User login
Bimekizumab Shows Long-term Benefits in PsA with Inadequate Response or Intolerance to TNFi
Key clinical point: Bimekizumab led to long-term improvements in efficacy outcomes and had a manageable safety profile in patients with active psoriatic arthritis (PsA) and prior inadequate response or intolerance to tumor necrosis factor inhibitors (TNFi-IR).
Major finding: At week 16, a significantly higher number of patients achieved ≥50% improvement in American College of Rheumatology response with bimekizumab vs placebo (43.4% vs 6.8%; P < .0001), with improvements sustained up to week 52 by >40% of patients receiving bimekizumab. No new safety signals were observed.
Study details: Findings are from the BE VITAL open-label extension study including 377 patients with active PsA and TNFi-IR who received bimekizumab or placebo for 16 weeks followed by only bimekizumab up to week 52.
Disclosures: This study was sponsored by UCB Pharma. Four authors reported being employees or shareholders of UCB Pharma. Several authors declared receiving research support or consulting fees or having other ties with various sources, including UCB Pharma.
Source: Coates LC, Landewe R, McInnes IB, et al. Bimekizumab treatment in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: 52-week safety and efficacy from the phase III BE COMPLETE study and its open-label extension BE VITAL. RMD Open. 2024;10:e003855 (Feb 22). doi: 10.1136/rmdopen-2023-003855 Source
Key clinical point: Bimekizumab led to long-term improvements in efficacy outcomes and had a manageable safety profile in patients with active psoriatic arthritis (PsA) and prior inadequate response or intolerance to tumor necrosis factor inhibitors (TNFi-IR).
Major finding: At week 16, a significantly higher number of patients achieved ≥50% improvement in American College of Rheumatology response with bimekizumab vs placebo (43.4% vs 6.8%; P < .0001), with improvements sustained up to week 52 by >40% of patients receiving bimekizumab. No new safety signals were observed.
Study details: Findings are from the BE VITAL open-label extension study including 377 patients with active PsA and TNFi-IR who received bimekizumab or placebo for 16 weeks followed by only bimekizumab up to week 52.
Disclosures: This study was sponsored by UCB Pharma. Four authors reported being employees or shareholders of UCB Pharma. Several authors declared receiving research support or consulting fees or having other ties with various sources, including UCB Pharma.
Source: Coates LC, Landewe R, McInnes IB, et al. Bimekizumab treatment in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: 52-week safety and efficacy from the phase III BE COMPLETE study and its open-label extension BE VITAL. RMD Open. 2024;10:e003855 (Feb 22). doi: 10.1136/rmdopen-2023-003855 Source
Key clinical point: Bimekizumab led to long-term improvements in efficacy outcomes and had a manageable safety profile in patients with active psoriatic arthritis (PsA) and prior inadequate response or intolerance to tumor necrosis factor inhibitors (TNFi-IR).
Major finding: At week 16, a significantly higher number of patients achieved ≥50% improvement in American College of Rheumatology response with bimekizumab vs placebo (43.4% vs 6.8%; P < .0001), with improvements sustained up to week 52 by >40% of patients receiving bimekizumab. No new safety signals were observed.
Study details: Findings are from the BE VITAL open-label extension study including 377 patients with active PsA and TNFi-IR who received bimekizumab or placebo for 16 weeks followed by only bimekizumab up to week 52.
Disclosures: This study was sponsored by UCB Pharma. Four authors reported being employees or shareholders of UCB Pharma. Several authors declared receiving research support or consulting fees or having other ties with various sources, including UCB Pharma.
Source: Coates LC, Landewe R, McInnes IB, et al. Bimekizumab treatment in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: 52-week safety and efficacy from the phase III BE COMPLETE study and its open-label extension BE VITAL. RMD Open. 2024;10:e003855 (Feb 22). doi: 10.1136/rmdopen-2023-003855 Source
Similar Incidences of MACE in Patients with PsA and RA
Key clinical point: The incidence rates of major adverse cardiovascular events (MACE) were similar in patients with psoriatic arthritis (PsA) and rheumatoid arthritis (RA), suggesting that both inflammatory disorders share a common atherogenic mechanism.
Major finding: After a total of 119,571 patient-years of follow-up, 6.7% of patients with RA or PsA developed MACE for the first time. The rates of MACE incidence (adjusted incidence rate ratio 0.96; P = .767) and MACE-free survival (P = .987) were comparable between patients with PsA and RA. Higher time-varying erythrocyte sedimentation rate, C-reactive protein levels, and exposure to glucocorticoids increased the risk for MACE in both patients with PsA and RA (all P < .05).
Study details: This population based retrospective cohort study included 13,905 patients with PsA (n = 1672) or RA (n = 12,233) who did not have any previous history of MACE.
Disclosures: This study did not receive any funding. The authors declared no conflicts of interest.
Source: Meng H, Lam SH, So H, Tam LS. Incidence and risk factors of major cardiovascular events in rheumatoid arthritis and psoriatic arthritis: A population-based cohort study. Semin Arthritis Rheum. 2024;65:152416 (Feb 17). Source
Key clinical point: The incidence rates of major adverse cardiovascular events (MACE) were similar in patients with psoriatic arthritis (PsA) and rheumatoid arthritis (RA), suggesting that both inflammatory disorders share a common atherogenic mechanism.
Major finding: After a total of 119,571 patient-years of follow-up, 6.7% of patients with RA or PsA developed MACE for the first time. The rates of MACE incidence (adjusted incidence rate ratio 0.96; P = .767) and MACE-free survival (P = .987) were comparable between patients with PsA and RA. Higher time-varying erythrocyte sedimentation rate, C-reactive protein levels, and exposure to glucocorticoids increased the risk for MACE in both patients with PsA and RA (all P < .05).
Study details: This population based retrospective cohort study included 13,905 patients with PsA (n = 1672) or RA (n = 12,233) who did not have any previous history of MACE.
Disclosures: This study did not receive any funding. The authors declared no conflicts of interest.
Source: Meng H, Lam SH, So H, Tam LS. Incidence and risk factors of major cardiovascular events in rheumatoid arthritis and psoriatic arthritis: A population-based cohort study. Semin Arthritis Rheum. 2024;65:152416 (Feb 17). Source
Key clinical point: The incidence rates of major adverse cardiovascular events (MACE) were similar in patients with psoriatic arthritis (PsA) and rheumatoid arthritis (RA), suggesting that both inflammatory disorders share a common atherogenic mechanism.
Major finding: After a total of 119,571 patient-years of follow-up, 6.7% of patients with RA or PsA developed MACE for the first time. The rates of MACE incidence (adjusted incidence rate ratio 0.96; P = .767) and MACE-free survival (P = .987) were comparable between patients with PsA and RA. Higher time-varying erythrocyte sedimentation rate, C-reactive protein levels, and exposure to glucocorticoids increased the risk for MACE in both patients with PsA and RA (all P < .05).
Study details: This population based retrospective cohort study included 13,905 patients with PsA (n = 1672) or RA (n = 12,233) who did not have any previous history of MACE.
Disclosures: This study did not receive any funding. The authors declared no conflicts of interest.
Source: Meng H, Lam SH, So H, Tam LS. Incidence and risk factors of major cardiovascular events in rheumatoid arthritis and psoriatic arthritis: A population-based cohort study. Semin Arthritis Rheum. 2024;65:152416 (Feb 17). Source
Ixekizumab Improves Distal Interphalangeal Joint Involvement and Adjacent Nail Psoriasis in PsA
Key clinical point: Ixekizumab was more effective than adalimumab in resolving distal interphalangeal (DIP) joint tenderness, swelling, and adjacent nail psoriasis in patients with psoriatic arthritis (PsA) and DIP joint involvement who nearly invariably had adjacent nail psoriasis.
Major finding: More than 96% of patients with PsA and simultaneous DIP joint involvement reported adjacent nail psoriasis in at least one digit in the finger unit. Ixekizumab vs adalimumab led to greater improvements in DIP involvement and adjacent nail psoriasis as early as week 12 (38.8% vs 28.4%; P < .0001), with improvements sustained up to week 52 (64.9% vs 57.5%; P = .0055).
Study details: This post hoc analysis of the SPIRIT-H2H study included 354 patients with PsA who had simultaneous DIP joint involvement and adjacent nail psoriasis and were randomly assigned to receive ixekizumab or adalimumab.
Disclosures: This study was supported by Eli Lilly and Company. Four authors declared being employees and shareholders of Eli Lilly and Company. The other authors reported ties with various sources, including Eli Lilly and Company.
Source: McGonagle D, Kavanaugh A, McInnes IB, et al. Association of the clinical components in the distal interphalangeal joint synovio-entheseal complex and subsequent response to ixekizumab or adalimumab in psoriatic arthritis. Rheumatology (Oxford). 2024 (Feb 10). doi: 10.1093/rheumatology/keae060 Source
Key clinical point: Ixekizumab was more effective than adalimumab in resolving distal interphalangeal (DIP) joint tenderness, swelling, and adjacent nail psoriasis in patients with psoriatic arthritis (PsA) and DIP joint involvement who nearly invariably had adjacent nail psoriasis.
Major finding: More than 96% of patients with PsA and simultaneous DIP joint involvement reported adjacent nail psoriasis in at least one digit in the finger unit. Ixekizumab vs adalimumab led to greater improvements in DIP involvement and adjacent nail psoriasis as early as week 12 (38.8% vs 28.4%; P < .0001), with improvements sustained up to week 52 (64.9% vs 57.5%; P = .0055).
Study details: This post hoc analysis of the SPIRIT-H2H study included 354 patients with PsA who had simultaneous DIP joint involvement and adjacent nail psoriasis and were randomly assigned to receive ixekizumab or adalimumab.
Disclosures: This study was supported by Eli Lilly and Company. Four authors declared being employees and shareholders of Eli Lilly and Company. The other authors reported ties with various sources, including Eli Lilly and Company.
Source: McGonagle D, Kavanaugh A, McInnes IB, et al. Association of the clinical components in the distal interphalangeal joint synovio-entheseal complex and subsequent response to ixekizumab or adalimumab in psoriatic arthritis. Rheumatology (Oxford). 2024 (Feb 10). doi: 10.1093/rheumatology/keae060 Source
Key clinical point: Ixekizumab was more effective than adalimumab in resolving distal interphalangeal (DIP) joint tenderness, swelling, and adjacent nail psoriasis in patients with psoriatic arthritis (PsA) and DIP joint involvement who nearly invariably had adjacent nail psoriasis.
Major finding: More than 96% of patients with PsA and simultaneous DIP joint involvement reported adjacent nail psoriasis in at least one digit in the finger unit. Ixekizumab vs adalimumab led to greater improvements in DIP involvement and adjacent nail psoriasis as early as week 12 (38.8% vs 28.4%; P < .0001), with improvements sustained up to week 52 (64.9% vs 57.5%; P = .0055).
Study details: This post hoc analysis of the SPIRIT-H2H study included 354 patients with PsA who had simultaneous DIP joint involvement and adjacent nail psoriasis and were randomly assigned to receive ixekizumab or adalimumab.
Disclosures: This study was supported by Eli Lilly and Company. Four authors declared being employees and shareholders of Eli Lilly and Company. The other authors reported ties with various sources, including Eli Lilly and Company.
Source: McGonagle D, Kavanaugh A, McInnes IB, et al. Association of the clinical components in the distal interphalangeal joint synovio-entheseal complex and subsequent response to ixekizumab or adalimumab in psoriatic arthritis. Rheumatology (Oxford). 2024 (Feb 10). doi: 10.1093/rheumatology/keae060 Source
Can AI Tool Improve Dx of Ear Infections?
TOPLINE:
Researchers have developed a tool that uses artificial intelligence (AI) to identify acute otitis media in children based on otoscopic videos. It may improve diagnosis of ear infections in primary care settings, the developers said.
METHODOLOGY:
- The developers relied on otoscopic videos of the tympanic membrane captured on smartphones connected to scopes.
- Their analysis focused on 1151 videos from 635 children, most younger than 3 years old, who were seen for sick or well visits at outpatient clinics in Pennsylvania from 2018 to 2023.
- The tool was trained to differentiate between patients who did and did not have acute otitis media.
TAKEAWAY:
- Out of an original pool of 1561 videos, 410 were excluded due to obstruction by cerumen. In the remaining videos, experts identified acute otitis media in 305 videos (26.5%) and no acute otitis media in 846 videos (73.5%).
- The tool achieved a sensitivity of 93.8% and specificity of 93.5%, with bulging of the tympanic membrane being the most indicative feature of acute otitis media, present in 100% of diagnosed cases, according to the researchers.
- Feedback from 60 parents was largely positive, with 80% wanting the tool to be used during future visits.
IN PRACTICE:
Based on the diagnostic accuracy of clinicians in other studies, “The algorithm exhibited higher accuracy than pediatricians, primary care physicians, and advance practice clinicians and, accordingly, could reasonably be used in these settings to aid with decisions regarding treatment,” the authors of the study wrote. “More accurate diagnosis of [acute otitis media] may help reduce unnecessary prescriptions of antimicrobials in young children,” they added.
