User login
Sodium vs Potassium for Lowering Blood Pressure?
A pair of dueling editorials in the journal Hypertension debate whether our focus should be on sodium or its often neglected partner, potassium.
A meta-analysis of 85 trials showed a consistent and linear. It may also depend on where you live and whether your concern is treating individuals or implementing effective food policy.
The Case for Sodium Restriction
Stephen Juraschek, MD, PhD, of the Beth Israel Deaconess Medical Center, Boston, Massachusetts, co-author of one editorial, told me in a zoom interview that he believes his side of the debate clearly has the stronger argument. Of the two cations in question, there has been infinitely more ink spilled about sodium.
Studies such as INTERSALT, the DASH diet, and TOHP may be the most well-known, but there are many, many intervention studies of sodium restriction’s effect on blood pressure. A meta-analysis of 85 trials of showed a consistent and linear relationship between sodium reduction and blood pressure. In contrast, the evidence base for potassium is more limited and less consistent. There are half as many trials with potassium, and its ability to lower blood pressure may depend on how much sodium is present in the diet.
An outlier in the sodium restriction evidence base is the PURE study, which suggested that extreme sodium restriction could increase cardiovascular mortality, but the trial suffered from two potential issues. First, it used a single spot urine specimen to measure sodium rather than the generally accepted more accurate 24-hour urine collection. A reanalysis of the TOHP study using a spot urine rather than a 24-hour urine collection changed the relationship between sodium intake and mortality and possibly explained the U-shaped association observed in PURE. Second, PURE was an observational cohort and was prone to confounding, or in this case, reverse causation. Why did people who consumed very little salt have an increased risk for cardiovascular disease? It is very possible that people with a high risk for cardiovascular disease were told to consume less salt to begin with. Hence B led to A rather than A leading to B.
The debate on sodium restriction has been bitter at times. Opposing camps formed, and people took sides in the “salt wars.” A group of researchers, termed the Jackson 6, met and decided to end the controversy by running a randomized trial in US prisons (having discounted the options of long-term care homes and military bases). They detailed their plan in an editorial in Hypertension. The study never came to fruition for two reasons: the obvious ethical problems of experimenting on prisoners and the revelation of undisclosed salt industry funding.
More recent studies have mercifully been more conventional. The SSaSS study, a randomized controlled trial of a salt substitute, provided the cardiovascular outcomes data that many were waiting for. And CARDIA-SSBP, a cross-over randomized trial recently presented at the American Heart Association meeting, showed that reducing dietary sodium was on par with medication when it came to lowering blood pressure.
For Dr. Juraschek, the evidence is clear: “If you were going to choose one, I would say the weight of the evidence is still really heavily on the sodium side.”
The Case for Potassium Supplementation
The evidence for salt restriction notwithstanding, Swapnil Hiremath, MD, MPH, from the University of Ottawa, Ontario, Canada, argued in his editorial that potassium supplementation has gotten short shrift. Though he admits the studies for potassium supplementation have been smaller and sometimes rely on observational evidence, the evidence is there. In the distal convoluted tubule, the sodium chloride cotransporter (NCC), aka the potassium switch, is turned on by low potassium levels and leads to sodium reabsorption by the kidney even in settings of high sodium intake (Figure). To nonnephrologists, renal physiology may be a black box. But if you quickly brush up on the mechanism of action of thiazide diuretics, the preceding descriptor will make more sense.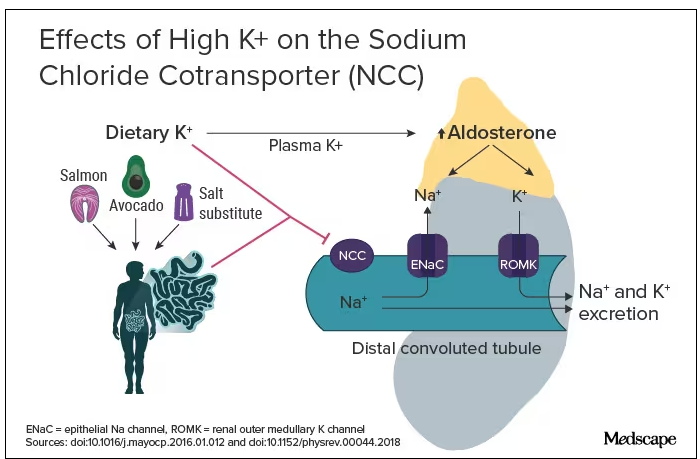
Dr. Hiremath points out that the DASH diet study also got patients to increase their potassium intake by eating more fruits and vegetables. Furthermore, the SSaSS study tested a salt substitute that was 25% potassium (and 75% sodium).
How much blood pressure lowering is due to sodium restriction vs potassium supplementation is a complex question because lowering sodium intake will invariably lead to more potassium intake. “It’s very hard to untangle the relationship,” Dr. Hiremath said in an interview. “It’s sort of synergistic but it’s not completely additive. It’s not as if you add four and four and get eight.” But he maintains there is more evidence regarding the benefit of potassium supplementation than many realize.
Realistic Diets and Taste Issues
“We know that increasing potassium, decreasing sodium is useful. The question is how do we do that?” says Dr. Hiremath. Should we encourage fruit and vegetable consumption in a healthy diet, give potassium supplements, or encourage the use of low-sodium salt substitutes?
Recommending a healthier diet with more fruits and vegetables is a no-brainer. But getting people to do it is hard. In a world where fruit is more expensive than junk food is, economic realities may drive food choice regardless of our best efforts. The 4700 mg of potassium in the DASH eating plan is the equivalent of eleven bananas daily; although not impossible, it would require a substantive shift in eating patterns for most people.
Given that we prescribe iron, vitamin B12, calcium, and vitamin D to patients who need them, why not potassium tablets to help with blood pressure? Granted, there are concerns about inducing hyperkalemia. Also, why not just prescribe a proven anti-hypertensive, such as ramipril, which has the added benefit of helping with renal protection or cardiac remodeling? Dr. Hiremath points out that patients are far less reluctant to take dietary supplements. Medication is something you take when sick. A supplement is seen as “natural” and “healthy” and might be more attractive to people resistant to prescription meds.
Another drawback of oral potassium supplementation is taste. In a Consumer Reports taste test, potassium chloride fared poorly. It was bitter and had a metallic aftertaste. At least one tester wouldn’t ever consume it again. Potassium citrate is slightly more palpable.
Salt substitutes, like the 75:25 ratio of sodium to potassium used in SSaSS, may be as high as you can go for potassium in any low-sodium salt alternative. If you go any higher than that, the taste will just turn people off, suggests Dr. Hiremath.
But SsaSS, which was done in China, may not be relevant to North America. In China, most sodium is added during cooking at home, and the consumption of processed foods is low. For the typical North American, roughly three quarters of the sodium eaten is added to their food by someone else; only about 15% is added during cooking at home or at the dinner table. If you aren’t someone who cooks, buying a salt substitute is probably not going to have much impact.
Given that reality, Dr. Juraschek thinks we need to target the sodium in processed foods. “There’s just so much sodium in so many products,” he says. “When you think about public policy, it’s most expeditious for there to be more regulation about how much is added to our food supply vs trying to get people to consume eight to 12 servings of fruit.”
No Salt War Here
Despite their different editorial takes, Dr. Hiremath and Dr. Juraschek largely agree on the broad strokes of the problem. This isn’t X (or Twitter) after all. Potassium supplementation may be useful in some parts of the world but may not address the underlying problem in countries where processed foods are the source of most dietary sodium.
The CARDIA-SSBP trial showed that a very low–sodium diet had the same blood pressure–lowering effect as a first-line antihypertensive, but most people will not be able to limit themselves to 500 mg of dietary sodium per day. In CARDIA-SSBP, just as in DASH, participants were provided with meals from study kitchens. They were not just told to eat less salt, which would almost certainly have failed.
“We should aim for stuff that is practical and doable rather than aim for stuff that cannot be done,” according to Dr. Hiremath. Whether that should be salt substitutes or policy change may depend on which part of the planet you live on.
One recent positive change may herald the beginning of a policy change, at least in the United States. In March 2023, the US Food and Drug Administration proposed a rule change to allow salt substitutes to be labeled as salt. This would make it easier for food manufacturers to swap out sodium chloride for a low-sodium alternative and reduce the amount of sodium in the US diet without having a large impact on taste and consumer uptake. Both Dr. Hiremath and Dr. Juraschek agree that it may not be enough on its own but that it’s a start.
Christopher Labos is a cardiologist with a degree in epidemiology. He spends most of his time doing things that he doesn’t get paid for, like research, teaching, and podcasting. Occasionally, he finds time to practice cardiology to pay the rent. He realizes that half of his research findings will be disproved in 5 years; he just doesn’t know which half. He is a regular contributor to the Montreal Gazette, CJAD radio, and CTV television in Montreal, and is host of the award-winning podcast The Body of Evidence.
A version of this article appeared on Medscape.com.
A pair of dueling editorials in the journal Hypertension debate whether our focus should be on sodium or its often neglected partner, potassium.
A meta-analysis of 85 trials showed a consistent and linear. It may also depend on where you live and whether your concern is treating individuals or implementing effective food policy.
The Case for Sodium Restriction
Stephen Juraschek, MD, PhD, of the Beth Israel Deaconess Medical Center, Boston, Massachusetts, co-author of one editorial, told me in a zoom interview that he believes his side of the debate clearly has the stronger argument. Of the two cations in question, there has been infinitely more ink spilled about sodium.
Studies such as INTERSALT, the DASH diet, and TOHP may be the most well-known, but there are many, many intervention studies of sodium restriction’s effect on blood pressure. A meta-analysis of 85 trials of showed a consistent and linear relationship between sodium reduction and blood pressure. In contrast, the evidence base for potassium is more limited and less consistent. There are half as many trials with potassium, and its ability to lower blood pressure may depend on how much sodium is present in the diet.
An outlier in the sodium restriction evidence base is the PURE study, which suggested that extreme sodium restriction could increase cardiovascular mortality, but the trial suffered from two potential issues. First, it used a single spot urine specimen to measure sodium rather than the generally accepted more accurate 24-hour urine collection. A reanalysis of the TOHP study using a spot urine rather than a 24-hour urine collection changed the relationship between sodium intake and mortality and possibly explained the U-shaped association observed in PURE. Second, PURE was an observational cohort and was prone to confounding, or in this case, reverse causation. Why did people who consumed very little salt have an increased risk for cardiovascular disease? It is very possible that people with a high risk for cardiovascular disease were told to consume less salt to begin with. Hence B led to A rather than A leading to B.
The debate on sodium restriction has been bitter at times. Opposing camps formed, and people took sides in the “salt wars.” A group of researchers, termed the Jackson 6, met and decided to end the controversy by running a randomized trial in US prisons (having discounted the options of long-term care homes and military bases). They detailed their plan in an editorial in Hypertension. The study never came to fruition for two reasons: the obvious ethical problems of experimenting on prisoners and the revelation of undisclosed salt industry funding.
More recent studies have mercifully been more conventional. The SSaSS study, a randomized controlled trial of a salt substitute, provided the cardiovascular outcomes data that many were waiting for. And CARDIA-SSBP, a cross-over randomized trial recently presented at the American Heart Association meeting, showed that reducing dietary sodium was on par with medication when it came to lowering blood pressure.
For Dr. Juraschek, the evidence is clear: “If you were going to choose one, I would say the weight of the evidence is still really heavily on the sodium side.”
