User login
Hospital Dermatology: Review of Research in 2024-2025
Hospital Dermatology: Review of Research in 2024-2025
Dermatologists play a central role in the care of hospitalized patients with skin disease. This review summarizes research from January 2024 to December 2025 on severe cutaneous adverse drug reactions, emerging infectious diseases, hidradenitis suppurativa (HS), and inpatient dermatology workforce issues. Key developments include improved recognition and management of drug reactions; updated diagnostic and prognostic tools for Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN); and guidance for emerging infections such as measles, dengue, mpox, orthopoxviruses, and resistant dermatophytes. Evidence-based strategies for HS aim to reduce unnecessary admissions and optimize care. Workforce challenges, including limited access, high call burden, and potential for artificial intelligence (AI)–assisted diagnosis, are also highlighted. These findings emphasize the critical contributions of dermatologists to hospital-based care and provide emerging evidence to guide clinical practice.
Dermatologists play a critical role in the care of hospitalized patients. Herein, we review the research developments between January 2024 and December 2025 most relevant to the care of hospitalized patients with skin disease, including severe cutaneous adverse reactions (SCARs), emerging and re-emerging infectious diseases, hidradenitis suppurativa (HS), and access to inpatient dermatology services.
Severe Cutaneous Adverse Drug Reactions
Severe cutaneous adverse drug reactions are among the most frequent reasons for inpatient dermatology consultation. A National Inpatient Sample study identified more than 160,000 cases of drug rash with eosinophilia and systemic symptoms (DRESS syndrome) between January 2016 and December 2020.1 The overall mortality rate was 2.0%, substantially lower than the rates of up to 10% reported in earlier studies.2 Case burden and mortality peaked during the fall months, possibly due to either increased use of antibiotics or increased viral infection or reactivation during these months.1
A retrospective cohort study of patients with probable or definite DRESS syndrome showed that, among 93 patients with at least 1 viral marker tested, human herpesvirus (HHV) reactivation was found in 42% (39/93), including HHV-6 (28%)(24/85), Epstein-Barr virus (17%)(15/87), and cytomegalovirus (20%)(18/89); furthermore, viral reactivation was associated with higher 1-year mortality (odds ratio, 3.9), dialysis initiation, flares of disease, and longer hospital stay (all P<.05).1 Multiple reactivations were associated with higher inpatient mortality and 1-year mortality; however, despite apparent prognostic importance, the role of screening for viral reactivation in DRESS syndrome is undefined.
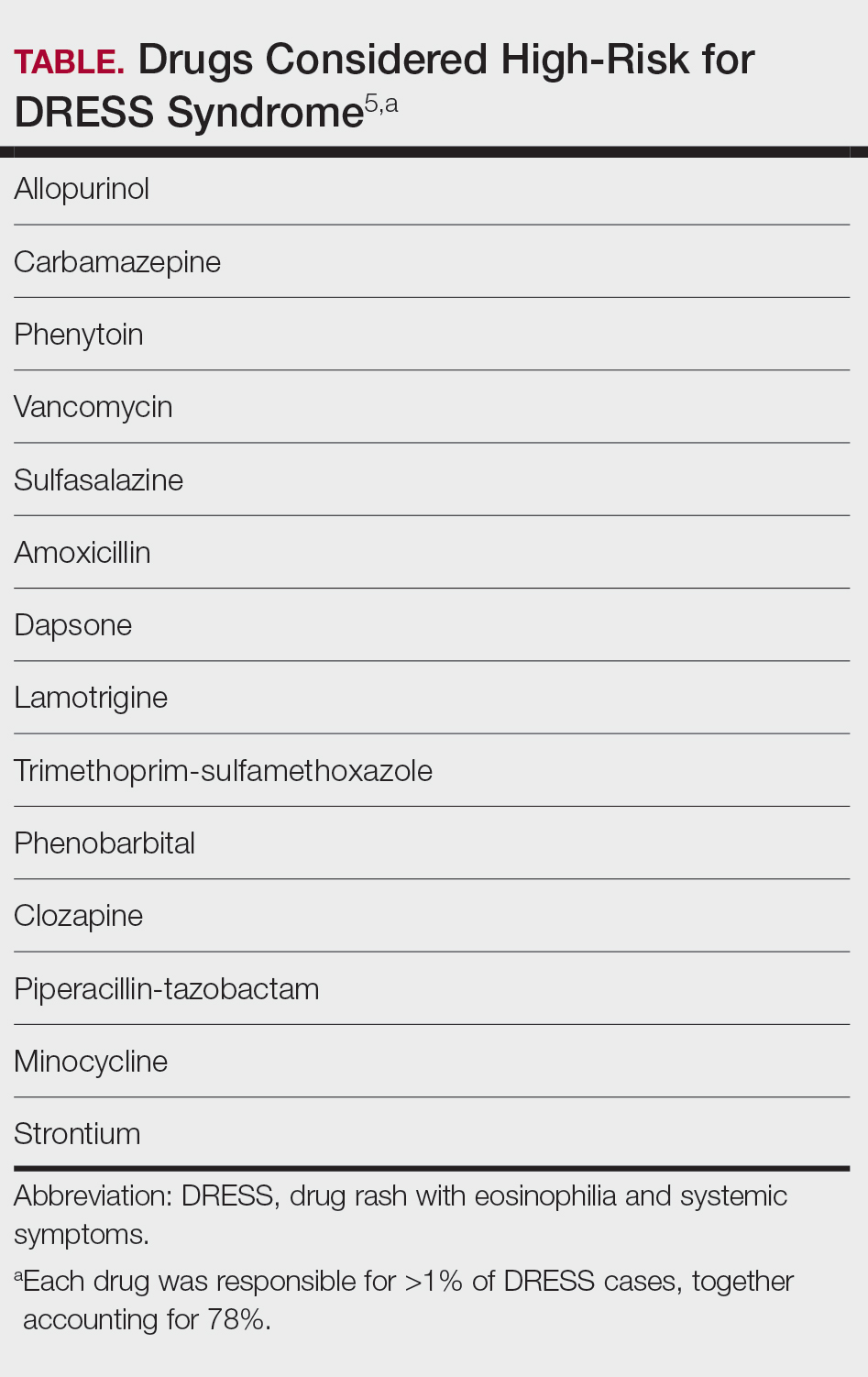
A systematic review of 610 studies including 2122 patients with DRESS syndrome demonstrated that, among 193 causal agents identified, 14 drugs accounted for more than 1% of cases each and therefore were considered high risk. Seventy-eight percent of cases were attributed to these 14 drugs (Table).5 A TriNetX Query study analyzed antibiotic exposures across SCARs and reported that sulfonamides (hazard ratio [HR], 7.5), aminoglycosides (HR, 3.7), and tetracyclines (HR, 1.7) were associated with an elevated risk for SCARs. Sulfonamides had the highest absolute incidence of SCARs, followed by cephalosporins and penicillins.6
A multicenter randomized clinical trial7 compared high-potency topical corticosteroids (clobetasol 30 g/d) to systemic corticosteroids (prednisone 0.5 mg/kg/d) for treatment of moderate DRESS syndrome. On day 30, 53.8% (14/26) of patients in the topical group had achieved remission of visceral involvement, compared to 72.0% (18/25) in the systemic group. Before day 30, 23.1% (6/26) of patients in the topical group worsened, necessitating transition to high-dose systemic steroids. When inpatient monitoring is available, low-dose systemic corticosteroids or high-potency topical steroids may be reasonable management strategies for moderate DRESS syndrome7; however, the frequent need for treatment intensification suggests limitations to this strategy.
Since prolonged courses of systemic steroids generally are necessary for management of DRESS syndrome, steroid-sparing options are needed. A retrospective case series examined interleukin 5 inhibition in patients with possible DRESS syndrome (Registry of Severe Cutaneous Adverse Reactions score ≥3). All patients demonstrated rapid eosinophil reduction within 1 to 3 days (mean [SD] time to resolution, 1.4 [0.9] days) after treatment with mepolizumab or benralizumab, with clinical improvement occurring at a mean (SD) of 16 (3.7) days (range, 13-21 days).8
A French cohort study of 1221 adult patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) reported in-hospital mortality of 19% and a total mortality of 34% at 1 year.9 Risk factors contributing to in-hospital mortality included age, history of/current diagnosis of cancer, dementia, and liver disease, while postdischarge mortality was associated with acute kidney injury and sepsis. Long-term complications included ophthalmologic and mood disorders.9
A new set of diagnostic criteria for SJS/TEN, known as the Niigata criteria,10 includes 3 main items: severe mucosal lesions in cutaneous-mucosal transition zones (eg, eyes, lips, vulva) or generalized erythema with necrotic lesions; fever of 38.5 °C or higher; and necrosis of the epidermis seen on histopathology. Because epidermal detachment involving 10% of the body surface area (BSA) is an important mortality risk predicter, SJS is defined as less than 10% BSA involvement, and TEN has been redefined as 10% or more BSA involvement (not ≥30%). A new prognostic score—clinical risk score for TEN (CRISTEN)—can be tabulated at the point of care without laboratory values. It was developed based on the 10 most important risk factors for death in a retrospective study of 382 patients, which included age 65 years or older; epidermal detachment involving 10% BSA or higher; an antibiotic as causative agent; systemic corticosteroid therapy before the onset of SJS/TEN; involvement of all 3 mucosal surfaces; and medical comorbidities such as renal impairment, diabetes, cardiac disease, active cancer, and bacterial infection.11
New potential therapeutic targets for SJS/TEN include PC111 (monoclonal antibody to Fas ligand), formyl peptide receptor 1 antagonists (which inhibit necroptosis induced by formyl peptide receptor 1–annexin A1 interaction), daratumumab (which depletes cytotoxic CD8-positive and CD38-positive T cells), and Janus kinase (JAK) inhibitors.10 Spatial proteomics showed marked enrichment of type I and type II interferon signatures as well as activation of signal transducer and activator of transcription 1. In vitro, tofacitinib reduced keratinocyte-directed cytotoxicity, and in vivo JAK inhibitors ameliorated disease severity in 2 TEN mouse models. Patients with TEN that was refractory to corticosteroid therapy received rescue treatment with JAK inhibitors and had re-epithelization within several days with marked reduction in levels of phosphorylated signal transducer and activator of transcription 1.12 Controlled studies are needed to assess the potential role of JAK inhibitors for SJS/TEN.
Emerging and Re-emerging Infectious Diseases
Dermatologists may encounter emerging or re-emerging infections, performing an essential public health role in the process. In 2025, a total of 2281 confirmed cases of measles had been reported across 45 of the United States.13 During the COVID-19 pandemic, measles vaccine coverage in the United States dropped to 93%—down from 95% to 97% prepandemic. Worldwide, 2022 saw an increase of 1.4 million measles cases (18% increase) and 41,200 excess deaths (43% increase) compared to the previous year. Complications of measles include pneumonia, blindness, otitis media, and encephalitis, with 1 in 5 (20%) unvaccinated people with measles in the United States requiring hospitalization.14 A vaccine coverage rate higher than 95% is needed to prevent community spread of disease. Since efforts to detect and rapidly isolate cases of measles are critical, dermatologists should consider measles in the differential of morbilliform eruptions with viral symptoms and ask about vaccination status.
Since 2023, dengue infection rates have tripled in the Americas, representing the highest levels recorded since tracking began in 1980. In 2024, there were more than 12 million cases, with approximately 8000 deaths reported. Ninety percent of cases occur in Argentina, Brazil, Colombia, and Mexico, but local transmission has been reported in Arizona, California, Florida, Hawaii, and Texas.15 The characteristic exanthem of dengue is diffuse erythema with islands of sparing.<
Unlike during the 2022 outbreak of mpox clade II, which predominantly impacted men who have sex with men, there now is an ongoing outbreak of mpox clades 1a and 1b in the Democratic Republic of the Congo and surrounding countries that more commonly affects children and heterosexual adults. It is also more transmissible and virulent. Cases of mpox clade I have been reported in several European countries and across the United States, mostly among travelers from areas of active transmission. Vaccination of at-risk individuals is considered effective; however, tecovirimat is not.16
Outbreaks of 2 emerging zoonotic orthopoxviruses recently have been reported. Buffalopox virus (BPXV) is transmitted via direct contact with the skin of infected cattle and buffalo as well as fomites and has been responsible for human cases in South Asia. Characteristics of BPXV include macules, umbilicated papules, vesicles, pustules, and eschars that evolve over several weeks, with a predilection for the hands and face. It can manifest with prodromal symptoms of fever, malaise, and lymphadenopathy.17 Borealpox virus (formerly known as Alaskapox) has similar manifestations. Its reservoir includes small mammals such as voles and shrews, but it also has been found in cats and dogs and has been responsible for at least one human fatality. Cidofovir may be an effective therapy for both BPXV and borealpox virus, and prior smallpox vaccination may provide protection.18 These outbreaks demonstrate the continued importance of research for more effective vaccines and therapies against smallpox and other orthopoxviruses.19 A recent review provided a detailed overview of the epidemiology, transmission, dermatologic findings, and management strategies associated with smallpox and other bioweapons.20
In 2023, a case was reported of a patient in a New York City hospital with tinea that was refractory to multiple rounds of topical antifungals, which called attention to the presence of Trichophyton indotineae in the United States.21 Since then, additional reports and case series have characterized the clinical presentation of T indotineae as widespread and atypical, refractory to traditional therapies, and most often encountered in travelers returning from Bangladesh or elsewhere in South Asia.22 The diagnosis should be confirmed via DNA testing of fungal culture. Itraconazole 100 to 200 mg/d is the antifungal therapy of choice.23
Other series have reported cases of tinea genitalis caused by Trichophyton mentagrophytes type VII seen predominately in sex workers and others engaging in high-risk sexual contact, highlighting the spread of dermatophytes through sexual activity.24-26 Lastly, it is important to culture pustules and consider atypical pathogens in patients with chronic folliculitis not responding to typical therapies such as tetracycline antibiotics. A case series reported the presence of pustules in the beard area of 7 men who have sex with men, with culture data showing Klebsiella aerogenes. Prolonged courses of fluoroquinolones were necessary for clearance.27
Reducing HS Admissions Through Evidence-Based Management
Hidradenitis suppurativa is a frequent cause of emergency department visits and hospital admissions. In an analysis of the Nationwide Readmissions Database, 17.8% (392/2204) of patients admitted to the hospital with HS were readmitted within 30 days, a number comparable to that of heart failure.28
Flaring HS can produce symptoms that mimic sepsis. A retrospective cohort study examining sepsislike features in HS showed that more than 50% (30/58) of those admitted to the hospital with an HS flare were misdiagnosed with sepsis, and more than 80% (53/64) of those patients received intravenous antibiotics.29 A National Inpatient Sample (January 2016-December 2018) study demonstrated minimal rates of true infection in patients admitted with HS flares,30 while patients with HS diagnosed as sepsis do not sustain the mortality expected from true sepsis. Improving recognition of HS and differentiation of the disease from true sepsis could decrease unnecessary antibiotic use, hospital admissions, and cost, underscoring the need for a framework to reliably and reproducibly distinguish sepsis from HS flare.31
While severe HS is difficult to manage, there may be a window of opportunity in which appropriate treatment of early disease may prevent progression and decrease inpatient utilization. A prospective cohort study of 335 biologic-naïve patients with mild to moderate HS (Hurley stages I and II) followed over a median of 2 years showed that active smoking, body mass index higher than 25, and the presence of disease in 2 or more anatomic areas were factors predictive of progression to severe disease.32
Despite high utilization of emergency and inpatient care, there has been no consensus on inpatient management of HS. A Delphi consensus exercise including 26 expert dermatologists reached consensus on 40 statements.33 Specific recommendations involve multidisciplinary care, including from a dermatologist; consideration of comorbid medical conditions; supportive care measures (wound care, pain control); evidence-based medical management, including initiation or adjustment of biologic therapies; targeted surgical intervention; nutritional support and maintenance of glycemic control; and attention to transitional care at discharge, including home health services, verification of insurance status, and timely outpatient dermatology follow-up.34 A retrospective review of 98 patients treated with intravenous ertapenem for a mean duration of 13 weeks demonstrated improvement in clinical and inflammatory markers.35 Patients with severe or treatment-refractory HS, including those admitted to the hospital, may benefit from initiation of this therapy in select circumstances.
Hospital Dermatology Workforce
Inpatient dermatology consultations are extremely valuable for improving diagnostic accuracy, reducing admissions for pseudocellulitis and inflammatory skin conditions, and keeping cancer patients on needed therapies.36-38 Despite this clear value added, a cross-sectional analysis of inpatient Medicare claims data from January 2013 to December 2019 found that the number of dermatologists performing more than 10 inpatient consults per year decreased from 356 to 281.39 Additionally, medical centers in which dermatology encounters occurred decreased from 239 to 157 during the same period. Ninety-eight percent of inpatient dermatologists were in metropolitan areas, with large regions lacking access to inpatient dermatology consultation altogether.39
A survey of Society for Pediatric Dermatology members similarly characterized the state of the pediatric dermatology workforce performing hospital consultation.40 Seventy-five percent reported a high call burden, defined as more than 11 days or nights per month, more than 1 weekend per month, and/or more than 5 hours per week seeing patients. Ninety-one percent of consultation services are based within academic institutions, reflecting disparities in access.40 A prospective cohort study of academic pediatric dermatologists reported that 310 curbside consultations were performed over 24 weeks; of these calls, 17% occurred during weeknights and 23% on weekends. None of these curbside interactions was reimbursed.41 These findings underscore the burden of uncompensated time a subset of pediatric dermatologists dedicates to inpatient consultations, highlighting the need for improved financial and administrative support and an increased number of physicians performing this role.
A survey study42 suggested that unfamiliarity with the inpatient setting, rather than medical knowledge, is the most important barrier to inpatient work among clinical dermatologists. Proposed interventions include resource guides (eg, hospital maps, pager numbers for key individuals, and protocols for urgent specimens). Reference guides and refresher courses may decrease gaps in knowledge or awareness among dermatologists in ambulatory practice.42 Another way to bolster the inpatient dermatology workforce may be to provide more guidance to qualified advanced practice providers to triage and address dermatologic emergencies.43
Artificial intelligence (AI) also has been explored as a tool for diagnosing complex dermatologic conditions. One study presented 15 published inpatient dermatology cases to 7 dermatologists. Participants were asked to formulate their top 3 differential diagnoses and were then shown AI-generated differentials and asked to submit a revised differential. Participants showed a diagnostic accuracy of 69% before seeing the AI-generated differential diagnosis and 79% after; however, in cases in which the AI differential was incorrect, diagnostic accuracy of the dermatologists decreased after being shown the AI model.44
Final Thoughts
This January 2024 to December 2025 review of research relevant to hospital dermatology highlights important developments and ongoing challenges in SCARs, emerging and re-emerging infectious diseases, HS, and the inpatient dermatology workforce. Dermatologists continue to play a critical role in the care of hospitalized patients with skin disease.
- Desai AD, Thomas C. Seasonal trends in drug reaction with eosinophilia and systemic symptoms. J Am Acad Dermatol. 2025;92:183-185.
- Wei BM, Fox LP, Kaffenberger BH, et al. Drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms. Part I. Epidemiology, pathogenesis, clinicopathological features, and prognosis. J Am Acad Dermatol. 2024;90:885-908. doi:10.1016/j.jaad.2023.02.072
- Chan LCE, Sultana R, Choo KJL, et al. Viral reactivation and clinical outcomes in drug reaction with eosinophilia and systemic symptoms (DRESS). Sci Rep. 2024;14:28492.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44
- Hansen E, Gallardo M, Yan A, et al. Risk assessment of drugs associated with DRESS syndrome based on publication frequency: a systematic review. J Am Acad Dermatol. 2024;91:962-966.
- Neubauer ZJK, Chan R, Singal A, et al. SCAR-ed by antibiotics: a retrospective cohort study of severe cutaneous adverse reactions (SCAR) relative risk. J Am Acad Dermatol. 2025;92:1143-1145.
- Ingen-Housz-Oro S, Guichard E, Milpied B, et al. Topical versus oral corticosteroids in moderate drug reaction with eosinophilia and systemic symptoms: a multicenter randomized clinical trial. J Am Acad Dermatol. 2024;91:544-547.
- Hijaz B, Nambudiri VE, Imadojemu S. IL-5 inhibitor treatment in drug reaction with eosinophilia and systemic symptoms. JAMA Dermatol. 2025;161:661-663.
- Bettuzzi T, Lebrun-Vignes B, Ingen-Housz-Oro S, et al. Incidence, in-hospital and long-term mortality, and sequelae of epidermal necrolysis in adults. JAMA Dermatol. 2024;160:1288-1296.
- Hama N, Aoki S, Chen CB, et al. Recent progress in Stevens-Johnson syndrome/toxic epidermal necrolysis: diagnostic criteria, pathogenesis and treatment. Br J Dermatol. 2024;192:9-18.
- Hama N, Sunaga Y, Ochiai H, et al. Development and validation of a novel score to predict mortality in Stevens-Johnson syndrome and toxic epidermal necrolysis: CRISTEN. J Allergy Clin Immunol Pract. 2023;11:3161-3168.e2.
- Nordmann TM, Anderton H, Hasegawa A, et al. Spatial proteomics identifies JAKi as treatment for a lethal skin disease. Nature. 2024;635:1001-1009.
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Updated January 7, 2026. Accessed January 12, 2026. https://www.cdc.gov/measles/data-research/
- Rubin R. Despite safe and effective vaccine, measles cases and deaths increased worldwide from 2021 to 2022. JAMA. 2024;331:188-189.
- Orrall A. Dengue cases in the Americas highest recorded. JAMA. 2025;333:452.
- Harris E. As mpox cases surge in Africa, WHO declares a global emergency-here’s what to know. JAMA. 2024;332:862-864.
- Burningham KM, Hinojosa T, Cavazos A, et al. Buffalopox: an emerging cutaneous disease in humans. J Eur Acad Dermatol Venereol. 2025;39:404-406.
- Parker ER. Emergence of Alaskapox infection: what dermatologists need to know. J Am Acad Dermatol. 2024;91:397-399.
- Gostin LO, Singaravelu S, Hynes N. Smallpox readiness: modern strategies against an ancient disease. JAMA. 2024;332:873-874.
- Osborne S, Kam O, Thacker S, et al. Review of category A bioweapons with cutaneous features: epidemiology, clinical presentation, and contemporary management strategies. J Am Acad Dermatol. 2025;93:165-175.
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field: first reported U.S. cases of tinea caused by Trichophyton indotineae - New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537.
- McKenna M. Why the rise of this drug-resistant fungus is raising international concern. JAMA. 2024;332:859-861.
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Luchsinger I, Bosshard PP, Kasper RS, et al. Tinea genitalis: a new entity of sexually transmitted infection? Case series and review of the literature. Sex Transm Infect. 2015;91:493-496.
- Khurana A, Sharath S, Sardana K, et al. Therapeutic updates on the management of tinea corporis or cruris in the era of Trichophyton indotineae: separating evidence from hype-a narrative review. Indian J Dermatol. 2023;68:525-540.
- Bérot V, Monsel G, Dauendorffer JN, et al; Groupe Infectiologie Dermatologique et Infections Sexuellement Transmissibles (GrIDIST) de la Société Française de Dermatologie. Klebsiella aerogenes-related facial folliculitis in men having sex with men: a hypothetical new STI?J Eur Acad Dermatol Venereol. 2025;39:E10-E12.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the Nationwide Readmissions Database. J Am Acad Dermatol. 2022;87:188-192.
- AbdelHameid D, Wang L, Mauskar MM, et al. Sepsis-like features in hidradenitis suppurativa flares requiring admission: a retrospective cohort study. J Am Acad Dermatol. 2024;90:1291-1294.
- Ehizogie E, Maghari I, Lo S, et al. Hidradenitis suppurativa, systemic inflammatory response syndrome and sepsis: a database study. Br J Dermatol. 2024;191:451-453.
- Maghari I, Abiad H, Griffin T, et al. Hidradenitis suppurativa (HS), systemic inflammatory response syndrome and sepsis, sepsis caused by HS: an empty systematic review. Br J Dermatol. 2024;191:449-450.
- Kjærsgaard Andersen R, Pedersen O, Eidsmo L, et al. Initial steps towards developing a predictive algorithm of disease progression for hidradenitis suppurativa (HS): results from a Cox proportional hazard regression analysis on disease progression among a cohort of 335 Danish patients with HS. Br J Dermatol. 2024;190:904-914.
- Needham M, Pichardo R, Alavi A, et al. Inpatient management of hidradenitis suppurativa: a Delphi consensus study. Cutis. 2024;113:251-254.
- Maskan Bermudez N, Elman SA, Kirsner RS, et al. Management of hidradenitis suppurativa in the inpatient setting: a clinical guide. Arch Dermatol Res. 2025;317:202.
- Nosrati A, Ch’en PY, Torpey ME, et al. Efficacy and durability of intravenous ertapenem therapy for recalcitrant hidradenitis suppurativa. JAMA Dermatol. 2024;160:312-318.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Hydol-Smith JA, Gallardo MA, Korman A, et al. The United States dermatology inpatient workforce between 2013 and 2019: a Medicare analysis reveals contraction of the workforce and vast access deserts-a cross-sectional analysis. Arch Dermatol Res. 2024;316:103.
- Pineider JL, Rangu SA, Shaw KS, et al. Pediatric consultative dermatology: a survey of the Society for Pediatric Dermatology workforce reveals shortcomings in existing practice models of pediatric dermatology consult services in the United States. Pediatr Dermatol. 2024;41:270-274.
- Puar NK, Canty KM, Newell BD, et al. An evaluation of pediatric dermatology curbside consultations in an academic center: a prospective cohort study. J Am Acad Dermatol. 2024;90:1258-1260.
- Lau CB, Smith GP. Strategies for improving dermatologist comfort and quality of patient care in inpatient settings: a cross-sectional survey study. Arch Dermatol Res. 2024;316:575.
- Hazim AH. Empowering advanced clinical practitioners in managing acute dermatological emergencies. Br J Nurs. 2024;33:448-455.
- Macklis P, Kaffenberger B, Kirven R, et al. Dermatology diagnostic accuracy is improved by artificial intelligence-generated differential diagnoses. Int J Dermatol. 2025;64:960-962.
Dermatologists play a central role in the care of hospitalized patients with skin disease. This review summarizes research from January 2024 to December 2025 on severe cutaneous adverse drug reactions, emerging infectious diseases, hidradenitis suppurativa (HS), and inpatient dermatology workforce issues. Key developments include improved recognition and management of drug reactions; updated diagnostic and prognostic tools for Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN); and guidance for emerging infections such as measles, dengue, mpox, orthopoxviruses, and resistant dermatophytes. Evidence-based strategies for HS aim to reduce unnecessary admissions and optimize care. Workforce challenges, including limited access, high call burden, and potential for artificial intelligence (AI)–assisted diagnosis, are also highlighted. These findings emphasize the critical contributions of dermatologists to hospital-based care and provide emerging evidence to guide clinical practice.
Dermatologists play a critical role in the care of hospitalized patients. Herein, we review the research developments between January 2024 and December 2025 most relevant to the care of hospitalized patients with skin disease, including severe cutaneous adverse reactions (SCARs), emerging and re-emerging infectious diseases, hidradenitis suppurativa (HS), and access to inpatient dermatology services.
Severe Cutaneous Adverse Drug Reactions
Severe cutaneous adverse drug reactions are among the most frequent reasons for inpatient dermatology consultation. A National Inpatient Sample study identified more than 160,000 cases of drug rash with eosinophilia and systemic symptoms (DRESS syndrome) between January 2016 and December 2020.1 The overall mortality rate was 2.0%, substantially lower than the rates of up to 10% reported in earlier studies.2 Case burden and mortality peaked during the fall months, possibly due to either increased use of antibiotics or increased viral infection or reactivation during these months.1
A retrospective cohort study of patients with probable or definite DRESS syndrome showed that, among 93 patients with at least 1 viral marker tested, human herpesvirus (HHV) reactivation was found in 42% (39/93), including HHV-6 (28%)(24/85), Epstein-Barr virus (17%)(15/87), and cytomegalovirus (20%)(18/89); furthermore, viral reactivation was associated with higher 1-year mortality (odds ratio, 3.9), dialysis initiation, flares of disease, and longer hospital stay (all P<.05).1 Multiple reactivations were associated with higher inpatient mortality and 1-year mortality; however, despite apparent prognostic importance, the role of screening for viral reactivation in DRESS syndrome is undefined.
A systematic review of 610 studies including 2122 patients with DRESS syndrome demonstrated that, among 193 causal agents identified, 14 drugs accounted for more than 1% of cases each and therefore were considered high risk. Seventy-eight percent of cases were attributed to these 14 drugs (Table).5 A TriNetX Query study analyzed antibiotic exposures across SCARs and reported that sulfonamides (hazard ratio [HR], 7.5), aminoglycosides (HR, 3.7), and tetracyclines (HR, 1.7) were associated with an elevated risk for SCARs. Sulfonamides had the highest absolute incidence of SCARs, followed by cephalosporins and penicillins.6
A multicenter randomized clinical trial7 compared high-potency topical corticosteroids (clobetasol 30 g/d) to systemic corticosteroids (prednisone 0.5 mg/kg/d) for treatment of moderate DRESS syndrome. On day 30, 53.8% (14/26) of patients in the topical group had achieved remission of visceral involvement, compared to 72.0% (18/25) in the systemic group. Before day 30, 23.1% (6/26) of patients in the topical group worsened, necessitating transition to high-dose systemic steroids. When inpatient monitoring is available, low-dose systemic corticosteroids or high-potency topical steroids may be reasonable management strategies for moderate DRESS syndrome7; however, the frequent need for treatment intensification suggests limitations to this strategy.
Since prolonged courses of systemic steroids generally are necessary for management of DRESS syndrome, steroid-sparing options are needed. A retrospective case series examined interleukin 5 inhibition in patients with possible DRESS syndrome (Registry of Severe Cutaneous Adverse Reactions score ≥3). All patients demonstrated rapid eosinophil reduction within 1 to 3 days (mean [SD] time to resolution, 1.4 [0.9] days) after treatment with mepolizumab or benralizumab, with clinical improvement occurring at a mean (SD) of 16 (3.7) days (range, 13-21 days).8
A French cohort study of 1221 adult patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) reported in-hospital mortality of 19% and a total mortality of 34% at 1 year.9 Risk factors contributing to in-hospital mortality included age, history of/current diagnosis of cancer, dementia, and liver disease, while postdischarge mortality was associated with acute kidney injury and sepsis. Long-term complications included ophthalmologic and mood disorders.9
A new set of diagnostic criteria for SJS/TEN, known as the Niigata criteria,10 includes 3 main items: severe mucosal lesions in cutaneous-mucosal transition zones (eg, eyes, lips, vulva) or generalized erythema with necrotic lesions; fever of 38.5 °C or higher; and necrosis of the epidermis seen on histopathology. Because epidermal detachment involving 10% of the body surface area (BSA) is an important mortality risk predicter, SJS is defined as less than 10% BSA involvement, and TEN has been redefined as 10% or more BSA involvement (not ≥30%). A new prognostic score—clinical risk score for TEN (CRISTEN)—can be tabulated at the point of care without laboratory values. It was developed based on the 10 most important risk factors for death in a retrospective study of 382 patients, which included age 65 years or older; epidermal detachment involving 10% BSA or higher; an antibiotic as causative agent; systemic corticosteroid therapy before the onset of SJS/TEN; involvement of all 3 mucosal surfaces; and medical comorbidities such as renal impairment, diabetes, cardiac disease, active cancer, and bacterial infection.11
New potential therapeutic targets for SJS/TEN include PC111 (monoclonal antibody to Fas ligand), formyl peptide receptor 1 antagonists (which inhibit necroptosis induced by formyl peptide receptor 1–annexin A1 interaction), daratumumab (which depletes cytotoxic CD8-positive and CD38-positive T cells), and Janus kinase (JAK) inhibitors.10 Spatial proteomics showed marked enrichment of type I and type II interferon signatures as well as activation of signal transducer and activator of transcription 1. In vitro, tofacitinib reduced keratinocyte-directed cytotoxicity, and in vivo JAK inhibitors ameliorated disease severity in 2 TEN mouse models. Patients with TEN that was refractory to corticosteroid therapy received rescue treatment with JAK inhibitors and had re-epithelization within several days with marked reduction in levels of phosphorylated signal transducer and activator of transcription 1.12 Controlled studies are needed to assess the potential role of JAK inhibitors for SJS/TEN.