Studies directly comparing the performance of the tool vs clinicians are still needed, however, according to an editorial accompanying the journal article.
“While the data from this study show the model’s accuracy (94%) is superior to historical accuracy of clinicians in diagnosing acute otitis media (84% or less), these data come from different studies not using the same definition for accuracy,” wrote Hojjat Salmasian, MD, MPH, PhD, and Lisa Biggs, MD, with Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania. “If we assume the model is confirmed to be highly accurate and free from bias, this model could truly transform care for patients with suspected acute otitis media.”
SOURCE:
Alejandro Hoberman, MD, with the University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania, was the corresponding author of the study. It was published online in JAMA Pediatrics .
LIMITATIONS:
The study used convenience sampling and did not include external validation of the tool. The researchers lacked information about participant demographics and the reason for their clinic visit.
DISCLOSURES:
Three authors of the study are listed as inventors on a patent for a tool to diagnose acute otitis media. Two authors with Dcipher Analytics disclosed fees from the University of Pittsburgh for their work on an application programming interface during the study. The research was supported by the Department of Pediatrics at the University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Researchers have developed a tool that uses artificial intelligence (AI) to identify acute otitis media in children based on otoscopic videos. It may improve diagnosis of ear infections in primary care settings, the developers said.
METHODOLOGY:
- The developers relied on otoscopic videos of the tympanic membrane captured on smartphones connected to scopes.
- Their analysis focused on 1151 videos from 635 children, most younger than 3 years old, who were seen for sick or well visits at outpatient clinics in Pennsylvania from 2018 to 2023.
- The tool was trained to differentiate between patients who did and did not have acute otitis media.
TAKEAWAY:
- Out of an original pool of 1561 videos, 410 were excluded due to obstruction by cerumen. In the remaining videos, experts identified acute otitis media in 305 videos (26.5%) and no acute otitis media in 846 videos (73.5%).
- The tool achieved a sensitivity of 93.8% and specificity of 93.5%, with bulging of the tympanic membrane being the most indicative feature of acute otitis media, present in 100% of diagnosed cases, according to the researchers.
- Feedback from 60 parents was largely positive, with 80% wanting the tool to be used during future visits.
IN PRACTICE:
Based on the diagnostic accuracy of clinicians in other studies, “The algorithm exhibited higher accuracy than pediatricians, primary care physicians, and advance practice clinicians and, accordingly, could reasonably be used in these settings to aid with decisions regarding treatment,” the authors of the study wrote. “More accurate diagnosis of [acute otitis media] may help reduce unnecessary prescriptions of antimicrobials in young children,” they added.
Studies directly comparing the performance of the tool vs clinicians are still needed, however, according to an editorial accompanying the journal article.
“While the data from this study show the model’s accuracy (94%) is superior to historical accuracy of clinicians in diagnosing acute otitis media (84% or less), these data come from different studies not using the same definition for accuracy,” wrote Hojjat Salmasian, MD, MPH, PhD, and Lisa Biggs, MD, with Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania. “If we assume the model is confirmed to be highly accurate and free from bias, this model could truly transform care for patients with suspected acute otitis media.”
SOURCE:
Alejandro Hoberman, MD, with the University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania, was the corresponding author of the study. It was published online in JAMA Pediatrics .
LIMITATIONS:
The study used convenience sampling and did not include external validation of the tool. The researchers lacked information about participant demographics and the reason for their clinic visit.
DISCLOSURES:
Three authors of the study are listed as inventors on a patent for a tool to diagnose acute otitis media. Two authors with Dcipher Analytics disclosed fees from the University of Pittsburgh for their work on an application programming interface during the study. The research was supported by the Department of Pediatrics at the University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Researchers have developed a tool that uses artificial intelligence (AI) to identify acute otitis media in children based on otoscopic videos. It may improve diagnosis of ear infections in primary care settings, the developers said.
METHODOLOGY:
- The developers relied on otoscopic videos of the tympanic membrane captured on smartphones connected to scopes.
- Their analysis focused on 1151 videos from 635 children, most younger than 3 years old, who were seen for sick or well visits at outpatient clinics in Pennsylvania from 2018 to 2023.
- The tool was trained to differentiate between patients who did and did not have acute otitis media.
TAKEAWAY:
- Out of an original pool of 1561 videos, 410 were excluded due to obstruction by cerumen. In the remaining videos, experts identified acute otitis media in 305 videos (26.5%) and no acute otitis media in 846 videos (73.5%).
- The tool achieved a sensitivity of 93.8% and specificity of 93.5%, with bulging of the tympanic membrane being the most indicative feature of acute otitis media, present in 100% of diagnosed cases, according to the researchers.
- Feedback from 60 parents was largely positive, with 80% wanting the tool to be used during future visits.
IN PRACTICE:
Based on the diagnostic accuracy of clinicians in other studies, “The algorithm exhibited higher accuracy than pediatricians, primary care physicians, and advance practice clinicians and, accordingly, could reasonably be used in these settings to aid with decisions regarding treatment,” the authors of the study wrote. “More accurate diagnosis of [acute otitis media] may help reduce unnecessary prescriptions of antimicrobials in young children,” they added.
Studies directly comparing the performance of the tool vs clinicians are still needed, however, according to an editorial accompanying the journal article.
“While the data from this study show the model’s accuracy (94%) is superior to historical accuracy of clinicians in diagnosing acute otitis media (84% or less), these data come from different studies not using the same definition for accuracy,” wrote Hojjat Salmasian, MD, MPH, PhD, and Lisa Biggs, MD, with Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania. “If we assume the model is confirmed to be highly accurate and free from bias, this model could truly transform care for patients with suspected acute otitis media.”
SOURCE:
Alejandro Hoberman, MD, with the University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania, was the corresponding author of the study. It was published online in JAMA Pediatrics .
LIMITATIONS:
The study used convenience sampling and did not include external validation of the tool. The researchers lacked information about participant demographics and the reason for their clinic visit.
DISCLOSURES:
Three authors of the study are listed as inventors on a patent for a tool to diagnose acute otitis media. Two authors with Dcipher Analytics disclosed fees from the University of Pittsburgh for their work on an application programming interface during the study. The research was supported by the Department of Pediatrics at the University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
When Should a Pediatrician Suspect a Rare Disease?
A wise medical precept is attributed to Theodore Woodward, MD (1914-2005): “When you hear hoofbeats, think of horses, not zebras.” Primary care pediatricians, however, often find themselves confronting so-called rare or uncommon diseases (“zebras”) in their offices. The pressing challenge is to know when to suspect them. How can one reconcile the need to dispel uncertainty with the use of diagnostic tests that can be costly and invasive? When can the desire to reassure parents mean delaying the detection of a potentially treatable condition?
“It may seem like wordplay, but it’s not uncommon to have a rare disease,” noted Alejandro Fainboim, MD, a specialist in rare diseases and head of the Multivalent Day Hospital at the Ricardo Gutiérrez Children’s Hospital in Buenos Aires, Argentina. “And pediatricians are the first line of defense in detecting these types of pathologies. To make the diagnosis, we have to consider them. And to consider them, we must be knowledgeable. That’s why sometimes, ignorance slows down the diagnosis,” Dr. Fainboim made his remarks during an online seminar organized for the press on the eve of Rare Disease Day, which is commemorated on February 29th.
There are more than 8000 rare diseases, which generally are defined as those affecting fewer than five people per 10,000. But collectively, one in every 13 people has one of these diseases, and one in every two diagnosed patients is a child. Dr. Fainboim emphasized that most of these rare diseases are severe or very severe, hereditary, degenerative, and potentially fatal. And although they are pediatric pathologies, some manifest later in adulthood.
“The major problem we pediatricians face is that we’re handed a model from adults to solve pediatric diseases. So, signs and symptoms are described that we won’t find early on, but we have to anticipate and learn to decode some that are hidden,” he remarked.
Diagnostic delays and repeated consultations with various doctors before identification are common. Dr. Fainboim added that in industrialized countries, the diagnosis of these diseases takes between 5 and 10 years, and in low-income countries, up to 30 years or more. However, “this has improved significantly in recent years,” he said.
Unnoticed Signs
Specialists who treat patients with rare diseases often feel that there were obvious signs that went unnoticed and should have aroused the suspicion of the primary care physician. An example is paroxysmal nocturnal hemoglobinuria, which affects 13-14 people per million inhabitants and can appear at any age, although the incidence is higher in the third decade of life.
“In my 50 years as a doctor, I’ve seen seven or eight patients with paroxysmal nocturnal hemoglobinuria,” said Elsa Nucifora, MD, a hematologist at the Italian Hospital of Buenos Aires, Argentina. But “the diagnosis is so easy” that doctors could make it if they “were to think instead of acting automatically because they’re in a hurry,” she added. The diagnosis should be considered “every time anemia occurs in a young person, with certain characteristics, instead of giving them iron like everyone else and ‘we’ll see later’…the diagnosis is in two or three steps, so it’s not complicated.”
Similar situations occur with more than 1000 neuromuscular diseases involving mutations in more than 600 genes, including spinal muscular atrophy and muscular dystrophies.
“What are the most common manifestations? The hypotonic infant, the child who walks late, who falls frequently, who can’t climb stairs, who later may have difficulty breathing, who loses strength: These are presentations often unrecognized by doctors not in the specialty,” said Alberto Dubrovsky, MD, director of the Department of Neurology and the Neuromuscular Diseases Unit at the Favaloro University Neuroscience Institute in Buenos Aires, Argentina, during the seminar. “And considering that these diseases are diagnosed based on genetic mutations that need to be known to search for and request them, we are faced with a truly complex scenario that requires subspecialization.”
In a study recently published in the Argentine Archives of Pediatrics, Dr. Dubrovsky and colleagues interviewed 112 families of Argentine patients with molecular diagnoses of spinal muscular atrophy types I, II, and III and found that in 75%-85% of cases, the first signs of the disease (such as hypotonia, developmental delay, inability to achieve bipedal standing, or frequent falls) were recognized by parents. For type I, the most severe and early onset, in only 17.5% of cases did a neonatologist or pediatrician first notice something. Of the 72 patients with types II and III, where routine checks are less frequent than in the first months of life, only one doctor detected the first signs of the disease before parents or other relatives.
In the same study, the median time elapsed between the first sign and confirmed molecular diagnosis was 2, 10, and 31.5 months for types I, II, and III, respectively. The delay “is primarily due to the lack of clinical suspicion on the part of the intervening physician, who often dismisses or misinterprets the signs reported by parents, as reflected in the alternative diagnoses invoked,” the authors wrote.
“I don’t even ask for suspicion of a specific rare disease because that requires specialization. What I ask for is a kind of recognition or realization that something is happening and then request a consultation with the specialist to ensure proper care,” said Dr. Dubrovsky.
In another study conducted among 70 Argentine patients under age 13 years who were diagnosed with Duchenne muscular dystrophy (one of the most severe forms of muscular dystrophy), 82% of the pediatricians who were initially consulted for any problem in motor agility that parents, other relatives, or teachers had detected dismissed the observation. “They’re told to wait, that it will mature a little more,” said Dr. Dubrovsky. This explains why the time to diagnosis in Argentina from the first signs is around 2 years. The delays are unfortunate because “today we have treatments capable of interfering with the disease’s progression slope, reducing its progression, or eventually stopping it,” he said.
“Do you mind that primary care pediatricians don’t notice or dismiss signs and symptoms strongly suggestive of one of these rare diseases? Does it frustrate you?” this news organization asked asked Dr. Dubrovsky. “Sometimes it does make me angry, but many times it’s understood that there can’t be highly trained specialists everywhere to realize and request diagnostic tests. One must consider the circumstances in each case, and that’s why we work in education,” he replied.
Rules and Experience
In an interview, Dr. Fainboim highlighted key factors that should prompt a pediatrician’s suspicion. One is common symptoms expressed in a more intense or complicated way or when many symptoms coexist in the same patient, even if each one separately is benign or not so severe.
Dr. Fainboim also recommended establishing a therapeutic alliance with parents. “We shouldn’t undermine what parents say, especially those who have other children and already know what normal child development is like. This is a very important milestone.
“We have to strengthen the suspicion clue, and for that, we rely on standards and our experience, which we keep refining. As Wilde said, experience is the sum of our mistakes. But there’s no universal answer. Not all families are the same. Not all diseases manifest in the same way. And unless there’s an imminent risk to life or function, one can wait and take the time to evaluate it. For example, if I have a child with slowed developmental milestones, what I have to do is teach how to stimulate them or send them for stimulation with another professional. And I observe the response to this initial basic treatment. If I see no response, the alarms start to grow louder,” said Dr. Fainboim.
Pablo Barvosa, MD, the principal physician in the outpatient area of the Juan P. Garrahan Pediatric Hospital in Buenos Aires, Argentina, and a member of the Working Group on Genetics and Rare Diseases of the Argentine Society of Pediatrics, told this news organization about other factors that should be considered for detecting these pathologies. Dr. Barvosa did not participate in the online seminar.
“Patients with rare diseases have common symptoms. What needs to be done is to prioritize those symptoms that behave abnormally, that have an unusual evolution compared with normal situations. For example, children who go into a coma after a fasting episode or after eating a certain food,” he said.
