User login
Large Subcutaneous Masses
The Diagnosis: Madelung Disease (Benign Symmetric Lipomatosis)
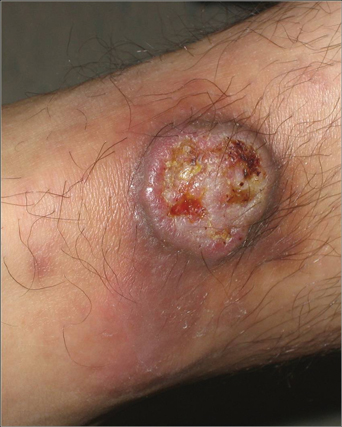
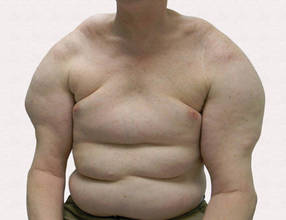
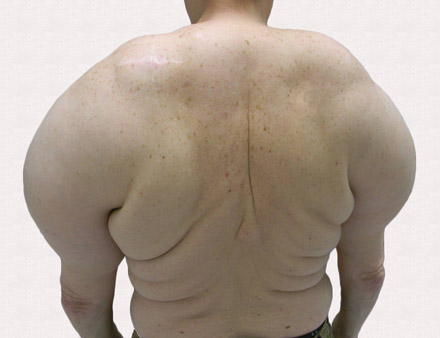
A 56-year-old man presented for evaluation of massive subcutaneous nodules the bilateral upper arms, shoulders, chest, abdomen, and lateral aspect of the proximal thighs (Figures 1 and 2) that developed over the last 12 to 18 months and continued to enlarge. In addition, he was beginning to develop symptoms of neuropathy of the bilateral hands. The patient had a long-standing history of alcohol abuse. Biopsies performed by the patient’s primary care physician revealed benign adipose tissue. He was referred to the dermatology clinic and subsequently diagnosed with Madelung disease.
 |
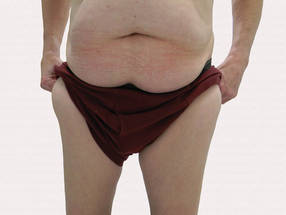 |
Madelung disease, also known as benign symmetric lipomatosis and Launois-Bensaude syndrome, is characterized by multiple large masses of nonencapsulated adipose tissue. These masses are symmetric and most prominent on the head, neck, trunk, and proximal extremities. Classically, a pseudoathletic appearance is described. Madelung disease most frequently affects men aged 30 to 60 years. In more than 90% of cases, it is associated with alcoholism.
In general, the masses of adipose tissue are asymptomatic. However, airway compression and dysphagia requiring surgical intervention has been reported in the otolaryngology literature.1 In addition, neuropathy develops in 84% of cases.2 Nerve biopsies from patients with Madelung disease have revealed a pattern of axonopathy that is distinct from alcohol-induced neuropathy.3 This neuropathy can involve sensory and motor nerves, with the most prominent findings being muscle weakness, tendon areflexia, interosseous muscle atrophy, vibratory sensation loss, and hypoesthesia. Furthermore, dysfunction of the autonomic nervous system can lead to segmental hyperhidrosis, gustatory sweating, and abnormal autonomic cardiovascular reflexes.2 Many patients who develop neuropathy will eventually become incapacitated.
The etiology of Madelung disease is not fully understood. There are several theories on the pathogenesis of this disease, most describing metabolic disturbances induced by alcohol. Specifically, studies have revealed chronic alcohol use causes numerous deletions in mitochondrial DNA.4,5 The mitochondrial DNA damage may explain both the resistance of the lipomatous masses to lipolysis and the nerve-related changes. Comparisons between human immunodeficiency virus/highly active antiretroviral therapy–associated lipodystrophy and Madelung disease lend credence to the metabolic disturbance theory and may help clarify the specific mechanisms involved.6
There have been no cases of spontaneous resolution, even in patients who stop consuming alcohol. For this reason, most patients are referred to a surgeon. Many surgeons prefer open excision for debulking large lipomatous masses, but this technique typically requires general anesthesia. For those in whom this treatment is not an appropriate option, liposuction can be considered with tumescent anesthesia.7 A combination of these surgical modalities also is an option.
Madelung disease is a distinctive disorder typically affecting chronic alcoholics. Recognition of this clinical entity is important, as severe neuropathy and airway compromise may ensue. Although surgical excision is an attractive option for cosmesis and airway compromise, the associated neuropathy can be extremely difficult to treat and can be quite debilitating.
1. Palacios E, Neitzschman HR, Nguyen J. Madelung disease: multiple symmetric lipomatosis. Ear Nose Throat J. 2014;93:94-96.
2. Enzi G, Angelini C, Negrin P, et al. Sensory, motor, and autonomic neuropathy in patients with multiple symmetric lipomatosis. Medicine (Baltimore). 1985;64:388-393.
3. Pollock M, Nicholson GI, Nukada H, et al. Neuropathy in multiple symmetric lipomatosis. Madelung’s disease. Brain. 1988;111:1157-1171.
4. Klopstock T, Naumann M, Schalke B, et al. Multiple symmetric lipomatosis: abnormalities in complex IV and multiple deletions in mitochondrial DNA. Neurology. 1994;44:862-866.
5. Mansouri A, Fromenty B, Berson A, et al. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J Hepatol. 1997;27:96-102.
6. Urso R, Gentile M. Are ‘buffalo hump’ syndrome, Madelung's disease and multiple symmetrical lipomatosis variants of the same dysmetabolism? AIDS. 2001;15:290-291.
7. Grassegger A, Häussler R, Schmalzl F. Tumescent liposuction in a patient with Launois-Bensaude syndrome and severe hepatopathy. Dermatol Surg. 2007;33:982-985.
The Diagnosis: Madelung Disease (Benign Symmetric Lipomatosis)
A 56-year-old man presented for evaluation of massive subcutaneous nodules the bilateral upper arms, shoulders, chest, abdomen, and lateral aspect of the proximal thighs (Figures 1 and 2) that developed over the last 12 to 18 months and continued to enlarge. In addition, he was beginning to develop symptoms of neuropathy of the bilateral hands. The patient had a long-standing history of alcohol abuse. Biopsies performed by the patient’s primary care physician revealed benign adipose tissue. He was referred to the dermatology clinic and subsequently diagnosed with Madelung disease.
|
|
Madelung disease, also known as benign symmetric lipomatosis and Launois-Bensaude syndrome, is characterized by multiple large masses of nonencapsulated adipose tissue. These masses are symmetric and most prominent on the head, neck, trunk, and proximal extremities. Classically, a pseudoathletic appearance is described. Madelung disease most frequently affects men aged 30 to 60 years. In more than 90% of cases, it is associated with alcoholism.
