User login
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
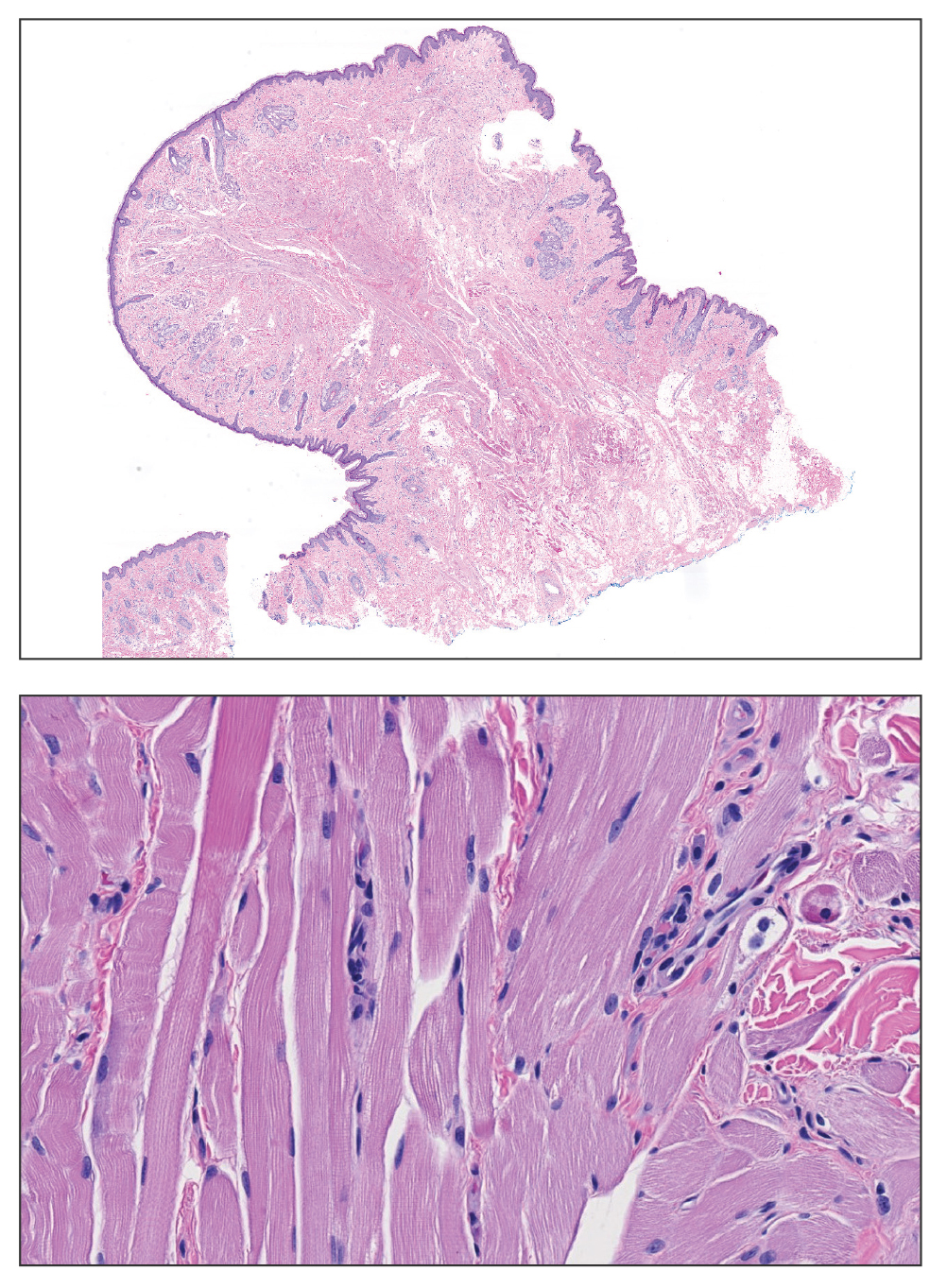
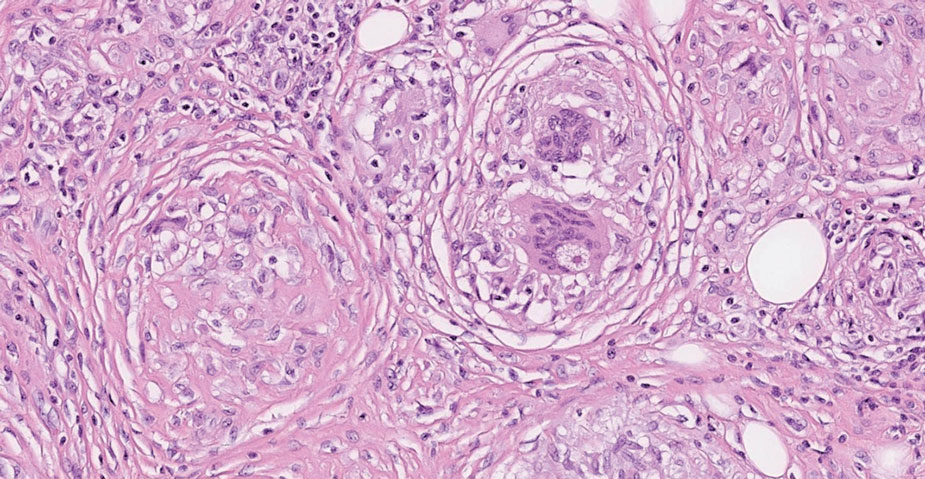
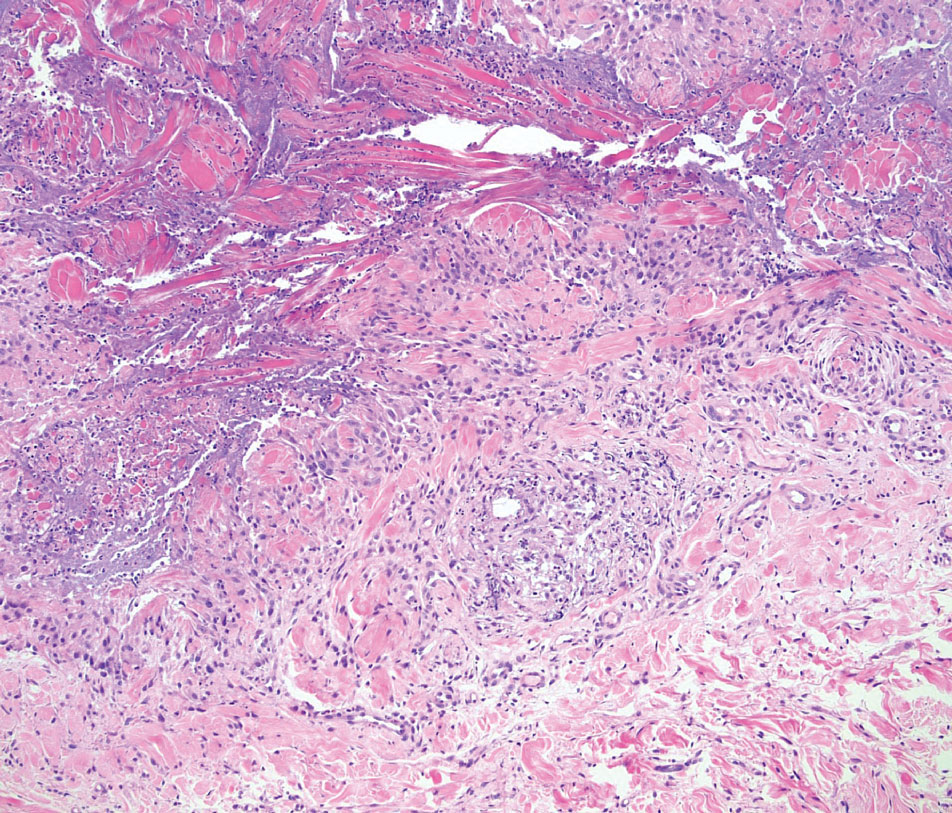
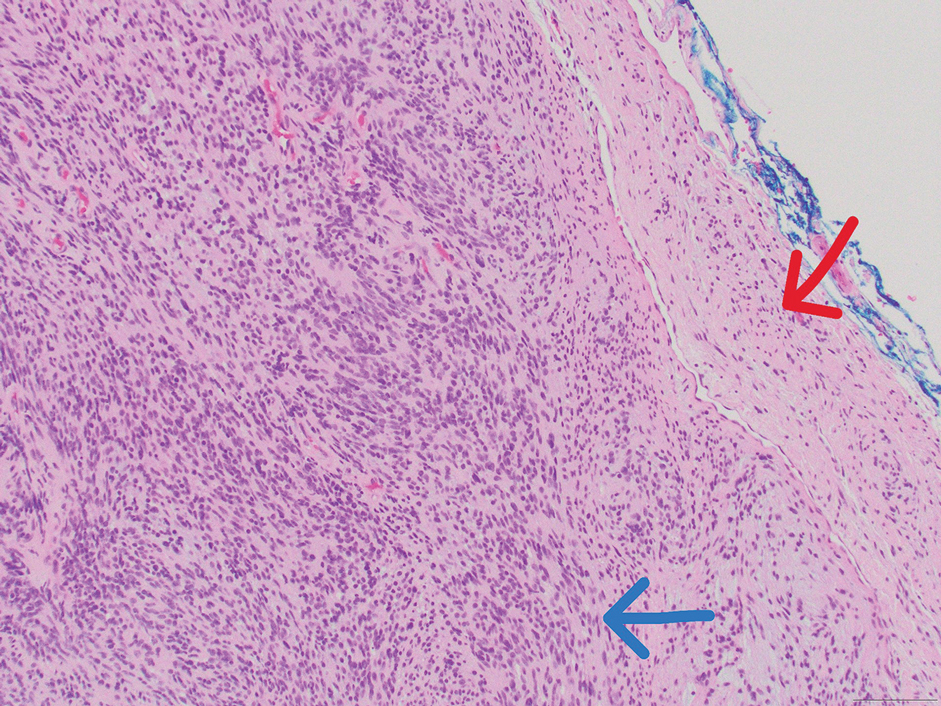
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
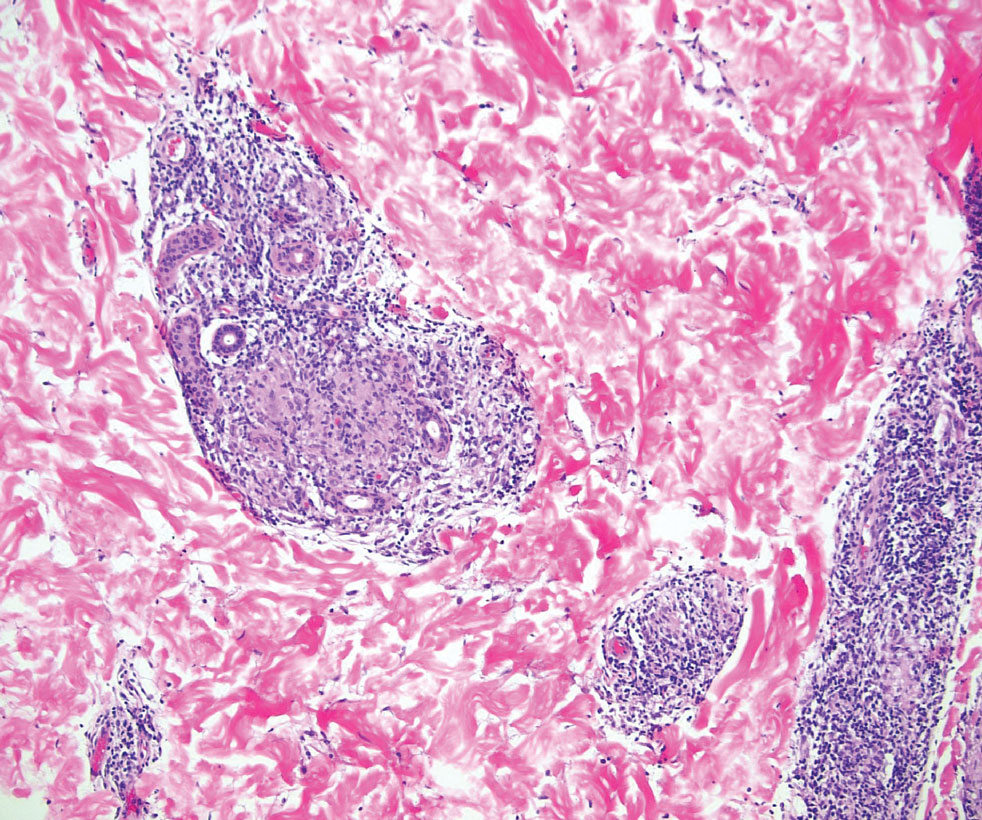
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
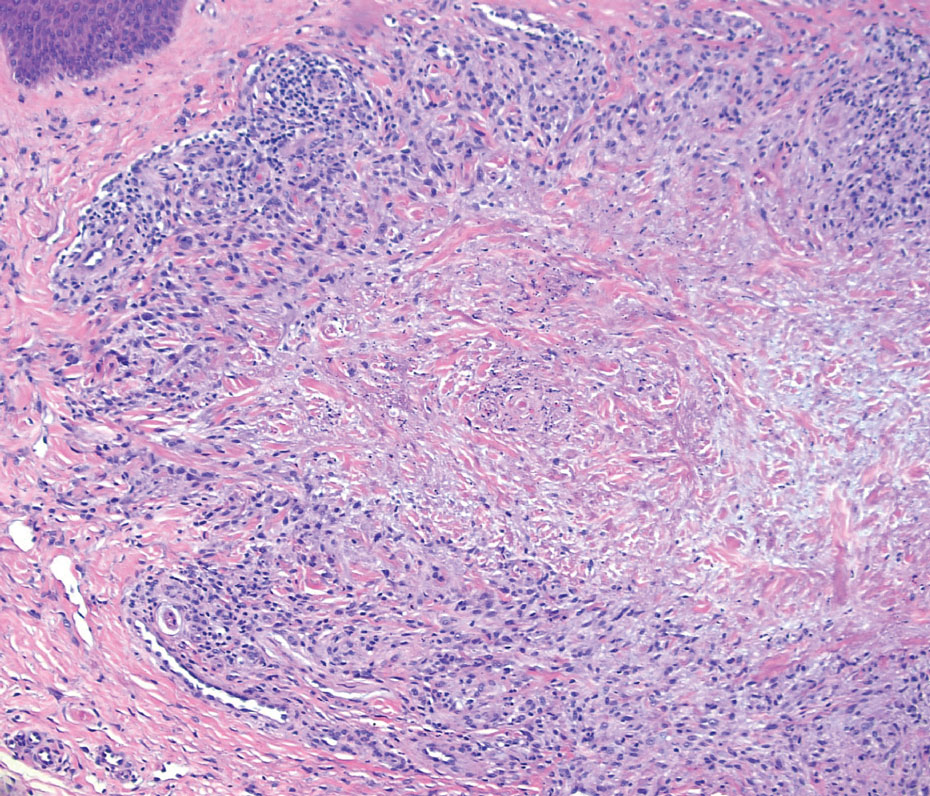
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
A 47-year-old man with a history of chronic kidney disease and bilateral clear cell renal cell carcinoma who was undergoing treatment with adjuvant pembrolizumab presented to the dermatology department with a scattered papulonodular eruption of several weeks’ duration. Physical examination revealed pink papules and nodules with coalescing erythema over the trunk and upper extremities, most pronounced on the right elbow (bottom [inset]). A 4-mm punch biopsy demonstrated dermal granulomatous inflammation. Special stains were negative for microorganisms. Computed tomography of the chest revealed a new subpleural nodule and new hilar lymphadenopathy.
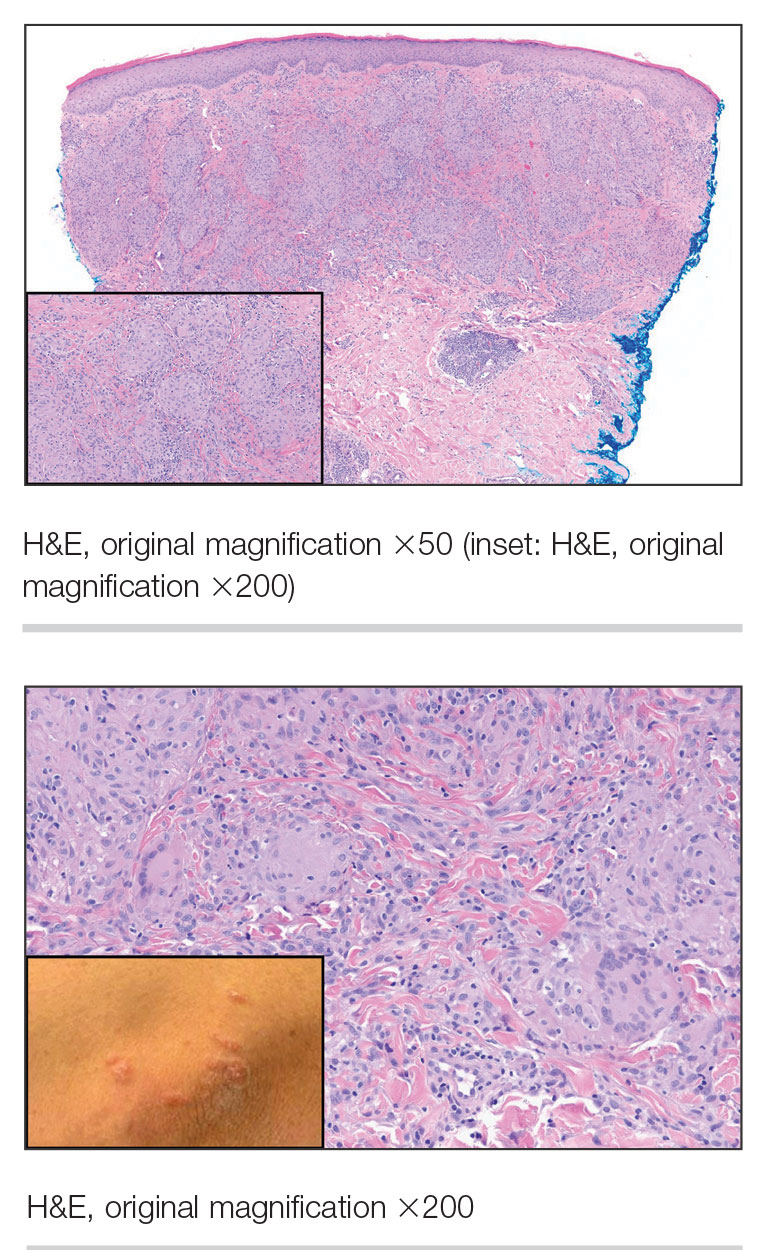

Solitary Papule on the Upper Back
Solitary Papule on the Upper Back
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
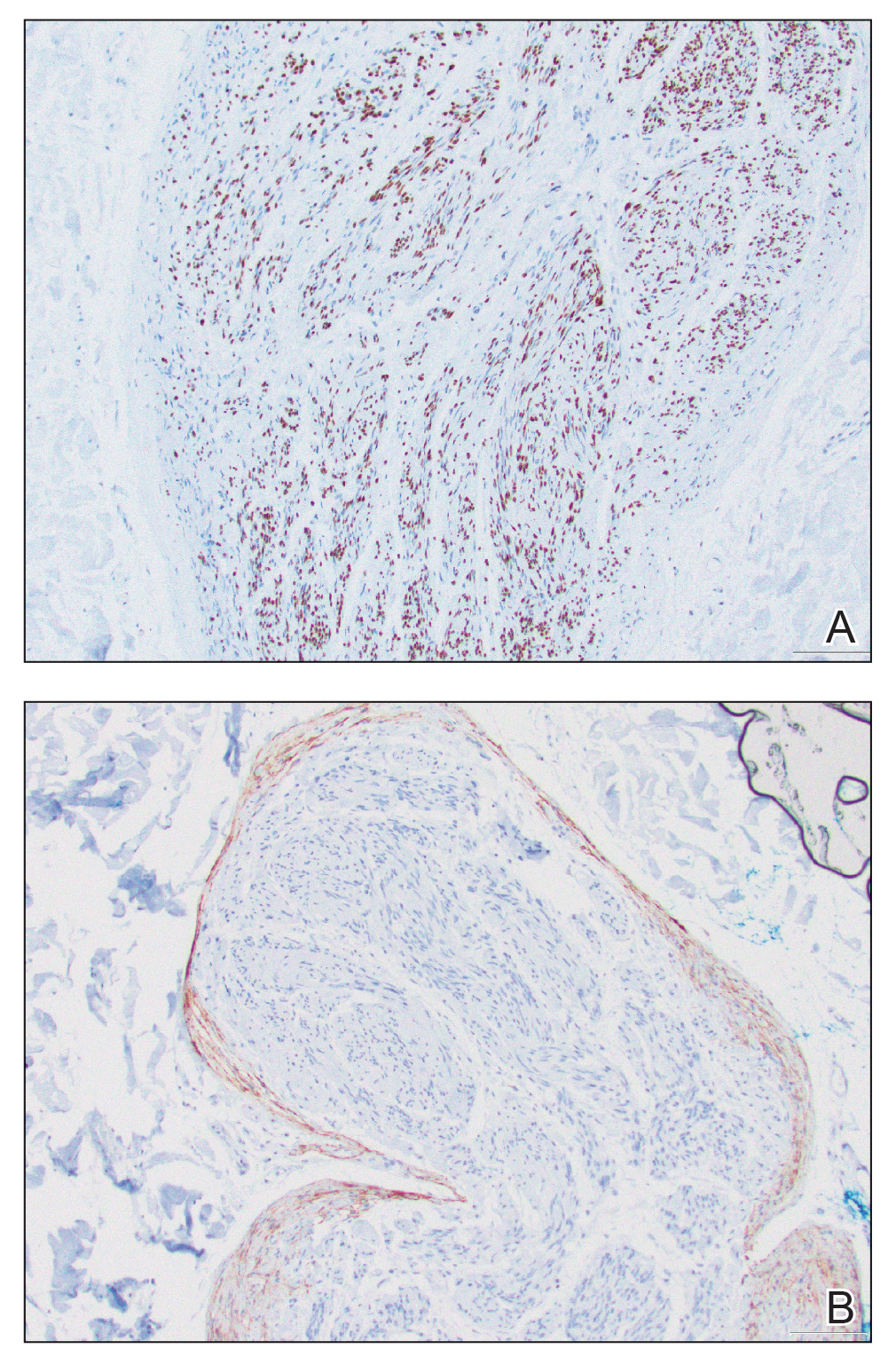
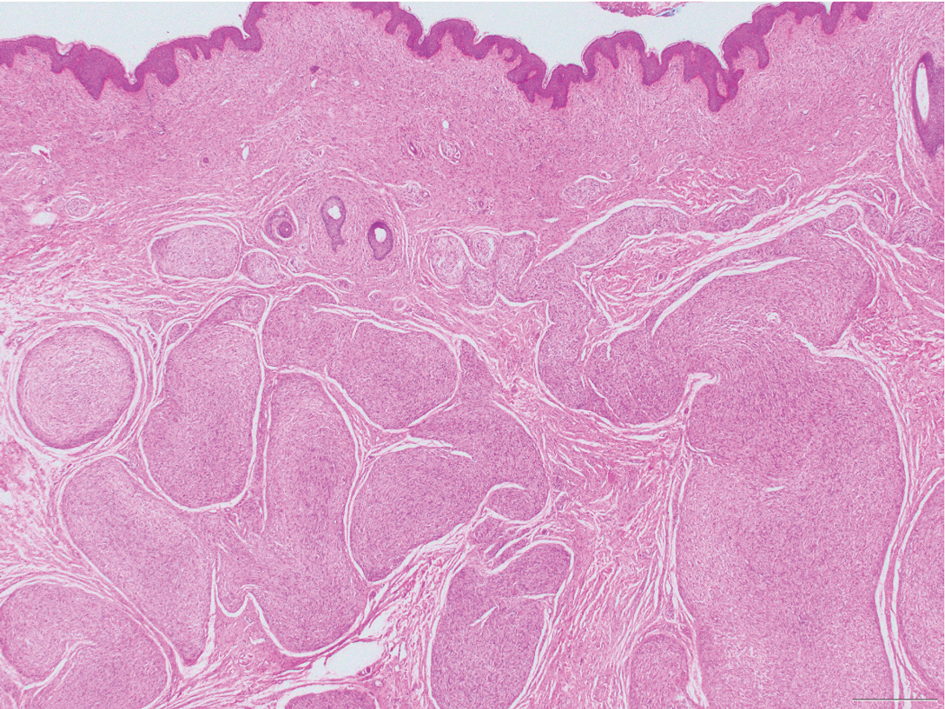
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
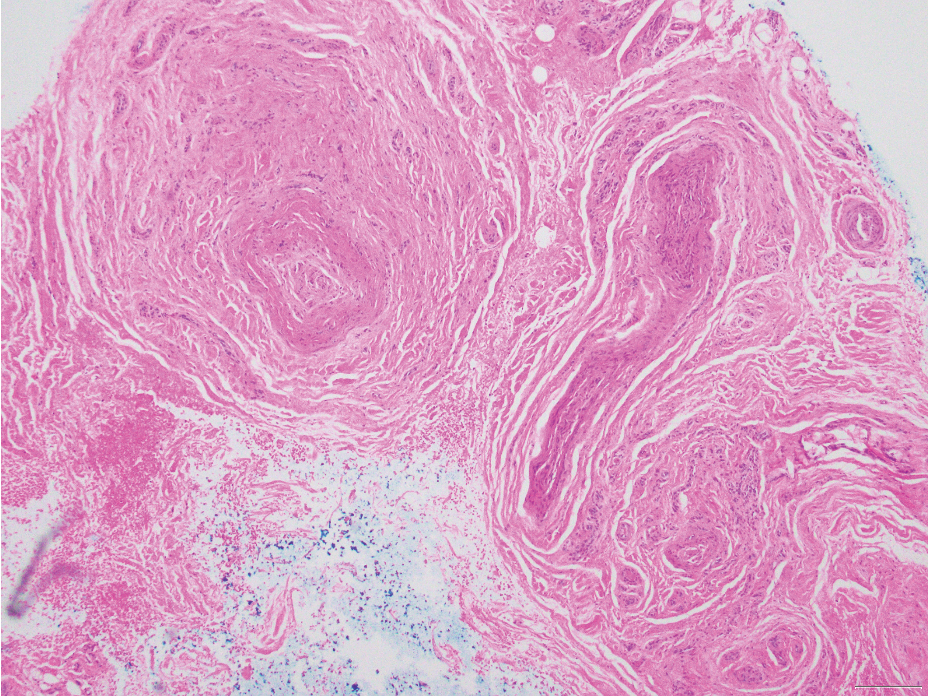
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
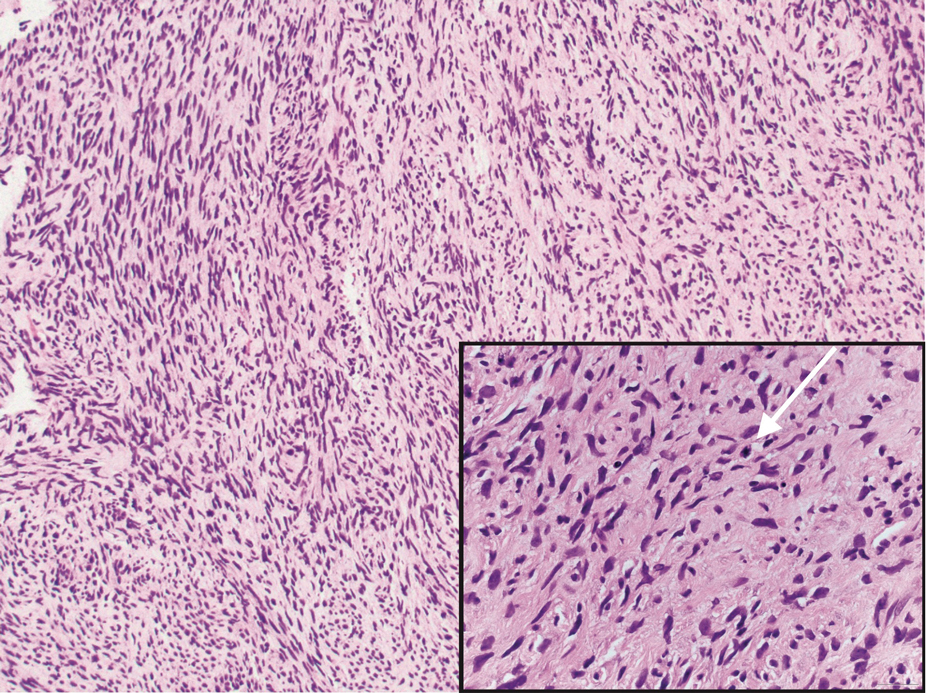
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
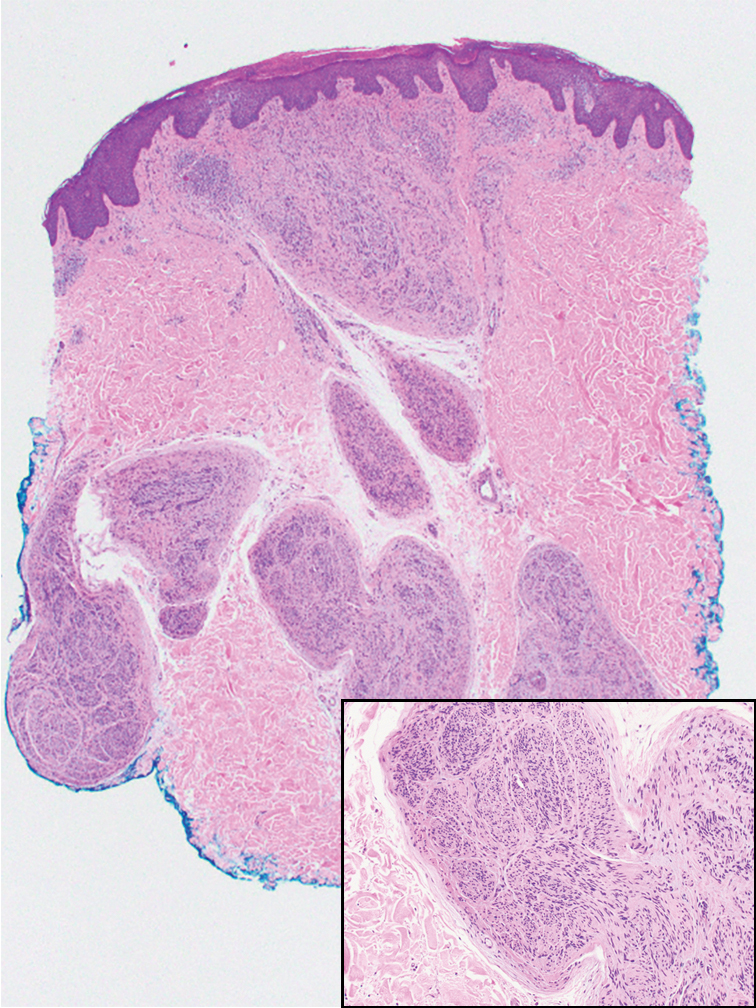
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
Solitary Papule on the Upper Back
Solitary Papule on the Upper Back
An 88-year-old woman presented to the dermatology clinic with an asymptomatic papule on the left upper back of unknown duration. The patient reported that her medical history was negative for eczematous dermatitis, hypertension, and osteoarthritis. Physical examination revealed a firm, well-circumscribed, flesh-colored, 6-mm papule with no overlying scale or ulceration. No other concerning lesions were noted on full skin examination. A punch biopsy of the papule was performed.