Dr. Barvosa also suggested considering when patients belong to certain communities where there is a lot of endogamy, due to the higher incidence of hereditary diseases. “Attention should be heightened when parents are cousins or relatives,” he pointed out.
“My view is that doctors should think more and better, be rational, sequential. If a disease is treated and resolved, but we find out that the child had 26 previous hospitalizations in the last 2 years, something is wrong. We have to look at the patient’s and family’s life histories. If a mother had 15 miscarriages, that’s a warning sign. We have to find a common thread. Be a sharp-witted pediatrician,” said Dr. Barvosa.
The suspicion and diagnosis of a rare disease can be devastating for families and painful for the professional, but even if there is no specific treatment, “something can always be done for patients,” he added.
And in certain circumstances, identifying a rare disease can reverse the ominous “stamp” of a wrong diagnosis. Dr. Barvosa commented on the case of a 7-year-old boy he attended at the hospital in 2014. The boy presented as quadriplegic, with no mobility in his limbs, and the parents were convinced he had that condition because he had fallen from the roof of the house. Although imaging techniques did not show a spinal injury, it was assumed to be a case of spinal cord injury without radiographic abnormality. But something caught Dr. Barvosa’s attention: The boy had well-developed abdominal muscles, as if he were an athlete. So, he requested an electromyogram, and the muscle was found to be in permanent contraction.
“The patient didn’t have a spinal cord injury: He had Isaacs’ syndrome,” said Dr. Barvosa. The syndrome also is known as acquired neuromyotonia, a rare condition of hyperexcitability of peripheral nerves that activate muscle fibers. “That is treated with anticonvulsants, such as phenytoin. Within a week, he was walking again, and shortly after, he was playing soccer. When I presented the case at a conference, I cried with emotion. That’s why the pediatrician must be insistent, be like the gadfly that stings in the ear” when there are clinical elements that don’t quite fit into a clear diagnosis, he added.
In recent publications, Dr. Dubrovsky has reported receiving fees for consultations or research from PTC, Sarepta, Biogen, Sanofi Genzyme, Takeda Avexis, Novartis, Raffo, and Roche. Dr. Nucifora has received fees from Jansen LATAM. Dr. Fainboim reported receiving fees from Sanofi. Dr. Barvosa has declared no relevant financial conflicts of interest. The webinar was organized by Urban Comunicaciones.
This story was translated from the Medscape Spanish edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
A wise medical precept is attributed to Theodore Woodward, MD (1914-2005): “When you hear hoofbeats, think of horses, not zebras.” Primary care pediatricians, however, often find themselves confronting so-called rare or uncommon diseases (“zebras”) in their offices. The pressing challenge is to know when to suspect them. How can one reconcile the need to dispel uncertainty with the use of diagnostic tests that can be costly and invasive? When can the desire to reassure parents mean delaying the detection of a potentially treatable condition?
“It may seem like wordplay, but it’s not uncommon to have a rare disease,” noted Alejandro Fainboim, MD, a specialist in rare diseases and head of the Multivalent Day Hospital at the Ricardo Gutiérrez Children’s Hospital in Buenos Aires, Argentina. “And pediatricians are the first line of defense in detecting these types of pathologies. To make the diagnosis, we have to consider them. And to consider them, we must be knowledgeable. That’s why sometimes, ignorance slows down the diagnosis,” Dr. Fainboim made his remarks during an online seminar organized for the press on the eve of Rare Disease Day, which is commemorated on February 29th.
There are more than 8000 rare diseases, which generally are defined as those affecting fewer than five people per 10,000. But collectively, one in every 13 people has one of these diseases, and one in every two diagnosed patients is a child. Dr. Fainboim emphasized that most of these rare diseases are severe or very severe, hereditary, degenerative, and potentially fatal. And although they are pediatric pathologies, some manifest later in adulthood.
“The major problem we pediatricians face is that we’re handed a model from adults to solve pediatric diseases. So, signs and symptoms are described that we won’t find early on, but we have to anticipate and learn to decode some that are hidden,” he remarked.
Diagnostic delays and repeated consultations with various doctors before identification are common. Dr. Fainboim added that in industrialized countries, the diagnosis of these diseases takes between 5 and 10 years, and in low-income countries, up to 30 years or more. However, “this has improved significantly in recent years,” he said.
Unnoticed Signs
Specialists who treat patients with rare diseases often feel that there were obvious signs that went unnoticed and should have aroused the suspicion of the primary care physician. An example is paroxysmal nocturnal hemoglobinuria, which affects 13-14 people per million inhabitants and can appear at any age, although the incidence is higher in the third decade of life.
“In my 50 years as a doctor, I’ve seen seven or eight patients with paroxysmal nocturnal hemoglobinuria,” said Elsa Nucifora, MD, a hematologist at the Italian Hospital of Buenos Aires, Argentina. But “the diagnosis is so easy” that doctors could make it if they “were to think instead of acting automatically because they’re in a hurry,” she added. The diagnosis should be considered “every time anemia occurs in a young person, with certain characteristics, instead of giving them iron like everyone else and ‘we’ll see later’…the diagnosis is in two or three steps, so it’s not complicated.”
Similar situations occur with more than 1000 neuromuscular diseases involving mutations in more than 600 genes, including spinal muscular atrophy and muscular dystrophies.
“What are the most common manifestations? The hypotonic infant, the child who walks late, who falls frequently, who can’t climb stairs, who later may have difficulty breathing, who loses strength: These are presentations often unrecognized by doctors not in the specialty,” said Alberto Dubrovsky, MD, director of the Department of Neurology and the Neuromuscular Diseases Unit at the Favaloro University Neuroscience Institute in Buenos Aires, Argentina, during the seminar. “And considering that these diseases are diagnosed based on genetic mutations that need to be known to search for and request them, we are faced with a truly complex scenario that requires subspecialization.”
In a study recently published in the Argentine Archives of Pediatrics, Dr. Dubrovsky and colleagues interviewed 112 families of Argentine patients with molecular diagnoses of spinal muscular atrophy types I, II, and III and found that in 75%-85% of cases, the first signs of the disease (such as hypotonia, developmental delay, inability to achieve bipedal standing, or frequent falls) were recognized by parents. For type I, the most severe and early onset, in only 17.5% of cases did a neonatologist or pediatrician first notice something. Of the 72 patients with types II and III, where routine checks are less frequent than in the first months of life, only one doctor detected the first signs of the disease before parents or other relatives.
In the same study, the median time elapsed between the first sign and confirmed molecular diagnosis was 2, 10, and 31.5 months for types I, II, and III, respectively. The delay “is primarily due to the lack of clinical suspicion on the part of the intervening physician, who often dismisses or misinterprets the signs reported by parents, as reflected in the alternative diagnoses invoked,” the authors wrote.
“I don’t even ask for suspicion of a specific rare disease because that requires specialization. What I ask for is a kind of recognition or realization that something is happening and then request a consultation with the specialist to ensure proper care,” said Dr. Dubrovsky.
In another study conducted among 70 Argentine patients under age 13 years who were diagnosed with Duchenne muscular dystrophy (one of the most severe forms of muscular dystrophy), 82% of the pediatricians who were initially consulted for any problem in motor agility that parents, other relatives, or teachers had detected dismissed the observation. “They’re told to wait, that it will mature a little more,” said Dr. Dubrovsky. This explains why the time to diagnosis in Argentina from the first signs is around 2 years. The delays are unfortunate because “today we have treatments capable of interfering with the disease’s progression slope, reducing its progression, or eventually stopping it,” he said.
“Do you mind that primary care pediatricians don’t notice or dismiss signs and symptoms strongly suggestive of one of these rare diseases? Does it frustrate you?” this news organization asked asked Dr. Dubrovsky. “Sometimes it does make me angry, but many times it’s understood that there can’t be highly trained specialists everywhere to realize and request diagnostic tests. One must consider the circumstances in each case, and that’s why we work in education,” he replied.
Rules and Experience
In an interview, Dr. Fainboim highlighted key factors that should prompt a pediatrician’s suspicion. One is common symptoms expressed in a more intense or complicated way or when many symptoms coexist in the same patient, even if each one separately is benign or not so severe.
Dr. Fainboim also recommended establishing a therapeutic alliance with parents. “We shouldn’t undermine what parents say, especially those who have other children and already know what normal child development is like. This is a very important milestone.
“We have to strengthen the suspicion clue, and for that, we rely on standards and our experience, which we keep refining. As Wilde said, experience is the sum of our mistakes. But there’s no universal answer. Not all families are the same. Not all diseases manifest in the same way. And unless there’s an imminent risk to life or function, one can wait and take the time to evaluate it. For example, if I have a child with slowed developmental milestones, what I have to do is teach how to stimulate them or send them for stimulation with another professional. And I observe the response to this initial basic treatment. If I see no response, the alarms start to grow louder,” said Dr. Fainboim.
Pablo Barvosa, MD, the principal physician in the outpatient area of the Juan P. Garrahan Pediatric Hospital in Buenos Aires, Argentina, and a member of the Working Group on Genetics and Rare Diseases of the Argentine Society of Pediatrics, told this news organization about other factors that should be considered for detecting these pathologies. Dr. Barvosa did not participate in the online seminar.
“Patients with rare diseases have common symptoms. What needs to be done is to prioritize those symptoms that behave abnormally, that have an unusual evolution compared with normal situations. For example, children who go into a coma after a fasting episode or after eating a certain food,” he said.
Dr. Barvosa also suggested considering when patients belong to certain communities where there is a lot of endogamy, due to the higher incidence of hereditary diseases. “Attention should be heightened when parents are cousins or relatives,” he pointed out.
“My view is that doctors should think more and better, be rational, sequential. If a disease is treated and resolved, but we find out that the child had 26 previous hospitalizations in the last 2 years, something is wrong. We have to look at the patient’s and family’s life histories. If a mother had 15 miscarriages, that’s a warning sign. We have to find a common thread. Be a sharp-witted pediatrician,” said Dr. Barvosa.
The suspicion and diagnosis of a rare disease can be devastating for families and painful for the professional, but even if there is no specific treatment, “something can always be done for patients,” he added.
And in certain circumstances, identifying a rare disease can reverse the ominous “stamp” of a wrong diagnosis. Dr. Barvosa commented on the case of a 7-year-old boy he attended at the hospital in 2014. The boy presented as quadriplegic, with no mobility in his limbs, and the parents were convinced he had that condition because he had fallen from the roof of the house. Although imaging techniques did not show a spinal injury, it was assumed to be a case of spinal cord injury without radiographic abnormality. But something caught Dr. Barvosa’s attention: The boy had well-developed abdominal muscles, as if he were an athlete. So, he requested an electromyogram, and the muscle was found to be in permanent contraction.
“The patient didn’t have a spinal cord injury: He had Isaacs’ syndrome,” said Dr. Barvosa. The syndrome also is known as acquired neuromyotonia, a rare condition of hyperexcitability of peripheral nerves that activate muscle fibers. “That is treated with anticonvulsants, such as phenytoin. Within a week, he was walking again, and shortly after, he was playing soccer. When I presented the case at a conference, I cried with emotion. That’s why the pediatrician must be insistent, be like the gadfly that stings in the ear” when there are clinical elements that don’t quite fit into a clear diagnosis, he added.
In recent publications, Dr. Dubrovsky has reported receiving fees for consultations or research from PTC, Sarepta, Biogen, Sanofi Genzyme, Takeda Avexis, Novartis, Raffo, and Roche. Dr. Nucifora has received fees from Jansen LATAM. Dr. Fainboim reported receiving fees from Sanofi. Dr. Barvosa has declared no relevant financial conflicts of interest. The webinar was organized by Urban Comunicaciones.
This story was translated from the Medscape Spanish edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
A wise medical precept is attributed to Theodore Woodward, MD (1914-2005): “When you hear hoofbeats, think of horses, not zebras.” Primary care pediatricians, however, often find themselves confronting so-called rare or uncommon diseases (“zebras”) in their offices. The pressing challenge is to know when to suspect them. How can one reconcile the need to dispel uncertainty with the use of diagnostic tests that can be costly and invasive? When can the desire to reassure parents mean delaying the detection of a potentially treatable condition?
“It may seem like wordplay, but it’s not uncommon to have a rare disease,” noted Alejandro Fainboim, MD, a specialist in rare diseases and head of the Multivalent Day Hospital at the Ricardo Gutiérrez Children’s Hospital in Buenos Aires, Argentina. “And pediatricians are the first line of defense in detecting these types of pathologies. To make the diagnosis, we have to consider them. And to consider them, we must be knowledgeable. That’s why sometimes, ignorance slows down the diagnosis,” Dr. Fainboim made his remarks during an online seminar organized for the press on the eve of Rare Disease Day, which is commemorated on February 29th.
There are more than 8000 rare diseases, which generally are defined as those affecting fewer than five people per 10,000. But collectively, one in every 13 people has one of these diseases, and one in every two diagnosed patients is a child. Dr. Fainboim emphasized that most of these rare diseases are severe or very severe, hereditary, degenerative, and potentially fatal. And although they are pediatric pathologies, some manifest later in adulthood.