In general, the masses of adipose tissue are asymptomatic. However, airway compression and dysphagia requiring surgical intervention has been reported in the otolaryngology literature.1 In addition, neuropathy develops in 84% of cases.2 Nerve biopsies from patients with Madelung disease have revealed a pattern of axonopathy that is distinct from alcohol-induced neuropathy.3 This neuropathy can involve sensory and motor nerves, with the most prominent findings being muscle weakness, tendon areflexia, interosseous muscle atrophy, vibratory sensation loss, and hypoesthesia. Furthermore, dysfunction of the autonomic nervous system can lead to segmental hyperhidrosis, gustatory sweating, and abnormal autonomic cardiovascular reflexes.2 Many patients who develop neuropathy will eventually become incapacitated.
The etiology of Madelung disease is not fully understood. There are several theories on the pathogenesis of this disease, most describing metabolic disturbances induced by alcohol. Specifically, studies have revealed chronic alcohol use causes numerous deletions in mitochondrial DNA.4,5 The mitochondrial DNA damage may explain both the resistance of the lipomatous masses to lipolysis and the nerve-related changes. Comparisons between human immunodeficiency virus/highly active antiretroviral therapy–associated lipodystrophy and Madelung disease lend credence to the metabolic disturbance theory and may help clarify the specific mechanisms involved.6
There have been no cases of spontaneous resolution, even in patients who stop consuming alcohol. For this reason, most patients are referred to a surgeon. Many surgeons prefer open excision for debulking large lipomatous masses, but this technique typically requires general anesthesia. For those in whom this treatment is not an appropriate option, liposuction can be considered with tumescent anesthesia.7 A combination of these surgical modalities also is an option.
Madelung disease is a distinctive disorder typically affecting chronic alcoholics. Recognition of this clinical entity is important, as severe neuropathy and airway compromise may ensue. Although surgical excision is an attractive option for cosmesis and airway compromise, the associated neuropathy can be extremely difficult to treat and can be quite debilitating.
The Diagnosis: Madelung Disease (Benign Symmetric Lipomatosis)
A 56-year-old man presented for evaluation of massive subcutaneous nodules the bilateral upper arms, shoulders, chest, abdomen, and lateral aspect of the proximal thighs (Figures 1 and 2) that developed over the last 12 to 18 months and continued to enlarge. In addition, he was beginning to develop symptoms of neuropathy of the bilateral hands. The patient had a long-standing history of alcohol abuse. Biopsies performed by the patient’s primary care physician revealed benign adipose tissue. He was referred to the dermatology clinic and subsequently diagnosed with Madelung disease.
|
|
Madelung disease, also known as benign symmetric lipomatosis and Launois-Bensaude syndrome, is characterized by multiple large masses of nonencapsulated adipose tissue. These masses are symmetric and most prominent on the head, neck, trunk, and proximal extremities. Classically, a pseudoathletic appearance is described. Madelung disease most frequently affects men aged 30 to 60 years. In more than 90% of cases, it is associated with alcoholism.
In general, the masses of adipose tissue are asymptomatic. However, airway compression and dysphagia requiring surgical intervention has been reported in the otolaryngology literature.1 In addition, neuropathy develops in 84% of cases.2 Nerve biopsies from patients with Madelung disease have revealed a pattern of axonopathy that is distinct from alcohol-induced neuropathy.3 This neuropathy can involve sensory and motor nerves, with the most prominent findings being muscle weakness, tendon areflexia, interosseous muscle atrophy, vibratory sensation loss, and hypoesthesia. Furthermore, dysfunction of the autonomic nervous system can lead to segmental hyperhidrosis, gustatory sweating, and abnormal autonomic cardiovascular reflexes.2 Many patients who develop neuropathy will eventually become incapacitated.
The etiology of Madelung disease is not fully understood. There are several theories on the pathogenesis of this disease, most describing metabolic disturbances induced by alcohol. Specifically, studies have revealed chronic alcohol use causes numerous deletions in mitochondrial DNA.4,5 The mitochondrial DNA damage may explain both the resistance of the lipomatous masses to lipolysis and the nerve-related changes. Comparisons between human immunodeficiency virus/highly active antiretroviral therapy–associated lipodystrophy and Madelung disease lend credence to the metabolic disturbance theory and may help clarify the specific mechanisms involved.6
There have been no cases of spontaneous resolution, even in patients who stop consuming alcohol. For this reason, most patients are referred to a surgeon. Many surgeons prefer open excision for debulking large lipomatous masses, but this technique typically requires general anesthesia. For those in whom this treatment is not an appropriate option, liposuction can be considered with tumescent anesthesia.7 A combination of these surgical modalities also is an option.
Madelung disease is a distinctive disorder typically affecting chronic alcoholics. Recognition of this clinical entity is important, as severe neuropathy and airway compromise may ensue. Although surgical excision is an attractive option for cosmesis and airway compromise, the associated neuropathy can be extremely difficult to treat and can be quite debilitating.
1. Palacios E, Neitzschman HR, Nguyen J. Madelung disease: multiple symmetric lipomatosis. Ear Nose Throat J. 2014;93:94-96.
2. Enzi G, Angelini C, Negrin P, et al. Sensory, motor, and autonomic neuropathy in patients with multiple symmetric lipomatosis. Medicine (Baltimore). 1985;64:388-393.
3. Pollock M, Nicholson GI, Nukada H, et al. Neuropathy in multiple symmetric lipomatosis. Madelung’s disease. Brain. 1988;111:1157-1171.
4. Klopstock T, Naumann M, Schalke B, et al. Multiple symmetric lipomatosis: abnormalities in complex IV and multiple deletions in mitochondrial DNA. Neurology. 1994;44:862-866.
5. Mansouri A, Fromenty B, Berson A, et al. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J Hepatol. 1997;27:96-102.
6. Urso R, Gentile M. Are ‘buffalo hump’ syndrome, Madelung's disease and multiple symmetrical lipomatosis variants of the same dysmetabolism? AIDS. 2001;15:290-291.
7. Grassegger A, Häussler R, Schmalzl F. Tumescent liposuction in a patient with Launois-Bensaude syndrome and severe hepatopathy. Dermatol Surg. 2007;33:982-985.
1. Palacios E, Neitzschman HR, Nguyen J. Madelung disease: multiple symmetric lipomatosis. Ear Nose Throat J. 2014;93:94-96.
2. Enzi G, Angelini C, Negrin P, et al. Sensory, motor, and autonomic neuropathy in patients with multiple symmetric lipomatosis. Medicine (Baltimore). 1985;64:388-393.
3. Pollock M, Nicholson GI, Nukada H, et al. Neuropathy in multiple symmetric lipomatosis. Madelung’s disease. Brain. 1988;111:1157-1171.
4. Klopstock T, Naumann M, Schalke B, et al. Multiple symmetric lipomatosis: abnormalities in complex IV and multiple deletions in mitochondrial DNA. Neurology. 1994;44:862-866.