Emerging and Re-emerging Infectious Diseases
Dermatologists may encounter emerging or re-emerging infections, performing an essential public health role in the process. In 2025, a total of 2281 confirmed cases of measles had been reported across 45 of the United States.13 During the COVID-19 pandemic, measles vaccine coverage in the United States dropped to 93%—down from 95% to 97% prepandemic. Worldwide, 2022 saw an increase of 1.4 million measles cases (18% increase) and 41,200 excess deaths (43% increase) compared to the previous year. Complications of measles include pneumonia, blindness, otitis media, and encephalitis, with 1 in 5 (20%) unvaccinated people with measles in the United States requiring hospitalization.14 A vaccine coverage rate higher than 95% is needed to prevent community spread of disease. Since efforts to detect and rapidly isolate cases of measles are critical, dermatologists should consider measles in the differential of morbilliform eruptions with viral symptoms and ask about vaccination status.
Since 2023, dengue infection rates have tripled in the Americas, representing the highest levels recorded since tracking began in 1980. In 2024, there were more than 12 million cases, with approximately 8000 deaths reported. Ninety percent of cases occur in Argentina, Brazil, Colombia, and Mexico, but local transmission has been reported in Arizona, California, Florida, Hawaii, and Texas.15 The characteristic exanthem of dengue is diffuse erythema with islands of sparing.<
Unlike during the 2022 outbreak of mpox clade II, which predominantly impacted men who have sex with men, there now is an ongoing outbreak of mpox clades 1a and 1b in the Democratic Republic of the Congo and surrounding countries that more commonly affects children and heterosexual adults. It is also more transmissible and virulent. Cases of mpox clade I have been reported in several European countries and across the United States, mostly among travelers from areas of active transmission. Vaccination of at-risk individuals is considered effective; however, tecovirimat is not.16
Outbreaks of 2 emerging zoonotic orthopoxviruses recently have been reported. Buffalopox virus (BPXV) is transmitted via direct contact with the skin of infected cattle and buffalo as well as fomites and has been responsible for human cases in South Asia. Characteristics of BPXV include macules, umbilicated papules, vesicles, pustules, and eschars that evolve over several weeks, with a predilection for the hands and face. It can manifest with prodromal symptoms of fever, malaise, and lymphadenopathy.17 Borealpox virus (formerly known as Alaskapox) has similar manifestations. Its reservoir includes small mammals such as voles and shrews, but it also has been found in cats and dogs and has been responsible for at least one human fatality. Cidofovir may be an effective therapy for both BPXV and borealpox virus, and prior smallpox vaccination may provide protection.18 These outbreaks demonstrate the continued importance of research for more effective vaccines and therapies against smallpox and other orthopoxviruses.19 A recent review provided a detailed overview of the epidemiology, transmission, dermatologic findings, and management strategies associated with smallpox and other bioweapons.20
In 2023, a case was reported of a patient in a New York City hospital with tinea that was refractory to multiple rounds of topical antifungals, which called attention to the presence of Trichophyton indotineae in the United States.21 Since then, additional reports and case series have characterized the clinical presentation of T indotineae as widespread and atypical, refractory to traditional therapies, and most often encountered in travelers returning from Bangladesh or elsewhere in South Asia.22 The diagnosis should be confirmed via DNA testing of fungal culture. Itraconazole 100 to 200 mg/d is the antifungal therapy of choice.23
Other series have reported cases of tinea genitalis caused by Trichophyton mentagrophytes type VII seen predominately in sex workers and others engaging in high-risk sexual contact, highlighting the spread of dermatophytes through sexual activity.24-26 Lastly, it is important to culture pustules and consider atypical pathogens in patients with chronic folliculitis not responding to typical therapies such as tetracycline antibiotics. A case series reported the presence of pustules in the beard area of 7 men who have sex with men, with culture data showing Klebsiella aerogenes. Prolonged courses of fluoroquinolones were necessary for clearance.27
Reducing HS Admissions Through Evidence-Based Management
Hidradenitis suppurativa is a frequent cause of emergency department visits and hospital admissions. In an analysis of the Nationwide Readmissions Database, 17.8% (392/2204) of patients admitted to the hospital with HS were readmitted within 30 days, a number comparable to that of heart failure.28
Flaring HS can produce symptoms that mimic sepsis. A retrospective cohort study examining sepsislike features in HS showed that more than 50% (30/58) of those admitted to the hospital with an HS flare were misdiagnosed with sepsis, and more than 80% (53/64) of those patients received intravenous antibiotics.29 A National Inpatient Sample (January 2016-December 2018) study demonstrated minimal rates of true infection in patients admitted with HS flares,30 while patients with HS diagnosed as sepsis do not sustain the mortality expected from true sepsis. Improving recognition of HS and differentiation of the disease from true sepsis could decrease unnecessary antibiotic use, hospital admissions, and cost, underscoring the need for a framework to reliably and reproducibly distinguish sepsis from HS flare.31
While severe HS is difficult to manage, there may be a window of opportunity in which appropriate treatment of early disease may prevent progression and decrease inpatient utilization. A prospective cohort study of 335 biologic-naïve patients with mild to moderate HS (Hurley stages I and II) followed over a median of 2 years showed that active smoking, body mass index higher than 25, and the presence of disease in 2 or more anatomic areas were factors predictive of progression to severe disease.32
Despite high utilization of emergency and inpatient care, there has been no consensus on inpatient management of HS. A Delphi consensus exercise including 26 expert dermatologists reached consensus on 40 statements.33 Specific recommendations involve multidisciplinary care, including from a dermatologist; consideration of comorbid medical conditions; supportive care measures (wound care, pain control); evidence-based medical management, including initiation or adjustment of biologic therapies; targeted surgical intervention; nutritional support and maintenance of glycemic control; and attention to transitional care at discharge, including home health services, verification of insurance status, and timely outpatient dermatology follow-up.34 A retrospective review of 98 patients treated with intravenous ertapenem for a mean duration of 13 weeks demonstrated improvement in clinical and inflammatory markers.35 Patients with severe or treatment-refractory HS, including those admitted to the hospital, may benefit from initiation of this therapy in select circumstances.
Hospital Dermatology Workforce
Inpatient dermatology consultations are extremely valuable for improving diagnostic accuracy, reducing admissions for pseudocellulitis and inflammatory skin conditions, and keeping cancer patients on needed therapies.36-38 Despite this clear value added, a cross-sectional analysis of inpatient Medicare claims data from January 2013 to December 2019 found that the number of dermatologists performing more than 10 inpatient consults per year decreased from 356 to 281.39 Additionally, medical centers in which dermatology encounters occurred decreased from 239 to 157 during the same period. Ninety-eight percent of inpatient dermatologists were in metropolitan areas, with large regions lacking access to inpatient dermatology consultation altogether.39
A survey of Society for Pediatric Dermatology members similarly characterized the state of the pediatric dermatology workforce performing hospital consultation.40 Seventy-five percent reported a high call burden, defined as more than 11 days or nights per month, more than 1 weekend per month, and/or more than 5 hours per week seeing patients. Ninety-one percent of consultation services are based within academic institutions, reflecting disparities in access.40 A prospective cohort study of academic pediatric dermatologists reported that 310 curbside consultations were performed over 24 weeks; of these calls, 17% occurred during weeknights and 23% on weekends. None of these curbside interactions was reimbursed.41 These findings underscore the burden of uncompensated time a subset of pediatric dermatologists dedicates to inpatient consultations, highlighting the need for improved financial and administrative support and an increased number of physicians performing this role.
A survey study42 suggested that unfamiliarity with the inpatient setting, rather than medical knowledge, is the most important barrier to inpatient work among clinical dermatologists. Proposed interventions include resource guides (eg, hospital maps, pager numbers for key individuals, and protocols for urgent specimens). Reference guides and refresher courses may decrease gaps in knowledge or awareness among dermatologists in ambulatory practice.42 Another way to bolster the inpatient dermatology workforce may be to provide more guidance to qualified advanced practice providers to triage and address dermatologic emergencies.43
Artificial intelligence (AI) also has been explored as a tool for diagnosing complex dermatologic conditions. One study presented 15 published inpatient dermatology cases to 7 dermatologists. Participants were asked to formulate their top 3 differential diagnoses and were then shown AI-generated differentials and asked to submit a revised differential. Participants showed a diagnostic accuracy of 69% before seeing the AI-generated differential diagnosis and 79% after; however, in cases in which the AI differential was incorrect, diagnostic accuracy of the dermatologists decreased after being shown the AI model.44
Final Thoughts
This January 2024 to December 2025 review of research relevant to hospital dermatology highlights important developments and ongoing challenges in SCARs, emerging and re-emerging infectious diseases, HS, and the inpatient dermatology workforce. Dermatologists continue to play a critical role in the care of hospitalized patients with skin disease.
Dermatologists play a central role in the care of hospitalized patients with skin disease. This review summarizes research from January 2024 to December 2025 on severe cutaneous adverse drug reactions, emerging infectious diseases, hidradenitis suppurativa (HS), and inpatient dermatology workforce issues. Key developments include improved recognition and management of drug reactions; updated diagnostic and prognostic tools for Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN); and guidance for emerging infections such as measles, dengue, mpox, orthopoxviruses, and resistant dermatophytes. Evidence-based strategies for HS aim to reduce unnecessary admissions and optimize care. Workforce challenges, including limited access, high call burden, and potential for artificial intelligence (AI)–assisted diagnosis, are also highlighted. These findings emphasize the critical contributions of dermatologists to hospital-based care and provide emerging evidence to guide clinical practice.
Dermatologists play a critical role in the care of hospitalized patients. Herein, we review the research developments between January 2024 and December 2025 most relevant to the care of hospitalized patients with skin disease, including severe cutaneous adverse reactions (SCARs), emerging and re-emerging infectious diseases, hidradenitis suppurativa (HS), and access to inpatient dermatology services.
Severe Cutaneous Adverse Drug Reactions
Severe cutaneous adverse drug reactions are among the most frequent reasons for inpatient dermatology consultation. A National Inpatient Sample study identified more than 160,000 cases of drug rash with eosinophilia and systemic symptoms (DRESS syndrome) between January 2016 and December 2020.1 The overall mortality rate was 2.0%, substantially lower than the rates of up to 10% reported in earlier studies.2 Case burden and mortality peaked during the fall months, possibly due to either increased use of antibiotics or increased viral infection or reactivation during these months.1
A retrospective cohort study of patients with probable or definite DRESS syndrome showed that, among 93 patients with at least 1 viral marker tested, human herpesvirus (HHV) reactivation was found in 42% (39/93), including HHV-6 (28%)(24/85), Epstein-Barr virus (17%)(15/87), and cytomegalovirus (20%)(18/89); furthermore, viral reactivation was associated with higher 1-year mortality (odds ratio, 3.9), dialysis initiation, flares of disease, and longer hospital stay (all P<.05).1 Multiple reactivations were associated with higher inpatient mortality and 1-year mortality; however, despite apparent prognostic importance, the role of screening for viral reactivation in DRESS syndrome is undefined.
A systematic review of 610 studies including 2122 patients with DRESS syndrome demonstrated that, among 193 causal agents identified, 14 drugs accounted for more than 1% of cases each and therefore were considered high risk. Seventy-eight percent of cases were attributed to these 14 drugs (Table).5 A TriNetX Query study analyzed antibiotic exposures across SCARs and reported that sulfonamides (hazard ratio [HR], 7.5), aminoglycosides (HR, 3.7), and tetracyclines (HR, 1.7) were associated with an elevated risk for SCARs. Sulfonamides had the highest absolute incidence of SCARs, followed by cephalosporins and penicillins.6
A multicenter randomized clinical trial7 compared high-potency topical corticosteroids (clobetasol 30 g/d) to systemic corticosteroids (prednisone 0.5 mg/kg/d) for treatment of moderate DRESS syndrome. On day 30, 53.8% (14/26) of patients in the topical group had achieved remission of visceral involvement, compared to 72.0% (18/25) in the systemic group. Before day 30, 23.1% (6/26) of patients in the topical group worsened, necessitating transition to high-dose systemic steroids. When inpatient monitoring is available, low-dose systemic corticosteroids or high-potency topical steroids may be reasonable management strategies for moderate DRESS syndrome7; however, the frequent need for treatment intensification suggests limitations to this strategy.
Since prolonged courses of systemic steroids generally are necessary for management of DRESS syndrome, steroid-sparing options are needed. A retrospective case series examined interleukin 5 inhibition in patients with possible DRESS syndrome (Registry of Severe Cutaneous Adverse Reactions score ≥3). All patients demonstrated rapid eosinophil reduction within 1 to 3 days (mean [SD] time to resolution, 1.4 [0.9] days) after treatment with mepolizumab or benralizumab, with clinical improvement occurring at a mean (SD) of 16 (3.7) days (range, 13-21 days).8
A French cohort study of 1221 adult patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) reported in-hospital mortality of 19% and a total mortality of 34% at 1 year.9 Risk factors contributing to in-hospital mortality included age, history of/current diagnosis of cancer, dementia, and liver disease, while postdischarge mortality was associated with acute kidney injury and sepsis. Long-term complications included ophthalmologic and mood disorders.9
A new set of diagnostic criteria for SJS/TEN, known as the Niigata criteria,10 includes 3 main items: severe mucosal lesions in cutaneous-mucosal transition zones (eg, eyes, lips, vulva) or generalized erythema with necrotic lesions; fever of 38.5 °C or higher; and necrosis of the epidermis seen on histopathology. Because epidermal detachment involving 10% of the body surface area (BSA) is an important mortality risk predicter, SJS is defined as less than 10% BSA involvement, and TEN has been redefined as 10% or more BSA involvement (not ≥30%). A new prognostic score—clinical risk score for TEN (CRISTEN)—can be tabulated at the point of care without laboratory values. It was developed based on the 10 most important risk factors for death in a retrospective study of 382 patients, which included age 65 years or older; epidermal detachment involving 10% BSA or higher; an antibiotic as causative agent; systemic corticosteroid therapy before the onset of SJS/TEN; involvement of all 3 mucosal surfaces; and medical comorbidities such as renal impairment, diabetes, cardiac disease, active cancer, and bacterial infection.11
New potential therapeutic targets for SJS/TEN include PC111 (monoclonal antibody to Fas ligand), formyl peptide receptor 1 antagonists (which inhibit necroptosis induced by formyl peptide receptor 1–annexin A1 interaction), daratumumab (which depletes cytotoxic CD8-positive and CD38-positive T cells), and Janus kinase (JAK) inhibitors.10 Spatial proteomics showed marked enrichment of type I and type II interferon signatures as well as activation of signal transducer and activator of transcription 1. In vitro, tofacitinib reduced keratinocyte-directed cytotoxicity, and in vivo JAK inhibitors ameliorated disease severity in 2 TEN mouse models. Patients with TEN that was refractory to corticosteroid therapy received rescue treatment with JAK inhibitors and had re-epithelization within several days with marked reduction in levels of phosphorylated signal transducer and activator of transcription 1.12 Controlled studies are needed to assess the potential role of JAK inhibitors for SJS/TEN.
Emerging and Re-emerging Infectious Diseases
Dermatologists may encounter emerging or re-emerging infections, performing an essential public health role in the process. In 2025, a total of 2281 confirmed cases of measles had been reported across 45 of the United States.13 During the COVID-19 pandemic, measles vaccine coverage in the United States dropped to 93%—down from 95% to 97% prepandemic. Worldwide, 2022 saw an increase of 1.4 million measles cases (18% increase) and 41,200 excess deaths (43% increase) compared to the previous year. Complications of measles include pneumonia, blindness, otitis media, and encephalitis, with 1 in 5 (20%) unvaccinated people with measles in the United States requiring hospitalization.14 A vaccine coverage rate higher than 95% is needed to prevent community spread of disease. Since efforts to detect and rapidly isolate cases of measles are critical, dermatologists should consider measles in the differential of morbilliform eruptions with viral symptoms and ask about vaccination status.
Since 2023, dengue infection rates have tripled in the Americas, representing the highest levels recorded since tracking began in 1980. In 2024, there were more than 12 million cases, with approximately 8000 deaths reported. Ninety percent of cases occur in Argentina, Brazil, Colombia, and Mexico, but local transmission has been reported in Arizona, California, Florida, Hawaii, and Texas.15 The characteristic exanthem of dengue is diffuse erythema with islands of sparing.<
Unlike during the 2022 outbreak of mpox clade II, which predominantly impacted men who have sex with men, there now is an ongoing outbreak of mpox clades 1a and 1b in the Democratic Republic of the Congo and surrounding countries that more commonly affects children and heterosexual adults. It is also more transmissible and virulent. Cases of mpox clade I have been reported in several European countries and across the United States, mostly among travelers from areas of active transmission. Vaccination of at-risk individuals is considered effective; however, tecovirimat is not.16
Outbreaks of 2 emerging zoonotic orthopoxviruses recently have been reported. Buffalopox virus (BPXV) is transmitted via direct contact with the skin of infected cattle and buffalo as well as fomites and has been responsible for human cases in South Asia. Characteristics of BPXV include macules, umbilicated papules, vesicles, pustules, and eschars that evolve over several weeks, with a predilection for the hands and face. It can manifest with prodromal symptoms of fever, malaise, and lymphadenopathy.17 Borealpox virus (formerly known as Alaskapox) has similar manifestations. Its reservoir includes small mammals such as voles and shrews, but it also has been found in cats and dogs and has been responsible for at least one human fatality. Cidofovir may be an effective therapy for both BPXV and borealpox virus, and prior smallpox vaccination may provide protection.18 These outbreaks demonstrate the continued importance of research for more effective vaccines and therapies against smallpox and other orthopoxviruses.19 A recent review provided a detailed overview of the epidemiology, transmission, dermatologic findings, and management strategies associated with smallpox and other bioweapons.20
In 2023, a case was reported of a patient in a New York City hospital with tinea that was refractory to multiple rounds of topical antifungals, which called attention to the presence of Trichophyton indotineae in the United States.21 Since then, additional reports and case series have characterized the clinical presentation of T indotineae as widespread and atypical, refractory to traditional therapies, and most often encountered in travelers returning from Bangladesh or elsewhere in South Asia.22 The diagnosis should be confirmed via DNA testing of fungal culture. Itraconazole 100 to 200 mg/d is the antifungal therapy of choice.23
Other series have reported cases of tinea genitalis caused by Trichophyton mentagrophytes type VII seen predominately in sex workers and others engaging in high-risk sexual contact, highlighting the spread of dermatophytes through sexual activity.24-26 Lastly, it is important to culture pustules and consider atypical pathogens in patients with chronic folliculitis not responding to typical therapies such as tetracycline antibiotics. A case series reported the presence of pustules in the beard area of 7 men who have sex with men, with culture data showing Klebsiella aerogenes. Prolonged courses of fluoroquinolones were necessary for clearance.27
Reducing HS Admissions Through Evidence-Based Management
Hidradenitis suppurativa is a frequent cause of emergency department visits and hospital admissions. In an analysis of the Nationwide Readmissions Database, 17.8% (392/2204) of patients admitted to the hospital with HS were readmitted within 30 days, a number comparable to that of heart failure.28
Flaring HS can produce symptoms that mimic sepsis. A retrospective cohort study examining sepsislike features in HS showed that more than 50% (30/58) of those admitted to the hospital with an HS flare were misdiagnosed with sepsis, and more than 80% (53/64) of those patients received intravenous antibiotics.29 A National Inpatient Sample (January 2016-December 2018) study demonstrated minimal rates of true infection in patients admitted with HS flares,30 while patients with HS diagnosed as sepsis do not sustain the mortality expected from true sepsis. Improving recognition of HS and differentiation of the disease from true sepsis could decrease unnecessary antibiotic use, hospital admissions, and cost, underscoring the need for a framework to reliably and reproducibly distinguish sepsis from HS flare.31
While severe HS is difficult to manage, there may be a window of opportunity in which appropriate treatment of early disease may prevent progression and decrease inpatient utilization. A prospective cohort study of 335 biologic-naïve patients with mild to moderate HS (Hurley stages I and II) followed over a median of 2 years showed that active smoking, body mass index higher than 25, and the presence of disease in 2 or more anatomic areas were factors predictive of progression to severe disease.32
Despite high utilization of emergency and inpatient care, there has been no consensus on inpatient management of HS. A Delphi consensus exercise including 26 expert dermatologists reached consensus on 40 statements.33 Specific recommendations involve multidisciplinary care, including from a dermatologist; consideration of comorbid medical conditions; supportive care measures (wound care, pain control); evidence-based medical management, including initiation or adjustment of biologic therapies; targeted surgical intervention; nutritional support and maintenance of glycemic control; and attention to transitional care at discharge, including home health services, verification of insurance status, and timely outpatient dermatology follow-up.34 A retrospective review of 98 patients treated with intravenous ertapenem for a mean duration of 13 weeks demonstrated improvement in clinical and inflammatory markers.35 Patients with severe or treatment-refractory HS, including those admitted to the hospital, may benefit from initiation of this therapy in select circumstances.
Hospital Dermatology Workforce
Inpatient dermatology consultations are extremely valuable for improving diagnostic accuracy, reducing admissions for pseudocellulitis and inflammatory skin conditions, and keeping cancer patients on needed therapies.36-38 Despite this clear value added, a cross-sectional analysis of inpatient Medicare claims data from January 2013 to December 2019 found that the number of dermatologists performing more than 10 inpatient consults per year decreased from 356 to 281.39 Additionally, medical centers in which dermatology encounters occurred decreased from 239 to 157 during the same period. Ninety-eight percent of inpatient dermatologists were in metropolitan areas, with large regions lacking access to inpatient dermatology consultation altogether.39
A survey of Society for Pediatric Dermatology members similarly characterized the state of the pediatric dermatology workforce performing hospital consultation.40 Seventy-five percent reported a high call burden, defined as more than 11 days or nights per month, more than 1 weekend per month, and/or more than 5 hours per week seeing patients. Ninety-one percent of consultation services are based within academic institutions, reflecting disparities in access.40 A prospective cohort study of academic pediatric dermatologists reported that 310 curbside consultations were performed over 24 weeks; of these calls, 17% occurred during weeknights and 23% on weekends. None of these curbside interactions was reimbursed.41 These findings underscore the burden of uncompensated time a subset of pediatric dermatologists dedicates to inpatient consultations, highlighting the need for improved financial and administrative support and an increased number of physicians performing this role.
A survey study42 suggested that unfamiliarity with the inpatient setting, rather than medical knowledge, is the most important barrier to inpatient work among clinical dermatologists. Proposed interventions include resource guides (eg, hospital maps, pager numbers for key individuals, and protocols for urgent specimens). Reference guides and refresher courses may decrease gaps in knowledge or awareness among dermatologists in ambulatory practice.42 Another way to bolster the inpatient dermatology workforce may be to provide more guidance to qualified advanced practice providers to triage and address dermatologic emergencies.43
Artificial intelligence (AI) also has been explored as a tool for diagnosing complex dermatologic conditions. One study presented 15 published inpatient dermatology cases to 7 dermatologists. Participants were asked to formulate their top 3 differential diagnoses and were then shown AI-generated differentials and asked to submit a revised differential. Participants showed a diagnostic accuracy of 69% before seeing the AI-generated differential diagnosis and 79% after; however, in cases in which the AI differential was incorrect, diagnostic accuracy of the dermatologists decreased after being shown the AI model.44
Final Thoughts
This January 2024 to December 2025 review of research relevant to hospital dermatology highlights important developments and ongoing challenges in SCARs, emerging and re-emerging infectious diseases, HS, and the inpatient dermatology workforce. Dermatologists continue to play a critical role in the care of hospitalized patients with skin disease.
- Desai AD, Thomas C. Seasonal trends in drug reaction with eosinophilia and systemic symptoms. J Am Acad Dermatol. 2025;92:183-185.
- Wei BM, Fox LP, Kaffenberger BH, et al. Drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms. Part I. Epidemiology, pathogenesis, clinicopathological features, and prognosis. J Am Acad Dermatol. 2024;90:885-908. doi:10.1016/j.jaad.2023.02.072
- Chan LCE, Sultana R, Choo KJL, et al. Viral reactivation and clinical outcomes in drug reaction with eosinophilia and systemic symptoms (DRESS). Sci Rep. 2024;14:28492.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44
- Hansen E, Gallardo M, Yan A, et al. Risk assessment of drugs associated with DRESS syndrome based on publication frequency: a systematic review. J Am Acad Dermatol. 2024;91:962-966.
- Neubauer ZJK, Chan R, Singal A, et al. SCAR-ed by antibiotics: a retrospective cohort study of severe cutaneous adverse reactions (SCAR) relative risk. J Am Acad Dermatol. 2025;92:1143-1145.
- Ingen-Housz-Oro S, Guichard E, Milpied B, et al. Topical versus oral corticosteroids in moderate drug reaction with eosinophilia and systemic symptoms: a multicenter randomized clinical trial. J Am Acad Dermatol. 2024;91:544-547.
- Hijaz B, Nambudiri VE, Imadojemu S. IL-5 inhibitor treatment in drug reaction with eosinophilia and systemic symptoms. JAMA Dermatol. 2025;161:661-663.
- Bettuzzi T, Lebrun-Vignes B, Ingen-Housz-Oro S, et al. Incidence, in-hospital and long-term mortality, and sequelae of epidermal necrolysis in adults. JAMA Dermatol. 2024;160:1288-1296.
- Hama N, Aoki S, Chen CB, et al. Recent progress in Stevens-Johnson syndrome/toxic epidermal necrolysis: diagnostic criteria, pathogenesis and treatment. Br J Dermatol. 2024;192:9-18.
- Hama N, Sunaga Y, Ochiai H, et al. Development and validation of a novel score to predict mortality in Stevens-Johnson syndrome and toxic epidermal necrolysis: CRISTEN. J Allergy Clin Immunol Pract. 2023;11:3161-3168.e2.
- Nordmann TM, Anderton H, Hasegawa A, et al. Spatial proteomics identifies JAKi as treatment for a lethal skin disease. Nature. 2024;635:1001-1009.
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Updated January 7, 2026. Accessed January 12, 2026. https://www.cdc.gov/measles/data-research/
- Rubin R. Despite safe and effective vaccine, measles cases and deaths increased worldwide from 2021 to 2022. JAMA. 2024;331:188-189.
- Orrall A. Dengue cases in the Americas highest recorded. JAMA. 2025;333:452.
- Harris E. As mpox cases surge in Africa, WHO declares a global emergency-here’s what to know. JAMA. 2024;332:862-864.
- Burningham KM, Hinojosa T, Cavazos A, et al. Buffalopox: an emerging cutaneous disease in humans. J Eur Acad Dermatol Venereol. 2025;39:404-406.
- Parker ER. Emergence of Alaskapox infection: what dermatologists need to know. J Am Acad Dermatol. 2024;91:397-399.
- Gostin LO, Singaravelu S, Hynes N. Smallpox readiness: modern strategies against an ancient disease. JAMA. 2024;332:873-874.
- Osborne S, Kam O, Thacker S, et al. Review of category A bioweapons with cutaneous features: epidemiology, clinical presentation, and contemporary management strategies. J Am Acad Dermatol. 2025;93:165-175.
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field: first reported U.S. cases of tinea caused by Trichophyton indotineae - New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537.
- McKenna M. Why the rise of this drug-resistant fungus is raising international concern. JAMA. 2024;332:859-861.
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Luchsinger I, Bosshard PP, Kasper RS, et al. Tinea genitalis: a new entity of sexually transmitted infection? Case series and review of the literature. Sex Transm Infect. 2015;91:493-496.
- Khurana A, Sharath S, Sardana K, et al. Therapeutic updates on the management of tinea corporis or cruris in the era of Trichophyton indotineae: separating evidence from hype-a narrative review. Indian J Dermatol. 2023;68:525-540.
- Bérot V, Monsel G, Dauendorffer JN, et al; Groupe Infectiologie Dermatologique et Infections Sexuellement Transmissibles (GrIDIST) de la Société Française de Dermatologie. Klebsiella aerogenes-related facial folliculitis in men having sex with men: a hypothetical new STI?J Eur Acad Dermatol Venereol. 2025;39:E10-E12.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the Nationwide Readmissions Database. J Am Acad Dermatol. 2022;87:188-192.
- AbdelHameid D, Wang L, Mauskar MM, et al. Sepsis-like features in hidradenitis suppurativa flares requiring admission: a retrospective cohort study. J Am Acad Dermatol. 2024;90:1291-1294.
- Ehizogie E, Maghari I, Lo S, et al. Hidradenitis suppurativa, systemic inflammatory response syndrome and sepsis: a database study. Br J Dermatol. 2024;191:451-453.
- Maghari I, Abiad H, Griffin T, et al. Hidradenitis suppurativa (HS), systemic inflammatory response syndrome and sepsis, sepsis caused by HS: an empty systematic review. Br J Dermatol. 2024;191:449-450.
- Kjærsgaard Andersen R, Pedersen O, Eidsmo L, et al. Initial steps towards developing a predictive algorithm of disease progression for hidradenitis suppurativa (HS): results from a Cox proportional hazard regression analysis on disease progression among a cohort of 335 Danish patients with HS. Br J Dermatol. 2024;190:904-914.
- Needham M, Pichardo R, Alavi A, et al. Inpatient management of hidradenitis suppurativa: a Delphi consensus study. Cutis. 2024;113:251-254.
- Maskan Bermudez N, Elman SA, Kirsner RS, et al. Management of hidradenitis suppurativa in the inpatient setting: a clinical guide. Arch Dermatol Res. 2025;317:202.
- Nosrati A, Ch’en PY, Torpey ME, et al. Efficacy and durability of intravenous ertapenem therapy for recalcitrant hidradenitis suppurativa. JAMA Dermatol. 2024;160:312-318.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Hydol-Smith JA, Gallardo MA, Korman A, et al. The United States dermatology inpatient workforce between 2013 and 2019: a Medicare analysis reveals contraction of the workforce and vast access deserts-a cross-sectional analysis. Arch Dermatol Res. 2024;316:103.
- Pineider JL, Rangu SA, Shaw KS, et al. Pediatric consultative dermatology: a survey of the Society for Pediatric Dermatology workforce reveals shortcomings in existing practice models of pediatric dermatology consult services in the United States. Pediatr Dermatol. 2024;41:270-274.
- Puar NK, Canty KM, Newell BD, et al. An evaluation of pediatric dermatology curbside consultations in an academic center: a prospective cohort study. J Am Acad Dermatol. 2024;90:1258-1260.
- Lau CB, Smith GP. Strategies for improving dermatologist comfort and quality of patient care in inpatient settings: a cross-sectional survey study. Arch Dermatol Res. 2024;316:575.
- Hazim AH. Empowering advanced clinical practitioners in managing acute dermatological emergencies. Br J Nurs. 2024;33:448-455.
- Macklis P, Kaffenberger B, Kirven R, et al. Dermatology diagnostic accuracy is improved by artificial intelligence-generated differential diagnoses. Int J Dermatol. 2025;64:960-962.
- Desai AD, Thomas C. Seasonal trends in drug reaction with eosinophilia and systemic symptoms. J Am Acad Dermatol. 2025;92:183-185.