Thick Yellow Plaques on the Eyelids
Thick Yellow Plaques on the Eyelids
THE DIAGNOSIS: Adult-Onset Asthma With Periocular Xanthogranuloma
In the context of pre-existing sinonasal disease and features consistent with adult-onset asthma, the constellation of clinical findings including linear periorbital yellow-orange plaques, imaging demonstrating extension of xanthogranulomatous lesions into the orbital fat, histopathologic features, and serologic abnormalities including elevated IgG4 levels supported a diagnosis of adult-onset asthma with periocular xanthogranuloma (AAPOX).
Adult-onset xanthogranuloma is a non–Langerhans cell histiocytosis (historically classified as type II) within the group of adult orbital xanthogranulomatous diseases resulting from infiltration and proliferation of histiocytes in the orbital and ocular adnexal structures and eyelids. Adult orbital xanthogranulomatous diseases are classified as 4 distinct conditions: adult-onset xanthogranuloma, AAPOX, Erdheim-Chester disease, and necrobiotic xanthogranuloma (NXG). Erdheim-Chester disease is the most severe among this group and often is fatal due to infiltration of the xanthogranulomas into multiple organ systems and tissues, including the long bones, heart, lungs, and retroperitoneum. Neurologic symptoms such as incoordination can occur. Adult-onset xanthogranuloma manifests as an isolated cutaneous lesion without systemic involvement. This entity often is self-limited and does not require aggressive treatment. Adult-onset asthma with periocular xanthogranuloma affects more males than females.1 Clinically, it manifests as bilateral yellow-orange, thickened, indurated eyelid plaques that can extend to the extraocular muscles or lacrimal glands. As the name suggests, this entity is associated with adult-onset asthma or rhinosinusitis as well as lymphadenopathy and extension into the orbital fat.1
When patients present with periorbital lesions and optic symptoms such as visual disturbances, tearing, and/ or a foreign body sensation in the eyes, a work-up should be performed to rule out infiltration of orbital adnexal structures and other organ systems, as AAPOX can be associated with IgG4-related disease. Histologically, adult xanthogranulomatous diseases are characterized by sheets of foamy histiocytes accompanied by variable numbers of lymphoid aggregates, plasma cells, and Touton giant cells. These infiltrating xanthoma cells appear as a garland or wreathlike nuclei surrounded by foamy cytoplasm. Oil-red O staining of frozen sections confirms the lipid content of the xanthoma cells.2 Immunohistochemically, the foamy histiocytes are strongly positive for CD68, CD163, and factor XIIIa but usually are negative for S100, CD1a and Birbeck granules.3 More distinctively, AAPOX is associated with prominent lymphoid aggregates containing reactive germinal centers.1
Given that AAPOX is a systemic multiorgan disease, local therapies such as surgical debulking or intralesional corticosteroids generally are insufficient to address the underlying pathology and therefore necessitate systemic, often multimodal, treatment within a multidisciplinary framework.1,4 Systemic corticosteroids remain first-line therapy, with steroid-sparing agents (eg, methotrexate, azathioprine, cyclophosphamide) used in refractory cases or to reduce steroid dependence.4,5 Rituximab has demonstrated efficacy in AAPOX, further highlighting the association between AAPOX and IgG4-related disease.5 Inebilizumab, a B-cell–depleting monoclonal antibody targeting CD19 and approved for the treatment of IgG4- related disease, represents a theoretically promising therapeutic option; however, additional studies are needed to establish its efficacy and safety in AAPOX.5
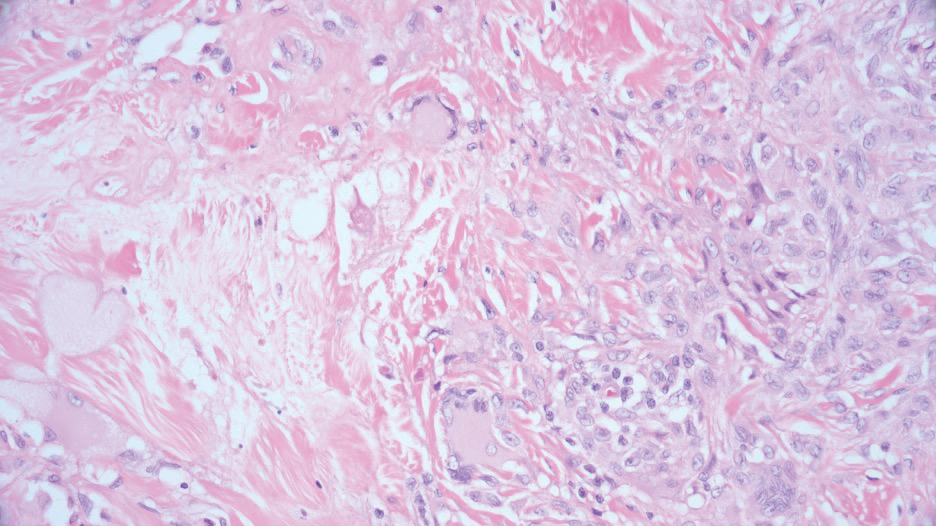
Necrobiotic xanthogranuloma is another xanthogranulomatous disorder that manifests as yellow-orange papules or nodules that gradually form infiltrative plaques. Scarring and ulceration can occur in 40% to 50% of patients.6 The most common site of involvement is the periorbital area, affecting 80% of cases, often resulting in ophthalmologic complications.6 Necrobiotic xanthogranuloma lesions also can involve the trunk, arms, and legs. Extracutaneous sites include the lungs, myocardium, larynx, pharynx, skeletal muscles, kidneys, ovaries, and intestines. The prognosis of NXG is poor due to associated hematologic malignancies such as multiple myeloma and lymphoma. About 80% of patients have a serum monoclonal gammopathy.6 Histologically, NXG shows features overlapping with other xanthogranulomatous disorders, such as foamy histiocytes, multinucleated giant cells, Touton giant cells, and nodular lymphocytic aggregates6,7; however, broad zones of necrobiosis are a distinct histologic finding in NXG that helps differentiate it from other xanthogranulomatous disorders (Figure 1).6
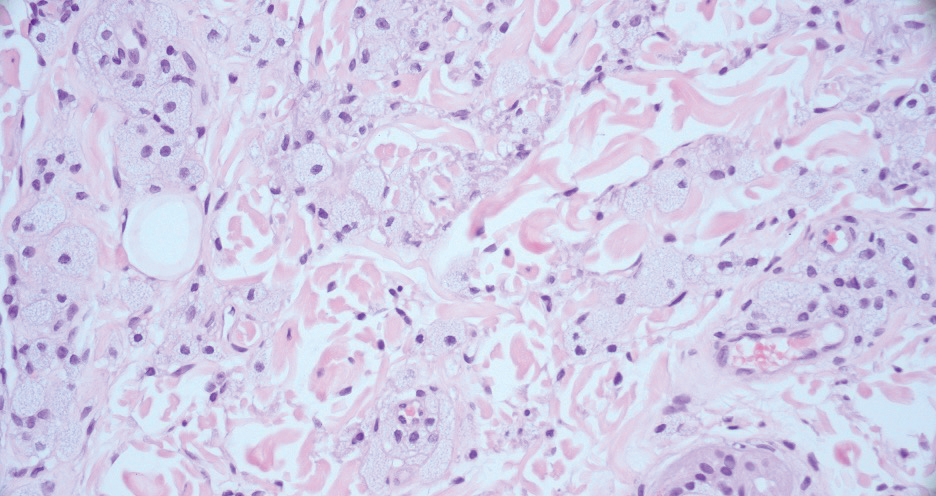
Xanthelasma manifests as yellow plaques on the medial upper and lower eyelids and lateral canthi resulting from accumulation of cholesterol-rich material in the skin, soft tissue, and sometimes the tendons. Fifty percent of patients have a primary or secondary lipid disorder such as familial dyslipidemia, thyroid disease, diabetes mellitus, or primary biliary cholangitis.8 Histologically, xanthelasmas demonstrate lipid-laden foamy histiocytes in the superficial dermis (Figure 2).8 Despite some clinical overlap in our case, the depth of orbital involvement and supportive systemic and histopathologic findings (including Touton giant cells) supported the diagnosis of AAPOX rather than xanthelasma.
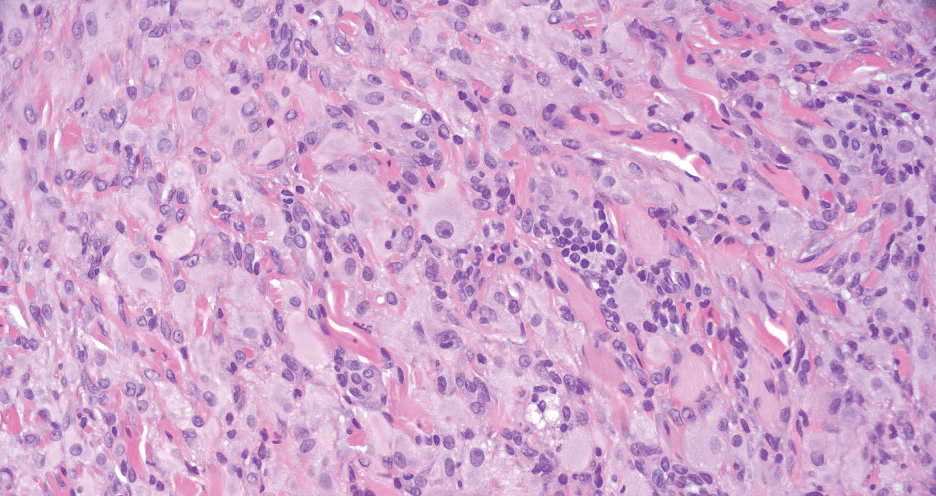
Juvenile xanthogranuloma manifests as solitary to multiple firm, yellow-orange papules or nodules on the face, neck, and upper torso. The lesions develop in early childhood, with 75% of lesions appearing in the first year of life, but rarely it may develop in adulthood.9 The most common extracutaneous manifestation involves ocular structures, most frequently the iris, followed by the lungs. Cutaneous lesions usually are asymptomatic and involute over the span of a few years. Ocular lesions can result in blindness, and juvenile xanthogranuloma also has been associated with neurofibromatosis type 1 and juvenile chronic myelogenous leukemia.9 The histopathology of juvenile xanthogranuloma often will show a dense histiocytic infiltrate in the dermis with blunting of the overlying rete ridges admixed with lymphocytes, plasma cells, and eosinophils. In the more mature phase, foam cells, foreign body giant cells, and Touton giant cells predominate. Touton giant cells have a garlandlike appearance (Figure 3).9
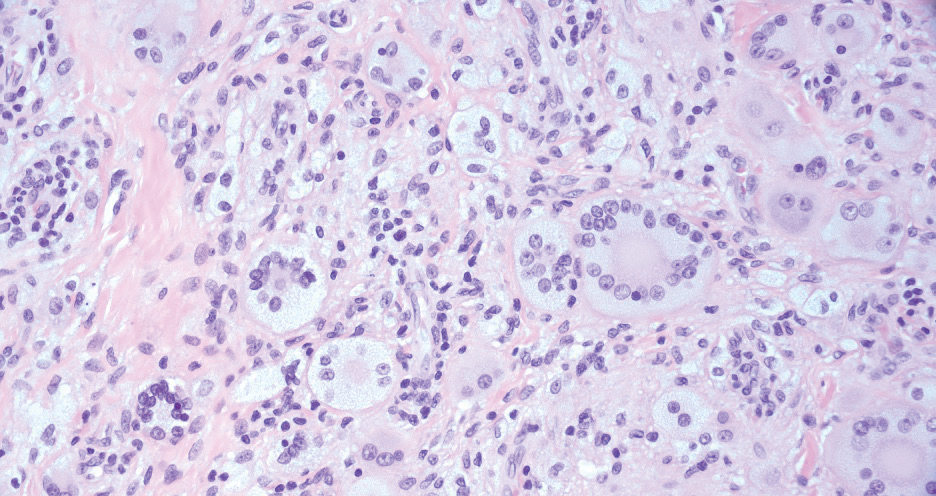
Reticulohistiocytoma (or solitary epithelioid histiocytoma) and multicentric reticulohistiocytosis are rare histiocytic proliferations. Multicentric reticulohistiocytosis refers to a systemic disease with arthropathy and multiple cutaneous histiocytic lesions located on acral sites and the face. Solitary reticulohistiocytoma manifests as papules or nodules found in many body locations, such as the trunk, arms, and legs. The lesions are uncommon on the face, which almost always is involved in multicentric reticulohistiocytosis. Solitary reticulohistiocytomas tend not to recur once excised and do not demonstrate systemic involvement. Histologically, the lesions demonstrate large eosinophilic epithelioid histiocytes with abundant glassy cytoplasm (Figure 4). Some of the epithelioid histiocytes are multinucleated, and immunophenotyping will show positivity for lysozyme, CD68, and CD163.10
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463. doi:10.1016 /j.det.2015.03.010
- Mandic` JJ, Bakula M, Šklebar LK, et al. Histiocytosis and adult-onset orbital xanthogranuloma in 2023: a review of the literature and mini case series. Int Ophthalmol. 2024;44:301. doi:10.1007/s10792-024-03181-y
- Campochiaro C, Tomelleri A, Cavalli G, et al. Erdheim-Chester disease. Eur J Intern Med. 2015;26:223-229. doi: 10.1016/j.ejim.2015.03.004
- Detiger SE, Hötte GJ, Verdijk RM, et al. Adult orbital xanthogranuloma: long-term follow-up of treated cases. Eye (Lond). 2023;37:2475-2481. doi: 10.1038/s41433-022-02357-z
- Stone JH, Khosroshahi A, Zhang W, et al. Inebilizumab for treatment of IgG4-related disease. N Engl J Med. 2025;392:1168-1177. doi:10.1056 /NEJMoa2409712
- Wood AJ, Wagner MVU, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279–284. doi:10.1001 /archdermatol.2008.583
- Schadt C, Jacobsen E. Necrobiotic xanthogranuloma. In: Connor RF, ed. UpToDate. Wolters Kluwer.
- Al Aboud AM, Shah SS, Blair K, et al. Xanthelasma palpebrarum. StatPearls [Internet]. StatPearls Publishing; 2024. Updated March 1, 2024. Accessed February 10, 2026. https://www.ncbi.nlm .nih.gov/books/NBK531501/
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated August 8, 2023. Accessed February 10, 2026. https://www.ncbi.nlm.nih .gov/books/NBK526103/
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528. doi:10.1097/00000478 -200604000-00014
THE DIAGNOSIS: Adult-Onset Asthma With Periocular Xanthogranuloma
In the context of pre-existing sinonasal disease and features consistent with adult-onset asthma, the constellation of clinical findings including linear periorbital yellow-orange plaques, imaging demonstrating extension of xanthogranulomatous lesions into the orbital fat, histopathologic features, and serologic abnormalities including elevated IgG4 levels supported a diagnosis of adult-onset asthma with periocular xanthogranuloma (AAPOX).
Adult-onset xanthogranuloma is a non–Langerhans cell histiocytosis (historically classified as type II) within the group of adult orbital xanthogranulomatous diseases resulting from infiltration and proliferation of histiocytes in the orbital and ocular adnexal structures and eyelids. Adult orbital xanthogranulomatous diseases are classified as 4 distinct conditions: adult-onset xanthogranuloma, AAPOX, Erdheim-Chester disease, and necrobiotic xanthogranuloma (NXG). Erdheim-Chester disease is the most severe among this group and often is fatal due to infiltration of the xanthogranulomas into multiple organ systems and tissues, including the long bones, heart, lungs, and retroperitoneum. Neurologic symptoms such as incoordination can occur. Adult-onset xanthogranuloma manifests as an isolated cutaneous lesion without systemic involvement. This entity often is self-limited and does not require aggressive treatment. Adult-onset asthma with periocular xanthogranuloma affects more males than females.1 Clinically, it manifests as bilateral yellow-orange, thickened, indurated eyelid plaques that can extend to the extraocular muscles or lacrimal glands. As the name suggests, this entity is associated with adult-onset asthma or rhinosinusitis as well as lymphadenopathy and extension into the orbital fat.1
When patients present with periorbital lesions and optic symptoms such as visual disturbances, tearing, and/ or a foreign body sensation in the eyes, a work-up should be performed to rule out infiltration of orbital adnexal structures and other organ systems, as AAPOX can be associated with IgG4-related disease. Histologically, adult xanthogranulomatous diseases are characterized by sheets of foamy histiocytes accompanied by variable numbers of lymphoid aggregates, plasma cells, and Touton giant cells. These infiltrating xanthoma cells appear as a garland or wreathlike nuclei surrounded by foamy cytoplasm. Oil-red O staining of frozen sections confirms the lipid content of the xanthoma cells.2 Immunohistochemically, the foamy histiocytes are strongly positive for CD68, CD163, and factor XIIIa but usually are negative for S100, CD1a and Birbeck granules.3 More distinctively, AAPOX is associated with prominent lymphoid aggregates containing reactive germinal centers.1
Given that AAPOX is a systemic multiorgan disease, local therapies such as surgical debulking or intralesional corticosteroids generally are insufficient to address the underlying pathology and therefore necessitate systemic, often multimodal, treatment within a multidisciplinary framework.1,4 Systemic corticosteroids remain first-line therapy, with steroid-sparing agents (eg, methotrexate, azathioprine, cyclophosphamide) used in refractory cases or to reduce steroid dependence.4,5 Rituximab has demonstrated efficacy in AAPOX, further highlighting the association between AAPOX and IgG4-related disease.5 Inebilizumab, a B-cell–depleting monoclonal antibody targeting CD19 and approved for the treatment of IgG4- related disease, represents a theoretically promising therapeutic option; however, additional studies are needed to establish its efficacy and safety in AAPOX.5
Necrobiotic xanthogranuloma is another xanthogranulomatous disorder that manifests as yellow-orange papules or nodules that gradually form infiltrative plaques. Scarring and ulceration can occur in 40% to 50% of patients.6 The most common site of involvement is the periorbital area, affecting 80% of cases, often resulting in ophthalmologic complications.6 Necrobiotic xanthogranuloma lesions also can involve the trunk, arms, and legs. Extracutaneous sites include the lungs, myocardium, larynx, pharynx, skeletal muscles, kidneys, ovaries, and intestines. The prognosis of NXG is poor due to associated hematologic malignancies such as multiple myeloma and lymphoma. About 80% of patients have a serum monoclonal gammopathy.6 Histologically, NXG shows features overlapping with other xanthogranulomatous disorders, such as foamy histiocytes, multinucleated giant cells, Touton giant cells, and nodular lymphocytic aggregates6,7; however, broad zones of necrobiosis are a distinct histologic finding in NXG that helps differentiate it from other xanthogranulomatous disorders (Figure 1).6
Xanthelasma manifests as yellow plaques on the medial upper and lower eyelids and lateral canthi resulting from accumulation of cholesterol-rich material in the skin, soft tissue, and sometimes the tendons. Fifty percent of patients have a primary or secondary lipid disorder such as familial dyslipidemia, thyroid disease, diabetes mellitus, or primary biliary cholangitis.8 Histologically, xanthelasmas demonstrate lipid-laden foamy histiocytes in the superficial dermis (Figure 2).8 Despite some clinical overlap in our case, the depth of orbital involvement and supportive systemic and histopathologic findings (including Touton giant cells) supported the diagnosis of AAPOX rather than xanthelasma.
Juvenile xanthogranuloma manifests as solitary to multiple firm, yellow-orange papules or nodules on the face, neck, and upper torso. The lesions develop in early childhood, with 75% of lesions appearing in the first year of life, but rarely it may develop in adulthood.9 The most common extracutaneous manifestation involves ocular structures, most frequently the iris, followed by the lungs. Cutaneous lesions usually are asymptomatic and involute over the span of a few years. Ocular lesions can result in blindness, and juvenile xanthogranuloma also has been associated with neurofibromatosis type 1 and juvenile chronic myelogenous leukemia.9 The histopathology of juvenile xanthogranuloma often will show a dense histiocytic infiltrate in the dermis with blunting of the overlying rete ridges admixed with lymphocytes, plasma cells, and eosinophils. In the more mature phase, foam cells, foreign body giant cells, and Touton giant cells predominate. Touton giant cells have a garlandlike appearance (Figure 3).9
Reticulohistiocytoma (or solitary epithelioid histiocytoma) and multicentric reticulohistiocytosis are rare histiocytic proliferations. Multicentric reticulohistiocytosis refers to a systemic disease with arthropathy and multiple cutaneous histiocytic lesions located on acral sites and the face. Solitary reticulohistiocytoma manifests as papules or nodules found in many body locations, such as the trunk, arms, and legs. The lesions are uncommon on the face, which almost always is involved in multicentric reticulohistiocytosis. Solitary reticulohistiocytomas tend not to recur once excised and do not demonstrate systemic involvement. Histologically, the lesions demonstrate large eosinophilic epithelioid histiocytes with abundant glassy cytoplasm (Figure 4). Some of the epithelioid histiocytes are multinucleated, and immunophenotyping will show positivity for lysozyme, CD68, and CD163.10
THE DIAGNOSIS: Adult-Onset Asthma With Periocular Xanthogranuloma
In the context of pre-existing sinonasal disease and features consistent with adult-onset asthma, the constellation of clinical findings including linear periorbital yellow-orange plaques, imaging demonstrating extension of xanthogranulomatous lesions into the orbital fat, histopathologic features, and serologic abnormalities including elevated IgG4 levels supported a diagnosis of adult-onset asthma with periocular xanthogranuloma (AAPOX).
Adult-onset xanthogranuloma is a non–Langerhans cell histiocytosis (historically classified as type II) within the group of adult orbital xanthogranulomatous diseases resulting from infiltration and proliferation of histiocytes in the orbital and ocular adnexal structures and eyelids. Adult orbital xanthogranulomatous diseases are classified as 4 distinct conditions: adult-onset xanthogranuloma, AAPOX, Erdheim-Chester disease, and necrobiotic xanthogranuloma (NXG). Erdheim-Chester disease is the most severe among this group and often is fatal due to infiltration of the xanthogranulomas into multiple organ systems and tissues, including the long bones, heart, lungs, and retroperitoneum. Neurologic symptoms such as incoordination can occur. Adult-onset xanthogranuloma manifests as an isolated cutaneous lesion without systemic involvement. This entity often is self-limited and does not require aggressive treatment. Adult-onset asthma with periocular xanthogranuloma affects more males than females.1 Clinically, it manifests as bilateral yellow-orange, thickened, indurated eyelid plaques that can extend to the extraocular muscles or lacrimal glands. As the name suggests, this entity is associated with adult-onset asthma or rhinosinusitis as well as lymphadenopathy and extension into the orbital fat.1
When patients present with periorbital lesions and optic symptoms such as visual disturbances, tearing, and/ or a foreign body sensation in the eyes, a work-up should be performed to rule out infiltration of orbital adnexal structures and other organ systems, as AAPOX can be associated with IgG4-related disease. Histologically, adult xanthogranulomatous diseases are characterized by sheets of foamy histiocytes accompanied by variable numbers of lymphoid aggregates, plasma cells, and Touton giant cells. These infiltrating xanthoma cells appear as a garland or wreathlike nuclei surrounded by foamy cytoplasm. Oil-red O staining of frozen sections confirms the lipid content of the xanthoma cells.2 Immunohistochemically, the foamy histiocytes are strongly positive for CD68, CD163, and factor XIIIa but usually are negative for S100, CD1a and Birbeck granules.3 More distinctively, AAPOX is associated with prominent lymphoid aggregates containing reactive germinal centers.1
Given that AAPOX is a systemic multiorgan disease, local therapies such as surgical debulking or intralesional corticosteroids generally are insufficient to address the underlying pathology and therefore necessitate systemic, often multimodal, treatment within a multidisciplinary framework.1,4 Systemic corticosteroids remain first-line therapy, with steroid-sparing agents (eg, methotrexate, azathioprine, cyclophosphamide) used in refractory cases or to reduce steroid dependence.4,5 Rituximab has demonstrated efficacy in AAPOX, further highlighting the association between AAPOX and IgG4-related disease.5 Inebilizumab, a B-cell–depleting monoclonal antibody targeting CD19 and approved for the treatment of IgG4- related disease, represents a theoretically promising therapeutic option; however, additional studies are needed to establish its efficacy and safety in AAPOX.5
Necrobiotic xanthogranuloma is another xanthogranulomatous disorder that manifests as yellow-orange papules or nodules that gradually form infiltrative plaques. Scarring and ulceration can occur in 40% to 50% of patients.6 The most common site of involvement is the periorbital area, affecting 80% of cases, often resulting in ophthalmologic complications.6 Necrobiotic xanthogranuloma lesions also can involve the trunk, arms, and legs. Extracutaneous sites include the lungs, myocardium, larynx, pharynx, skeletal muscles, kidneys, ovaries, and intestines. The prognosis of NXG is poor due to associated hematologic malignancies such as multiple myeloma and lymphoma. About 80% of patients have a serum monoclonal gammopathy.6 Histologically, NXG shows features overlapping with other xanthogranulomatous disorders, such as foamy histiocytes, multinucleated giant cells, Touton giant cells, and nodular lymphocytic aggregates6,7; however, broad zones of necrobiosis are a distinct histologic finding in NXG that helps differentiate it from other xanthogranulomatous disorders (Figure 1).6
Xanthelasma manifests as yellow plaques on the medial upper and lower eyelids and lateral canthi resulting from accumulation of cholesterol-rich material in the skin, soft tissue, and sometimes the tendons. Fifty percent of patients have a primary or secondary lipid disorder such as familial dyslipidemia, thyroid disease, diabetes mellitus, or primary biliary cholangitis.8 Histologically, xanthelasmas demonstrate lipid-laden foamy histiocytes in the superficial dermis (Figure 2).8 Despite some clinical overlap in our case, the depth of orbital involvement and supportive systemic and histopathologic findings (including Touton giant cells) supported the diagnosis of AAPOX rather than xanthelasma.
Juvenile xanthogranuloma manifests as solitary to multiple firm, yellow-orange papules or nodules on the face, neck, and upper torso. The lesions develop in early childhood, with 75% of lesions appearing in the first year of life, but rarely it may develop in adulthood.9 The most common extracutaneous manifestation involves ocular structures, most frequently the iris, followed by the lungs. Cutaneous lesions usually are asymptomatic and involute over the span of a few years. Ocular lesions can result in blindness, and juvenile xanthogranuloma also has been associated with neurofibromatosis type 1 and juvenile chronic myelogenous leukemia.9 The histopathology of juvenile xanthogranuloma often will show a dense histiocytic infiltrate in the dermis with blunting of the overlying rete ridges admixed with lymphocytes, plasma cells, and eosinophils. In the more mature phase, foam cells, foreign body giant cells, and Touton giant cells predominate. Touton giant cells have a garlandlike appearance (Figure 3).9
Reticulohistiocytoma (or solitary epithelioid histiocytoma) and multicentric reticulohistiocytosis are rare histiocytic proliferations. Multicentric reticulohistiocytosis refers to a systemic disease with arthropathy and multiple cutaneous histiocytic lesions located on acral sites and the face. Solitary reticulohistiocytoma manifests as papules or nodules found in many body locations, such as the trunk, arms, and legs. The lesions are uncommon on the face, which almost always is involved in multicentric reticulohistiocytosis. Solitary reticulohistiocytomas tend not to recur once excised and do not demonstrate systemic involvement. Histologically, the lesions demonstrate large eosinophilic epithelioid histiocytes with abundant glassy cytoplasm (Figure 4). Some of the epithelioid histiocytes are multinucleated, and immunophenotyping will show positivity for lysozyme, CD68, and CD163.10
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463. doi:10.1016 /j.det.2015.03.010
- Mandic` JJ, Bakula M, Šklebar LK, et al. Histiocytosis and adult-onset orbital xanthogranuloma in 2023: a review of the literature and mini case series. Int Ophthalmol. 2024;44:301. doi:10.1007/s10792-024-03181-y
- Campochiaro C, Tomelleri A, Cavalli G, et al. Erdheim-Chester disease. Eur J Intern Med. 2015;26:223-229. doi: 10.1016/j.ejim.2015.03.004
- Detiger SE, Hötte GJ, Verdijk RM, et al. Adult orbital xanthogranuloma: long-term follow-up of treated cases. Eye (Lond). 2023;37:2475-2481. doi: 10.1038/s41433-022-02357-z
- Stone JH, Khosroshahi A, Zhang W, et al. Inebilizumab for treatment of IgG4-related disease. N Engl J Med. 2025;392:1168-1177. doi:10.1056 /NEJMoa2409712
- Wood AJ, Wagner MVU, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279–284. doi:10.1001 /archdermatol.2008.583
- Schadt C, Jacobsen E. Necrobiotic xanthogranuloma. In: Connor RF, ed. UpToDate. Wolters Kluwer.