The Case for Potassium Supplementation
The evidence for salt restriction notwithstanding, Swapnil Hiremath, MD, MPH, from the University of Ottawa, Ontario, Canada, argued in his editorial that potassium supplementation has gotten short shrift. Though he admits the studies for potassium supplementation have been smaller and sometimes rely on observational evidence, the evidence is there. In the distal convoluted tubule, the sodium chloride cotransporter (NCC), aka the potassium switch, is turned on by low potassium levels and leads to sodium reabsorption by the kidney even in settings of high sodium intake (Figure). To nonnephrologists, renal physiology may be a black box. But if you quickly brush up on the mechanism of action of thiazide diuretics, the preceding descriptor will make more sense.
Dr. Hiremath points out that the DASH diet study also got patients to increase their potassium intake by eating more fruits and vegetables. Furthermore, the SSaSS study tested a salt substitute that was 25% potassium (and 75% sodium).
How much blood pressure lowering is due to sodium restriction vs potassium supplementation is a complex question because lowering sodium intake will invariably lead to more potassium intake. “It’s very hard to untangle the relationship,” Dr. Hiremath said in an interview. “It’s sort of synergistic but it’s not completely additive. It’s not as if you add four and four and get eight.” But he maintains there is more evidence regarding the benefit of potassium supplementation than many realize.
Realistic Diets and Taste Issues
“We know that increasing potassium, decreasing sodium is useful. The question is how do we do that?” says Dr. Hiremath. Should we encourage fruit and vegetable consumption in a healthy diet, give potassium supplements, or encourage the use of low-sodium salt substitutes?
Recommending a healthier diet with more fruits and vegetables is a no-brainer. But getting people to do it is hard. In a world where fruit is more expensive than junk food is, economic realities may drive food choice regardless of our best efforts. The 4700 mg of potassium in the DASH eating plan is the equivalent of eleven bananas daily; although not impossible, it would require a substantive shift in eating patterns for most people.
Given that we prescribe iron, vitamin B12, calcium, and vitamin D to patients who need them, why not potassium tablets to help with blood pressure? Granted, there are concerns about inducing hyperkalemia. Also, why not just prescribe a proven anti-hypertensive, such as ramipril, which has the added benefit of helping with renal protection or cardiac remodeling? Dr. Hiremath points out that patients are far less reluctant to take dietary supplements. Medication is something you take when sick. A supplement is seen as “natural” and “healthy” and might be more attractive to people resistant to prescription meds.
Another drawback of oral potassium supplementation is taste. In a Consumer Reports taste test, potassium chloride fared poorly. It was bitter and had a metallic aftertaste. At least one tester wouldn’t ever consume it again. Potassium citrate is slightly more palpable.
Salt substitutes, like the 75:25 ratio of sodium to potassium used in SSaSS, may be as high as you can go for potassium in any low-sodium salt alternative. If you go any higher than that, the taste will just turn people off, suggests Dr. Hiremath.
But SsaSS, which was done in China, may not be relevant to North America. In China, most sodium is added during cooking at home, and the consumption of processed foods is low. For the typical North American, roughly three quarters of the sodium eaten is added to their food by someone else; only about 15% is added during cooking at home or at the dinner table. If you aren’t someone who cooks, buying a salt substitute is probably not going to have much impact.
Given that reality, Dr. Juraschek thinks we need to target the sodium in processed foods. “There’s just so much sodium in so many products,” he says. “When you think about public policy, it’s most expeditious for there to be more regulation about how much is added to our food supply vs trying to get people to consume eight to 12 servings of fruit.”
No Salt War Here
Despite their different editorial takes, Dr. Hiremath and Dr. Juraschek largely agree on the broad strokes of the problem. This isn’t X (or Twitter) after all. Potassium supplementation may be useful in some parts of the world but may not address the underlying problem in countries where processed foods are the source of most dietary sodium.
The CARDIA-SSBP trial showed that a very low–sodium diet had the same blood pressure–lowering effect as a first-line antihypertensive, but most people will not be able to limit themselves to 500 mg of dietary sodium per day. In CARDIA-SSBP, just as in DASH, participants were provided with meals from study kitchens. They were not just told to eat less salt, which would almost certainly have failed.
“We should aim for stuff that is practical and doable rather than aim for stuff that cannot be done,” according to Dr. Hiremath. Whether that should be salt substitutes or policy change may depend on which part of the planet you live on.
One recent positive change may herald the beginning of a policy change, at least in the United States. In March 2023, the US Food and Drug Administration proposed a rule change to allow salt substitutes to be labeled as salt. This would make it easier for food manufacturers to swap out sodium chloride for a low-sodium alternative and reduce the amount of sodium in the US diet without having a large impact on taste and consumer uptake. Both Dr. Hiremath and Dr. Juraschek agree that it may not be enough on its own but that it’s a start.
Christopher Labos is a cardiologist with a degree in epidemiology. He spends most of his time doing things that he doesn’t get paid for, like research, teaching, and podcasting. Occasionally, he finds time to practice cardiology to pay the rent. He realizes that half of his research findings will be disproved in 5 years; he just doesn’t know which half. He is a regular contributor to the Montreal Gazette, CJAD radio, and CTV television in Montreal, and is host of the award-winning podcast The Body of Evidence.
A version of this article appeared on Medscape.com.
A pair of dueling editorials in the journal Hypertension debate whether our focus should be on sodium or its often neglected partner, potassium.
A meta-analysis of 85 trials showed a consistent and linear. It may also depend on where you live and whether your concern is treating individuals or implementing effective food policy.
The Case for Sodium Restriction
Stephen Juraschek, MD, PhD, of the Beth Israel Deaconess Medical Center, Boston, Massachusetts, co-author of one editorial, told me in a zoom interview that he believes his side of the debate clearly has the stronger argument. Of the two cations in question, there has been infinitely more ink spilled about sodium.
Studies such as INTERSALT, the DASH diet, and TOHP may be the most well-known, but there are many, many intervention studies of sodium restriction’s effect on blood pressure. A meta-analysis of 85 trials of showed a consistent and linear relationship between sodium reduction and blood pressure. In contrast, the evidence base for potassium is more limited and less consistent. There are half as many trials with potassium, and its ability to lower blood pressure may depend on how much sodium is present in the diet.
An outlier in the sodium restriction evidence base is the PURE study, which suggested that extreme sodium restriction could increase cardiovascular mortality, but the trial suffered from two potential issues. First, it used a single spot urine specimen to measure sodium rather than the generally accepted more accurate 24-hour urine collection. A reanalysis of the TOHP study using a spot urine rather than a 24-hour urine collection changed the relationship between sodium intake and mortality and possibly explained the U-shaped association observed in PURE. Second, PURE was an observational cohort and was prone to confounding, or in this case, reverse causation. Why did people who consumed very little salt have an increased risk for cardiovascular disease? It is very possible that people with a high risk for cardiovascular disease were told to consume less salt to begin with. Hence B led to A rather than A leading to B.
The debate on sodium restriction has been bitter at times. Opposing camps formed, and people took sides in the “salt wars.” A group of researchers, termed the Jackson 6, met and decided to end the controversy by running a randomized trial in US prisons (having discounted the options of long-term care homes and military bases). They detailed their plan in an editorial in Hypertension. The study never came to fruition for two reasons: the obvious ethical problems of experimenting on prisoners and the revelation of undisclosed salt industry funding.
More recent studies have mercifully been more conventional. The SSaSS study, a randomized controlled trial of a salt substitute, provided the cardiovascular outcomes data that many were waiting for. And CARDIA-SSBP, a cross-over randomized trial recently presented at the American Heart Association meeting, showed that reducing dietary sodium was on par with medication when it came to lowering blood pressure.
For Dr. Juraschek, the evidence is clear: “If you were going to choose one, I would say the weight of the evidence is still really heavily on the sodium side.”
The Case for Potassium Supplementation
The evidence for salt restriction notwithstanding, Swapnil Hiremath, MD, MPH, from the University of Ottawa, Ontario, Canada, argued in his editorial that potassium supplementation has gotten short shrift. Though he admits the studies for potassium supplementation have been smaller and sometimes rely on observational evidence, the evidence is there. In the distal convoluted tubule, the sodium chloride cotransporter (NCC), aka the potassium switch, is turned on by low potassium levels and leads to sodium reabsorption by the kidney even in settings of high sodium intake (Figure). To nonnephrologists, renal physiology may be a black box. But if you quickly brush up on the mechanism of action of thiazide diuretics, the preceding descriptor will make more sense.
Dr. Hiremath points out that the DASH diet study also got patients to increase their potassium intake by eating more fruits and vegetables. Furthermore, the SSaSS study tested a salt substitute that was 25% potassium (and 75% sodium).
How much blood pressure lowering is due to sodium restriction vs potassium supplementation is a complex question because lowering sodium intake will invariably lead to more potassium intake. “It’s very hard to untangle the relationship,” Dr. Hiremath said in an interview. “It’s sort of synergistic but it’s not completely additive. It’s not as if you add four and four and get eight.” But he maintains there is more evidence regarding the benefit of potassium supplementation than many realize.
Realistic Diets and Taste Issues
“We know that increasing potassium, decreasing sodium is useful. The question is how do we do that?” says Dr. Hiremath. Should we encourage fruit and vegetable consumption in a healthy diet, give potassium supplements, or encourage the use of low-sodium salt substitutes?
Recommending a healthier diet with more fruits and vegetables is a no-brainer. But getting people to do it is hard. In a world where fruit is more expensive than junk food is, economic realities may drive food choice regardless of our best efforts. The 4700 mg of potassium in the DASH eating plan is the equivalent of eleven bananas daily; although not impossible, it would require a substantive shift in eating patterns for most people.
Given that we prescribe iron, vitamin B12, calcium, and vitamin D to patients who need them, why not potassium tablets to help with blood pressure? Granted, there are concerns about inducing hyperkalemia. Also, why not just prescribe a proven anti-hypertensive, such as ramipril, which has the added benefit of helping with renal protection or cardiac remodeling? Dr. Hiremath points out that patients are far less reluctant to take dietary supplements. Medication is something you take when sick. A supplement is seen as “natural” and “healthy” and might be more attractive to people resistant to prescription meds.
Another drawback of oral potassium supplementation is taste. In a Consumer Reports taste test, potassium chloride fared poorly. It was bitter and had a metallic aftertaste. At least one tester wouldn’t ever consume it again. Potassium citrate is slightly more palpable.
Salt substitutes, like the 75:25 ratio of sodium to potassium used in SSaSS, may be as high as you can go for potassium in any low-sodium salt alternative. If you go any higher than that, the taste will just turn people off, suggests Dr. Hiremath.
But SsaSS, which was done in China, may not be relevant to North America. In China, most sodium is added during cooking at home, and the consumption of processed foods is low. For the typical North American, roughly three quarters of the sodium eaten is added to their food by someone else; only about 15% is added during cooking at home or at the dinner table. If you aren’t someone who cooks, buying a salt substitute is probably not going to have much impact.
Given that reality, Dr. Juraschek thinks we need to target the sodium in processed foods. “There’s just so much sodium in so many products,” he says. “When you think about public policy, it’s most expeditious for there to be more regulation about how much is added to our food supply vs trying to get people to consume eight to 12 servings of fruit.”
No Salt War Here
Despite their different editorial takes, Dr. Hiremath and Dr. Juraschek largely agree on the broad strokes of the problem. This isn’t X (or Twitter) after all. Potassium supplementation may be useful in some parts of the world but may not address the underlying problem in countries where processed foods are the source of most dietary sodium.
The CARDIA-SSBP trial showed that a very low–sodium diet had the same blood pressure–lowering effect as a first-line antihypertensive, but most people will not be able to limit themselves to 500 mg of dietary sodium per day. In CARDIA-SSBP, just as in DASH, participants were provided with meals from study kitchens. They were not just told to eat less salt, which would almost certainly have failed.