5. Mansouri A, Fromenty B, Berson A, et al. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J Hepatol. 1997;27:96-102.
6. Urso R, Gentile M. Are ‘buffalo hump’ syndrome, Madelung's disease and multiple symmetrical lipomatosis variants of the same dysmetabolism? AIDS. 2001;15:290-291.
7. Grassegger A, Häussler R, Schmalzl F. Tumescent liposuction in a patient with Launois-Bensaude syndrome and severe hepatopathy. Dermatol Surg. 2007;33:982-985.
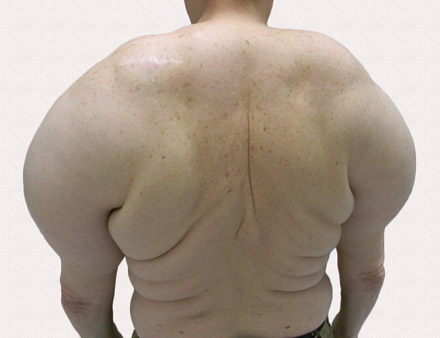
A 56-year-old man presented for evaluation of massive subcutaneous nodules on the bilateral upper arms, shoulders, chest, abdomen, and lateral aspect of the proximal thighs that developed over the last 12 to 18 months and continued to enlarge. His medical history was remarkable for alcoholism, hyperlipidemia, and hypertension.
Cutaneous Manifestations of Cocaine Use
The Diagnosis: Levamisole-Induced Cutaneous Vasculopathy
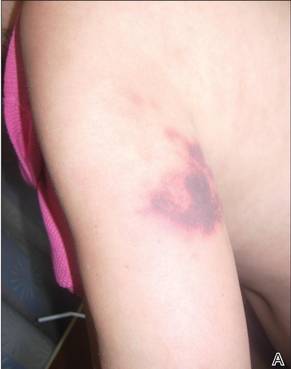
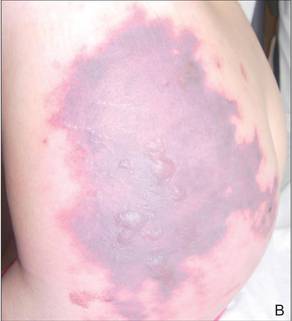
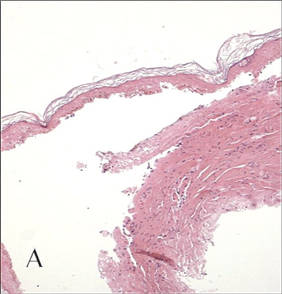
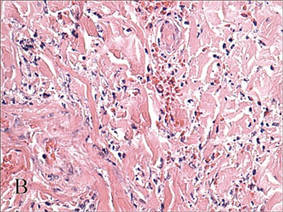
In our patient, tender stellate purpura and occasional bullae were present on the ears, arms and legs, groin, and buttocks (Figure 1). Histopathologic examination revealed subepidermal detachment, perivascular neutrophilic infiltrate, and red blood cell extravasation, consistent with early leukocyctoclastic vasculitis (Figure 2).
|
Levamisole-induced vasculopathy is a condition related primarily to cocaine use. Levamisole is an immunomodulatory agent, historically used as a disease-modifying antirheumatic drug for rheumatoid arthritis and as adjuvant chemotherapy for various types of cancer. However, levamisole for human use was banned from US and Canadian markets in 1999 and 2003, respectively, due to increased risk for agranulocytosis, retiform purpura, and epilepsy.1 Currently, veterinarians use levamisole as an anthelminthic agent to deworm house and farm animals. In Europe, pediatric nephrologists use it as a steroid-sparing agent in children with steroid-dependent nephritic syndrome.
Over the last decade, levamisole has increasingly been used as a cocaine adulterant or bulking agent. This contaminant closely resembles cocaine physically and is theorized to prolong or attenuate cocaine’s “high.” Approximately 69% of cocaine sampled by the US Drug Enforcement Administration is adulterated with levamisole.2 Similarly, levamisole-contaminated cocaine also has been found in Europe, Australia, and other parts of the world. Potential complications include vasculitis, thromboembolism, neutropenia, and agranulocytosis.3
Levamisole-induced vasculopathy appears to affect cocaine users of all ages, ethnicities, and genders. Cocaine can be smoked, snorted, or injected. In nearly all reported cases, patients characteristically present with hemorrhagic bullae of the bilateral ear helix, cheeks, or nasal tip. Any body site can be affected with retiform purpura or necrotic bullae. Along with skin lesions, arthralgia is commonly reported, as are constitutional symptoms (eg, fever, night sweats, weight loss, malaise)4; oral mucosal involvement also has been reported.5 Laboratory investigation can reveal neutropenia, positive antineutrophil cytoplasmic antibodies (ANCAs) in the perinuclear or cytoplasmic pattern, positive proteinase 3, and negative or mildly elevated antimyeloperoxidase.3-5 Acute renal injury and pulmonary hemorrhage are other potentially serious copmlications.4 Antihuman neutrophil elastase antibody testing can help distinguish levamisole-induced vasculopathy from other forms of immune-mediated vasculitis and will be negative in immune-mediated vasculitides such as Churg-Strauss syndrome (allergic granulomatosis), Wegener granulomatosis (granulomatosis with polyangiitis), and polyarteritis nodosa.6 On histology, microvascular thrombosis or leukocytoclastic vasculitis can both, or individually, be seen. Epidermal necrosis, dermal hemorrhage, and endothelial hyperplasia have all been noted in skin biopsied from necrotic bullae.
|
Levamisole’s short half-life (approximately 5–6 hours) makes it difficult to detect on routine blood draws. An astute physician suspecting this diagnosis on initial presentation can ask for levamisole detection on urine toxicology screening.7 Urine samples also can be sent for testing with gas chromatography–mass spectrometry, though this test may only be available at major research centers.8
Differential diagnosis of levamisole toxicity includes different types of vasculitides such as cryoglobulinemia (positive serum IgM and IgG cryoglobulins; possible hepatitis C infection), Wegener granulomatosis (cytoplasmic ANCA positive; associated with upper and lower respiratory tract inflammation, glomerulonephritis), Churg-Strauss syndrome (perinuclear ANCA positive; associated with asthma and eosinophilia), and polyarteritis nodosa (medium vessel involvement only; associated with livedo reticularis, subcutaneous nodules, ulcers).9 Necrotic lesions also may raise the possibility of warfarin necrosis, heparin necrosis, or cholesterol emboli. Cholesterol embolism most frequently presents with small vessel vasculitis and necrosis of distal extremities such as the toes. With large areas of skin involvement and bullae, erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis also should be considered.9
Definitive treatment of this condition requires complete and immediate cessation of cocaine use. Levamisole also has been found as a contaminant in heroin.1 Thus, it may be prudent to recommend heroin avoidance to the patient to prevent recurrences. Management of acute levamisole-induced vasculopathy is primarily symptomatic. Some patients with severe neutropenia at risk for infection have been treated with granulocyte colony-stimulating factor, while others have only required pain control, usually with nonsteroidal anti-inflammatory drugs.10 Oral prednisone and colchicine also have been used with reported success.5
Given the increasing incidence of levamisole toxicity and public health implications, clinicians should be aware of this association and the classic clinical and laboratory findings.