- Al Aboud AM, Shah SS, Blair K, et al. Xanthelasma palpebrarum. StatPearls [Internet]. StatPearls Publishing; 2024. Updated March 1, 2024. Accessed February 10, 2026. https://www.ncbi.nlm .nih.gov/books/NBK531501/
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated August 8, 2023. Accessed February 10, 2026. https://www.ncbi.nlm.nih .gov/books/NBK526103/
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528. doi:10.1097/00000478 -200604000-00014
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463. doi:10.1016 /j.det.2015.03.010
- Mandic` JJ, Bakula M, Šklebar LK, et al. Histiocytosis and adult-onset orbital xanthogranuloma in 2023: a review of the literature and mini case series. Int Ophthalmol. 2024;44:301. doi:10.1007/s10792-024-03181-y
- Campochiaro C, Tomelleri A, Cavalli G, et al. Erdheim-Chester disease. Eur J Intern Med. 2015;26:223-229. doi: 10.1016/j.ejim.2015.03.004
- Detiger SE, Hötte GJ, Verdijk RM, et al. Adult orbital xanthogranuloma: long-term follow-up of treated cases. Eye (Lond). 2023;37:2475-2481. doi: 10.1038/s41433-022-02357-z
- Stone JH, Khosroshahi A, Zhang W, et al. Inebilizumab for treatment of IgG4-related disease. N Engl J Med. 2025;392:1168-1177. doi:10.1056 /NEJMoa2409712
- Wood AJ, Wagner MVU, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279–284. doi:10.1001 /archdermatol.2008.583
- Schadt C, Jacobsen E. Necrobiotic xanthogranuloma. In: Connor RF, ed. UpToDate. Wolters Kluwer.
- Al Aboud AM, Shah SS, Blair K, et al. Xanthelasma palpebrarum. StatPearls [Internet]. StatPearls Publishing; 2024. Updated March 1, 2024. Accessed February 10, 2026. https://www.ncbi.nlm .nih.gov/books/NBK531501/
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated August 8, 2023. Accessed February 10, 2026. https://www.ncbi.nlm.nih .gov/books/NBK526103/
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528. doi:10.1097/00000478 -200604000-00014
Thick Yellow Plaques on the Eyelids
Thick Yellow Plaques on the Eyelids
A 54-year-old man presented to the dermatology department for evaluation of enlarging lesions on the eyelids of 18 months’ duration causing tearing, dryness, and heaviness. The patient’s medical history was positive for hyperlipidemia, chronic rhinosinusitis, and mild asthma diagnosed in adulthood. A review of systems was negative for bone pain, polyuria, polydipsia, dysuria, hematuria, decreased coordination, chest pain, palpitations, abdominal pain, easy bruising, and jaundice. Laboratory testing revealed elevated IgG4 levels and a slight increase in gamma globulins on serum protein electrophoresis, with no evidence of paraproteinemia. Liver and kidney function test results were within normal limits. Magnetic resonance imaging of the orbits revealed bilateral superolateral intraorbital masses within the extraconal fat that were displacing the superior and lateral rectus muscles. No intraconal masses were identified. Due to bilateral eyelid ptosis, the patient underwent debulking by oculoplastic surgery, and the tissue was submitted for histologic examination.
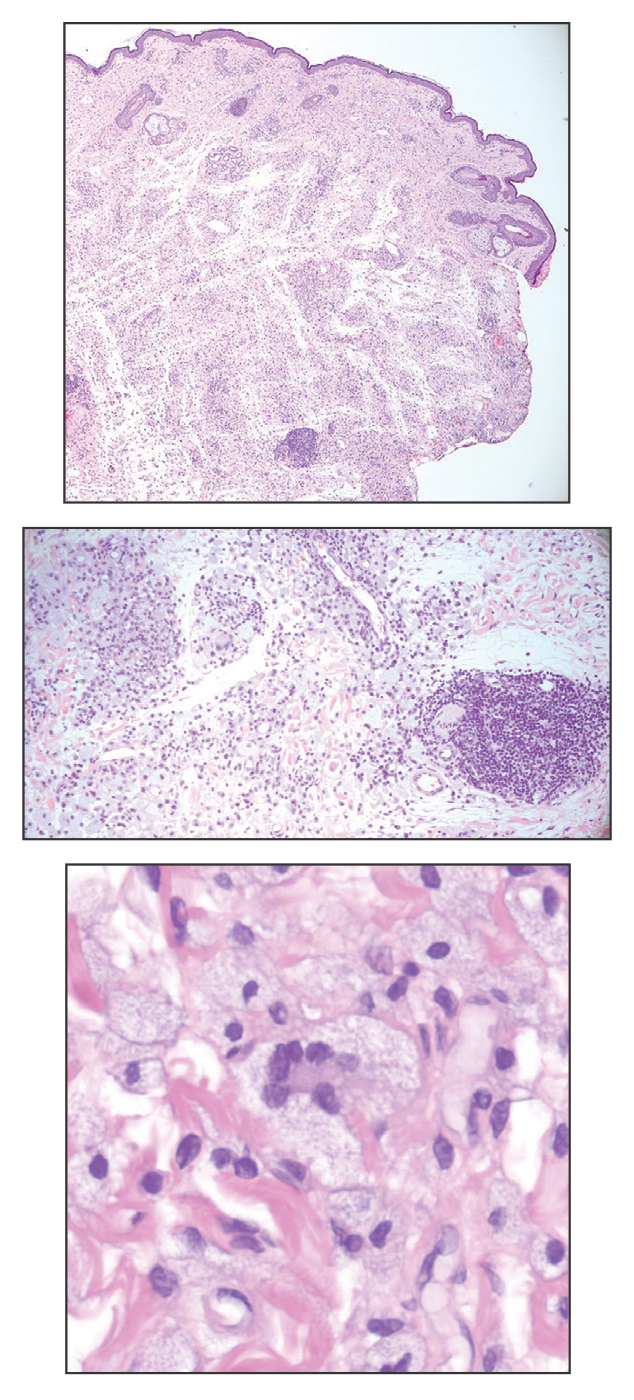

Verrucous Nodule on the Cheek
Verrucous Nodule on the Cheek
THE DIAGNOSIS: Pilomatrix Carcinoma
Histopathology revealed poorly circumscribed dermal nodules composed of large pleomorphic and highly atypical basaloid cells as well as increased mitoses. Foci of central necrosis admixed with keratinized cells containing pale eosinophilic cytoplasm and faint nuclear outlines without nuclei also were present. Immunohistochemistry for p63 was positive, while adipophilin, BerEP4, cytokeratin 20, and carcinoembryonic antigen were negative. Tumor cells also demonstrated strong and diffuse nuclear and cytoplasmic β-catenin staining, leading to a diagnosis of pilomatrix carcinoma (PC). The tumor was treated with Mohs micrographic surgery, and the patient was subsequently lost to follow-up.
Pilomatrix carcinoma, historically known as calcifying epitheliocarcinoma of Malherbe, is a rare, locally aggressive, low-grade adnexal tumor of germinative hair follicle matrix cell origin. Similar to its benign pilomatrixoma counterpart, malignant PC manifests as a firm, nontender, asymptomatic nodule most commonly (but not exclusively) manifesting in the head and neck region; however, in contrast to benign pilomatrixoma, PC is a rapidly growing tumor with a high rate of local recurrence after surgical excision and has the potential to become metastatic.1
Pilomatrix carcinoma occurs most often in the fifth through seventh decades of life, with a male-to-female ratio of approximately 1.3:1.1 Due to its rarity, PC management guidelines are not well defined. Histologically, PC will show asymmetry, poor circumscription, and an infiltrative growth pattern at low power. Pilomatrix carcinoma is further characterized by the presence of nodules of atypical basaloid cells demonstrating pleomorphism and nuclear hyperchromatism, increased mitotic index, and the presence of ghost cells (Figure 1).2 Ghost cells are evidence of matrical differentiation. The transition from basaloid to ghost cells may be abrupt. Intralesional calcification is possible but less common.2,3 The tumor nodules can be surrounded by a dense desmoplastic stroma with a predominantly lymphohistiocytic infiltrate.2 Immunohistochemical stains that support a PC diagnosis include lymphoid enhancer-binding factor 1 (LEF1), Ki-67, β-catenin, and p53. Although not specific for malignancy, nuclear LEF1 helps confirm matrical (hair matrix) differentiation.4 Pilomatrix carcinomas show a markedly elevated Ki-67 proliferation marker, reflecting high mitotic activity.5 While benign pilomatricoma may show patchy or minimal p53 staining, PC can demonstrate diffuse strong p53 positivity, consistent with the p53 pathway dysregulation seen in malignant matrical neoplasms.6 Most classically, PC stains strongly positive for nuclear and cytoplasmic β-catenin. Aberrant β-catenin disrupting normal Wnt/β-catenin/Tcf-Lef pathway regulation, which ultimately promotes cellular differentiation and division, is proposed to play a role in tumorigenesis.6,7
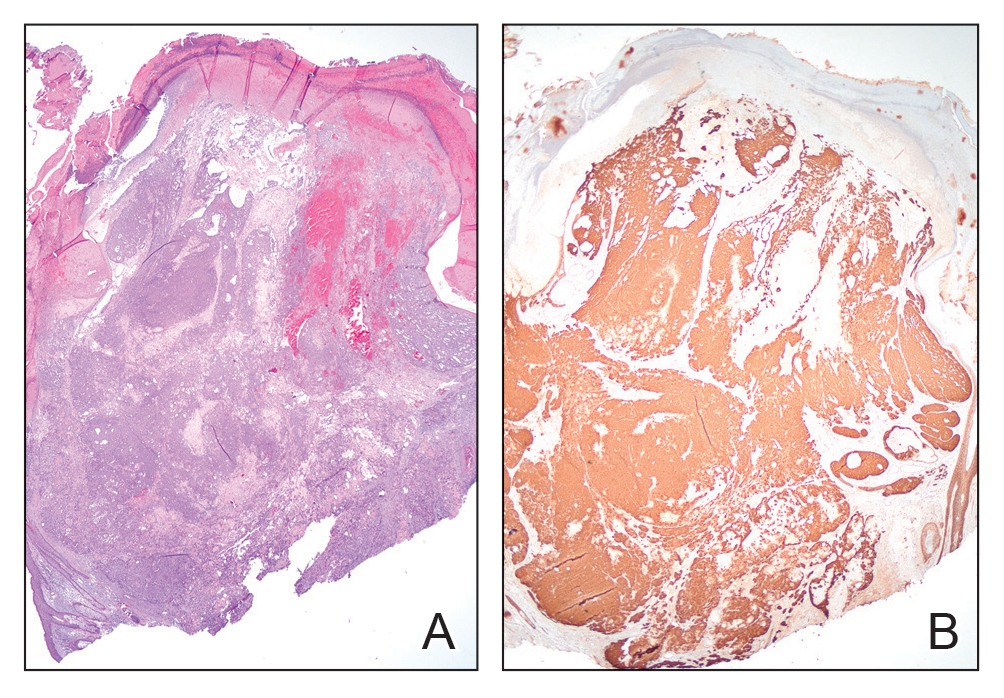
The differential diagnoses for PC include basal cell carcinoma (BCC), Merkel cell carcinoma, moderately differentiated squamous cell carcinoma, and porocarcinoma. Basal cell carcinoma is a common tumor occurring on the head and neck regions that typically manifests as a slow-growing, flesh-colored, pink or pigmented papule, plaque, or nodule. Spontaneous bleeding or ulceration can sometimes occur. Basal cell carcinoma has various histologic subtypes, with tumors potentially exhibiting more than one histologic pattern. Common features of BCC include basaloid nodules arising from the epidermis, peripheral palisading, clefting artifacts, and a myxoid stroma (Figure 2).8 These features help distinguish BCC from PC histologically, although there is a rare matrical BCC subtype with a handful of reported cases expressing features of both.9 Staining can be a helpful differentiator as pancellular staining for LEF1, and β-catenin is exclusively observed in the pilomatrixoma and PC, in contrast to BCC, which shows staining confined to focal germinative matrix cell nests.10
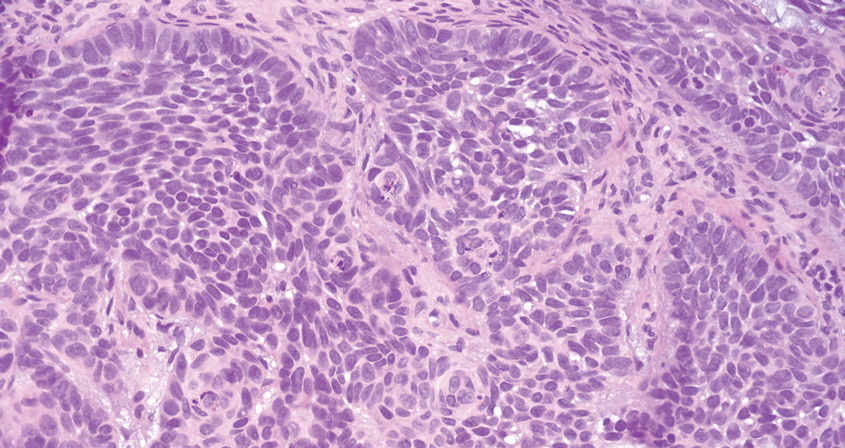
Squamous cell carcinoma (SCC) also commonly manifests clinically in the head and neck region and is associated with sun damage. Squamous cell carcinoma can be histologically graded based on cellular differentiation, from well differentiated to poorly differentiated subtypes. Moderately differentiated SCC is characterized histologically by reduced keratinization, frequent loss of intercellular bridges, and enlarged pleomorphic cells demonstrating a high degree of atypia and frequent abnormal mitoses (Figure 3).11 Similar to PC, moderately differentiated SCC also may comprise basaloid cells but lacks shadow cells. Further distinction from PC can be made through immunohistochemistry. Expression of p63, p40, MNF116, and CK903 expression help identify the squamous origin of the tumor and are useful in the diagnosis of less-differentiated SCC.12 In addition, SCC does not show matrical differentiation (ghost cells).
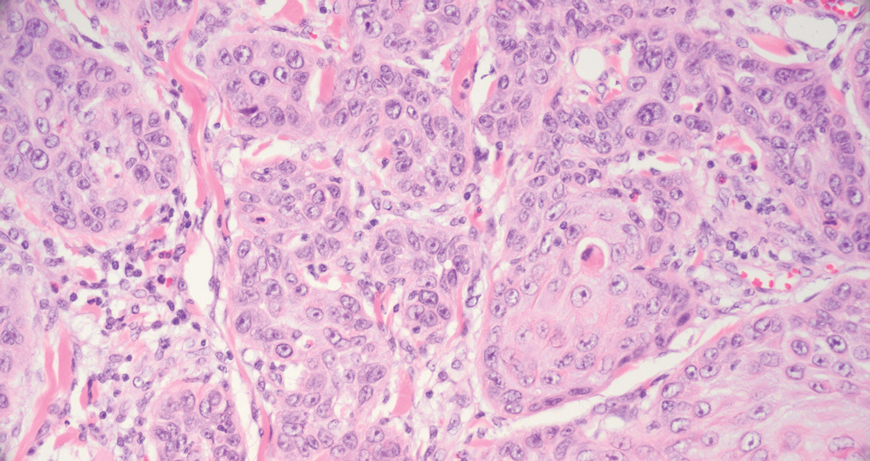
Merkel cell carcinoma is a rare and aggressive skin cancer that manifests as a rapidly growing, sometimes ulcerating nodule or plaque with a predilection for sun‐exposed areas of the skin. Merkel cell carcinoma is characterized by neuroendocrine differentiation. The gold standard diagnostic modalities are histopathology and immunohistochemistry. Characteristic histopathologic findings include diffuse atypical blue cells with large nuclei, minimal cytoplasm, and frequent mitoses (Figure 4).13,14 Staining with cytokeratin 20 and neuroendocrine markers such as synaptophysin and chromogranin A on immunohistochemistry supports the diagnosis, as does positive AE1/3; neuron-specific enolase and epithelial membrane antigen; and negative S100, carcinoembryonic antigen, and leukocyte common antigen staining.13,14
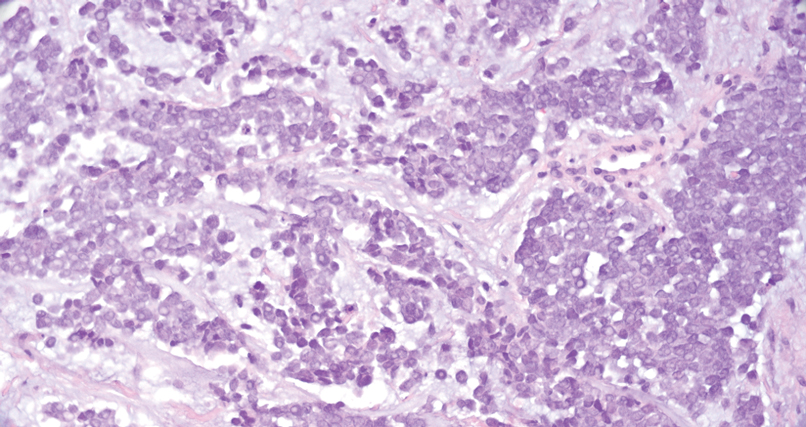
Porocarcinoma is a rare malignant growth arising from the cutaneous intraepidermal ducts of the sweat glands. Porocarcinomas may originate from benign eccrine poromas, but the etiology remains poorly understood. Clinically, porocarcinoma manifests as a flesh-colored, erythematous, or violaceous firm, single, dome-shaped papule or nodule that can ulcerate and may be asymptomatic, itchy, or painful.15 Porocarcinoma poses a diagnostic challenge due to the variability of both its clinical presentation and histopathologic findings. The histology often resembles that of cutaneous squamous cell carcinoma or poroma. On hematoxylin and eosin staining, porocarcinoma is characterized by poromatous basaloid cells with cytologic atypia and ductal differentiation. Common histopathologic features include formation of mature ducts lined with cuboidal epithelial cells, foci of necrosis, intracytoplasmic lumina, and squamous differentiation (Figure 5).15 Carcinoembryonic antigen and epithelial membrane antigen immunohistochemical staining to identify ductal structures may help to distinguish porocarcinoma from other tumors. Cluster of Differentiation 117/c-KIT, cytokeratin 19, and BerEP4 positivity also have been shown to be useful in diagnosing porocarcinoma. CD117/c-KIT highlights eccrine ductal differentiation16; CK19 supports adnexal ductal differentiation and often is increased in malignant poroid neoplasms17; and BerEP4, although classically used for BCC diagnosis, also may be positive in porocarcinoma, particularly in ductal areas, and can support the diagnosis.18
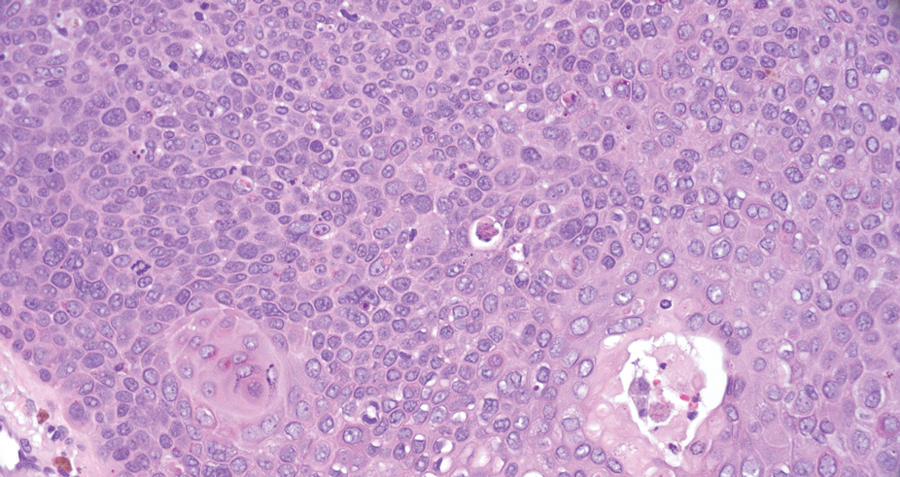
- Toffoli L, Bazzacco G, Conforti C, et al. Pilomatrix carcinoma: report of two cases of the head and review of the literature. Curr Oncol. 2023;30:1426-1438. doi:10.3390/curroncol30020109
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.E2. doi:10.1016/j.jaad.2014.02.042
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38. doi:10.1111/cup.13046
- Reymundo-Jiménez A, Martos-Cabrera L, Muñoz-Hernández P, et al. Usefulness of LEF-1 immunostaining for the diagnosis of matricoma. Actas Dermosifiliogr. 2022;113:T907-T910. doi:10.1016/j.ad.2022.08.003
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498. doi:10.1002/1097-0142(19930415)71:8<2491 ::aid-cncr2820710811>3.0.co;2-i
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding β-catenin. J Cutan Pathol. 2005;32:148-157. doi:10.1111/j.0303-6987.2005.00267.x
- Abula A, Ma SQ, Wang S, et al. Case report: Pilomatrix carcinoma with PDL1 expression and CDKN2A aberrant. Front Immunol. 2024;15. doi:10.3389/fimmu.2024.1337400
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kanitakis J, Ducroux E, Hoelt P, et al. Basal-cell carcinoma with matrical differentiation: report of a new case in a renal-transplant recipient and literature review. Am J Dermatopathol. 2018;40:E115-E118. doi:10.1097 /DAD.0000000000001146
- White C, Farsi M, Esguerra D, et al. Not your average skin cancer: a rare case of pilomatrix carcinoma. J Clin Aesthet Dermatol. 2020; 13:40-42.
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2010;2011:210813. doi:10.1155/2011/210813
- Balas¸escu E, Gheorghe AC, Moroianu A, et al. Role of immunohistochemistry in the diagnosis and staging of cutaneous squamouscell carcinomas (review). Exp Ther Med. 2022;23:383. doi:10.3892 /etm.2022.11308
- Zhang Z, Shi W, Zhang R. Facial Merkel cell carcinoma in a 92-year-old man: a case report. Clin Case Rep. 2024;12:E9523. doi:10.1002/ccr3.9523
- Rapini R. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Miyamoto K, Yanagi T, Maeda T, et al. Diagnosis and management of porocarcinoma. Cancers. 2022;14:5232. doi:10.3390 /cancers14215232
- Goto K. Immunohistochemistry for CD117 (KIT) is effective in distinguishing cutaneous adnexal tumors with apocrine/eccrine or sebaceous differentiation from other epithelial tumors of the skin. J Cutan Pathol. 2015;42:480-488. doi:10.1111/cup.12492
- Requena L, Sangüeza O. General principles for the histopathologic diagnosis of neoplasms with eccrine and apocrine differentiation. Classification and histopathologic criteria for eccrine and apocrine differentiation. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:19-24. doi:10.1007/978- 3-319-45704-8_2
- Huet P, Dandurand M, Pignodel C, et al. Metastasizing eccrine porocarcinoma: report of a case and review of the literature. J Am Acad Dermatol. 1996;35:860-864. doi:10.1016/s0190-9622(96)90105-x
THE DIAGNOSIS: Pilomatrix Carcinoma
Histopathology revealed poorly circumscribed dermal nodules composed of large pleomorphic and highly atypical basaloid cells as well as increased mitoses. Foci of central necrosis admixed with keratinized cells containing pale eosinophilic cytoplasm and faint nuclear outlines without nuclei also were present. Immunohistochemistry for p63 was positive, while adipophilin, BerEP4, cytokeratin 20, and carcinoembryonic antigen were negative. Tumor cells also demonstrated strong and diffuse nuclear and cytoplasmic β-catenin staining, leading to a diagnosis of pilomatrix carcinoma (PC). The tumor was treated with Mohs micrographic surgery, and the patient was subsequently lost to follow-up.
Pilomatrix carcinoma, historically known as calcifying epitheliocarcinoma of Malherbe, is a rare, locally aggressive, low-grade adnexal tumor of germinative hair follicle matrix cell origin. Similar to its benign pilomatrixoma counterpart, malignant PC manifests as a firm, nontender, asymptomatic nodule most commonly (but not exclusively) manifesting in the head and neck region; however, in contrast to benign pilomatrixoma, PC is a rapidly growing tumor with a high rate of local recurrence after surgical excision and has the potential to become metastatic.1
Pilomatrix carcinoma occurs most often in the fifth through seventh decades of life, with a male-to-female ratio of approximately 1.3:1.1 Due to its rarity, PC management guidelines are not well defined. Histologically, PC will show asymmetry, poor circumscription, and an infiltrative growth pattern at low power. Pilomatrix carcinoma is further characterized by the presence of nodules of atypical basaloid cells demonstrating pleomorphism and nuclear hyperchromatism, increased mitotic index, and the presence of ghost cells (Figure 1).2 Ghost cells are evidence of matrical differentiation. The transition from basaloid to ghost cells may be abrupt. Intralesional calcification is possible but less common.2,3 The tumor nodules can be surrounded by a dense desmoplastic stroma with a predominantly lymphohistiocytic infiltrate.2 Immunohistochemical stains that support a PC diagnosis include lymphoid enhancer-binding factor 1 (LEF1), Ki-67, β-catenin, and p53. Although not specific for malignancy, nuclear LEF1 helps confirm matrical (hair matrix) differentiation.4 Pilomatrix carcinomas show a markedly elevated Ki-67 proliferation marker, reflecting high mitotic activity.5 While benign pilomatricoma may show patchy or minimal p53 staining, PC can demonstrate diffuse strong p53 positivity, consistent with the p53 pathway dysregulation seen in malignant matrical neoplasms.6 Most classically, PC stains strongly positive for nuclear and cytoplasmic β-catenin. Aberrant β-catenin disrupting normal Wnt/β-catenin/Tcf-Lef pathway regulation, which ultimately promotes cellular differentiation and division, is proposed to play a role in tumorigenesis.6,7
The differential diagnoses for PC include basal cell carcinoma (BCC), Merkel cell carcinoma, moderately differentiated squamous cell carcinoma, and porocarcinoma. Basal cell carcinoma is a common tumor occurring on the head and neck regions that typically manifests as a slow-growing, flesh-colored, pink or pigmented papule, plaque, or nodule. Spontaneous bleeding or ulceration can sometimes occur. Basal cell carcinoma has various histologic subtypes, with tumors potentially exhibiting more than one histologic pattern. Common features of BCC include basaloid nodules arising from the epidermis, peripheral palisading, clefting artifacts, and a myxoid stroma (Figure 2).8 These features help distinguish BCC from PC histologically, although there is a rare matrical BCC subtype with a handful of reported cases expressing features of both.9 Staining can be a helpful differentiator as pancellular staining for LEF1, and β-catenin is exclusively observed in the pilomatrixoma and PC, in contrast to BCC, which shows staining confined to focal germinative matrix cell nests.10
Squamous cell carcinoma (SCC) also commonly manifests clinically in the head and neck region and is associated with sun damage. Squamous cell carcinoma can be histologically graded based on cellular differentiation, from well differentiated to poorly differentiated subtypes. Moderately differentiated SCC is characterized histologically by reduced keratinization, frequent loss of intercellular bridges, and enlarged pleomorphic cells demonstrating a high degree of atypia and frequent abnormal mitoses (Figure 3).11 Similar to PC, moderately differentiated SCC also may comprise basaloid cells but lacks shadow cells. Further distinction from PC can be made through immunohistochemistry. Expression of p63, p40, MNF116, and CK903 expression help identify the squamous origin of the tumor and are useful in the diagnosis of less-differentiated SCC.12 In addition, SCC does not show matrical differentiation (ghost cells).
Merkel cell carcinoma is a rare and aggressive skin cancer that manifests as a rapidly growing, sometimes ulcerating nodule or plaque with a predilection for sun‐exposed areas of the skin. Merkel cell carcinoma is characterized by neuroendocrine differentiation. The gold standard diagnostic modalities are histopathology and immunohistochemistry. Characteristic histopathologic findings include diffuse atypical blue cells with large nuclei, minimal cytoplasm, and frequent mitoses (Figure 4).13,14 Staining with cytokeratin 20 and neuroendocrine markers such as synaptophysin and chromogranin A on immunohistochemistry supports the diagnosis, as does positive AE1/3; neuron-specific enolase and epithelial membrane antigen; and negative S100, carcinoembryonic antigen, and leukocyte common antigen staining.13,14
Porocarcinoma is a rare malignant growth arising from the cutaneous intraepidermal ducts of the sweat glands. Porocarcinomas may originate from benign eccrine poromas, but the etiology remains poorly understood. Clinically, porocarcinoma manifests as a flesh-colored, erythematous, or violaceous firm, single, dome-shaped papule or nodule that can ulcerate and may be asymptomatic, itchy, or painful.15 Porocarcinoma poses a diagnostic challenge due to the variability of both its clinical presentation and histopathologic findings. The histology often resembles that of cutaneous squamous cell carcinoma or poroma. On hematoxylin and eosin staining, porocarcinoma is characterized by poromatous basaloid cells with cytologic atypia and ductal differentiation. Common histopathologic features include formation of mature ducts lined with cuboidal epithelial cells, foci of necrosis, intracytoplasmic lumina, and squamous differentiation (Figure 5).15 Carcinoembryonic antigen and epithelial membrane antigen immunohistochemical staining to identify ductal structures may help to distinguish porocarcinoma from other tumors. Cluster of Differentiation 117/c-KIT, cytokeratin 19, and BerEP4 positivity also have been shown to be useful in diagnosing porocarcinoma. CD117/c-KIT highlights eccrine ductal differentiation16; CK19 supports adnexal ductal differentiation and often is increased in malignant poroid neoplasms17; and BerEP4, although classically used for BCC diagnosis, also may be positive in porocarcinoma, particularly in ductal areas, and can support the diagnosis.18
THE DIAGNOSIS: Pilomatrix Carcinoma
Histopathology revealed poorly circumscribed dermal nodules composed of large pleomorphic and highly atypical basaloid cells as well as increased mitoses. Foci of central necrosis admixed with keratinized cells containing pale eosinophilic cytoplasm and faint nuclear outlines without nuclei also were present. Immunohistochemistry for p63 was positive, while adipophilin, BerEP4, cytokeratin 20, and carcinoembryonic antigen were negative. Tumor cells also demonstrated strong and diffuse nuclear and cytoplasmic β-catenin staining, leading to a diagnosis of pilomatrix carcinoma (PC). The tumor was treated with Mohs micrographic surgery, and the patient was subsequently lost to follow-up.