- Wei BM, Fox LP, Kaffenberger BH, et al. Drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms. Part I. Epidemiology, pathogenesis, clinicopathological features, and prognosis. J Am Acad Dermatol. 2024;90:885-908. doi:10.1016/j.jaad.2023.02.072
- Chan LCE, Sultana R, Choo KJL, et al. Viral reactivation and clinical outcomes in drug reaction with eosinophilia and systemic symptoms (DRESS). Sci Rep. 2024;14:28492.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44
- Hansen E, Gallardo M, Yan A, et al. Risk assessment of drugs associated with DRESS syndrome based on publication frequency: a systematic review. J Am Acad Dermatol. 2024;91:962-966.
- Neubauer ZJK, Chan R, Singal A, et al. SCAR-ed by antibiotics: a retrospective cohort study of severe cutaneous adverse reactions (SCAR) relative risk. J Am Acad Dermatol. 2025;92:1143-1145.
- Ingen-Housz-Oro S, Guichard E, Milpied B, et al. Topical versus oral corticosteroids in moderate drug reaction with eosinophilia and systemic symptoms: a multicenter randomized clinical trial. J Am Acad Dermatol. 2024;91:544-547.
- Hijaz B, Nambudiri VE, Imadojemu S. IL-5 inhibitor treatment in drug reaction with eosinophilia and systemic symptoms. JAMA Dermatol. 2025;161:661-663.
- Bettuzzi T, Lebrun-Vignes B, Ingen-Housz-Oro S, et al. Incidence, in-hospital and long-term mortality, and sequelae of epidermal necrolysis in adults. JAMA Dermatol. 2024;160:1288-1296.
- Hama N, Aoki S, Chen CB, et al. Recent progress in Stevens-Johnson syndrome/toxic epidermal necrolysis: diagnostic criteria, pathogenesis and treatment. Br J Dermatol. 2024;192:9-18.
- Hama N, Sunaga Y, Ochiai H, et al. Development and validation of a novel score to predict mortality in Stevens-Johnson syndrome and toxic epidermal necrolysis: CRISTEN. J Allergy Clin Immunol Pract. 2023;11:3161-3168.e2.
- Nordmann TM, Anderton H, Hasegawa A, et al. Spatial proteomics identifies JAKi as treatment for a lethal skin disease. Nature. 2024;635:1001-1009.
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Updated January 7, 2026. Accessed January 12, 2026. https://www.cdc.gov/measles/data-research/
- Rubin R. Despite safe and effective vaccine, measles cases and deaths increased worldwide from 2021 to 2022. JAMA. 2024;331:188-189.
- Orrall A. Dengue cases in the Americas highest recorded. JAMA. 2025;333:452.
- Harris E. As mpox cases surge in Africa, WHO declares a global emergency-here’s what to know. JAMA. 2024;332:862-864.
- Burningham KM, Hinojosa T, Cavazos A, et al. Buffalopox: an emerging cutaneous disease in humans. J Eur Acad Dermatol Venereol. 2025;39:404-406.
- Parker ER. Emergence of Alaskapox infection: what dermatologists need to know. J Am Acad Dermatol. 2024;91:397-399.
- Gostin LO, Singaravelu S, Hynes N. Smallpox readiness: modern strategies against an ancient disease. JAMA. 2024;332:873-874.
- Osborne S, Kam O, Thacker S, et al. Review of category A bioweapons with cutaneous features: epidemiology, clinical presentation, and contemporary management strategies. J Am Acad Dermatol. 2025;93:165-175.
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field: first reported U.S. cases of tinea caused by Trichophyton indotineae - New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537.
- McKenna M. Why the rise of this drug-resistant fungus is raising international concern. JAMA. 2024;332:859-861.
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Luchsinger I, Bosshard PP, Kasper RS, et al. Tinea genitalis: a new entity of sexually transmitted infection? Case series and review of the literature. Sex Transm Infect. 2015;91:493-496.
- Khurana A, Sharath S, Sardana K, et al. Therapeutic updates on the management of tinea corporis or cruris in the era of Trichophyton indotineae: separating evidence from hype-a narrative review. Indian J Dermatol. 2023;68:525-540.
- Bérot V, Monsel G, Dauendorffer JN, et al; Groupe Infectiologie Dermatologique et Infections Sexuellement Transmissibles (GrIDIST) de la Société Française de Dermatologie. Klebsiella aerogenes-related facial folliculitis in men having sex with men: a hypothetical new STI?J Eur Acad Dermatol Venereol. 2025;39:E10-E12.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the Nationwide Readmissions Database. J Am Acad Dermatol. 2022;87:188-192.
- AbdelHameid D, Wang L, Mauskar MM, et al. Sepsis-like features in hidradenitis suppurativa flares requiring admission: a retrospective cohort study. J Am Acad Dermatol. 2024;90:1291-1294.
- Ehizogie E, Maghari I, Lo S, et al. Hidradenitis suppurativa, systemic inflammatory response syndrome and sepsis: a database study. Br J Dermatol. 2024;191:451-453.
- Maghari I, Abiad H, Griffin T, et al. Hidradenitis suppurativa (HS), systemic inflammatory response syndrome and sepsis, sepsis caused by HS: an empty systematic review. Br J Dermatol. 2024;191:449-450.
- Kjærsgaard Andersen R, Pedersen O, Eidsmo L, et al. Initial steps towards developing a predictive algorithm of disease progression for hidradenitis suppurativa (HS): results from a Cox proportional hazard regression analysis on disease progression among a cohort of 335 Danish patients with HS. Br J Dermatol. 2024;190:904-914.
- Needham M, Pichardo R, Alavi A, et al. Inpatient management of hidradenitis suppurativa: a Delphi consensus study. Cutis. 2024;113:251-254.
- Maskan Bermudez N, Elman SA, Kirsner RS, et al. Management of hidradenitis suppurativa in the inpatient setting: a clinical guide. Arch Dermatol Res. 2025;317:202.
- Nosrati A, Ch’en PY, Torpey ME, et al. Efficacy and durability of intravenous ertapenem therapy for recalcitrant hidradenitis suppurativa. JAMA Dermatol. 2024;160:312-318.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Hydol-Smith JA, Gallardo MA, Korman A, et al. The United States dermatology inpatient workforce between 2013 and 2019: a Medicare analysis reveals contraction of the workforce and vast access deserts-a cross-sectional analysis. Arch Dermatol Res. 2024;316:103.
- Pineider JL, Rangu SA, Shaw KS, et al. Pediatric consultative dermatology: a survey of the Society for Pediatric Dermatology workforce reveals shortcomings in existing practice models of pediatric dermatology consult services in the United States. Pediatr Dermatol. 2024;41:270-274.
- Puar NK, Canty KM, Newell BD, et al. An evaluation of pediatric dermatology curbside consultations in an academic center: a prospective cohort study. J Am Acad Dermatol. 2024;90:1258-1260.
- Lau CB, Smith GP. Strategies for improving dermatologist comfort and quality of patient care in inpatient settings: a cross-sectional survey study. Arch Dermatol Res. 2024;316:575.
- Hazim AH. Empowering advanced clinical practitioners in managing acute dermatological emergencies. Br J Nurs. 2024;33:448-455.
- Macklis P, Kaffenberger B, Kirven R, et al. Dermatology diagnostic accuracy is improved by artificial intelligence-generated differential diagnoses. Int J Dermatol. 2025;64:960-962.
Hospital Dermatology: Review of Research in 2024-2025
Hospital Dermatology: Review of Research in 2024-2025
Practice Points
- In suspected drug reaction with eosinophilia and systemic symptoms, discontinue the offending drug; test for human herpesvirus 6, Epstein-Barr virus, and cytomegalovirus when available; and treat moderate cases with low-dose corticosteroids. Reserve interleukin 5 inhibitors for refractory disease.
- For Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN), apply Niigata diagnostic criteria and clinical risk score for TEN, refer patients with 10% or more body surface area detachment to higher-level or burn care, and consider targeted therapies for refractory cases.
- When assessing infectious rashes, consider measles, dengue, mpox, orthopoxviruses, and resistant dermatophytes. Review the patient’s vaccination and travel history, isolate suspected measles cases, and confirm atypical tinea with culture or DNA testing.
- To reduce unnecessary admissions and optimize care for hidradenitis suppurativa, avoid misdiagnosing flares as sepsis, implement multidisciplinary protocols, consider selective intravenous antibiotics, and support expanded inpatient dermatology coverage.
Dermatology Immediate Care: A Game Changer for the Health Care System?
Dermatology Immediate Care: A Game Changer for the Health Care System?
Emergency departments (EDs) and immediate care (IC) facilities often do not have prompt dermatologic care available for triage and treatment. Many EDs do not have staff dermatologists on call, instead relying on input from other specialists or quick outpatient dermatology appointments. It can be challenging to obtain a prompt appointment with a board-certified dermatologist, which is preferred for complex cases such as severe drug reactions or infection. In the United States, there are few well-established IC centers equipped to address dermatologic needs. The orthopedic specialty has modeled a concept that has led to the establishment of orthopedic urgent care/IC in many larger institutions,1 and many private practice clinics serve their communities as well. We present a rationale for why a similar IC concept for dermatology would be beneficial, particularly within a large institution or health system.
Dermatology Consultation Changes Disease Management
There is diagnostic and therapeutic utility in dermatology evaluation. In a prospective study of 591 patients who were either hospitalized or evaluated in an ED/urgent care setting, treatment was changed in more than 60% of cases when dermatology consultation was utilized.2 In another prospective review of 691 cases on an inpatient service, dermatology consultation resulted in treatment changes more than 80% of the time.3
Cellulitis has been a particularly well-studied diagnosis. Dermatologists often change the diagnosis of cellulitis in the hospital setting and reduce antibiotic exposure. In a prospective cohort study of 116 patients, 33.6% had their diagnosis of cellulitis changed to pseudocellulitis following evaluation by the dermatologist; of 34 patients who had started antibiotic therapy, 82.4% were recommended to discontinue the treatment, and all 39 patients with pseudocellulitis had a proven stable clinical course at 1-month follow-up.4 In another trial, 175 patients with presumed cellulitis were given standard management (provided by the medicine inpatient team) either alone or with the addition of dermatology consultation. Duration of antibiotic treatment (including intravenous therapy) was reduced when dermatology was consulted. Two weeks after discharge, patients who had dermatology consultations demonstrated greater clinical improvement.5
Improving ED and IC Access to Dermatology
Emergency department and IC teams across the United States work tirelessly to meet the demands of patients presenting with medically urgent conditions. In a study examining 861 ED cases, dermatology made up only 9.5% of specialist consultations, and in the opinion of the on-call dermatology resident, 51.0% (439/861) of cases warranted ED-level care.6
Data from the 2021 National Hospital Ambulatory Medical Care Survey showed that the mean wait time to see a physician, nurse, or physician assistant in an ED was 37.5 minutes, but wait times could range from less than 15 minutes to more than 6 hours.7 According to a study of 35,849 ED visits at nonfederal hospitals in the United States, only 47.7% of EDs admitted more than 90% of their patients within 6 hours.8 Moreover, perceived wait times in the ED have been shown to greatly impact patient satisfaction. Two predictors of perceived wait time include appropriate assessment of emergency level and the feeling of being forgotten.9 In a study of 2377 ED visits with primary dermatologic diagnoses, only 5.5% led to admission.10 This suggests many patients who come to the ED for dermatologic needs do not require inpatient hospital care. In these cases, patients with primary dermatologic concerns may experience longer ED wait times, as higher acuity or emergency cases take precedence. Studies also have shown that more vulnerable populations are utilizing ED visits most for primary dermatologic concerns.10,11 This includes individuals of lower income and/or those with Medicaid/Medicare or those without insurance.11 Predictors of high ED use for dermatologic concerns include prior frequent use of the ED (for nondermatologic concerns) instead of outpatient care, income below the poverty level, and lack of insurance; older individuals (>65 years) also were found to use the ED more frequently for dermatologic concerns when compared to younger individuals.10
Importantly, there is a great need for urgent dermatology consultation for pediatric patients. A single-institution study showed that over a 36-month period, there were 347 pediatric dermatology consultations from the pediatric ED mostly for children aged 0 days to 5 years; nearly half of these consultations required outpatient clinic follow-up.12 However, dermatology outpatient follow-up can be difficult to obtain, especially for vulnerable groups. In a study of 611 dermatology clinics, patients with Medicaid were shown to have longer wait times and less success in obtaining dermatology appointments compared to those with Medicare or private insurance.13 Only about 30% of private dermatology practices accept Medicaid patients, likely pushing these patients toward utilization of emergency services for dermatologic concerns.13,14
There is a clear role for a dermatology IC in our health care system, and the concept already has been identified and trialed in several institutions. At Oregon Health and Science University (Portland, Oregon), a retrospective chart review of patients with diagnoses of Morgellons disease and neurotic excoriations seen in dermatology urgent care between 2018 and 2020 showed an 88% decrease in annual rates of health care visits and a 77% decrease in ED visits after dermatology services were engaged compared to before the opening of the dermatology urgent care.15 Another study showed that uninsured or self-pay patients were more than 14 times more likely to access dermatology urgent care than to schedule a routine clinic appointment, suggesting that there is a barrier to making outpatient dermatologic appointments for uninsured patients. An urgent access model may facilitate the ability of underinsured patients to access care.16
Improving Dermatology Access for Other Specialties
Needs for dermatologic care are encountered in many other specialties. Having direct access to immediate dermatologic treatment is best for patients and may avoid inpatient care and trips to the ED for consultation access. Ideally, a dermatology IC would allow direct care to be provided alongside the oncology outpatient team. New immunologic therapies (cytotoxic T-lymphocyte–associated protein 4 and programmed cell death protein 1/programmed death-ligand 1 treatments) can cause dermatologic reactions in more than 40% of patients.17 Paraneoplastic syndromes can manifest with cutaneous symptoms, as can acute graft-vs-host-disease.18 In a study at Memorial Sloan Kettering (New York, New York) analyzing 426 same-day outpatient dermatology consultations, 17% of patients experienced interruptions in their cancer therapy, but 83% responded quickly to dermatologic treatment and resumed oncologic therapy—19% of them at a reduced dose.19 This is an important demonstration of prompt dermatologic consultation in an outpatient setting reducing interruptions to anticancer therapy. The heterogeneity of the cutaneous reactions seen from oncologic and immunomodulatory medications is profound, with more than 140 different types of skin-specific reactions.20
Solid-organ transplant recipients also could benefit from urgent access to dermatology services. These patients are at a much higher risk for skin cancers, and a study showed that those who receive referrals to dermatology are seen sooner after transplantation (5.6 years) than those who self-refer (7.2 years). Importantly, annual skin cancer screenings are recommended to begin 1 year after transplantation.21
Direct access to dermatology care could benefit patients with complicated rheumatologic conditions who present with skin findings; for example, patients with lupus erythematosus or dermatomyositis can have a spectrum of disease ranging from skin-predominant to systemic manifestations. Identification and treatment of such diseases require collaboration between dermatologists and rheumatologists.22 Likewise, a study of a joint rheumatology-dermatology clinic for psoriatic arthritis showed that a multidisciplinary approach to management leads to decreased time for patients to obtain proper rheumatologic and dermatologic examination and a faster time to diagnosis; however, such multidisciplinary clinic models and approaches to care often are found only at large university-based hospitals.23 In a patient population for whom time to diagnosis is crucial to avoid permanent changes such as joint destruction, a dermatology IC could fill this role in community hospitals and clinics. A dermatology IC also can serve patients with specific diagnoses who would benefit from more direct access to care; for example, in 2017 there were 131,430 ED visits for hidradenitis suppurativa (HS) in the United States. While HS is not uncommon, it usually is underdiagnosed because it can be challenging to differentiate from an uncomplicated abscess. Emergency department visits often are utilized for first-time presentations as well as flares of HS. In these situations, ED doctors can provide palliative treatment, but prompt referrals to dermatologists should be made for disease management to decrease recurrence.24
Final Thoughts
A huge caveat to the dermatology urgent care system is determining what is deemed “urgent.” We propose starting with a referral-based system only from other physicians (including IC and urgent care) rather than having patients walk in directly. Ideally, as support and staff increases, the availability can increase as well. In our institution, we suggested half-day clinics staffed by varying physicians, with compensation models similar to an ED or IC physician rather than by productivity. Each group considering this kind of addition to patient care will need to assess these points in building an IC for dermatology. The University of Pennsylvania’s (Philadelphia, Pennsylvania) system of rapid-access clinics to facilitate access to care for patients requiring urgent appointments may function as a model for future similar clinics.25 Creating a specialized IC/urgent care is not a novel concept. Orthopedic urgent care centers have increased greatly in the past decade, reducing ED burden for musculoskeletal complaints. In a study evaluating the utility of orthopedic urgent care settings, time to see an orthopedic specialist and cost were both greatly reduced with this system.1 The same has been shown in same-day access ophthalmology clinics, which are organized similarly to an urgent care.26
In 2021, there were 107.4 million treat-and-release visits to the ED in the United States for a total cost of $80.3 billion.27 This emphasizes the need to consider care models that not only provide excellent clinical care and treat the most acute diagnoses promptly and accurately but also reduce overall costs. While this may be convoluted for other specialties given the difficulty of having patients self-triage, dermatologic concerns are similar to orthopedic concerns for the patient to decipher the etiology of the concern. As in orthopedics, a dermatology IC could function similarly, increasing access, decreasing ED and IC wait times, saving overall health care spending, and allowing underserved and publicly insured individuals to have improved, prompt care.
- Anderson TJ, Althausen PL. The role of dedicated musculoskeletal urgent care centers in reducing cost and improving access to orthopaedic care. J Orthop Trauma. 2016;30:S3-S6.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Grillo E, Vañó-Galván S, Jiménez-Gómez N, et al. Dermatologic emergencies: descriptive analysis of 861 patients in a tertiary care teaching hospital. Actas Dermosifiliogr. 2013;104:316-324.
- National Center for Health Statistics. National Hospital Ambulatory Medical Care Survey, 2021. Accessed September 23, 2025. https://www.cdc.gov/nchs/data/nhamcs/web_tables/2021-nhamcs-ed-web-tables-508.pdf
- Horwitz LI, Green J, Bradley EH. US emergency department performance on wait time and length of visit. Ann Emerg Med. 2010;55:133-141.
- Spechbach H, Rochat J, Gaspoz JM, et al. Patients’ time perception in the waiting room of an ambulatory emergency unit: a cross-sectional study. BMC Emerg Med. 2019;19:41.
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483.
- Chen CL, Fitzpatrick L, Kamel H. Who uses the emergency department for dermatologic care? a statewide analysis. J Am Acad Dermatol. 2014;71:308-313.
- Moon AT, Castelo-Soccio L, Yan AC. Emergency department utilization of pediatric dermatology (PD) consultations. J Am Acad Dermatol. 2016;74:1173-1177.
- Creadore A, Desai S, Li SJ, et al. Insurance acceptance, appointment wait time, and dermatologist access across practice types in the US. JAMA Dermatol. 2021;157:181-188.
- Mazmudar RS, Gupta N, Desai BJ, et al. Dermatologist appointment access and waiting times: a comparative study of insurance types. J Am Acad Dermatol. 2020;83:1468-1470.
- Johnson J, Cutler B, Latour E, et al. Dermatology urgent care model reduces costs and healthcare utilization for psychodermatology patients-a retrospective chart review. Dermatol Online J. 2022;28:5.
- Wintringham JA, Strock DM, Perkins-Holtsclaw K, et al. Dermatology in the urgent care setting: a retrospective review of patients seen in an urgent access dermatology clinic. J Am Acad Dermatol. 2023;89:1271-1273.
- Yoo MJ, Long B, Brady WJ, et al. Immune checkpoint inhibitors: an emergency medicine focused review. Am J Emerg Med. 2021;50:335-344.
- Merlo G, Cozzani E, Canale F, et al. Cutaneous manifestations of hematologic malignancies the experience of an Italian dermatology department. Hematol Oncol. 2019;37:285-290.
- Barrios D, Phillips G, Freites-Martinez A, et al. Outpatient dermatology consultations for oncology patients with acute dermatologic adverse events impact anticancer therapy interruption: a retrospective study.J Eur Acad Dermatol Venereol. 2020;34:1340-1347.
- Salah S, Kerob D, Pages Laurent C, et al. Evaluation of anticancer therapy-related dermatologic adverse events: insights from Food and Drug Administration’s Adverse Event Reporting System dataset. J Am Acad Dermatol. 2024;91:863-871. doi:10.1016/j.jaad.2024.07.1456
- Shope C, Andrews L, Girvin A, et al. Referrals to dermatology following solid organ transplant. J Am Acad Dermatol. 2023;88:1159-1160. doi:10.1016/j.jaad.2022.11.052
- Werth VP, Askanase AD, Lundberg IE. Importance of collaboration of dermatology and rheumatology to advance the field for lupus and dermatomyositis. Int J Womens Dermatol. 2021;7:583-587.
- Ziob J, Behning C, Brossart P, et al. Specialized dermatological-rheumatological patient management improves diagnostic outcome and patient journey in psoriasis and psoriatic arthritis: a four-year analysis. BMC Rheumatol. 2021;5:1-8. doi:10.1186/s41927-021-00217-z
- Okun MM, Flamm A, Werley EB, et al. Hidradenitis suppurativa: diagnosis and management in the emergency department. J Emerg Med. 2022;63:636-644.
- Jayakumar KL, Samimi SS, Vittorio CC, et al. Expediting patient appointments with dermatology rapid access clinics. Dermatol Online J. 2018;24:13030/qt2zv07510.
- Singman EL, Smith K, Mehta R, et al. Cost and visit duration of same-day access at an academic ophthalmology department vs emergency department. JAMA Ophthalmol. 2019;137:729-735. doi:10.1001/jamaophthalmol.2019.0864
- Roemer M. Costs of treat-and-release emergency department visits in the United States, 2021. Agency for Healthcare Research and Quality. Published September 2024. Accessed September 16, 2025. https://hcup-us.ahrq.gov/reports/statbriefs/sb311-ED-visit-costs-2021.pdf
Emergency departments (EDs) and immediate care (IC) facilities often do not have prompt dermatologic care available for triage and treatment. Many EDs do not have staff dermatologists on call, instead relying on input from other specialists or quick outpatient dermatology appointments. It can be challenging to obtain a prompt appointment with a board-certified dermatologist, which is preferred for complex cases such as severe drug reactions or infection. In the United States, there are few well-established IC centers equipped to address dermatologic needs. The orthopedic specialty has modeled a concept that has led to the establishment of orthopedic urgent care/IC in many larger institutions,1 and many private practice clinics serve their communities as well. We present a rationale for why a similar IC concept for dermatology would be beneficial, particularly within a large institution or health system.
Dermatology Consultation Changes Disease Management
There is diagnostic and therapeutic utility in dermatology evaluation. In a prospective study of 591 patients who were either hospitalized or evaluated in an ED/urgent care setting, treatment was changed in more than 60% of cases when dermatology consultation was utilized.2 In another prospective review of 691 cases on an inpatient service, dermatology consultation resulted in treatment changes more than 80% of the time.3
Cellulitis has been a particularly well-studied diagnosis. Dermatologists often change the diagnosis of cellulitis in the hospital setting and reduce antibiotic exposure. In a prospective cohort study of 116 patients, 33.6% had their diagnosis of cellulitis changed to pseudocellulitis following evaluation by the dermatologist; of 34 patients who had started antibiotic therapy, 82.4% were recommended to discontinue the treatment, and all 39 patients with pseudocellulitis had a proven stable clinical course at 1-month follow-up.4 In another trial, 175 patients with presumed cellulitis were given standard management (provided by the medicine inpatient team) either alone or with the addition of dermatology consultation. Duration of antibiotic treatment (including intravenous therapy) was reduced when dermatology was consulted. Two weeks after discharge, patients who had dermatology consultations demonstrated greater clinical improvement.5
Improving ED and IC Access to Dermatology
Emergency department and IC teams across the United States work tirelessly to meet the demands of patients presenting with medically urgent conditions. In a study examining 861 ED cases, dermatology made up only 9.5% of specialist consultations, and in the opinion of the on-call dermatology resident, 51.0% (439/861) of cases warranted ED-level care.6
Data from the 2021 National Hospital Ambulatory Medical Care Survey showed that the mean wait time to see a physician, nurse, or physician assistant in an ED was 37.5 minutes, but wait times could range from less than 15 minutes to more than 6 hours.7 According to a study of 35,849 ED visits at nonfederal hospitals in the United States, only 47.7% of EDs admitted more than 90% of their patients within 6 hours.8 Moreover, perceived wait times in the ED have been shown to greatly impact patient satisfaction. Two predictors of perceived wait time include appropriate assessment of emergency level and the feeling of being forgotten.9 In a study of 2377 ED visits with primary dermatologic diagnoses, only 5.5% led to admission.10 This suggests many patients who come to the ED for dermatologic needs do not require inpatient hospital care. In these cases, patients with primary dermatologic concerns may experience longer ED wait times, as higher acuity or emergency cases take precedence. Studies also have shown that more vulnerable populations are utilizing ED visits most for primary dermatologic concerns.10,11 This includes individuals of lower income and/or those with Medicaid/Medicare or those without insurance.11 Predictors of high ED use for dermatologic concerns include prior frequent use of the ED (for nondermatologic concerns) instead of outpatient care, income below the poverty level, and lack of insurance; older individuals (>65 years) also were found to use the ED more frequently for dermatologic concerns when compared to younger individuals.10
Importantly, there is a great need for urgent dermatology consultation for pediatric patients. A single-institution study showed that over a 36-month period, there were 347 pediatric dermatology consultations from the pediatric ED mostly for children aged 0 days to 5 years; nearly half of these consultations required outpatient clinic follow-up.12 However, dermatology outpatient follow-up can be difficult to obtain, especially for vulnerable groups. In a study of 611 dermatology clinics, patients with Medicaid were shown to have longer wait times and less success in obtaining dermatology appointments compared to those with Medicare or private insurance.13 Only about 30% of private dermatology practices accept Medicaid patients, likely pushing these patients toward utilization of emergency services for dermatologic concerns.13,14
There is a clear role for a dermatology IC in our health care system, and the concept already has been identified and trialed in several institutions. At Oregon Health and Science University (Portland, Oregon), a retrospective chart review of patients with diagnoses of Morgellons disease and neurotic excoriations seen in dermatology urgent care between 2018 and 2020 showed an 88% decrease in annual rates of health care visits and a 77% decrease in ED visits after dermatology services were engaged compared to before the opening of the dermatology urgent care.15 Another study showed that uninsured or self-pay patients were more than 14 times more likely to access dermatology urgent care than to schedule a routine clinic appointment, suggesting that there is a barrier to making outpatient dermatologic appointments for uninsured patients. An urgent access model may facilitate the ability of underinsured patients to access care.16
Improving Dermatology Access for Other Specialties
Needs for dermatologic care are encountered in many other specialties. Having direct access to immediate dermatologic treatment is best for patients and may avoid inpatient care and trips to the ED for consultation access. Ideally, a dermatology IC would allow direct care to be provided alongside the oncology outpatient team. New immunologic therapies (cytotoxic T-lymphocyte–associated protein 4 and programmed cell death protein 1/programmed death-ligand 1 treatments) can cause dermatologic reactions in more than 40% of patients.17 Paraneoplastic syndromes can manifest with cutaneous symptoms, as can acute graft-vs-host-disease.18 In a study at Memorial Sloan Kettering (New York, New York) analyzing 426 same-day outpatient dermatology consultations, 17% of patients experienced interruptions in their cancer therapy, but 83% responded quickly to dermatologic treatment and resumed oncologic therapy—19% of them at a reduced dose.19 This is an important demonstration of prompt dermatologic consultation in an outpatient setting reducing interruptions to anticancer therapy. The heterogeneity of the cutaneous reactions seen from oncologic and immunomodulatory medications is profound, with more than 140 different types of skin-specific reactions.20
Solid-organ transplant recipients also could benefit from urgent access to dermatology services. These patients are at a much higher risk for skin cancers, and a study showed that those who receive referrals to dermatology are seen sooner after transplantation (5.6 years) than those who self-refer (7.2 years). Importantly, annual skin cancer screenings are recommended to begin 1 year after transplantation.21
Direct access to dermatology care could benefit patients with complicated rheumatologic conditions who present with skin findings; for example, patients with lupus erythematosus or dermatomyositis can have a spectrum of disease ranging from skin-predominant to systemic manifestations. Identification and treatment of such diseases require collaboration between dermatologists and rheumatologists.22 Likewise, a study of a joint rheumatology-dermatology clinic for psoriatic arthritis showed that a multidisciplinary approach to management leads to decreased time for patients to obtain proper rheumatologic and dermatologic examination and a faster time to diagnosis; however, such multidisciplinary clinic models and approaches to care often are found only at large university-based hospitals.23 In a patient population for whom time to diagnosis is crucial to avoid permanent changes such as joint destruction, a dermatology IC could fill this role in community hospitals and clinics. A dermatology IC also can serve patients with specific diagnoses who would benefit from more direct access to care; for example, in 2017 there were 131,430 ED visits for hidradenitis suppurativa (HS) in the United States. While HS is not uncommon, it usually is underdiagnosed because it can be challenging to differentiate from an uncomplicated abscess. Emergency department visits often are utilized for first-time presentations as well as flares of HS. In these situations, ED doctors can provide palliative treatment, but prompt referrals to dermatologists should be made for disease management to decrease recurrence.24
Final Thoughts
A huge caveat to the dermatology urgent care system is determining what is deemed “urgent.” We propose starting with a referral-based system only from other physicians (including IC and urgent care) rather than having patients walk in directly. Ideally, as support and staff increases, the availability can increase as well. In our institution, we suggested half-day clinics staffed by varying physicians, with compensation models similar to an ED or IC physician rather than by productivity. Each group considering this kind of addition to patient care will need to assess these points in building an IC for dermatology. The University of Pennsylvania’s (Philadelphia, Pennsylvania) system of rapid-access clinics to facilitate access to care for patients requiring urgent appointments may function as a model for future similar clinics.25 Creating a specialized IC/urgent care is not a novel concept. Orthopedic urgent care centers have increased greatly in the past decade, reducing ED burden for musculoskeletal complaints. In a study evaluating the utility of orthopedic urgent care settings, time to see an orthopedic specialist and cost were both greatly reduced with this system.1 The same has been shown in same-day access ophthalmology clinics, which are organized similarly to an urgent care.26
In 2021, there were 107.4 million treat-and-release visits to the ED in the United States for a total cost of $80.3 billion.27 This emphasizes the need to consider care models that not only provide excellent clinical care and treat the most acute diagnoses promptly and accurately but also reduce overall costs. While this may be convoluted for other specialties given the difficulty of having patients self-triage, dermatologic concerns are similar to orthopedic concerns for the patient to decipher the etiology of the concern. As in orthopedics, a dermatology IC could function similarly, increasing access, decreasing ED and IC wait times, saving overall health care spending, and allowing underserved and publicly insured individuals to have improved, prompt care.
Emergency departments (EDs) and immediate care (IC) facilities often do not have prompt dermatologic care available for triage and treatment. Many EDs do not have staff dermatologists on call, instead relying on input from other specialists or quick outpatient dermatology appointments. It can be challenging to obtain a prompt appointment with a board-certified dermatologist, which is preferred for complex cases such as severe drug reactions or infection. In the United States, there are few well-established IC centers equipped to address dermatologic needs. The orthopedic specialty has modeled a concept that has led to the establishment of orthopedic urgent care/IC in many larger institutions,1 and many private practice clinics serve their communities as well. We present a rationale for why a similar IC concept for dermatology would be beneficial, particularly within a large institution or health system.