“The major problem we pediatricians face is that we’re handed a model from adults to solve pediatric diseases. So, signs and symptoms are described that we won’t find early on, but we have to anticipate and learn to decode some that are hidden,” he remarked.
Diagnostic delays and repeated consultations with various doctors before identification are common. Dr. Fainboim added that in industrialized countries, the diagnosis of these diseases takes between 5 and 10 years, and in low-income countries, up to 30 years or more. However, “this has improved significantly in recent years,” he said.
Unnoticed Signs
Specialists who treat patients with rare diseases often feel that there were obvious signs that went unnoticed and should have aroused the suspicion of the primary care physician. An example is paroxysmal nocturnal hemoglobinuria, which affects 13-14 people per million inhabitants and can appear at any age, although the incidence is higher in the third decade of life.
“In my 50 years as a doctor, I’ve seen seven or eight patients with paroxysmal nocturnal hemoglobinuria,” said Elsa Nucifora, MD, a hematologist at the Italian Hospital of Buenos Aires, Argentina. But “the diagnosis is so easy” that doctors could make it if they “were to think instead of acting automatically because they’re in a hurry,” she added. The diagnosis should be considered “every time anemia occurs in a young person, with certain characteristics, instead of giving them iron like everyone else and ‘we’ll see later’…the diagnosis is in two or three steps, so it’s not complicated.”
Similar situations occur with more than 1000 neuromuscular diseases involving mutations in more than 600 genes, including spinal muscular atrophy and muscular dystrophies.
“What are the most common manifestations? The hypotonic infant, the child who walks late, who falls frequently, who can’t climb stairs, who later may have difficulty breathing, who loses strength: These are presentations often unrecognized by doctors not in the specialty,” said Alberto Dubrovsky, MD, director of the Department of Neurology and the Neuromuscular Diseases Unit at the Favaloro University Neuroscience Institute in Buenos Aires, Argentina, during the seminar. “And considering that these diseases are diagnosed based on genetic mutations that need to be known to search for and request them, we are faced with a truly complex scenario that requires subspecialization.”
In a study recently published in the Argentine Archives of Pediatrics, Dr. Dubrovsky and colleagues interviewed 112 families of Argentine patients with molecular diagnoses of spinal muscular atrophy types I, II, and III and found that in 75%-85% of cases, the first signs of the disease (such as hypotonia, developmental delay, inability to achieve bipedal standing, or frequent falls) were recognized by parents. For type I, the most severe and early onset, in only 17.5% of cases did a neonatologist or pediatrician first notice something. Of the 72 patients with types II and III, where routine checks are less frequent than in the first months of life, only one doctor detected the first signs of the disease before parents or other relatives.
In the same study, the median time elapsed between the first sign and confirmed molecular diagnosis was 2, 10, and 31.5 months for types I, II, and III, respectively. The delay “is primarily due to the lack of clinical suspicion on the part of the intervening physician, who often dismisses or misinterprets the signs reported by parents, as reflected in the alternative diagnoses invoked,” the authors wrote.
“I don’t even ask for suspicion of a specific rare disease because that requires specialization. What I ask for is a kind of recognition or realization that something is happening and then request a consultation with the specialist to ensure proper care,” said Dr. Dubrovsky.
In another study conducted among 70 Argentine patients under age 13 years who were diagnosed with Duchenne muscular dystrophy (one of the most severe forms of muscular dystrophy), 82% of the pediatricians who were initially consulted for any problem in motor agility that parents, other relatives, or teachers had detected dismissed the observation. “They’re told to wait, that it will mature a little more,” said Dr. Dubrovsky. This explains why the time to diagnosis in Argentina from the first signs is around 2 years. The delays are unfortunate because “today we have treatments capable of interfering with the disease’s progression slope, reducing its progression, or eventually stopping it,” he said.
“Do you mind that primary care pediatricians don’t notice or dismiss signs and symptoms strongly suggestive of one of these rare diseases? Does it frustrate you?” this news organization asked asked Dr. Dubrovsky. “Sometimes it does make me angry, but many times it’s understood that there can’t be highly trained specialists everywhere to realize and request diagnostic tests. One must consider the circumstances in each case, and that’s why we work in education,” he replied.
Rules and Experience
In an interview, Dr. Fainboim highlighted key factors that should prompt a pediatrician’s suspicion. One is common symptoms expressed in a more intense or complicated way or when many symptoms coexist in the same patient, even if each one separately is benign or not so severe.
Dr. Fainboim also recommended establishing a therapeutic alliance with parents. “We shouldn’t undermine what parents say, especially those who have other children and already know what normal child development is like. This is a very important milestone.
“We have to strengthen the suspicion clue, and for that, we rely on standards and our experience, which we keep refining. As Wilde said, experience is the sum of our mistakes. But there’s no universal answer. Not all families are the same. Not all diseases manifest in the same way. And unless there’s an imminent risk to life or function, one can wait and take the time to evaluate it. For example, if I have a child with slowed developmental milestones, what I have to do is teach how to stimulate them or send them for stimulation with another professional. And I observe the response to this initial basic treatment. If I see no response, the alarms start to grow louder,” said Dr. Fainboim.
Pablo Barvosa, MD, the principal physician in the outpatient area of the Juan P. Garrahan Pediatric Hospital in Buenos Aires, Argentina, and a member of the Working Group on Genetics and Rare Diseases of the Argentine Society of Pediatrics, told this news organization about other factors that should be considered for detecting these pathologies. Dr. Barvosa did not participate in the online seminar.
“Patients with rare diseases have common symptoms. What needs to be done is to prioritize those symptoms that behave abnormally, that have an unusual evolution compared with normal situations. For example, children who go into a coma after a fasting episode or after eating a certain food,” he said.
Dr. Barvosa also suggested considering when patients belong to certain communities where there is a lot of endogamy, due to the higher incidence of hereditary diseases. “Attention should be heightened when parents are cousins or relatives,” he pointed out.
“My view is that doctors should think more and better, be rational, sequential. If a disease is treated and resolved, but we find out that the child had 26 previous hospitalizations in the last 2 years, something is wrong. We have to look at the patient’s and family’s life histories. If a mother had 15 miscarriages, that’s a warning sign. We have to find a common thread. Be a sharp-witted pediatrician,” said Dr. Barvosa.
The suspicion and diagnosis of a rare disease can be devastating for families and painful for the professional, but even if there is no specific treatment, “something can always be done for patients,” he added.
And in certain circumstances, identifying a rare disease can reverse the ominous “stamp” of a wrong diagnosis. Dr. Barvosa commented on the case of a 7-year-old boy he attended at the hospital in 2014. The boy presented as quadriplegic, with no mobility in his limbs, and the parents were convinced he had that condition because he had fallen from the roof of the house. Although imaging techniques did not show a spinal injury, it was assumed to be a case of spinal cord injury without radiographic abnormality. But something caught Dr. Barvosa’s attention: The boy had well-developed abdominal muscles, as if he were an athlete. So, he requested an electromyogram, and the muscle was found to be in permanent contraction.
“The patient didn’t have a spinal cord injury: He had Isaacs’ syndrome,” said Dr. Barvosa. The syndrome also is known as acquired neuromyotonia, a rare condition of hyperexcitability of peripheral nerves that activate muscle fibers. “That is treated with anticonvulsants, such as phenytoin. Within a week, he was walking again, and shortly after, he was playing soccer. When I presented the case at a conference, I cried with emotion. That’s why the pediatrician must be insistent, be like the gadfly that stings in the ear” when there are clinical elements that don’t quite fit into a clear diagnosis, he added.
In recent publications, Dr. Dubrovsky has reported receiving fees for consultations or research from PTC, Sarepta, Biogen, Sanofi Genzyme, Takeda Avexis, Novartis, Raffo, and Roche. Dr. Nucifora has received fees from Jansen LATAM. Dr. Fainboim reported receiving fees from Sanofi. Dr. Barvosa has declared no relevant financial conflicts of interest. The webinar was organized by Urban Comunicaciones.
This story was translated from the Medscape Spanish edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Treating Pediatric Vitiligo: Consensus Statement Provides Recommendations
TOPLINE:
METHODOLOGY:
- While half of all vitiligo cases manifest within the initial two decades of life, no guidelines specifically address the management of vitiligo in children, adolescents, and young adults with vitiligo.
- A protocol was established to formulate consensus recommendations addressing questions related to pediatric vitiligo.
- Overall, 50 articles on topical corticosteroids and/or topical calcineurin inhibitors, five on topical Janus kinase inhibitors, and two each on pseudocatalase and microdermabrasion were included.
- The participants recorded their agreement levels with the formulated statements, using a 5-point Likert scale.
TAKEAWAY:
- TCIs, TCSs, JAK inhibitors, and phototherapy, specifically narrowband ultraviolet (UV)-B light therapy, are mainstay treatments; the combination of UV-B light and topical therapy may enhance initial repigmentation.
- Long-term monitoring for skin cancers is advised, and short outdoor UV exposure is suggested for pediatric patients.
- TCIs, such as tacrolimus and pimecrolimus, are recommended as first-line therapy, particularly on the face, applied twice daily for ≥ 3 months; continued use for 6-12 additional months is recommended if repigmentation is observed.
- The choice of TCS class depends on the site and planned usage duration. Short-term use or overlap with TCIs is recommended because of the risk for atrophy with long-term TCS use. Class 5-6 agents are another option.
- For areas with thin skin, TCSs can be considered second-line treatments.
- Topical JAK inhibitors, specifically topical 1.5% ruxolitinib cream, are recommended for patients aged ≥ 12 years, as first- or second-line therapy. Limitation to 10% body surface area is recommended to minimize systemic absorption. Limited evidence exists for children aged < 12 years.
IN PRACTICE:
“Effective therapy requires a focus on long-term therapeutic interventions to maximize the local gain and retention of pigmentation with a trial period of twice-weekly application. Counseling should include discussion of the chronicity of vitiligo and the need for long-term care,” the authors wrote.
LIMITATIONS:
Some of the recommendations were opinion-based because of the scarcity of evidence-based literature.
SOURCE:
The consensus statement was published on March 13 in JAMA Dermatology.
DISCLOSURES:
This work was supported by grants from Vitiligo Research Foundation and Incyte Pharmaceuticals. The majority of authors disclosed financial relationships outside this work; several reported no disclosures.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- While half of all vitiligo cases manifest within the initial two decades of life, no guidelines specifically address the management of vitiligo in children, adolescents, and young adults with vitiligo.
- A protocol was established to formulate consensus recommendations addressing questions related to pediatric vitiligo.
- Overall, 50 articles on topical corticosteroids and/or topical calcineurin inhibitors, five on topical Janus kinase inhibitors, and two each on pseudocatalase and microdermabrasion were included.
- The participants recorded their agreement levels with the formulated statements, using a 5-point Likert scale.
TAKEAWAY:
- TCIs, TCSs, JAK inhibitors, and phototherapy, specifically narrowband ultraviolet (UV)-B light therapy, are mainstay treatments; the combination of UV-B light and topical therapy may enhance initial repigmentation.
- Long-term monitoring for skin cancers is advised, and short outdoor UV exposure is suggested for pediatric patients.
- TCIs, such as tacrolimus and pimecrolimus, are recommended as first-line therapy, particularly on the face, applied twice daily for ≥ 3 months; continued use for 6-12 additional months is recommended if repigmentation is observed.
- The choice of TCS class depends on the site and planned usage duration. Short-term use or overlap with TCIs is recommended because of the risk for atrophy with long-term TCS use. Class 5-6 agents are another option.
- For areas with thin skin, TCSs can be considered second-line treatments.
- Topical JAK inhibitors, specifically topical 1.5% ruxolitinib cream, are recommended for patients aged ≥ 12 years, as first- or second-line therapy. Limitation to 10% body surface area is recommended to minimize systemic absorption. Limited evidence exists for children aged < 12 years.
IN PRACTICE:
“Effective therapy requires a focus on long-term therapeutic interventions to maximize the local gain and retention of pigmentation with a trial period of twice-weekly application. Counseling should include discussion of the chronicity of vitiligo and the need for long-term care,” the authors wrote.
LIMITATIONS:
Some of the recommendations were opinion-based because of the scarcity of evidence-based literature.
SOURCE:
The consensus statement was published on March 13 in JAMA Dermatology.
DISCLOSURES:
This work was supported by grants from Vitiligo Research Foundation and Incyte Pharmaceuticals. The majority of authors disclosed financial relationships outside this work; several reported no disclosures.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- While half of all vitiligo cases manifest within the initial two decades of life, no guidelines specifically address the management of vitiligo in children, adolescents, and young adults with vitiligo.
- A protocol was established to formulate consensus recommendations addressing questions related to pediatric vitiligo.
- Overall, 50 articles on topical corticosteroids and/or topical calcineurin inhibitors, five on topical Janus kinase inhibitors, and two each on pseudocatalase and microdermabrasion were included.
- The participants recorded their agreement levels with the formulated statements, using a 5-point Likert scale.
TAKEAWAY:
- TCIs, TCSs, JAK inhibitors, and phototherapy, specifically narrowband ultraviolet (UV)-B light therapy, are mainstay treatments; the combination of UV-B light and topical therapy may enhance initial repigmentation.