1. Aberastury MN, Silva WH, Vaccarezza MM, et al. Epilepsia partialis continua associated with levamisole. Pediatr Neurol. 2011;44:385-388.
2. Nationwide public health alert issued concerning life-threatening risk posed by cocaine laced with veterinary anti-parasite drug [press release]. Rockville, MD: Substance Abuse and Mental Health Services Administration; September 21, 2009. http://beta.samhsa.gov/newsroom/press-announcements/200909211245. Accessed October 9, 2014.
3. Lee KC, Culpepper K, Kessler M. Levamisole-induced thrombosis: literature review and pertinent laboratory findings. J Am Acad Dermatol. 2011;65:e128-e129.
4. McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasm antibody-associated disease. Clin J Am Soc Nephrol. 2011;6:2799-2805.
5. Poon SH, Baliog CR Jr, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leucopenia. Semin Arthritis Rheum. 2011;41:434-444.
6. Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
7. Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
8. Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2001;35:545-550.
9. Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
10. Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
The Diagnosis: Levamisole-Induced Cutaneous Vasculopathy
In our patient, tender stellate purpura and occasional bullae were present on the ears, arms and legs, groin, and buttocks (Figure 1). Histopathologic examination revealed subepidermal detachment, perivascular neutrophilic infiltrate, and red blood cell extravasation, consistent with early leukocyctoclastic vasculitis (Figure 2).
|
Levamisole-induced vasculopathy is a condition related primarily to cocaine use. Levamisole is an immunomodulatory agent, historically used as a disease-modifying antirheumatic drug for rheumatoid arthritis and as adjuvant chemotherapy for various types of cancer. However, levamisole for human use was banned from US and Canadian markets in 1999 and 2003, respectively, due to increased risk for agranulocytosis, retiform purpura, and epilepsy.1 Currently, veterinarians use levamisole as an anthelminthic agent to deworm house and farm animals. In Europe, pediatric nephrologists use it as a steroid-sparing agent in children with steroid-dependent nephritic syndrome.
Over the last decade, levamisole has increasingly been used as a cocaine adulterant or bulking agent. This contaminant closely resembles cocaine physically and is theorized to prolong or attenuate cocaine’s “high.” Approximately 69% of cocaine sampled by the US Drug Enforcement Administration is adulterated with levamisole.2 Similarly, levamisole-contaminated cocaine also has been found in Europe, Australia, and other parts of the world. Potential complications include vasculitis, thromboembolism, neutropenia, and agranulocytosis.3
Levamisole-induced vasculopathy appears to affect cocaine users of all ages, ethnicities, and genders. Cocaine can be smoked, snorted, or injected. In nearly all reported cases, patients characteristically present with hemorrhagic bullae of the bilateral ear helix, cheeks, or nasal tip. Any body site can be affected with retiform purpura or necrotic bullae. Along with skin lesions, arthralgia is commonly reported, as are constitutional symptoms (eg, fever, night sweats, weight loss, malaise)4; oral mucosal involvement also has been reported.5 Laboratory investigation can reveal neutropenia, positive antineutrophil cytoplasmic antibodies (ANCAs) in the perinuclear or cytoplasmic pattern, positive proteinase 3, and negative or mildly elevated antimyeloperoxidase.3-5 Acute renal injury and pulmonary hemorrhage are other potentially serious copmlications.4 Antihuman neutrophil elastase antibody testing can help distinguish levamisole-induced vasculopathy from other forms of immune-mediated vasculitis and will be negative in immune-mediated vasculitides such as Churg-Strauss syndrome (allergic granulomatosis), Wegener granulomatosis (granulomatosis with polyangiitis), and polyarteritis nodosa.6 On histology, microvascular thrombosis or leukocytoclastic vasculitis can both, or individually, be seen. Epidermal necrosis, dermal hemorrhage, and endothelial hyperplasia have all been noted in skin biopsied from necrotic bullae.
|
Levamisole’s short half-life (approximately 5–6 hours) makes it difficult to detect on routine blood draws. An astute physician suspecting this diagnosis on initial presentation can ask for levamisole detection on urine toxicology screening.7 Urine samples also can be sent for testing with gas chromatography–mass spectrometry, though this test may only be available at major research centers.8
Differential diagnosis of levamisole toxicity includes different types of vasculitides such as cryoglobulinemia (positive serum IgM and IgG cryoglobulins; possible hepatitis C infection), Wegener granulomatosis (cytoplasmic ANCA positive; associated with upper and lower respiratory tract inflammation, glomerulonephritis), Churg-Strauss syndrome (perinuclear ANCA positive; associated with asthma and eosinophilia), and polyarteritis nodosa (medium vessel involvement only; associated with livedo reticularis, subcutaneous nodules, ulcers).9 Necrotic lesions also may raise the possibility of warfarin necrosis, heparin necrosis, or cholesterol emboli. Cholesterol embolism most frequently presents with small vessel vasculitis and necrosis of distal extremities such as the toes. With large areas of skin involvement and bullae, erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis also should be considered.9
Definitive treatment of this condition requires complete and immediate cessation of cocaine use. Levamisole also has been found as a contaminant in heroin.1 Thus, it may be prudent to recommend heroin avoidance to the patient to prevent recurrences. Management of acute levamisole-induced vasculopathy is primarily symptomatic. Some patients with severe neutropenia at risk for infection have been treated with granulocyte colony-stimulating factor, while others have only required pain control, usually with nonsteroidal anti-inflammatory drugs.10 Oral prednisone and colchicine also have been used with reported success.5
Given the increasing incidence of levamisole toxicity and public health implications, clinicians should be aware of this association and the classic clinical and laboratory findings.
The Diagnosis: Levamisole-Induced Cutaneous Vasculopathy
In our patient, tender stellate purpura and occasional bullae were present on the ears, arms and legs, groin, and buttocks (Figure 1). Histopathologic examination revealed subepidermal detachment, perivascular neutrophilic infiltrate, and red blood cell extravasation, consistent with early leukocyctoclastic vasculitis (Figure 2).
|
Levamisole-induced vasculopathy is a condition related primarily to cocaine use. Levamisole is an immunomodulatory agent, historically used as a disease-modifying antirheumatic drug for rheumatoid arthritis and as adjuvant chemotherapy for various types of cancer. However, levamisole for human use was banned from US and Canadian markets in 1999 and 2003, respectively, due to increased risk for agranulocytosis, retiform purpura, and epilepsy.1 Currently, veterinarians use levamisole as an anthelminthic agent to deworm house and farm animals. In Europe, pediatric nephrologists use it as a steroid-sparing agent in children with steroid-dependent nephritic syndrome.