Dermatology Consultation Changes Disease Management
There is diagnostic and therapeutic utility in dermatology evaluation. In a prospective study of 591 patients who were either hospitalized or evaluated in an ED/urgent care setting, treatment was changed in more than 60% of cases when dermatology consultation was utilized.2 In another prospective review of 691 cases on an inpatient service, dermatology consultation resulted in treatment changes more than 80% of the time.3
Cellulitis has been a particularly well-studied diagnosis. Dermatologists often change the diagnosis of cellulitis in the hospital setting and reduce antibiotic exposure. In a prospective cohort study of 116 patients, 33.6% had their diagnosis of cellulitis changed to pseudocellulitis following evaluation by the dermatologist; of 34 patients who had started antibiotic therapy, 82.4% were recommended to discontinue the treatment, and all 39 patients with pseudocellulitis had a proven stable clinical course at 1-month follow-up.4 In another trial, 175 patients with presumed cellulitis were given standard management (provided by the medicine inpatient team) either alone or with the addition of dermatology consultation. Duration of antibiotic treatment (including intravenous therapy) was reduced when dermatology was consulted. Two weeks after discharge, patients who had dermatology consultations demonstrated greater clinical improvement.5
Improving ED and IC Access to Dermatology
Emergency department and IC teams across the United States work tirelessly to meet the demands of patients presenting with medically urgent conditions. In a study examining 861 ED cases, dermatology made up only 9.5% of specialist consultations, and in the opinion of the on-call dermatology resident, 51.0% (439/861) of cases warranted ED-level care.6
Data from the 2021 National Hospital Ambulatory Medical Care Survey showed that the mean wait time to see a physician, nurse, or physician assistant in an ED was 37.5 minutes, but wait times could range from less than 15 minutes to more than 6 hours.7 According to a study of 35,849 ED visits at nonfederal hospitals in the United States, only 47.7% of EDs admitted more than 90% of their patients within 6 hours.8 Moreover, perceived wait times in the ED have been shown to greatly impact patient satisfaction. Two predictors of perceived wait time include appropriate assessment of emergency level and the feeling of being forgotten.9 In a study of 2377 ED visits with primary dermatologic diagnoses, only 5.5% led to admission.10 This suggests many patients who come to the ED for dermatologic needs do not require inpatient hospital care. In these cases, patients with primary dermatologic concerns may experience longer ED wait times, as higher acuity or emergency cases take precedence. Studies also have shown that more vulnerable populations are utilizing ED visits most for primary dermatologic concerns.10,11 This includes individuals of lower income and/or those with Medicaid/Medicare or those without insurance.11 Predictors of high ED use for dermatologic concerns include prior frequent use of the ED (for nondermatologic concerns) instead of outpatient care, income below the poverty level, and lack of insurance; older individuals (>65 years) also were found to use the ED more frequently for dermatologic concerns when compared to younger individuals.10
Importantly, there is a great need for urgent dermatology consultation for pediatric patients. A single-institution study showed that over a 36-month period, there were 347 pediatric dermatology consultations from the pediatric ED mostly for children aged 0 days to 5 years; nearly half of these consultations required outpatient clinic follow-up.12 However, dermatology outpatient follow-up can be difficult to obtain, especially for vulnerable groups. In a study of 611 dermatology clinics, patients with Medicaid were shown to have longer wait times and less success in obtaining dermatology appointments compared to those with Medicare or private insurance.13 Only about 30% of private dermatology practices accept Medicaid patients, likely pushing these patients toward utilization of emergency services for dermatologic concerns.13,14
There is a clear role for a dermatology IC in our health care system, and the concept already has been identified and trialed in several institutions. At Oregon Health and Science University (Portland, Oregon), a retrospective chart review of patients with diagnoses of Morgellons disease and neurotic excoriations seen in dermatology urgent care between 2018 and 2020 showed an 88% decrease in annual rates of health care visits and a 77% decrease in ED visits after dermatology services were engaged compared to before the opening of the dermatology urgent care.15 Another study showed that uninsured or self-pay patients were more than 14 times more likely to access dermatology urgent care than to schedule a routine clinic appointment, suggesting that there is a barrier to making outpatient dermatologic appointments for uninsured patients. An urgent access model may facilitate the ability of underinsured patients to access care.16
Improving Dermatology Access for Other Specialties
Needs for dermatologic care are encountered in many other specialties. Having direct access to immediate dermatologic treatment is best for patients and may avoid inpatient care and trips to the ED for consultation access. Ideally, a dermatology IC would allow direct care to be provided alongside the oncology outpatient team. New immunologic therapies (cytotoxic T-lymphocyte–associated protein 4 and programmed cell death protein 1/programmed death-ligand 1 treatments) can cause dermatologic reactions in more than 40% of patients.17 Paraneoplastic syndromes can manifest with cutaneous symptoms, as can acute graft-vs-host-disease.18 In a study at Memorial Sloan Kettering (New York, New York) analyzing 426 same-day outpatient dermatology consultations, 17% of patients experienced interruptions in their cancer therapy, but 83% responded quickly to dermatologic treatment and resumed oncologic therapy—19% of them at a reduced dose.19 This is an important demonstration of prompt dermatologic consultation in an outpatient setting reducing interruptions to anticancer therapy. The heterogeneity of the cutaneous reactions seen from oncologic and immunomodulatory medications is profound, with more than 140 different types of skin-specific reactions.20
Solid-organ transplant recipients also could benefit from urgent access to dermatology services. These patients are at a much higher risk for skin cancers, and a study showed that those who receive referrals to dermatology are seen sooner after transplantation (5.6 years) than those who self-refer (7.2 years). Importantly, annual skin cancer screenings are recommended to begin 1 year after transplantation.21
Direct access to dermatology care could benefit patients with complicated rheumatologic conditions who present with skin findings; for example, patients with lupus erythematosus or dermatomyositis can have a spectrum of disease ranging from skin-predominant to systemic manifestations. Identification and treatment of such diseases require collaboration between dermatologists and rheumatologists.22 Likewise, a study of a joint rheumatology-dermatology clinic for psoriatic arthritis showed that a multidisciplinary approach to management leads to decreased time for patients to obtain proper rheumatologic and dermatologic examination and a faster time to diagnosis; however, such multidisciplinary clinic models and approaches to care often are found only at large university-based hospitals.23 In a patient population for whom time to diagnosis is crucial to avoid permanent changes such as joint destruction, a dermatology IC could fill this role in community hospitals and clinics. A dermatology IC also can serve patients with specific diagnoses who would benefit from more direct access to care; for example, in 2017 there were 131,430 ED visits for hidradenitis suppurativa (HS) in the United States. While HS is not uncommon, it usually is underdiagnosed because it can be challenging to differentiate from an uncomplicated abscess. Emergency department visits often are utilized for first-time presentations as well as flares of HS. In these situations, ED doctors can provide palliative treatment, but prompt referrals to dermatologists should be made for disease management to decrease recurrence.24
Final Thoughts
A huge caveat to the dermatology urgent care system is determining what is deemed “urgent.” We propose starting with a referral-based system only from other physicians (including IC and urgent care) rather than having patients walk in directly. Ideally, as support and staff increases, the availability can increase as well. In our institution, we suggested half-day clinics staffed by varying physicians, with compensation models similar to an ED or IC physician rather than by productivity. Each group considering this kind of addition to patient care will need to assess these points in building an IC for dermatology. The University of Pennsylvania’s (Philadelphia, Pennsylvania) system of rapid-access clinics to facilitate access to care for patients requiring urgent appointments may function as a model for future similar clinics.25 Creating a specialized IC/urgent care is not a novel concept. Orthopedic urgent care centers have increased greatly in the past decade, reducing ED burden for musculoskeletal complaints. In a study evaluating the utility of orthopedic urgent care settings, time to see an orthopedic specialist and cost were both greatly reduced with this system.1 The same has been shown in same-day access ophthalmology clinics, which are organized similarly to an urgent care.26
In 2021, there were 107.4 million treat-and-release visits to the ED in the United States for a total cost of $80.3 billion.27 This emphasizes the need to consider care models that not only provide excellent clinical care and treat the most acute diagnoses promptly and accurately but also reduce overall costs. While this may be convoluted for other specialties given the difficulty of having patients self-triage, dermatologic concerns are similar to orthopedic concerns for the patient to decipher the etiology of the concern. As in orthopedics, a dermatology IC could function similarly, increasing access, decreasing ED and IC wait times, saving overall health care spending, and allowing underserved and publicly insured individuals to have improved, prompt care.
- Anderson TJ, Althausen PL. The role of dedicated musculoskeletal urgent care centers in reducing cost and improving access to orthopaedic care. J Orthop Trauma. 2016;30:S3-S6.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Grillo E, Vañó-Galván S, Jiménez-Gómez N, et al. Dermatologic emergencies: descriptive analysis of 861 patients in a tertiary care teaching hospital. Actas Dermosifiliogr. 2013;104:316-324.
- National Center for Health Statistics. National Hospital Ambulatory Medical Care Survey, 2021. Accessed September 23, 2025. https://www.cdc.gov/nchs/data/nhamcs/web_tables/2021-nhamcs-ed-web-tables-508.pdf
- Horwitz LI, Green J, Bradley EH. US emergency department performance on wait time and length of visit. Ann Emerg Med. 2010;55:133-141.
- Spechbach H, Rochat J, Gaspoz JM, et al. Patients’ time perception in the waiting room of an ambulatory emergency unit: a cross-sectional study. BMC Emerg Med. 2019;19:41.
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483.
- Chen CL, Fitzpatrick L, Kamel H. Who uses the emergency department for dermatologic care? a statewide analysis. J Am Acad Dermatol. 2014;71:308-313.
- Moon AT, Castelo-Soccio L, Yan AC. Emergency department utilization of pediatric dermatology (PD) consultations. J Am Acad Dermatol. 2016;74:1173-1177.
- Creadore A, Desai S, Li SJ, et al. Insurance acceptance, appointment wait time, and dermatologist access across practice types in the US. JAMA Dermatol. 2021;157:181-188.
- Mazmudar RS, Gupta N, Desai BJ, et al. Dermatologist appointment access and waiting times: a comparative study of insurance types. J Am Acad Dermatol. 2020;83:1468-1470.
- Johnson J, Cutler B, Latour E, et al. Dermatology urgent care model reduces costs and healthcare utilization for psychodermatology patients-a retrospective chart review. Dermatol Online J. 2022;28:5.
- Wintringham JA, Strock DM, Perkins-Holtsclaw K, et al. Dermatology in the urgent care setting: a retrospective review of patients seen in an urgent access dermatology clinic. J Am Acad Dermatol. 2023;89:1271-1273.
- Yoo MJ, Long B, Brady WJ, et al. Immune checkpoint inhibitors: an emergency medicine focused review. Am J Emerg Med. 2021;50:335-344.
- Merlo G, Cozzani E, Canale F, et al. Cutaneous manifestations of hematologic malignancies the experience of an Italian dermatology department. Hematol Oncol. 2019;37:285-290.
- Barrios D, Phillips G, Freites-Martinez A, et al. Outpatient dermatology consultations for oncology patients with acute dermatologic adverse events impact anticancer therapy interruption: a retrospective study.J Eur Acad Dermatol Venereol. 2020;34:1340-1347.
- Salah S, Kerob D, Pages Laurent C, et al. Evaluation of anticancer therapy-related dermatologic adverse events: insights from Food and Drug Administration’s Adverse Event Reporting System dataset. J Am Acad Dermatol. 2024;91:863-871. doi:10.1016/j.jaad.2024.07.1456
- Shope C, Andrews L, Girvin A, et al. Referrals to dermatology following solid organ transplant. J Am Acad Dermatol. 2023;88:1159-1160. doi:10.1016/j.jaad.2022.11.052
- Werth VP, Askanase AD, Lundberg IE. Importance of collaboration of dermatology and rheumatology to advance the field for lupus and dermatomyositis. Int J Womens Dermatol. 2021;7:583-587.
- Ziob J, Behning C, Brossart P, et al. Specialized dermatological-rheumatological patient management improves diagnostic outcome and patient journey in psoriasis and psoriatic arthritis: a four-year analysis. BMC Rheumatol. 2021;5:1-8. doi:10.1186/s41927-021-00217-z
- Okun MM, Flamm A, Werley EB, et al. Hidradenitis suppurativa: diagnosis and management in the emergency department. J Emerg Med. 2022;63:636-644.
- Jayakumar KL, Samimi SS, Vittorio CC, et al. Expediting patient appointments with dermatology rapid access clinics. Dermatol Online J. 2018;24:13030/qt2zv07510.
- Singman EL, Smith K, Mehta R, et al. Cost and visit duration of same-day access at an academic ophthalmology department vs emergency department. JAMA Ophthalmol. 2019;137:729-735. doi:10.1001/jamaophthalmol.2019.0864
- Roemer M. Costs of treat-and-release emergency department visits in the United States, 2021. Agency for Healthcare Research and Quality. Published September 2024. Accessed September 16, 2025. https://hcup-us.ahrq.gov/reports/statbriefs/sb311-ED-visit-costs-2021.pdf
- Anderson TJ, Althausen PL. The role of dedicated musculoskeletal urgent care centers in reducing cost and improving access to orthopaedic care. J Orthop Trauma. 2016;30:S3-S6.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Grillo E, Vañó-Galván S, Jiménez-Gómez N, et al. Dermatologic emergencies: descriptive analysis of 861 patients in a tertiary care teaching hospital. Actas Dermosifiliogr. 2013;104:316-324.
- National Center for Health Statistics. National Hospital Ambulatory Medical Care Survey, 2021. Accessed September 23, 2025. https://www.cdc.gov/nchs/data/nhamcs/web_tables/2021-nhamcs-ed-web-tables-508.pdf
- Horwitz LI, Green J, Bradley EH. US emergency department performance on wait time and length of visit. Ann Emerg Med. 2010;55:133-141.
- Spechbach H, Rochat J, Gaspoz JM, et al. Patients’ time perception in the waiting room of an ambulatory emergency unit: a cross-sectional study. BMC Emerg Med. 2019;19:41.
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483.
- Chen CL, Fitzpatrick L, Kamel H. Who uses the emergency department for dermatologic care? a statewide analysis. J Am Acad Dermatol. 2014;71:308-313.
- Moon AT, Castelo-Soccio L, Yan AC. Emergency department utilization of pediatric dermatology (PD) consultations. J Am Acad Dermatol. 2016;74:1173-1177.
- Creadore A, Desai S, Li SJ, et al. Insurance acceptance, appointment wait time, and dermatologist access across practice types in the US. JAMA Dermatol. 2021;157:181-188.
- Mazmudar RS, Gupta N, Desai BJ, et al. Dermatologist appointment access and waiting times: a comparative study of insurance types. J Am Acad Dermatol. 2020;83:1468-1470.
- Johnson J, Cutler B, Latour E, et al. Dermatology urgent care model reduces costs and healthcare utilization for psychodermatology patients-a retrospective chart review. Dermatol Online J. 2022;28:5.
- Wintringham JA, Strock DM, Perkins-Holtsclaw K, et al. Dermatology in the urgent care setting: a retrospective review of patients seen in an urgent access dermatology clinic. J Am Acad Dermatol. 2023;89:1271-1273.
- Yoo MJ, Long B, Brady WJ, et al. Immune checkpoint inhibitors: an emergency medicine focused review. Am J Emerg Med. 2021;50:335-344.
- Merlo G, Cozzani E, Canale F, et al. Cutaneous manifestations of hematologic malignancies the experience of an Italian dermatology department. Hematol Oncol. 2019;37:285-290.
- Barrios D, Phillips G, Freites-Martinez A, et al. Outpatient dermatology consultations for oncology patients with acute dermatologic adverse events impact anticancer therapy interruption: a retrospective study.J Eur Acad Dermatol Venereol. 2020;34:1340-1347.
- Salah S, Kerob D, Pages Laurent C, et al. Evaluation of anticancer therapy-related dermatologic adverse events: insights from Food and Drug Administration’s Adverse Event Reporting System dataset. J Am Acad Dermatol. 2024;91:863-871. doi:10.1016/j.jaad.2024.07.1456
- Shope C, Andrews L, Girvin A, et al. Referrals to dermatology following solid organ transplant. J Am Acad Dermatol. 2023;88:1159-1160. doi:10.1016/j.jaad.2022.11.052
- Werth VP, Askanase AD, Lundberg IE. Importance of collaboration of dermatology and rheumatology to advance the field for lupus and dermatomyositis. Int J Womens Dermatol. 2021;7:583-587.
- Ziob J, Behning C, Brossart P, et al. Specialized dermatological-rheumatological patient management improves diagnostic outcome and patient journey in psoriasis and psoriatic arthritis: a four-year analysis. BMC Rheumatol. 2021;5:1-8. doi:10.1186/s41927-021-00217-z
- Okun MM, Flamm A, Werley EB, et al. Hidradenitis suppurativa: diagnosis and management in the emergency department. J Emerg Med. 2022;63:636-644.
- Jayakumar KL, Samimi SS, Vittorio CC, et al. Expediting patient appointments with dermatology rapid access clinics. Dermatol Online J. 2018;24:13030/qt2zv07510.
- Singman EL, Smith K, Mehta R, et al. Cost and visit duration of same-day access at an academic ophthalmology department vs emergency department. JAMA Ophthalmol. 2019;137:729-735. doi:10.1001/jamaophthalmol.2019.0864
- Roemer M. Costs of treat-and-release emergency department visits in the United States, 2021. Agency for Healthcare Research and Quality. Published September 2024. Accessed September 16, 2025. https://hcup-us.ahrq.gov/reports/statbriefs/sb311-ED-visit-costs-2021.pdf
Dermatology Immediate Care: A Game Changer for the Health Care System?
Dermatology Immediate Care: A Game Changer for the Health Care System?
Practice Points
- Emergency departments and most immediate care (IC) centers often lack prompt access to board-certified dermatologists.
- A dermatology urgent care/IC model may shorten wait times, improve access for vulnerable patients and pediatric populations, and reduce unnecessary hospital admissions and costs.
- Increased access to dermatology benefits other specialties by enabling multidisciplinary care leading to faster diagnosis and treatment.
- A staged referral-first dermatology IC pilot with defined staffing and triage rules is a practical path to demonstrate value and scale the service.

Scurvy in Hospitalized Patients
Scurvy in Hospitalized Patients
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
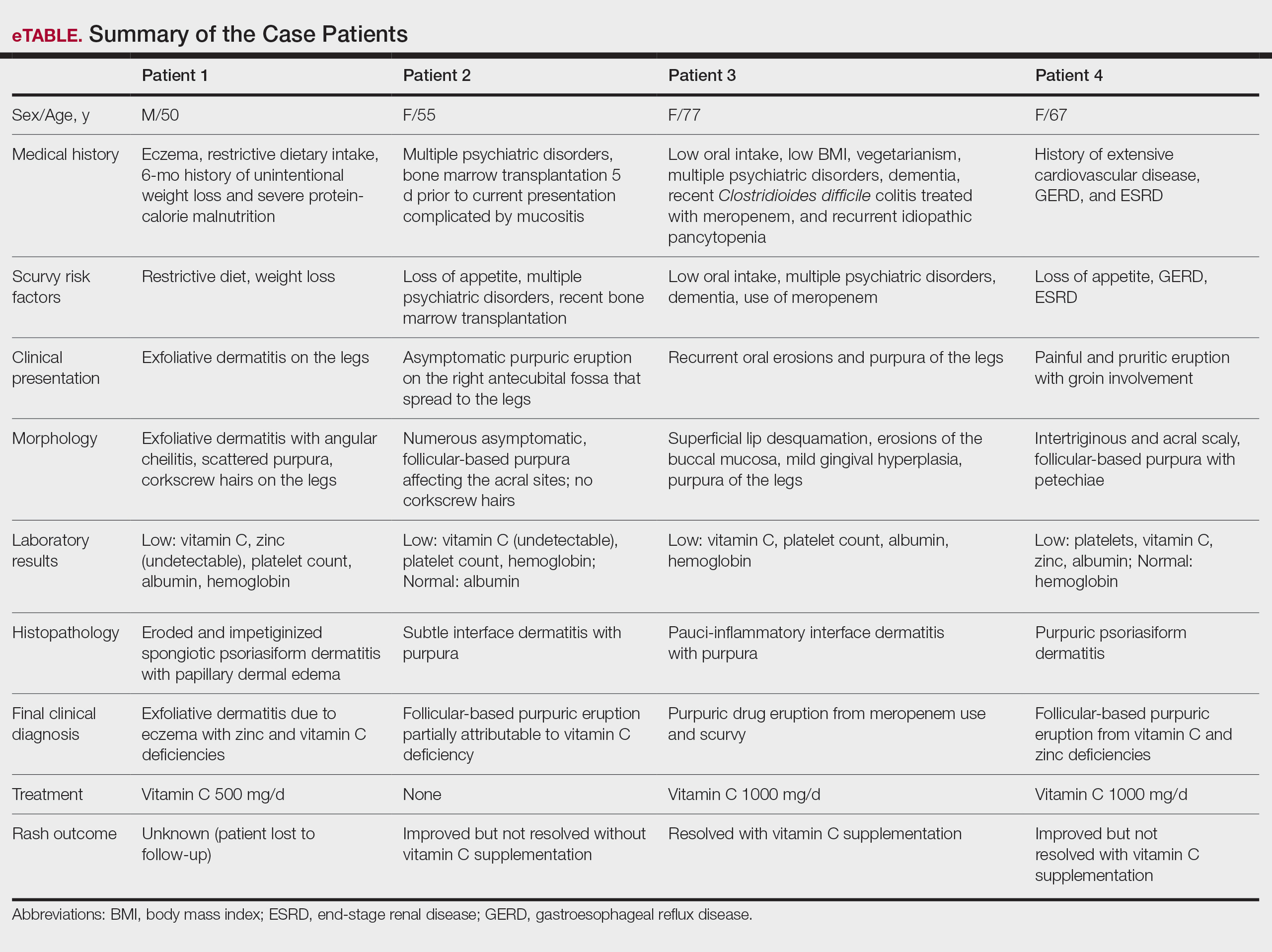
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
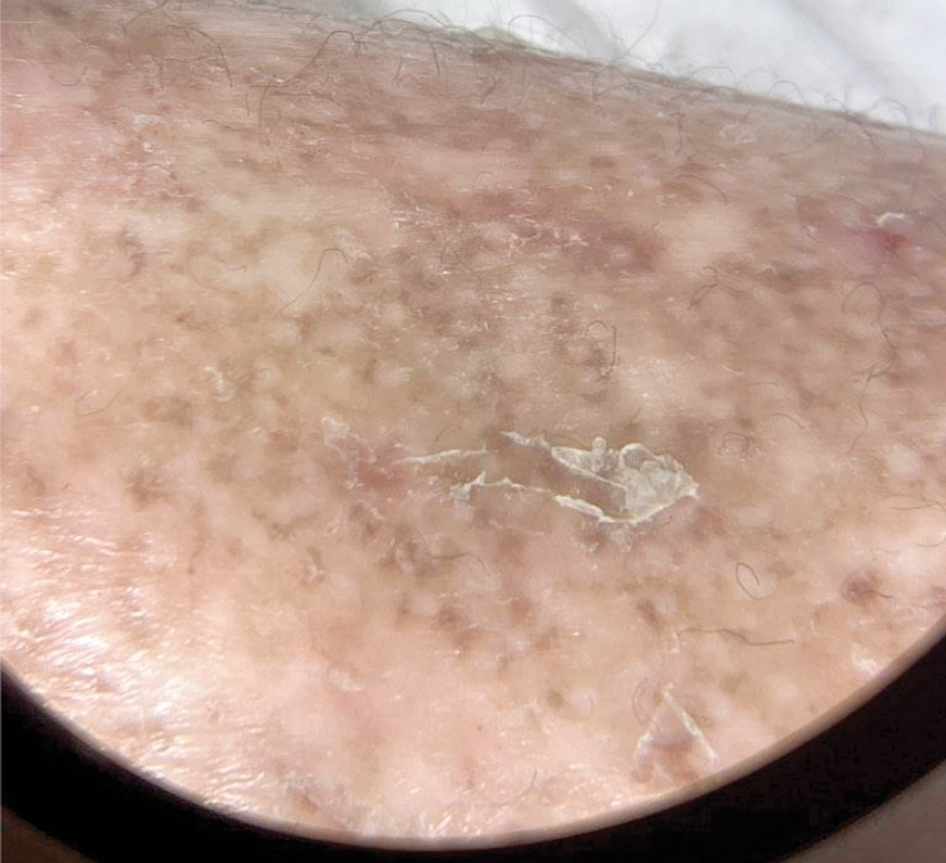
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
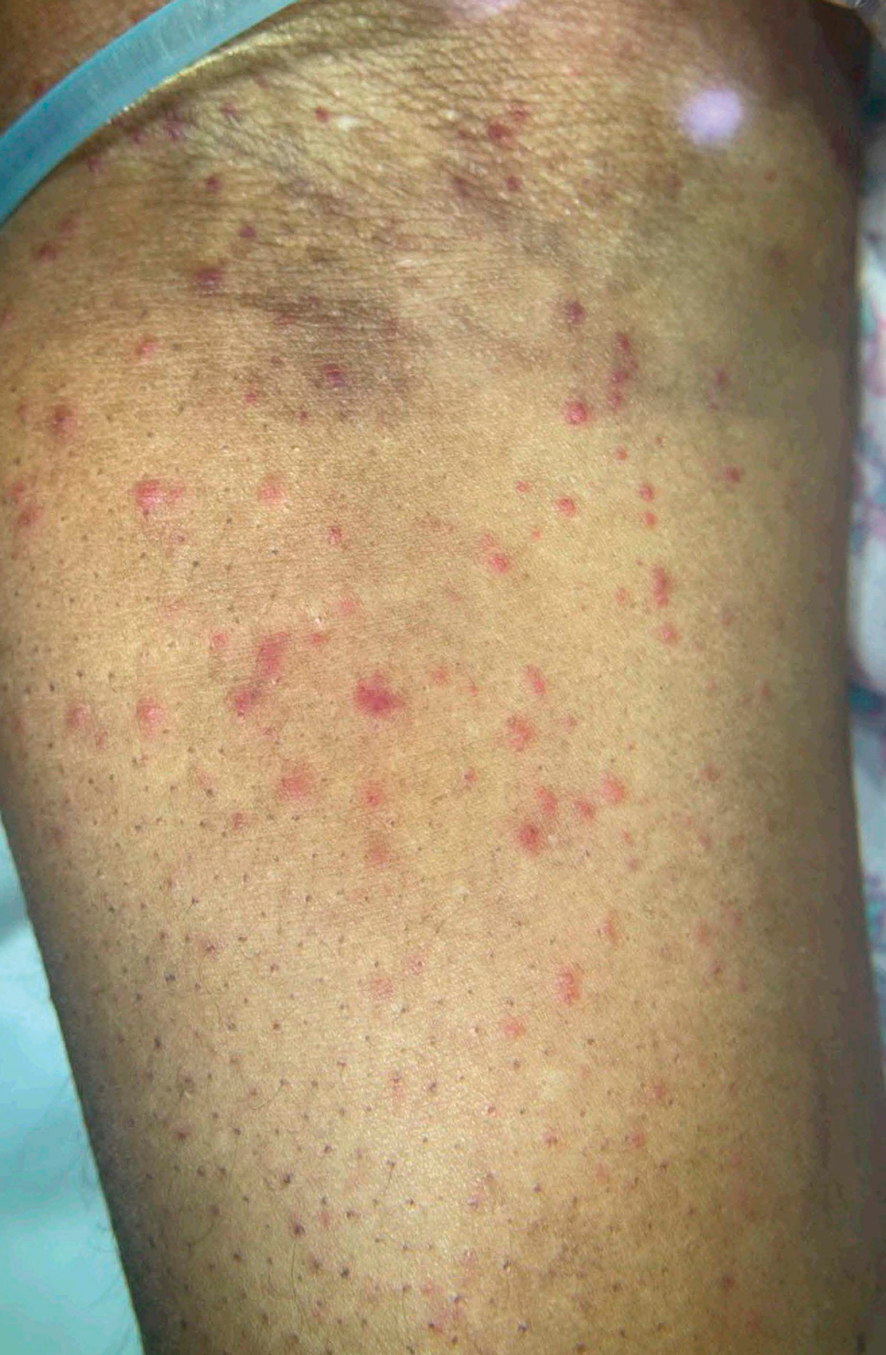
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
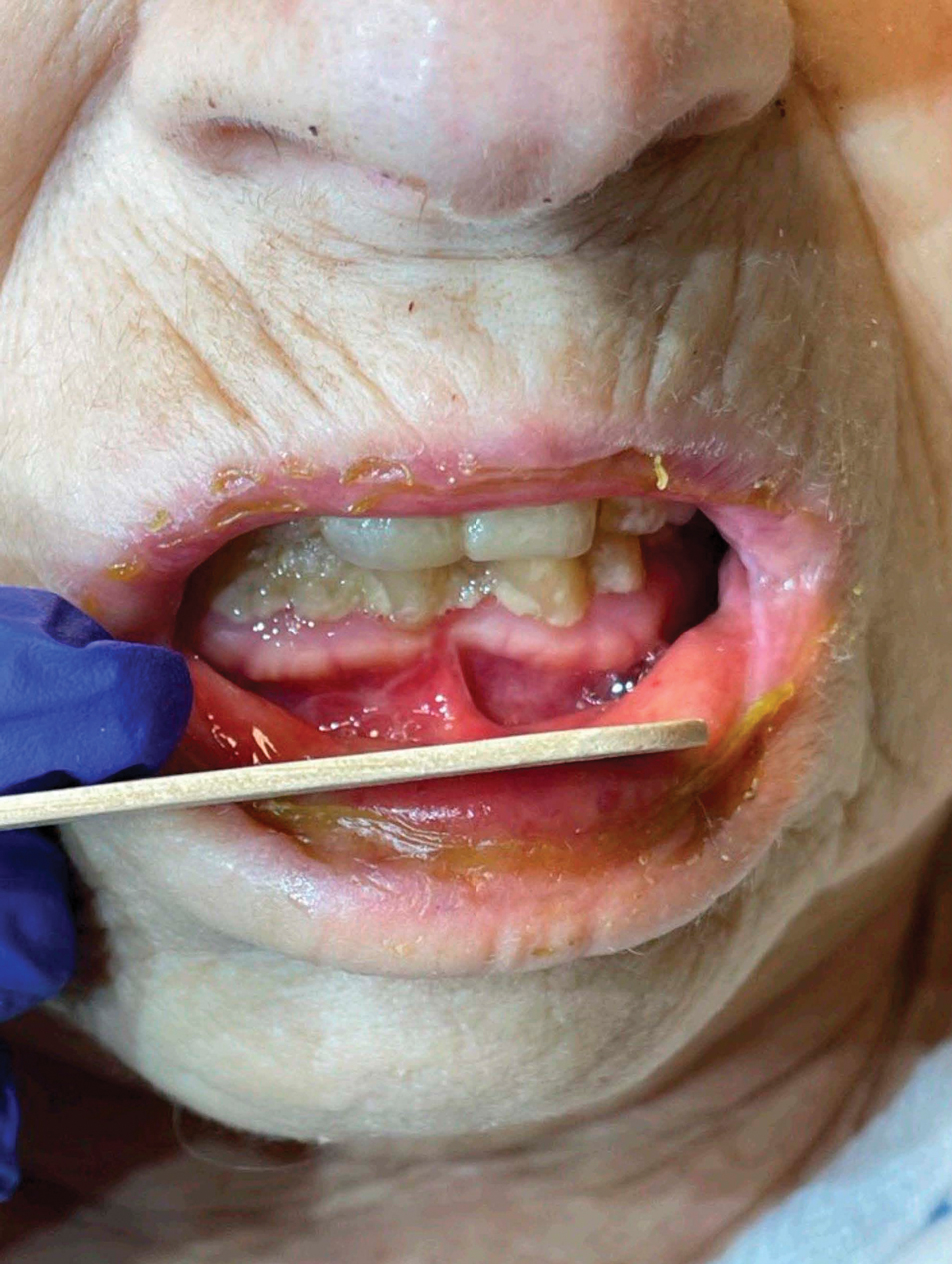
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
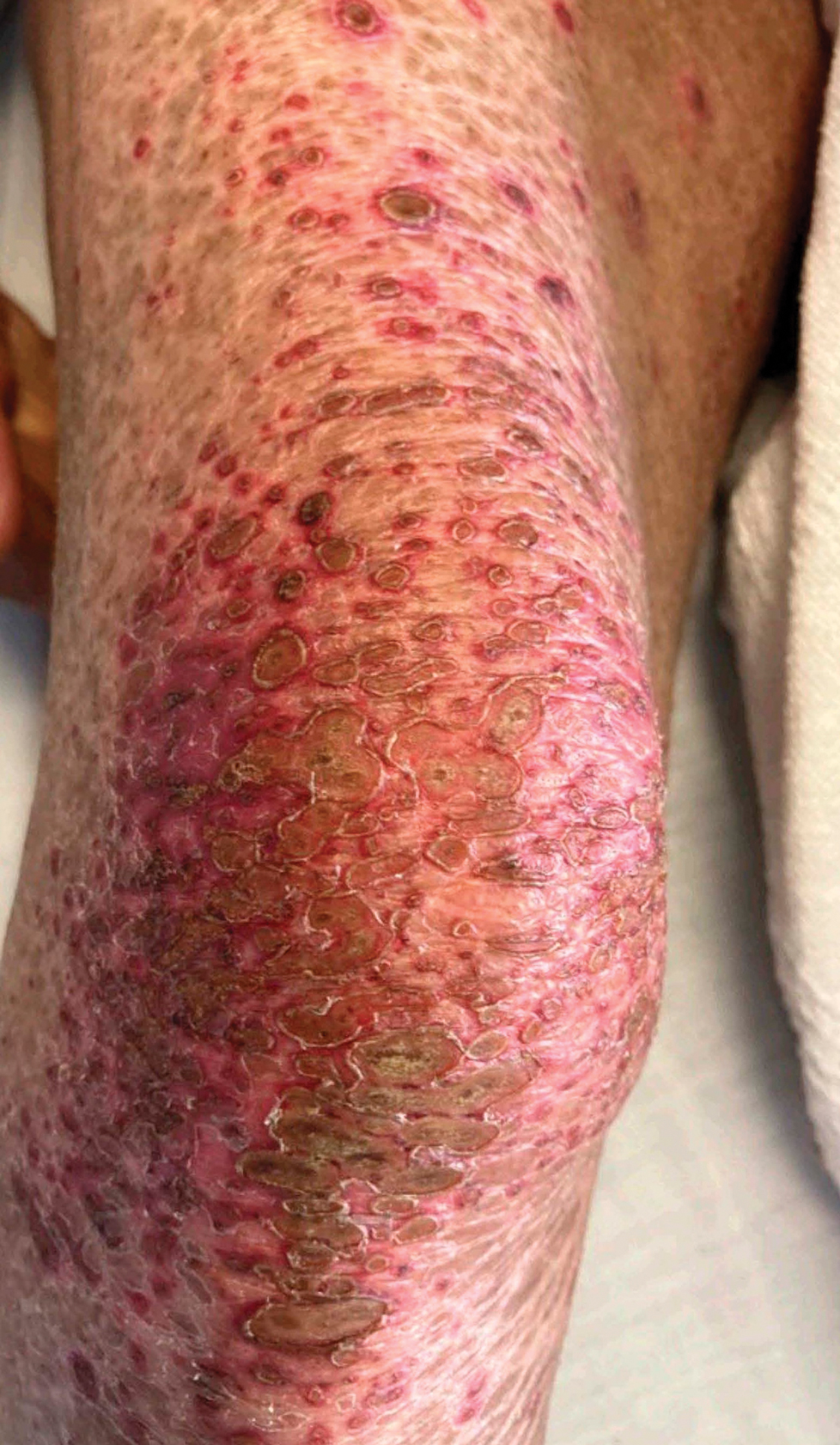
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
Scurvy in Hospitalized Patients
Scurvy in Hospitalized Patients
PRACTICE POINTS
- Clinicians should maintain a high index of suspicion for vitamin C deficiency/scurvy in hospitalized patients with purpura who have multiple medical comorbidities and risk factors.