Pilomatrix carcinoma, historically known as calcifying epitheliocarcinoma of Malherbe, is a rare, locally aggressive, low-grade adnexal tumor of germinative hair follicle matrix cell origin. Similar to its benign pilomatrixoma counterpart, malignant PC manifests as a firm, nontender, asymptomatic nodule most commonly (but not exclusively) manifesting in the head and neck region; however, in contrast to benign pilomatrixoma, PC is a rapidly growing tumor with a high rate of local recurrence after surgical excision and has the potential to become metastatic.1
Pilomatrix carcinoma occurs most often in the fifth through seventh decades of life, with a male-to-female ratio of approximately 1.3:1.1 Due to its rarity, PC management guidelines are not well defined. Histologically, PC will show asymmetry, poor circumscription, and an infiltrative growth pattern at low power. Pilomatrix carcinoma is further characterized by the presence of nodules of atypical basaloid cells demonstrating pleomorphism and nuclear hyperchromatism, increased mitotic index, and the presence of ghost cells (Figure 1).2 Ghost cells are evidence of matrical differentiation. The transition from basaloid to ghost cells may be abrupt. Intralesional calcification is possible but less common.2,3 The tumor nodules can be surrounded by a dense desmoplastic stroma with a predominantly lymphohistiocytic infiltrate.2 Immunohistochemical stains that support a PC diagnosis include lymphoid enhancer-binding factor 1 (LEF1), Ki-67, β-catenin, and p53. Although not specific for malignancy, nuclear LEF1 helps confirm matrical (hair matrix) differentiation.4 Pilomatrix carcinomas show a markedly elevated Ki-67 proliferation marker, reflecting high mitotic activity.5 While benign pilomatricoma may show patchy or minimal p53 staining, PC can demonstrate diffuse strong p53 positivity, consistent with the p53 pathway dysregulation seen in malignant matrical neoplasms.6 Most classically, PC stains strongly positive for nuclear and cytoplasmic β-catenin. Aberrant β-catenin disrupting normal Wnt/β-catenin/Tcf-Lef pathway regulation, which ultimately promotes cellular differentiation and division, is proposed to play a role in tumorigenesis.6,7
The differential diagnoses for PC include basal cell carcinoma (BCC), Merkel cell carcinoma, moderately differentiated squamous cell carcinoma, and porocarcinoma. Basal cell carcinoma is a common tumor occurring on the head and neck regions that typically manifests as a slow-growing, flesh-colored, pink or pigmented papule, plaque, or nodule. Spontaneous bleeding or ulceration can sometimes occur. Basal cell carcinoma has various histologic subtypes, with tumors potentially exhibiting more than one histologic pattern. Common features of BCC include basaloid nodules arising from the epidermis, peripheral palisading, clefting artifacts, and a myxoid stroma (Figure 2).8 These features help distinguish BCC from PC histologically, although there is a rare matrical BCC subtype with a handful of reported cases expressing features of both.9 Staining can be a helpful differentiator as pancellular staining for LEF1, and β-catenin is exclusively observed in the pilomatrixoma and PC, in contrast to BCC, which shows staining confined to focal germinative matrix cell nests.10
Squamous cell carcinoma (SCC) also commonly manifests clinically in the head and neck region and is associated with sun damage. Squamous cell carcinoma can be histologically graded based on cellular differentiation, from well differentiated to poorly differentiated subtypes. Moderately differentiated SCC is characterized histologically by reduced keratinization, frequent loss of intercellular bridges, and enlarged pleomorphic cells demonstrating a high degree of atypia and frequent abnormal mitoses (Figure 3).11 Similar to PC, moderately differentiated SCC also may comprise basaloid cells but lacks shadow cells. Further distinction from PC can be made through immunohistochemistry. Expression of p63, p40, MNF116, and CK903 expression help identify the squamous origin of the tumor and are useful in the diagnosis of less-differentiated SCC.12 In addition, SCC does not show matrical differentiation (ghost cells).
Merkel cell carcinoma is a rare and aggressive skin cancer that manifests as a rapidly growing, sometimes ulcerating nodule or plaque with a predilection for sun‐exposed areas of the skin. Merkel cell carcinoma is characterized by neuroendocrine differentiation. The gold standard diagnostic modalities are histopathology and immunohistochemistry. Characteristic histopathologic findings include diffuse atypical blue cells with large nuclei, minimal cytoplasm, and frequent mitoses (Figure 4).13,14 Staining with cytokeratin 20 and neuroendocrine markers such as synaptophysin and chromogranin A on immunohistochemistry supports the diagnosis, as does positive AE1/3; neuron-specific enolase and epithelial membrane antigen; and negative S100, carcinoembryonic antigen, and leukocyte common antigen staining.13,14
Porocarcinoma is a rare malignant growth arising from the cutaneous intraepidermal ducts of the sweat glands. Porocarcinomas may originate from benign eccrine poromas, but the etiology remains poorly understood. Clinically, porocarcinoma manifests as a flesh-colored, erythematous, or violaceous firm, single, dome-shaped papule or nodule that can ulcerate and may be asymptomatic, itchy, or painful.15 Porocarcinoma poses a diagnostic challenge due to the variability of both its clinical presentation and histopathologic findings. The histology often resembles that of cutaneous squamous cell carcinoma or poroma. On hematoxylin and eosin staining, porocarcinoma is characterized by poromatous basaloid cells with cytologic atypia and ductal differentiation. Common histopathologic features include formation of mature ducts lined with cuboidal epithelial cells, foci of necrosis, intracytoplasmic lumina, and squamous differentiation (Figure 5).15 Carcinoembryonic antigen and epithelial membrane antigen immunohistochemical staining to identify ductal structures may help to distinguish porocarcinoma from other tumors. Cluster of Differentiation 117/c-KIT, cytokeratin 19, and BerEP4 positivity also have been shown to be useful in diagnosing porocarcinoma. CD117/c-KIT highlights eccrine ductal differentiation16; CK19 supports adnexal ductal differentiation and often is increased in malignant poroid neoplasms17; and BerEP4, although classically used for BCC diagnosis, also may be positive in porocarcinoma, particularly in ductal areas, and can support the diagnosis.18
- Toffoli L, Bazzacco G, Conforti C, et al. Pilomatrix carcinoma: report of two cases of the head and review of the literature. Curr Oncol. 2023;30:1426-1438. doi:10.3390/curroncol30020109
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.E2. doi:10.1016/j.jaad.2014.02.042
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38. doi:10.1111/cup.13046
- Reymundo-Jiménez A, Martos-Cabrera L, Muñoz-Hernández P, et al. Usefulness of LEF-1 immunostaining for the diagnosis of matricoma. Actas Dermosifiliogr. 2022;113:T907-T910. doi:10.1016/j.ad.2022.08.003
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498. doi:10.1002/1097-0142(19930415)71:8<2491 ::aid-cncr2820710811>3.0.co;2-i
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding β-catenin. J Cutan Pathol. 2005;32:148-157. doi:10.1111/j.0303-6987.2005.00267.x
- Abula A, Ma SQ, Wang S, et al. Case report: Pilomatrix carcinoma with PDL1 expression and CDKN2A aberrant. Front Immunol. 2024;15. doi:10.3389/fimmu.2024.1337400
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kanitakis J, Ducroux E, Hoelt P, et al. Basal-cell carcinoma with matrical differentiation: report of a new case in a renal-transplant recipient and literature review. Am J Dermatopathol. 2018;40:E115-E118. doi:10.1097 /DAD.0000000000001146
- White C, Farsi M, Esguerra D, et al. Not your average skin cancer: a rare case of pilomatrix carcinoma. J Clin Aesthet Dermatol. 2020; 13:40-42.
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2010;2011:210813. doi:10.1155/2011/210813
- Balas¸escu E, Gheorghe AC, Moroianu A, et al. Role of immunohistochemistry in the diagnosis and staging of cutaneous squamouscell carcinomas (review). Exp Ther Med. 2022;23:383. doi:10.3892 /etm.2022.11308
- Zhang Z, Shi W, Zhang R. Facial Merkel cell carcinoma in a 92-year-old man: a case report. Clin Case Rep. 2024;12:E9523. doi:10.1002/ccr3.9523
- Rapini R. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Miyamoto K, Yanagi T, Maeda T, et al. Diagnosis and management of porocarcinoma. Cancers. 2022;14:5232. doi:10.3390 /cancers14215232
- Goto K. Immunohistochemistry for CD117 (KIT) is effective in distinguishing cutaneous adnexal tumors with apocrine/eccrine or sebaceous differentiation from other epithelial tumors of the skin. J Cutan Pathol. 2015;42:480-488. doi:10.1111/cup.12492
- Requena L, Sangüeza O. General principles for the histopathologic diagnosis of neoplasms with eccrine and apocrine differentiation. Classification and histopathologic criteria for eccrine and apocrine differentiation. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:19-24. doi:10.1007/978- 3-319-45704-8_2
- Huet P, Dandurand M, Pignodel C, et al. Metastasizing eccrine porocarcinoma: report of a case and review of the literature. J Am Acad Dermatol. 1996;35:860-864. doi:10.1016/s0190-9622(96)90105-x
- Toffoli L, Bazzacco G, Conforti C, et al. Pilomatrix carcinoma: report of two cases of the head and review of the literature. Curr Oncol. 2023;30:1426-1438. doi:10.3390/curroncol30020109
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.E2. doi:10.1016/j.jaad.2014.02.042
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38. doi:10.1111/cup.13046
- Reymundo-Jiménez A, Martos-Cabrera L, Muñoz-Hernández P, et al. Usefulness of LEF-1 immunostaining for the diagnosis of matricoma. Actas Dermosifiliogr. 2022;113:T907-T910. doi:10.1016/j.ad.2022.08.003
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498. doi:10.1002/1097-0142(19930415)71:8<2491 ::aid-cncr2820710811>3.0.co;2-i
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding β-catenin. J Cutan Pathol. 2005;32:148-157. doi:10.1111/j.0303-6987.2005.00267.x
- Abula A, Ma SQ, Wang S, et al. Case report: Pilomatrix carcinoma with PDL1 expression and CDKN2A aberrant. Front Immunol. 2024;15. doi:10.3389/fimmu.2024.1337400
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kanitakis J, Ducroux E, Hoelt P, et al. Basal-cell carcinoma with matrical differentiation: report of a new case in a renal-transplant recipient and literature review. Am J Dermatopathol. 2018;40:E115-E118. doi:10.1097 /DAD.0000000000001146
- White C, Farsi M, Esguerra D, et al. Not your average skin cancer: a rare case of pilomatrix carcinoma. J Clin Aesthet Dermatol. 2020; 13:40-42.
- Yanofsky VR, Mercer SE, Phelps RG. Histopathological variants of cutaneous squamous cell carcinoma: a review. J Skin Cancer. 2010;2011:210813. doi:10.1155/2011/210813
- Balas¸escu E, Gheorghe AC, Moroianu A, et al. Role of immunohistochemistry in the diagnosis and staging of cutaneous squamouscell carcinomas (review). Exp Ther Med. 2022;23:383. doi:10.3892 /etm.2022.11308
- Zhang Z, Shi W, Zhang R. Facial Merkel cell carcinoma in a 92-year-old man: a case report. Clin Case Rep. 2024;12:E9523. doi:10.1002/ccr3.9523
- Rapini R. Practical Dermatopathology. 3rd ed. Elsevier; 2021.
- Miyamoto K, Yanagi T, Maeda T, et al. Diagnosis and management of porocarcinoma. Cancers. 2022;14:5232. doi:10.3390 /cancers14215232
- Goto K. Immunohistochemistry for CD117 (KIT) is effective in distinguishing cutaneous adnexal tumors with apocrine/eccrine or sebaceous differentiation from other epithelial tumors of the skin. J Cutan Pathol. 2015;42:480-488. doi:10.1111/cup.12492
- Requena L, Sangüeza O. General principles for the histopathologic diagnosis of neoplasms with eccrine and apocrine differentiation. Classification and histopathologic criteria for eccrine and apocrine differentiation. In: Requena L, Sangüeza O, eds. Cutaneous Adnexal Neoplasms. Springer International Publishing; 2017:19-24. doi:10.1007/978- 3-319-45704-8_2
- Huet P, Dandurand M, Pignodel C, et al. Metastasizing eccrine porocarcinoma: report of a case and review of the literature. J Am Acad Dermatol. 1996;35:860-864. doi:10.1016/s0190-9622(96)90105-x
Verrucous Nodule on the Cheek
Verrucous Nodule on the Cheek
A 73-year-old man presented to the dermatology department for evaluation of an asymptomatic verrucous brown nodule on the right superior malar cheek of a few months’ duration. The patient reported a history of hyperlipidemia and hypertension and no prior treatment at the site of the nodule. A biopsy of the lesion was performed.
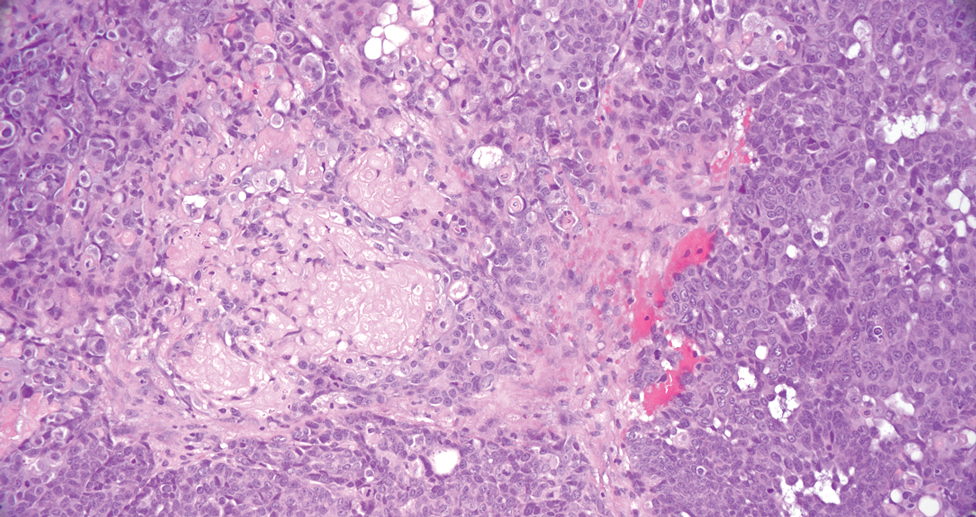

Mobile Tender Papule on the Scalp
Mobile Tender Papule on the Scalp
The Diagnosis: Spiradenocylindroma
T he biopsy results confirmed the diagnosis of spiradenocylindroma with negative margins. At 6-week follow-up, the patient had no signs of recurrence. Spiradenocylindroma is a benign hybrid neoplasm consisting of histologically intermixed areas representing the spectrum of morphology between spiradenoma and cylindromas.1,2 Both spiradenoma and cylindroma comprise 2 distinct populations of dark and pale basaloid cells.2,3 The spiradenomatous areas of the spiradenocylindroma are arranged in large, well-circumscribed collections of small, darkly staining cells with interspersed lymphocytes and a thin basement membrane surrounding spiradenocylindroma component.2,3 The spiradenocylindroma regions also may contain tubular structures dilated by hemorrhage.2 In contrast, the cylindromatous regions have a jigsaw-puzzle configuration of polygonal tumor nests containing peripherally palisading dark cells and central pale cells, surrounded by a thick basement membrane (top quiz image).2,3
Clinically, sporadic spiradenocylindromas may resemble other lesions, manifesting as a papule or nodule with coloration ranging from gray-blue to salmon pink along with arborizing telangiectasias.4,5 Although spiradenocylindromas typically are found on the head, neck, and trunk, they also have been reported in the kidney, vulva, anus, and rectum.2,6,7 Not only are spiradenocylindromas clinically indistinct from other adnexal growths, but they also share some features with basal cell carcinomas (BCCs) and amelanotic melanomas.8 Features of arborizing telangiectasias on a papule may resemble BCC, requiring histopathology for a definitive diagnosis.
Spiradenocylindromas classically are associated with Brooke-Spiegler syndrome, a rare, autosomal-dominant genodermatosis caused by a germline mutation in the cylindromatosis lysine 63 deubiquitinase tumor-suppressor gene.5 Patients develop adnexal neoplasms of the folliculosebaceous-apocrine unit, including spiradenomas, cylindromas, and trichoepitheliomas.5 Rarely, malignant transformation to spiradenocylindrocarcinoma has been reported.9 Features of malignant transformation include loss of the 2-cell population, cytologic atypia, increased mitotic activity, and loss of intratumoral lymphocytes.10
Trichoepitheliomas are benign, firm, flesh-colored papules to nodules that commonly are found on the mid face but may appear on the scalp, neck, and upper trunk.5-11 Trichoepitheliomas are closely related to spiradenomas and cylindromas; the familial form, multiple familial trichoepitheliomas, exists on a spectrum with Brooke-Spiegler syndrome.3,11 Multiple familial trichoepithelioma is characterized by multiple trichoepitheliomas without accompanying spiradenomas, cylindromas, or spiradenocylindromas.3 On histopathology, trichoepitheliomas are distinguished by cribriform clusters or nests of basaloid follicular germinative cells with bulbar differentiation, known as papillary mesenchymal bodies, surrounded by an adherent stroma (eFigure 1).3,5,11 In addition to follicular bulbar differentiation, trichoepitheliomas are surrounded by an adherent cellular stroma without the retraction artifact around tumor islands seen in BCC, although artifactual clefts may occur within the stroma.11 In contrast, spiradenocylindromas do not demonstrate keratin cysts or artifactual clefts within the stroma.
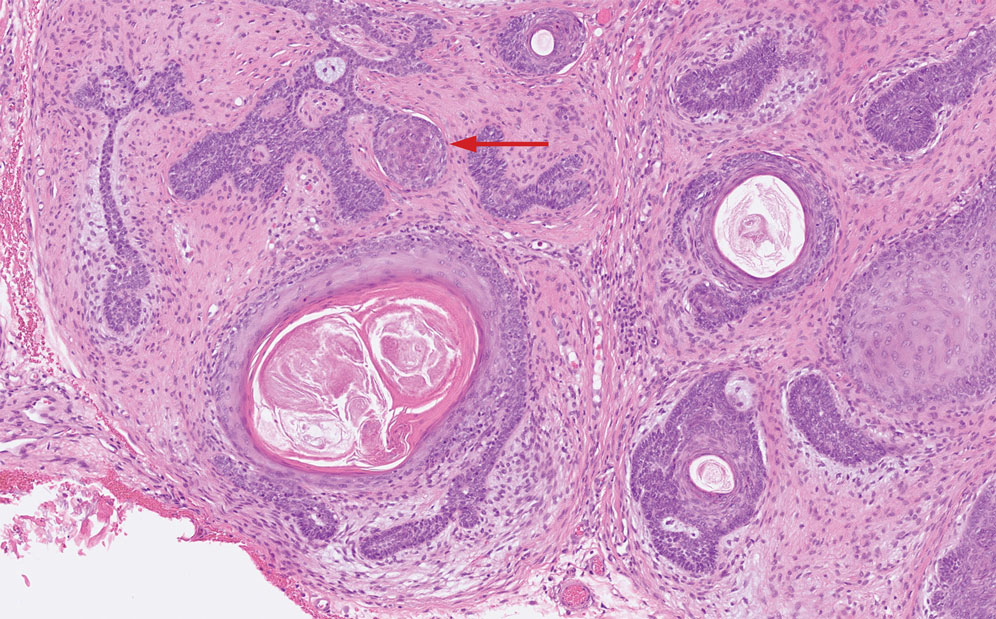
Trichilemmal cysts, or pilar cysts, are benign adnexal neoplasms derived from the outer root sheath at the isthmus.12-14 Approximately 90% of pilar cysts are found on the scalp and 2% of trichilemmal cysts may progress to a proliferating trichilemmal cyst, which is locally aggressive and contains an expanding buckled epithelium within the cyst space.12,14 Clinically, trichilemmal cysts are slow-growing, smooth, round, mobile nodules without a central punctum.12,13 On histopathology, the cyst wall contains peripherally palisading basal cells and maturing cells showing no intercellular bridging (eFigure 2). As the cells mature, they swell with pale cytoplasm and abruptly keratinize without a granular layer, a process known as trichilemmal keratinization.12-14 Additionally, cholesterol clefts are common in the keratinous lumen, and about 25% of cysts contain calcifications.13,14 The broadly basophilic spiradenocylindromas sharply contrast the abundant eosinophilic keratin of trichilemmal cysts.
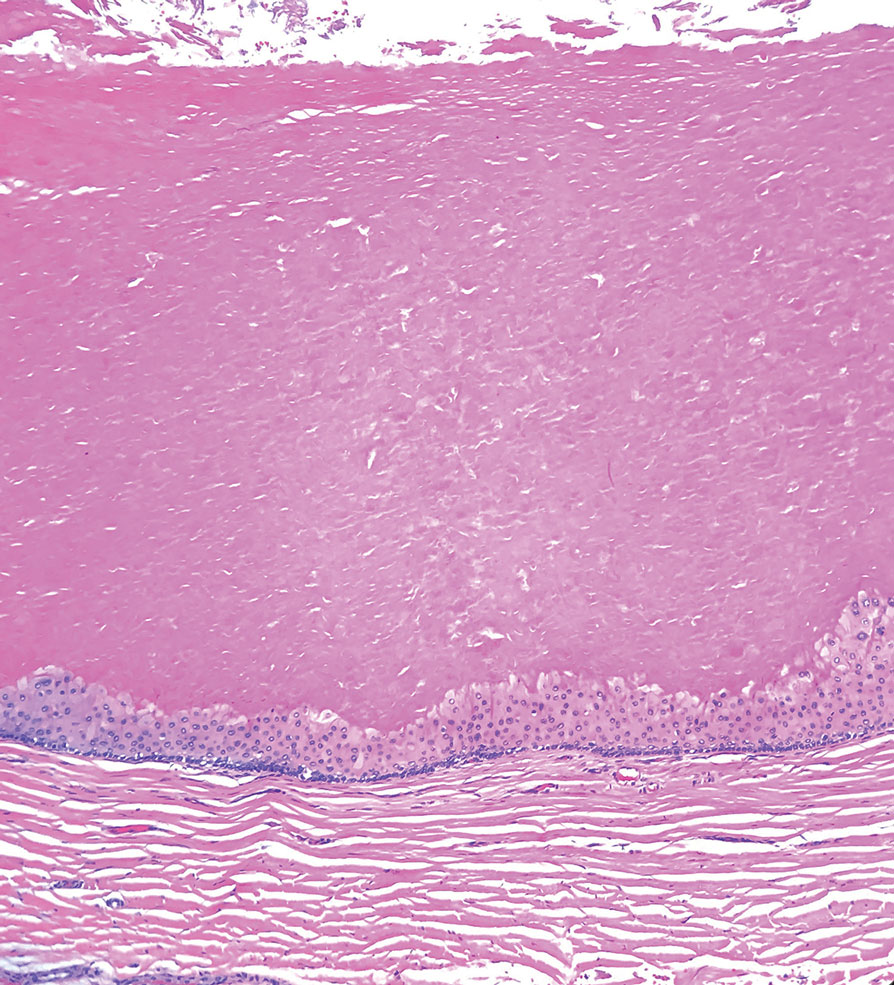
Basal cell carcinoma is a slow-growing, locally destructive neoplasm that develops due to chronic sun exposure; thus, BCCs commonly arise on exposed areas of the face, head, neck, arms, and legs.15 Nodular BCC is the most common subtype and typically manifests as a shiny pearly papule or nodule with a smooth surface, rolled borders, and arborizing telangiectasias.16 On histopathology, nodular BCCs demonstrate nests or nodules of basaloid keratinocytes with peripheral palisading and retraction artifact between the tumor and stroma (eFigure 3).15,16 A lack of retraction artifact, cystic dilation of tubular structures, jigsaw molding of nests, and a distinct 2-cell population distinguish spiradenocylindroma from BCC. Of note, in rare instances BCCs also may display a thick fibrous stroma, similar to the stroma of cylindromas.15
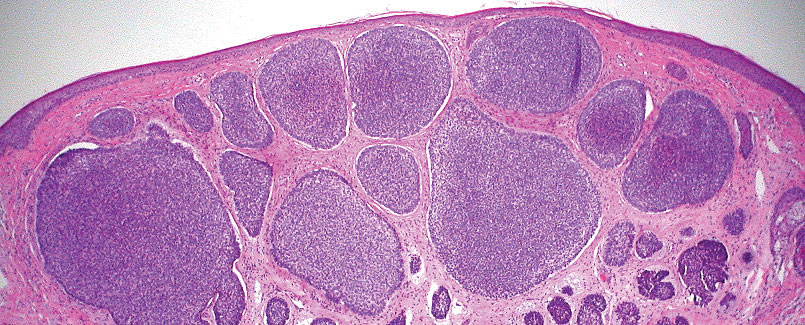
Amelanotic melanoma is a variant of melanoma characterized by little to no pigment. Any of the 4 classic subtypes of melanoma (nodular, superficial spreading, lentigo maligna, acral lentiginous) can be amelanotic.17 Clinically, amelanotic melanomas can vary greatly, manifesting as erythematous macules, dermal plaques, or papulonodular lesions, often with scaling.18 On histopathology, findings common to all melanomas include cellular atypia, mitoses, pagetoid spread, and pleomorphism (eFigure 4).18,19 Immunohistochemistry is an important method to distinguish melanoma from other melanocytic proliferations and to aid in the assessment of Breslow depth. Markers include SOX10 (sex-determining region Y-box transcription factor 10), S100, and MART-1 (melanoma antigen recognized by T cells 1/melan-A).19,20 Expression of PRAME (preferentially expressed antigen in melanoma) often is positive but is not necessary for diagnosis.21 Histologically, the atypical pleomorphic cells of melanoma are markedly distinct from both spiradenomas and cylindromas.
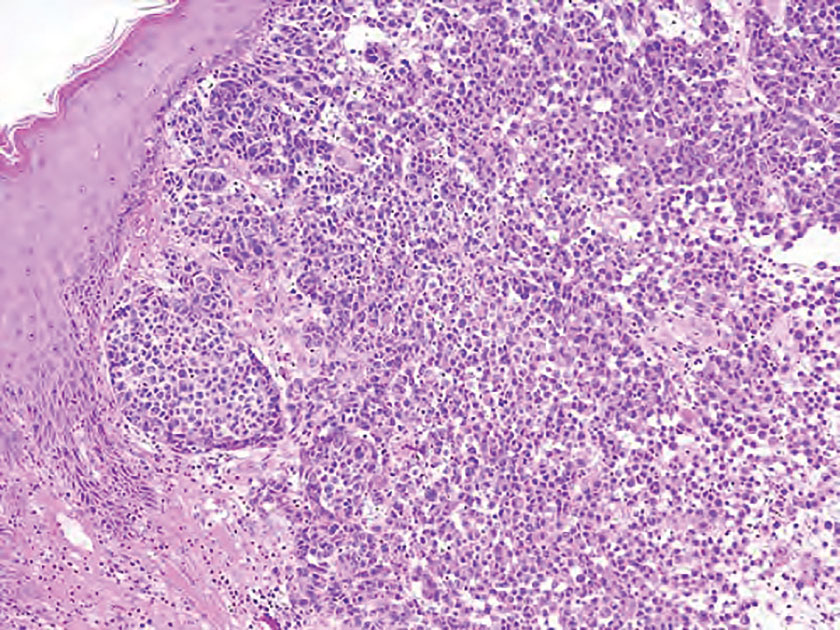
- Soyer HP, Kerl H, Ott A. Spiradenocylindroma—more than a coincidence? Am J Dermatopathol. 1998;20:315-317.
- Michal M, Lamovec J, Mukenˇ snabl P, et al. Spiradenocylindromas of the skin: tumors with morphological features of spiradenoma and cylindroma in the same lesion: report of 12 cases. Pathol Int. 1999;49:419-425.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bostan E, Boynuyogun E, Gokoz O, et al. Hybrid tumor “spiradenocylindroma” with unusual dermoscopic features. An Bras Dermatol. 2023;98:382-384.
- Pinho AC, Gouveia MJ, Gameiro AR, et al. Brooke-Spiegler syndrome—an underrecognized cause of multiple familial scalp tumors: report of a new germline mutation. J Dermatol Case Rep. 2015;9:67-70.
- Ströbel P, Zettl A, Ren Z, et al. Spiradenocylindroma of the kidney: clinical and genetic findings suggesting a role of somatic mutation of the CYLD1 gene in the oncogenesis of an unusual renal neoplasm. Am J Surg Pathol. 2002;26:119-124.