Over the last decade, levamisole has increasingly been used as a cocaine adulterant or bulking agent. This contaminant closely resembles cocaine physically and is theorized to prolong or attenuate cocaine’s “high.” Approximately 69% of cocaine sampled by the US Drug Enforcement Administration is adulterated with levamisole.2 Similarly, levamisole-contaminated cocaine also has been found in Europe, Australia, and other parts of the world. Potential complications include vasculitis, thromboembolism, neutropenia, and agranulocytosis.3
Levamisole-induced vasculopathy appears to affect cocaine users of all ages, ethnicities, and genders. Cocaine can be smoked, snorted, or injected. In nearly all reported cases, patients characteristically present with hemorrhagic bullae of the bilateral ear helix, cheeks, or nasal tip. Any body site can be affected with retiform purpura or necrotic bullae. Along with skin lesions, arthralgia is commonly reported, as are constitutional symptoms (eg, fever, night sweats, weight loss, malaise)4; oral mucosal involvement also has been reported.5 Laboratory investigation can reveal neutropenia, positive antineutrophil cytoplasmic antibodies (ANCAs) in the perinuclear or cytoplasmic pattern, positive proteinase 3, and negative or mildly elevated antimyeloperoxidase.3-5 Acute renal injury and pulmonary hemorrhage are other potentially serious copmlications.4 Antihuman neutrophil elastase antibody testing can help distinguish levamisole-induced vasculopathy from other forms of immune-mediated vasculitis and will be negative in immune-mediated vasculitides such as Churg-Strauss syndrome (allergic granulomatosis), Wegener granulomatosis (granulomatosis with polyangiitis), and polyarteritis nodosa.6 On histology, microvascular thrombosis or leukocytoclastic vasculitis can both, or individually, be seen. Epidermal necrosis, dermal hemorrhage, and endothelial hyperplasia have all been noted in skin biopsied from necrotic bullae.
|
Levamisole’s short half-life (approximately 5–6 hours) makes it difficult to detect on routine blood draws. An astute physician suspecting this diagnosis on initial presentation can ask for levamisole detection on urine toxicology screening.7 Urine samples also can be sent for testing with gas chromatography–mass spectrometry, though this test may only be available at major research centers.8
Differential diagnosis of levamisole toxicity includes different types of vasculitides such as cryoglobulinemia (positive serum IgM and IgG cryoglobulins; possible hepatitis C infection), Wegener granulomatosis (cytoplasmic ANCA positive; associated with upper and lower respiratory tract inflammation, glomerulonephritis), Churg-Strauss syndrome (perinuclear ANCA positive; associated with asthma and eosinophilia), and polyarteritis nodosa (medium vessel involvement only; associated with livedo reticularis, subcutaneous nodules, ulcers).9 Necrotic lesions also may raise the possibility of warfarin necrosis, heparin necrosis, or cholesterol emboli. Cholesterol embolism most frequently presents with small vessel vasculitis and necrosis of distal extremities such as the toes. With large areas of skin involvement and bullae, erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis also should be considered.9
Definitive treatment of this condition requires complete and immediate cessation of cocaine use. Levamisole also has been found as a contaminant in heroin.1 Thus, it may be prudent to recommend heroin avoidance to the patient to prevent recurrences. Management of acute levamisole-induced vasculopathy is primarily symptomatic. Some patients with severe neutropenia at risk for infection have been treated with granulocyte colony-stimulating factor, while others have only required pain control, usually with nonsteroidal anti-inflammatory drugs.10 Oral prednisone and colchicine also have been used with reported success.5
Given the increasing incidence of levamisole toxicity and public health implications, clinicians should be aware of this association and the classic clinical and laboratory findings.
1. Aberastury MN, Silva WH, Vaccarezza MM, et al. Epilepsia partialis continua associated with levamisole. Pediatr Neurol. 2011;44:385-388.
2. Nationwide public health alert issued concerning life-threatening risk posed by cocaine laced with veterinary anti-parasite drug [press release]. Rockville, MD: Substance Abuse and Mental Health Services Administration; September 21, 2009. http://beta.samhsa.gov/newsroom/press-announcements/200909211245. Accessed October 9, 2014.
3. Lee KC, Culpepper K, Kessler M. Levamisole-induced thrombosis: literature review and pertinent laboratory findings. J Am Acad Dermatol. 2011;65:e128-e129.
4. McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasm antibody-associated disease. Clin J Am Soc Nephrol. 2011;6:2799-2805.
5. Poon SH, Baliog CR Jr, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leucopenia. Semin Arthritis Rheum. 2011;41:434-444.
6. Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
7. Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
8. Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2001;35:545-550.
9. Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
10. Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
1. Aberastury MN, Silva WH, Vaccarezza MM, et al. Epilepsia partialis continua associated with levamisole. Pediatr Neurol. 2011;44:385-388.
2. Nationwide public health alert issued concerning life-threatening risk posed by cocaine laced with veterinary anti-parasite drug [press release]. Rockville, MD: Substance Abuse and Mental Health Services Administration; September 21, 2009. http://beta.samhsa.gov/newsroom/press-announcements/200909211245. Accessed October 9, 2014.
3. Lee KC, Culpepper K, Kessler M. Levamisole-induced thrombosis: literature review and pertinent laboratory findings. J Am Acad Dermatol. 2011;65:e128-e129.
4. McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasm antibody-associated disease. Clin J Am Soc Nephrol. 2011;6:2799-2805.
5. Poon SH, Baliog CR Jr, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leucopenia. Semin Arthritis Rheum. 2011;41:434-444.
6. Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
7. Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
8. Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2001;35:545-550.
9. Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
10. Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
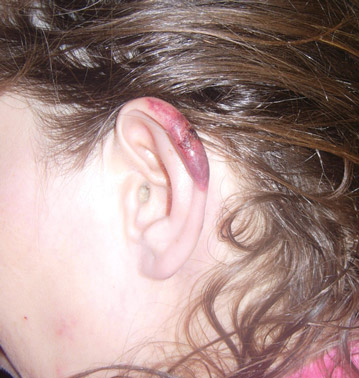
A 43-year-old woman presented to the emergency department with painful skin lesions of 1 day’s duration. Physical examination revealed tender stellate purpura and occasional bullae on the ears, arms and legs, groin, and buttocks. Laboratory results revealed neutropenia and positive lupus anticoagulant; antineutrophil cytoplasmic antibody, antinuclear antibody, and double-stranded DNA antibodies were all negative. Urine toxicology was positive for cocaine and opioids. An incisional biopsy of the left arm was performed.