- A low platelet count may mask underlying vitamin C deficiency, and patients may have concurrent deficiencies in other nutrients such as zinc.

A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.

Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
PRACTICE POINTS
- Hemophagocytic lymphohistiocytosis (HLH) is a complex, life-threatening immunologic condition that is associated with various diagnostic tools.
- Physicians who care for patients with HLH should know that skin findings are not uncommon but are largely nonspecific and can be a direct result of HLH itself, systemic complications, or the underlying etiology of the condition.
- There is a myriad of cutaneous findings that can manifest in adult patients with HLH. Awareness of HLH-associated dermatologic conditions and available diagnostic tools among multidisciplinary teams will aid in diagnosis.
Hospital Dermatology: Review of Research in 2023-2024
Inpatient consultative dermatology has advanced as a subspecialty and increasingly gained recognition in recent years. Since its founding in 2009, the Society of Dermatology Hospitalists has fostered research and education in hospital dermatology. Last year, we reviewed the 2022-2023 literature with a focus on developments in severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.1 In this review, we highlight 3 areas of interest from the 2023-2024 literature: severe cutaneous adverse drug reactions, skin and soft tissue infections, and autoimmune blistering diseases (AIBDs).
Severe Cutaneous Adverse Drug Reactions
Adverse drug reactions are among the most common diagnoses encountered by inpatient dermatology consultants.2,3 Severe cutaneous adverse drug reactions are associated with substantial morbidity and mortality. Efforts to characterize these conditions and standardize their diagnosis and management continue to be a major focus of ongoing research.
A single-center retrospective analysis of 102 cases of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome evaluated differences in clinical manifestations depending on the culprit drug, offering insights into the heterogeneity of DRESS syndrome and the potential for diagnostic uncertainty.4 The shortest median latency was observed in a case caused by penicillin and cephalosporins (12 and 18 days, respectively), while DRESS syndrome secondary to allopurinol had the longest median latency (36 days). Nonsteroidal anti-inflammatory drug–induced DRESS syndrome was associated with the shortest hospital stay (6.5 days), while cephalosporin and vancomycin cases had the highest mortality rates.4
In the first international Delphi consensus study on the diagnostic workup, severity assessment, and management of DRESS syndrome, 54 dermatology and/or allergy experts reached consensus on 93 statements.5 Specific recommendations included basic evaluation with complete blood count with differential, kidney and liver function parameters, and electrocardiogram for all patients with suspected DRESS syndrome, with additional complementary workup considered in patients with evidence of specific organ damage and/or severe disease. In the proposed DRESS syndrome severity grading scheme, laboratory values that reached consensus for inclusion were hemoglobin, neutrophil, and platelet counts and creatinine, transaminases, and alkaline phosphatase levels. Although treatment of DRESS syndrome should be based on assessed disease severity, treatment with corticosteroids should be initiated in all patients with confirmed DRESS syndrome. Cyclosporine, antibodies interfering with the IL-5 axis, and intravenous immunoglobulins can be considered in patients with corticosteroid-refractory DRESS syndrome, and antiviral treatment can be considered in patients with a high serum cytomegalovirus viral load. Regularly following up with laboratory evaluation of involved organs; screening for autoantibodies, thyroid dysfunction, and steroid adverse effects; and offering of psychological support also were consensus recommendations.5
Identifying causative agents in drug hypersensitivity reactions remains challenging. A retrospective cohort study of 48 patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) highlighted the need for a systematic unbiased approach to identifying culprit drugs. Using the RegiSCAR database and algorithm for drug causality for epidermal necrolysis to analyze the cohort, more than half of causative agents were determined to be different from those initially identified by the treating physicians. Nine additional suspected culprit drugs were identified, while 43 drugs initially identified as allergens were exonerated.6
Etiology-associated definitions for blistering reactions in children have been proposed to replace the existing terms Stevens-Johnson syndrome, toxic epidermal necrolysis, and others.7 Investigators in a recent study reclassified cases of SJS and TEN as reactive infectious mucocutaneous eruption (RIME) or drug-induced epidermal necrolysis (DEN), respectively. In RIME cases, Mycoplasma pneumoniae was the most commonly identified trigger, and in DEN cases, anticonvulsants were the most common class of culprit medications. Cases of RIME were less severe and were most often treated with antibiotics, whereas patients with DEN were more likely to receive supportive care, corticosteroids, intravenous immunoglobulins, and other immunosuppressive therapies.7
In addition to causing acute devastating mucocutaneous complications, SJS and TEN have long-lasting effects that require ongoing care. In a cohort of 6552 incident SJS/TEN cases over an 11-year period, survivors of SJS/TEN endured a mean loss of 9.4 years in life expectancy and excess health care expenditures of $3752 per year compared with age- and sex-matched controls. Patients with more severe disease, comorbid malignancy, diabetes, end-stage renal disease, or SJS/TEN sequelae experienced greater loss in life expectancy and lifetime health care expenditures.8 Separately, a qualitative study investigating the psychological impact of SJS/TEN in pediatric patients described sequelae including night terrors, posttraumatic stress disorder, depression, and anxiety for many years after the acute phase. Many patients reported a desire for increased support for their physical and emotional needs following hospital discharge.9
Skin and Soft Tissue Infections: Diagnosis, Management, and Prevention
Dermatology consultation has been shown to be a cost-effective intervention to improve outcomes in hospitalized patients with skin and soft tissue infections.10,11 In particular, cellulitis frequently is misdiagnosed, leading to unnecessary antibiotic use, hospitalizations, and major health care expenditures.12 Recognizing this challenge, researchers have worked to develop objective tools to improve diagnostic accuracy. In a large prospective prognostic validation study, Pulia et al13 found that thermal imaging alone or in combination with the ALT-70 prediction model (asymmetry, leukocytosis, tachycardia, and age ≥70 years) could be used successfully to reduce overdiagnosis of cellulitis. Both thermal imaging and the ALT-70 prediction model demonstrated robust sensitivity (93.5% and 98.8%, respectively) but low specificity (38.4% and 22.0%, respectively, and 53.9% when combined).13
In a systematic review, Kovacs et al14 analyzed case reports of pseudocellulitis caused by chemotherapeutic medications. Of the 81 cases selected, 58 (71.6%) were associated with gemcitabine, with the remaining 23 (28.4%) attributed to pemetrexed. Within this group, two-thirds of the patients received antibiotic treatment prior to receiving the correct diagnosis, and 36% experienced interruptions to their oncologic therapies. In contrast to infectious cellulitis, which tends to be unilateral and associated with elevated erythrocyte sedimentation rate or C-reactive protein, most chemotherapy-induced pseudocellulitis cases occurred bilaterally on the lower extremities, while erythrocyte sedimentation rate and C-reactive protein seldom were elevated.14
Necrotizing soft tissue infections (NSTIs) are severe life-threatening conditions characterized by widespread tissue destruction, signs of systemic toxicity, hemodynamic collapse, organ failure, and high mortality. Surgical inspection along with intraoperative tissue culture is the gold standard for diagnosis. Early detection, prompt surgical intervention, and appropriate antibiotic treatment are essential to reduce mortality and improve outcomes.15 A retrospective study of patients with surgically confirmed NSTIs assessed the incidence and risk factors for recurrence within 1 year following an initial NSTI of the lower extremity. Among 93 included patients, 32 (34.4%) had recurrence within 1 year, and more than half of recurrences occurred in the first 3 months (median, 66 days). The comparison of patients with and without recurrence showed similar proportions of antibiotic prophylaxis use after the first NSTI. There was significantly less compression therapy use (33.3% vs 62.3%; P=.13) and more negative pressure wound therapy use (83.3% vs 63.3%; P=.03) in the recurrence group, though the authors acknowledged that factors such as severity of pain and size of soft tissue defect may have affected the decisions for compression and negative pressure wound therapy.16
Residents of nursing homes are a particularly vulnerable population at high risk for health care–associated infections due to older age and a higher likelihood of having wounds, indwelling medical devices, and/or coexisting conditions.17 One cluster-randomized trial compared universal decolonization with routine-care bathing practices in nursing homes (N=28,956 residents). Decolonization entailed the use of chlorhexidine for all routine bathing and showering and administration of nasal povidone-iodine twice daily for the first 5 days after admission and then twice daily for 5 days every other week. Transfer to a hospital due to infection decreased from 62.9% to 52.2% with decolonization, for a difference in risk ratio of 16.6% (P<.001) compared with routine care. Additionally, the difference in risk ratio of the secondary end point (transfer to a hospital for any reason) was 14.6%. The number needed to treat was 9.7 to prevent 1 infection-related hospitalization and 8.9 to prevent 1 hospitalization for any reason.17
Autoimmune Blistering Diseases
Although rare, AIBDs are potentially life-threatening cutaneous diseases that often require inpatient management. While corticosteroids remain the mainstay of initial AIBD management, rituximab is now well recognized as the steroid-sparing treatment of choice for patients with moderate to severe pemphigus. In a long-term follow-up study of Ritux 318—the trial that led to the US Food and Drug Administration approval of rituximab in the treatment of moderate to severe pemphigus vulgaris—researchers assessed the long-term efficacy and safety of rituximab as a first-line treatment in patients with pemphigus.19 The 5- and 7-year disease-free survival rates without corticosteroid therapy for patients treated with rituximab were 76.7% and 72.1%, respectively, compared with 35.3% and 35.3% in those treated with prednisone alone (P<.001). Fewer serious adverse events were reported in those treated with rituximab plus prednisone compared with those treated with prednisone alone. None of the patients who maintained complete remission off corticosteroid therapy received any additional maintenance infusions of rituximab after the end of the Ritux 3 regimen (1 g of rituximab at day 0 and day 14, then 500 mg at months 12 and 18).19
By contrast, treatment of severe bullous pemphigoid (BP) often is less clear-cut, as no single therapeutic option has been shown to be superior to other immunomodulatory and immunosuppressive regimens, and the medical comorbidities of elderly patients with BP can be limiting. Fortunately, newer therapies with favorable safety profiles have emerged in recent years. In a multicenter retrospective study, 100 patients with BP received omalizumab after previously failing to respond to at least one alternative therapy. Disease control was obtained after a median of 10 days, and complete remission was achieved in 77% of patients in a median time of 3 months.20 In a multicenter retrospective cohort study of 146 patients with BP treated with dupilumab following the atopic dermatitis dosing schedule (one 600-mg dose followed by 300 mg every 2 weeks), disease control was achieved in a median of 14 days, while complete remission was achieved in 35.6% of patients, with 8.9% relapsing during the observation period.21 A retrospective case series of 30 patients with BP treated with dupilumab with maintenance dosing frequency tailored to individual patient response showed complete remission or marked response in 76.7% (23/30) of patients.22 A phase 2/3 randomized controlled trial of dupilumab in BP is currently ongoing (ClinicalTrials.gov identifier NCT04206553).
Pemphigoid gestationis is a rare autoimmune subepidermal bullous dermatosis of pregnancy that may be difficult to distinguish clinically from polymorphic eruption of pregnancy but confers notably different maternal and fetal risks. Researchers developed and validated a scoring system using clinical factors—history of pemphigoid gestationis, primigravidae, timing of rash onset, and specific clinical examination findings—that was able to differentiate between the 2 diseases with 79% sensitivity, 95% specificity, and an area under the curve of 0.93 without the need for advanced immunologic testing.23
Final Thoughts
Highlights of the literature from 2023-2024 demonstrate advancements in hospital-based dermatology as well as ongoing challenges. This year’s review emphasizes key developments in severe cutaneous adverse drug reactions, skin and soft tissue infections, and AIBDs. Continued expansion of knowledge in these areas and others informs patient care and demonstrates the value of dermatologic expertise in the inpatient setting.
- Berk-Krauss J, Micheletti RG. Hospital dermatology: review of research in 2022-2023. Cutis. 2023;112:236-239.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480.
- Blumenthal KG, Alvarez-Arango S, Kroshinsky D, et al. Drug reaction eosinophilia and systemic symptoms: clinical phenotypic patterns according to causative drug. J Am Acad Dermatol. 2024;90:1240-1242.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44.
- Li DJ, Velasquez GA, Romar GA, et al. Assessment of need for improved identification of a culprit drug in Stevens-Johnson syndrome/toxic epidermal necrolysis. JAMA Dermatol. 2023;159:830-836.
- Martinez-Cabriales S, Coulombe J, Aaron M, et al. Preliminary summary and reclassification of cases from the Pediatric Research of Management in Stevens-Johnson syndrome and Epidermonecrolysis (PROMISE) study: a North American, multisite retrospective cohort. J Am Acad Dermatol. 2024;90:635-637.
- Chiu YM, Chiu HY. Lifetime risk, life expectancy, loss-of-life expectancy and lifetime healthcare expenditure for Stevens-Johnson syndrome/toxic epidermal necrolysis in Taiwan: follow-up of a nationwide cohort from 2008 to 2019. Br J Dermatol. 2023;189:553-560.
- Phillips C, Russell E, McNiven A, et al. A qualitative study of psychological morbidity in paediatric survivors of Stevens-Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol. 2024;191:293-295.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Pulia MS, Schwei RJ, Alexandridis R, et al. Validation of thermal imaging and the ALT-70 prediction model to differentiate cellulitis from pseudocellulitis. JAMA Dermatol. 2024;160:511-517.
- Kovacs LD, O’Donoghue M, Cogen AL. Chemotherapy-induced pseudocellulitis without prior radiation exposure: a systematic review. JAMA Dermatol. 2023;159:870-874.
- Yildiz H, Yombi JC. Necrotizing soft-tissue infections. comment. N Engl J Med. 2018;378:970.
- Traineau H, Charpentier C, Lepeule R, et al. First-year recurrence rate of skin and soft tissue infections following an initial necrotizing soft tissue infection of the lower extremities: a retrospective cohort study of 93 patients. J Am Acad Dermatol. 2023;88:1360-1363.
- Miller LG, McKinnell JA, Singh RD, et al. Decolonization in nursing homes to prevent infection and hospitalization. N Engl J Med. 2023;389:1766-1777.
- Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al; French Study Group on Autoimmune Bullous Skin Diseases. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-2040.
- Tedbirt B, Maho-Vaillant M, Houivet E, et al; French Reference Center for Autoimmune Blistering Diseases MALIBUL. Sustained remission without corticosteroids among patients with pemphigus who had rituximab as first-line therapy: follow-up of the Ritux 3 Trial. JAMA Dermatol. 2024;160:290-296.
- Chebani R, Lombart F, Chaby G, et al; French Study Group on Autoimmune Bullous Diseases. Omalizumab in the treatment of bullous pemphigoid resistant to first-line therapy: a French national multicentre retrospective study of 100 patients. Br J Dermatol. 2024;190:258-265.
- Zhao L, Wang Q, Liang G, et al. Evaluation of dupilumab in patients with bullous pemphigoid. JAMA Dermatol. 2023;159:953-960.
- Miller AC, Temiz LA, Adjei S, et al. Treatment of bullous pemphigoid with dupilumab: a case series of 30 patients. J Drugs Dermatol. 2024;23:E144-E148.
- Xie F, Davis DMR, Baban F, et al. Development and multicenter international validation of a diagnostic tool to differentiate between pemphigoid gestationis and polymorphic eruption of pregnancy. J Am Acad Dermatol. 2023;89:106-113.
Inpatient consultative dermatology has advanced as a subspecialty and increasingly gained recognition in recent years. Since its founding in 2009, the Society of Dermatology Hospitalists has fostered research and education in hospital dermatology. Last year, we reviewed the 2022-2023 literature with a focus on developments in severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.1 In this review, we highlight 3 areas of interest from the 2023-2024 literature: severe cutaneous adverse drug reactions, skin and soft tissue infections, and autoimmune blistering diseases (AIBDs).
Severe Cutaneous Adverse Drug Reactions
Adverse drug reactions are among the most common diagnoses encountered by inpatient dermatology consultants.2,3 Severe cutaneous adverse drug reactions are associated with substantial morbidity and mortality. Efforts to characterize these conditions and standardize their diagnosis and management continue to be a major focus of ongoing research.
A single-center retrospective analysis of 102 cases of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome evaluated differences in clinical manifestations depending on the culprit drug, offering insights into the heterogeneity of DRESS syndrome and the potential for diagnostic uncertainty.4 The shortest median latency was observed in a case caused by penicillin and cephalosporins (12 and 18 days, respectively), while DRESS syndrome secondary to allopurinol had the longest median latency (36 days). Nonsteroidal anti-inflammatory drug–induced DRESS syndrome was associated with the shortest hospital stay (6.5 days), while cephalosporin and vancomycin cases had the highest mortality rates.4
In the first international Delphi consensus study on the diagnostic workup, severity assessment, and management of DRESS syndrome, 54 dermatology and/or allergy experts reached consensus on 93 statements.5 Specific recommendations included basic evaluation with complete blood count with differential, kidney and liver function parameters, and electrocardiogram for all patients with suspected DRESS syndrome, with additional complementary workup considered in patients with evidence of specific organ damage and/or severe disease. In the proposed DRESS syndrome severity grading scheme, laboratory values that reached consensus for inclusion were hemoglobin, neutrophil, and platelet counts and creatinine, transaminases, and alkaline phosphatase levels. Although treatment of DRESS syndrome should be based on assessed disease severity, treatment with corticosteroids should be initiated in all patients with confirmed DRESS syndrome. Cyclosporine, antibodies interfering with the IL-5 axis, and intravenous immunoglobulins can be considered in patients with corticosteroid-refractory DRESS syndrome, and antiviral treatment can be considered in patients with a high serum cytomegalovirus viral load. Regularly following up with laboratory evaluation of involved organs; screening for autoantibodies, thyroid dysfunction, and steroid adverse effects; and offering of psychological support also were consensus recommendations.5
Identifying causative agents in drug hypersensitivity reactions remains challenging. A retrospective cohort study of 48 patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) highlighted the need for a systematic unbiased approach to identifying culprit drugs. Using the RegiSCAR database and algorithm for drug causality for epidermal necrolysis to analyze the cohort, more than half of causative agents were determined to be different from those initially identified by the treating physicians. Nine additional suspected culprit drugs were identified, while 43 drugs initially identified as allergens were exonerated.6
Etiology-associated definitions for blistering reactions in children have been proposed to replace the existing terms Stevens-Johnson syndrome, toxic epidermal necrolysis, and others.7 Investigators in a recent study reclassified cases of SJS and TEN as reactive infectious mucocutaneous eruption (RIME) or drug-induced epidermal necrolysis (DEN), respectively. In RIME cases, Mycoplasma pneumoniae was the most commonly identified trigger, and in DEN cases, anticonvulsants were the most common class of culprit medications. Cases of RIME were less severe and were most often treated with antibiotics, whereas patients with DEN were more likely to receive supportive care, corticosteroids, intravenous immunoglobulins, and other immunosuppressive therapies.7
In addition to causing acute devastating mucocutaneous complications, SJS and TEN have long-lasting effects that require ongoing care. In a cohort of 6552 incident SJS/TEN cases over an 11-year period, survivors of SJS/TEN endured a mean loss of 9.4 years in life expectancy and excess health care expenditures of $3752 per year compared with age- and sex-matched controls. Patients with more severe disease, comorbid malignancy, diabetes, end-stage renal disease, or SJS/TEN sequelae experienced greater loss in life expectancy and lifetime health care expenditures.8 Separately, a qualitative study investigating the psychological impact of SJS/TEN in pediatric patients described sequelae including night terrors, posttraumatic stress disorder, depression, and anxiety for many years after the acute phase. Many patients reported a desire for increased support for their physical and emotional needs following hospital discharge.9
Skin and Soft Tissue Infections: Diagnosis, Management, and Prevention
Dermatology consultation has been shown to be a cost-effective intervention to improve outcomes in hospitalized patients with skin and soft tissue infections.10,11 In particular, cellulitis frequently is misdiagnosed, leading to unnecessary antibiotic use, hospitalizations, and major health care expenditures.12 Recognizing this challenge, researchers have worked to develop objective tools to improve diagnostic accuracy. In a large prospective prognostic validation study, Pulia et al13 found that thermal imaging alone or in combination with the ALT-70 prediction model (asymmetry, leukocytosis, tachycardia, and age ≥70 years) could be used successfully to reduce overdiagnosis of cellulitis. Both thermal imaging and the ALT-70 prediction model demonstrated robust sensitivity (93.5% and 98.8%, respectively) but low specificity (38.4% and 22.0%, respectively, and 53.9% when combined).13
In a systematic review, Kovacs et al14 analyzed case reports of pseudocellulitis caused by chemotherapeutic medications. Of the 81 cases selected, 58 (71.6%) were associated with gemcitabine, with the remaining 23 (28.4%) attributed to pemetrexed. Within this group, two-thirds of the patients received antibiotic treatment prior to receiving the correct diagnosis, and 36% experienced interruptions to their oncologic therapies. In contrast to infectious cellulitis, which tends to be unilateral and associated with elevated erythrocyte sedimentation rate or C-reactive protein, most chemotherapy-induced pseudocellulitis cases occurred bilaterally on the lower extremities, while erythrocyte sedimentation rate and C-reactive protein seldom were elevated.14
Necrotizing soft tissue infections (NSTIs) are severe life-threatening conditions characterized by widespread tissue destruction, signs of systemic toxicity, hemodynamic collapse, organ failure, and high mortality. Surgical inspection along with intraoperative tissue culture is the gold standard for diagnosis. Early detection, prompt surgical intervention, and appropriate antibiotic treatment are essential to reduce mortality and improve outcomes.15 A retrospective study of patients with surgically confirmed NSTIs assessed the incidence and risk factors for recurrence within 1 year following an initial NSTI of the lower extremity. Among 93 included patients, 32 (34.4%) had recurrence within 1 year, and more than half of recurrences occurred in the first 3 months (median, 66 days). The comparison of patients with and without recurrence showed similar proportions of antibiotic prophylaxis use after the first NSTI. There was significantly less compression therapy use (33.3% vs 62.3%; P=.13) and more negative pressure wound therapy use (83.3% vs 63.3%; P=.03) in the recurrence group, though the authors acknowledged that factors such as severity of pain and size of soft tissue defect may have affected the decisions for compression and negative pressure wound therapy.16
Residents of nursing homes are a particularly vulnerable population at high risk for health care–associated infections due to older age and a higher likelihood of having wounds, indwelling medical devices, and/or coexisting conditions.17 One cluster-randomized trial compared universal decolonization with routine-care bathing practices in nursing homes (N=28,956 residents). Decolonization entailed the use of chlorhexidine for all routine bathing and showering and administration of nasal povidone-iodine twice daily for the first 5 days after admission and then twice daily for 5 days every other week. Transfer to a hospital due to infection decreased from 62.9% to 52.2% with decolonization, for a difference in risk ratio of 16.6% (P<.001) compared with routine care. Additionally, the difference in risk ratio of the secondary end point (transfer to a hospital for any reason) was 14.6%. The number needed to treat was 9.7 to prevent 1 infection-related hospitalization and 8.9 to prevent 1 hospitalization for any reason.17
Autoimmune Blistering Diseases
Although rare, AIBDs are potentially life-threatening cutaneous diseases that often require inpatient management. While corticosteroids remain the mainstay of initial AIBD management, rituximab is now well recognized as the steroid-sparing treatment of choice for patients with moderate to severe pemphigus. In a long-term follow-up study of Ritux 318—the trial that led to the US Food and Drug Administration approval of rituximab in the treatment of moderate to severe pemphigus vulgaris—researchers assessed the long-term efficacy and safety of rituximab as a first-line treatment in patients with pemphigus.19 The 5- and 7-year disease-free survival rates without corticosteroid therapy for patients treated with rituximab were 76.7% and 72.1%, respectively, compared with 35.3% and 35.3% in those treated with prednisone alone (P<.001). Fewer serious adverse events were reported in those treated with rituximab plus prednisone compared with those treated with prednisone alone. None of the patients who maintained complete remission off corticosteroid therapy received any additional maintenance infusions of rituximab after the end of the Ritux 3 regimen (1 g of rituximab at day 0 and day 14, then 500 mg at months 12 and 18).19
By contrast, treatment of severe bullous pemphigoid (BP) often is less clear-cut, as no single therapeutic option has been shown to be superior to other immunomodulatory and immunosuppressive regimens, and the medical comorbidities of elderly patients with BP can be limiting. Fortunately, newer therapies with favorable safety profiles have emerged in recent years. In a multicenter retrospective study, 100 patients with BP received omalizumab after previously failing to respond to at least one alternative therapy. Disease control was obtained after a median of 10 days, and complete remission was achieved in 77% of patients in a median time of 3 months.20 In a multicenter retrospective cohort study of 146 patients with BP treated with dupilumab following the atopic dermatitis dosing schedule (one 600-mg dose followed by 300 mg every 2 weeks), disease control was achieved in a median of 14 days, while complete remission was achieved in 35.6% of patients, with 8.9% relapsing during the observation period.21 A retrospective case series of 30 patients with BP treated with dupilumab with maintenance dosing frequency tailored to individual patient response showed complete remission or marked response in 76.7% (23/30) of patients.22 A phase 2/3 randomized controlled trial of dupilumab in BP is currently ongoing (ClinicalTrials.gov identifier NCT04206553).
Pemphigoid gestationis is a rare autoimmune subepidermal bullous dermatosis of pregnancy that may be difficult to distinguish clinically from polymorphic eruption of pregnancy but confers notably different maternal and fetal risks. Researchers developed and validated a scoring system using clinical factors—history of pemphigoid gestationis, primigravidae, timing of rash onset, and specific clinical examination findings—that was able to differentiate between the 2 diseases with 79% sensitivity, 95% specificity, and an area under the curve of 0.93 without the need for advanced immunologic testing.23
Final Thoughts
Highlights of the literature from 2023-2024 demonstrate advancements in hospital-based dermatology as well as ongoing challenges. This year’s review emphasizes key developments in severe cutaneous adverse drug reactions, skin and soft tissue infections, and AIBDs. Continued expansion of knowledge in these areas and others informs patient care and demonstrates the value of dermatologic expertise in the inpatient setting.
Inpatient consultative dermatology has advanced as a subspecialty and increasingly gained recognition in recent years. Since its founding in 2009, the Society of Dermatology Hospitalists has fostered research and education in hospital dermatology. Last year, we reviewed the 2022-2023 literature with a focus on developments in severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.1 In this review, we highlight 3 areas of interest from the 2023-2024 literature: severe cutaneous adverse drug reactions, skin and soft tissue infections, and autoimmune blistering diseases (AIBDs).
Severe Cutaneous Adverse Drug Reactions
Adverse drug reactions are among the most common diagnoses encountered by inpatient dermatology consultants.2,3 Severe cutaneous adverse drug reactions are associated with substantial morbidity and mortality. Efforts to characterize these conditions and standardize their diagnosis and management continue to be a major focus of ongoing research.