“We should aim for stuff that is practical and doable rather than aim for stuff that cannot be done,” according to Dr. Hiremath. Whether that should be salt substitutes or policy change may depend on which part of the planet you live on.
One recent positive change may herald the beginning of a policy change, at least in the United States. In March 2023, the US Food and Drug Administration proposed a rule change to allow salt substitutes to be labeled as salt. This would make it easier for food manufacturers to swap out sodium chloride for a low-sodium alternative and reduce the amount of sodium in the US diet without having a large impact on taste and consumer uptake. Both Dr. Hiremath and Dr. Juraschek agree that it may not be enough on its own but that it’s a start.
Christopher Labos is a cardiologist with a degree in epidemiology. He spends most of his time doing things that he doesn’t get paid for, like research, teaching, and podcasting. Occasionally, he finds time to practice cardiology to pay the rent. He realizes that half of his research findings will be disproved in 5 years; he just doesn’t know which half. He is a regular contributor to the Montreal Gazette, CJAD radio, and CTV television in Montreal, and is host of the award-winning podcast The Body of Evidence.
A version of this article appeared on Medscape.com.
Colchicine May Benefit Patients With Diabetes and Recent MI
TOPLINE:
A daily low dose of colchicine significantly reduces ischemic cardiovascular events in patients with type 2 diabetes (T2D) and a recent myocardial infarction (MI).
METHODOLOGY:
- After an MI, patients with vs without T2D have a higher risk for another cardiovascular event.
- The Colchicine Cardiovascular Outcomes Trial (COLCOT), a randomized, double-blinded trial, found a lower risk for ischemic cardiovascular events with 0.5 mg colchicine taken daily vs placebo, initiated within 30 days of an MI.
- Researchers conducted a prespecified subgroup analysis of 959 adult patients with T2D (mean age, 62.4 years; 22.2% women) in COLCOT (462 patients in colchicine and 497 patients in placebo groups).
- The primary efficacy endpoint was a composite of cardiovascular death, resuscitated cardiac arrest, MI, stroke, or urgent hospitalization for angina requiring coronary revascularization within a median 23 months.
- The patients were taking a variety of appropriate medications, including aspirin and another antiplatelet agent and a statin (98%-99%) and metformin (75%-76%).
TAKEAWAY:
- The risk for the primary endpoint was reduced by 35% in patients with T2D who received colchicine than in those who received placebo (hazard ratio, 0.65; P = .03).
- The primary endpoint event rate per 100 patient-months was significantly lower in the colchicine group than in the placebo group (rate ratio, 0.53; P = .01).
- The frequencies of adverse events were similar in both the treatment and placebo groups (14.6% and 12.8%, respectively; P = .41), with gastrointestinal adverse events being the most common.
- In COLCOT, patients with T2D had a 1.86-fold higher risk for a primary endpoint cardiovascular event, but there was no significant difference in the primary endpoint between those with and without T2D on colchicine.
IN PRACTICE:
“Patients with both T2D and a recent MI derive a large benefit from inflammation-reducing therapy with colchicine,” the authors noted.
SOURCE:
This study, led by François Roubille, University Hospital of Montpellier, France, was published online on January 5, 2024, in Diabetes Care.
LIMITATIONS:
Patients were not stratified at inclusion for the presence of diabetes. Also, the study did not evaluate the role of glycated hemoglobin and low-density lipoprotein cholesterol, as well as the effects of different glucose-lowering medications or possible hypoglycemic episodes.
DISCLOSURES:
The COLCOT study was funded by the Government of Quebec, the Canadian Institutes of Health Research, and philanthropic foundations. Coauthors Jean-Claude Tardif and Wolfgang Koenig declared receiving research grants, honoraria, advisory board fees, and lecture fees from pharmaceutical companies, as well as having other ties with various sources.
A version of this article appeared on Medscape.com.
TOPLINE:
A daily low dose of colchicine significantly reduces ischemic cardiovascular events in patients with type 2 diabetes (T2D) and a recent myocardial infarction (MI).
METHODOLOGY:
- After an MI, patients with vs without T2D have a higher risk for another cardiovascular event.
- The Colchicine Cardiovascular Outcomes Trial (COLCOT), a randomized, double-blinded trial, found a lower risk for ischemic cardiovascular events with 0.5 mg colchicine taken daily vs placebo, initiated within 30 days of an MI.
- Researchers conducted a prespecified subgroup analysis of 959 adult patients with T2D (mean age, 62.4 years; 22.2% women) in COLCOT (462 patients in colchicine and 497 patients in placebo groups).
- The primary efficacy endpoint was a composite of cardiovascular death, resuscitated cardiac arrest, MI, stroke, or urgent hospitalization for angina requiring coronary revascularization within a median 23 months.
- The patients were taking a variety of appropriate medications, including aspirin and another antiplatelet agent and a statin (98%-99%) and metformin (75%-76%).
TAKEAWAY:
- The risk for the primary endpoint was reduced by 35% in patients with T2D who received colchicine than in those who received placebo (hazard ratio, 0.65; P = .03).
- The primary endpoint event rate per 100 patient-months was significantly lower in the colchicine group than in the placebo group (rate ratio, 0.53; P = .01).
- The frequencies of adverse events were similar in both the treatment and placebo groups (14.6% and 12.8%, respectively; P = .41), with gastrointestinal adverse events being the most common.
- In COLCOT, patients with T2D had a 1.86-fold higher risk for a primary endpoint cardiovascular event, but there was no significant difference in the primary endpoint between those with and without T2D on colchicine.
IN PRACTICE:
“Patients with both T2D and a recent MI derive a large benefit from inflammation-reducing therapy with colchicine,” the authors noted.
SOURCE:
This study, led by François Roubille, University Hospital of Montpellier, France, was published online on January 5, 2024, in Diabetes Care.
LIMITATIONS:
Patients were not stratified at inclusion for the presence of diabetes. Also, the study did not evaluate the role of glycated hemoglobin and low-density lipoprotein cholesterol, as well as the effects of different glucose-lowering medications or possible hypoglycemic episodes.
DISCLOSURES:
The COLCOT study was funded by the Government of Quebec, the Canadian Institutes of Health Research, and philanthropic foundations. Coauthors Jean-Claude Tardif and Wolfgang Koenig declared receiving research grants, honoraria, advisory board fees, and lecture fees from pharmaceutical companies, as well as having other ties with various sources.
A version of this article appeared on Medscape.com.
TOPLINE:
A daily low dose of colchicine significantly reduces ischemic cardiovascular events in patients with type 2 diabetes (T2D) and a recent myocardial infarction (MI).
METHODOLOGY:
- After an MI, patients with vs without T2D have a higher risk for another cardiovascular event.
- The Colchicine Cardiovascular Outcomes Trial (COLCOT), a randomized, double-blinded trial, found a lower risk for ischemic cardiovascular events with 0.5 mg colchicine taken daily vs placebo, initiated within 30 days of an MI.
- Researchers conducted a prespecified subgroup analysis of 959 adult patients with T2D (mean age, 62.4 years; 22.2% women) in COLCOT (462 patients in colchicine and 497 patients in placebo groups).
- The primary efficacy endpoint was a composite of cardiovascular death, resuscitated cardiac arrest, MI, stroke, or urgent hospitalization for angina requiring coronary revascularization within a median 23 months.
- The patients were taking a variety of appropriate medications, including aspirin and another antiplatelet agent and a statin (98%-99%) and metformin (75%-76%).
TAKEAWAY:
- The risk for the primary endpoint was reduced by 35% in patients with T2D who received colchicine than in those who received placebo (hazard ratio, 0.65; P = .03).
- The primary endpoint event rate per 100 patient-months was significantly lower in the colchicine group than in the placebo group (rate ratio, 0.53; P = .01).
- The frequencies of adverse events were similar in both the treatment and placebo groups (14.6% and 12.8%, respectively; P = .41), with gastrointestinal adverse events being the most common.
- In COLCOT, patients with T2D had a 1.86-fold higher risk for a primary endpoint cardiovascular event, but there was no significant difference in the primary endpoint between those with and without T2D on colchicine.
IN PRACTICE:
“Patients with both T2D and a recent MI derive a large benefit from inflammation-reducing therapy with colchicine,” the authors noted.
SOURCE:
This study, led by François Roubille, University Hospital of Montpellier, France, was published online on January 5, 2024, in Diabetes Care.
LIMITATIONS:
Patients were not stratified at inclusion for the presence of diabetes. Also, the study did not evaluate the role of glycated hemoglobin and low-density lipoprotein cholesterol, as well as the effects of different glucose-lowering medications or possible hypoglycemic episodes.
DISCLOSURES:
The COLCOT study was funded by the Government of Quebec, the Canadian Institutes of Health Research, and philanthropic foundations. Coauthors Jean-Claude Tardif and Wolfgang Koenig declared receiving research grants, honoraria, advisory board fees, and lecture fees from pharmaceutical companies, as well as having other ties with various sources.
A version of this article appeared on Medscape.com.
FDA Expands Dupilumab for EoE to Younger Children
The US Food and Drug Administration (FDA) has approved dupilumab (Dupixent, Regeneron/Sanofi) for the treatment of eosinophilic esophagitis (EoE) in children aged 1-11 years and weighing ≥ 15 kg. It is the first and only medicine approved to treat these patients.
, as reported by this news organization.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway.
EoE KIDS Trial
The FDA approval of dupilumab for younger children is based on results from the phase 3 randomized, double-blind, placebo-controlled EoE KIDS trial, which had two parts.
Part A was a 16-week double-blind treatment period that evaluated the safety and efficacy of dupilumab in a tiered weight-based dosing schema.
At 16 weeks, 66% of children who received higher dose dupilumab at tiered dosing regimens based on weight achieved histologic disease remission (six or fewer eosinophils/high power field), which was the primary endpoint, compared with only 3% of children who received placebo.
In addition, a greater decrease in the proportion of days with one or more signs of EoE according to the Pediatric EoE Sign/Symptom Questionnaire caregiver version (PESQ-C) was observed in children treated with dupilumab at 16 weeks compared placebo.
Part B was a 36-week extended active treatment period in which eligible children from Part A in the dupilumab group continued to receive their dose level and those in the placebo group in Part A switched to active treatment.
Histologic remission was sustained at week 52 in 53% of children treated with dupilumab in Parts A and B. Histologic remission was also achieved at week 52 in 53% of children who switched to dupilumab from placebo in Part B.
The safety profile of dupilumab observed through 16 weeks in these children was generally in line to that seen through 24 weeks in persons aged 12 years or older with EoE.
The most common adverse events (≥ 2%) more frequently observed with dupilumab than with placebo were injection site reactions, upper respiratory tract infections, arthralgia, and herpes viral infections. In EoE KIDS Part B, one case of helminth infection was reported in the dupilumab arm.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has approved dupilumab (Dupixent, Regeneron/Sanofi) for the treatment of eosinophilic esophagitis (EoE) in children aged 1-11 years and weighing ≥ 15 kg. It is the first and only medicine approved to treat these patients.
, as reported by this news organization.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway.
EoE KIDS Trial
The FDA approval of dupilumab for younger children is based on results from the phase 3 randomized, double-blind, placebo-controlled EoE KIDS trial, which had two parts.
Part A was a 16-week double-blind treatment period that evaluated the safety and efficacy of dupilumab in a tiered weight-based dosing schema.
At 16 weeks, 66% of children who received higher dose dupilumab at tiered dosing regimens based on weight achieved histologic disease remission (six or fewer eosinophils/high power field), which was the primary endpoint, compared with only 3% of children who received placebo.
In addition, a greater decrease in the proportion of days with one or more signs of EoE according to the Pediatric EoE Sign/Symptom Questionnaire caregiver version (PESQ-C) was observed in children treated with dupilumab at 16 weeks compared placebo.