Hairs With an Irregular Shape
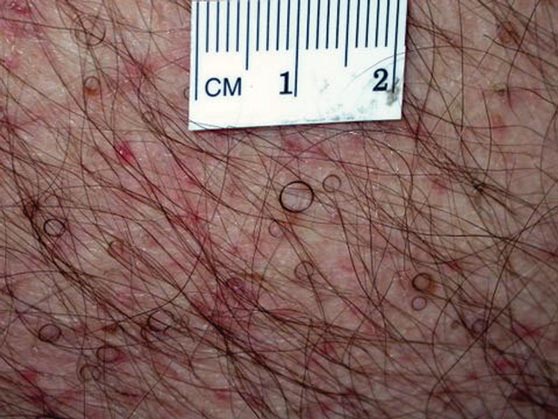
The Diagnosis: Circle Hairs
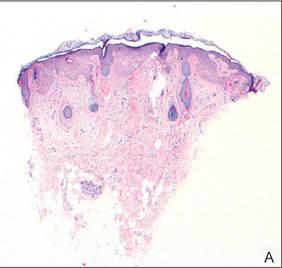
The patient’s hairs were visualized under dermoscopy (Figure 1). A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (Figure 2). The patient was diagnosed with circle hairs.
Circle hairs were first described in 1963.1 These peculiar hairs grow in a circular horizontal distribution beneath the stratum corneum and are considered benign incidental findings. Their exact cause is unknown. If taken out and unrolled, their length and diameter tends to be smaller than surrounding hairs. It has been hypothesized that they are the result of hairs that lack the size necessary to perforate the stratum corneum.2 Others propose that they are vestigial remains that once had a part in preserving body heat.3 Circle hairs tend to grow in elderly, hairy, and obese males, predominantly on the torso and thighs.2,4
It is important to distinguish between circle hairs and rolled hairs. Rolled hairs may be found on the surface or beneath the stratum corneum and are associated with inflammation and keratinization abnormalities.2 If taken together, these latter findings can help differentiate between the two. The importance stands in recognizing that both circle hairs and rolled hairs are benign; however, rolled hairs can be related to other skin disorders that need additional treatment.

 |
 |
| Figure 2. A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (A and B)(both Verhoeff-van Gieson, original magnifications ×40). |
1. Adatto R. Poils en spirale (poils enroules). Dermatologica. 1963;127:145-147.
2. Smith JB, Hogan DJ. Circle hairs are not rolled hairs. J Am Acad Dermatol. 1996;35:634-635.
3. Contreras-Ruiz J, Duran-McKinster C, Tamayo-Sanchez L, et al. Circle hairs: a clinical curiosity. J Eur Acad Dermatol Venereol. 2000;14:495-497.
4. Levit F, Scott MJ Jr. Circle hairs. J Am Acad Dermatol. 1983;8:423-425.
The Diagnosis: Circle Hairs
The patient’s hairs were visualized under dermoscopy (Figure 1). A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (Figure 2). The patient was diagnosed with circle hairs.
Circle hairs were first described in 1963.1 These peculiar hairs grow in a circular horizontal distribution beneath the stratum corneum and are considered benign incidental findings. Their exact cause is unknown. If taken out and unrolled, their length and diameter tends to be smaller than surrounding hairs. It has been hypothesized that they are the result of hairs that lack the size necessary to perforate the stratum corneum.2 Others propose that they are vestigial remains that once had a part in preserving body heat.3 Circle hairs tend to grow in elderly, hairy, and obese males, predominantly on the torso and thighs.2,4
It is important to distinguish between circle hairs and rolled hairs. Rolled hairs may be found on the surface or beneath the stratum corneum and are associated with inflammation and keratinization abnormalities.2 If taken together, these latter findings can help differentiate between the two. The importance stands in recognizing that both circle hairs and rolled hairs are benign; however, rolled hairs can be related to other skin disorders that need additional treatment.
|
|
| Figure 2. A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (A and B)(both Verhoeff-van Gieson, original magnifications ×40). |
The Diagnosis: Circle Hairs
The patient’s hairs were visualized under dermoscopy (Figure 1). A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (Figure 2). The patient was diagnosed with circle hairs.
Circle hairs were first described in 1963.1 These peculiar hairs grow in a circular horizontal distribution beneath the stratum corneum and are considered benign incidental findings. Their exact cause is unknown. If taken out and unrolled, their length and diameter tends to be smaller than surrounding hairs. It has been hypothesized that they are the result of hairs that lack the size necessary to perforate the stratum corneum.2 Others propose that they are vestigial remains that once had a part in preserving body heat.3 Circle hairs tend to grow in elderly, hairy, and obese males, predominantly on the torso and thighs.2,4
It is important to distinguish between circle hairs and rolled hairs. Rolled hairs may be found on the surface or beneath the stratum corneum and are associated with inflammation and keratinization abnormalities.2 If taken together, these latter findings can help differentiate between the two. The importance stands in recognizing that both circle hairs and rolled hairs are benign; however, rolled hairs can be related to other skin disorders that need additional treatment.
|
|
| Figure 2. A skin biopsy showed a terminal hair in a horizontal distribution that was located beneath the stratum corneum (A and B)(both Verhoeff-van Gieson, original magnifications ×40). |
1. Adatto R. Poils en spirale (poils enroules). Dermatologica. 1963;127:145-147.
2. Smith JB, Hogan DJ. Circle hairs are not rolled hairs. J Am Acad Dermatol. 1996;35:634-635.
3. Contreras-Ruiz J, Duran-McKinster C, Tamayo-Sanchez L, et al. Circle hairs: a clinical curiosity. J Eur Acad Dermatol Venereol. 2000;14:495-497.
4. Levit F, Scott MJ Jr. Circle hairs. J Am Acad Dermatol. 1983;8:423-425.
1. Adatto R. Poils en spirale (poils enroules). Dermatologica. 1963;127:145-147.
2. Smith JB, Hogan DJ. Circle hairs are not rolled hairs. J Am Acad Dermatol. 1996;35:634-635.
3. Contreras-Ruiz J, Duran-McKinster C, Tamayo-Sanchez L, et al. Circle hairs: a clinical curiosity. J Eur Acad Dermatol Venereol. 2000;14:495-497.