- Long-term monitoring for skin cancers is advised, and short outdoor UV exposure is suggested for pediatric patients.
- TCIs, such as tacrolimus and pimecrolimus, are recommended as first-line therapy, particularly on the face, applied twice daily for ≥ 3 months; continued use for 6-12 additional months is recommended if repigmentation is observed.
- The choice of TCS class depends on the site and planned usage duration. Short-term use or overlap with TCIs is recommended because of the risk for atrophy with long-term TCS use. Class 5-6 agents are another option.
- For areas with thin skin, TCSs can be considered second-line treatments.
- Topical JAK inhibitors, specifically topical 1.5% ruxolitinib cream, are recommended for patients aged ≥ 12 years, as first- or second-line therapy. Limitation to 10% body surface area is recommended to minimize systemic absorption. Limited evidence exists for children aged < 12 years.
IN PRACTICE:
“Effective therapy requires a focus on long-term therapeutic interventions to maximize the local gain and retention of pigmentation with a trial period of twice-weekly application. Counseling should include discussion of the chronicity of vitiligo and the need for long-term care,” the authors wrote.
LIMITATIONS:
Some of the recommendations were opinion-based because of the scarcity of evidence-based literature.
SOURCE:
The consensus statement was published on March 13 in JAMA Dermatology.
DISCLOSURES:
This work was supported by grants from Vitiligo Research Foundation and Incyte Pharmaceuticals. The majority of authors disclosed financial relationships outside this work; several reported no disclosures.
A version of this article appeared on Medscape.com.
Medtronic’s Duet EDMS Catheter Tubing Under Class I Recall
If this happens, potential harm to patients may include infections, cerebrospinal fluid (CSF) leakage, overdrainage of CSF, and abnormality of the ventricles. Uncontrolled overdrainage of CSF could lead to neurological injury or death if the disconnection is undetected.
The Food and Drug Administration has identified this as a Class I recall — the most serious type — due to the risk for serious injury or death. To date, there have been 26 reported injuries and no deaths related to this issue.
The recall includes 45,176 devices distributed in the United States between May 3, 2021, and January 9, 2024, with model numbers 46913, 46914, 46915, 46916, and 46917.
The Duet EDMS is used for temporary CSF drainage or sampling in patients who have surgery for open descending thoracic aortic aneurysm (TAA) or descending thoraco-abdominal aortic aneurysm (TAAA) or patients who have TAA/TAAA repair surgery and develop symptoms such as paraplegia.
Medtronic has sent an urgent medical device recall letter to all affected customers asking them to identify, quarantine, and return any unused recalled products.
Customers are also advised to check all Duet EDMS components for damage and ensure that all connections are secure and leak-free.
If a patient is currently connected to an impacted Duet EDMS and a leak or disconnection is detected, the device should be changed to a new alternative device utilizing a sterile technique.
It is not recommended that a Duet system device that is connected to a patient and working as intended be removed or replaced.
Customers in the United States with questions about this recall should contact Medtronic at 1-800-874-5797.
A version of this article appeared on Medscape.com.
If this happens, potential harm to patients may include infections, cerebrospinal fluid (CSF) leakage, overdrainage of CSF, and abnormality of the ventricles. Uncontrolled overdrainage of CSF could lead to neurological injury or death if the disconnection is undetected.
The Food and Drug Administration has identified this as a Class I recall — the most serious type — due to the risk for serious injury or death. To date, there have been 26 reported injuries and no deaths related to this issue.
The recall includes 45,176 devices distributed in the United States between May 3, 2021, and January 9, 2024, with model numbers 46913, 46914, 46915, 46916, and 46917.
The Duet EDMS is used for temporary CSF drainage or sampling in patients who have surgery for open descending thoracic aortic aneurysm (TAA) or descending thoraco-abdominal aortic aneurysm (TAAA) or patients who have TAA/TAAA repair surgery and develop symptoms such as paraplegia.
Medtronic has sent an urgent medical device recall letter to all affected customers asking them to identify, quarantine, and return any unused recalled products.
Customers are also advised to check all Duet EDMS components for damage and ensure that all connections are secure and leak-free.
If a patient is currently connected to an impacted Duet EDMS and a leak or disconnection is detected, the device should be changed to a new alternative device utilizing a sterile technique.
It is not recommended that a Duet system device that is connected to a patient and working as intended be removed or replaced.
Customers in the United States with questions about this recall should contact Medtronic at 1-800-874-5797.
A version of this article appeared on Medscape.com.
If this happens, potential harm to patients may include infections, cerebrospinal fluid (CSF) leakage, overdrainage of CSF, and abnormality of the ventricles. Uncontrolled overdrainage of CSF could lead to neurological injury or death if the disconnection is undetected.
The Food and Drug Administration has identified this as a Class I recall — the most serious type — due to the risk for serious injury or death. To date, there have been 26 reported injuries and no deaths related to this issue.
The recall includes 45,176 devices distributed in the United States between May 3, 2021, and January 9, 2024, with model numbers 46913, 46914, 46915, 46916, and 46917.
The Duet EDMS is used for temporary CSF drainage or sampling in patients who have surgery for open descending thoracic aortic aneurysm (TAA) or descending thoraco-abdominal aortic aneurysm (TAAA) or patients who have TAA/TAAA repair surgery and develop symptoms such as paraplegia.
Medtronic has sent an urgent medical device recall letter to all affected customers asking them to identify, quarantine, and return any unused recalled products.
Customers are also advised to check all Duet EDMS components for damage and ensure that all connections are secure and leak-free.
If a patient is currently connected to an impacted Duet EDMS and a leak or disconnection is detected, the device should be changed to a new alternative device utilizing a sterile technique.
It is not recommended that a Duet system device that is connected to a patient and working as intended be removed or replaced.
Customers in the United States with questions about this recall should contact Medtronic at 1-800-874-5797.
A version of this article appeared on Medscape.com.
Vitamin D Supplements May Be a Double-Edged Sword
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Imagine, if you will, the great Cathedral of Our Lady of Correlation. You walk through the majestic oak doors depicting the link between ice cream sales and shark attacks, past the rose window depicting the cardiovascular benefits of red wine, and down the aisles frescoed in dramatic images showing how Facebook usage is associated with less life satisfaction. And then you reach the altar, the holy of holies where, emblazoned in shimmering pyrite, you see the patron saint of this church: vitamin D.
Yes, if you’ve watched this space, then you know that I have little truck with the wildly popular supplement. In all of clinical research, I believe that there is no molecule with stronger data for correlation and weaker data for causation.
Low serum vitamin D levels have been linked to higher risks for heart disease, cancer, falls, COVID, dementia, C diff, and others. And yet, when we do randomized trials of vitamin D supplementation — the thing that can prove that the low level was causally linked to the outcome of interest — we get negative results.
Trials aren’t perfect, of course, and we’ll talk in a moment about a big one that had some issues. But we are at a point where we need to either be vitamin D apologists, saying, “Forget what those lying RCTs tell you and buy this supplement” — an $800 million-a-year industry, by the way — or conclude that vitamin D levels are a convenient marker of various lifestyle factors that are associated with better outcomes: markers of exercise, getting outside, eating a varied diet.
Or perhaps vitamin D supplements have real effects. It’s just that the beneficial effects are matched by the harmful ones. Stay tuned.
The Women’s Health Initiative remains among the largest randomized trials of vitamin D and calcium supplementation ever conducted — and a major contributor to the negative outcomes of vitamin D trials.
But if you dig into the inclusion and exclusion criteria for this trial, you’ll find that individuals were allowed to continue taking vitamins and supplements while they were in the trial, regardless of their randomization status. In fact, the majority took supplements at baseline, and more took supplements over time.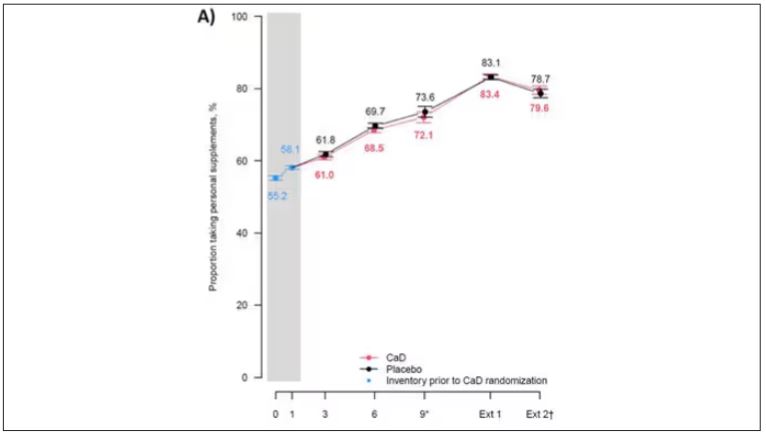
That means, of course, that people in the placebo group, who were getting sugar pills instead of vitamin D and calcium, may have been taking vitamin D and calcium on the side. That would certainly bias the results of the trial toward the null, which is what the primary analyses showed. To wit, the original analysis of the Women’s Health Initiative trial showed no effect of randomization to vitamin D supplementation on improving cancer or cardiovascular outcomes.
But the Women’s Health Initiative trial started 30 years ago. Today, with the benefit of decades of follow-up, we can re-investigate — and perhaps re-litigate — those findings, courtesy of this study, “Long-Term Effect of Randomization to Calcium and Vitamin D Supplementation on Health in Older Women” appearing in Annals of Internal Medicine.
Dr Cynthia Thomson, of the Mel and Enid Zuckerman College of Public Health at the University of Arizona, and colleagues led this updated analysis focused on two findings that had been hinted at, but not statistically confirmed, in other vitamin D studies: a potential for the supplement to reduce the risk for cancer, and a potential for it to increase the risk for heart disease.
The randomized trial itself only lasted 7 years. What we are seeing in this analysis of 36,282 women is outcomes that happened at any time from randomization to the end of 2023 — around 20 years after the randomization to supplementation stopped. But, the researchers would argue, that’s probably okay. Cancer and heart disease take time to develop; we see lung cancer long after people stop smoking. So a history of consistent vitamin D supplementation may indeed be protective — or harmful.
Here are the top-line results. Those randomized to vitamin D and calcium supplementation had a 7% reduction in the rate of death from cancer, driven primarily by a reduction in colorectal cancer. This was statistically significant. Also statistically significant? Those randomized to supplementation had a 6% increase in the rate of death from cardiovascular disease. Put those findings together and what do you get? Stone-cold nothing, in terms of overall mortality.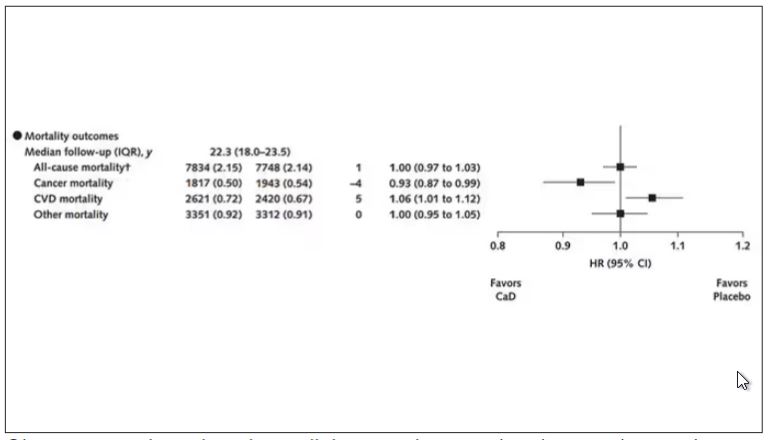
Okay, you say, but what about all that supplementation that was happening outside of the context of the trial, biasing our results toward the null?
The researchers finally clue us in.
First of all, I’ll tell you that, yes, people who were supplementing outside of the trial had higher baseline vitamin D levels — a median of 54.5 nmol/L vs 32.8 nmol/L. This may be because they were supplementing with vitamin D, but it could also be because people who take supplements tend to do other healthy things — another correlation to add to the great cathedral.