Part B was a 36-week extended active treatment period in which eligible children from Part A in the dupilumab group continued to receive their dose level and those in the placebo group in Part A switched to active treatment.
Histologic remission was sustained at week 52 in 53% of children treated with dupilumab in Parts A and B. Histologic remission was also achieved at week 52 in 53% of children who switched to dupilumab from placebo in Part B.
The safety profile of dupilumab observed through 16 weeks in these children was generally in line to that seen through 24 weeks in persons aged 12 years or older with EoE.
The most common adverse events (≥ 2%) more frequently observed with dupilumab than with placebo were injection site reactions, upper respiratory tract infections, arthralgia, and herpes viral infections. In EoE KIDS Part B, one case of helminth infection was reported in the dupilumab arm.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has approved dupilumab (Dupixent, Regeneron/Sanofi) for the treatment of eosinophilic esophagitis (EoE) in children aged 1-11 years and weighing ≥ 15 kg. It is the first and only medicine approved to treat these patients.
, as reported by this news organization.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway.
EoE KIDS Trial
The FDA approval of dupilumab for younger children is based on results from the phase 3 randomized, double-blind, placebo-controlled EoE KIDS trial, which had two parts.
Part A was a 16-week double-blind treatment period that evaluated the safety and efficacy of dupilumab in a tiered weight-based dosing schema.
At 16 weeks, 66% of children who received higher dose dupilumab at tiered dosing regimens based on weight achieved histologic disease remission (six or fewer eosinophils/high power field), which was the primary endpoint, compared with only 3% of children who received placebo.
In addition, a greater decrease in the proportion of days with one or more signs of EoE according to the Pediatric EoE Sign/Symptom Questionnaire caregiver version (PESQ-C) was observed in children treated with dupilumab at 16 weeks compared placebo.
Part B was a 36-week extended active treatment period in which eligible children from Part A in the dupilumab group continued to receive their dose level and those in the placebo group in Part A switched to active treatment.
Histologic remission was sustained at week 52 in 53% of children treated with dupilumab in Parts A and B. Histologic remission was also achieved at week 52 in 53% of children who switched to dupilumab from placebo in Part B.
The safety profile of dupilumab observed through 16 weeks in these children was generally in line to that seen through 24 weeks in persons aged 12 years or older with EoE.
The most common adverse events (≥ 2%) more frequently observed with dupilumab than with placebo were injection site reactions, upper respiratory tract infections, arthralgia, and herpes viral infections. In EoE KIDS Part B, one case of helminth infection was reported in the dupilumab arm.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
Robitussin Cough Syrup Recalled Nationwide Due to Fungus Concerns
The company that makes Robitussin syrups did not specify which microorganisms may be in the products. The recall announcement from the global consumer health products company Haleon stated that the contamination could lead to fungal infections or the presence of fungi or yeasts in a person’s blood. So far, the company has not received any reports of people being sickened by the recalled products.
The recall applies to bottles of Robitussin Honey CF Max Day and Robitussin Honey CF Max Nighttime. Both varieties are for adults. Affected products were sold nationwide and have specific lot numbers printed at the bottom of the back of the bottles. Consumers can view the lot numbers on the FDA’s recall webpage.
People with weakened immune systems have a higher risk of life-threatening health problems due to the cough syrup, the company warned.
“In non-immunocompromised consumers, the population most likely to use the product, life-threatening infections are not likely to occur,” the recall notice from Haleon stated. “However, the occurrence of an infection that may necessitate medical intervention cannot be completely ruled out.”
People who have affected products should stop using them immediately. The company asked that anyone with the products email Haleon at mystory.us@haleon.com, or call the company at 800-245-1040 Monday through Friday from 8 a.m. to 6 p.m. Eastern time.
A version of this article appeared on WebMD.com.
The company that makes Robitussin syrups did not specify which microorganisms may be in the products. The recall announcement from the global consumer health products company Haleon stated that the contamination could lead to fungal infections or the presence of fungi or yeasts in a person’s blood. So far, the company has not received any reports of people being sickened by the recalled products.
The recall applies to bottles of Robitussin Honey CF Max Day and Robitussin Honey CF Max Nighttime. Both varieties are for adults. Affected products were sold nationwide and have specific lot numbers printed at the bottom of the back of the bottles. Consumers can view the lot numbers on the FDA’s recall webpage.
People with weakened immune systems have a higher risk of life-threatening health problems due to the cough syrup, the company warned.
“In non-immunocompromised consumers, the population most likely to use the product, life-threatening infections are not likely to occur,” the recall notice from Haleon stated. “However, the occurrence of an infection that may necessitate medical intervention cannot be completely ruled out.”
People who have affected products should stop using them immediately. The company asked that anyone with the products email Haleon at mystory.us@haleon.com, or call the company at 800-245-1040 Monday through Friday from 8 a.m. to 6 p.m. Eastern time.
A version of this article appeared on WebMD.com.
The company that makes Robitussin syrups did not specify which microorganisms may be in the products. The recall announcement from the global consumer health products company Haleon stated that the contamination could lead to fungal infections or the presence of fungi or yeasts in a person’s blood. So far, the company has not received any reports of people being sickened by the recalled products.
The recall applies to bottles of Robitussin Honey CF Max Day and Robitussin Honey CF Max Nighttime. Both varieties are for adults. Affected products were sold nationwide and have specific lot numbers printed at the bottom of the back of the bottles. Consumers can view the lot numbers on the FDA’s recall webpage.
People with weakened immune systems have a higher risk of life-threatening health problems due to the cough syrup, the company warned.
“In non-immunocompromised consumers, the population most likely to use the product, life-threatening infections are not likely to occur,” the recall notice from Haleon stated. “However, the occurrence of an infection that may necessitate medical intervention cannot be completely ruled out.”
People who have affected products should stop using them immediately. The company asked that anyone with the products email Haleon at mystory.us@haleon.com, or call the company at 800-245-1040 Monday through Friday from 8 a.m. to 6 p.m. Eastern time.
A version of this article appeared on WebMD.com.
The Breakthrough Drug Whose Full Promise Remains Unrealized
Celebrating a Decade of Sofosbuvir for Hepatitis C
Prior to 2013, the backbone of hepatitis C virus (HCV) therapy was pegylated interferon (PEG) in combination with ribavirin (RBV). This year-long therapy was associated with significant side effects and abysmal cure rates. Although efficacy improved with the addition of first-generation protease inhibitors, cure rates remained suboptimal and treatment side effects continued to be significant.
Clinicians and patients needed better options and looked to the drug pipeline with hope. However, even among the most optimistic, the idea that HCV therapy could evolve into an all-oral option seemed a relative pipe dream.
The Sofosbuvir Revolution Begins
The Liver Meeting held in 2013 changed everything.
Several presentations featured compelling data with sofosbuvir, a new polymerase inhibitor that, when combined with RBV, offered an all-oral option to patients with genotypes 2 and 3, as well as improved efficacy for patients with genotypes 1, 4, 5, and 6 when it was combined with 12 weeks of PEG/RBV.
However, the glass ceiling of HCV care was truly shattered with the randomized COSMOS trial, a late-breaker abstract that revealed 12-week functional cure rates in patients receiving sofosbuvir in combination with the protease inhibitor simeprevir.
This phase 2a trial in treatment-naive and -experienced genotype 1 patients with and without cirrhosis showed that an all-oral option was not only viable for the most common strain of HCV but was also safe and efficacious, even in difficult-to-treat populations.
On December 6, 2013, the US Food and Drug Administration (FDA) approved sofosbuvir for the treatment of HCV, ushering in a new era of therapy.
Guidelines quickly changed to advocate for both expansive HCV screening and generous treatment. Yet, as this more permissive approach was being recommended, the high price tag and large anticipated volume of those seeking prescriptions were setting off alarms. The drug cost triggered extensive restrictions based on degree of fibrosis, sobriety, and provider type in an effort to prevent immediate healthcare expenditures.
Given its high cost, rules restricting a patient to only one course of sofosbuvir-based therapy also surfaced. Although treatment with first-generation protease inhibitors carried a hefty price of $161,813.49 per sustained virologic response (SVR), compared with $66,000-$100,000 for 12 weeks of all-oral therapy, its uptake was low and limited by side effects and comorbid conditions. All-oral treatment appeared to have few medical barriers, leading payers to find ways to slow utilization. These restrictions are now gradually being eliminated.
Because of high SVR rates and few contraindications to therapy, most patients who gained access to treatment achieved cure. This included patients who had previously not responded to treatment and prioritized those with more advanced disease.
This quickly led to a significant shift in the population in need of treatment. Prior to 2013, many patients with HCV had advanced disease and did not respond to prior treatment options. After uptake of all-oral therapy, individuals in need were typically treatment naive without advanced disease.
This shift also added new psychosocial dimensions, as many of the newly infected individuals were struggling with active substance abuse. HCV treatment providers needed to change, with increasing recruitment of advanced practice providers, primary care physicians, and addiction medication specialists.
Progress, but Far From Reaching Targets
Fast-forward to 2023.
Ten years after FDA approval, 13.2 million individuals infected with HCV have been treated globally, 82% with sofosbuvir-based regimens and most in lower-middle-income countries. This is absolutely cause for celebration, but not complacency.
In 2016, the World Health Assembly adopted a resolution of elimination of viral hepatitis by 2030. The World Health Organization (WHO) defined elimination of HCV as 90% reduction in new cases of infection, 90% diagnosis of those infected, 80% of eligible individuals treated, and 65% reduction of deaths by 2030.
Despite all the success thus far, the CDA Foundation estimates that the WHO elimination targets will not be achieved until after the year 2050. They also note that in 2020, over 50 million individuals were infected with HCV, of which only 20% were diagnosed and 1% annually treated.
The HCV care cascade, by which the patient journeys from screening to cure, is complicated, and a one-size-fits-all solution is not possible. Reflex testing (an automatic transition to HCV polymerase chain reaction [PCR] testing in the lab for those who are HCV antibody positive) has significantly improved diagnosis. However, communicating these results and linking a patient to curative therapy remain significant obstacles.
Models and real-life experience show that multiple strategies can be successful. They include leveraging the electronic medical record, simplified treatment algorithms, test-and-treat strategies (screening high-risk populations with a point-of-care test that allows treatment initiation at the same visit), and co-localizing HCV screening and treatment with addiction services and relinkage programs (finding those who are already diagnosed and linking them to treatment).
In addition, focusing on populations at high risk for HCV infection — such as people who inject drugs, men who have sex with men, and incarcerated individuals — allows for better resource utilization.
Though daunting, HCV elimination is not impossible. There are several examples of success, including in the countries of Georgia and Iceland. Although, comparatively, the United States remains behind the curve, the White House has asked Congress for $11 billion to fund HCV elimination domestically.
As we await action at the national level, clinicians are reminded that there are several things we can do in caring for patients with HCV:
- A one-time HCV screening is recommended in all individuals aged 18 or older, including pregnant people with each pregnancy.
- HCV antibody testing with reflex to PCR should be used as the screening test.
- Pan-genotypic all-oral therapy is recommended for patients with HCV. Cure rates are greater than 95%, and there are few contraindications to treatment.
- Most people are eligible for simplified treatment algorithms that allow minimal on-treatment monitoring.
Without increased screening and linkage to curative therapy, we will not meet the WHO goals for HCV elimination.
Dr. Reau is chief of the hepatology section at Rush University Medical Center in Chicago and a regular contributor to this news organization. She serves as editor of Clinical Liver Disease, a multimedia review journal, and recently as a member of HCVGuidelines.org, a web-based resource from the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America, as well as educational chair of the AASLD hepatitis C special interest group. She continues to have an active role in the hepatology interest group of the World Gastroenterology Organisation and the American Liver Foundation at the regional and national levels. She disclosed ties with AbbVie, Gilead, Arbutus, Intercept, and Salix.
