed with lymphocytes, plasma cells, and eosinophils along with a proliferation of thin- and thick-walled blood vessels in the superficial as well as deep dermis (bottom quiz image).

Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