To get a better view of the real effects of randomization, the authors restricted the analysis to just those who did not use outside supplements. If vitamin D supplements help, then these are the people they should help. This group had about a 11% reduction in the incidence of cancer — statistically significant — and a 7% reduction in cancer mortality that did not meet the bar for statistical significance.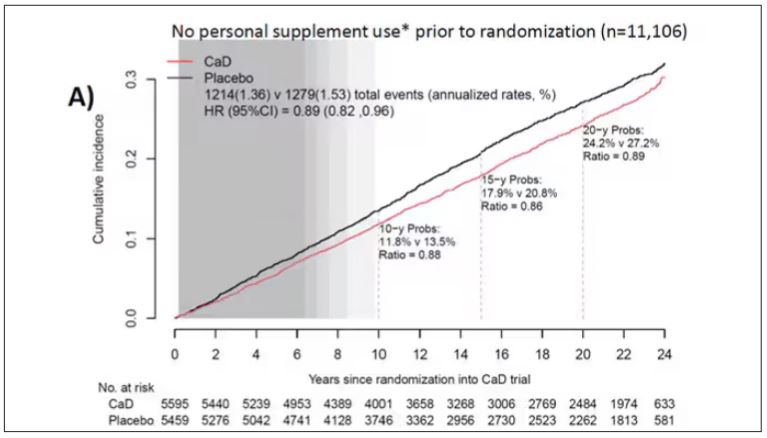
There was no increase in cardiovascular disease among this group. But this small effect on cancer was nowhere near enough to significantly reduce the rate of all-cause mortality.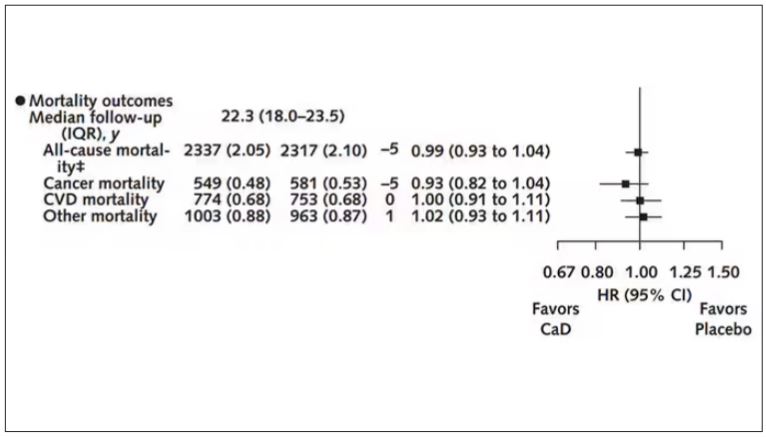
Among those using supplements, vitamin D supplementation didn’t really move the needle on any outcome.
I know what you’re thinking: How many of these women were vitamin D deficient when we got started? These results may simply be telling us that people who have normal vitamin D levels are fine to go without supplementation.
Nearly three fourths of women who were not taking supplements entered the trial with vitamin D levels below the 50 nmol/L cutoff that the authors suggest would qualify for deficiency. Around half of those who used supplements were deficient. And yet, frustratingly, I could not find data on the effect of randomization to supplementation stratified by baseline vitamin D level. I even reached out to Dr Thomson to ask about this. She replied, “We did not stratify on baseline values because the numbers are too small statistically to test this.” Sorry.
In the meantime, I can tell you that for your “average woman,” vitamin D supplementation likely has no effect on mortality. It might modestly reduce the risk for certain cancers while increasing the risk for heart disease (probably through coronary calcification). So, there might be some room for personalization here. Perhaps women with a strong family history of cancer or other risk factors would do better with supplements, and those with a high risk for heart disease would do worse. Seems like a strategy that could be tested in a clinical trial. But maybe we could ask the participants to give up their extracurricular supplement use before they enter the trial. F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his book, How Medicine Works and When It Doesn’t, is available now.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Imagine, if you will, the great Cathedral of Our Lady of Correlation. You walk through the majestic oak doors depicting the link between ice cream sales and shark attacks, past the rose window depicting the cardiovascular benefits of red wine, and down the aisles frescoed in dramatic images showing how Facebook usage is associated with less life satisfaction. And then you reach the altar, the holy of holies where, emblazoned in shimmering pyrite, you see the patron saint of this church: vitamin D.
Yes, if you’ve watched this space, then you know that I have little truck with the wildly popular supplement. In all of clinical research, I believe that there is no molecule with stronger data for correlation and weaker data for causation.
Low serum vitamin D levels have been linked to higher risks for heart disease, cancer, falls, COVID, dementia, C diff, and others. And yet, when we do randomized trials of vitamin D supplementation — the thing that can prove that the low level was causally linked to the outcome of interest — we get negative results.
Trials aren’t perfect, of course, and we’ll talk in a moment about a big one that had some issues. But we are at a point where we need to either be vitamin D apologists, saying, “Forget what those lying RCTs tell you and buy this supplement” — an $800 million-a-year industry, by the way — or conclude that vitamin D levels are a convenient marker of various lifestyle factors that are associated with better outcomes: markers of exercise, getting outside, eating a varied diet.
Or perhaps vitamin D supplements have real effects. It’s just that the beneficial effects are matched by the harmful ones. Stay tuned.
The Women’s Health Initiative remains among the largest randomized trials of vitamin D and calcium supplementation ever conducted — and a major contributor to the negative outcomes of vitamin D trials.
But if you dig into the inclusion and exclusion criteria for this trial, you’ll find that individuals were allowed to continue taking vitamins and supplements while they were in the trial, regardless of their randomization status. In fact, the majority took supplements at baseline, and more took supplements over time.
That means, of course, that people in the placebo group, who were getting sugar pills instead of vitamin D and calcium, may have been taking vitamin D and calcium on the side. That would certainly bias the results of the trial toward the null, which is what the primary analyses showed. To wit, the original analysis of the Women’s Health Initiative trial showed no effect of randomization to vitamin D supplementation on improving cancer or cardiovascular outcomes.
But the Women’s Health Initiative trial started 30 years ago. Today, with the benefit of decades of follow-up, we can re-investigate — and perhaps re-litigate — those findings, courtesy of this study, “Long-Term Effect of Randomization to Calcium and Vitamin D Supplementation on Health in Older Women” appearing in Annals of Internal Medicine.
Dr Cynthia Thomson, of the Mel and Enid Zuckerman College of Public Health at the University of Arizona, and colleagues led this updated analysis focused on two findings that had been hinted at, but not statistically confirmed, in other vitamin D studies: a potential for the supplement to reduce the risk for cancer, and a potential for it to increase the risk for heart disease.
The randomized trial itself only lasted 7 years. What we are seeing in this analysis of 36,282 women is outcomes that happened at any time from randomization to the end of 2023 — around 20 years after the randomization to supplementation stopped. But, the researchers would argue, that’s probably okay. Cancer and heart disease take time to develop; we see lung cancer long after people stop smoking. So a history of consistent vitamin D supplementation may indeed be protective — or harmful.
Here are the top-line results. Those randomized to vitamin D and calcium supplementation had a 7% reduction in the rate of death from cancer, driven primarily by a reduction in colorectal cancer. This was statistically significant. Also statistically significant? Those randomized to supplementation had a 6% increase in the rate of death from cardiovascular disease. Put those findings together and what do you get? Stone-cold nothing, in terms of overall mortality.
Okay, you say, but what about all that supplementation that was happening outside of the context of the trial, biasing our results toward the null?
The researchers finally clue us in.
First of all, I’ll tell you that, yes, people who were supplementing outside of the trial had higher baseline vitamin D levels — a median of 54.5 nmol/L vs 32.8 nmol/L. This may be because they were supplementing with vitamin D, but it could also be because people who take supplements tend to do other healthy things — another correlation to add to the great cathedral.
To get a better view of the real effects of randomization, the authors restricted the analysis to just those who did not use outside supplements. If vitamin D supplements help, then these are the people they should help. This group had about a 11% reduction in the incidence of cancer — statistically significant — and a 7% reduction in cancer mortality that did not meet the bar for statistical significance.
There was no increase in cardiovascular disease among this group. But this small effect on cancer was nowhere near enough to significantly reduce the rate of all-cause mortality.
Among those using supplements, vitamin D supplementation didn’t really move the needle on any outcome.
I know what you’re thinking: How many of these women were vitamin D deficient when we got started? These results may simply be telling us that people who have normal vitamin D levels are fine to go without supplementation.
Nearly three fourths of women who were not taking supplements entered the trial with vitamin D levels below the 50 nmol/L cutoff that the authors suggest would qualify for deficiency. Around half of those who used supplements were deficient. And yet, frustratingly, I could not find data on the effect of randomization to supplementation stratified by baseline vitamin D level. I even reached out to Dr Thomson to ask about this. She replied, “We did not stratify on baseline values because the numbers are too small statistically to test this.” Sorry.
In the meantime, I can tell you that for your “average woman,” vitamin D supplementation likely has no effect on mortality. It might modestly reduce the risk for certain cancers while increasing the risk for heart disease (probably through coronary calcification). So, there might be some room for personalization here. Perhaps women with a strong family history of cancer or other risk factors would do better with supplements, and those with a high risk for heart disease would do worse. Seems like a strategy that could be tested in a clinical trial. But maybe we could ask the participants to give up their extracurricular supplement use before they enter the trial. F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his book, How Medicine Works and When It Doesn’t, is available now.
This transcript has been edited for clarity.
Welcome to Impact Factor, your weekly dose of commentary on a new medical study. I’m Dr F. Perry Wilson of the Yale School of Medicine.
Imagine, if you will, the great Cathedral of Our Lady of Correlation. You walk through the majestic oak doors depicting the link between ice cream sales and shark attacks, past the rose window depicting the cardiovascular benefits of red wine, and down the aisles frescoed in dramatic images showing how Facebook usage is associated with less life satisfaction. And then you reach the altar, the holy of holies where, emblazoned in shimmering pyrite, you see the patron saint of this church: vitamin D.
Yes, if you’ve watched this space, then you know that I have little truck with the wildly popular supplement. In all of clinical research, I believe that there is no molecule with stronger data for correlation and weaker data for causation.
Low serum vitamin D levels have been linked to higher risks for heart disease, cancer, falls, COVID, dementia, C diff, and others. And yet, when we do randomized trials of vitamin D supplementation — the thing that can prove that the low level was causally linked to the outcome of interest — we get negative results.
Trials aren’t perfect, of course, and we’ll talk in a moment about a big one that had some issues. But we are at a point where we need to either be vitamin D apologists, saying, “Forget what those lying RCTs tell you and buy this supplement” — an $800 million-a-year industry, by the way — or conclude that vitamin D levels are a convenient marker of various lifestyle factors that are associated with better outcomes: markers of exercise, getting outside, eating a varied diet.
Or perhaps vitamin D supplements have real effects. It’s just that the beneficial effects are matched by the harmful ones. Stay tuned.
The Women’s Health Initiative remains among the largest randomized trials of vitamin D and calcium supplementation ever conducted — and a major contributor to the negative outcomes of vitamin D trials.
But if you dig into the inclusion and exclusion criteria for this trial, you’ll find that individuals were allowed to continue taking vitamins and supplements while they were in the trial, regardless of their randomization status. In fact, the majority took supplements at baseline, and more took supplements over time.
That means, of course, that people in the placebo group, who were getting sugar pills instead of vitamin D and calcium, may have been taking vitamin D and calcium on the side. That would certainly bias the results of the trial toward the null, which is what the primary analyses showed. To wit, the original analysis of the Women’s Health Initiative trial showed no effect of randomization to vitamin D supplementation on improving cancer or cardiovascular outcomes.
But the Women’s Health Initiative trial started 30 years ago. Today, with the benefit of decades of follow-up, we can re-investigate — and perhaps re-litigate — those findings, courtesy of this study, “Long-Term Effect of Randomization to Calcium and Vitamin D Supplementation on Health in Older Women” appearing in Annals of Internal Medicine.
Dr Cynthia Thomson, of the Mel and Enid Zuckerman College of Public Health at the University of Arizona, and colleagues led this updated analysis focused on two findings that had been hinted at, but not statistically confirmed, in other vitamin D studies: a potential for the supplement to reduce the risk for cancer, and a potential for it to increase the risk for heart disease.
The randomized trial itself only lasted 7 years. What we are seeing in this analysis of 36,282 women is outcomes that happened at any time from randomization to the end of 2023 — around 20 years after the randomization to supplementation stopped. But, the researchers would argue, that’s probably okay. Cancer and heart disease take time to develop; we see lung cancer long after people stop smoking. So a history of consistent vitamin D supplementation may indeed be protective — or harmful.
Here are the top-line results. Those randomized to vitamin D and calcium supplementation had a 7% reduction in the rate of death from cancer, driven primarily by a reduction in colorectal cancer. This was statistically significant. Also statistically significant? Those randomized to supplementation had a 6% increase in the rate of death from cardiovascular disease. Put those findings together and what do you get? Stone-cold nothing, in terms of overall mortality.
Okay, you say, but what about all that supplementation that was happening outside of the context of the trial, biasing our results toward the null?
The researchers finally clue us in.
First of all, I’ll tell you that, yes, people who were supplementing outside of the trial had higher baseline vitamin D levels — a median of 54.5 nmol/L vs 32.8 nmol/L. This may be because they were supplementing with vitamin D, but it could also be because people who take supplements tend to do other healthy things — another correlation to add to the great cathedral.
To get a better view of the real effects of randomization, the authors restricted the analysis to just those who did not use outside supplements. If vitamin D supplements help, then these are the people they should help. This group had about a 11% reduction in the incidence of cancer — statistically significant — and a 7% reduction in cancer mortality that did not meet the bar for statistical significance.
There was no increase in cardiovascular disease among this group. But this small effect on cancer was nowhere near enough to significantly reduce the rate of all-cause mortality.
Among those using supplements, vitamin D supplementation didn’t really move the needle on any outcome.
I know what you’re thinking: How many of these women were vitamin D deficient when we got started? These results may simply be telling us that people who have normal vitamin D levels are fine to go without supplementation.