A version of this article appeared on Medscape.com.
Celebrating a Decade of Sofosbuvir for Hepatitis C
Celebrating a Decade of Sofosbuvir for Hepatitis C
Prior to 2013, the backbone of hepatitis C virus (HCV) therapy was pegylated interferon (PEG) in combination with ribavirin (RBV). This year-long therapy was associated with significant side effects and abysmal cure rates. Although efficacy improved with the addition of first-generation protease inhibitors, cure rates remained suboptimal and treatment side effects continued to be significant.
Clinicians and patients needed better options and looked to the drug pipeline with hope. However, even among the most optimistic, the idea that HCV therapy could evolve into an all-oral option seemed a relative pipe dream.
The Sofosbuvir Revolution Begins
The Liver Meeting held in 2013 changed everything.
Several presentations featured compelling data with sofosbuvir, a new polymerase inhibitor that, when combined with RBV, offered an all-oral option to patients with genotypes 2 and 3, as well as improved efficacy for patients with genotypes 1, 4, 5, and 6 when it was combined with 12 weeks of PEG/RBV.
However, the glass ceiling of HCV care was truly shattered with the randomized COSMOS trial, a late-breaker abstract that revealed 12-week functional cure rates in patients receiving sofosbuvir in combination with the protease inhibitor simeprevir.
This phase 2a trial in treatment-naive and -experienced genotype 1 patients with and without cirrhosis showed that an all-oral option was not only viable for the most common strain of HCV but was also safe and efficacious, even in difficult-to-treat populations.
On December 6, 2013, the US Food and Drug Administration (FDA) approved sofosbuvir for the treatment of HCV, ushering in a new era of therapy.
Guidelines quickly changed to advocate for both expansive HCV screening and generous treatment. Yet, as this more permissive approach was being recommended, the high price tag and large anticipated volume of those seeking prescriptions were setting off alarms. The drug cost triggered extensive restrictions based on degree of fibrosis, sobriety, and provider type in an effort to prevent immediate healthcare expenditures.
Given its high cost, rules restricting a patient to only one course of sofosbuvir-based therapy also surfaced. Although treatment with first-generation protease inhibitors carried a hefty price of $161,813.49 per sustained virologic response (SVR), compared with $66,000-$100,000 for 12 weeks of all-oral therapy, its uptake was low and limited by side effects and comorbid conditions. All-oral treatment appeared to have few medical barriers, leading payers to find ways to slow utilization. These restrictions are now gradually being eliminated.
Because of high SVR rates and few contraindications to therapy, most patients who gained access to treatment achieved cure. This included patients who had previously not responded to treatment and prioritized those with more advanced disease.
This quickly led to a significant shift in the population in need of treatment. Prior to 2013, many patients with HCV had advanced disease and did not respond to prior treatment options. After uptake of all-oral therapy, individuals in need were typically treatment naive without advanced disease.
This shift also added new psychosocial dimensions, as many of the newly infected individuals were struggling with active substance abuse. HCV treatment providers needed to change, with increasing recruitment of advanced practice providers, primary care physicians, and addiction medication specialists.
Progress, but Far From Reaching Targets
Fast-forward to 2023.
Ten years after FDA approval, 13.2 million individuals infected with HCV have been treated globally, 82% with sofosbuvir-based regimens and most in lower-middle-income countries. This is absolutely cause for celebration, but not complacency.
In 2016, the World Health Assembly adopted a resolution of elimination of viral hepatitis by 2030. The World Health Organization (WHO) defined elimination of HCV as 90% reduction in new cases of infection, 90% diagnosis of those infected, 80% of eligible individuals treated, and 65% reduction of deaths by 2030.
Despite all the success thus far, the CDA Foundation estimates that the WHO elimination targets will not be achieved until after the year 2050. They also note that in 2020, over 50 million individuals were infected with HCV, of which only 20% were diagnosed and 1% annually treated.
The HCV care cascade, by which the patient journeys from screening to cure, is complicated, and a one-size-fits-all solution is not possible. Reflex testing (an automatic transition to HCV polymerase chain reaction [PCR] testing in the lab for those who are HCV antibody positive) has significantly improved diagnosis. However, communicating these results and linking a patient to curative therapy remain significant obstacles.
Models and real-life experience show that multiple strategies can be successful. They include leveraging the electronic medical record, simplified treatment algorithms, test-and-treat strategies (screening high-risk populations with a point-of-care test that allows treatment initiation at the same visit), and co-localizing HCV screening and treatment with addiction services and relinkage programs (finding those who are already diagnosed and linking them to treatment).
In addition, focusing on populations at high risk for HCV infection — such as people who inject drugs, men who have sex with men, and incarcerated individuals — allows for better resource utilization.
Though daunting, HCV elimination is not impossible. There are several examples of success, including in the countries of Georgia and Iceland. Although, comparatively, the United States remains behind the curve, the White House has asked Congress for $11 billion to fund HCV elimination domestically.
As we await action at the national level, clinicians are reminded that there are several things we can do in caring for patients with HCV:
- A one-time HCV screening is recommended in all individuals aged 18 or older, including pregnant people with each pregnancy.
- HCV antibody testing with reflex to PCR should be used as the screening test.
- Pan-genotypic all-oral therapy is recommended for patients with HCV. Cure rates are greater than 95%, and there are few contraindications to treatment.
- Most people are eligible for simplified treatment algorithms that allow minimal on-treatment monitoring.
Without increased screening and linkage to curative therapy, we will not meet the WHO goals for HCV elimination.
Dr. Reau is chief of the hepatology section at Rush University Medical Center in Chicago and a regular contributor to this news organization. She serves as editor of Clinical Liver Disease, a multimedia review journal, and recently as a member of HCVGuidelines.org, a web-based resource from the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America, as well as educational chair of the AASLD hepatitis C special interest group. She continues to have an active role in the hepatology interest group of the World Gastroenterology Organisation and the American Liver Foundation at the regional and national levels. She disclosed ties with AbbVie, Gilead, Arbutus, Intercept, and Salix.
A version of this article appeared on Medscape.com.
Prior to 2013, the backbone of hepatitis C virus (HCV) therapy was pegylated interferon (PEG) in combination with ribavirin (RBV). This year-long therapy was associated with significant side effects and abysmal cure rates. Although efficacy improved with the addition of first-generation protease inhibitors, cure rates remained suboptimal and treatment side effects continued to be significant.
Clinicians and patients needed better options and looked to the drug pipeline with hope. However, even among the most optimistic, the idea that HCV therapy could evolve into an all-oral option seemed a relative pipe dream.
The Sofosbuvir Revolution Begins
The Liver Meeting held in 2013 changed everything.
Several presentations featured compelling data with sofosbuvir, a new polymerase inhibitor that, when combined with RBV, offered an all-oral option to patients with genotypes 2 and 3, as well as improved efficacy for patients with genotypes 1, 4, 5, and 6 when it was combined with 12 weeks of PEG/RBV.
However, the glass ceiling of HCV care was truly shattered with the randomized COSMOS trial, a late-breaker abstract that revealed 12-week functional cure rates in patients receiving sofosbuvir in combination with the protease inhibitor simeprevir.
This phase 2a trial in treatment-naive and -experienced genotype 1 patients with and without cirrhosis showed that an all-oral option was not only viable for the most common strain of HCV but was also safe and efficacious, even in difficult-to-treat populations.
On December 6, 2013, the US Food and Drug Administration (FDA) approved sofosbuvir for the treatment of HCV, ushering in a new era of therapy.
Guidelines quickly changed to advocate for both expansive HCV screening and generous treatment. Yet, as this more permissive approach was being recommended, the high price tag and large anticipated volume of those seeking prescriptions were setting off alarms. The drug cost triggered extensive restrictions based on degree of fibrosis, sobriety, and provider type in an effort to prevent immediate healthcare expenditures.
Given its high cost, rules restricting a patient to only one course of sofosbuvir-based therapy also surfaced. Although treatment with first-generation protease inhibitors carried a hefty price of $161,813.49 per sustained virologic response (SVR), compared with $66,000-$100,000 for 12 weeks of all-oral therapy, its uptake was low and limited by side effects and comorbid conditions. All-oral treatment appeared to have few medical barriers, leading payers to find ways to slow utilization. These restrictions are now gradually being eliminated.
Because of high SVR rates and few contraindications to therapy, most patients who gained access to treatment achieved cure. This included patients who had previously not responded to treatment and prioritized those with more advanced disease.
This quickly led to a significant shift in the population in need of treatment. Prior to 2013, many patients with HCV had advanced disease and did not respond to prior treatment options. After uptake of all-oral therapy, individuals in need were typically treatment naive without advanced disease.
This shift also added new psychosocial dimensions, as many of the newly infected individuals were struggling with active substance abuse. HCV treatment providers needed to change, with increasing recruitment of advanced practice providers, primary care physicians, and addiction medication specialists.
Progress, but Far From Reaching Targets
Fast-forward to 2023.
Ten years after FDA approval, 13.2 million individuals infected with HCV have been treated globally, 82% with sofosbuvir-based regimens and most in lower-middle-income countries. This is absolutely cause for celebration, but not complacency.
In 2016, the World Health Assembly adopted a resolution of elimination of viral hepatitis by 2030. The World Health Organization (WHO) defined elimination of HCV as 90% reduction in new cases of infection, 90% diagnosis of those infected, 80% of eligible individuals treated, and 65% reduction of deaths by 2030.
Despite all the success thus far, the CDA Foundation estimates that the WHO elimination targets will not be achieved until after the year 2050. They also note that in 2020, over 50 million individuals were infected with HCV, of which only 20% were diagnosed and 1% annually treated.
The HCV care cascade, by which the patient journeys from screening to cure, is complicated, and a one-size-fits-all solution is not possible. Reflex testing (an automatic transition to HCV polymerase chain reaction [PCR] testing in the lab for those who are HCV antibody positive) has significantly improved diagnosis. However, communicating these results and linking a patient to curative therapy remain significant obstacles.
Models and real-life experience show that multiple strategies can be successful. They include leveraging the electronic medical record, simplified treatment algorithms, test-and-treat strategies (screening high-risk populations with a point-of-care test that allows treatment initiation at the same visit), and co-localizing HCV screening and treatment with addiction services and relinkage programs (finding those who are already diagnosed and linking them to treatment).
In addition, focusing on populations at high risk for HCV infection — such as people who inject drugs, men who have sex with men, and incarcerated individuals — allows for better resource utilization.
Though daunting, HCV elimination is not impossible. There are several examples of success, including in the countries of Georgia and Iceland. Although, comparatively, the United States remains behind the curve, the White House has asked Congress for $11 billion to fund HCV elimination domestically.
As we await action at the national level, clinicians are reminded that there are several things we can do in caring for patients with HCV:
- A one-time HCV screening is recommended in all individuals aged 18 or older, including pregnant people with each pregnancy.
- HCV antibody testing with reflex to PCR should be used as the screening test.
- Pan-genotypic all-oral therapy is recommended for patients with HCV. Cure rates are greater than 95%, and there are few contraindications to treatment.
- Most people are eligible for simplified treatment algorithms that allow minimal on-treatment monitoring.
Without increased screening and linkage to curative therapy, we will not meet the WHO goals for HCV elimination.
Dr. Reau is chief of the hepatology section at Rush University Medical Center in Chicago and a regular contributor to this news organization. She serves as editor of Clinical Liver Disease, a multimedia review journal, and recently as a member of HCVGuidelines.org, a web-based resource from the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America, as well as educational chair of the AASLD hepatitis C special interest group. She continues to have an active role in the hepatology interest group of the World Gastroenterology Organisation and the American Liver Foundation at the regional and national levels. She disclosed ties with AbbVie, Gilead, Arbutus, Intercept, and Salix.