A single-center retrospective analysis of 102 cases of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome evaluated differences in clinical manifestations depending on the culprit drug, offering insights into the heterogeneity of DRESS syndrome and the potential for diagnostic uncertainty.4 The shortest median latency was observed in a case caused by penicillin and cephalosporins (12 and 18 days, respectively), while DRESS syndrome secondary to allopurinol had the longest median latency (36 days). Nonsteroidal anti-inflammatory drug–induced DRESS syndrome was associated with the shortest hospital stay (6.5 days), while cephalosporin and vancomycin cases had the highest mortality rates.4
In the first international Delphi consensus study on the diagnostic workup, severity assessment, and management of DRESS syndrome, 54 dermatology and/or allergy experts reached consensus on 93 statements.5 Specific recommendations included basic evaluation with complete blood count with differential, kidney and liver function parameters, and electrocardiogram for all patients with suspected DRESS syndrome, with additional complementary workup considered in patients with evidence of specific organ damage and/or severe disease. In the proposed DRESS syndrome severity grading scheme, laboratory values that reached consensus for inclusion were hemoglobin, neutrophil, and platelet counts and creatinine, transaminases, and alkaline phosphatase levels. Although treatment of DRESS syndrome should be based on assessed disease severity, treatment with corticosteroids should be initiated in all patients with confirmed DRESS syndrome. Cyclosporine, antibodies interfering with the IL-5 axis, and intravenous immunoglobulins can be considered in patients with corticosteroid-refractory DRESS syndrome, and antiviral treatment can be considered in patients with a high serum cytomegalovirus viral load. Regularly following up with laboratory evaluation of involved organs; screening for autoantibodies, thyroid dysfunction, and steroid adverse effects; and offering of psychological support also were consensus recommendations.5
Identifying causative agents in drug hypersensitivity reactions remains challenging. A retrospective cohort study of 48 patients with Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) highlighted the need for a systematic unbiased approach to identifying culprit drugs. Using the RegiSCAR database and algorithm for drug causality for epidermal necrolysis to analyze the cohort, more than half of causative agents were determined to be different from those initially identified by the treating physicians. Nine additional suspected culprit drugs were identified, while 43 drugs initially identified as allergens were exonerated.6
Etiology-associated definitions for blistering reactions in children have been proposed to replace the existing terms Stevens-Johnson syndrome, toxic epidermal necrolysis, and others.7 Investigators in a recent study reclassified cases of SJS and TEN as reactive infectious mucocutaneous eruption (RIME) or drug-induced epidermal necrolysis (DEN), respectively. In RIME cases, Mycoplasma pneumoniae was the most commonly identified trigger, and in DEN cases, anticonvulsants were the most common class of culprit medications. Cases of RIME were less severe and were most often treated with antibiotics, whereas patients with DEN were more likely to receive supportive care, corticosteroids, intravenous immunoglobulins, and other immunosuppressive therapies.7
In addition to causing acute devastating mucocutaneous complications, SJS and TEN have long-lasting effects that require ongoing care. In a cohort of 6552 incident SJS/TEN cases over an 11-year period, survivors of SJS/TEN endured a mean loss of 9.4 years in life expectancy and excess health care expenditures of $3752 per year compared with age- and sex-matched controls. Patients with more severe disease, comorbid malignancy, diabetes, end-stage renal disease, or SJS/TEN sequelae experienced greater loss in life expectancy and lifetime health care expenditures.8 Separately, a qualitative study investigating the psychological impact of SJS/TEN in pediatric patients described sequelae including night terrors, posttraumatic stress disorder, depression, and anxiety for many years after the acute phase. Many patients reported a desire for increased support for their physical and emotional needs following hospital discharge.9
Skin and Soft Tissue Infections: Diagnosis, Management, and Prevention
Dermatology consultation has been shown to be a cost-effective intervention to improve outcomes in hospitalized patients with skin and soft tissue infections.10,11 In particular, cellulitis frequently is misdiagnosed, leading to unnecessary antibiotic use, hospitalizations, and major health care expenditures.12 Recognizing this challenge, researchers have worked to develop objective tools to improve diagnostic accuracy. In a large prospective prognostic validation study, Pulia et al13 found that thermal imaging alone or in combination with the ALT-70 prediction model (asymmetry, leukocytosis, tachycardia, and age ≥70 years) could be used successfully to reduce overdiagnosis of cellulitis. Both thermal imaging and the ALT-70 prediction model demonstrated robust sensitivity (93.5% and 98.8%, respectively) but low specificity (38.4% and 22.0%, respectively, and 53.9% when combined).13
In a systematic review, Kovacs et al14 analyzed case reports of pseudocellulitis caused by chemotherapeutic medications. Of the 81 cases selected, 58 (71.6%) were associated with gemcitabine, with the remaining 23 (28.4%) attributed to pemetrexed. Within this group, two-thirds of the patients received antibiotic treatment prior to receiving the correct diagnosis, and 36% experienced interruptions to their oncologic therapies. In contrast to infectious cellulitis, which tends to be unilateral and associated with elevated erythrocyte sedimentation rate or C-reactive protein, most chemotherapy-induced pseudocellulitis cases occurred bilaterally on the lower extremities, while erythrocyte sedimentation rate and C-reactive protein seldom were elevated.14
Necrotizing soft tissue infections (NSTIs) are severe life-threatening conditions characterized by widespread tissue destruction, signs of systemic toxicity, hemodynamic collapse, organ failure, and high mortality. Surgical inspection along with intraoperative tissue culture is the gold standard for diagnosis. Early detection, prompt surgical intervention, and appropriate antibiotic treatment are essential to reduce mortality and improve outcomes.15 A retrospective study of patients with surgically confirmed NSTIs assessed the incidence and risk factors for recurrence within 1 year following an initial NSTI of the lower extremity. Among 93 included patients, 32 (34.4%) had recurrence within 1 year, and more than half of recurrences occurred in the first 3 months (median, 66 days). The comparison of patients with and without recurrence showed similar proportions of antibiotic prophylaxis use after the first NSTI. There was significantly less compression therapy use (33.3% vs 62.3%; P=.13) and more negative pressure wound therapy use (83.3% vs 63.3%; P=.03) in the recurrence group, though the authors acknowledged that factors such as severity of pain and size of soft tissue defect may have affected the decisions for compression and negative pressure wound therapy.16
Residents of nursing homes are a particularly vulnerable population at high risk for health care–associated infections due to older age and a higher likelihood of having wounds, indwelling medical devices, and/or coexisting conditions.17 One cluster-randomized trial compared universal decolonization with routine-care bathing practices in nursing homes (N=28,956 residents). Decolonization entailed the use of chlorhexidine for all routine bathing and showering and administration of nasal povidone-iodine twice daily for the first 5 days after admission and then twice daily for 5 days every other week. Transfer to a hospital due to infection decreased from 62.9% to 52.2% with decolonization, for a difference in risk ratio of 16.6% (P<.001) compared with routine care. Additionally, the difference in risk ratio of the secondary end point (transfer to a hospital for any reason) was 14.6%. The number needed to treat was 9.7 to prevent 1 infection-related hospitalization and 8.9 to prevent 1 hospitalization for any reason.17
Autoimmune Blistering Diseases
Although rare, AIBDs are potentially life-threatening cutaneous diseases that often require inpatient management. While corticosteroids remain the mainstay of initial AIBD management, rituximab is now well recognized as the steroid-sparing treatment of choice for patients with moderate to severe pemphigus. In a long-term follow-up study of Ritux 318—the trial that led to the US Food and Drug Administration approval of rituximab in the treatment of moderate to severe pemphigus vulgaris—researchers assessed the long-term efficacy and safety of rituximab as a first-line treatment in patients with pemphigus.19 The 5- and 7-year disease-free survival rates without corticosteroid therapy for patients treated with rituximab were 76.7% and 72.1%, respectively, compared with 35.3% and 35.3% in those treated with prednisone alone (P<.001). Fewer serious adverse events were reported in those treated with rituximab plus prednisone compared with those treated with prednisone alone. None of the patients who maintained complete remission off corticosteroid therapy received any additional maintenance infusions of rituximab after the end of the Ritux 3 regimen (1 g of rituximab at day 0 and day 14, then 500 mg at months 12 and 18).19
By contrast, treatment of severe bullous pemphigoid (BP) often is less clear-cut, as no single therapeutic option has been shown to be superior to other immunomodulatory and immunosuppressive regimens, and the medical comorbidities of elderly patients with BP can be limiting. Fortunately, newer therapies with favorable safety profiles have emerged in recent years. In a multicenter retrospective study, 100 patients with BP received omalizumab after previously failing to respond to at least one alternative therapy. Disease control was obtained after a median of 10 days, and complete remission was achieved in 77% of patients in a median time of 3 months.20 In a multicenter retrospective cohort study of 146 patients with BP treated with dupilumab following the atopic dermatitis dosing schedule (one 600-mg dose followed by 300 mg every 2 weeks), disease control was achieved in a median of 14 days, while complete remission was achieved in 35.6% of patients, with 8.9% relapsing during the observation period.21 A retrospective case series of 30 patients with BP treated with dupilumab with maintenance dosing frequency tailored to individual patient response showed complete remission or marked response in 76.7% (23/30) of patients.22 A phase 2/3 randomized controlled trial of dupilumab in BP is currently ongoing (ClinicalTrials.gov identifier NCT04206553).
Pemphigoid gestationis is a rare autoimmune subepidermal bullous dermatosis of pregnancy that may be difficult to distinguish clinically from polymorphic eruption of pregnancy but confers notably different maternal and fetal risks. Researchers developed and validated a scoring system using clinical factors—history of pemphigoid gestationis, primigravidae, timing of rash onset, and specific clinical examination findings—that was able to differentiate between the 2 diseases with 79% sensitivity, 95% specificity, and an area under the curve of 0.93 without the need for advanced immunologic testing.23
Final Thoughts
Highlights of the literature from 2023-2024 demonstrate advancements in hospital-based dermatology as well as ongoing challenges. This year’s review emphasizes key developments in severe cutaneous adverse drug reactions, skin and soft tissue infections, and AIBDs. Continued expansion of knowledge in these areas and others informs patient care and demonstrates the value of dermatologic expertise in the inpatient setting.
- Berk-Krauss J, Micheletti RG. Hospital dermatology: review of research in 2022-2023. Cutis. 2023;112:236-239.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480.
- Blumenthal KG, Alvarez-Arango S, Kroshinsky D, et al. Drug reaction eosinophilia and systemic symptoms: clinical phenotypic patterns according to causative drug. J Am Acad Dermatol. 2024;90:1240-1242.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44.
- Li DJ, Velasquez GA, Romar GA, et al. Assessment of need for improved identification of a culprit drug in Stevens-Johnson syndrome/toxic epidermal necrolysis. JAMA Dermatol. 2023;159:830-836.
- Martinez-Cabriales S, Coulombe J, Aaron M, et al. Preliminary summary and reclassification of cases from the Pediatric Research of Management in Stevens-Johnson syndrome and Epidermonecrolysis (PROMISE) study: a North American, multisite retrospective cohort. J Am Acad Dermatol. 2024;90:635-637.
- Chiu YM, Chiu HY. Lifetime risk, life expectancy, loss-of-life expectancy and lifetime healthcare expenditure for Stevens-Johnson syndrome/toxic epidermal necrolysis in Taiwan: follow-up of a nationwide cohort from 2008 to 2019. Br J Dermatol. 2023;189:553-560.
- Phillips C, Russell E, McNiven A, et al. A qualitative study of psychological morbidity in paediatric survivors of Stevens-Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol. 2024;191:293-295.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Pulia MS, Schwei RJ, Alexandridis R, et al. Validation of thermal imaging and the ALT-70 prediction model to differentiate cellulitis from pseudocellulitis. JAMA Dermatol. 2024;160:511-517.
- Kovacs LD, O’Donoghue M, Cogen AL. Chemotherapy-induced pseudocellulitis without prior radiation exposure: a systematic review. JAMA Dermatol. 2023;159:870-874.
- Yildiz H, Yombi JC. Necrotizing soft-tissue infections. comment. N Engl J Med. 2018;378:970.
- Traineau H, Charpentier C, Lepeule R, et al. First-year recurrence rate of skin and soft tissue infections following an initial necrotizing soft tissue infection of the lower extremities: a retrospective cohort study of 93 patients. J Am Acad Dermatol. 2023;88:1360-1363.
- Miller LG, McKinnell JA, Singh RD, et al. Decolonization in nursing homes to prevent infection and hospitalization. N Engl J Med. 2023;389:1766-1777.
- Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al; French Study Group on Autoimmune Bullous Skin Diseases. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-2040.
- Tedbirt B, Maho-Vaillant M, Houivet E, et al; French Reference Center for Autoimmune Blistering Diseases MALIBUL. Sustained remission without corticosteroids among patients with pemphigus who had rituximab as first-line therapy: follow-up of the Ritux 3 Trial. JAMA Dermatol. 2024;160:290-296.
- Chebani R, Lombart F, Chaby G, et al; French Study Group on Autoimmune Bullous Diseases. Omalizumab in the treatment of bullous pemphigoid resistant to first-line therapy: a French national multicentre retrospective study of 100 patients. Br J Dermatol. 2024;190:258-265.
- Zhao L, Wang Q, Liang G, et al. Evaluation of dupilumab in patients with bullous pemphigoid. JAMA Dermatol. 2023;159:953-960.
- Miller AC, Temiz LA, Adjei S, et al. Treatment of bullous pemphigoid with dupilumab: a case series of 30 patients. J Drugs Dermatol. 2024;23:E144-E148.
- Xie F, Davis DMR, Baban F, et al. Development and multicenter international validation of a diagnostic tool to differentiate between pemphigoid gestationis and polymorphic eruption of pregnancy. J Am Acad Dermatol. 2023;89:106-113.
- Berk-Krauss J, Micheletti RG. Hospital dermatology: review of research in 2022-2023. Cutis. 2023;112:236-239.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480.
- Blumenthal KG, Alvarez-Arango S, Kroshinsky D, et al. Drug reaction eosinophilia and systemic symptoms: clinical phenotypic patterns according to causative drug. J Am Acad Dermatol. 2024;90:1240-1242.
- Brüggen MC, Walsh S, Ameri MM, et al. Management of adult patients with drug reaction with eosinophilia and systemic symptoms: a Delphi-based international consensus. JAMA Dermatol. 2024;160:37-44.
- Li DJ, Velasquez GA, Romar GA, et al. Assessment of need for improved identification of a culprit drug in Stevens-Johnson syndrome/toxic epidermal necrolysis. JAMA Dermatol. 2023;159:830-836.
- Martinez-Cabriales S, Coulombe J, Aaron M, et al. Preliminary summary and reclassification of cases from the Pediatric Research of Management in Stevens-Johnson syndrome and Epidermonecrolysis (PROMISE) study: a North American, multisite retrospective cohort. J Am Acad Dermatol. 2024;90:635-637.
- Chiu YM, Chiu HY. Lifetime risk, life expectancy, loss-of-life expectancy and lifetime healthcare expenditure for Stevens-Johnson syndrome/toxic epidermal necrolysis in Taiwan: follow-up of a nationwide cohort from 2008 to 2019. Br J Dermatol. 2023;189:553-560.
- Phillips C, Russell E, McNiven A, et al. A qualitative study of psychological morbidity in paediatric survivors of Stevens-Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol. 2024;191:293-295.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Pulia MS, Schwei RJ, Alexandridis R, et al. Validation of thermal imaging and the ALT-70 prediction model to differentiate cellulitis from pseudocellulitis. JAMA Dermatol. 2024;160:511-517.
- Kovacs LD, O’Donoghue M, Cogen AL. Chemotherapy-induced pseudocellulitis without prior radiation exposure: a systematic review. JAMA Dermatol. 2023;159:870-874.
- Yildiz H, Yombi JC. Necrotizing soft-tissue infections. comment. N Engl J Med. 2018;378:970.
- Traineau H, Charpentier C, Lepeule R, et al. First-year recurrence rate of skin and soft tissue infections following an initial necrotizing soft tissue infection of the lower extremities: a retrospective cohort study of 93 patients. J Am Acad Dermatol. 2023;88:1360-1363.
- Miller LG, McKinnell JA, Singh RD, et al. Decolonization in nursing homes to prevent infection and hospitalization. N Engl J Med. 2023;389:1766-1777.
- Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al; French Study Group on Autoimmune Bullous Skin Diseases. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-2040.
- Tedbirt B, Maho-Vaillant M, Houivet E, et al; French Reference Center for Autoimmune Blistering Diseases MALIBUL. Sustained remission without corticosteroids among patients with pemphigus who had rituximab as first-line therapy: follow-up of the Ritux 3 Trial. JAMA Dermatol. 2024;160:290-296.
- Chebani R, Lombart F, Chaby G, et al; French Study Group on Autoimmune Bullous Diseases. Omalizumab in the treatment of bullous pemphigoid resistant to first-line therapy: a French national multicentre retrospective study of 100 patients. Br J Dermatol. 2024;190:258-265.
- Zhao L, Wang Q, Liang G, et al. Evaluation of dupilumab in patients with bullous pemphigoid. JAMA Dermatol. 2023;159:953-960.
- Miller AC, Temiz LA, Adjei S, et al. Treatment of bullous pemphigoid with dupilumab: a case series of 30 patients. J Drugs Dermatol. 2024;23:E144-E148.
- Xie F, Davis DMR, Baban F, et al. Development and multicenter international validation of a diagnostic tool to differentiate between pemphigoid gestationis and polymorphic eruption of pregnancy. J Am Acad Dermatol. 2023;89:106-113.
Practice Points
- An international Delphi study reached consensus on 93 statements regarding workup, severity assessment, and management of DRESS syndrome.
- In nursing homes, universal decolonization with chlorhexidine and nasal iodophor greatly reduced the risk for hospital transfers due to infection compared to routine care.
- Rituximab as the first-line therapy for pemphigus vulgaris is associated with long-term sustained complete remission without corticosteroid therapy.
- Dupilumab and omalizumab are emerging safe and effective treatment options for bullous pemphigoid.
Inpatient Management of Hidradenitis Suppurativa: A Delphi Consensus Study
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Practice Points
- Given the increase in hospital-based care for hidradenitis suppurativa (HS) and the lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial.
- Our Delphi study yielded 40 statements that reached consensus covering a range of patient care issues (eg, appropriate inpatient subspecialists [care team]), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition to outpatient management (transitional care).
- These recommendations serve as an important resource for providers caring for inpatients with HS and represent a successful collaboration between inpatient dermatology and HS experts.
E-Consults in Dermatology: A Retrospective Analysis
Dermatologic conditions affect approximately one-third of individuals in the United States.1,2 Nearly 1 in 4 physician office visits in the United States are for skin conditions, and less than one-third of these visits are with dermatologists. Although many of these patients may prefer to see a dermatologist for their concerns, they may not be able to access specialist care.3 The limited supply and urban-focused distribution of dermatologists along with reduced acceptance of state-funded insurance plans and long appointment wait times all pose considerable challenges to individuals seeking dermatologic care.2 Electronic consultations (e-consults) have emerged as a promising solution to overcoming these barriers while providing high-quality dermatologic care to a large diverse patient population.2,4 Although e-consults can be of service to all dermatology patients, this modality may be especially beneficial to underserved populations, such as the uninsured and Medicaid patients—groups that historically have experienced limited access to dermatology care due to the low reimbursement rates and high administrative burdens accompanying care delivery.4 This limited access leads to inequity in care, as timely access to dermatology is associated with improved diagnostic accuracy and disease outcomes.3 E-consult implementation can facilitate timely access for these underserved populations and bypass additional barriers to care such as lack of transportation or time off work. Prior e-consult studies have demonstrated relatively high numbers of Medicaid patients utilizing e-consult services.3,5
Although in-person visits remain the gold standard for diagnosis and treatment of dermatologic conditions, e-consults placed by primary care providers (PCPs) can improve access and help triage patients who require in-person dermatology visits.6 In this study, we conducted a retrospective chart review to characterize the e-consults requested of the dermatology department at a large tertiary care medical center in Winston-Salem, North Carolina.
Methods
The electronic health record (EHR) of Atrium Health Wake Forest Baptist (Winston-Salem, North Carolina) was screened for eligible patients from January 1, 2020, to May 31, 2021. Patients—both adult (aged ≥18 years) and pediatric (aged <18 years)—were included if they underwent a dermatology e-consult within this time frame. Provider notes in the medical records were reviewed to determine the nature of the lesion, how long the dermatologist took to complete the e-consult, whether an in-person appointment was recommended, and whether the patient was seen by dermatology within 90 days of the e-consult. Institutional review board approval was obtained.
For each e-consult, the PCP obtained clinical photographs of the lesion in question either through the EHR mobile application or by having patients upload their own photographs directly to their medical records. The referring PCP then completed a brief template regarding the patient’s clinical question and medical history and then sent the completed information to the consulting dermatologist’s EHR inbox. From there, the dermatologist could view the clinical question, documented photographs, and patient medical record to create a brief consult note with recommendations. The note was then sent back via EHR to the PCP to follow up with the patient. Patients were not charged for the e-consult.

Results
Two hundred fifty-four dermatology e-consults were requested by providers at the study center (eTable), which included 252 unique patients (2 patients had 2 separate e-consults regarding different clinical questions). The median time for completion of the e-consult—from submission of the PCP’s e-consult request to dermatologist completion—was 0.37 days. Fifty-six patients (22.0%) were recommended for an in-person appointment (Figure), 33 (58.9%) of whom ultimately scheduled the in-person appointment, and the median length of time between the completion of the e-consult and the in-person appointment was 16.5 days. The remaining 198 patients (78.0%) were not triaged to receive an in-person appointment following the e-consult,but 2 patients (8.7%) were ultimately seen in-person anyway via other referral pathways, with a median length of 33 days between e-consult completion and the in-person appointment. One hundred seventy-six patients (69.8%) avoided an in-person dermatology visit, although 38 (21.6%) of those patients were fewer than 90 days out from their e-consults at the time of data collection. The 254 e-consults included patients from 50 different zip codes, 49 (98.0%) of which were in North Carolina.
 resulted in reduced frequencies of in-person dermatology appointments.")
Comment
An e-consult is an asynchronous telehealth modality through which PCPs can request specialty evaluation to provide diagnostic and therapeutic guidance, facilitate PCP-specialist coordination of care, and increase access to specialty care with reduced wait times.7,8 Increased care access is especially important, as specialty referral can decrease overall health care expenditure; however, the demand for specialists often exceeds the availability.8 Our e-consult program drastically reduced the time from patients’ initial presentation at their PCP’s office to dermatologist recommendations for treatment or need for in-person dermatology follow-up.
In our analysis, patients were of different racial, ethnic, and socioeconomic backgrounds and lived across a variety of zip codes, predominantly in central and western North Carolina. Almost three-quarters of the patients resided in zip codes where the average income was less than the North Carolina median household income ($66,196).9 Additionally, 82 patients (32.3%) were uninsured or on Medicaid (eTable). These economically disadvantaged patient populations historically have had limited access to dermatologic care.4 One study showed that privately insured individuals were accepted as new patients by dermatologists 91% of the time compared to a 29.8% acceptance rate for publicly insured individuals.10 Uninsured and Medicaid patients also have to wait 34% longer for an appointment compared to individuals with Medicare or private insurance.2 Considering these patients may already be at an economic disadvantage when it comes to seeing and paying for dermatologic services, e-consults may reduce patient travel and appointment expenses while increasing access to specialty care. Based on a 2020 study, each e-consult generates an estimated savings of $80 out-of-pocket per patient per avoided in-person visit.11
In our study, the most common condition for an e-consult in both adult and pediatric patients was rash, which is consistent with prior e-consult studies.5,11 We found that most e-consult patients were not recommended for an in-person dermatology visit, and for those who were recommended to have an in-person visit, the wait time was reduced (Figure). These results corroborate that e-consults may be used as an important triage tool for determining whether a specialist appointment is indicated as well as a public health tool, as timely evaluation is associated with better dermatologic health care outcomes.3 However, the number of patients who did not present for an in-person appointment in our study may be overestimated, as 38 patients’ (21.6%) e-consults were conducted fewer than 90 days before our data collection. Although none of these patients had been seen in person, it is possible they requested an in-person visit after their medical records were reviewed for this study. Additionally, it is possible patients sought care from outside providers not documented in the EHR.
With regard to the payment model for the e-consult program, Atrium Health Wake Forest Baptist initially piloted the e-consult system through a partnership with the American Academy of Medical Colleges’ Project CORE: Coordinating Optimal Referral Experiences (https://www.aamc.org/what-we-do/mission-areas/health-care/project-core). Grant funding through Project CORE allowed both the referring PCP and the specialist completing the e-consult to each receive approximately 0.5 relative value units in payment for each consult completed. Based on early adoption successes, the institution has created additional internal funding to support the continued expansion of the e-consult system and is incentivized to continue funding, as proper utilization of e-consults improves patient access to timely specialist care, avoids no-shows or last-minute cancellations for specialist appointments, and decreases back-door access to specialist care through the emergency department and urgent care facilities.5 Although 0.5 relative value units is not equivalent compensation to an in-person office visit, our study showed that e-consults can be completed much more quickly and efficiently and do not utilize nursing staff or other office resources.
Conclusion
E-consults are an effective telehealth modality that can increase patients’ access to dermatologic specialty care.
Acknowledgments—The authors thank the Wake Forest University School of Medicine Department of Medical Education and Department of Dermatology (Winston-Salem, North Carolina) for their contributions to this research study as well as the Wake Forest Clinical and Translational Science Institute (Winston-Salem, North Carolina) for their help extracting EHR data.
- Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
- Naka F, Lu J, Porto A, et al. Impact of dermatology econsults on access to care and skin cancer screening in underserved populations: a model for teledermatology services in community health centers. J Am Acad Dermatol. 2018;78:293-302.
- Mulcahy A, Mehrotra A, Edison K, et al. Variation in dermatologist visits by sociodemographic characteristics. J Am Acad Dermatol. 2017;76:918-924.
- Yang X, Barbieri JS, Kovarik CL. Cost analysis of a store-and-forward teledermatology consult system in Philadelphia. J Am Acad Dermatol. 2019;81:758-764.
- Wang RF, Trinidad J, Lawrence J, et al. Improved patient access and outcomes with the integration of an econsult program (teledermatology) within a large academic medical center. J Am Acad Dermatol. 2020;83:1633-1638.
- Lee KJ, Finnane A, Soyer HP. Recent trends in teledermatology and teledermoscopy. Dermatol Pract Concept. 2018;8:214-223.
- Parikh PJ, Mowrey C, Gallimore J, et al. Evaluating e-consultation implementations based on use and time-line across various specialties. Int J Med Inform. 2017;108:42-48.
- Wasfy JH, Rao SK, Kalwani N, et al. Longer-term impact of cardiology e-consults. Am Heart J. 2016;173:86-93.
- United States Census Bureau. QuickFacts: North Carolina; United States. Accessed February 26, 2024. https://www.census.gov/quickfacts/fact/table/NC,US/PST045222
- Alghothani L, Jacks SK, Vander Horst A, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
- Seiger K, Hawryluk EB, Kroshinsky D, et al. Pediatric dermatology econsults: reduced wait times and dermatology office visits. Pediatr Dermatol. 2020;37:804-810.
Dermatologic conditions affect approximately one-third of individuals in the United States.1,2 Nearly 1 in 4 physician office visits in the United States are for skin conditions, and less than one-third of these visits are with dermatologists. Although many of these patients may prefer to see a dermatologist for their concerns, they may not be able to access specialist care.3 The limited supply and urban-focused distribution of dermatologists along with reduced acceptance of state-funded insurance plans and long appointment wait times all pose considerable challenges to individuals seeking dermatologic care.2 Electronic consultations (e-consults) have emerged as a promising solution to overcoming these barriers while providing high-quality dermatologic care to a large diverse patient population.2,4 Although e-consults can be of service to all dermatology patients, this modality may be especially beneficial to underserved populations, such as the uninsured and Medicaid patients—groups that historically have experienced limited access to dermatology care due to the low reimbursement rates and high administrative burdens accompanying care delivery.4 This limited access leads to inequity in care, as timely access to dermatology is associated with improved diagnostic accuracy and disease outcomes.3 E-consult implementation can facilitate timely access for these underserved populations and bypass additional barriers to care such as lack of transportation or time off work. Prior e-consult studies have demonstrated relatively high numbers of Medicaid patients utilizing e-consult services.3,5
Although in-person visits remain the gold standard for diagnosis and treatment of dermatologic conditions, e-consults placed by primary care providers (PCPs) can improve access and help triage patients who require in-person dermatology visits.6 In this study, we conducted a retrospective chart review to characterize the e-consults requested of the dermatology department at a large tertiary care medical center in Winston-Salem, North Carolina.
Methods
The electronic health record (EHR) of Atrium Health Wake Forest Baptist (Winston-Salem, North Carolina) was screened for eligible patients from January 1, 2020, to May 31, 2021. Patients—both adult (aged ≥18 years) and pediatric (aged <18 years)—were included if they underwent a dermatology e-consult within this time frame. Provider notes in the medical records were reviewed to determine the nature of the lesion, how long the dermatologist took to complete the e-consult, whether an in-person appointment was recommended, and whether the patient was seen by dermatology within 90 days of the e-consult. Institutional review board approval was obtained.
For each e-consult, the PCP obtained clinical photographs of the lesion in question either through the EHR mobile application or by having patients upload their own photographs directly to their medical records. The referring PCP then completed a brief template regarding the patient’s clinical question and medical history and then sent the completed information to the consulting dermatologist’s EHR inbox. From there, the dermatologist could view the clinical question, documented photographs, and patient medical record to create a brief consult note with recommendations. The note was then sent back via EHR to the PCP to follow up with the patient. Patients were not charged for the e-consult.
Results
Two hundred fifty-four dermatology e-consults were requested by providers at the study center (eTable), which included 252 unique patients (2 patients had 2 separate e-consults regarding different clinical questions). The median time for completion of the e-consult—from submission of the PCP’s e-consult request to dermatologist completion—was 0.37 days. Fifty-six patients (22.0%) were recommended for an in-person appointment (Figure), 33 (58.9%) of whom ultimately scheduled the in-person appointment, and the median length of time between the completion of the e-consult and the in-person appointment was 16.5 days. The remaining 198 patients (78.0%) were not triaged to receive an in-person appointment following the e-consult,but 2 patients (8.7%) were ultimately seen in-person anyway via other referral pathways, with a median length of 33 days between e-consult completion and the in-person appointment. One hundred seventy-six patients (69.8%) avoided an in-person dermatology visit, although 38 (21.6%) of those patients were fewer than 90 days out from their e-consults at the time of data collection. The 254 e-consults included patients from 50 different zip codes, 49 (98.0%) of which were in North Carolina.
Comment
An e-consult is an asynchronous telehealth modality through which PCPs can request specialty evaluation to provide diagnostic and therapeutic guidance, facilitate PCP-specialist coordination of care, and increase access to specialty care with reduced wait times.7,8 Increased care access is especially important, as specialty referral can decrease overall health care expenditure; however, the demand for specialists often exceeds the availability.8 Our e-consult program drastically reduced the time from patients’ initial presentation at their PCP’s office to dermatologist recommendations for treatment or need for in-person dermatology follow-up.
In our analysis, patients were of different racial, ethnic, and socioeconomic backgrounds and lived across a variety of zip codes, predominantly in central and western North Carolina. Almost three-quarters of the patients resided in zip codes where the average income was less than the North Carolina median household income ($66,196).9 Additionally, 82 patients (32.3%) were uninsured or on Medicaid (eTable). These economically disadvantaged patient populations historically have had limited access to dermatologic care.4 One study showed that privately insured individuals were accepted as new patients by dermatologists 91% of the time compared to a 29.8% acceptance rate for publicly insured individuals.10 Uninsured and Medicaid patients also have to wait 34% longer for an appointment compared to individuals with Medicare or private insurance.2 Considering these patients may already be at an economic disadvantage when it comes to seeing and paying for dermatologic services, e-consults may reduce patient travel and appointment expenses while increasing access to specialty care. Based on a 2020 study, each e-consult generates an estimated savings of $80 out-of-pocket per patient per avoided in-person visit.11
In our study, the most common condition for an e-consult in both adult and pediatric patients was rash, which is consistent with prior e-consult studies.5,11 We found that most e-consult patients were not recommended for an in-person dermatology visit, and for those who were recommended to have an in-person visit, the wait time was reduced (Figure). These results corroborate that e-consults may be used as an important triage tool for determining whether a specialist appointment is indicated as well as a public health tool, as timely evaluation is associated with better dermatologic health care outcomes.3 However, the number of patients who did not present for an in-person appointment in our study may be overestimated, as 38 patients’ (21.6%) e-consults were conducted fewer than 90 days before our data collection. Although none of these patients had been seen in person, it is possible they requested an in-person visit after their medical records were reviewed for this study. Additionally, it is possible patients sought care from outside providers not documented in the EHR.