- Kacerovska D, Szepe P, Vanecek T, et al. Spiradenocylindroma-like basaloid carcinoma of the anus and rectum: case report, including HPV studies and analysis of the CYLD gene mutations. Am J Dermatopathol. 2008;30:472-476.
- Silvestri F, Maida P, Venturi F, et al. Scalp spiradenocylindroma: a challenging dermoscopic diagnosis. Dermatol Ther. 2020;33:E14307.
- Held L, Ruetten A, Saggini A, et al. Metaplastic spiradenocarcinoma: report of two cases with sarcomatous differentiation. J Cutan Pathol. 2021;48:384-389.
- Płachta I, Kleibert M, Czarnecka AM, et al. Current diagnosis and treatment options for cutaneous adnexal neoplasms with apocrine and eccrine differentiation. Int J Mol Sci. 2021;22:5077.
- Johnson H, Robles M, Kamino H, et al. Trichoepithelioma. Dermatol Online J. 2008;14:5.
- He P, Cui LG, Wang JR, et al. Trichilemmal cyst: clinical and sonographic feature. J Ultrasound Med. 2019;38:91-96.
- Liu M, Han H, Zheng Y, et al. Pilar cyst on the dorsum of hand: a case report and review of literature. Medicine (United States). 2020;99:E21519.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. In: Archives of Pathology and Laboratory Medicine. Vol 141. College of American Pathologists; 2017:1490-1502.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Kaizer-Salk KA, Herten RJ, Ragsdale BD, et al. Amelanotic melanoma: a unique case study and review of the literature. BMJ Case Rep. 2018:bcr2017222751.
- Silva TS, de Araujo LR, Faro GB de A, et al. Nodular amelanotic melanoma. An Bras Dermatol. 2019;94:497-498.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321.
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
The Diagnosis: Spiradenocylindroma
T he biopsy results confirmed the diagnosis of spiradenocylindroma with negative margins. At 6-week follow-up, the patient had no signs of recurrence. Spiradenocylindroma is a benign hybrid neoplasm consisting of histologically intermixed areas representing the spectrum of morphology between spiradenoma and cylindromas.1,2 Both spiradenoma and cylindroma comprise 2 distinct populations of dark and pale basaloid cells.2,3 The spiradenomatous areas of the spiradenocylindroma are arranged in large, well-circumscribed collections of small, darkly staining cells with interspersed lymphocytes and a thin basement membrane surrounding spiradenocylindroma component.2,3 The spiradenocylindroma regions also may contain tubular structures dilated by hemorrhage.2 In contrast, the cylindromatous regions have a jigsaw-puzzle configuration of polygonal tumor nests containing peripherally palisading dark cells and central pale cells, surrounded by a thick basement membrane (top quiz image).2,3
Clinically, sporadic spiradenocylindromas may resemble other lesions, manifesting as a papule or nodule with coloration ranging from gray-blue to salmon pink along with arborizing telangiectasias.4,5 Although spiradenocylindromas typically are found on the head, neck, and trunk, they also have been reported in the kidney, vulva, anus, and rectum.2,6,7 Not only are spiradenocylindromas clinically indistinct from other adnexal growths, but they also share some features with basal cell carcinomas (BCCs) and amelanotic melanomas.8 Features of arborizing telangiectasias on a papule may resemble BCC, requiring histopathology for a definitive diagnosis.
Spiradenocylindromas classically are associated with Brooke-Spiegler syndrome, a rare, autosomal-dominant genodermatosis caused by a germline mutation in the cylindromatosis lysine 63 deubiquitinase tumor-suppressor gene.5 Patients develop adnexal neoplasms of the folliculosebaceous-apocrine unit, including spiradenomas, cylindromas, and trichoepitheliomas.5 Rarely, malignant transformation to spiradenocylindrocarcinoma has been reported.9 Features of malignant transformation include loss of the 2-cell population, cytologic atypia, increased mitotic activity, and loss of intratumoral lymphocytes.10
Trichoepitheliomas are benign, firm, flesh-colored papules to nodules that commonly are found on the mid face but may appear on the scalp, neck, and upper trunk.5-11 Trichoepitheliomas are closely related to spiradenomas and cylindromas; the familial form, multiple familial trichoepitheliomas, exists on a spectrum with Brooke-Spiegler syndrome.3,11 Multiple familial trichoepithelioma is characterized by multiple trichoepitheliomas without accompanying spiradenomas, cylindromas, or spiradenocylindromas.3 On histopathology, trichoepitheliomas are distinguished by cribriform clusters or nests of basaloid follicular germinative cells with bulbar differentiation, known as papillary mesenchymal bodies, surrounded by an adherent stroma (eFigure 1).3,5,11 In addition to follicular bulbar differentiation, trichoepitheliomas are surrounded by an adherent cellular stroma without the retraction artifact around tumor islands seen in BCC, although artifactual clefts may occur within the stroma.11 In contrast, spiradenocylindromas do not demonstrate keratin cysts or artifactual clefts within the stroma.
Trichilemmal cysts, or pilar cysts, are benign adnexal neoplasms derived from the outer root sheath at the isthmus.12-14 Approximately 90% of pilar cysts are found on the scalp and 2% of trichilemmal cysts may progress to a proliferating trichilemmal cyst, which is locally aggressive and contains an expanding buckled epithelium within the cyst space.12,14 Clinically, trichilemmal cysts are slow-growing, smooth, round, mobile nodules without a central punctum.12,13 On histopathology, the cyst wall contains peripherally palisading basal cells and maturing cells showing no intercellular bridging (eFigure 2). As the cells mature, they swell with pale cytoplasm and abruptly keratinize without a granular layer, a process known as trichilemmal keratinization.12-14 Additionally, cholesterol clefts are common in the keratinous lumen, and about 25% of cysts contain calcifications.13,14 The broadly basophilic spiradenocylindromas sharply contrast the abundant eosinophilic keratin of trichilemmal cysts.
Basal cell carcinoma is a slow-growing, locally destructive neoplasm that develops due to chronic sun exposure; thus, BCCs commonly arise on exposed areas of the face, head, neck, arms, and legs.15 Nodular BCC is the most common subtype and typically manifests as a shiny pearly papule or nodule with a smooth surface, rolled borders, and arborizing telangiectasias.16 On histopathology, nodular BCCs demonstrate nests or nodules of basaloid keratinocytes with peripheral palisading and retraction artifact between the tumor and stroma (eFigure 3).15,16 A lack of retraction artifact, cystic dilation of tubular structures, jigsaw molding of nests, and a distinct 2-cell population distinguish spiradenocylindroma from BCC. Of note, in rare instances BCCs also may display a thick fibrous stroma, similar to the stroma of cylindromas.15
Amelanotic melanoma is a variant of melanoma characterized by little to no pigment. Any of the 4 classic subtypes of melanoma (nodular, superficial spreading, lentigo maligna, acral lentiginous) can be amelanotic.17 Clinically, amelanotic melanomas can vary greatly, manifesting as erythematous macules, dermal plaques, or papulonodular lesions, often with scaling.18 On histopathology, findings common to all melanomas include cellular atypia, mitoses, pagetoid spread, and pleomorphism (eFigure 4).18,19 Immunohistochemistry is an important method to distinguish melanoma from other melanocytic proliferations and to aid in the assessment of Breslow depth. Markers include SOX10 (sex-determining region Y-box transcription factor 10), S100, and MART-1 (melanoma antigen recognized by T cells 1/melan-A).19,20 Expression of PRAME (preferentially expressed antigen in melanoma) often is positive but is not necessary for diagnosis.21 Histologically, the atypical pleomorphic cells of melanoma are markedly distinct from both spiradenomas and cylindromas.
The Diagnosis: Spiradenocylindroma
T he biopsy results confirmed the diagnosis of spiradenocylindroma with negative margins. At 6-week follow-up, the patient had no signs of recurrence. Spiradenocylindroma is a benign hybrid neoplasm consisting of histologically intermixed areas representing the spectrum of morphology between spiradenoma and cylindromas.1,2 Both spiradenoma and cylindroma comprise 2 distinct populations of dark and pale basaloid cells.2,3 The spiradenomatous areas of the spiradenocylindroma are arranged in large, well-circumscribed collections of small, darkly staining cells with interspersed lymphocytes and a thin basement membrane surrounding spiradenocylindroma component.2,3 The spiradenocylindroma regions also may contain tubular structures dilated by hemorrhage.2 In contrast, the cylindromatous regions have a jigsaw-puzzle configuration of polygonal tumor nests containing peripherally palisading dark cells and central pale cells, surrounded by a thick basement membrane (top quiz image).2,3
Clinically, sporadic spiradenocylindromas may resemble other lesions, manifesting as a papule or nodule with coloration ranging from gray-blue to salmon pink along with arborizing telangiectasias.4,5 Although spiradenocylindromas typically are found on the head, neck, and trunk, they also have been reported in the kidney, vulva, anus, and rectum.2,6,7 Not only are spiradenocylindromas clinically indistinct from other adnexal growths, but they also share some features with basal cell carcinomas (BCCs) and amelanotic melanomas.8 Features of arborizing telangiectasias on a papule may resemble BCC, requiring histopathology for a definitive diagnosis.
Spiradenocylindromas classically are associated with Brooke-Spiegler syndrome, a rare, autosomal-dominant genodermatosis caused by a germline mutation in the cylindromatosis lysine 63 deubiquitinase tumor-suppressor gene.5 Patients develop adnexal neoplasms of the folliculosebaceous-apocrine unit, including spiradenomas, cylindromas, and trichoepitheliomas.5 Rarely, malignant transformation to spiradenocylindrocarcinoma has been reported.9 Features of malignant transformation include loss of the 2-cell population, cytologic atypia, increased mitotic activity, and loss of intratumoral lymphocytes.10
Trichoepitheliomas are benign, firm, flesh-colored papules to nodules that commonly are found on the mid face but may appear on the scalp, neck, and upper trunk.5-11 Trichoepitheliomas are closely related to spiradenomas and cylindromas; the familial form, multiple familial trichoepitheliomas, exists on a spectrum with Brooke-Spiegler syndrome.3,11 Multiple familial trichoepithelioma is characterized by multiple trichoepitheliomas without accompanying spiradenomas, cylindromas, or spiradenocylindromas.3 On histopathology, trichoepitheliomas are distinguished by cribriform clusters or nests of basaloid follicular germinative cells with bulbar differentiation, known as papillary mesenchymal bodies, surrounded by an adherent stroma (eFigure 1).3,5,11 In addition to follicular bulbar differentiation, trichoepitheliomas are surrounded by an adherent cellular stroma without the retraction artifact around tumor islands seen in BCC, although artifactual clefts may occur within the stroma.11 In contrast, spiradenocylindromas do not demonstrate keratin cysts or artifactual clefts within the stroma.
Trichilemmal cysts, or pilar cysts, are benign adnexal neoplasms derived from the outer root sheath at the isthmus.12-14 Approximately 90% of pilar cysts are found on the scalp and 2% of trichilemmal cysts may progress to a proliferating trichilemmal cyst, which is locally aggressive and contains an expanding buckled epithelium within the cyst space.12,14 Clinically, trichilemmal cysts are slow-growing, smooth, round, mobile nodules without a central punctum.12,13 On histopathology, the cyst wall contains peripherally palisading basal cells and maturing cells showing no intercellular bridging (eFigure 2). As the cells mature, they swell with pale cytoplasm and abruptly keratinize without a granular layer, a process known as trichilemmal keratinization.12-14 Additionally, cholesterol clefts are common in the keratinous lumen, and about 25% of cysts contain calcifications.13,14 The broadly basophilic spiradenocylindromas sharply contrast the abundant eosinophilic keratin of trichilemmal cysts.
Basal cell carcinoma is a slow-growing, locally destructive neoplasm that develops due to chronic sun exposure; thus, BCCs commonly arise on exposed areas of the face, head, neck, arms, and legs.15 Nodular BCC is the most common subtype and typically manifests as a shiny pearly papule or nodule with a smooth surface, rolled borders, and arborizing telangiectasias.16 On histopathology, nodular BCCs demonstrate nests or nodules of basaloid keratinocytes with peripheral palisading and retraction artifact between the tumor and stroma (eFigure 3).15,16 A lack of retraction artifact, cystic dilation of tubular structures, jigsaw molding of nests, and a distinct 2-cell population distinguish spiradenocylindroma from BCC. Of note, in rare instances BCCs also may display a thick fibrous stroma, similar to the stroma of cylindromas.15
Amelanotic melanoma is a variant of melanoma characterized by little to no pigment. Any of the 4 classic subtypes of melanoma (nodular, superficial spreading, lentigo maligna, acral lentiginous) can be amelanotic.17 Clinically, amelanotic melanomas can vary greatly, manifesting as erythematous macules, dermal plaques, or papulonodular lesions, often with scaling.18 On histopathology, findings common to all melanomas include cellular atypia, mitoses, pagetoid spread, and pleomorphism (eFigure 4).18,19 Immunohistochemistry is an important method to distinguish melanoma from other melanocytic proliferations and to aid in the assessment of Breslow depth. Markers include SOX10 (sex-determining region Y-box transcription factor 10), S100, and MART-1 (melanoma antigen recognized by T cells 1/melan-A).19,20 Expression of PRAME (preferentially expressed antigen in melanoma) often is positive but is not necessary for diagnosis.21 Histologically, the atypical pleomorphic cells of melanoma are markedly distinct from both spiradenomas and cylindromas.
- Soyer HP, Kerl H, Ott A. Spiradenocylindroma—more than a coincidence? Am J Dermatopathol. 1998;20:315-317.
- Michal M, Lamovec J, Mukenˇ snabl P, et al. Spiradenocylindromas of the skin: tumors with morphological features of spiradenoma and cylindroma in the same lesion: report of 12 cases. Pathol Int. 1999;49:419-425.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bostan E, Boynuyogun E, Gokoz O, et al. Hybrid tumor “spiradenocylindroma” with unusual dermoscopic features. An Bras Dermatol. 2023;98:382-384.
- Pinho AC, Gouveia MJ, Gameiro AR, et al. Brooke-Spiegler syndrome—an underrecognized cause of multiple familial scalp tumors: report of a new germline mutation. J Dermatol Case Rep. 2015;9:67-70.
- Ströbel P, Zettl A, Ren Z, et al. Spiradenocylindroma of the kidney: clinical and genetic findings suggesting a role of somatic mutation of the CYLD1 gene in the oncogenesis of an unusual renal neoplasm. Am J Surg Pathol. 2002;26:119-124.
- Kacerovska D, Szepe P, Vanecek T, et al. Spiradenocylindroma-like basaloid carcinoma of the anus and rectum: case report, including HPV studies and analysis of the CYLD gene mutations. Am J Dermatopathol. 2008;30:472-476.
- Silvestri F, Maida P, Venturi F, et al. Scalp spiradenocylindroma: a challenging dermoscopic diagnosis. Dermatol Ther. 2020;33:E14307.
- Held L, Ruetten A, Saggini A, et al. Metaplastic spiradenocarcinoma: report of two cases with sarcomatous differentiation. J Cutan Pathol. 2021;48:384-389.
- Płachta I, Kleibert M, Czarnecka AM, et al. Current diagnosis and treatment options for cutaneous adnexal neoplasms with apocrine and eccrine differentiation. Int J Mol Sci. 2021;22:5077.
- Johnson H, Robles M, Kamino H, et al. Trichoepithelioma. Dermatol Online J. 2008;14:5.
- He P, Cui LG, Wang JR, et al. Trichilemmal cyst: clinical and sonographic feature. J Ultrasound Med. 2019;38:91-96.
- Liu M, Han H, Zheng Y, et al. Pilar cyst on the dorsum of hand: a case report and review of literature. Medicine (United States). 2020;99:E21519.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. In: Archives of Pathology and Laboratory Medicine. Vol 141. College of American Pathologists; 2017:1490-1502.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Kaizer-Salk KA, Herten RJ, Ragsdale BD, et al. Amelanotic melanoma: a unique case study and review of the literature. BMJ Case Rep. 2018:bcr2017222751.
- Silva TS, de Araujo LR, Faro GB de A, et al. Nodular amelanotic melanoma. An Bras Dermatol. 2019;94:497-498.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321.
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Soyer HP, Kerl H, Ott A. Spiradenocylindroma—more than a coincidence? Am J Dermatopathol. 1998;20:315-317.
- Michal M, Lamovec J, Mukenˇ snabl P, et al. Spiradenocylindromas of the skin: tumors with morphological features of spiradenoma and cylindroma in the same lesion: report of 12 cases. Pathol Int. 1999;49:419-425.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bostan E, Boynuyogun E, Gokoz O, et al. Hybrid tumor “spiradenocylindroma” with unusual dermoscopic features. An Bras Dermatol. 2023;98:382-384.
- Pinho AC, Gouveia MJ, Gameiro AR, et al. Brooke-Spiegler syndrome—an underrecognized cause of multiple familial scalp tumors: report of a new germline mutation. J Dermatol Case Rep. 2015;9:67-70.
- Ströbel P, Zettl A, Ren Z, et al. Spiradenocylindroma of the kidney: clinical and genetic findings suggesting a role of somatic mutation of the CYLD1 gene in the oncogenesis of an unusual renal neoplasm. Am J Surg Pathol. 2002;26:119-124.
- Kacerovska D, Szepe P, Vanecek T, et al. Spiradenocylindroma-like basaloid carcinoma of the anus and rectum: case report, including HPV studies and analysis of the CYLD gene mutations. Am J Dermatopathol. 2008;30:472-476.
- Silvestri F, Maida P, Venturi F, et al. Scalp spiradenocylindroma: a challenging dermoscopic diagnosis. Dermatol Ther. 2020;33:E14307.
- Held L, Ruetten A, Saggini A, et al. Metaplastic spiradenocarcinoma: report of two cases with sarcomatous differentiation. J Cutan Pathol. 2021;48:384-389.
- Płachta I, Kleibert M, Czarnecka AM, et al. Current diagnosis and treatment options for cutaneous adnexal neoplasms with apocrine and eccrine differentiation. Int J Mol Sci. 2021;22:5077.
- Johnson H, Robles M, Kamino H, et al. Trichoepithelioma. Dermatol Online J. 2008;14:5.
- He P, Cui LG, Wang JR, et al. Trichilemmal cyst: clinical and sonographic feature. J Ultrasound Med. 2019;38:91-96.
- Liu M, Han H, Zheng Y, et al. Pilar cyst on the dorsum of hand: a case report and review of literature. Medicine (United States). 2020;99:E21519.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128.
- Stanoszek LM, Wang GY, Harms PW. Histologic mimics of basal cell carcinoma. In: Archives of Pathology and Laboratory Medicine. Vol 141. College of American Pathologists; 2017:1490-1502.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Kaizer-Salk KA, Herten RJ, Ragsdale BD, et al. Amelanotic melanoma: a unique case study and review of the literature. BMJ Case Rep. 2018:bcr2017222751.
- Silva TS, de Araujo LR, Faro GB de A, et al. Nodular amelanotic melanoma. An Bras Dermatol. 2019;94:497-498.
- Bobos M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital J Dermatol Venereol. 2021;156:300-321.
- Ohsie SJ, Sarantopoulos GP, Cochran AJ, et al. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35:433-444.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
Mobile Tender Papule on the Scalp
Mobile Tender Papule on the Scalp
A 73-year-old man presented to the plastic surgery department with a single, progressively enlarging nodule on the scalp of 1 year’s duration. Dermatologic examination revealed a 0.8-cm, soft, mobile, gray-blue, dome-shaped papule on the left postauricular scalp that was tender to palpation. There was no central punctum, and the patient denied any history of drainage or odor. He had no personal or family history of similar lesions. An excisional biopsy of the papule was performed.
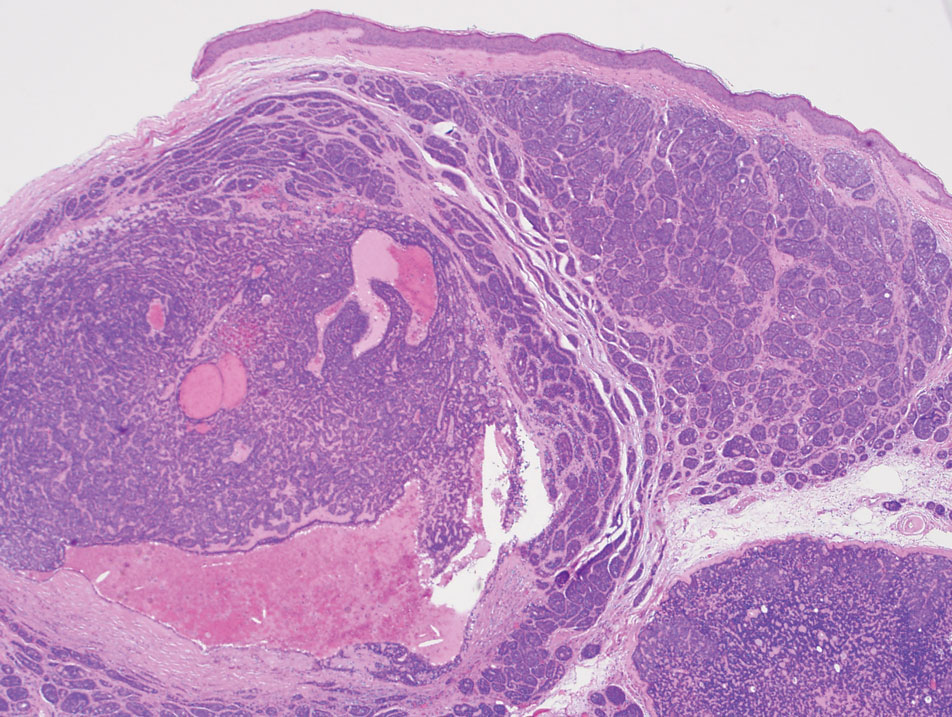
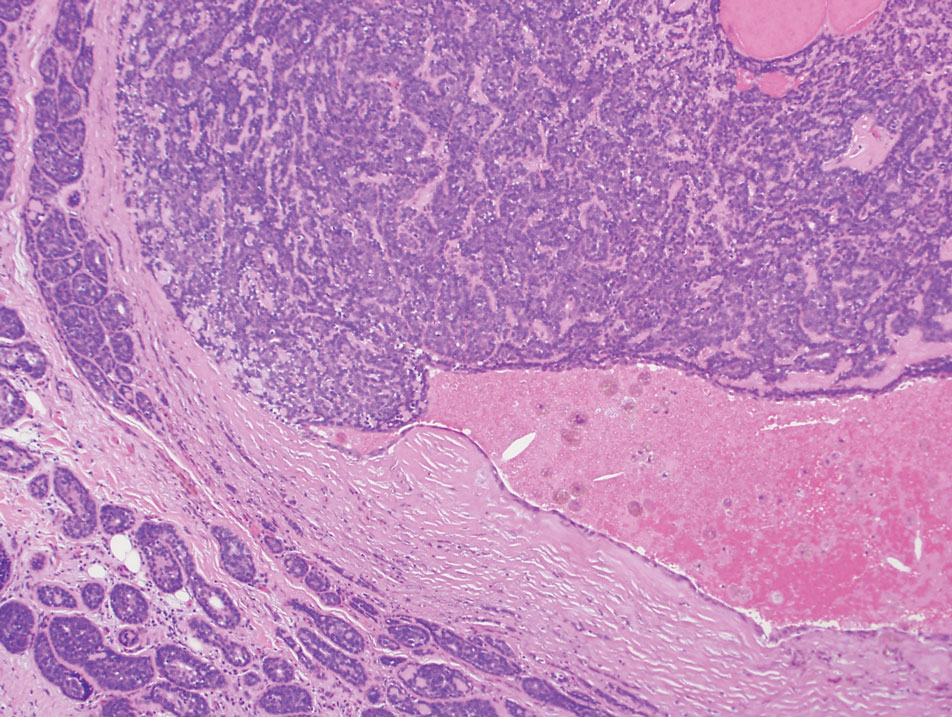
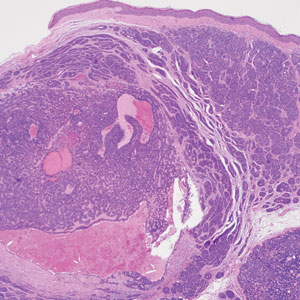
Growing Nodule on the Parietal Scalp
Growing Nodule on the Parietal Scalp
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.

Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).

Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
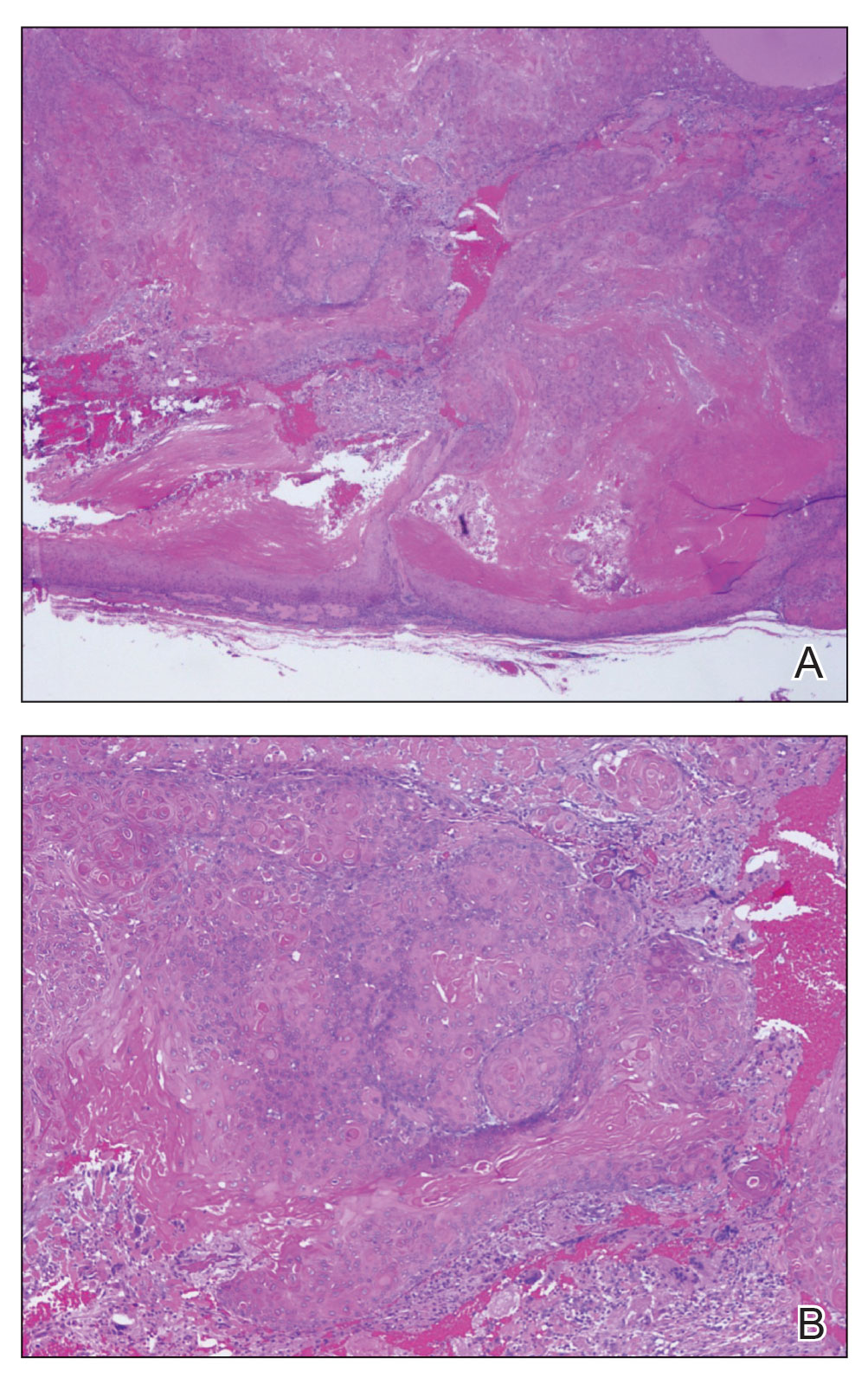
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
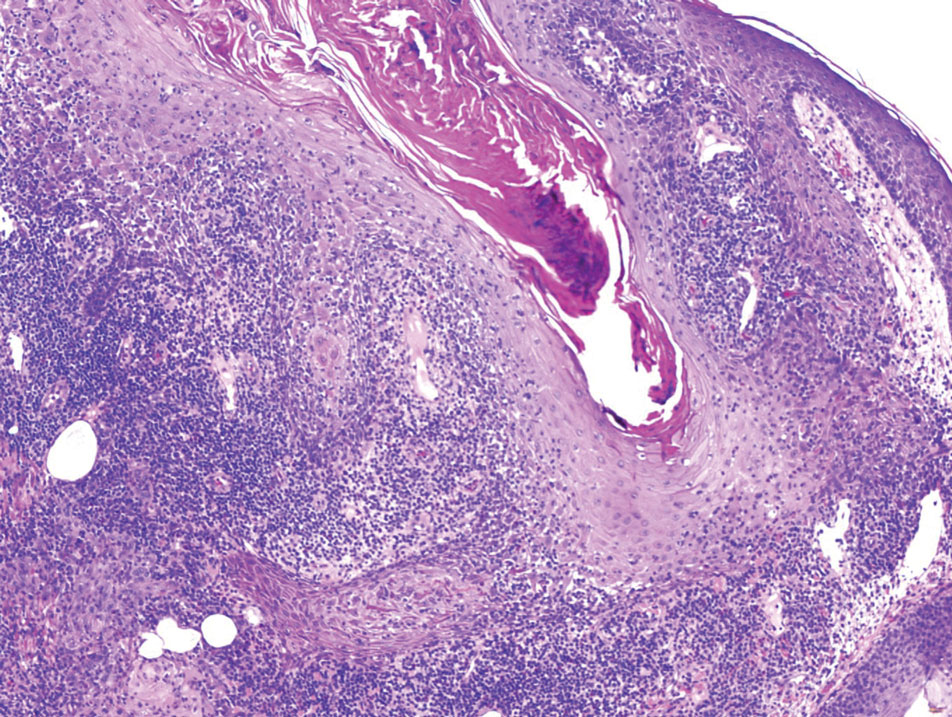
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
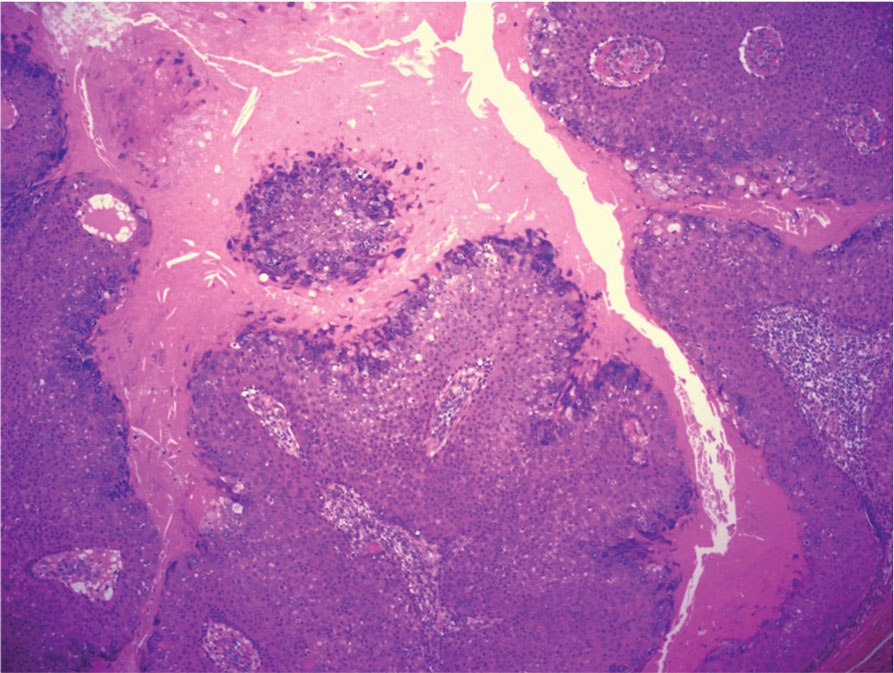
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.
Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).
Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.
Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).
Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
Growing Nodule on the Parietal Scalp
Growing Nodule on the Parietal Scalp
A 38-year-old woman with no notable medical history presented to the dermatology department with a firm enlarging nodule on the scalp of many years’ duration. The patient noted there was no drainage or bleeding. Physical examination revealed a mobile, 2.5-cm, subcutaneous nodule on the right parietal medial scalp. An excisional biopsy was performed.
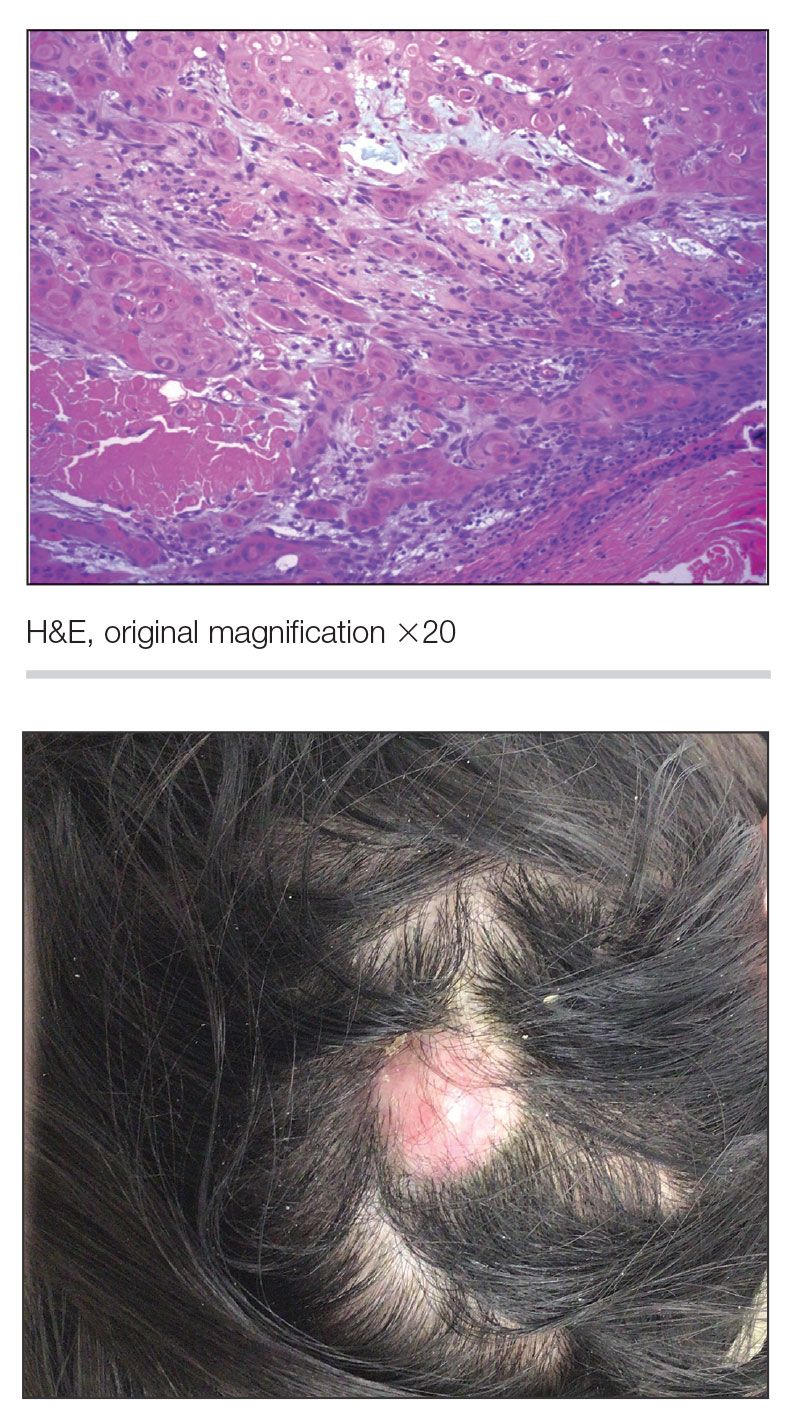

Smooth Symmetric Plaques on the Face, Trunk, and Extremities
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
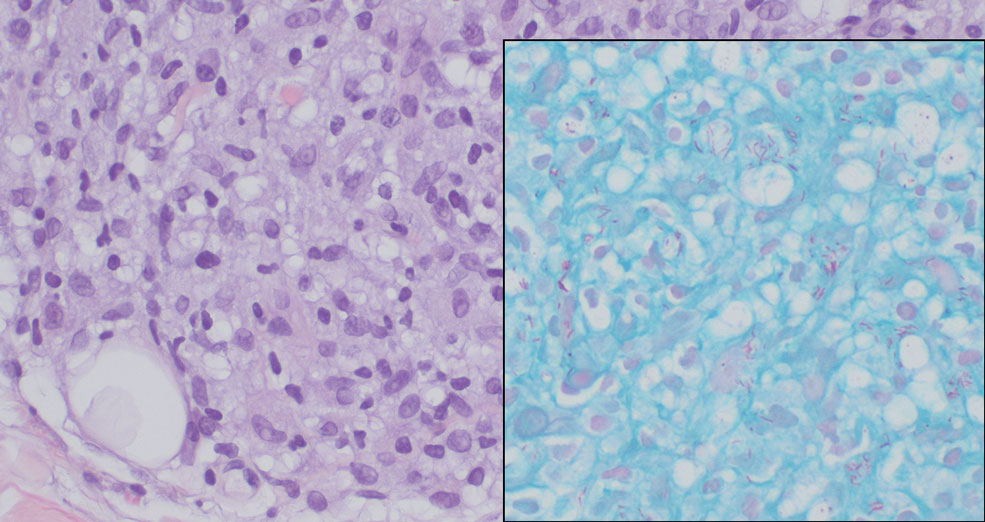
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
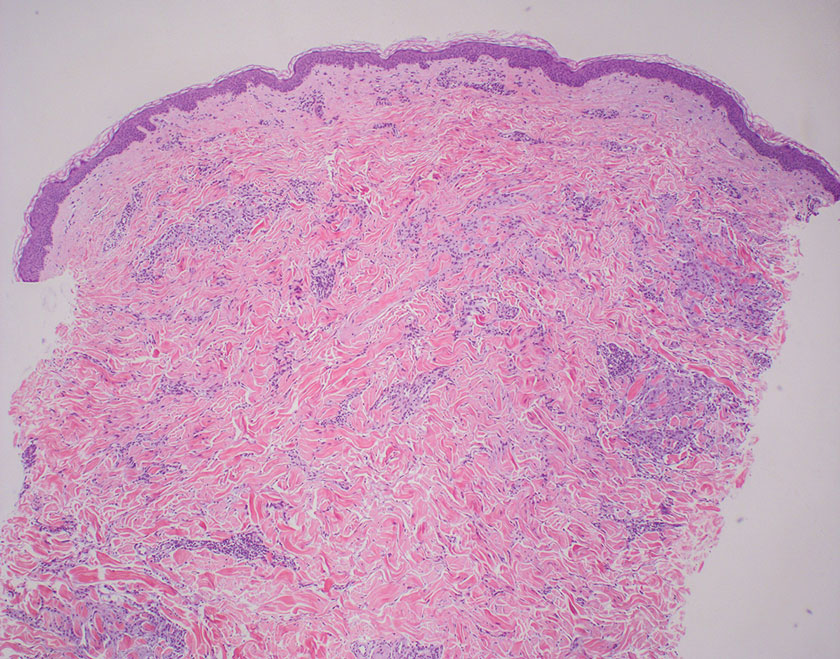
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
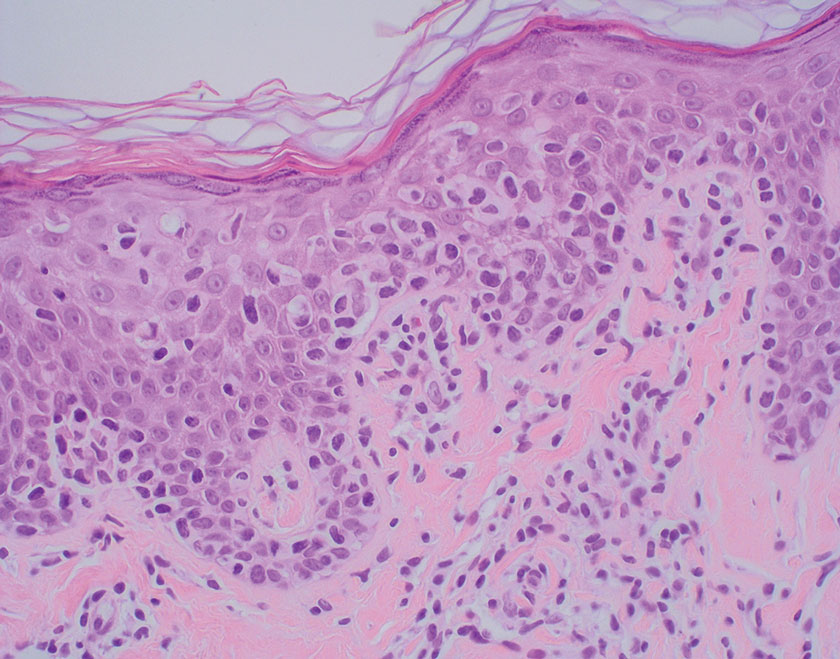
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
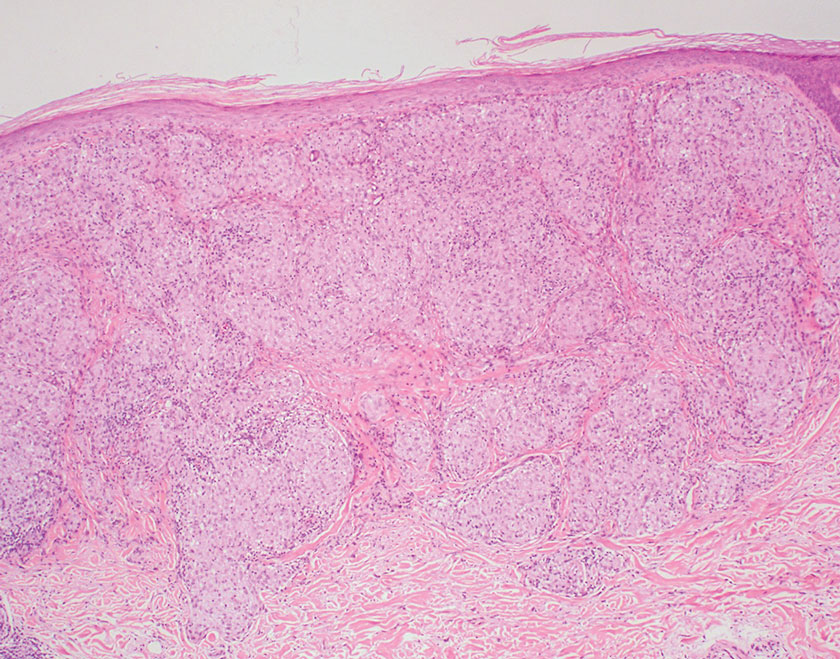
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
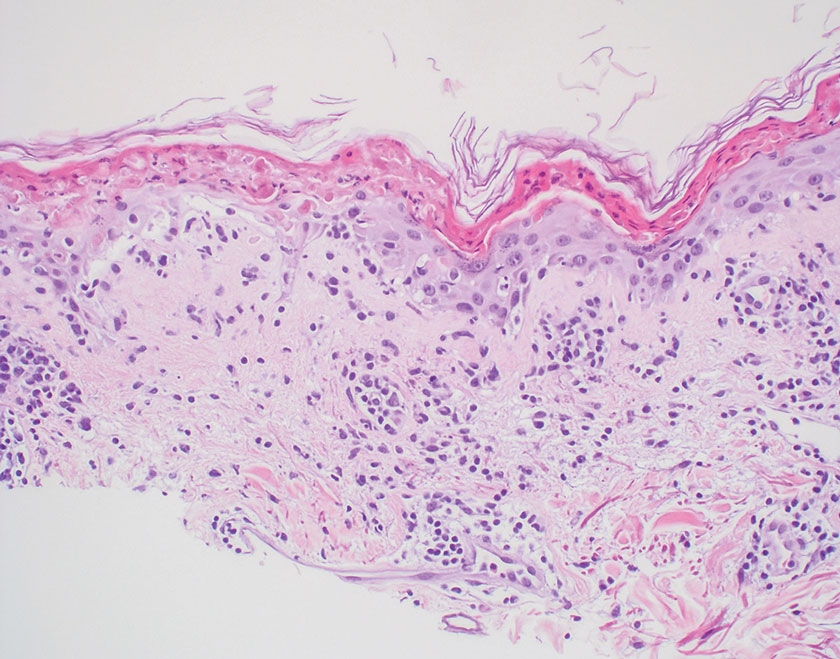
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
A 44-year-old woman presented to the dermatology clinic with a widespread red, itchy, bumpy rash of 1 year’s duration. Physical examination revealed smooth, coalescing, erythematous and edematous plaques on the face (notably the forehead, malar cheeks, and nose), back, arms, and legs. Several plaques on the back had central hypopigmentation. The patient also reported numbness and weakness in the fingers and toes, and hypoesthesia within the lesions was noted. A biopsy of one of the lesions on the left ventral forearm was performed.
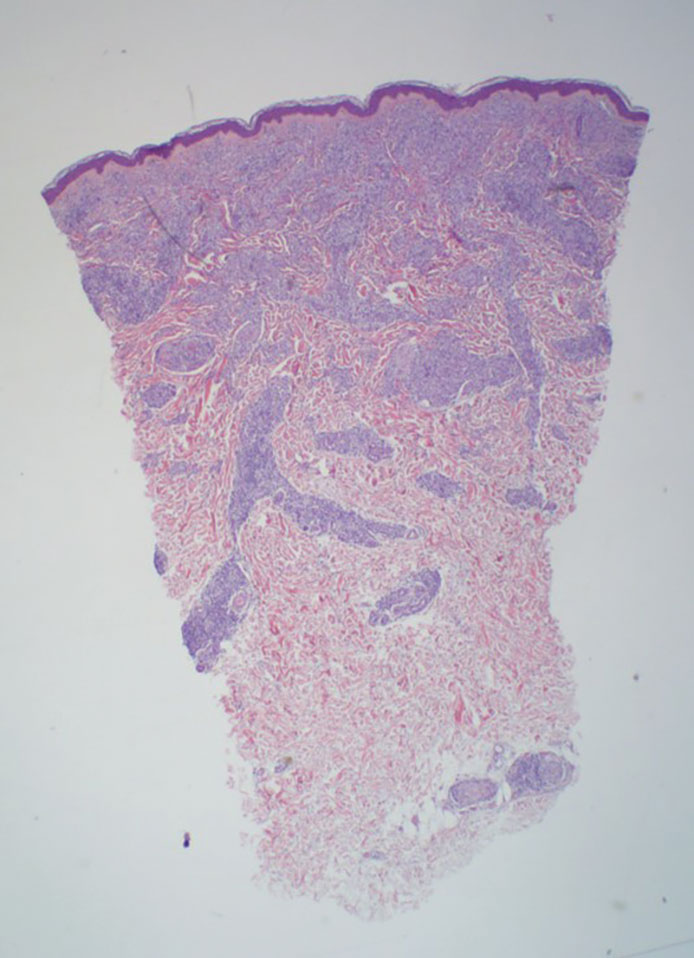

Nonhealing Friable Nodule on the Distal Edge of the Toe
Nonhealing Friable Nodule on the Distal Edge of the Toe
THE DIAGNOSIS: Squamoid Eccrine Ductal Carcinoma
Immunohistochemical staining of the biopsy specimen showed neoplastic aggregates that were diffusely positive for pancytokeratin and strongly positive for cytokeratin (CK) 5/6. Epithelial membrane antigen (EMA) and CK7 also were positive, CAM 5.2 was partially positive, and carcinoembryonic antigen (CEA) was focally positive (periluminal); S100 was negative. Given the histologic findings of irregular infiltrative cords and stranding exhibiting ductal differentiation in a fibrotic stroma in combination with the staining pattern, a diagnosis of squamous eccrine ductal carcinoma (SEDC) was made.
Squamoid eccrine ductal carcinoma is a rare primary cutaneous tumor with aggressive features that can be confused both clinically and histologically with squamous cell carcinoma (SCC). Histologically, SEDC is a biphasic tumor. If a shallow histologic specimen is obtained, it may be indistinguishable from a well-differentiated SCC (Figure 1). A deeper biopsy reveals irregular infiltrative cords and strands exhibiting ductal differentiation in a fibrotic stroma.1
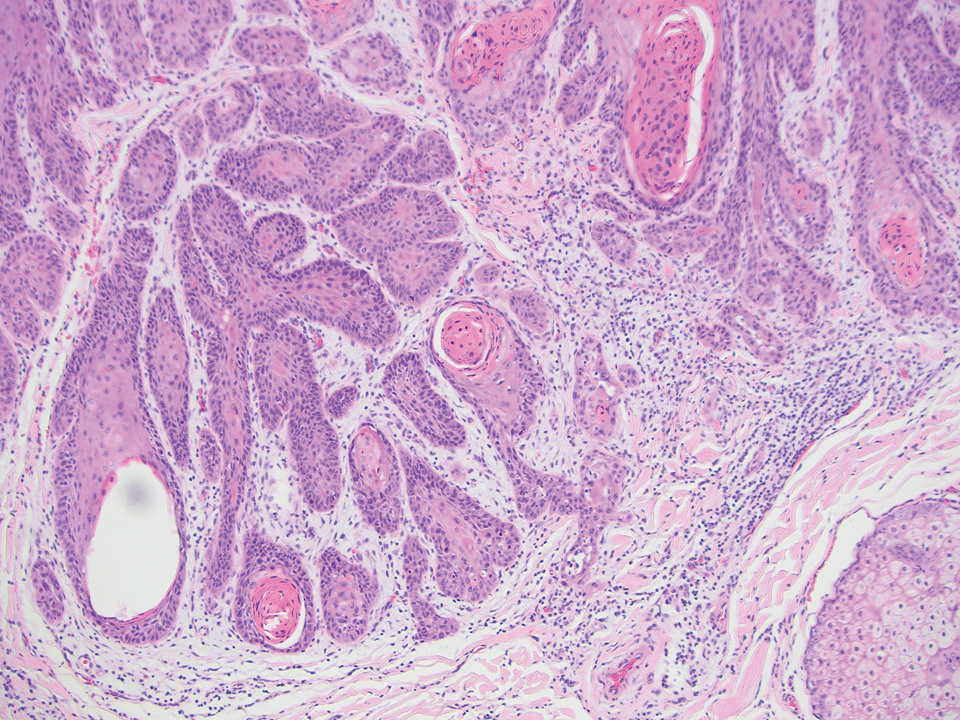
The immunohistochemical staining pattern of SEDC is similar to that of SCC, showing diffuse staining with pancytokeratin (AE1/AE3), CK 5/6, CK7, p63, and EMA. What distinguishes SEDC from SCC is that CEA highlights areas of glandular differentiation. An additional histologic feature seen commonly with SEDC is perineural invasion.
The etiology of SEDC remains controversial; although it originally was considered an aggressive variant of SCC along the same continuum as adenosquamous carcinoma, the fifth edition of the WHO Classification of Skin Tumors2 has categorized SEDC as an adnexal neoplasm. Our patient demonstrated an atypical presentation of this tumor, which has been most commonly described in the literature as manifesting on the head, neck, or upper extremities in older adults.3 Mohs micrographic surgery is the recommended treatment for this aggressive tumor.3
The differential diagnosis for SEDC includes microcystic adnexal carcinoma, porocarcinoma, and eccrine syringofibroadenoma. Microcystic adnexal carcinoma is a rare, low-grade tumor of the sweat glands that typically manifests as a firm pink papule or plaque in the head and neck region. Microscopically, it demonstrates cords of basaloid cells in a paisley-tie tadpole pattern with a dense pink to red stroma and horn cysts (Figure 2). Histologic differential diagnoses include syringoma, morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and trichoadenoma. Carcinoembryonic antigen stains positive in microcystic adnexal carcinoma, which helps distinguish it from basal cell carcinoma and SCC. Surgical excision or Mohs surgery are recommended for management.4
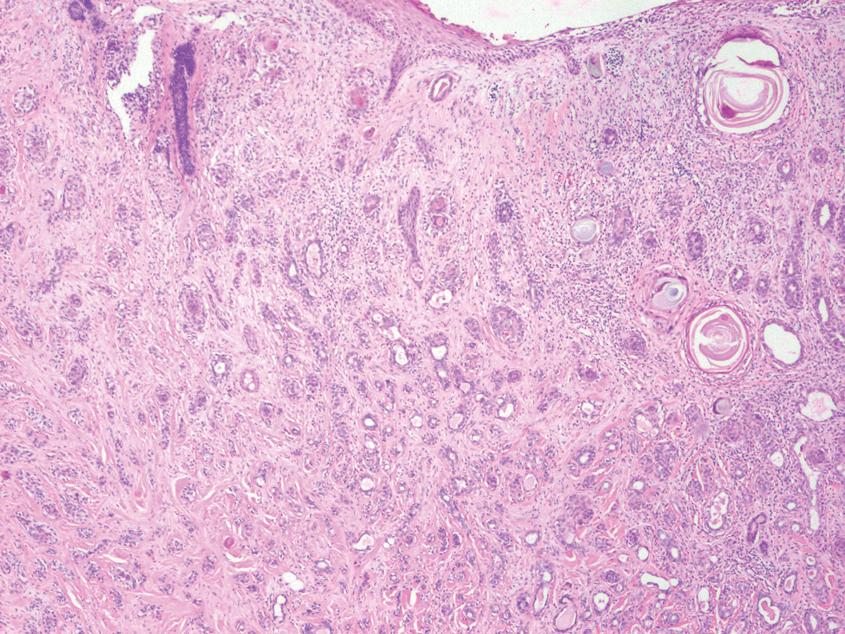
Porocarcinoma is a malignant skin tumor that originates from the intraepidermal sweat gland ducts. It also has been proposed that porocarcinoma develops from benign eccrine poroma. Porocarcinoma often is seen in elderly individuals, with a predilection for the lower extremities. Porocarcinoma demonstrates diverse clinical and histopathologic features, which can make diagnosis challenging. Histopathologically, porocarcinoma has an infiltrative growth pattern, with large basaloid epithelial cells that demonstrate ductal differentiation, cytologic atypia, increased mitotic activity, and tumor necrosis (Figure 3). Some porocarcinomas may exhibit squamous-cell, spindle-cell, or clear-cell differentiation. Neoplastic cells stain positive for CEA, EMA, and CD117, which can assist in distinguishing porocarcinoma from cutaneous SCC.5
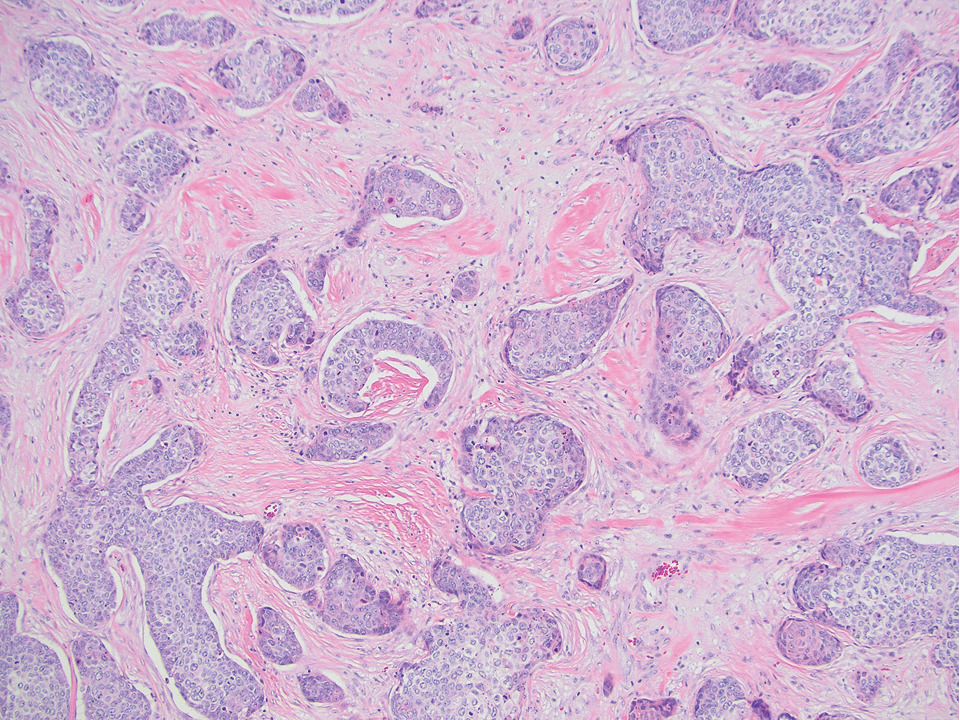
Eccrine syringofibroadenoma is an unusual benign cutaneous adnexal tumor that manifests mostly in individuals aged 40 years or older. It develops as single or multiple lesions that usually affect the lower extremities. Histologically, eccrine syringofibroadenoma demonstrates unique findings of anastomosing ducts and monomorphous epithelial cells within a fibrovascular stroma (Figure 4). On immunohistochemistry, it stains positive for EMA, CEA, high-molecular-weight kininogen, and filaggrin.6 Periodic acid–Schiff staining also is positive.