Nearly three fourths of women who were not taking supplements entered the trial with vitamin D levels below the 50 nmol/L cutoff that the authors suggest would qualify for deficiency. Around half of those who used supplements were deficient. And yet, frustratingly, I could not find data on the effect of randomization to supplementation stratified by baseline vitamin D level. I even reached out to Dr Thomson to ask about this. She replied, “We did not stratify on baseline values because the numbers are too small statistically to test this.” Sorry.
In the meantime, I can tell you that for your “average woman,” vitamin D supplementation likely has no effect on mortality. It might modestly reduce the risk for certain cancers while increasing the risk for heart disease (probably through coronary calcification). So, there might be some room for personalization here. Perhaps women with a strong family history of cancer or other risk factors would do better with supplements, and those with a high risk for heart disease would do worse. Seems like a strategy that could be tested in a clinical trial. But maybe we could ask the participants to give up their extracurricular supplement use before they enter the trial. F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his book, How Medicine Works and When It Doesn’t, is available now.
Long-Term Calcium and Vitamin D: Cancer Deaths Down, CVD Deaths Up in Older Women?
Some doctors may be scratching their heads over a new analysis reporting that combined calcium and vitamin D (CaD) supplements appear to be associated with a slight 6% increase in cardiovascular (CVD) mortality, a slight 7% decrease in cancer risk, and no effect on osteoporotic fracture in postmenopausal women.
The study, in Annals of Internal Medicine, found no effect of supplementation on all-cause mortality.
The findings emerged from an analysis of more than 20 years’ follow-up data on a randomized trial in postmenopausal women conducted as part of the Women’s Health Initiative (WHI).
Cynthia A. Thomson, PhD, RD, first author and cancer prevention scientist at the Arizona Cancer Center and a professor of health promotion sciences at the University of Arizona in Tucson said the findings recommend individualized assessment of the need for supplements for older women as they consider them in hopes of preventing fractures.

“Evaluate your patients individually and understand that there are some who may benefit from supplementation, for example, in terms of reducing colorectal cancer mortality,” Dr. Thomson said in an interview. The approach should be nuanced. “If you check the adequacy of vitamin D and calcium in their diets, supplementation may not be needed.” She added that supplementation is best considered in the context of a woman’s overall health profile, including risk factors for fracture, heart disease, and cancer, especially colorectal cancer (CRC).
Study Details
The investigators conducted postintervention follow-up of the WHI’s 7-year multicenter randomized intervention trial of CaD vs placebo.
Since existing evidence of long-term health outcomes was limited, the trial, begun in 1999 and closed in 2005, enrolled 36,282 postmenopausal women (mean age 62) with no history of breast or colorectal cancer. They were randomly assigned 1:1 to supplementation with 1000 mg of calcium carbonate (400 mg elemental calcium) plus 400 IU of vitamin D3 daily or placebo, taken twice daily in half doses.
Study outcomes were incidence of CRC, total and invasive breast cancer; disease-specific and all-cause mortality; total CVD; and hip fracture measured through December 2020, with analyses stratified by personal supplement usage.
Cancer. CaD was associated with reduced incident total cancer, CRC, and invasive breast cancer — notably among participants not taking CaD before randomization. Cancer incidence estimates varied widely, the authors noted, when stratified by supplement use before randomization. Noting that CaD seemed to have more cancer-related impact in those without prior supplementation, the authors suggested supplementation may affect cancer biology primarily by augmenting nutrient insufficiency.
An estimated 7% reduction in cancer mortality was observed after a median cumulative follow-up of 22.3 years: 1817 vs 1943 deaths (hazard ratio, 0.93; 95% CI, 0.87-0.99).
CVD. An estimated 6% increase in CVD mortality was seen in the CaD group: 2621 vs 2420 deaths (HR, 1.06; 95% CI, 1.01-1.12). Pretrial supplement users were found to be at higher CVD risk.
Hip fracture. No effect on hip fracture risk was measured, but the authors cautioned that hip fracture and CVD outcomes were available only for a subset of participants, and the effects of calcium alone vs vitamin D alone vs the combination could not be disentangled.
In a small subgroup analysis, some CaD users were seen to respond in terms of bone mineral density but since only 4 of the study’s 40 sites collected such information, the study was underpowered to examine the effect. ”Many other studies, however, show a response to supplementation in women who already have bone mineral deficits,” Dr. Thomson said.
The Calcification Question
One of the possible mechanisms of harm is that high-dose calcium supplements can increase the rate of blood coagulation and promote vascular calcification, said Emma Laing, PhD, RD, director of dietetics at the University of Georgia in Athens and a spokesperson for the Chicago-based Academy of Nutrition and Dietetics.

“Other factors that should be considered when determining a patient’s CVD risk are race, genetic predisposition, medical and social history, response to stress, and lifestyle behaviors, as well as the length of time supplements have been consumed,” added Dr. Laing, who was not involved in the WHI analysis.
“We asked ourselves if CaD supplements might contribute to calcification of the coronary arteries, since some believe this to be the case, although the literature is mixed,” said Dr. Thomson.
“So we did a shorter ancillary study in a small sample of several hundred [women] to see if there was any increase in calcification” and no difference was seen on imaging across the two arms. “However, women who were already on supplements before entering the study seemed to be at higher CVD risk,” she said.
Added study coauthor JoAnn E. Manson, MD, DrPH, chief of the division of preventive medicine at Brigham and Women’s Hospital and professor of women’s health at Harvard Medical School, both in Boston: “With no increase or decrease in coronary artery calcium at the end of the trial, we don’t believe starting or continuing calcium/vitamin D supplements should require screening for coronary artery disease.”

Some randomized trials and systematic reviews, however, have observed an increased risk of CVD in healthy patients on calcium supplements, with one Korean meta-analysis reporting a 15% increase in CVD risk in healthy postmenopausal women taking calcium supplements. Another meta-analysis found a link between calcium supplements and a greater risk of various cardiovascular outcomes, especially myocardial infarction.
Vitamin D Supplementation
As for vitamin D only supplementation, an updated meta-analysis including more than 83,000 individuals showed that it confers no cardiovascular protection and is therefore not indicated for this purpose.
Practice Considerations
Offering an outsider’s perspective, Sarah G. Candler, MD, MPH, an internist in Houston specializing in primary care for older high-risk adults, said: “Unfortunately, this latest study continues the trend of creating more questions than answers. If the adverse outcome of CVD death is a result of supplementation, it is unclear if this is due to the vitamin D, the calcium, or both. And it is unclear if this is dose dependent, time dependent, or due to concurrent risk factors unique to certain populations.

“It is recommended that patients at risk of osteoporosis based on age, sex, medications, and lifestyle be screened for osteoporosis and treated accordingly, including supplementation with CaD,” Dr. Candler said. “It remains unclear whether supplementation with CaD in the absence of osteoporosis and osteopenia is net beneficial or harmful, and at this time I would not recommend it to my patients.”
Added Dr. Manson: “The very small increase seen in cardiovascular mortality wouldn’t be a reason to discontinue supplementation among women who have been advised by their healthcare providers to take these supplements for bone health or other purposes.
“Among those at usual risk of fracture, we recommend trying to obtain adequate calcium and vitamin D from food sources first and to use supplements only for the purpose of filling gaps in intake,” Dr. Manson continued. Overall, the findings support the national recommended dietary allowances for daily calcium intake of 1200 mg and daily vitamin D intake of 600-800 IU among postmenopausal women for maintenance of bone health, she said.
While a 2022 study found that vitamin D supplementation alone did not prevent fractures in healthy adults, other research has shown that a calcium/vitamin D combination is more likely to protect the skeleton.
“Patients at risk for fractures will probably benefit from calcium and/or vitamin D supplementation if they do not meet dietary intake requirements, have malabsorption syndromes, are taking medications that affect nutrient absorption, or if they are older and not regularly exposed to sunlight,” said Dr. Laing. “A combination of biochemical, imaging, functional, and dietary intake data can help determine if a supplement is warranted.”
She stressed that additional research is needed in more diverse populations before changing practice guidelines. “However, doctors should continue to weigh the risks and benefits of prescribing supplements for each patient.”
The WHI program is funded by the National Heart, Lung, and Blood Institute. Dr. Thomson disclosed no competing interests. Dr. Manson reported a relationship with Mars Edge. Multiple authors reported grant support from government funding agencies. The outside commentators had no relevant competing interests to disclose.
Some doctors may be scratching their heads over a new analysis reporting that combined calcium and vitamin D (CaD) supplements appear to be associated with a slight 6% increase in cardiovascular (CVD) mortality, a slight 7% decrease in cancer risk, and no effect on osteoporotic fracture in postmenopausal women.
The study, in Annals of Internal Medicine, found no effect of supplementation on all-cause mortality.
The findings emerged from an analysis of more than 20 years’ follow-up data on a randomized trial in postmenopausal women conducted as part of the Women’s Health Initiative (WHI).
Cynthia A. Thomson, PhD, RD, first author and cancer prevention scientist at the Arizona Cancer Center and a professor of health promotion sciences at the University of Arizona in Tucson said the findings recommend individualized assessment of the need for supplements for older women as they consider them in hopes of preventing fractures.
“Evaluate your patients individually and understand that there are some who may benefit from supplementation, for example, in terms of reducing colorectal cancer mortality,” Dr. Thomson said in an interview. The approach should be nuanced. “If you check the adequacy of vitamin D and calcium in their diets, supplementation may not be needed.” She added that supplementation is best considered in the context of a woman’s overall health profile, including risk factors for fracture, heart disease, and cancer, especially colorectal cancer (CRC).
Study Details
The investigators conducted postintervention follow-up of the WHI’s 7-year multicenter randomized intervention trial of CaD vs placebo.
Since existing evidence of long-term health outcomes was limited, the trial, begun in 1999 and closed in 2005, enrolled 36,282 postmenopausal women (mean age 62) with no history of breast or colorectal cancer. They were randomly assigned 1:1 to supplementation with 1000 mg of calcium carbonate (400 mg elemental calcium) plus 400 IU of vitamin D3 daily or placebo, taken twice daily in half doses.
Study outcomes were incidence of CRC, total and invasive breast cancer; disease-specific and all-cause mortality; total CVD; and hip fracture measured through December 2020, with analyses stratified by personal supplement usage.
Cancer. CaD was associated with reduced incident total cancer, CRC, and invasive breast cancer — notably among participants not taking CaD before randomization. Cancer incidence estimates varied widely, the authors noted, when stratified by supplement use before randomization. Noting that CaD seemed to have more cancer-related impact in those without prior supplementation, the authors suggested supplementation may affect cancer biology primarily by augmenting nutrient insufficiency.
An estimated 7% reduction in cancer mortality was observed after a median cumulative follow-up of 22.3 years: 1817 vs 1943 deaths (hazard ratio, 0.93; 95% CI, 0.87-0.99).
CVD. An estimated 6% increase in CVD mortality was seen in the CaD group: 2621 vs 2420 deaths (HR, 1.06; 95% CI, 1.01-1.12). Pretrial supplement users were found to be at higher CVD risk.
Hip fracture. No effect on hip fracture risk was measured, but the authors cautioned that hip fracture and CVD outcomes were available only for a subset of participants, and the effects of calcium alone vs vitamin D alone vs the combination could not be disentangled.
In a small subgroup analysis, some CaD users were seen to respond in terms of bone mineral density but since only 4 of the study’s 40 sites collected such information, the study was underpowered to examine the effect. ”Many other studies, however, show a response to supplementation in women who already have bone mineral deficits,” Dr. Thomson said.
The Calcification Question
One of the possible mechanisms of harm is that high-dose calcium supplements can increase the rate of blood coagulation and promote vascular calcification, said Emma Laing, PhD, RD, director of dietetics at the University of Georgia in Athens and a spokesperson for the Chicago-based Academy of Nutrition and Dietetics.
“Other factors that should be considered when determining a patient’s CVD risk are race, genetic predisposition, medical and social history, response to stress, and lifestyle behaviors, as well as the length of time supplements have been consumed,” added Dr. Laing, who was not involved in the WHI analysis.
“We asked ourselves if CaD supplements might contribute to calcification of the coronary arteries, since some believe this to be the case, although the literature is mixed,” said Dr. Thomson.
“So we did a shorter ancillary study in a small sample of several hundred [women] to see if there was any increase in calcification” and no difference was seen on imaging across the two arms. “However, women who were already on supplements before entering the study seemed to be at higher CVD risk,” she said.
Added study coauthor JoAnn E. Manson, MD, DrPH, chief of the division of preventive medicine at Brigham and Women’s Hospital and professor of women’s health at Harvard Medical School, both in Boston: “With no increase or decrease in coronary artery calcium at the end of the trial, we don’t believe starting or continuing calcium/vitamin D supplements should require screening for coronary artery disease.”
Some randomized trials and systematic reviews, however, have observed an increased risk of CVD in healthy patients on calcium supplements, with one Korean meta-analysis reporting a 15% increase in CVD risk in healthy postmenopausal women taking calcium supplements. Another meta-analysis found a link between calcium supplements and a greater risk of various cardiovascular outcomes, especially myocardial infarction.
Vitamin D Supplementation
As for vitamin D only supplementation, an updated meta-analysis including more than 83,000 individuals showed that it confers no cardiovascular protection and is therefore not indicated for this purpose.