With regard to the payment model for the e-consult program, Atrium Health Wake Forest Baptist initially piloted the e-consult system through a partnership with the American Academy of Medical Colleges’ Project CORE: Coordinating Optimal Referral Experiences (https://www.aamc.org/what-we-do/mission-areas/health-care/project-core). Grant funding through Project CORE allowed both the referring PCP and the specialist completing the e-consult to each receive approximately 0.5 relative value units in payment for each consult completed. Based on early adoption successes, the institution has created additional internal funding to support the continued expansion of the e-consult system and is incentivized to continue funding, as proper utilization of e-consults improves patient access to timely specialist care, avoids no-shows or last-minute cancellations for specialist appointments, and decreases back-door access to specialist care through the emergency department and urgent care facilities.5 Although 0.5 relative value units is not equivalent compensation to an in-person office visit, our study showed that e-consults can be completed much more quickly and efficiently and do not utilize nursing staff or other office resources.
Conclusion
E-consults are an effective telehealth modality that can increase patients’ access to dermatologic specialty care.
Acknowledgments—The authors thank the Wake Forest University School of Medicine Department of Medical Education and Department of Dermatology (Winston-Salem, North Carolina) for their contributions to this research study as well as the Wake Forest Clinical and Translational Science Institute (Winston-Salem, North Carolina) for their help extracting EHR data.
Dermatologic conditions affect approximately one-third of individuals in the United States.1,2 Nearly 1 in 4 physician office visits in the United States are for skin conditions, and less than one-third of these visits are with dermatologists. Although many of these patients may prefer to see a dermatologist for their concerns, they may not be able to access specialist care.3 The limited supply and urban-focused distribution of dermatologists along with reduced acceptance of state-funded insurance plans and long appointment wait times all pose considerable challenges to individuals seeking dermatologic care.2 Electronic consultations (e-consults) have emerged as a promising solution to overcoming these barriers while providing high-quality dermatologic care to a large diverse patient population.2,4 Although e-consults can be of service to all dermatology patients, this modality may be especially beneficial to underserved populations, such as the uninsured and Medicaid patients—groups that historically have experienced limited access to dermatology care due to the low reimbursement rates and high administrative burdens accompanying care delivery.4 This limited access leads to inequity in care, as timely access to dermatology is associated with improved diagnostic accuracy and disease outcomes.3 E-consult implementation can facilitate timely access for these underserved populations and bypass additional barriers to care such as lack of transportation or time off work. Prior e-consult studies have demonstrated relatively high numbers of Medicaid patients utilizing e-consult services.3,5
Although in-person visits remain the gold standard for diagnosis and treatment of dermatologic conditions, e-consults placed by primary care providers (PCPs) can improve access and help triage patients who require in-person dermatology visits.6 In this study, we conducted a retrospective chart review to characterize the e-consults requested of the dermatology department at a large tertiary care medical center in Winston-Salem, North Carolina.
Methods
The electronic health record (EHR) of Atrium Health Wake Forest Baptist (Winston-Salem, North Carolina) was screened for eligible patients from January 1, 2020, to May 31, 2021. Patients—both adult (aged ≥18 years) and pediatric (aged <18 years)—were included if they underwent a dermatology e-consult within this time frame. Provider notes in the medical records were reviewed to determine the nature of the lesion, how long the dermatologist took to complete the e-consult, whether an in-person appointment was recommended, and whether the patient was seen by dermatology within 90 days of the e-consult. Institutional review board approval was obtained.
For each e-consult, the PCP obtained clinical photographs of the lesion in question either through the EHR mobile application or by having patients upload their own photographs directly to their medical records. The referring PCP then completed a brief template regarding the patient’s clinical question and medical history and then sent the completed information to the consulting dermatologist’s EHR inbox. From there, the dermatologist could view the clinical question, documented photographs, and patient medical record to create a brief consult note with recommendations. The note was then sent back via EHR to the PCP to follow up with the patient. Patients were not charged for the e-consult.
Results
Two hundred fifty-four dermatology e-consults were requested by providers at the study center (eTable), which included 252 unique patients (2 patients had 2 separate e-consults regarding different clinical questions). The median time for completion of the e-consult—from submission of the PCP’s e-consult request to dermatologist completion—was 0.37 days. Fifty-six patients (22.0%) were recommended for an in-person appointment (Figure), 33 (58.9%) of whom ultimately scheduled the in-person appointment, and the median length of time between the completion of the e-consult and the in-person appointment was 16.5 days. The remaining 198 patients (78.0%) were not triaged to receive an in-person appointment following the e-consult,but 2 patients (8.7%) were ultimately seen in-person anyway via other referral pathways, with a median length of 33 days between e-consult completion and the in-person appointment. One hundred seventy-six patients (69.8%) avoided an in-person dermatology visit, although 38 (21.6%) of those patients were fewer than 90 days out from their e-consults at the time of data collection. The 254 e-consults included patients from 50 different zip codes, 49 (98.0%) of which were in North Carolina.
Comment
An e-consult is an asynchronous telehealth modality through which PCPs can request specialty evaluation to provide diagnostic and therapeutic guidance, facilitate PCP-specialist coordination of care, and increase access to specialty care with reduced wait times.7,8 Increased care access is especially important, as specialty referral can decrease overall health care expenditure; however, the demand for specialists often exceeds the availability.8 Our e-consult program drastically reduced the time from patients’ initial presentation at their PCP’s office to dermatologist recommendations for treatment or need for in-person dermatology follow-up.
In our analysis, patients were of different racial, ethnic, and socioeconomic backgrounds and lived across a variety of zip codes, predominantly in central and western North Carolina. Almost three-quarters of the patients resided in zip codes where the average income was less than the North Carolina median household income ($66,196).9 Additionally, 82 patients (32.3%) were uninsured or on Medicaid (eTable). These economically disadvantaged patient populations historically have had limited access to dermatologic care.4 One study showed that privately insured individuals were accepted as new patients by dermatologists 91% of the time compared to a 29.8% acceptance rate for publicly insured individuals.10 Uninsured and Medicaid patients also have to wait 34% longer for an appointment compared to individuals with Medicare or private insurance.2 Considering these patients may already be at an economic disadvantage when it comes to seeing and paying for dermatologic services, e-consults may reduce patient travel and appointment expenses while increasing access to specialty care. Based on a 2020 study, each e-consult generates an estimated savings of $80 out-of-pocket per patient per avoided in-person visit.11
In our study, the most common condition for an e-consult in both adult and pediatric patients was rash, which is consistent with prior e-consult studies.5,11 We found that most e-consult patients were not recommended for an in-person dermatology visit, and for those who were recommended to have an in-person visit, the wait time was reduced (Figure). These results corroborate that e-consults may be used as an important triage tool for determining whether a specialist appointment is indicated as well as a public health tool, as timely evaluation is associated with better dermatologic health care outcomes.3 However, the number of patients who did not present for an in-person appointment in our study may be overestimated, as 38 patients’ (21.6%) e-consults were conducted fewer than 90 days before our data collection. Although none of these patients had been seen in person, it is possible they requested an in-person visit after their medical records were reviewed for this study. Additionally, it is possible patients sought care from outside providers not documented in the EHR.
With regard to the payment model for the e-consult program, Atrium Health Wake Forest Baptist initially piloted the e-consult system through a partnership with the American Academy of Medical Colleges’ Project CORE: Coordinating Optimal Referral Experiences (https://www.aamc.org/what-we-do/mission-areas/health-care/project-core). Grant funding through Project CORE allowed both the referring PCP and the specialist completing the e-consult to each receive approximately 0.5 relative value units in payment for each consult completed. Based on early adoption successes, the institution has created additional internal funding to support the continued expansion of the e-consult system and is incentivized to continue funding, as proper utilization of e-consults improves patient access to timely specialist care, avoids no-shows or last-minute cancellations for specialist appointments, and decreases back-door access to specialist care through the emergency department and urgent care facilities.5 Although 0.5 relative value units is not equivalent compensation to an in-person office visit, our study showed that e-consults can be completed much more quickly and efficiently and do not utilize nursing staff or other office resources.
Conclusion
E-consults are an effective telehealth modality that can increase patients’ access to dermatologic specialty care.
Acknowledgments—The authors thank the Wake Forest University School of Medicine Department of Medical Education and Department of Dermatology (Winston-Salem, North Carolina) for their contributions to this research study as well as the Wake Forest Clinical and Translational Science Institute (Winston-Salem, North Carolina) for their help extracting EHR data.
- Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
- Naka F, Lu J, Porto A, et al. Impact of dermatology econsults on access to care and skin cancer screening in underserved populations: a model for teledermatology services in community health centers. J Am Acad Dermatol. 2018;78:293-302.
- Mulcahy A, Mehrotra A, Edison K, et al. Variation in dermatologist visits by sociodemographic characteristics. J Am Acad Dermatol. 2017;76:918-924.
- Yang X, Barbieri JS, Kovarik CL. Cost analysis of a store-and-forward teledermatology consult system in Philadelphia. J Am Acad Dermatol. 2019;81:758-764.
- Wang RF, Trinidad J, Lawrence J, et al. Improved patient access and outcomes with the integration of an econsult program (teledermatology) within a large academic medical center. J Am Acad Dermatol. 2020;83:1633-1638.
- Lee KJ, Finnane A, Soyer HP. Recent trends in teledermatology and teledermoscopy. Dermatol Pract Concept. 2018;8:214-223.
- Parikh PJ, Mowrey C, Gallimore J, et al. Evaluating e-consultation implementations based on use and time-line across various specialties. Int J Med Inform. 2017;108:42-48.
- Wasfy JH, Rao SK, Kalwani N, et al. Longer-term impact of cardiology e-consults. Am Heart J. 2016;173:86-93.
- United States Census Bureau. QuickFacts: North Carolina; United States. Accessed February 26, 2024. https://www.census.gov/quickfacts/fact/table/NC,US/PST045222
- Alghothani L, Jacks SK, Vander Horst A, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
- Seiger K, Hawryluk EB, Kroshinsky D, et al. Pediatric dermatology econsults: reduced wait times and dermatology office visits. Pediatr Dermatol. 2020;37:804-810.
- Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527-1534.
- Naka F, Lu J, Porto A, et al. Impact of dermatology econsults on access to care and skin cancer screening in underserved populations: a model for teledermatology services in community health centers. J Am Acad Dermatol. 2018;78:293-302.
- Mulcahy A, Mehrotra A, Edison K, et al. Variation in dermatologist visits by sociodemographic characteristics. J Am Acad Dermatol. 2017;76:918-924.
- Yang X, Barbieri JS, Kovarik CL. Cost analysis of a store-and-forward teledermatology consult system in Philadelphia. J Am Acad Dermatol. 2019;81:758-764.
- Wang RF, Trinidad J, Lawrence J, et al. Improved patient access and outcomes with the integration of an econsult program (teledermatology) within a large academic medical center. J Am Acad Dermatol. 2020;83:1633-1638.
- Lee KJ, Finnane A, Soyer HP. Recent trends in teledermatology and teledermoscopy. Dermatol Pract Concept. 2018;8:214-223.
- Parikh PJ, Mowrey C, Gallimore J, et al. Evaluating e-consultation implementations based on use and time-line across various specialties. Int J Med Inform. 2017;108:42-48.
- Wasfy JH, Rao SK, Kalwani N, et al. Longer-term impact of cardiology e-consults. Am Heart J. 2016;173:86-93.
- United States Census Bureau. QuickFacts: North Carolina; United States. Accessed February 26, 2024. https://www.census.gov/quickfacts/fact/table/NC,US/PST045222
- Alghothani L, Jacks SK, Vander Horst A, et al. Disparities in access to dermatologic care according to insurance type. Arch Dermatol. 2012;148:956-957.
- Seiger K, Hawryluk EB, Kroshinsky D, et al. Pediatric dermatology econsults: reduced wait times and dermatology office visits. Pediatr Dermatol. 2020;37:804-810.
Practice Points
- Most electronic consult patients may be able to avoid in-person dermatology appointments.
- E-consults can increase patient access to dermatologic specialty care.
Hospital Dermatology: Review of Research in 2022-2023
Dermatologists improve the diagnostic accuracy and quality of care of patients in the hospital setting. They help shorten the length of stay, improve outpatient follow-up, and reduce the rate of hospital readmission.1 Medicare beneficiaries hospitalized with skin conditions at institutions with a dermatology hospitalist—a provider with a specialty interest in inpatient dermatology—have 24% lower odds of risk-adjusted 30-day mortality and 12% lower odds of risk-adjusted 30-day readmissions.2
In the last year, research among the dermatology hospitalist community has actively contributed to our understanding of challenging inpatient skin diseases and has identified new ways in which dermatologists can contribute to the care of hospitalized patients. In this review, we highlight 4 areas of focus from the published literature in 2022-2023—severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.
Severe Cutaneous Adverse Reactions: Old and New
Severe cutaneous adverse reactions to medications frequently are encountered in the inpatient setting. Dermatology hospitalists are well positioned to phenotype these reactions, drawing insights that aid in identifying, characterizing, risk stratifying, and managing these conditions, which have considerable morbidity and mortality.
A recent 20-year retrospective review of cases of acute generalized exanthematous pustulosis (N=340) across 10 academic systems—the largest to date—improves our understanding of the features of this rare entity.3 The authors found that acute generalized exanthematous pustulosis most often is triggered by β-lactam and other antibiotics (75.5%) and is accompanied by fever (49.7%), neutrophilia (85.1%), and eosinophilia (52.1%). Kidney and liver involvement occur in less than 10% of cases, and mortality rates are low but not zero, with an all-cause 30-day mortality rate of 3.5%.3
In a multi-institutional retrospective study of 68 patients diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, Sharma et al4 developed a scoring system to identify those at greatest risk for DRESS recurrence. Variables associated with recurrence including younger age, female sex, and features considered atypical for DRESS syndrome—nonmorbilliform rash; absence of facial edema; antinuclear antibody positivity; medication class other than antibiotic, antigout, or antiseizure—were used to develop a “ReDRESS” score. This predictive model had a sensitivity of 73% and specificity of 83% for predicting DRESS recurrence.4
Another case series characterized SCoRCH (sudden conjunctivitis, lymphopenia, sunburnlike rash, and hemodynamic changes), a newly described hypersensitivity reaction to trimethoprim-sulfamethoxazole.5 The onset of this reaction typically occurs 4 to 11 days after initiation of trimethoprim-sulfamethoxazole but can occur as quickly as 1 day following re-exposure. Patients are systemically ill with fever, hypotension, tachycardia, acute renal insufficiency, and transaminitis, and they have a diffuse sunburnlike erythema without scale, facial edema, and conjunctivitis. It is thought this distinct hypersensitivity reaction may be mediated by IL-6, which has a role in triggering a sepsislike physiology, with vasodilation, hypotension, and edema.5
A systematic review and meta-analysis found that sulfonamides remain the most prominent cause of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).6 A case-control study described SJS/TEN presentations triggered by Mycoplasma, advocating for routine Mycoplasma screening, especially in patients without a clear medication culprit. Mycoplasma-induced cases carried statistically lower rates of mortality (0%) compared with medication-induced cases (22.5%).7 Another prospective open-label study evaluated SJS/TEN management by randomizing 25 patients to receive either combination therapy with methylprednisolone plus a tumor necrosis factor α inhibitor or methylprednisolone alone.8 Anti–tumor necrosis factor therapy was associated with a shorter length of initial steroid treatment and duration of the acute stage, hospitalization, and time to re-epithelialization8; however, as in a prior randomized unblinded trial,9 there was no difference in mortality between the 2 groups.
There is limited high-quality evidence to support the use of any systemic immunomodulator to decrease SJS/TEN–related mortality.10 A Cochrane systematic review highlighted the many limitations of the available data due to variations in presentation, assessment, and management.11 Because SJS/TEN is rare, powering studies based on mortality is infeasible; the authors calculated that 2872 participants were needed to detect a 50% mortality reduction among those with SCORTEN (severity-of-illness score for TEN) scores of 0 to 1.11 Therefore, collaborative efforts using appropriate outcomes measures (eg, time to re-epithelialization, length of hospital stay), standardized terminology and dosing regimens, and adaptive trial designs are needed. Consensus-derived assessment and treatment protocols could help account for variation, ensure consistency in treatment, and enable head-to-head comparisons. Members of the Society of Dermatology Hospitalists are working on efforts to standardize terminology and validate outcomes measures needed for future studies.12
Supportive Oncodermatology: A New Frontier
With the advent of immune checkpoint inhibitors (ICIs) for a growing number of cancers, dermatologists have become critical to identifying and managing cutaneous immune-related adverse events (cirAEs). Recent findings have demonstrated that dermatology input improves patient outcomes, not only regarding the treatment of dermatoses but also by augmenting cancer-related survival. One group found that patients with cirAEs who were evaluated by a dermatologist had improved progression-free (hazard ratio, 0.69; 95% CI, 0.54-0.87; P=.002) and overall survival rates (hazard ratio, 0.62; 95% CI, 0.45-0.84; P=.002), controlling for cirAE severity, age, sex, cancer type, and ICI subtype. Patients who were under the care of a dermatologist also were more likely to resume ICI therapy following an interruption (odds ratio, 10.52; 95% CI, 5.15-21.48; P<.001).13 Dermatologists help to optimize skin-directed and targeted therapies, such as dupilumab, minimizing exposure to systemic immunosuppression in these complex patients.14
Supportive oncodermatologists also have made important observations on how cirAEs relate to other adverse events and prognosis. A review of 628 patients found that almost half of those with cirAEs had co-occurring noncutaneous immune-related adverse events, most commonly pulmonary. Psoriasiform eruptions were most frequently associated with noncutaneous immune-related adverse events, and cutaneous reactions frequently preceded the development of systemic manifestations, serving as a clinical biomarker to provide prognostic information.15 A review of 95 patients found that spongiotic and lichenoid interface reactions were associated with decreased mortality rates, whereas vacuolar interface and perivascular dermatitis were associated with increased mortality.16
As with severe cutaneous adverse events, dermatology input has been critical for accurately phenotyping and risk stratifying these novel reactions. The dermatologist’s skill set is necessary for optimizing skin-directed and targeted therapies while minimizing systemic immunosuppression, thereby improving patient outcomes with respect to rash, cancer response, and survival.
The Cost of Inpatient Skin Disease
Hospitalizations account for approximately half of all health care expenditures, and hospital readmission, seen as a measure of the quality of health care delivery, can double this cost.17 Identifying and developing protocols for addressing patients with complex chronic inflammatory disorders is one strategy for improving outcomes and reducing financial burden. Inpatient dermatologists have identified hidradenitis suppurativa as one disease that can benefit from early intervention by dermatologists in the hospital, with its 30-day (17.8%) and 180-day (48.6%) readmission rates being comparable to those of heart failure.18
Following an index emergency department (ED) visit, 17.2% (3484/20,269) of patients with HS have at least 1 return ED visit within 30 days, while only 2.4% (483/20,269) have a dermatology visit within the same time frame.19 Understanding the risk factors for hospital readmission and ED utilization, including severity of illness, the presence of medical comorbidities, health coverage under Medicaid, and receipt of opioids, can allow dermatologists to anticipate those at greatest risk.19 Opportunities exist for cross-specialty interventions to anticipate and address modifiable risk factors. Shorter time to dermatology outpatient follow-up leads to improved clinic attendance and may help reduce ED utilization and hospital readmission.20
Teledermatology: Leveraging Inpatient Expertise
Although the benefit of inpatient dermatologic care is substantial, access to that care is finite. Following the COVID-19 pandemic, there is an increased acceptance of telemedicine and the long-term role it can play in leveraging dermatologic expertise, including meeting the increasing demand for inpatient dermatology care in rural and resource-poor communities.21
Recent studies conducted by dermatology hospitalists have illustrated the value of asynchronous store-and-forward technology in settings lacking access to consultative dermatology.22,23 Stephens et al22 found that expanding provider-to-provider electronic consultation (e-consultation) capacity to an inpatient rehabilitation facility resulted in completed consultations within 1.5 days compared with a 7- to 14-day wait time for patients attending an in-person urgent access dermatology clinic. In another study, the implementation of asynchronous dermatology e-consultations for immunobullous diseases, vasculitis, and herpes zoster resulted in a change in diagnosis 86% of the time, accompanied by at least 1 new systemic or topical therapy recommendation.23
Researchers also identified ways in which teledermatology can be inelegant and proposed specific supplemental data to aid in diagnosis. A review of 126 inpatient e-consultations demonstrated limitations related to the diagnosis of skin and soft-tissue infections. In two-thirds to three-quarters of cases, potentially useful descriptive information was missing, and in 70% (88/126), images were not appropriately focused. The authors developed a detailed checklist to help primary medical teams focus their differential diagnoses.24 A recent pilot study found that supplementation of clinical information with a standardized questionnaire and thermal images improved the accuracy of cellulitis diagnosis. Using this method, there was no difference in accuracy between dermatology hospitalists and other board-certified dermatologists, supporting the notion that any dermatologist can fulfill this need successfully, even without specific inpatient experience.25 Due to the high incidence and cost of cellulitis and related hospital admissions,26 such an intervention could have a considerable financial and patient safety impact.
Final Thoughts
This last year brought many changes to the health care landscape, the recession of a global pandemic, and an increasingly complex health care delivery system. Inpatient dermatologists met these challenges by providing high-quality dermatologic care and practice-modifying research in the areas of severe cutaneous adverse reactions, supportive oncodermatology, hospital readmission, telemedicine, and more, demonstrating the value of dermatologic expertise in the hospital setting.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Puri P, Pollock BD, Yousif M, et al. Association of Society of Dermatology hospitalist institutions with improved outcomes in Medicare beneficiaries hospitalized for skin disease. J Am Acad Dermatol. 2023;88:1372-1375.
- Creadore A, Desai S, Alloo A, et al. Clinical characteristics, disease course, and outcomes of patients with acute generalized exanthematous pustulosis in the US. JAMA Dermatol. 2022;158:176-183.
- Sharma AN, Murphy K, Shwe S, et al. Predicting DRESS syndrome recurrence—the ReDRESS score. JAMA Dermatol. 2022;158:1445-1447.
- Brian M, Rose EK, Mauskar MM, et al. Sudden conjunctivitis, lymphopenia, and rash combined with hemodynamic changes (SCoRCH) after trimethoprim-sulfamethoxazole use: a case series study of a hypersensitivity reaction. JAMA Dermatol. 2023;159:73-78.
- Lee EY, Knox C, Phillips EJ. Worldwide prevalence of antibiotic-associated Stevens-Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. JAMA Dermatol. 2023;159:384-392.
- Liew YCC, Choo KJL, Oh CC, et al. Mycoplasma-induced Stevens-Johnson syndrome/toxic epidermal necrolysis: case-control analysis of a cohort managed in a specialized center. J Am Acad Dermatol. 2022;86:811-817.
- Ao S, Gao X, Zhan J, et al. Inhibition of tumor necrosis factor improves conventional steroid therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis in a cohort of patients. J Am Acad Dermatol. 2022;86:1236-1245.
- Wang CW, Yang LY, Chen CB, et al; the Taiwan Severe Cutaneous Adverse Reaction (TSCAR) Consortium. Randomized, controlled trial of TNF-α antagonist in CTL-mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Han JJ, Creadore A, Seminario-Vidal L, et al. Medical management of Stevens-Johnson syndrome/toxic epidermal necrolysis among North American dermatologists. J Am Acad Dermatol. 2022;87:429-431.
- Noe MH, Micheletti RG. Systemic interventions for treatment of Stevens-Johnson syndrome/toxic epidermal necrolysis: summary of a Cochrane review. JAMA Dermatol. 2022;158:1436-1437.
- Waters M, Dobry A, Le ST, et al. Development of a skin-directed scoring system for Stevens-Johnson syndrome and epidermal necrolysis: a Delphi consensus exercise. JAMA Dermatol. 2023;159:772-777.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Said JT, Elman SA, Perez-Chada LM, et al. Treatment of immune checkpoint inhibitor-mediated psoriasis: a systematic review. J Am Acad Dermatol. 2022;87:399-400.
- Asdourian MS, Shah N, Jacoby TV, et al. Evaluating patterns of co-occurrence between cutaneous and noncutaneous immune-related adverse events after immune checkpoint inhibitor therapy. J Am Acad Dermatol. 2023;88:246-249.
- Hirotsu KE, Scott MKD, Marquez C, et al. Histologic subtype of cutaneous immune-related adverse events predicts overall survival in patients receiving immune checkpoint inhibitors. J Am Acad Dermatol. 2022;87:651-653.
- Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160:1074-1081.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192.
- Wang CX, Buss JL, Keller M, et al. Factors associated with dermatologic follow-up vs emergency department return in patients with hidradenitis suppurativa after an initial emergency department visit. JAMA Dermatol. 2022;158:1378-1386.
- Zakaria A, Chang AY, Kim-Lim P, et al. Predictors of postdischarge follow-up attendance among hospitalized dermatology patients: disparities and potential interventions. J Am Acad Dermatol. 2022;87:186-188.
- Arnold JD, Yoon S, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2019;80:425-432. doi:10.1016/j.jaad.2018.06.070
- Stephens MR, Das S, Smith GP. Utilization and outcomes of an asynchronous teledermatology pilot for an inpatient rehabilitation hospital. J Am Acad Dermatol. 2022;87:421-423.
- Ortiz C, Khosravi H, Kettering C, et al. Concordance data for inpatient asynchronous eDermatology consultation for immunobullous disease, zoster, and vasculitis. J Am Acad Dermatol. 2022;86:918-920.
- Salle R, Hua C, Mongereau M, et al. Challenges and limitations of teledermatology for skin and soft-tissue infections: a real-world study of an expert center. J Am Acad Dermatol. 2023;88:457-459.
- Creadore A, Manjaly P, Tkachenko E, et al. The utility of augmented teledermatology to improve dermatologist diagnosis of cellulitis: a cross-sectional study. Arch Dermatol Res. 2023;315:1347-1353.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
Dermatologists improve the diagnostic accuracy and quality of care of patients in the hospital setting. They help shorten the length of stay, improve outpatient follow-up, and reduce the rate of hospital readmission.1 Medicare beneficiaries hospitalized with skin conditions at institutions with a dermatology hospitalist—a provider with a specialty interest in inpatient dermatology—have 24% lower odds of risk-adjusted 30-day mortality and 12% lower odds of risk-adjusted 30-day readmissions.2
In the last year, research among the dermatology hospitalist community has actively contributed to our understanding of challenging inpatient skin diseases and has identified new ways in which dermatologists can contribute to the care of hospitalized patients. In this review, we highlight 4 areas of focus from the published literature in 2022-2023—severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.
Severe Cutaneous Adverse Reactions: Old and New
Severe cutaneous adverse reactions to medications frequently are encountered in the inpatient setting. Dermatology hospitalists are well positioned to phenotype these reactions, drawing insights that aid in identifying, characterizing, risk stratifying, and managing these conditions, which have considerable morbidity and mortality.
A recent 20-year retrospective review of cases of acute generalized exanthematous pustulosis (N=340) across 10 academic systems—the largest to date—improves our understanding of the features of this rare entity.3 The authors found that acute generalized exanthematous pustulosis most often is triggered by β-lactam and other antibiotics (75.5%) and is accompanied by fever (49.7%), neutrophilia (85.1%), and eosinophilia (52.1%). Kidney and liver involvement occur in less than 10% of cases, and mortality rates are low but not zero, with an all-cause 30-day mortality rate of 3.5%.3
In a multi-institutional retrospective study of 68 patients diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, Sharma et al4 developed a scoring system to identify those at greatest risk for DRESS recurrence. Variables associated with recurrence including younger age, female sex, and features considered atypical for DRESS syndrome—nonmorbilliform rash; absence of facial edema; antinuclear antibody positivity; medication class other than antibiotic, antigout, or antiseizure—were used to develop a “ReDRESS” score. This predictive model had a sensitivity of 73% and specificity of 83% for predicting DRESS recurrence.4
Another case series characterized SCoRCH (sudden conjunctivitis, lymphopenia, sunburnlike rash, and hemodynamic changes), a newly described hypersensitivity reaction to trimethoprim-sulfamethoxazole.5 The onset of this reaction typically occurs 4 to 11 days after initiation of trimethoprim-sulfamethoxazole but can occur as quickly as 1 day following re-exposure. Patients are systemically ill with fever, hypotension, tachycardia, acute renal insufficiency, and transaminitis, and they have a diffuse sunburnlike erythema without scale, facial edema, and conjunctivitis. It is thought this distinct hypersensitivity reaction may be mediated by IL-6, which has a role in triggering a sepsislike physiology, with vasodilation, hypotension, and edema.5
A systematic review and meta-analysis found that sulfonamides remain the most prominent cause of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).6 A case-control study described SJS/TEN presentations triggered by Mycoplasma, advocating for routine Mycoplasma screening, especially in patients without a clear medication culprit. Mycoplasma-induced cases carried statistically lower rates of mortality (0%) compared with medication-induced cases (22.5%).7 Another prospective open-label study evaluated SJS/TEN management by randomizing 25 patients to receive either combination therapy with methylprednisolone plus a tumor necrosis factor α inhibitor or methylprednisolone alone.8 Anti–tumor necrosis factor therapy was associated with a shorter length of initial steroid treatment and duration of the acute stage, hospitalization, and time to re-epithelialization8; however, as in a prior randomized unblinded trial,9 there was no difference in mortality between the 2 groups.
There is limited high-quality evidence to support the use of any systemic immunomodulator to decrease SJS/TEN–related mortality.10 A Cochrane systematic review highlighted the many limitations of the available data due to variations in presentation, assessment, and management.11 Because SJS/TEN is rare, powering studies based on mortality is infeasible; the authors calculated that 2872 participants were needed to detect a 50% mortality reduction among those with SCORTEN (severity-of-illness score for TEN) scores of 0 to 1.11 Therefore, collaborative efforts using appropriate outcomes measures (eg, time to re-epithelialization, length of hospital stay), standardized terminology and dosing regimens, and adaptive trial designs are needed. Consensus-derived assessment and treatment protocols could help account for variation, ensure consistency in treatment, and enable head-to-head comparisons. Members of the Society of Dermatology Hospitalists are working on efforts to standardize terminology and validate outcomes measures needed for future studies.12
Supportive Oncodermatology: A New Frontier
With the advent of immune checkpoint inhibitors (ICIs) for a growing number of cancers, dermatologists have become critical to identifying and managing cutaneous immune-related adverse events (cirAEs). Recent findings have demonstrated that dermatology input improves patient outcomes, not only regarding the treatment of dermatoses but also by augmenting cancer-related survival. One group found that patients with cirAEs who were evaluated by a dermatologist had improved progression-free (hazard ratio, 0.69; 95% CI, 0.54-0.87; P=.002) and overall survival rates (hazard ratio, 0.62; 95% CI, 0.45-0.84; P=.002), controlling for cirAE severity, age, sex, cancer type, and ICI subtype. Patients who were under the care of a dermatologist also were more likely to resume ICI therapy following an interruption (odds ratio, 10.52; 95% CI, 5.15-21.48; P<.001).13 Dermatologists help to optimize skin-directed and targeted therapies, such as dupilumab, minimizing exposure to systemic immunosuppression in these complex patients.14
Supportive oncodermatologists also have made important observations on how cirAEs relate to other adverse events and prognosis. A review of 628 patients found that almost half of those with cirAEs had co-occurring noncutaneous immune-related adverse events, most commonly pulmonary. Psoriasiform eruptions were most frequently associated with noncutaneous immune-related adverse events, and cutaneous reactions frequently preceded the development of systemic manifestations, serving as a clinical biomarker to provide prognostic information.15 A review of 95 patients found that spongiotic and lichenoid interface reactions were associated with decreased mortality rates, whereas vacuolar interface and perivascular dermatitis were associated with increased mortality.16
As with severe cutaneous adverse events, dermatology input has been critical for accurately phenotyping and risk stratifying these novel reactions. The dermatologist’s skill set is necessary for optimizing skin-directed and targeted therapies while minimizing systemic immunosuppression, thereby improving patient outcomes with respect to rash, cancer response, and survival.