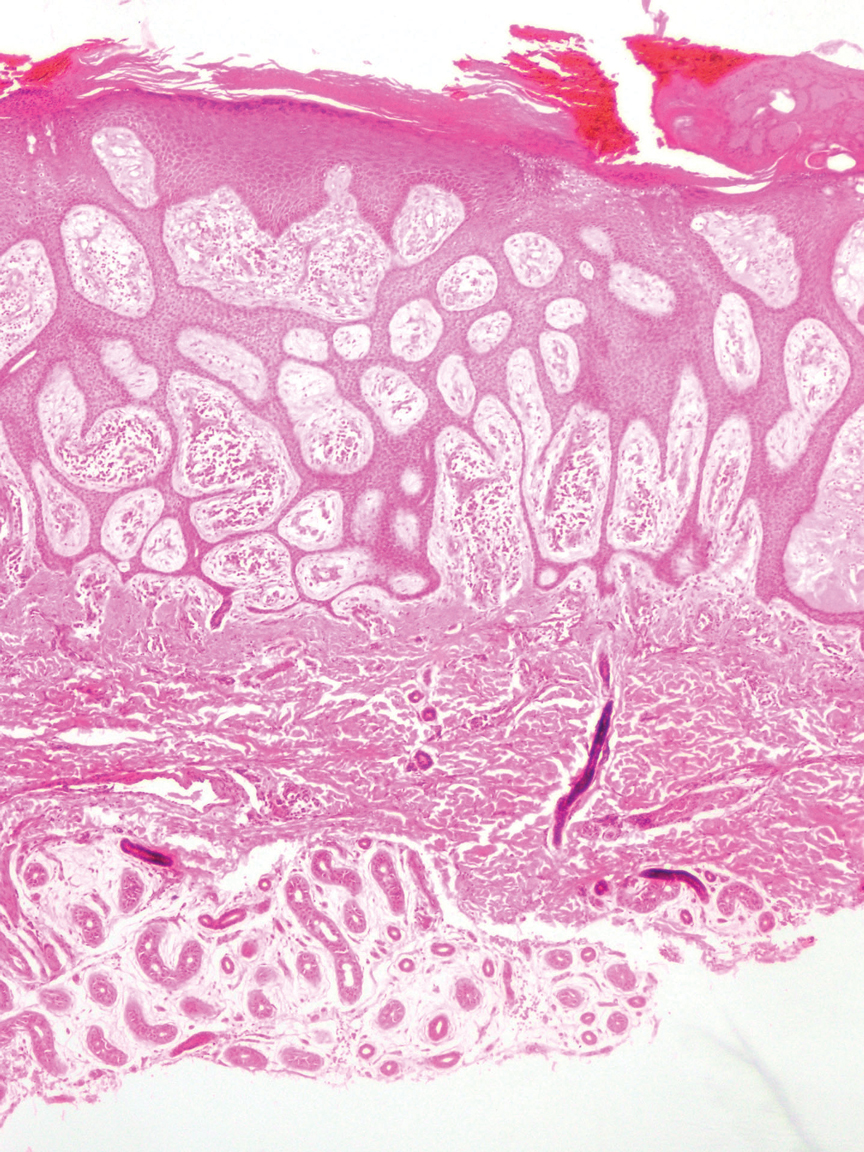
- Svoboda SA, Rush PS, Garofola CJ, et al. Squamoid eccrine ductal carcinoma. Cutis. 2021;107:E5-E9. doi:10.12788/cutis.0280
- WHO Classification of Tumours Editorial Board. Skin tumours. 5th ed. Lyon (France): International Agency for Research on Cancer; 2023.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760. doi:10.1097/PAS.0000000000000599
- Zito PM, Mazzoni T. Microcystic adnexal carcinoma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated April 24, 2023. Accessed August 3, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557857/
- Tsiogka A, Koumaki D, Kyriazopoulou M, et al. Eccrine porocarcinoma: a review of the literature. Diagnostics (Basel). 2023;13:8. doi:10.3390/diagnostics13081431
- Ko EJ, Park KY, Kwon HJ, et al. Eccrine syringofibroadenoma in a patient with long-standing exfoliative dermatitis. Ann Dermatol. 2016;28:765-768. doi:10.5021/ad.2016.28.6.765
THE DIAGNOSIS: Squamoid Eccrine Ductal Carcinoma
Immunohistochemical staining of the biopsy specimen showed neoplastic aggregates that were diffusely positive for pancytokeratin and strongly positive for cytokeratin (CK) 5/6. Epithelial membrane antigen (EMA) and CK7 also were positive, CAM 5.2 was partially positive, and carcinoembryonic antigen (CEA) was focally positive (periluminal); S100 was negative. Given the histologic findings of irregular infiltrative cords and stranding exhibiting ductal differentiation in a fibrotic stroma in combination with the staining pattern, a diagnosis of squamous eccrine ductal carcinoma (SEDC) was made.
Squamoid eccrine ductal carcinoma is a rare primary cutaneous tumor with aggressive features that can be confused both clinically and histologically with squamous cell carcinoma (SCC). Histologically, SEDC is a biphasic tumor. If a shallow histologic specimen is obtained, it may be indistinguishable from a well-differentiated SCC (Figure 1). A deeper biopsy reveals irregular infiltrative cords and strands exhibiting ductal differentiation in a fibrotic stroma.1
The immunohistochemical staining pattern of SEDC is similar to that of SCC, showing diffuse staining with pancytokeratin (AE1/AE3), CK 5/6, CK7, p63, and EMA. What distinguishes SEDC from SCC is that CEA highlights areas of glandular differentiation. An additional histologic feature seen commonly with SEDC is perineural invasion.
The etiology of SEDC remains controversial; although it originally was considered an aggressive variant of SCC along the same continuum as adenosquamous carcinoma, the fifth edition of the WHO Classification of Skin Tumors2 has categorized SEDC as an adnexal neoplasm. Our patient demonstrated an atypical presentation of this tumor, which has been most commonly described in the literature as manifesting on the head, neck, or upper extremities in older adults.3 Mohs micrographic surgery is the recommended treatment for this aggressive tumor.3
The differential diagnosis for SEDC includes microcystic adnexal carcinoma, porocarcinoma, and eccrine syringofibroadenoma. Microcystic adnexal carcinoma is a rare, low-grade tumor of the sweat glands that typically manifests as a firm pink papule or plaque in the head and neck region. Microscopically, it demonstrates cords of basaloid cells in a paisley-tie tadpole pattern with a dense pink to red stroma and horn cysts (Figure 2). Histologic differential diagnoses include syringoma, morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and trichoadenoma. Carcinoembryonic antigen stains positive in microcystic adnexal carcinoma, which helps distinguish it from basal cell carcinoma and SCC. Surgical excision or Mohs surgery are recommended for management.4
Porocarcinoma is a malignant skin tumor that originates from the intraepidermal sweat gland ducts. It also has been proposed that porocarcinoma develops from benign eccrine poroma. Porocarcinoma often is seen in elderly individuals, with a predilection for the lower extremities. Porocarcinoma demonstrates diverse clinical and histopathologic features, which can make diagnosis challenging. Histopathologically, porocarcinoma has an infiltrative growth pattern, with large basaloid epithelial cells that demonstrate ductal differentiation, cytologic atypia, increased mitotic activity, and tumor necrosis (Figure 3). Some porocarcinomas may exhibit squamous-cell, spindle-cell, or clear-cell differentiation. Neoplastic cells stain positive for CEA, EMA, and CD117, which can assist in distinguishing porocarcinoma from cutaneous SCC.5
Eccrine syringofibroadenoma is an unusual benign cutaneous adnexal tumor that manifests mostly in individuals aged 40 years or older. It develops as single or multiple lesions that usually affect the lower extremities. Histologically, eccrine syringofibroadenoma demonstrates unique findings of anastomosing ducts and monomorphous epithelial cells within a fibrovascular stroma (Figure 4). On immunohistochemistry, it stains positive for EMA, CEA, high-molecular-weight kininogen, and filaggrin.6 Periodic acid–Schiff staining also is positive.
THE DIAGNOSIS: Squamoid Eccrine Ductal Carcinoma
Immunohistochemical staining of the biopsy specimen showed neoplastic aggregates that were diffusely positive for pancytokeratin and strongly positive for cytokeratin (CK) 5/6. Epithelial membrane antigen (EMA) and CK7 also were positive, CAM 5.2 was partially positive, and carcinoembryonic antigen (CEA) was focally positive (periluminal); S100 was negative. Given the histologic findings of irregular infiltrative cords and stranding exhibiting ductal differentiation in a fibrotic stroma in combination with the staining pattern, a diagnosis of squamous eccrine ductal carcinoma (SEDC) was made.
Squamoid eccrine ductal carcinoma is a rare primary cutaneous tumor with aggressive features that can be confused both clinically and histologically with squamous cell carcinoma (SCC). Histologically, SEDC is a biphasic tumor. If a shallow histologic specimen is obtained, it may be indistinguishable from a well-differentiated SCC (Figure 1). A deeper biopsy reveals irregular infiltrative cords and strands exhibiting ductal differentiation in a fibrotic stroma.1
The immunohistochemical staining pattern of SEDC is similar to that of SCC, showing diffuse staining with pancytokeratin (AE1/AE3), CK 5/6, CK7, p63, and EMA. What distinguishes SEDC from SCC is that CEA highlights areas of glandular differentiation. An additional histologic feature seen commonly with SEDC is perineural invasion.
The etiology of SEDC remains controversial; although it originally was considered an aggressive variant of SCC along the same continuum as adenosquamous carcinoma, the fifth edition of the WHO Classification of Skin Tumors2 has categorized SEDC as an adnexal neoplasm. Our patient demonstrated an atypical presentation of this tumor, which has been most commonly described in the literature as manifesting on the head, neck, or upper extremities in older adults.3 Mohs micrographic surgery is the recommended treatment for this aggressive tumor.3
The differential diagnosis for SEDC includes microcystic adnexal carcinoma, porocarcinoma, and eccrine syringofibroadenoma. Microcystic adnexal carcinoma is a rare, low-grade tumor of the sweat glands that typically manifests as a firm pink papule or plaque in the head and neck region. Microscopically, it demonstrates cords of basaloid cells in a paisley-tie tadpole pattern with a dense pink to red stroma and horn cysts (Figure 2). Histologic differential diagnoses include syringoma, morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and trichoadenoma. Carcinoembryonic antigen stains positive in microcystic adnexal carcinoma, which helps distinguish it from basal cell carcinoma and SCC. Surgical excision or Mohs surgery are recommended for management.4
Porocarcinoma is a malignant skin tumor that originates from the intraepidermal sweat gland ducts. It also has been proposed that porocarcinoma develops from benign eccrine poroma. Porocarcinoma often is seen in elderly individuals, with a predilection for the lower extremities. Porocarcinoma demonstrates diverse clinical and histopathologic features, which can make diagnosis challenging. Histopathologically, porocarcinoma has an infiltrative growth pattern, with large basaloid epithelial cells that demonstrate ductal differentiation, cytologic atypia, increased mitotic activity, and tumor necrosis (Figure 3). Some porocarcinomas may exhibit squamous-cell, spindle-cell, or clear-cell differentiation. Neoplastic cells stain positive for CEA, EMA, and CD117, which can assist in distinguishing porocarcinoma from cutaneous SCC.5
Eccrine syringofibroadenoma is an unusual benign cutaneous adnexal tumor that manifests mostly in individuals aged 40 years or older. It develops as single or multiple lesions that usually affect the lower extremities. Histologically, eccrine syringofibroadenoma demonstrates unique findings of anastomosing ducts and monomorphous epithelial cells within a fibrovascular stroma (Figure 4). On immunohistochemistry, it stains positive for EMA, CEA, high-molecular-weight kininogen, and filaggrin.6 Periodic acid–Schiff staining also is positive.
- Svoboda SA, Rush PS, Garofola CJ, et al. Squamoid eccrine ductal carcinoma. Cutis. 2021;107:E5-E9. doi:10.12788/cutis.0280
- WHO Classification of Tumours Editorial Board. Skin tumours. 5th ed. Lyon (France): International Agency for Research on Cancer; 2023.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760. doi:10.1097/PAS.0000000000000599
- Zito PM, Mazzoni T. Microcystic adnexal carcinoma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated April 24, 2023. Accessed August 3, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557857/
- Tsiogka A, Koumaki D, Kyriazopoulou M, et al. Eccrine porocarcinoma: a review of the literature. Diagnostics (Basel). 2023;13:8. doi:10.3390/diagnostics13081431
- Ko EJ, Park KY, Kwon HJ, et al. Eccrine syringofibroadenoma in a patient with long-standing exfoliative dermatitis. Ann Dermatol. 2016;28:765-768. doi:10.5021/ad.2016.28.6.765
- Svoboda SA, Rush PS, Garofola CJ, et al. Squamoid eccrine ductal carcinoma. Cutis. 2021;107:E5-E9. doi:10.12788/cutis.0280
- WHO Classification of Tumours Editorial Board. Skin tumours. 5th ed. Lyon (France): International Agency for Research on Cancer; 2023.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760. doi:10.1097/PAS.0000000000000599
- Zito PM, Mazzoni T. Microcystic adnexal carcinoma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated April 24, 2023. Accessed August 3, 2025. https://www.ncbi.nlm.nih.gov/books/NBK557857/
- Tsiogka A, Koumaki D, Kyriazopoulou M, et al. Eccrine porocarcinoma: a review of the literature. Diagnostics (Basel). 2023;13:8. doi:10.3390/diagnostics13081431
- Ko EJ, Park KY, Kwon HJ, et al. Eccrine syringofibroadenoma in a patient with long-standing exfoliative dermatitis. Ann Dermatol. 2016;28:765-768. doi:10.5021/ad.2016.28.6.765
Nonhealing Friable Nodule on the Distal Edge of the Toe
Nonhealing Friable Nodule on the Distal Edge of the Toe
A 37-year-old woman with no notable medical history presented to the dermatology clinic with a nonhealing wound on the left fifth toe of 10 month’s duration. The patient reported that the wound developed after burning the toe on an indoor space heater. Physical examination revealed a friable pink papule with a hemorrhagic crust. A biopsy of the lesion was performed.
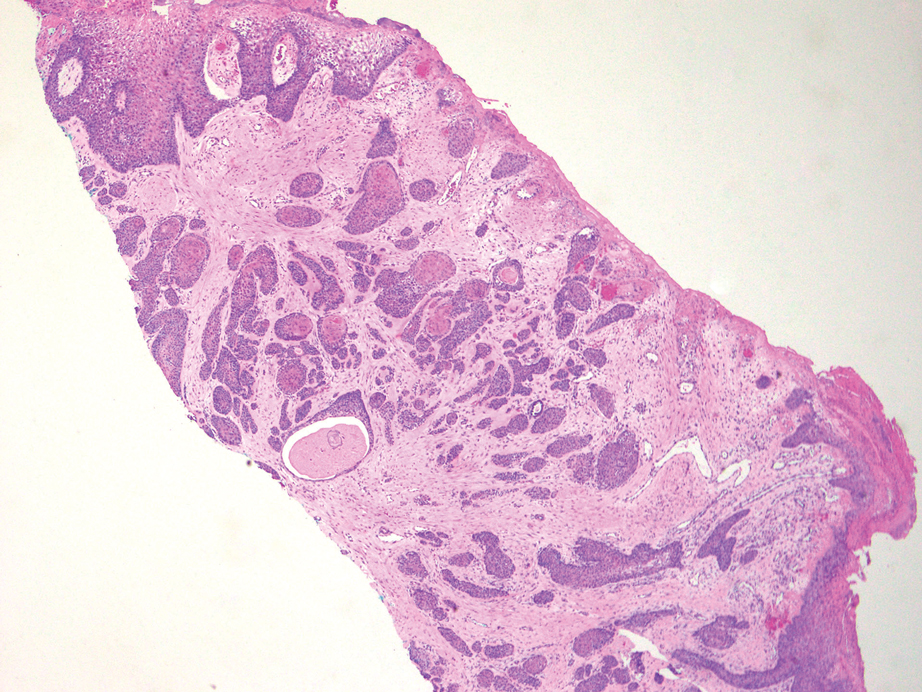
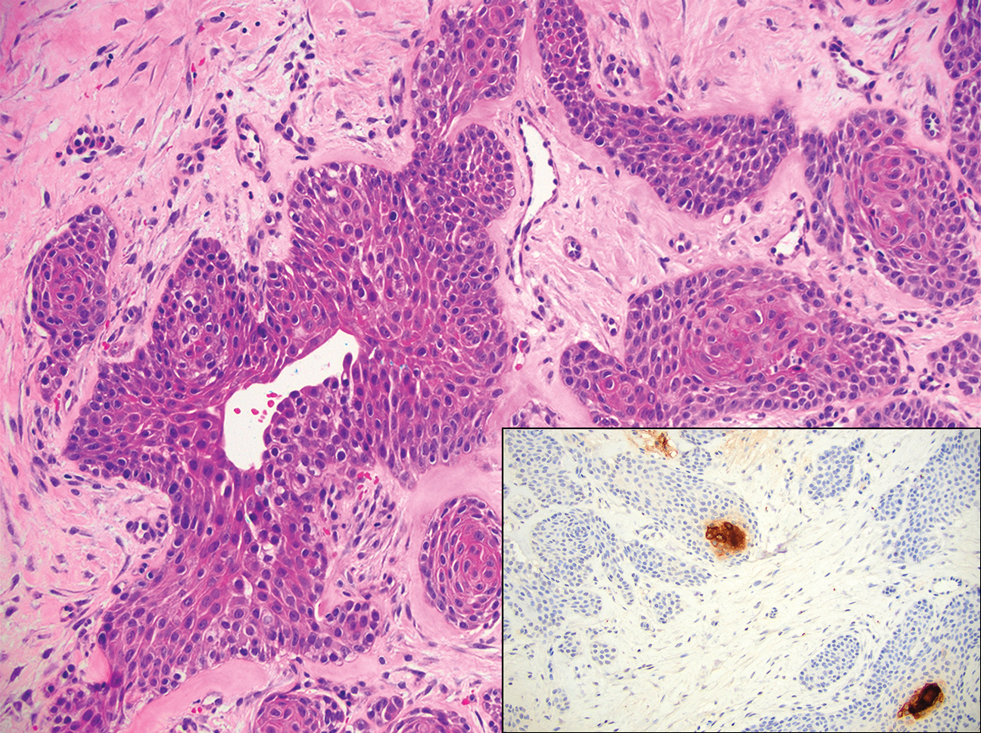

Scattered Umbilicated Papules on the Cheek, Neck, and Arms
Scattered Umbilicated Papules on the Cheek, Neck, and Arms
THE DIAGNOSIS: Mpox Virus
The histopathologic features of mpox virus infection may vary depending on the stage of evolution; findings include ballooning degeneration with multinucleated keratinocytes, acanthosis, spongiosis, a neutrophil-rich inflammatory infiltrate, and eosinophilic intracytoplasmic (Guarnieri) inclusion bodies (quiz image inset [arrows]). Prominent neutrophil exocytosis also has been described and may be a characteristic feature in the pustular stage.1,2 A pattern of interface dermatitis also has been observed on histopathology.3 In our patient, the diagnosis of mpox initially was made by clinical and histopathologic correlation and exclusion of other entities in the differential diagnosis. The diagnosis subsequently was confirmed by real-time polymerase chain reaction. The patient received treatment with tecovirimat, but lesions progressed over the following 6 weeks. He subsequently died due to sepsis and multiorgan failure secondary to AIDS.
Mpox is a zoonotic, double-stranded DNA virus of the genus Orthopoxvirus in the family Poxviridae.4 It is transmitted to humans via direct contact with infected animals, most commonly small mammals such as monkeys, squirrels, and rodents. Mpox also may be transmitted between humans through direct contact with bodily fluids, skin and mucosal lesions, respiratory droplets, or fomites. Mpox infection typically begins with a nonspecific flulike prodrome after a 5- to 21-day incubation period, followed by skin lesions of variable morphology affecting any region of the body. Clinically, mpox lesions have been reported to evolve through macular, papular, and vesiculopustular phases, followed by resolution with crusting. Lesions may occur anywhere on the body but frequently manifest on the face then spread centrifugally across the body, with various phases observed simultaneously.5 A worldwide outbreak in 2022 involved larger numbers of cases in nonendemic areas, primarily due to skin-to-skin contact, with predominant anal and genital localization of the lesions as well as fewer prodromal symptoms.6
The differential diagnosis of crusted and umbilicated papules includes disseminated herpesvirus infection, molluscum contagiosum, disseminated cryptococcosis, and histoplasmosis. Additional causative organisms to consider include Penicillium, Mycobacterium tuberculosis and nontuberculous mycobacteria, as well as Sporothrix schenckii.
Herpesvirus infections may have similar clinical and histopathologic findings to mpox. Histopathologically, herpes simplex virus (HSV) and varicella zoster virus (VZV) are essentially identical; both demonstrate ballooning and reticular epidermal degeneration, chromatin condensation, nuclear degeneration, multinucleated keratinocytes with steel-gray nuclei, and prominent epidermal acantholysis with an inflammatory infiltrate (Figure 1). However, involvement of folliculosebaceous units may favor a diagnosis of VZV. Immunohistochemical staining can further differentiate between HSV and VZV.7 While mpox may have features that overlap with both HSV and VZV, including ballooning degeneration and multinucleated keratinocytes with nuclear degeneration, acantholysis is a less commonly reported feature of mpox, and mpox virus infection is characterized by intracytoplasmic (Guarnieri) inclusion bodies rather than the intranuclear inclusion bodies of HSV and VZV.2,5 The presence of Guarnieri bodies in mpox may further help to distinguish mpox from HSV infection on routine histology.
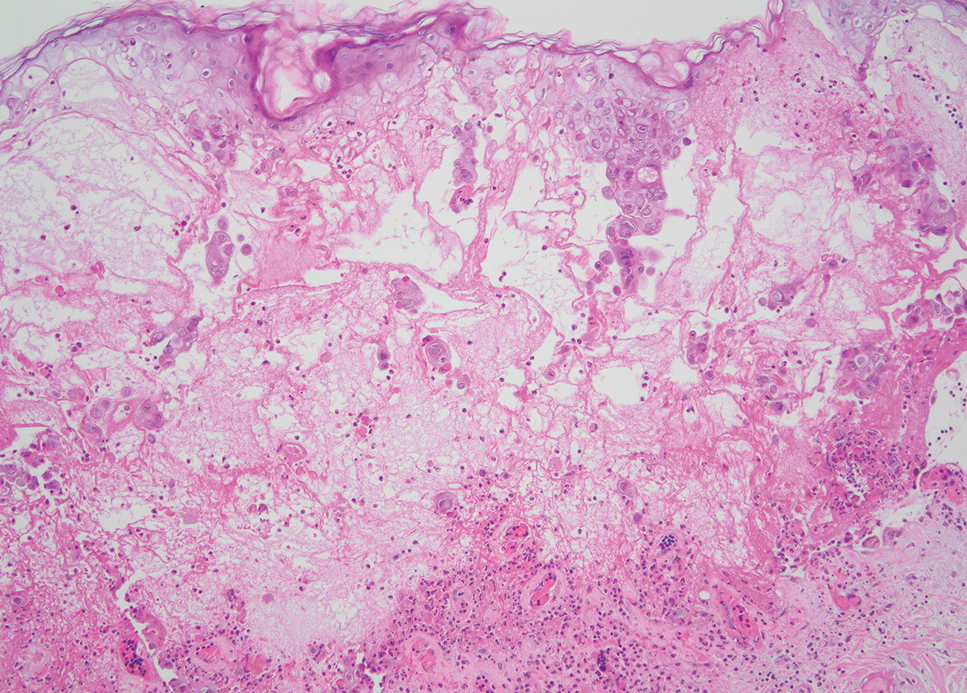
Molluscum contagiosum infection typically manifests as multiple umbilicated papules at sites of inoculation. Large lesions may be seen in the setting of immunosuppression; however, they usually do not progress to vesicular, pustular, or crusted morphologies. Histopathology demonstrates a cup-shaped invagination of the epidermis into the dermis and proliferative rete ridges that descend downward and encircle the dermis with large eosinophilic intracytoplasmic inclusion (Henderson-Patterson) bodies (Figure 2).8
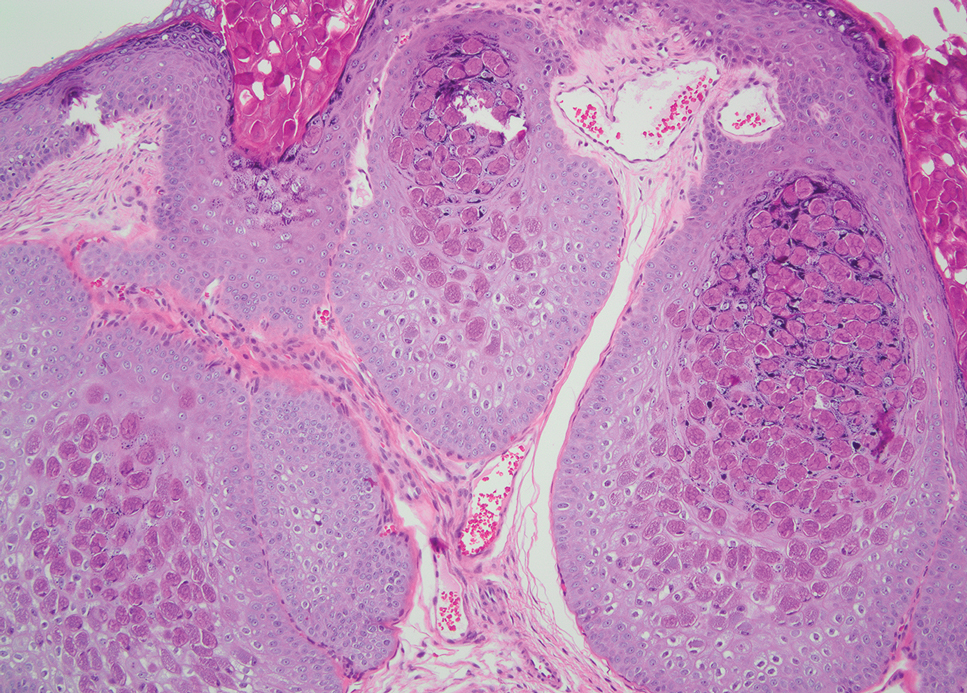
Disseminated cryptococcus infection is caused by the invasive fungus Cryptococcus neoformans and is characterized by meningitis along with fever, malaise, headache, neck stiffness, photophobia, nausea, vomiting, pneumonia with cough and dyspnea, and skin rash, most commonly in immunocompromised individuals.9 Skin lesions are a sign of disseminated infection and can manifest as umbilicated or molluscumlike lesions. Histopathology of cryptococcosis demonstrates a granulomatous dermal infiltrate with neutrophils and pleomorphic yeasts measuring 4 µm to 6 µm with refringent capsules.10 Staining with Grocott methenamine silver and/or mucicarmine for yeast capsules can help to identify organisms (Figure 3).
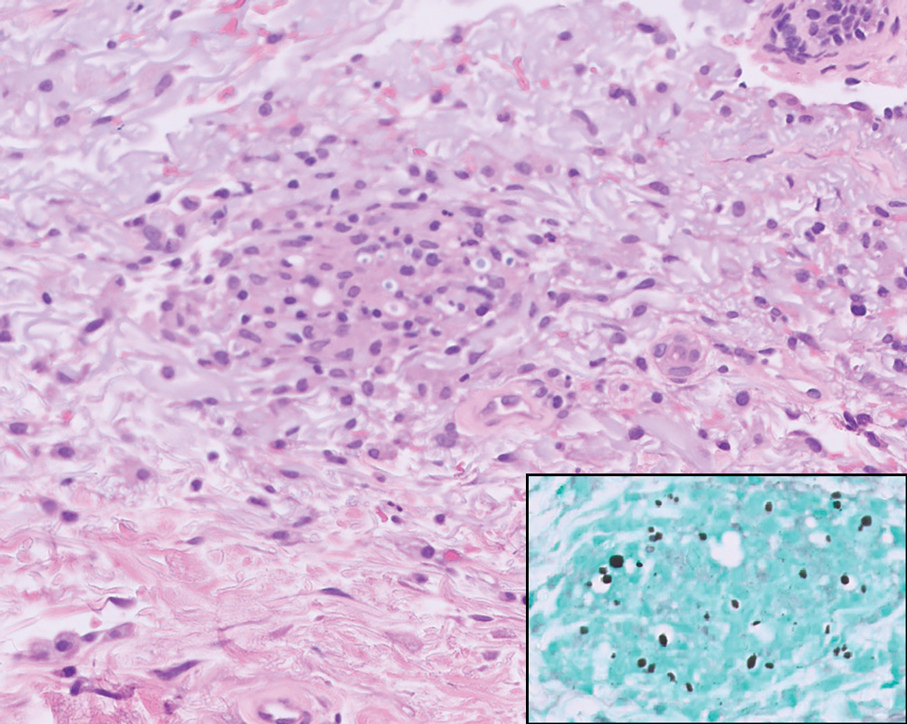
Cutaneous histoplasmosis is caused by Histoplasma capsulatum, a dimorphic fungus that can lead to pulmonary, cutaneous, and disseminated disease, often in immunocompromised patients.11 Cutaneous disease may manifest with molluscumlike or verrucous papules and plaques. Histopathologic examination reveals diffuse suppurative and granulomatous infiltrates with foamy histiocytes and multinucleated giant cells, containing intracellular and extracellular yeasts measuring 1µm to 5µm, surrounded by a clear halo visible with Grocott methenamine silver stain (Figure 4).
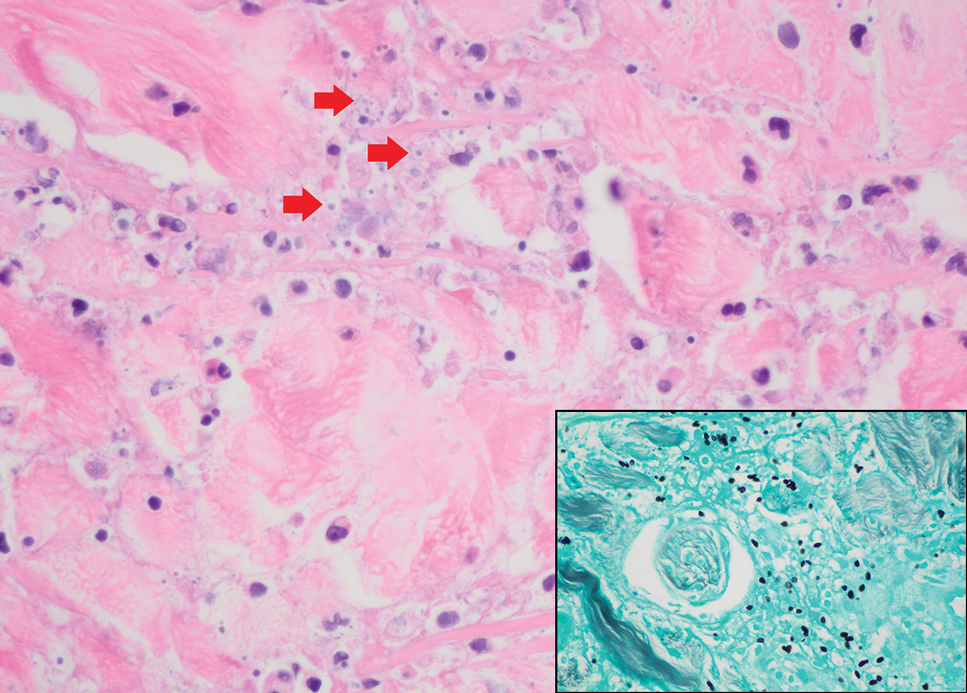
×600). Grocott methenamine silver staining highlights numerous intracellular yeasts (inset, original magnification ×600).