A version of this article appeared on Medscape.com.
Shelf Life for Opioid Overdose Drug Naloxone Extended
At the request of the US Food and Drug Administration (FDA), Emergent BioSolutions has extended the shelf life of the rapid opioid overdose reversal agent, naloxone (4 mg) nasal spray (Narcan), from 3 to 4 years.
Naloxone is “an important tool” in addressing opioid overdoses, and this extension supports the FDA’s “efforts to ensure more OTC naloxone products remain available to the public,” Marta Sokolowska, PhD, with the FDA Center for Drug Evaluation and Research, said in a statement.
Naloxone nasal spray was first approved by the FDA in 2015 as a prescription drug. Last spring, the agency approved the drug for over-the-counter use.
“The shelf life of products that were produced and distributed prior to this announcement is not affected and remains unchanged. Prescribers, patients, and caregivers are advised to continue to abide by the expiration date printed on each product’s packaging and within the product’s labeling,” the FDA advised.
“FDA’s request for this shelf-life extension is a testament to the agency’s continuing progress toward implementing the FDA Overdose Prevention Framework, which provides our vision to undertake impactful, creative actions to encourage harm reduction and innovation in reducing controlled substance-related overdoses and deaths,” the agency said.
According to the US Centers for Disease Control and Prevention, from 1999 to 2021, nearly 645,000 people died from an overdose involving any opioid, including prescription and illicit opioids.
A version of this article appeared on Medscape.com.
At the request of the US Food and Drug Administration (FDA), Emergent BioSolutions has extended the shelf life of the rapid opioid overdose reversal agent, naloxone (4 mg) nasal spray (Narcan), from 3 to 4 years.
Naloxone is “an important tool” in addressing opioid overdoses, and this extension supports the FDA’s “efforts to ensure more OTC naloxone products remain available to the public,” Marta Sokolowska, PhD, with the FDA Center for Drug Evaluation and Research, said in a statement.
Naloxone nasal spray was first approved by the FDA in 2015 as a prescription drug. Last spring, the agency approved the drug for over-the-counter use.
“The shelf life of products that were produced and distributed prior to this announcement is not affected and remains unchanged. Prescribers, patients, and caregivers are advised to continue to abide by the expiration date printed on each product’s packaging and within the product’s labeling,” the FDA advised.
“FDA’s request for this shelf-life extension is a testament to the agency’s continuing progress toward implementing the FDA Overdose Prevention Framework, which provides our vision to undertake impactful, creative actions to encourage harm reduction and innovation in reducing controlled substance-related overdoses and deaths,” the agency said.
According to the US Centers for Disease Control and Prevention, from 1999 to 2021, nearly 645,000 people died from an overdose involving any opioid, including prescription and illicit opioids.
A version of this article appeared on Medscape.com.
At the request of the US Food and Drug Administration (FDA), Emergent BioSolutions has extended the shelf life of the rapid opioid overdose reversal agent, naloxone (4 mg) nasal spray (Narcan), from 3 to 4 years.
Naloxone is “an important tool” in addressing opioid overdoses, and this extension supports the FDA’s “efforts to ensure more OTC naloxone products remain available to the public,” Marta Sokolowska, PhD, with the FDA Center for Drug Evaluation and Research, said in a statement.
Naloxone nasal spray was first approved by the FDA in 2015 as a prescription drug. Last spring, the agency approved the drug for over-the-counter use.
“The shelf life of products that were produced and distributed prior to this announcement is not affected and remains unchanged. Prescribers, patients, and caregivers are advised to continue to abide by the expiration date printed on each product’s packaging and within the product’s labeling,” the FDA advised.
“FDA’s request for this shelf-life extension is a testament to the agency’s continuing progress toward implementing the FDA Overdose Prevention Framework, which provides our vision to undertake impactful, creative actions to encourage harm reduction and innovation in reducing controlled substance-related overdoses and deaths,” the agency said.
According to the US Centers for Disease Control and Prevention, from 1999 to 2021, nearly 645,000 people died from an overdose involving any opioid, including prescription and illicit opioids.
A version of this article appeared on Medscape.com.
Corticosteroid Injections Don’t Move Blood Sugar for Most
TOPLINE:
Intra-articular corticosteroid (IACS) injections pose a minimal risk of accelerating diabetes for most people, despite temporarily elevating blood glucose levels, according to a study published in Clinical Diabetes.
METHODOLOGY:
- Almost half of Americans with diabetes have arthritis, so glycemic control is a concern for many receiving IACS injections.
- IACS injections are known to cause short-term hyperglycemia, but their long-term effects on glycemic control are not well studied.
- For the retrospective cohort study, researchers at Mayo Clinic in Rochester, Minnesota, used electronic health records from 1169 adults who had received an IACS injection in one large joint between 2012 and 2018.
- They analyzed data on A1C levels for study participants from 18 months before and after the injections.
- Researchers assessed if participants had a greater-than-expected (defined as an increase of more than 0.5% above expected) concentration of A1C after the injection, and examined rates of diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome in the 30 days following an injection.
TAKEAWAY:
- Nearly 16% of people experienced a greater-than-expected A1C level after receiving an injection.
- A1C levels rose by an average of 1.2% in the greater-than-expected group, but decreased by an average of 0.2% in the average group.
- One patient had an episode of severe hyperglycemia that was linked to the injection.
- A baseline level of A1C above 8% was the only factor associated with a greater-than-expected increase in the marker after an IACS injection.
IN PRACTICE:
“Although most patients do not experience an increase in A1C after IACS, clinicians should counsel patients with suboptimally controlled diabetes about risks of further hyperglycemia after IACS administration,” the researchers wrote.
SOURCE:
The study was led by Terin T. Sytsma, MD, of Mayo Clinic in Rochester, Minnesota.
LIMITATIONS:
The study was retrospective and could not establish causation. In addition, the population was of residents from one county in Minnesota, and was not racially or ethnically diverse. Details about the injection, such as location and total dose, were not available. The study also did not include a control group.
DISCLOSURES:
The study was funded by Mayo Clinic and the National Center for Advancing Translational Sciences. The authors reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
TOPLINE:
Intra-articular corticosteroid (IACS) injections pose a minimal risk of accelerating diabetes for most people, despite temporarily elevating blood glucose levels, according to a study published in Clinical Diabetes.
METHODOLOGY:
- Almost half of Americans with diabetes have arthritis, so glycemic control is a concern for many receiving IACS injections.
- IACS injections are known to cause short-term hyperglycemia, but their long-term effects on glycemic control are not well studied.
- For the retrospective cohort study, researchers at Mayo Clinic in Rochester, Minnesota, used electronic health records from 1169 adults who had received an IACS injection in one large joint between 2012 and 2018.
- They analyzed data on A1C levels for study participants from 18 months before and after the injections.
- Researchers assessed if participants had a greater-than-expected (defined as an increase of more than 0.5% above expected) concentration of A1C after the injection, and examined rates of diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome in the 30 days following an injection.
TAKEAWAY:
- Nearly 16% of people experienced a greater-than-expected A1C level after receiving an injection.
- A1C levels rose by an average of 1.2% in the greater-than-expected group, but decreased by an average of 0.2% in the average group.
- One patient had an episode of severe hyperglycemia that was linked to the injection.
- A baseline level of A1C above 8% was the only factor associated with a greater-than-expected increase in the marker after an IACS injection.
IN PRACTICE:
“Although most patients do not experience an increase in A1C after IACS, clinicians should counsel patients with suboptimally controlled diabetes about risks of further hyperglycemia after IACS administration,” the researchers wrote.
SOURCE:
The study was led by Terin T. Sytsma, MD, of Mayo Clinic in Rochester, Minnesota.
LIMITATIONS:
The study was retrospective and could not establish causation. In addition, the population was of residents from one county in Minnesota, and was not racially or ethnically diverse. Details about the injection, such as location and total dose, were not available. The study also did not include a control group.
DISCLOSURES:
The study was funded by Mayo Clinic and the National Center for Advancing Translational Sciences. The authors reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
TOPLINE:
Intra-articular corticosteroid (IACS) injections pose a minimal risk of accelerating diabetes for most people, despite temporarily elevating blood glucose levels, according to a study published in Clinical Diabetes.
METHODOLOGY:
- Almost half of Americans with diabetes have arthritis, so glycemic control is a concern for many receiving IACS injections.
- IACS injections are known to cause short-term hyperglycemia, but their long-term effects on glycemic control are not well studied.
- For the retrospective cohort study, researchers at Mayo Clinic in Rochester, Minnesota, used electronic health records from 1169 adults who had received an IACS injection in one large joint between 2012 and 2018.
- They analyzed data on A1C levels for study participants from 18 months before and after the injections.
- Researchers assessed if participants had a greater-than-expected (defined as an increase of more than 0.5% above expected) concentration of A1C after the injection, and examined rates of diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome in the 30 days following an injection.
TAKEAWAY:
- Nearly 16% of people experienced a greater-than-expected A1C level after receiving an injection.
- A1C levels rose by an average of 1.2% in the greater-than-expected group, but decreased by an average of 0.2% in the average group.
- One patient had an episode of severe hyperglycemia that was linked to the injection.
- A baseline level of A1C above 8% was the only factor associated with a greater-than-expected increase in the marker after an IACS injection.
IN PRACTICE:
“Although most patients do not experience an increase in A1C after IACS, clinicians should counsel patients with suboptimally controlled diabetes about risks of further hyperglycemia after IACS administration,” the researchers wrote.
SOURCE:
The study was led by Terin T. Sytsma, MD, of Mayo Clinic in Rochester, Minnesota.
LIMITATIONS:
The study was retrospective and could not establish causation. In addition, the population was of residents from one county in Minnesota, and was not racially or ethnically diverse. Details about the injection, such as location and total dose, were not available. The study also did not include a control group.
DISCLOSURES:
The study was funded by Mayo Clinic and the National Center for Advancing Translational Sciences. The authors reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
No Compelling Evidence of Pancreatic Cancer Risk With GLP-1s
TOPLINE:
New data provide no support for an increased risk for pancreatic cancer with use of a glucagon-like peptide-1 receptor agonist (GLP-1 RA) for up to 7 years, although longer-term data are needed, researchers said.
METHODOLOGY:
- Some studies have raised concern about a possible increased risk for pancreatitis and pancreatic cancer in patients taking a GLP-1 RA.
- Investigators behind this population-based cohort study assessed the association of GLP-1 RA treatment with pancreatic cancer incidence over a median of 7 years in 543,595 adults (mean age, 59.9 years; 51% women) with type 2 diabetes.
- Treatment with basal insulin was used as an active comparator.
- The analyses accounted for major confounding factors and time-related biases and adjusted for treatment with other glucose-lowering medications and a history of pancreatitis.
TAKEAWAY:
- During the study period, 33,377 patients (6.1%) used GLP-1 RAs and 106,849 (19.7%) used basal insulin, with 1665 of all patients diagnosed with pancreatic cancer.
- There was no evidence that GLP-1 RA use increased pancreatic cancer risk compared with basal insulin.
- The estimated hazard ratio (HR) for pancreatic cancer associated with incremental use of one defined daily dose per day of GLP-1 RA compared with basal insulin in years 5-7 was 0.50 (95% CI, 0.15-1.71).
- New-user and prevalent new-user analyses showed HRs from year 5 onward following initiation of a GLP-1 RA vs basal insulin was 0.52 (95% CI, 0.19-1.41) and 0.75 (95% CI, 0.37-1.53), respectively.