4. Levit F, Scott MJ Jr. Circle hairs. J Am Acad Dermatol. 1983;8:423-425.
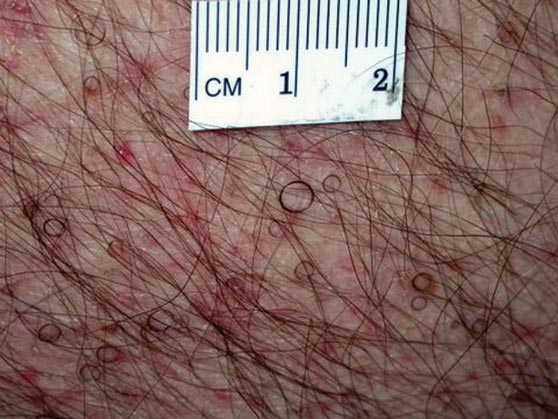
A 74-year-old man was evaluated for numerous peculiar hairs on the back that had been present for several years. He reported no other dermatologic concerns. The patient was obese and led a sedentary lifestyle, spending most of the day sitting or lying down. Physical examination revealed a hairy back with many irregularly shaped hairs.
Hypopigmented Facial Papules on the Cheeks
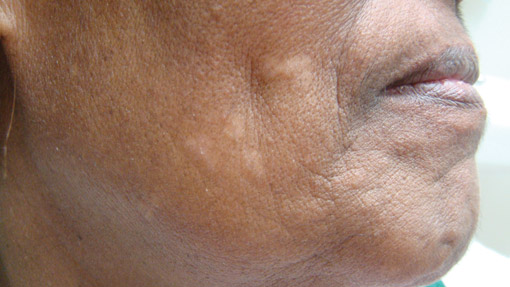
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
 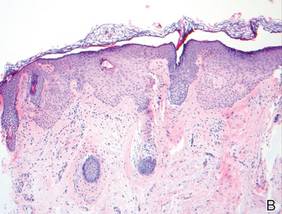 Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
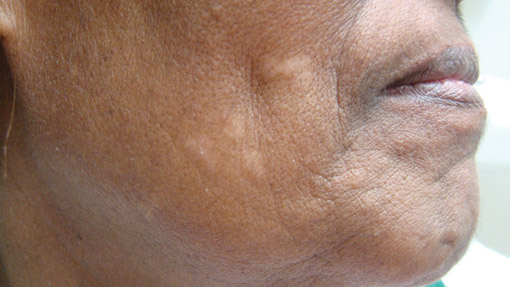
A 73-year-old woman presented with multiple mildly pruritic, hypopigmented, thin papules involving both cheeks of 5 months’ duration. The patient had no improvement with ketoconazole cream 2% and hydrocortisone cream 1% used daily for 1 month for presumed tinea versicolor. Physical examination revealed 10 ill-defined, 2- to 5-mm, round and oval, smooth hypopigmented, slightly raised papules located on the lower aspect of both cheeks.
What Is Your Diagnosis? Cutaneous B-cell Lymphoma
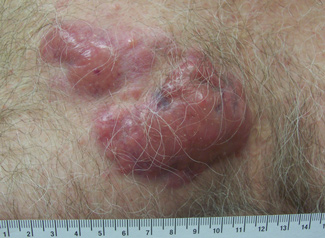
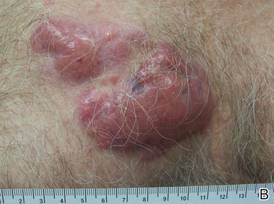
A 59-year-old white man presented with 2 large erythematous lesions on the right side of the chest wall that had gradually progressed over the last 1.5 years. The patient denied any fever, night sweats, fatigue, unintentional weight loss, or loss of appetite. Physical examination revealed 2 large, well-circumscribed, nearly contiguous, firm, erythematous tumors. One tumor measured 7.5×4.5 cm and the other measured 4×3.5 cm.
The Diagnosis: Cutaneous B-cell Lymphoma
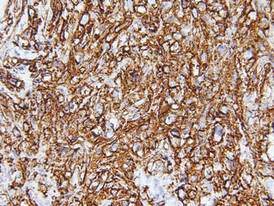
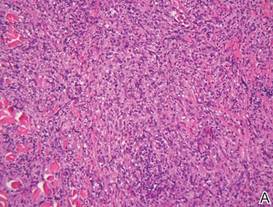
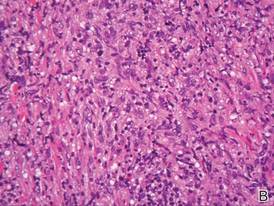
Biopsies from the right side of the chest wall (Figure 1) revealed an atypical dense and diffuse lymphocytic infiltrate throughout the dermis. There was extensive crush artifact throughout the specimen. However, the findings were consistent with cutaneous B-cell lymphoma (CBCL), and the diffuse large B-cell type was favored (Figure 2). Atypical lymphocytes stained positively for antibodies against CD20 (Figure 3), CD79a, and BCL-6, and stained negatively for antibodies against MUM-1 and BCL-2. Although flow cytometry revealed no definitive immunophenotypic lymphoma population, polymerase chain reaction analysis revealed a monoclonal immunoglobulin heavy chain gene rearrangement. Computed tomography (CT) scans of the chest, abdomen, and pelvis were unremarkable. A preliminary diagnosis of primary CBCL (PCBCL) was formulated. Diffuse large B-cell lymphoma (DLBCL) and follicle center lymphoma subtypes were each considered, which triggered further workup to rule out systemic involvement.
|
|
 |
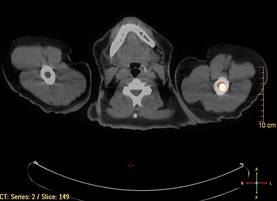 |

A bone marrow biopsy from the posterior iliac crest revealed normocellular bone marrow with normal trilineage hematopoiesis. However, whole-body staging with positron emission tomography (PET)–CT scanning revealed osseous disease in the left proximal humerus (Figure 4) as well as a slightly hypermetabolic right axillary lymph node. Magnetic resonance imaging of the brain showed no evidence of intracranial disease. Because of the apparent systemic involvement, stage IV non-Hodgkin lymphoma (DLBCL) became the new suspected diagnosis. The patient was started on the first of 6 cycles of chemotherapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), and the skin lesions quickly dissipated and flattened. A faint pink discoloration remained over a slightly indented area. A repeat PET-CT scan following 4 cycles of R-CHOP chemotherapy also confirmed a complete response to therapy.
In general, CBCL tends to affect adults and presents as relatively firm and plum-colored papules, nodules, tumors, or plaques, which can be either fast or slow growing. Cutaneous B-cell lymphoma may be primary or secondary to systemic involvement. Primary CBCL refers to a group of non-Hodgkin lymphomas that initially present in the skin with no evidence of extracutaneous involvement at the time of diagnosis.1,2 Secondary CBCL (SCBCL) refers to cutaneous disease that occurs secondary to systemic B-cell lymphoma. Detecting systemic involvement and distinguishing between PCBCL and SCBCL is valuable in determining prognosis and therapeutic options, as subtypes of PCBCL often have an improved prognosis and may be treated with local irradiation.