Practice Considerations
Offering an outsider’s perspective, Sarah G. Candler, MD, MPH, an internist in Houston specializing in primary care for older high-risk adults, said: “Unfortunately, this latest study continues the trend of creating more questions than answers. If the adverse outcome of CVD death is a result of supplementation, it is unclear if this is due to the vitamin D, the calcium, or both. And it is unclear if this is dose dependent, time dependent, or due to concurrent risk factors unique to certain populations.
“It is recommended that patients at risk of osteoporosis based on age, sex, medications, and lifestyle be screened for osteoporosis and treated accordingly, including supplementation with CaD,” Dr. Candler said. “It remains unclear whether supplementation with CaD in the absence of osteoporosis and osteopenia is net beneficial or harmful, and at this time I would not recommend it to my patients.”
Added Dr. Manson: “The very small increase seen in cardiovascular mortality wouldn’t be a reason to discontinue supplementation among women who have been advised by their healthcare providers to take these supplements for bone health or other purposes.
“Among those at usual risk of fracture, we recommend trying to obtain adequate calcium and vitamin D from food sources first and to use supplements only for the purpose of filling gaps in intake,” Dr. Manson continued. Overall, the findings support the national recommended dietary allowances for daily calcium intake of 1200 mg and daily vitamin D intake of 600-800 IU among postmenopausal women for maintenance of bone health, she said.
While a 2022 study found that vitamin D supplementation alone did not prevent fractures in healthy adults, other research has shown that a calcium/vitamin D combination is more likely to protect the skeleton.
“Patients at risk for fractures will probably benefit from calcium and/or vitamin D supplementation if they do not meet dietary intake requirements, have malabsorption syndromes, are taking medications that affect nutrient absorption, or if they are older and not regularly exposed to sunlight,” said Dr. Laing. “A combination of biochemical, imaging, functional, and dietary intake data can help determine if a supplement is warranted.”
She stressed that additional research is needed in more diverse populations before changing practice guidelines. “However, doctors should continue to weigh the risks and benefits of prescribing supplements for each patient.”
The WHI program is funded by the National Heart, Lung, and Blood Institute. Dr. Thomson disclosed no competing interests. Dr. Manson reported a relationship with Mars Edge. Multiple authors reported grant support from government funding agencies. The outside commentators had no relevant competing interests to disclose.
Some doctors may be scratching their heads over a new analysis reporting that combined calcium and vitamin D (CaD) supplements appear to be associated with a slight 6% increase in cardiovascular (CVD) mortality, a slight 7% decrease in cancer risk, and no effect on osteoporotic fracture in postmenopausal women.
The study, in Annals of Internal Medicine, found no effect of supplementation on all-cause mortality.
The findings emerged from an analysis of more than 20 years’ follow-up data on a randomized trial in postmenopausal women conducted as part of the Women’s Health Initiative (WHI).
Cynthia A. Thomson, PhD, RD, first author and cancer prevention scientist at the Arizona Cancer Center and a professor of health promotion sciences at the University of Arizona in Tucson said the findings recommend individualized assessment of the need for supplements for older women as they consider them in hopes of preventing fractures.
“Evaluate your patients individually and understand that there are some who may benefit from supplementation, for example, in terms of reducing colorectal cancer mortality,” Dr. Thomson said in an interview. The approach should be nuanced. “If you check the adequacy of vitamin D and calcium in their diets, supplementation may not be needed.” She added that supplementation is best considered in the context of a woman’s overall health profile, including risk factors for fracture, heart disease, and cancer, especially colorectal cancer (CRC).
Study Details
The investigators conducted postintervention follow-up of the WHI’s 7-year multicenter randomized intervention trial of CaD vs placebo.
Since existing evidence of long-term health outcomes was limited, the trial, begun in 1999 and closed in 2005, enrolled 36,282 postmenopausal women (mean age 62) with no history of breast or colorectal cancer. They were randomly assigned 1:1 to supplementation with 1000 mg of calcium carbonate (400 mg elemental calcium) plus 400 IU of vitamin D3 daily or placebo, taken twice daily in half doses.
Study outcomes were incidence of CRC, total and invasive breast cancer; disease-specific and all-cause mortality; total CVD; and hip fracture measured through December 2020, with analyses stratified by personal supplement usage.
Cancer. CaD was associated with reduced incident total cancer, CRC, and invasive breast cancer — notably among participants not taking CaD before randomization. Cancer incidence estimates varied widely, the authors noted, when stratified by supplement use before randomization. Noting that CaD seemed to have more cancer-related impact in those without prior supplementation, the authors suggested supplementation may affect cancer biology primarily by augmenting nutrient insufficiency.
An estimated 7% reduction in cancer mortality was observed after a median cumulative follow-up of 22.3 years: 1817 vs 1943 deaths (hazard ratio, 0.93; 95% CI, 0.87-0.99).
CVD. An estimated 6% increase in CVD mortality was seen in the CaD group: 2621 vs 2420 deaths (HR, 1.06; 95% CI, 1.01-1.12). Pretrial supplement users were found to be at higher CVD risk.
Hip fracture. No effect on hip fracture risk was measured, but the authors cautioned that hip fracture and CVD outcomes were available only for a subset of participants, and the effects of calcium alone vs vitamin D alone vs the combination could not be disentangled.
In a small subgroup analysis, some CaD users were seen to respond in terms of bone mineral density but since only 4 of the study’s 40 sites collected such information, the study was underpowered to examine the effect. ”Many other studies, however, show a response to supplementation in women who already have bone mineral deficits,” Dr. Thomson said.
The Calcification Question
One of the possible mechanisms of harm is that high-dose calcium supplements can increase the rate of blood coagulation and promote vascular calcification, said Emma Laing, PhD, RD, director of dietetics at the University of Georgia in Athens and a spokesperson for the Chicago-based Academy of Nutrition and Dietetics.
“Other factors that should be considered when determining a patient’s CVD risk are race, genetic predisposition, medical and social history, response to stress, and lifestyle behaviors, as well as the length of time supplements have been consumed,” added Dr. Laing, who was not involved in the WHI analysis.
“We asked ourselves if CaD supplements might contribute to calcification of the coronary arteries, since some believe this to be the case, although the literature is mixed,” said Dr. Thomson.
“So we did a shorter ancillary study in a small sample of several hundred [women] to see if there was any increase in calcification” and no difference was seen on imaging across the two arms. “However, women who were already on supplements before entering the study seemed to be at higher CVD risk,” she said.
Added study coauthor JoAnn E. Manson, MD, DrPH, chief of the division of preventive medicine at Brigham and Women’s Hospital and professor of women’s health at Harvard Medical School, both in Boston: “With no increase or decrease in coronary artery calcium at the end of the trial, we don’t believe starting or continuing calcium/vitamin D supplements should require screening for coronary artery disease.”
Some randomized trials and systematic reviews, however, have observed an increased risk of CVD in healthy patients on calcium supplements, with one Korean meta-analysis reporting a 15% increase in CVD risk in healthy postmenopausal women taking calcium supplements. Another meta-analysis found a link between calcium supplements and a greater risk of various cardiovascular outcomes, especially myocardial infarction.
Vitamin D Supplementation
As for vitamin D only supplementation, an updated meta-analysis including more than 83,000 individuals showed that it confers no cardiovascular protection and is therefore not indicated for this purpose.
Practice Considerations
Offering an outsider’s perspective, Sarah G. Candler, MD, MPH, an internist in Houston specializing in primary care for older high-risk adults, said: “Unfortunately, this latest study continues the trend of creating more questions than answers. If the adverse outcome of CVD death is a result of supplementation, it is unclear if this is due to the vitamin D, the calcium, or both. And it is unclear if this is dose dependent, time dependent, or due to concurrent risk factors unique to certain populations.
“It is recommended that patients at risk of osteoporosis based on age, sex, medications, and lifestyle be screened for osteoporosis and treated accordingly, including supplementation with CaD,” Dr. Candler said. “It remains unclear whether supplementation with CaD in the absence of osteoporosis and osteopenia is net beneficial or harmful, and at this time I would not recommend it to my patients.”
Added Dr. Manson: “The very small increase seen in cardiovascular mortality wouldn’t be a reason to discontinue supplementation among women who have been advised by their healthcare providers to take these supplements for bone health or other purposes.
“Among those at usual risk of fracture, we recommend trying to obtain adequate calcium and vitamin D from food sources first and to use supplements only for the purpose of filling gaps in intake,” Dr. Manson continued. Overall, the findings support the national recommended dietary allowances for daily calcium intake of 1200 mg and daily vitamin D intake of 600-800 IU among postmenopausal women for maintenance of bone health, she said.
While a 2022 study found that vitamin D supplementation alone did not prevent fractures in healthy adults, other research has shown that a calcium/vitamin D combination is more likely to protect the skeleton.
“Patients at risk for fractures will probably benefit from calcium and/or vitamin D supplementation if they do not meet dietary intake requirements, have malabsorption syndromes, are taking medications that affect nutrient absorption, or if they are older and not regularly exposed to sunlight,” said Dr. Laing. “A combination of biochemical, imaging, functional, and dietary intake data can help determine if a supplement is warranted.”
She stressed that additional research is needed in more diverse populations before changing practice guidelines. “However, doctors should continue to weigh the risks and benefits of prescribing supplements for each patient.”
The WHI program is funded by the National Heart, Lung, and Blood Institute. Dr. Thomson disclosed no competing interests. Dr. Manson reported a relationship with Mars Edge. Multiple authors reported grant support from government funding agencies. The outside commentators had no relevant competing interests to disclose.
FROM ANNALS OF INTERNAL MEDICINE
Meta-analysis Identifies Unique Risk Factors of Triple-Negative Breast Cancer
Key clinical point: The risk factors for overall breast cancer (such as parity, menopausal hormone therapy use, and alcohol consumption) did not increase the risk for triple-negative breast cancer (TNBC), which had a distinct risk factor profile.
Major finding: Parity, menopausal hormone therapy use, alcohol consumption, smoking, and higher body mass index were not significantly associated with TNBC risk (all P > .05); instead, family history (odds ratio [OR] 1.55; P < .001), longer duration of oral contraceptive use (OR 1.29; P < .001), and higher breast density (OR 2.19; P < .001) were significantly associated with an increased risk for TNBC.
Study details: This meta-analysis evaluated the association between TNBC incidence and established BC risk factors using data from 33 studies.
Disclosures: This study was supported by grants from the American Cancer Society and Royal College of Surgeons in Ireland - Medical University of Bahrain (RCSI-MUB Bahrain). The authors declared no conflicts of interest.
Source: Kumar N, Ehsan S, Banerjee S, et al. The unique risk factor profile of triple negative breast cancer: A comprehensive meta-analysis. J Natl Cancer Inst. 2024 (Mar 5). Doi: 10.1093/jnci/djae056 Source
Key clinical point: The risk factors for overall breast cancer (such as parity, menopausal hormone therapy use, and alcohol consumption) did not increase the risk for triple-negative breast cancer (TNBC), which had a distinct risk factor profile.
Major finding: Parity, menopausal hormone therapy use, alcohol consumption, smoking, and higher body mass index were not significantly associated with TNBC risk (all P > .05); instead, family history (odds ratio [OR] 1.55; P < .001), longer duration of oral contraceptive use (OR 1.29; P < .001), and higher breast density (OR 2.19; P < .001) were significantly associated with an increased risk for TNBC.
Study details: This meta-analysis evaluated the association between TNBC incidence and established BC risk factors using data from 33 studies.
Disclosures: This study was supported by grants from the American Cancer Society and Royal College of Surgeons in Ireland - Medical University of Bahrain (RCSI-MUB Bahrain). The authors declared no conflicts of interest.
Source: Kumar N, Ehsan S, Banerjee S, et al. The unique risk factor profile of triple negative breast cancer: A comprehensive meta-analysis. J Natl Cancer Inst. 2024 (Mar 5). Doi: 10.1093/jnci/djae056 Source
Key clinical point: The risk factors for overall breast cancer (such as parity, menopausal hormone therapy use, and alcohol consumption) did not increase the risk for triple-negative breast cancer (TNBC), which had a distinct risk factor profile.
Major finding: Parity, menopausal hormone therapy use, alcohol consumption, smoking, and higher body mass index were not significantly associated with TNBC risk (all P > .05); instead, family history (odds ratio [OR] 1.55; P < .001), longer duration of oral contraceptive use (OR 1.29; P < .001), and higher breast density (OR 2.19; P < .001) were significantly associated with an increased risk for TNBC.
Study details: This meta-analysis evaluated the association between TNBC incidence and established BC risk factors using data from 33 studies.
Disclosures: This study was supported by grants from the American Cancer Society and Royal College of Surgeons in Ireland - Medical University of Bahrain (RCSI-MUB Bahrain). The authors declared no conflicts of interest.
Source: Kumar N, Ehsan S, Banerjee S, et al. The unique risk factor profile of triple negative breast cancer: A comprehensive meta-analysis. J Natl Cancer Inst. 2024 (Mar 5). Doi: 10.1093/jnci/djae056 Source