IN PRACTICE:
Using several analytical approaches, these findings do not suggest an increase in pancreatic cancer incidence over 7 years following the start of GLP-1 RA treatment, according to the investigation. “However, monitoring for pancreatic cancer risk beyond 7 years following initiation of treatment is still required,” the authors wrote.
SOURCE:
The study, with first author Rachel Dankner, MD, MPH, Gertner Institute for Epidemiology and Health Policy Research, Sheba Medical Center, Israel, was published online on January 4, 2024, in JAMA Network Open.
LIMITATIONS:
Data on the exact type of GLP-1 RA were not available. The analyses accounted for history of pancreatitis but not alcohol use or exposure to pesticides/chemicals. Because of the risk for bias due to reverse causation, an emphasis was placed on drug effects several years after their initiation. However, this reduced the number of pancreatic cancer cases available and led to estimated HRs with wider CIs.
DISCLOSURES:
The study received no specific funding. The authors disclosed no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
New data provide no support for an increased risk for pancreatic cancer with use of a glucagon-like peptide-1 receptor agonist (GLP-1 RA) for up to 7 years, although longer-term data are needed, researchers said.
METHODOLOGY:
- Some studies have raised concern about a possible increased risk for pancreatitis and pancreatic cancer in patients taking a GLP-1 RA.
- Investigators behind this population-based cohort study assessed the association of GLP-1 RA treatment with pancreatic cancer incidence over a median of 7 years in 543,595 adults (mean age, 59.9 years; 51% women) with type 2 diabetes.
- Treatment with basal insulin was used as an active comparator.
- The analyses accounted for major confounding factors and time-related biases and adjusted for treatment with other glucose-lowering medications and a history of pancreatitis.
TAKEAWAY:
- During the study period, 33,377 patients (6.1%) used GLP-1 RAs and 106,849 (19.7%) used basal insulin, with 1665 of all patients diagnosed with pancreatic cancer.
- There was no evidence that GLP-1 RA use increased pancreatic cancer risk compared with basal insulin.
- The estimated hazard ratio (HR) for pancreatic cancer associated with incremental use of one defined daily dose per day of GLP-1 RA compared with basal insulin in years 5-7 was 0.50 (95% CI, 0.15-1.71).
- New-user and prevalent new-user analyses showed HRs from year 5 onward following initiation of a GLP-1 RA vs basal insulin was 0.52 (95% CI, 0.19-1.41) and 0.75 (95% CI, 0.37-1.53), respectively.
IN PRACTICE:
Using several analytical approaches, these findings do not suggest an increase in pancreatic cancer incidence over 7 years following the start of GLP-1 RA treatment, according to the investigation. “However, monitoring for pancreatic cancer risk beyond 7 years following initiation of treatment is still required,” the authors wrote.
SOURCE:
The study, with first author Rachel Dankner, MD, MPH, Gertner Institute for Epidemiology and Health Policy Research, Sheba Medical Center, Israel, was published online on January 4, 2024, in JAMA Network Open.
LIMITATIONS:
Data on the exact type of GLP-1 RA were not available. The analyses accounted for history of pancreatitis but not alcohol use or exposure to pesticides/chemicals. Because of the risk for bias due to reverse causation, an emphasis was placed on drug effects several years after their initiation. However, this reduced the number of pancreatic cancer cases available and led to estimated HRs with wider CIs.
DISCLOSURES:
The study received no specific funding. The authors disclosed no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
New data provide no support for an increased risk for pancreatic cancer with use of a glucagon-like peptide-1 receptor agonist (GLP-1 RA) for up to 7 years, although longer-term data are needed, researchers said.
METHODOLOGY:
- Some studies have raised concern about a possible increased risk for pancreatitis and pancreatic cancer in patients taking a GLP-1 RA.
- Investigators behind this population-based cohort study assessed the association of GLP-1 RA treatment with pancreatic cancer incidence over a median of 7 years in 543,595 adults (mean age, 59.9 years; 51% women) with type 2 diabetes.
- Treatment with basal insulin was used as an active comparator.
- The analyses accounted for major confounding factors and time-related biases and adjusted for treatment with other glucose-lowering medications and a history of pancreatitis.
TAKEAWAY:
- During the study period, 33,377 patients (6.1%) used GLP-1 RAs and 106,849 (19.7%) used basal insulin, with 1665 of all patients diagnosed with pancreatic cancer.
- There was no evidence that GLP-1 RA use increased pancreatic cancer risk compared with basal insulin.
- The estimated hazard ratio (HR) for pancreatic cancer associated with incremental use of one defined daily dose per day of GLP-1 RA compared with basal insulin in years 5-7 was 0.50 (95% CI, 0.15-1.71).
- New-user and prevalent new-user analyses showed HRs from year 5 onward following initiation of a GLP-1 RA vs basal insulin was 0.52 (95% CI, 0.19-1.41) and 0.75 (95% CI, 0.37-1.53), respectively.
IN PRACTICE:
Using several analytical approaches, these findings do not suggest an increase in pancreatic cancer incidence over 7 years following the start of GLP-1 RA treatment, according to the investigation. “However, monitoring for pancreatic cancer risk beyond 7 years following initiation of treatment is still required,” the authors wrote.
SOURCE:
The study, with first author Rachel Dankner, MD, MPH, Gertner Institute for Epidemiology and Health Policy Research, Sheba Medical Center, Israel, was published online on January 4, 2024, in JAMA Network Open.
LIMITATIONS:
Data on the exact type of GLP-1 RA were not available. The analyses accounted for history of pancreatitis but not alcohol use or exposure to pesticides/chemicals. Because of the risk for bias due to reverse causation, an emphasis was placed on drug effects several years after their initiation. However, this reduced the number of pancreatic cancer cases available and led to estimated HRs with wider CIs.
DISCLOSURES:
The study received no specific funding. The authors disclosed no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
Hypocalcemia Risk Warning Added to Osteoporosis Drug
The US Food and Drug Administration (FDA) has added a boxed warning to the label of the osteoporosis drug denosumab (Prolia) about increased risk for severe hypocalcemia in patients with advanced chronic kidney disease (CKD).
Denosumab is a monoclonal antibody, indicated for the treatment of postmenopausal women with osteoporosis who are at increased risk for fracture for whom other treatments aren’t effective or can’t be tolerated. It’s also indicated to increase bone mass in men with osteoporosis at high risk for fracture, treat glucocorticoid-induced osteoporosis in men and women at high risk for fracture, increase bone mass in men at high risk for fracture receiving androgen-deprivation therapy for nonmetastatic prostate cancer, and increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
This new warning updates a November 2022 alert based on preliminary evidence for a “substantial risk” for hypocalcemia in patients with CKD on dialysis.
Upon further examination of the data from two trials including more than 500,000 denosumab-treated women with CKD, the FDA concluded that severe hypocalcemia appears to be more common in those with CKD who also have mineral and bone disorder (CKD-MBD). And, for patients with advanced CKD taking denosumab, “severe hypocalcemia resulted in serious harm, including hospitalization, life-threatening events, and death.”
Most of the severe hypocalcemia events occurred 2-10 weeks after denosumab injection, with the greatest risk during weeks 2-5.
The new warning advises healthcare professionals to assess patients’ kidney function before prescribing denosumab, and for those with advanced CKD, “consider the risk of severe hypocalcemia with Prolia in the context of other available treatments for osteoporosis.”
If the drug is still being considered for those patients for initial or continued use, calcium blood levels should be checked, and patients should be evaluated for CKD-MBD. Prior to prescribing denosumab in these patients, CKD-MBD should be properly managed, hypocalcemia corrected, and patients supplemented with calcium and activated vitamin D to decrease the risk for severe hypocalcemia and associated complications.
“Treatment with denosumab in patients with advanced CKD, including those on dialysis, and particularly patients with diagnosed CKD-MBD should involve a health care provider with expertise in the diagnosis and management of CKD-MBD,” the FDA advises.
Once denosumab is administered, close monitoring of blood calcium levels and prompt hypocalcemia management is essential to prevent complications including seizures or arrythmias. Patients should be advised to promptly report symptoms that could be consistent with hypocalcemia, including confusion, seizures, irregular heartbeat, fainting, muscle spasms or weakness, face twitching, tingling, or numbness anywhere in the body.
In 2022, an estimated 2.2 million Prolia prefilled syringes were sold by the manufacturer to US healthcare settings.
A version of this article appeared on Medscape.com.
The US Food and Drug Administration (FDA) has added a boxed warning to the label of the osteoporosis drug denosumab (Prolia) about increased risk for severe hypocalcemia in patients with advanced chronic kidney disease (CKD).
Denosumab is a monoclonal antibody, indicated for the treatment of postmenopausal women with osteoporosis who are at increased risk for fracture for whom other treatments aren’t effective or can’t be tolerated. It’s also indicated to increase bone mass in men with osteoporosis at high risk for fracture, treat glucocorticoid-induced osteoporosis in men and women at high risk for fracture, increase bone mass in men at high risk for fracture receiving androgen-deprivation therapy for nonmetastatic prostate cancer, and increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
This new warning updates a November 2022 alert based on preliminary evidence for a “substantial risk” for hypocalcemia in patients with CKD on dialysis.
Upon further examination of the data from two trials including more than 500,000 denosumab-treated women with CKD, the FDA concluded that severe hypocalcemia appears to be more common in those with CKD who also have mineral and bone disorder (CKD-MBD). And, for patients with advanced CKD taking denosumab, “severe hypocalcemia resulted in serious harm, including hospitalization, life-threatening events, and death.”
Most of the severe hypocalcemia events occurred 2-10 weeks after denosumab injection, with the greatest risk during weeks 2-5.
The new warning advises healthcare professionals to assess patients’ kidney function before prescribing denosumab, and for those with advanced CKD, “consider the risk of severe hypocalcemia with Prolia in the context of other available treatments for osteoporosis.”
If the drug is still being considered for those patients for initial or continued use, calcium blood levels should be checked, and patients should be evaluated for CKD-MBD. Prior to prescribing denosumab in these patients, CKD-MBD should be properly managed, hypocalcemia corrected, and patients supplemented with calcium and activated vitamin D to decrease the risk for severe hypocalcemia and associated complications.
“Treatment with denosumab in patients with advanced CKD, including those on dialysis, and particularly patients with diagnosed CKD-MBD should involve a health care provider with expertise in the diagnosis and management of CKD-MBD,” the FDA advises.
Once denosumab is administered, close monitoring of blood calcium levels and prompt hypocalcemia management is essential to prevent complications including seizures or arrythmias. Patients should be advised to promptly report symptoms that could be consistent with hypocalcemia, including confusion, seizures, irregular heartbeat, fainting, muscle spasms or weakness, face twitching, tingling, or numbness anywhere in the body.
In 2022, an estimated 2.2 million Prolia prefilled syringes were sold by the manufacturer to US healthcare settings.
A version of this article appeared on Medscape.com.
The US Food and Drug Administration (FDA) has added a boxed warning to the label of the osteoporosis drug denosumab (Prolia) about increased risk for severe hypocalcemia in patients with advanced chronic kidney disease (CKD).
Denosumab is a monoclonal antibody, indicated for the treatment of postmenopausal women with osteoporosis who are at increased risk for fracture for whom other treatments aren’t effective or can’t be tolerated. It’s also indicated to increase bone mass in men with osteoporosis at high risk for fracture, treat glucocorticoid-induced osteoporosis in men and women at high risk for fracture, increase bone mass in men at high risk for fracture receiving androgen-deprivation therapy for nonmetastatic prostate cancer, and increase bone mass in women at high risk for fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
This new warning updates a November 2022 alert based on preliminary evidence for a “substantial risk” for hypocalcemia in patients with CKD on dialysis.