The Cost of Inpatient Skin Disease
Hospitalizations account for approximately half of all health care expenditures, and hospital readmission, seen as a measure of the quality of health care delivery, can double this cost.17 Identifying and developing protocols for addressing patients with complex chronic inflammatory disorders is one strategy for improving outcomes and reducing financial burden. Inpatient dermatologists have identified hidradenitis suppurativa as one disease that can benefit from early intervention by dermatologists in the hospital, with its 30-day (17.8%) and 180-day (48.6%) readmission rates being comparable to those of heart failure.18
Following an index emergency department (ED) visit, 17.2% (3484/20,269) of patients with HS have at least 1 return ED visit within 30 days, while only 2.4% (483/20,269) have a dermatology visit within the same time frame.19 Understanding the risk factors for hospital readmission and ED utilization, including severity of illness, the presence of medical comorbidities, health coverage under Medicaid, and receipt of opioids, can allow dermatologists to anticipate those at greatest risk.19 Opportunities exist for cross-specialty interventions to anticipate and address modifiable risk factors. Shorter time to dermatology outpatient follow-up leads to improved clinic attendance and may help reduce ED utilization and hospital readmission.20
Teledermatology: Leveraging Inpatient Expertise
Although the benefit of inpatient dermatologic care is substantial, access to that care is finite. Following the COVID-19 pandemic, there is an increased acceptance of telemedicine and the long-term role it can play in leveraging dermatologic expertise, including meeting the increasing demand for inpatient dermatology care in rural and resource-poor communities.21
Recent studies conducted by dermatology hospitalists have illustrated the value of asynchronous store-and-forward technology in settings lacking access to consultative dermatology.22,23 Stephens et al22 found that expanding provider-to-provider electronic consultation (e-consultation) capacity to an inpatient rehabilitation facility resulted in completed consultations within 1.5 days compared with a 7- to 14-day wait time for patients attending an in-person urgent access dermatology clinic. In another study, the implementation of asynchronous dermatology e-consultations for immunobullous diseases, vasculitis, and herpes zoster resulted in a change in diagnosis 86% of the time, accompanied by at least 1 new systemic or topical therapy recommendation.23
Researchers also identified ways in which teledermatology can be inelegant and proposed specific supplemental data to aid in diagnosis. A review of 126 inpatient e-consultations demonstrated limitations related to the diagnosis of skin and soft-tissue infections. In two-thirds to three-quarters of cases, potentially useful descriptive information was missing, and in 70% (88/126), images were not appropriately focused. The authors developed a detailed checklist to help primary medical teams focus their differential diagnoses.24 A recent pilot study found that supplementation of clinical information with a standardized questionnaire and thermal images improved the accuracy of cellulitis diagnosis. Using this method, there was no difference in accuracy between dermatology hospitalists and other board-certified dermatologists, supporting the notion that any dermatologist can fulfill this need successfully, even without specific inpatient experience.25 Due to the high incidence and cost of cellulitis and related hospital admissions,26 such an intervention could have a considerable financial and patient safety impact.
Final Thoughts
This last year brought many changes to the health care landscape, the recession of a global pandemic, and an increasingly complex health care delivery system. Inpatient dermatologists met these challenges by providing high-quality dermatologic care and practice-modifying research in the areas of severe cutaneous adverse reactions, supportive oncodermatology, hospital readmission, telemedicine, and more, demonstrating the value of dermatologic expertise in the hospital setting.
Dermatologists improve the diagnostic accuracy and quality of care of patients in the hospital setting. They help shorten the length of stay, improve outpatient follow-up, and reduce the rate of hospital readmission.1 Medicare beneficiaries hospitalized with skin conditions at institutions with a dermatology hospitalist—a provider with a specialty interest in inpatient dermatology—have 24% lower odds of risk-adjusted 30-day mortality and 12% lower odds of risk-adjusted 30-day readmissions.2
In the last year, research among the dermatology hospitalist community has actively contributed to our understanding of challenging inpatient skin diseases and has identified new ways in which dermatologists can contribute to the care of hospitalized patients. In this review, we highlight 4 areas of focus from the published literature in 2022-2023—severe cutaneous adverse reactions, supportive oncodermatology, cost of inpatient services, and teledermatology.
Severe Cutaneous Adverse Reactions: Old and New
Severe cutaneous adverse reactions to medications frequently are encountered in the inpatient setting. Dermatology hospitalists are well positioned to phenotype these reactions, drawing insights that aid in identifying, characterizing, risk stratifying, and managing these conditions, which have considerable morbidity and mortality.
A recent 20-year retrospective review of cases of acute generalized exanthematous pustulosis (N=340) across 10 academic systems—the largest to date—improves our understanding of the features of this rare entity.3 The authors found that acute generalized exanthematous pustulosis most often is triggered by β-lactam and other antibiotics (75.5%) and is accompanied by fever (49.7%), neutrophilia (85.1%), and eosinophilia (52.1%). Kidney and liver involvement occur in less than 10% of cases, and mortality rates are low but not zero, with an all-cause 30-day mortality rate of 3.5%.3
In a multi-institutional retrospective study of 68 patients diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, Sharma et al4 developed a scoring system to identify those at greatest risk for DRESS recurrence. Variables associated with recurrence including younger age, female sex, and features considered atypical for DRESS syndrome—nonmorbilliform rash; absence of facial edema; antinuclear antibody positivity; medication class other than antibiotic, antigout, or antiseizure—were used to develop a “ReDRESS” score. This predictive model had a sensitivity of 73% and specificity of 83% for predicting DRESS recurrence.4
Another case series characterized SCoRCH (sudden conjunctivitis, lymphopenia, sunburnlike rash, and hemodynamic changes), a newly described hypersensitivity reaction to trimethoprim-sulfamethoxazole.5 The onset of this reaction typically occurs 4 to 11 days after initiation of trimethoprim-sulfamethoxazole but can occur as quickly as 1 day following re-exposure. Patients are systemically ill with fever, hypotension, tachycardia, acute renal insufficiency, and transaminitis, and they have a diffuse sunburnlike erythema without scale, facial edema, and conjunctivitis. It is thought this distinct hypersensitivity reaction may be mediated by IL-6, which has a role in triggering a sepsislike physiology, with vasodilation, hypotension, and edema.5
A systematic review and meta-analysis found that sulfonamides remain the most prominent cause of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).6 A case-control study described SJS/TEN presentations triggered by Mycoplasma, advocating for routine Mycoplasma screening, especially in patients without a clear medication culprit. Mycoplasma-induced cases carried statistically lower rates of mortality (0%) compared with medication-induced cases (22.5%).7 Another prospective open-label study evaluated SJS/TEN management by randomizing 25 patients to receive either combination therapy with methylprednisolone plus a tumor necrosis factor α inhibitor or methylprednisolone alone.8 Anti–tumor necrosis factor therapy was associated with a shorter length of initial steroid treatment and duration of the acute stage, hospitalization, and time to re-epithelialization8; however, as in a prior randomized unblinded trial,9 there was no difference in mortality between the 2 groups.
There is limited high-quality evidence to support the use of any systemic immunomodulator to decrease SJS/TEN–related mortality.10 A Cochrane systematic review highlighted the many limitations of the available data due to variations in presentation, assessment, and management.11 Because SJS/TEN is rare, powering studies based on mortality is infeasible; the authors calculated that 2872 participants were needed to detect a 50% mortality reduction among those with SCORTEN (severity-of-illness score for TEN) scores of 0 to 1.11 Therefore, collaborative efforts using appropriate outcomes measures (eg, time to re-epithelialization, length of hospital stay), standardized terminology and dosing regimens, and adaptive trial designs are needed. Consensus-derived assessment and treatment protocols could help account for variation, ensure consistency in treatment, and enable head-to-head comparisons. Members of the Society of Dermatology Hospitalists are working on efforts to standardize terminology and validate outcomes measures needed for future studies.12
Supportive Oncodermatology: A New Frontier
With the advent of immune checkpoint inhibitors (ICIs) for a growing number of cancers, dermatologists have become critical to identifying and managing cutaneous immune-related adverse events (cirAEs). Recent findings have demonstrated that dermatology input improves patient outcomes, not only regarding the treatment of dermatoses but also by augmenting cancer-related survival. One group found that patients with cirAEs who were evaluated by a dermatologist had improved progression-free (hazard ratio, 0.69; 95% CI, 0.54-0.87; P=.002) and overall survival rates (hazard ratio, 0.62; 95% CI, 0.45-0.84; P=.002), controlling for cirAE severity, age, sex, cancer type, and ICI subtype. Patients who were under the care of a dermatologist also were more likely to resume ICI therapy following an interruption (odds ratio, 10.52; 95% CI, 5.15-21.48; P<.001).13 Dermatologists help to optimize skin-directed and targeted therapies, such as dupilumab, minimizing exposure to systemic immunosuppression in these complex patients.14
Supportive oncodermatologists also have made important observations on how cirAEs relate to other adverse events and prognosis. A review of 628 patients found that almost half of those with cirAEs had co-occurring noncutaneous immune-related adverse events, most commonly pulmonary. Psoriasiform eruptions were most frequently associated with noncutaneous immune-related adverse events, and cutaneous reactions frequently preceded the development of systemic manifestations, serving as a clinical biomarker to provide prognostic information.15 A review of 95 patients found that spongiotic and lichenoid interface reactions were associated with decreased mortality rates, whereas vacuolar interface and perivascular dermatitis were associated with increased mortality.16
As with severe cutaneous adverse events, dermatology input has been critical for accurately phenotyping and risk stratifying these novel reactions. The dermatologist’s skill set is necessary for optimizing skin-directed and targeted therapies while minimizing systemic immunosuppression, thereby improving patient outcomes with respect to rash, cancer response, and survival.
The Cost of Inpatient Skin Disease
Hospitalizations account for approximately half of all health care expenditures, and hospital readmission, seen as a measure of the quality of health care delivery, can double this cost.17 Identifying and developing protocols for addressing patients with complex chronic inflammatory disorders is one strategy for improving outcomes and reducing financial burden. Inpatient dermatologists have identified hidradenitis suppurativa as one disease that can benefit from early intervention by dermatologists in the hospital, with its 30-day (17.8%) and 180-day (48.6%) readmission rates being comparable to those of heart failure.18
Following an index emergency department (ED) visit, 17.2% (3484/20,269) of patients with HS have at least 1 return ED visit within 30 days, while only 2.4% (483/20,269) have a dermatology visit within the same time frame.19 Understanding the risk factors for hospital readmission and ED utilization, including severity of illness, the presence of medical comorbidities, health coverage under Medicaid, and receipt of opioids, can allow dermatologists to anticipate those at greatest risk.19 Opportunities exist for cross-specialty interventions to anticipate and address modifiable risk factors. Shorter time to dermatology outpatient follow-up leads to improved clinic attendance and may help reduce ED utilization and hospital readmission.20
Teledermatology: Leveraging Inpatient Expertise
Although the benefit of inpatient dermatologic care is substantial, access to that care is finite. Following the COVID-19 pandemic, there is an increased acceptance of telemedicine and the long-term role it can play in leveraging dermatologic expertise, including meeting the increasing demand for inpatient dermatology care in rural and resource-poor communities.21
Recent studies conducted by dermatology hospitalists have illustrated the value of asynchronous store-and-forward technology in settings lacking access to consultative dermatology.22,23 Stephens et al22 found that expanding provider-to-provider electronic consultation (e-consultation) capacity to an inpatient rehabilitation facility resulted in completed consultations within 1.5 days compared with a 7- to 14-day wait time for patients attending an in-person urgent access dermatology clinic. In another study, the implementation of asynchronous dermatology e-consultations for immunobullous diseases, vasculitis, and herpes zoster resulted in a change in diagnosis 86% of the time, accompanied by at least 1 new systemic or topical therapy recommendation.23
Researchers also identified ways in which teledermatology can be inelegant and proposed specific supplemental data to aid in diagnosis. A review of 126 inpatient e-consultations demonstrated limitations related to the diagnosis of skin and soft-tissue infections. In two-thirds to three-quarters of cases, potentially useful descriptive information was missing, and in 70% (88/126), images were not appropriately focused. The authors developed a detailed checklist to help primary medical teams focus their differential diagnoses.24 A recent pilot study found that supplementation of clinical information with a standardized questionnaire and thermal images improved the accuracy of cellulitis diagnosis. Using this method, there was no difference in accuracy between dermatology hospitalists and other board-certified dermatologists, supporting the notion that any dermatologist can fulfill this need successfully, even without specific inpatient experience.25 Due to the high incidence and cost of cellulitis and related hospital admissions,26 such an intervention could have a considerable financial and patient safety impact.
Final Thoughts
This last year brought many changes to the health care landscape, the recession of a global pandemic, and an increasingly complex health care delivery system. Inpatient dermatologists met these challenges by providing high-quality dermatologic care and practice-modifying research in the areas of severe cutaneous adverse reactions, supportive oncodermatology, hospital readmission, telemedicine, and more, demonstrating the value of dermatologic expertise in the hospital setting.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Puri P, Pollock BD, Yousif M, et al. Association of Society of Dermatology hospitalist institutions with improved outcomes in Medicare beneficiaries hospitalized for skin disease. J Am Acad Dermatol. 2023;88:1372-1375.
- Creadore A, Desai S, Alloo A, et al. Clinical characteristics, disease course, and outcomes of patients with acute generalized exanthematous pustulosis in the US. JAMA Dermatol. 2022;158:176-183.
- Sharma AN, Murphy K, Shwe S, et al. Predicting DRESS syndrome recurrence—the ReDRESS score. JAMA Dermatol. 2022;158:1445-1447.
- Brian M, Rose EK, Mauskar MM, et al. Sudden conjunctivitis, lymphopenia, and rash combined with hemodynamic changes (SCoRCH) after trimethoprim-sulfamethoxazole use: a case series study of a hypersensitivity reaction. JAMA Dermatol. 2023;159:73-78.
- Lee EY, Knox C, Phillips EJ. Worldwide prevalence of antibiotic-associated Stevens-Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. JAMA Dermatol. 2023;159:384-392.
- Liew YCC, Choo KJL, Oh CC, et al. Mycoplasma-induced Stevens-Johnson syndrome/toxic epidermal necrolysis: case-control analysis of a cohort managed in a specialized center. J Am Acad Dermatol. 2022;86:811-817.
- Ao S, Gao X, Zhan J, et al. Inhibition of tumor necrosis factor improves conventional steroid therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis in a cohort of patients. J Am Acad Dermatol. 2022;86:1236-1245.
- Wang CW, Yang LY, Chen CB, et al; the Taiwan Severe Cutaneous Adverse Reaction (TSCAR) Consortium. Randomized, controlled trial of TNF-α antagonist in CTL-mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Han JJ, Creadore A, Seminario-Vidal L, et al. Medical management of Stevens-Johnson syndrome/toxic epidermal necrolysis among North American dermatologists. J Am Acad Dermatol. 2022;87:429-431.
- Noe MH, Micheletti RG. Systemic interventions for treatment of Stevens-Johnson syndrome/toxic epidermal necrolysis: summary of a Cochrane review. JAMA Dermatol. 2022;158:1436-1437.
- Waters M, Dobry A, Le ST, et al. Development of a skin-directed scoring system for Stevens-Johnson syndrome and epidermal necrolysis: a Delphi consensus exercise. JAMA Dermatol. 2023;159:772-777.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Said JT, Elman SA, Perez-Chada LM, et al. Treatment of immune checkpoint inhibitor-mediated psoriasis: a systematic review. J Am Acad Dermatol. 2022;87:399-400.
- Asdourian MS, Shah N, Jacoby TV, et al. Evaluating patterns of co-occurrence between cutaneous and noncutaneous immune-related adverse events after immune checkpoint inhibitor therapy. J Am Acad Dermatol. 2023;88:246-249.
- Hirotsu KE, Scott MKD, Marquez C, et al. Histologic subtype of cutaneous immune-related adverse events predicts overall survival in patients receiving immune checkpoint inhibitors. J Am Acad Dermatol. 2022;87:651-653.
- Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160:1074-1081.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192.
- Wang CX, Buss JL, Keller M, et al. Factors associated with dermatologic follow-up vs emergency department return in patients with hidradenitis suppurativa after an initial emergency department visit. JAMA Dermatol. 2022;158:1378-1386.
- Zakaria A, Chang AY, Kim-Lim P, et al. Predictors of postdischarge follow-up attendance among hospitalized dermatology patients: disparities and potential interventions. J Am Acad Dermatol. 2022;87:186-188.
- Arnold JD, Yoon S, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2019;80:425-432. doi:10.1016/j.jaad.2018.06.070
- Stephens MR, Das S, Smith GP. Utilization and outcomes of an asynchronous teledermatology pilot for an inpatient rehabilitation hospital. J Am Acad Dermatol. 2022;87:421-423.
- Ortiz C, Khosravi H, Kettering C, et al. Concordance data for inpatient asynchronous eDermatology consultation for immunobullous disease, zoster, and vasculitis. J Am Acad Dermatol. 2022;86:918-920.
- Salle R, Hua C, Mongereau M, et al. Challenges and limitations of teledermatology for skin and soft-tissue infections: a real-world study of an expert center. J Am Acad Dermatol. 2023;88:457-459.
- Creadore A, Manjaly P, Tkachenko E, et al. The utility of augmented teledermatology to improve dermatologist diagnosis of cellulitis: a cross-sectional study. Arch Dermatol Res. 2023;315:1347-1353.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Puri P, Pollock BD, Yousif M, et al. Association of Society of Dermatology hospitalist institutions with improved outcomes in Medicare beneficiaries hospitalized for skin disease. J Am Acad Dermatol. 2023;88:1372-1375.
- Creadore A, Desai S, Alloo A, et al. Clinical characteristics, disease course, and outcomes of patients with acute generalized exanthematous pustulosis in the US. JAMA Dermatol. 2022;158:176-183.
- Sharma AN, Murphy K, Shwe S, et al. Predicting DRESS syndrome recurrence—the ReDRESS score. JAMA Dermatol. 2022;158:1445-1447.
- Brian M, Rose EK, Mauskar MM, et al. Sudden conjunctivitis, lymphopenia, and rash combined with hemodynamic changes (SCoRCH) after trimethoprim-sulfamethoxazole use: a case series study of a hypersensitivity reaction. JAMA Dermatol. 2023;159:73-78.
- Lee EY, Knox C, Phillips EJ. Worldwide prevalence of antibiotic-associated Stevens-Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. JAMA Dermatol. 2023;159:384-392.
- Liew YCC, Choo KJL, Oh CC, et al. Mycoplasma-induced Stevens-Johnson syndrome/toxic epidermal necrolysis: case-control analysis of a cohort managed in a specialized center. J Am Acad Dermatol. 2022;86:811-817.
- Ao S, Gao X, Zhan J, et al. Inhibition of tumor necrosis factor improves conventional steroid therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis in a cohort of patients. J Am Acad Dermatol. 2022;86:1236-1245.
- Wang CW, Yang LY, Chen CB, et al; the Taiwan Severe Cutaneous Adverse Reaction (TSCAR) Consortium. Randomized, controlled trial of TNF-α antagonist in CTL-mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Han JJ, Creadore A, Seminario-Vidal L, et al. Medical management of Stevens-Johnson syndrome/toxic epidermal necrolysis among North American dermatologists. J Am Acad Dermatol. 2022;87:429-431.
- Noe MH, Micheletti RG. Systemic interventions for treatment of Stevens-Johnson syndrome/toxic epidermal necrolysis: summary of a Cochrane review. JAMA Dermatol. 2022;158:1436-1437.
- Waters M, Dobry A, Le ST, et al. Development of a skin-directed scoring system for Stevens-Johnson syndrome and epidermal necrolysis: a Delphi consensus exercise. JAMA Dermatol. 2023;159:772-777.
- Jacoby TV, Shah N, Asdourian MS, et al. Dermatology evaluation for cutaneous immune-related adverse events is associated with improved survival in cancer patients treated with checkpoint inhibition. J Am Acad Dermatol. 2023;88:711-714.
- Said JT, Elman SA, Perez-Chada LM, et al. Treatment of immune checkpoint inhibitor-mediated psoriasis: a systematic review. J Am Acad Dermatol. 2022;87:399-400.
- Asdourian MS, Shah N, Jacoby TV, et al. Evaluating patterns of co-occurrence between cutaneous and noncutaneous immune-related adverse events after immune checkpoint inhibitor therapy. J Am Acad Dermatol. 2023;88:246-249.
- Hirotsu KE, Scott MKD, Marquez C, et al. Histologic subtype of cutaneous immune-related adverse events predicts overall survival in patients receiving immune checkpoint inhibitors. J Am Acad Dermatol. 2022;87:651-653.
- Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160:1074-1081.
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192.
- Wang CX, Buss JL, Keller M, et al. Factors associated with dermatologic follow-up vs emergency department return in patients with hidradenitis suppurativa after an initial emergency department visit. JAMA Dermatol. 2022;158:1378-1386.
- Zakaria A, Chang AY, Kim-Lim P, et al. Predictors of postdischarge follow-up attendance among hospitalized dermatology patients: disparities and potential interventions. J Am Acad Dermatol. 2022;87:186-188.
- Arnold JD, Yoon S, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2019;80:425-432. doi:10.1016/j.jaad.2018.06.070
- Stephens MR, Das S, Smith GP. Utilization and outcomes of an asynchronous teledermatology pilot for an inpatient rehabilitation hospital. J Am Acad Dermatol. 2022;87:421-423.
- Ortiz C, Khosravi H, Kettering C, et al. Concordance data for inpatient asynchronous eDermatology consultation for immunobullous disease, zoster, and vasculitis. J Am Acad Dermatol. 2022;86:918-920.
- Salle R, Hua C, Mongereau M, et al. Challenges and limitations of teledermatology for skin and soft-tissue infections: a real-world study of an expert center. J Am Acad Dermatol. 2023;88:457-459.
- Creadore A, Manjaly P, Tkachenko E, et al. The utility of augmented teledermatology to improve dermatologist diagnosis of cellulitis: a cross-sectional study. Arch Dermatol Res. 2023;315:1347-1353.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis. JAMA Dermatol. 2017;153:141-146.
Practice Points
- A severe hypersensitivity reaction to trimethoprim-sulfamethoxazole—sudden conjunctivitis, lymphopenia, sunburnlike rash, and hemodynamic changes (SCoRCH)—has been described.
- Patients experiencing cutaneous reactions to immune checkpoint inhibitors have improved progression-free and overall survival rates if evaluated by a dermatologist who can optimize skin-directed and targeted therapies.
- Interventions, including shorter time to dermatology outpatient follow-up, are needed to reduce emergency department utilization by patients with hidradenitis suppurativa.
- Asynchronous store-and-forward dermatology e-consultation is effective for immunobullous diseases, vasculitis, herpes zoster, and cellulitis, demonstrating the utility of teledermatology in the inpatient setting, particularly when standardized data capture tools are used.
Palliative Care: Utilization Patterns in Inpatient Dermatology
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).

Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
, acute pain management, recreation therapy (RT), or pastoral care consultations during hospitalization.")
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).
Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).
Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
Practice Points
- Although severe dermatologic disease negatively impacts patients’ quality of life, palliative care may be underutilized in this population.
- Palliative care should be an integral part of caring for patients who are admitted to the hospital with serious dermatologic illnesses.
Epidermal Growth Factor Receptor Inhibitor–Induced Symmetrical Drug-Related Intertriginous and Flexural Exanthema: Should You Discontinue the Offending Agent?
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.

Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.

Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.

Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.
Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.
Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.
Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
Epidermal growth factor receptor (EGFR) inhibitors cause numerous cutaneous adverse events (AEs), including papulopustular eruptions, paronychia, acral fissures, xerosis, alopecia, and trichomegaly.1 Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon type IV hypersensitivity reaction reported most commonly in association with β-lactam antibiotics and other medications.2 Treatment of SDRIFE generally involves withdrawing the inciting medication; however, in SDRIFE secondary to oncologic therapies, medication withdrawal may not be feasible or desirable. We present 2 cases of SDRIFE secondary to EGFR inhibitors in which treatment was continued alongside supportive skin-directed therapies. We also review the literature.
Case Reports
Patient 1—A 65-year-old man with stage IV non–small cell lung cancer presented to the dermatology clinic with an eruption of 2 months’ duration that began in the periumbilical area and spread to the perianal area within 2 weeks of starting treatment with lazertinib and amivantamab. Physical examination was notable for Common Terminology Criteria for Adverse Events (CTCAE) Grade 2 periumbilical erythema and erosions as well as symmetric red-brown patches with linear erosions in the gluteal cleft (Figure 1) and Grade 2 facial papulopustular rash. Herpes simplex virus polymerase chain reaction and bacterial culture were negative. A skin biopsy from the left buttock revealed dermal edema and a perivascular lymphocytic infiltrate compatible with SDRIFE. Triamcinolone ointment 0.1% twice daily was initiated, then uptitrated to betamethasone ointment 0.05% twice daily with moderate improvement. The patient had a treatment interruption due to malignancy complications, at which time his skin improved, with recurrence of the eruption after treatment re-initiation. He resumed skin-directed treatment and was maintained on betamethasone ointment 0.05% and tacrolimus ointment 0.1% twice daily on alternating days. This treatment was continued for 4 months before the patient died from complications of the malignancy.
Patient 2—A 68-year-old woman with stage IV lung adenocarcinoma presented to the dermatology clinic with a rash of 3 weeks’ duration. Treatment with osimertinib was initiated 8 months prior to presentation, and there were no recent medication changes. Physical examination revealed CTCAE Grade 2 erythematous patches in the inguinal folds (Figure 2A), inframammary folds (Figure 2B), and on the nasal tip, as well as Grade 2 paronychia. The patient was managed with hydrocortisone cream 1% twice daily, and osimertinib was continued. At follow-up 4 weeks later, the erythema had faded to hyperpigmentation in affected areas with resolution of symptoms. No further treatment was required.
Comment
Supportive oncodermatologists and dermatology hospitalists should be aware of SDRIFE as an uncommon but increasingly recognized cutaneous AE of EGFR inhibitors. Other cases of SDRIFE secondary to EGFR inhibition are described in the Table.2-5 Although SDRIFE typically is treated by discontinuation of the offending agent, in all reported cases of EGFR inhibitor–associated SDRIFE the rash was CTCAE Grade 2, meaning that it did not interfere with instrumental activities of daily living. In 5 of 6 cases, EGFR therapy was continued while skin-directed therapies were used for symptom management.
Presentation of SDRIFE—Symmetrical drug-related intertriginous and flexural exanthema is characterized by a symmetric, sharply demarcated erythema in the inguinal, gluteal, or perianal area with at least 1 other flexural localization involved in the absence of systemic signs. It is observed most frequently at initial exposure or re-exposure to a medication. Onset typically is within a few hours to a few days after exposure to a medication.6 Interestingly, in this case series, half of reported SDRIFE cases developed 8 months or more after EGFR inhibitor initiation.
Pathophysiology of SDRIFE—The mechanism of SDRIFE has not been clearly elucidated; it generally is accepted to be a delayed-type hypersensitivity drug reaction, though other proposed pathophysiologic mechanisms for the distribution of SDRIFE include recall phenomenon or predisposing anatomic factors such as temperature, humidity, and apocrine or eccrine gland density.6,7 Epidermal growth factor receptor plays a critical role in regulating differentiation and proliferation of epidermal keratinocytes, hair follicles, and the sweat gland apparatus. Additionally, it has been hypothesized that EGFR inhibitor use may affect the microflora of the skin and that EGFR inhibitors directly affect the immune system, as demonstrated in an experiment showing EGFR inhibitor–treated mice had enhanced skin inflammation and contact hypersensitivity responses.8 How these disparate mechanisms may interact to produce SDRIFE and the reason for the notably delayed presentation of SDRIFE in half of the cases we reviewed is not known. Other delayed cutaneous AEs of EGFR inhibitor therapy, such as paronychia, are thought to be secondary to development of skin fragility and decreased keratinocyte proliferation with secondary infection.1 It is conceivable that a combination of proliferative, immunologic, and microbiome-related factors may each be playing a role in EGFR inhibitor–related SDRIFE.
Dermatology Inpatient Considerations—As seen in our cases, dermatologists can play a valuable role in diagnosing, grading, and managing cutaneous AEs associated with the administration of oncologic therapies. The array of cutaneous AEs has grown as cancer treatment options have expanded from conventional antimetabolite agents to kinase inhibitors and immune checkpoint inhibitors. Dermatologists may play an important role in differentiating the etiology of a skin finding (eg, infectious vs inflammatory) and can identify serious or dose-limiting reactions, such as Stevens-Johnson syndrome or drug reaction with eosinophilia and systemic symptoms (DRESS). If cutaneous AEs appear to occur secondary to administration of a chemotherapeutic agent, use of the National Cancer Institute CTCAE should be employed. For certain AEs (eg, alopecia, acneiform rashes, bullous dermatitis), specific grading has been developed based on a combination of body surface area involved, psychosocial impact, symptoms, and other associated morbidity.9
In management of chemotherapy-associated cutaneous AEs, dermatologists are likely to be the members of the health care team most comfortable with prescribing high-potency anti-inflammatory topical medications. Dermatologic consultation for management of cutaneous AEs has been shown to both reduce the need for systemic immunosuppression and limit interruptions in oncologic treatment.10
Conclusion
Epidermal growth factor receptor inhibitors commonly are prescribed for colorectal cancer, non–small cell lung cancer, and squamous cell carcinoma of the head and neck. They are associated with a variety of cutaneous AEs, including acneiform eruptions, paronychia, and xerosis, which rarely necessitate stopping EGFR inhibitor therapy. Our cases support an approach to managing EGFR inhibitor–related SDRIFE that does not involve discontinuation of the offending agent. Further studies are needed on the best supportive topical and systemic regimens for EGFR inhibitor–associated SDRIFE.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol. 2007;56:317-326.
- Coppola R, Santo B, Silipigni S, et al. Symmetrical drug-related intertriginous and flexural exanthema and acneiform eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:331-332.
- Yalici-Armagan B, Ayanoglu BT, Demirdag HG. Targeted tumour therapy induced papulopustular rash and other dermatologic side effects: a retrospective study. Cutan Ocul Toxicol. 2019;38:261-266.
- Copps B, Lacroix JP, Sasseville D. Symmetrical drug-related intertriginous and flexural exanthema secondary to epidermal growth factor receptor inhibitor gefitinib. JAAD Case Rep. 2020;6:172-175.
- Coppola R, Santo B, Ramella S, et al. Novel skin toxicity of epidermal growth factor receptor inhibitors: a case of intertrigo-like eruption in a patient with metastatic colorectal cancer treated with cetuximab. Clin Cancer Investig J. 2021;10:91-92.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Mascia F, Mariani V, Girolomoni G, et al. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303-312.
- National Cancer Institute (U.S.). Common Terminology Criteria for Adverse Events: (CTCAE), Version 5.0. US Department of Health and Human Services; 2017. Accessed December 16, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf
- Chen ST, Molina GE, Lo JA, et al. Dermatology consultation reduces interruption of oncologic management among hospitalized patients with immune-related adverse events: a retrospective cohort study. J Am Acad Dermatol. 2020;82:994-996.
Practice Points
- Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) is an uncommon but increasingly recognized cutaneous adverse event (AE) of epidermal growth factor receptor (EGFR) inhibitors.
- Epidermal growth factor receptor inhibitor–associated SDRIFE may be approached similarly to other EGFR inhibitor–related cutaneous AEs in that it may not require discontinuation of the offending agent.