Spreading cutaneous lesions in an immunocompromised individual may be the presentation of multiple infectious etiologies. With the recent rise in mpox cases occurring in nonendemic areas, clinicians should be aware of the spectrum of clinical findings that may occur. Notably, more than one infection may be present in severely immunocompromised individuals, as seen in our patient with chronic orolabial HSV-2 and acute mpox infection. Thorough clinical, histopathologic, and laboratory investigations are necessary for timely diagnosis, appropriate treatment, and exclusion of other life-threatening conditions.
- Moltrasio C, Boggio FL, Romagnuolo M, et al. Monkeypox: a histopathological and transmission electron microscopy study. Microorganisms. 2023;11:1781-1793. doi:10.3390/microorganisms11071781
- Ortins-Pina A, Hegemann B, Saggini A, et al. Histopathological features of human mpox: report of two cases and review of the literature. J Cutan Pathol. 2023;50:706-710. doi:10.1111/cup.14398
- Chalali F, Merlant M, Truong A, et al. Histological features associated with human mpox virus infection in 2022 outbreak in a nonendemic country. Clin Infect Dis. 21;76:1132-1135. doi:10.1093/cid/ciac856.
- Mpox (monkeypox). World Health Organization. https://www.who.int/health-topics/monkeypox/#tab=tab_1. Accessed August 6, 2025.
- Petersen E, Kantele A, Koopmans M, et al. Human monkeypox: epidemiologic and clinical characteristics, diagnosis, and prevention. Infect Dis Clin North Am. 2019;33:1027-1043. doi:10.1016/j.idc.2019.03.001
- Philpott D, Hughes CM, Alroy KA, et al. Epidemiologic and clinical characteristics of monkeypox cases — United States, May 17–July 22, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1018-1022. doi:10.15585 /mmwr.mm7132e3
- Nikkels AF, Debrus S, Sadzot-Delvaux C, et al. Comparative immunohistochemical study of herpes simplex and varicella-zoster infections. Virchows Arch A Pathol Anat Histopathol. 1993;422:121-126. doi:10.1007 /BF01607163
- Badri T, Gandhi GR. Molluscum Contagiosum. StatPearls [Internet]. StatPearls Publishing; 2025. Updated March 27, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK441898/
- Mada PK, Jamil RT, Alam MU. Cryptococcus. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 7, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK431060/
- Hayashida MZ, Seque CA, Pasin VP, et al. Disseminated cryptococcosis with skin lesions: report of a case series. An Bras Dermatol. 2017;92:69-72. doi:10.1590/abd1806-4841.20176343
- Mustari AP, Rao S, Keshavamurthy V, et al. Dermoscopic evaluation of cutaneous histoplasmosis. Indian J Dermatol Venereol Leprol. 2023;19:1-4. doi:10.25259/IJDVL_889_2022
THE DIAGNOSIS: Mpox Virus
The histopathologic features of mpox virus infection may vary depending on the stage of evolution; findings include ballooning degeneration with multinucleated keratinocytes, acanthosis, spongiosis, a neutrophil-rich inflammatory infiltrate, and eosinophilic intracytoplasmic (Guarnieri) inclusion bodies (quiz image inset [arrows]). Prominent neutrophil exocytosis also has been described and may be a characteristic feature in the pustular stage.1,2 A pattern of interface dermatitis also has been observed on histopathology.3 In our patient, the diagnosis of mpox initially was made by clinical and histopathologic correlation and exclusion of other entities in the differential diagnosis. The diagnosis subsequently was confirmed by real-time polymerase chain reaction. The patient received treatment with tecovirimat, but lesions progressed over the following 6 weeks. He subsequently died due to sepsis and multiorgan failure secondary to AIDS.
Mpox is a zoonotic, double-stranded DNA virus of the genus Orthopoxvirus in the family Poxviridae.4 It is transmitted to humans via direct contact with infected animals, most commonly small mammals such as monkeys, squirrels, and rodents. Mpox also may be transmitted between humans through direct contact with bodily fluids, skin and mucosal lesions, respiratory droplets, or fomites. Mpox infection typically begins with a nonspecific flulike prodrome after a 5- to 21-day incubation period, followed by skin lesions of variable morphology affecting any region of the body. Clinically, mpox lesions have been reported to evolve through macular, papular, and vesiculopustular phases, followed by resolution with crusting. Lesions may occur anywhere on the body but frequently manifest on the face then spread centrifugally across the body, with various phases observed simultaneously.5 A worldwide outbreak in 2022 involved larger numbers of cases in nonendemic areas, primarily due to skin-to-skin contact, with predominant anal and genital localization of the lesions as well as fewer prodromal symptoms.6
The differential diagnosis of crusted and umbilicated papules includes disseminated herpesvirus infection, molluscum contagiosum, disseminated cryptococcosis, and histoplasmosis. Additional causative organisms to consider include Penicillium, Mycobacterium tuberculosis and nontuberculous mycobacteria, as well as Sporothrix schenckii.
Herpesvirus infections may have similar clinical and histopathologic findings to mpox. Histopathologically, herpes simplex virus (HSV) and varicella zoster virus (VZV) are essentially identical; both demonstrate ballooning and reticular epidermal degeneration, chromatin condensation, nuclear degeneration, multinucleated keratinocytes with steel-gray nuclei, and prominent epidermal acantholysis with an inflammatory infiltrate (Figure 1). However, involvement of folliculosebaceous units may favor a diagnosis of VZV. Immunohistochemical staining can further differentiate between HSV and VZV.7 While mpox may have features that overlap with both HSV and VZV, including ballooning degeneration and multinucleated keratinocytes with nuclear degeneration, acantholysis is a less commonly reported feature of mpox, and mpox virus infection is characterized by intracytoplasmic (Guarnieri) inclusion bodies rather than the intranuclear inclusion bodies of HSV and VZV.2,5 The presence of Guarnieri bodies in mpox may further help to distinguish mpox from HSV infection on routine histology.
Molluscum contagiosum infection typically manifests as multiple umbilicated papules at sites of inoculation. Large lesions may be seen in the setting of immunosuppression; however, they usually do not progress to vesicular, pustular, or crusted morphologies. Histopathology demonstrates a cup-shaped invagination of the epidermis into the dermis and proliferative rete ridges that descend downward and encircle the dermis with large eosinophilic intracytoplasmic inclusion (Henderson-Patterson) bodies (Figure 2).8
Disseminated cryptococcus infection is caused by the invasive fungus Cryptococcus neoformans and is characterized by meningitis along with fever, malaise, headache, neck stiffness, photophobia, nausea, vomiting, pneumonia with cough and dyspnea, and skin rash, most commonly in immunocompromised individuals.9 Skin lesions are a sign of disseminated infection and can manifest as umbilicated or molluscumlike lesions. Histopathology of cryptococcosis demonstrates a granulomatous dermal infiltrate with neutrophils and pleomorphic yeasts measuring 4 µm to 6 µm with refringent capsules.10 Staining with Grocott methenamine silver and/or mucicarmine for yeast capsules can help to identify organisms (Figure 3).
Cutaneous histoplasmosis is caused by Histoplasma capsulatum, a dimorphic fungus that can lead to pulmonary, cutaneous, and disseminated disease, often in immunocompromised patients.11 Cutaneous disease may manifest with molluscumlike or verrucous papules and plaques. Histopathologic examination reveals diffuse suppurative and granulomatous infiltrates with foamy histiocytes and multinucleated giant cells, containing intracellular and extracellular yeasts measuring 1µm to 5µm, surrounded by a clear halo visible with Grocott methenamine silver stain (Figure 4).
×600). Grocott methenamine silver staining highlights numerous intracellular yeasts (inset, original magnification ×600).
Spreading cutaneous lesions in an immunocompromised individual may be the presentation of multiple infectious etiologies. With the recent rise in mpox cases occurring in nonendemic areas, clinicians should be aware of the spectrum of clinical findings that may occur. Notably, more than one infection may be present in severely immunocompromised individuals, as seen in our patient with chronic orolabial HSV-2 and acute mpox infection. Thorough clinical, histopathologic, and laboratory investigations are necessary for timely diagnosis, appropriate treatment, and exclusion of other life-threatening conditions.
THE DIAGNOSIS: Mpox Virus
The histopathologic features of mpox virus infection may vary depending on the stage of evolution; findings include ballooning degeneration with multinucleated keratinocytes, acanthosis, spongiosis, a neutrophil-rich inflammatory infiltrate, and eosinophilic intracytoplasmic (Guarnieri) inclusion bodies (quiz image inset [arrows]). Prominent neutrophil exocytosis also has been described and may be a characteristic feature in the pustular stage.1,2 A pattern of interface dermatitis also has been observed on histopathology.3 In our patient, the diagnosis of mpox initially was made by clinical and histopathologic correlation and exclusion of other entities in the differential diagnosis. The diagnosis subsequently was confirmed by real-time polymerase chain reaction. The patient received treatment with tecovirimat, but lesions progressed over the following 6 weeks. He subsequently died due to sepsis and multiorgan failure secondary to AIDS.
Mpox is a zoonotic, double-stranded DNA virus of the genus Orthopoxvirus in the family Poxviridae.4 It is transmitted to humans via direct contact with infected animals, most commonly small mammals such as monkeys, squirrels, and rodents. Mpox also may be transmitted between humans through direct contact with bodily fluids, skin and mucosal lesions, respiratory droplets, or fomites. Mpox infection typically begins with a nonspecific flulike prodrome after a 5- to 21-day incubation period, followed by skin lesions of variable morphology affecting any region of the body. Clinically, mpox lesions have been reported to evolve through macular, papular, and vesiculopustular phases, followed by resolution with crusting. Lesions may occur anywhere on the body but frequently manifest on the face then spread centrifugally across the body, with various phases observed simultaneously.5 A worldwide outbreak in 2022 involved larger numbers of cases in nonendemic areas, primarily due to skin-to-skin contact, with predominant anal and genital localization of the lesions as well as fewer prodromal symptoms.6
The differential diagnosis of crusted and umbilicated papules includes disseminated herpesvirus infection, molluscum contagiosum, disseminated cryptococcosis, and histoplasmosis. Additional causative organisms to consider include Penicillium, Mycobacterium tuberculosis and nontuberculous mycobacteria, as well as Sporothrix schenckii.
Herpesvirus infections may have similar clinical and histopathologic findings to mpox. Histopathologically, herpes simplex virus (HSV) and varicella zoster virus (VZV) are essentially identical; both demonstrate ballooning and reticular epidermal degeneration, chromatin condensation, nuclear degeneration, multinucleated keratinocytes with steel-gray nuclei, and prominent epidermal acantholysis with an inflammatory infiltrate (Figure 1). However, involvement of folliculosebaceous units may favor a diagnosis of VZV. Immunohistochemical staining can further differentiate between HSV and VZV.7 While mpox may have features that overlap with both HSV and VZV, including ballooning degeneration and multinucleated keratinocytes with nuclear degeneration, acantholysis is a less commonly reported feature of mpox, and mpox virus infection is characterized by intracytoplasmic (Guarnieri) inclusion bodies rather than the intranuclear inclusion bodies of HSV and VZV.2,5 The presence of Guarnieri bodies in mpox may further help to distinguish mpox from HSV infection on routine histology.
Molluscum contagiosum infection typically manifests as multiple umbilicated papules at sites of inoculation. Large lesions may be seen in the setting of immunosuppression; however, they usually do not progress to vesicular, pustular, or crusted morphologies. Histopathology demonstrates a cup-shaped invagination of the epidermis into the dermis and proliferative rete ridges that descend downward and encircle the dermis with large eosinophilic intracytoplasmic inclusion (Henderson-Patterson) bodies (Figure 2).8
Disseminated cryptococcus infection is caused by the invasive fungus Cryptococcus neoformans and is characterized by meningitis along with fever, malaise, headache, neck stiffness, photophobia, nausea, vomiting, pneumonia with cough and dyspnea, and skin rash, most commonly in immunocompromised individuals.9 Skin lesions are a sign of disseminated infection and can manifest as umbilicated or molluscumlike lesions. Histopathology of cryptococcosis demonstrates a granulomatous dermal infiltrate with neutrophils and pleomorphic yeasts measuring 4 µm to 6 µm with refringent capsules.10 Staining with Grocott methenamine silver and/or mucicarmine for yeast capsules can help to identify organisms (Figure 3).
Cutaneous histoplasmosis is caused by Histoplasma capsulatum, a dimorphic fungus that can lead to pulmonary, cutaneous, and disseminated disease, often in immunocompromised patients.11 Cutaneous disease may manifest with molluscumlike or verrucous papules and plaques. Histopathologic examination reveals diffuse suppurative and granulomatous infiltrates with foamy histiocytes and multinucleated giant cells, containing intracellular and extracellular yeasts measuring 1µm to 5µm, surrounded by a clear halo visible with Grocott methenamine silver stain (Figure 4).
×600). Grocott methenamine silver staining highlights numerous intracellular yeasts (inset, original magnification ×600).
Spreading cutaneous lesions in an immunocompromised individual may be the presentation of multiple infectious etiologies. With the recent rise in mpox cases occurring in nonendemic areas, clinicians should be aware of the spectrum of clinical findings that may occur. Notably, more than one infection may be present in severely immunocompromised individuals, as seen in our patient with chronic orolabial HSV-2 and acute mpox infection. Thorough clinical, histopathologic, and laboratory investigations are necessary for timely diagnosis, appropriate treatment, and exclusion of other life-threatening conditions.
- Moltrasio C, Boggio FL, Romagnuolo M, et al. Monkeypox: a histopathological and transmission electron microscopy study. Microorganisms. 2023;11:1781-1793. doi:10.3390/microorganisms11071781
- Ortins-Pina A, Hegemann B, Saggini A, et al. Histopathological features of human mpox: report of two cases and review of the literature. J Cutan Pathol. 2023;50:706-710. doi:10.1111/cup.14398
- Chalali F, Merlant M, Truong A, et al. Histological features associated with human mpox virus infection in 2022 outbreak in a nonendemic country. Clin Infect Dis. 21;76:1132-1135. doi:10.1093/cid/ciac856.
- Mpox (monkeypox). World Health Organization. https://www.who.int/health-topics/monkeypox/#tab=tab_1. Accessed August 6, 2025.
- Petersen E, Kantele A, Koopmans M, et al. Human monkeypox: epidemiologic and clinical characteristics, diagnosis, and prevention. Infect Dis Clin North Am. 2019;33:1027-1043. doi:10.1016/j.idc.2019.03.001
- Philpott D, Hughes CM, Alroy KA, et al. Epidemiologic and clinical characteristics of monkeypox cases — United States, May 17–July 22, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1018-1022. doi:10.15585 /mmwr.mm7132e3
- Nikkels AF, Debrus S, Sadzot-Delvaux C, et al. Comparative immunohistochemical study of herpes simplex and varicella-zoster infections. Virchows Arch A Pathol Anat Histopathol. 1993;422:121-126. doi:10.1007 /BF01607163
- Badri T, Gandhi GR. Molluscum Contagiosum. StatPearls [Internet]. StatPearls Publishing; 2025. Updated March 27, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK441898/
- Mada PK, Jamil RT, Alam MU. Cryptococcus. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 7, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK431060/
- Hayashida MZ, Seque CA, Pasin VP, et al. Disseminated cryptococcosis with skin lesions: report of a case series. An Bras Dermatol. 2017;92:69-72. doi:10.1590/abd1806-4841.20176343
- Mustari AP, Rao S, Keshavamurthy V, et al. Dermoscopic evaluation of cutaneous histoplasmosis. Indian J Dermatol Venereol Leprol. 2023;19:1-4. doi:10.25259/IJDVL_889_2022
- Moltrasio C, Boggio FL, Romagnuolo M, et al. Monkeypox: a histopathological and transmission electron microscopy study. Microorganisms. 2023;11:1781-1793. doi:10.3390/microorganisms11071781
- Ortins-Pina A, Hegemann B, Saggini A, et al. Histopathological features of human mpox: report of two cases and review of the literature. J Cutan Pathol. 2023;50:706-710. doi:10.1111/cup.14398
- Chalali F, Merlant M, Truong A, et al. Histological features associated with human mpox virus infection in 2022 outbreak in a nonendemic country. Clin Infect Dis. 21;76:1132-1135. doi:10.1093/cid/ciac856.
- Mpox (monkeypox). World Health Organization. https://www.who.int/health-topics/monkeypox/#tab=tab_1. Accessed August 6, 2025.
- Petersen E, Kantele A, Koopmans M, et al. Human monkeypox: epidemiologic and clinical characteristics, diagnosis, and prevention. Infect Dis Clin North Am. 2019;33:1027-1043. doi:10.1016/j.idc.2019.03.001
- Philpott D, Hughes CM, Alroy KA, et al. Epidemiologic and clinical characteristics of monkeypox cases — United States, May 17–July 22, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:1018-1022. doi:10.15585 /mmwr.mm7132e3
- Nikkels AF, Debrus S, Sadzot-Delvaux C, et al. Comparative immunohistochemical study of herpes simplex and varicella-zoster infections. Virchows Arch A Pathol Anat Histopathol. 1993;422:121-126. doi:10.1007 /BF01607163
- Badri T, Gandhi GR. Molluscum Contagiosum. StatPearls [Internet]. StatPearls Publishing; 2025. Updated March 27, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK441898/
- Mada PK, Jamil RT, Alam MU. Cryptococcus. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 7, 2023. Accessed August 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK431060/
- Hayashida MZ, Seque CA, Pasin VP, et al. Disseminated cryptococcosis with skin lesions: report of a case series. An Bras Dermatol. 2017;92:69-72. doi:10.1590/abd1806-4841.20176343
- Mustari AP, Rao S, Keshavamurthy V, et al. Dermoscopic evaluation of cutaneous histoplasmosis. Indian J Dermatol Venereol Leprol. 2023;19:1-4. doi:10.25259/IJDVL_889_2022
Scattered Umbilicated Papules on the Cheek, Neck, and Arms
Scattered Umbilicated Papules on the Cheek, Neck, and Arms
A 42-year-old man with a history of multidrug-resistant HIV/AIDS presented to the emergency department for evaluation of pruritic, scattered, umbilicated papules on the left cheek, neck, and arms of 3 days’ duration. The patient’s most recent CD4+ T-cell count 6 weeks prior to the development of the rash was 1 cell/mm3. He was noncompliant with antiretroviral therapy. He reported that the lesions had progressed rapidly, starting on the face and extending down the neck and arms. Physical examination revealed scattered umbilicated and centrally crusted papules and plaques on the left cheek, neck, and arms. Erosions involving the oral mucosa also were noted, which the patient reported had been present for several weeks. An oral swab was positive for herpes simplex virus 2 on polymerase chain reaction. A shave biopsy of a lesion from the left cheek was performed.
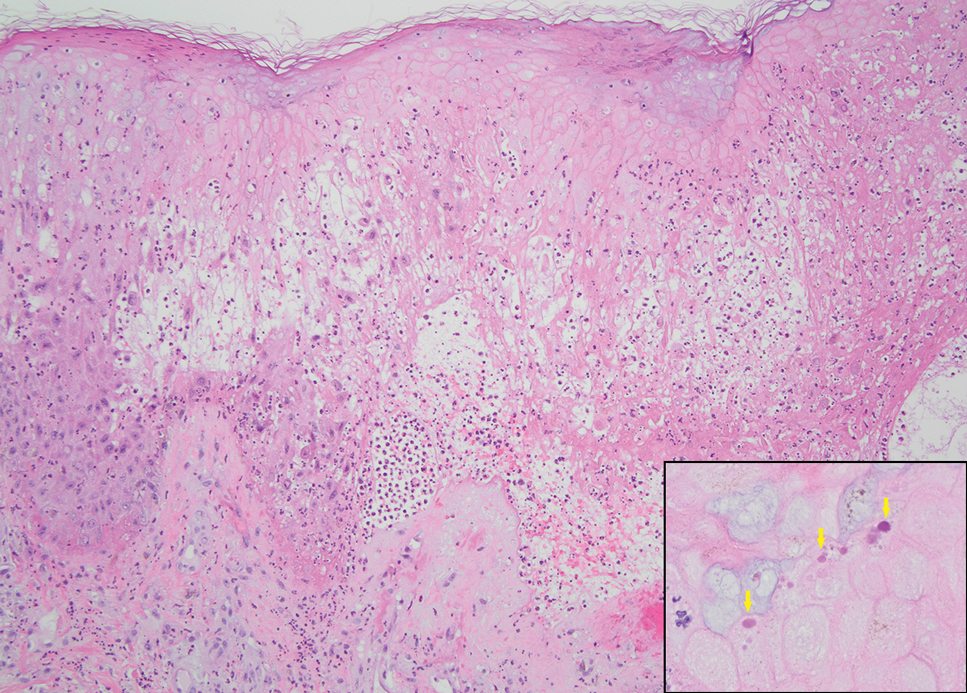

Exophytic Papule on the Chin of a Child
Exophytic Papule on the Chin of a Child
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
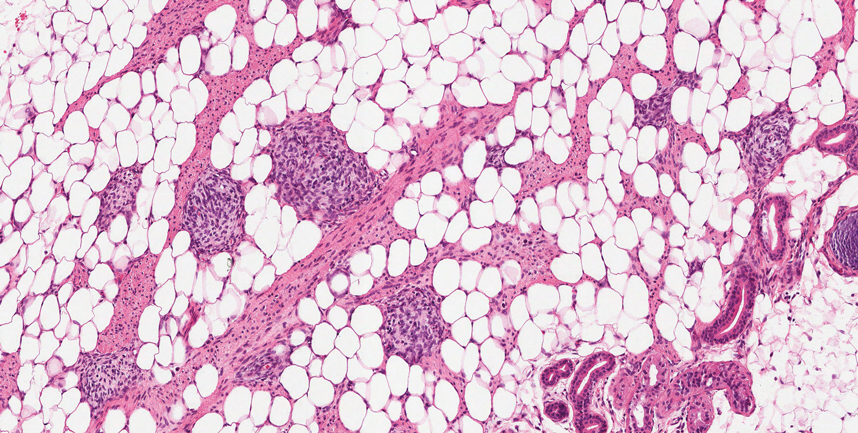
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
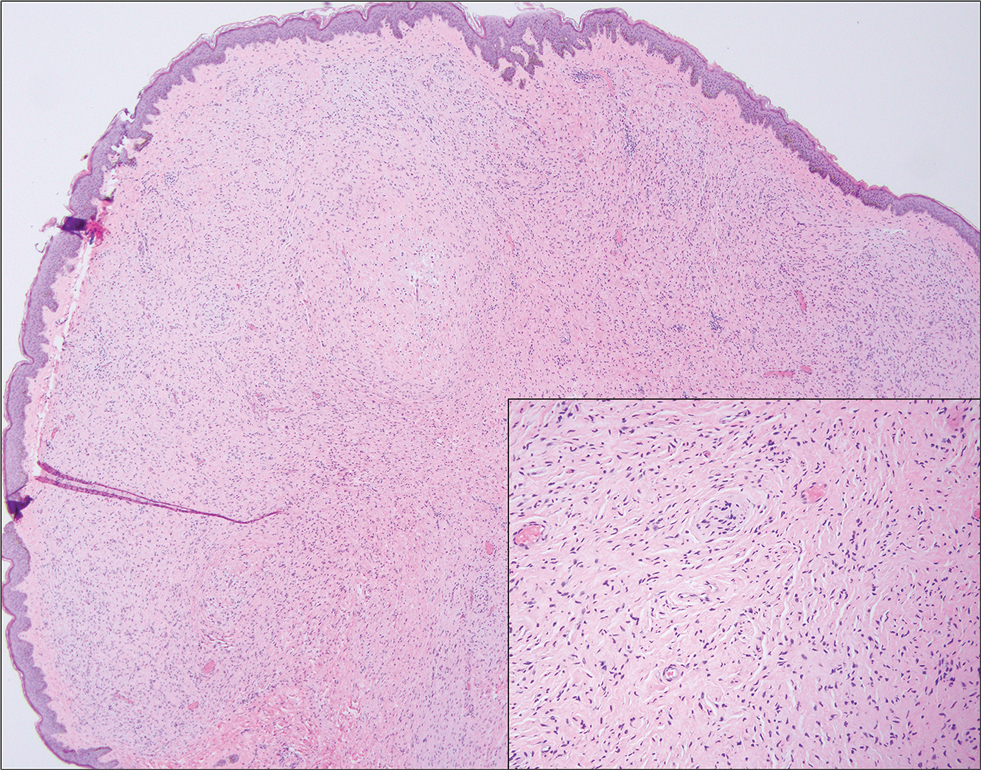
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
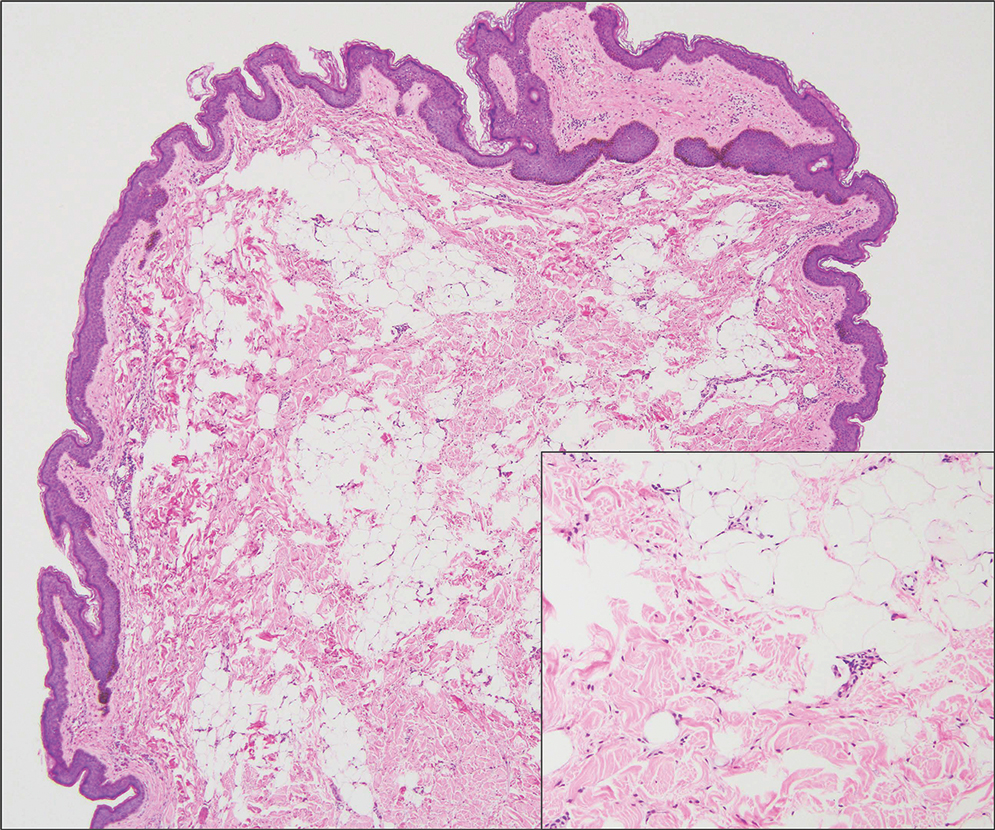
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
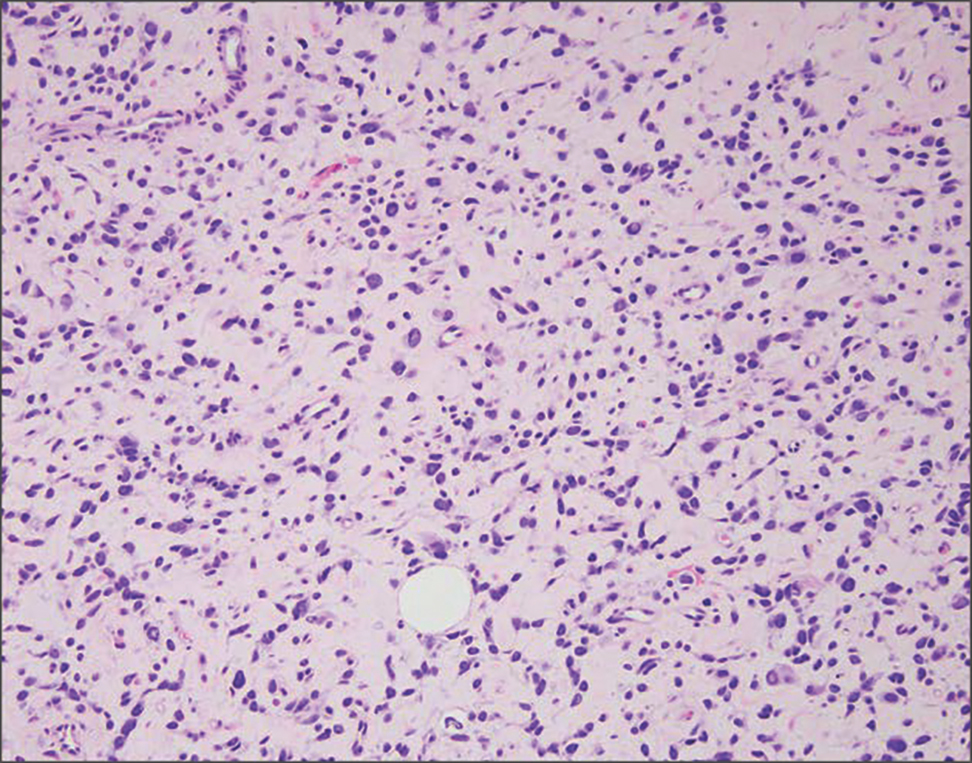
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
Exophytic Papule on the Chin of a Child
Exophytic Papule on the Chin of a Child
A 3-year-old boy presented to the dermatology department for evaluation of an asymptomatic papule on the chin that had been present since birth. His medical history was otherwise unremarkable. Physical examination revealed a 4×2-mm, flesh-colored, exophytic papule on the midline chin. An excisional biopsy was performed.