Upon further examination of the data from two trials including more than 500,000 denosumab-treated women with CKD, the FDA concluded that severe hypocalcemia appears to be more common in those with CKD who also have mineral and bone disorder (CKD-MBD). And, for patients with advanced CKD taking denosumab, “severe hypocalcemia resulted in serious harm, including hospitalization, life-threatening events, and death.”
Most of the severe hypocalcemia events occurred 2-10 weeks after denosumab injection, with the greatest risk during weeks 2-5.
The new warning advises healthcare professionals to assess patients’ kidney function before prescribing denosumab, and for those with advanced CKD, “consider the risk of severe hypocalcemia with Prolia in the context of other available treatments for osteoporosis.”
If the drug is still being considered for those patients for initial or continued use, calcium blood levels should be checked, and patients should be evaluated for CKD-MBD. Prior to prescribing denosumab in these patients, CKD-MBD should be properly managed, hypocalcemia corrected, and patients supplemented with calcium and activated vitamin D to decrease the risk for severe hypocalcemia and associated complications.
“Treatment with denosumab in patients with advanced CKD, including those on dialysis, and particularly patients with diagnosed CKD-MBD should involve a health care provider with expertise in the diagnosis and management of CKD-MBD,” the FDA advises.
Once denosumab is administered, close monitoring of blood calcium levels and prompt hypocalcemia management is essential to prevent complications including seizures or arrythmias. Patients should be advised to promptly report symptoms that could be consistent with hypocalcemia, including confusion, seizures, irregular heartbeat, fainting, muscle spasms or weakness, face twitching, tingling, or numbness anywhere in the body.
In 2022, an estimated 2.2 million Prolia prefilled syringes were sold by the manufacturer to US healthcare settings.
A version of this article appeared on Medscape.com.
Continued Caution Needed Combining Nitrates With ED Drugs
New research supports continued caution in prescribing a phosphodiesterase-5 inhibitor (PDE5i) to treat erectile dysfunction (ED) in men with heart disease using nitrate medications.
In a large Swedish population study of men with stable coronary artery disease (CAD), the combined use of a PDE5i and nitrates was associated with a higher risk for cardiovascular (CV) morbidity and mortality.
“According to current recommendations, PDE5i are contraindicated in patients taking organic nitrates; however, in clinical practice, both are commonly prescribed, and concomitant use has increased,” first author Ylva Trolle Lagerros, MD, PhD, with Karolinska Institutet, Stockholm, Sweden, told this news organization.
and weigh the benefits of the medication against the possible increased risk for cardiovascular morbidity and mortality given by this combination,” Dr. Lagerros said.
The study was published online in the Journal of the American College of Cardiology (JACC).
The researchers used the Swedish Patient Register and the Prescribed Drug Register to assess the association between PDE5i treatment and CV outcomes in men with stable CAD treated with nitrate medication.
Among 55,777 men with a history of previous myocardial infarction (MI) or coronary revascularization who had filled at least two nitrate prescriptions (sublingual, oral, or both), 5710 also had at least two filled prescriptions of a PDE5i.
In multivariate-adjusted analysis, the combined use of PDE5i treatment with nitrates was associated with an increased relative risk for all studied outcomes, including all-cause mortality, CV and non-CV mortality, MI, heart failure, cardiac revascularization (hazard ratio), and major adverse cardiovascular events.
However, the number of events 28 days following a PDE5i prescription fill was “few, with lower incidence rates than in subjects taking nitrates only, indicating a low immediate risk for any event,” the authors noted in their article.
‘Common Bedfellows’
In a JACC editorial, Glenn N. Levine, MD, with Baylor College of Medicine, Houston, Texas, noted that, “ED and CAD are unfortunate, and all too common, bedfellows. But, as with most relationships, assuming proper precautions and care, they can coexist together for many years, perhaps even a lifetime.”
Dr. Levine noted that PDE5is are “reasonably safe” in most patients with stable CAD and only mild angina if not on chronic nitrate therapy. For those on chronic oral nitrate therapy, the use of PDE5is should continue to be regarded as “ill-advised at best and generally contraindicated.”
In some patients on oral nitrate therapy who want to use a PDE5i, particularly those who have undergone revascularization and have minimal or no angina, Dr. Levine said it may be reasonable to initiate a several-week trial of the nitrate therapy (or on a different class of antianginal therapy) and assess if the patient remains relatively angina-free.
In those patients with just rare exertional angina at generally higher levels of activity or those prescribed sublingual nitroglycerin “just in case,” it may be reasonable to prescribe PDE5i after a “clear and detailed” discussion with the patient of the risks for temporarily combining PDE5i and sublingual nitroglycerin.
Dr. Levine said these patients should be instructed not to take nitroglycerin within 24 hours of using a shorter-acting PDE5i and within 48 hours of using the longer-acting PDE5i tadalafil.
They should also be told to call 9-1-1 if angina develops during sexual intercourse and does not resolve upon cessation of such sexual activity, as well as to make medical personnel aware that they have recently used a PDE5i.
The study was funded by Region Stockholm, the Center for Innovative Medicine, and Karolinska Institutet. The researchers and editorial writer had declared no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
New research supports continued caution in prescribing a phosphodiesterase-5 inhibitor (PDE5i) to treat erectile dysfunction (ED) in men with heart disease using nitrate medications.
In a large Swedish population study of men with stable coronary artery disease (CAD), the combined use of a PDE5i and nitrates was associated with a higher risk for cardiovascular (CV) morbidity and mortality.
“According to current recommendations, PDE5i are contraindicated in patients taking organic nitrates; however, in clinical practice, both are commonly prescribed, and concomitant use has increased,” first author Ylva Trolle Lagerros, MD, PhD, with Karolinska Institutet, Stockholm, Sweden, told this news organization.
and weigh the benefits of the medication against the possible increased risk for cardiovascular morbidity and mortality given by this combination,” Dr. Lagerros said.
The study was published online in the Journal of the American College of Cardiology (JACC).
The researchers used the Swedish Patient Register and the Prescribed Drug Register to assess the association between PDE5i treatment and CV outcomes in men with stable CAD treated with nitrate medication.
Among 55,777 men with a history of previous myocardial infarction (MI) or coronary revascularization who had filled at least two nitrate prescriptions (sublingual, oral, or both), 5710 also had at least two filled prescriptions of a PDE5i.
In multivariate-adjusted analysis, the combined use of PDE5i treatment with nitrates was associated with an increased relative risk for all studied outcomes, including all-cause mortality, CV and non-CV mortality, MI, heart failure, cardiac revascularization (hazard ratio), and major adverse cardiovascular events.
However, the number of events 28 days following a PDE5i prescription fill was “few, with lower incidence rates than in subjects taking nitrates only, indicating a low immediate risk for any event,” the authors noted in their article.
‘Common Bedfellows’
In a JACC editorial, Glenn N. Levine, MD, with Baylor College of Medicine, Houston, Texas, noted that, “ED and CAD are unfortunate, and all too common, bedfellows. But, as with most relationships, assuming proper precautions and care, they can coexist together for many years, perhaps even a lifetime.”
Dr. Levine noted that PDE5is are “reasonably safe” in most patients with stable CAD and only mild angina if not on chronic nitrate therapy. For those on chronic oral nitrate therapy, the use of PDE5is should continue to be regarded as “ill-advised at best and generally contraindicated.”
In some patients on oral nitrate therapy who want to use a PDE5i, particularly those who have undergone revascularization and have minimal or no angina, Dr. Levine said it may be reasonable to initiate a several-week trial of the nitrate therapy (or on a different class of antianginal therapy) and assess if the patient remains relatively angina-free.
In those patients with just rare exertional angina at generally higher levels of activity or those prescribed sublingual nitroglycerin “just in case,” it may be reasonable to prescribe PDE5i after a “clear and detailed” discussion with the patient of the risks for temporarily combining PDE5i and sublingual nitroglycerin.
Dr. Levine said these patients should be instructed not to take nitroglycerin within 24 hours of using a shorter-acting PDE5i and within 48 hours of using the longer-acting PDE5i tadalafil.
They should also be told to call 9-1-1 if angina develops during sexual intercourse and does not resolve upon cessation of such sexual activity, as well as to make medical personnel aware that they have recently used a PDE5i.
The study was funded by Region Stockholm, the Center for Innovative Medicine, and Karolinska Institutet. The researchers and editorial writer had declared no relevant conflicts of interest.
A version of this article appeared on Medscape.com.
New research supports continued caution in prescribing a phosphodiesterase-5 inhibitor (PDE5i) to treat erectile dysfunction (ED) in men with heart disease using nitrate medications.
In a large Swedish population study of men with stable coronary artery disease (CAD), the combined use of a PDE5i and nitrates was associated with a higher risk for cardiovascular (CV) morbidity and mortality.
“According to current recommendations, PDE5i are contraindicated in patients taking organic nitrates; however, in clinical practice, both are commonly prescribed, and concomitant use has increased,” first author Ylva Trolle Lagerros, MD, PhD, with Karolinska Institutet, Stockholm, Sweden, told this news organization.
and weigh the benefits of the medication against the possible increased risk for cardiovascular morbidity and mortality given by this combination,” Dr. Lagerros said.
The study was published online in the Journal of the American College of Cardiology (JACC).
The researchers used the Swedish Patient Register and the Prescribed Drug Register to assess the association between PDE5i treatment and CV outcomes in men with stable CAD treated with nitrate medication.
Among 55,777 men with a history of previous myocardial infarction (MI) or coronary revascularization who had filled at least two nitrate prescriptions (sublingual, oral, or both), 5710 also had at least two filled prescriptions of a PDE5i.
In multivariate-adjusted analysis, the combined use of PDE5i treatment with nitrates was associated with an increased relative risk for all studied outcomes, including all-cause mortality, CV and non-CV mortality, MI, heart failure, cardiac revascularization (hazard ratio), and major adverse cardiovascular events.
However, the number of events 28 days following a PDE5i prescription fill was “few, with lower incidence rates than in subjects taking nitrates only, indicating a low immediate risk for any event,” the authors noted in their article.
‘Common Bedfellows’
In a JACC editorial, Glenn N. Levine, MD, with Baylor College of Medicine, Houston, Texas, noted that, “ED and CAD are unfortunate, and all too common, bedfellows. But, as with most relationships, assuming proper precautions and care, they can coexist together for many years, perhaps even a lifetime.”
Dr. Levine noted that PDE5is are “reasonably safe” in most patients with stable CAD and only mild angina if not on chronic nitrate therapy. For those on chronic oral nitrate therapy, the use of PDE5is should continue to be regarded as “ill-advised at best and generally contraindicated.”
In some patients on oral nitrate therapy who want to use a PDE5i, particularly those who have undergone revascularization and have minimal or no angina, Dr. Levine said it may be reasonable to initiate a several-week trial of the nitrate therapy (or on a different class of antianginal therapy) and assess if the patient remains relatively angina-free.
In those patients with just rare exertional angina at generally higher levels of activity or those prescribed sublingual nitroglycerin “just in case,” it may be reasonable to prescribe PDE5i after a “clear and detailed” discussion with the patient of the risks for temporarily combining PDE5i and sublingual nitroglycerin.
Dr. Levine said these patients should be instructed not to take nitroglycerin within 24 hours of using a shorter-acting PDE5i and within 48 hours of using the longer-acting PDE5i tadalafil.
They should also be told to call 9-1-1 if angina develops during sexual intercourse and does not resolve upon cessation of such sexual activity, as well as to make medical personnel aware that they have recently used a PDE5i.
The study was funded by Region Stockholm, the Center for Innovative Medicine, and Karolinska Institutet. The researchers and editorial writer had declared no relevant conflicts of interest.
A version of this article appeared on Medscape.com.