The initial staging techniques that are preferred for cutaneous lymphomas have been debated.3-5 For cutaneous lymphomas, except mycosis fungoides and Sézary syndrome, the International Society for Cutaneous Lymphomas and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer recommends obtaining a complete blood cell count with differential; complete metabolic studies including lactate dehydrogenase; and imaging studies of the chest, abdomen, and pelvis. Bone marrow biopsies and imaging studies of the neck or whole-body PET-CT scanning also may be useful depending on the clinical scenario.3 Although a more limited workup may be sufficient for PCBCLs such as primary cutaneous marginal zone lymphoma,5 a bone marrow biopsy is recommended for cases of primary cutaneous DLBCL (leg type).3 Senff et al5 supported the use of a bone marrow biopsy in the evaluation of follicle center lymphomas first presenting in the skin, though this method is controversial. In our patient, the laboratory results; bone marrow biopsy; and CT scan of the chest, abdomen, and pelvis failed to suggest extracutaneous disease, while the PET-CT scan revealed systemic involvement.
The differential diagnosis of CBCL includes cutaneous lymphoid hyperplasia (pseudolymphoma), which may be the result of insults such as arthropod bites, stings, vaccinations, or trauma. The clinical presentation, histology, and results of molecular studies and immunohistochemistry are essential in differentiating benign versus malignant processes.6 Lymphomas are expected to be larger and more persistent than benign processes, demonstrating an atypical lymphocytic infiltrate and monoclonality; immunohistochemistry will aid in the distinction between B-cell and T-cell processes and can delineate the type of B-cell lymphoma. Histology for CBCL typically reveals an atypical lymphocytic infiltrate showing a CD20+ and CD79a+ immunophenotype. Staining for antibodies against BCL-2, BCL-6, CD10, and MUM-1 also plays an important role in the diagnosis of cutaneous lymphoma and determining where the lesion(s) falls within the classification schemes. For example, to differentiate between primary cutaneous lymphoma subtypes, BCL-2 negativity and BCL-6 positivity in the context of a CD20+ and CD79a+ immunophenotype supports a follicle center lymphoma or a DLBCL (non–leg type). By contrast, CD20, CD79a, BCL-2, and MUM-1 positivity would favor a DLBCL (leg type).7
The natural history and therapeutic options differ greatly between subtypes of CBCL. For example, the prognosis of primary cutaneous follicle center lymphoma is generally favorable with a 5-year disease-specific survival rate of roughly 95%, and radiation therapy is recommended as a first-line therapy for localized disease.2,8 Conversely, primary cutaneous DLBCL (leg type) frequently spreads to extracutaneous sites8 and carries a much lower estimated 5-year disease-specific survival rate of 55%.2 Chemotherapy with R-CHOP is typically included in initial therapy for primary cutaneous DLBCL (leg type).8 The prognosis of systemic B-cell lymphomas also is highly variable and may depend on the type of B-cell lymphoma, the stage of disease at diagnosis, histologic and immunologic characteristics, and the therapy received. Wright et al9 reported that patients with systemic germinal center B cell–like DLBCL had a 5-year survival rate of 62%, whereas patients with activated B cell–like variants of DLBCL had a 5-year survival rate of 26%. Expression of CD40 may be a favorable prognostic factor following treatment with systemic chemotherapy in patients with DLBCL,10 whereas FOXP1 protein overexpression is correlated with poor disease-specific survival in certain DLBCL phenotypes.11
Although it is uncertain whether the cutaneous lesions preceded systemic disease in our patient, the cutaneous lesions could be arbitrarily classified as secondary because extracutaneous disease was discovered within 6 months of the initial diagnosis.1 However, classifying the CBCL as primary or secondary did not alter the course of treatment in our patient, as the presumed systemic disease necessitated treatment with systemic chemotherapy; both PCBCLs that develop systemic involvement and SCBCLs (primary extracutaneous disease) usually are treated with systemic chemotherapy. Our case highlights the importance of whole-body staging, as PET-CT scanning changed the course of care by detecting osseous involvement, necessitating systemic therapy as opposed to local radiation therapy alone. A multidisciplinary team with a focus on the diagnosis and management of cutaneous lymphomas helped streamline our patient’s laboratory testing and imaging studies, diagnostic and therapeutic decision making, and treatment implementation. Open channels and frequent opportunities for communication among dermatologists, dermatopathologists, medical oncologists, hematopathologists, radiologists, and radiation oncologists are needed to optimize and coordinate care for patients with cutaneous lymphoma who require transdisciplinary care.
Acknowledgement—The authors would like to thank Henry Koon, MD (hematology/oncology), for his input and expertise.
1. Willemze R, Kerl H, Sterry W, et al. EORTC classification for primary cutaneous lymphomas: a proposal from the cutaneous lymphoma study group of the European organization for research and treatment of cancer. Blood. 1997;90:354-371.
2. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
3. Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
4. Quereux G, Frot AS, Brocard A, et al. Routine bone marrow biopsy in the initial evaluation of primary cutaneous B-cell lymphoma does not appear justified. Eur J Dermatol. 2009;19:216-220.
5. Senff NJ, Kluin-Nelemans HC, Willemze R. Results of bone marrow examination in 275 patients with histological features that suggest an indolent type of cutaneous B-cell lymphoma. Br J Haematol. 2008;142:52-56.
6. Gilliam AC, Wood GS. Cutaneous lymphoid hyperplasias. Semin Cutan Med Surg. 2000;19:133-141.
7. Burg G, Kempf W, Cozzio A, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32:647-674.
8. Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112:1600-1609.
9. Wright G, Tan B, Rosenwald A, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9991-9996.
10. Rydström K, Linderoth J, Nyman H, et al. CD40 is a potential marker of favorable prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Leuk Lymphoma. 2010;51:1643-1648.
11. Hoeller S, Schneider A, Haralambieva E, et al. FOXP1 protein overexpression is associated with inferior outcome in nodal diffuse large B-cell lymphomas with non-gerzminal centre phenotype, independent of gains and structural aberrations at 3p14.1. Histopathology. 2010;57:73-80.
A 59-year-old white man presented with 2 large erythematous lesions on the right side of the chest wall that had gradually progressed over the last 1.5 years. The patient denied any fever, night sweats, fatigue, unintentional weight loss, or loss of appetite. Physical examination revealed 2 large, well-circumscribed, nearly contiguous, firm, erythematous tumors. One tumor measured 7.5×4.5 cm and the other measured 4×3.5 cm.
The Diagnosis: Cutaneous B-cell Lymphoma
Biopsies from the right side of the chest wall (Figure 1) revealed an atypical dense and diffuse lymphocytic infiltrate throughout the dermis. There was extensive crush artifact throughout the specimen. However, the findings were consistent with cutaneous B-cell lymphoma (CBCL), and the diffuse large B-cell type was favored (Figure 2). Atypical lymphocytes stained positively for antibodies against CD20 (Figure 3), CD79a, and BCL-6, and stained negatively for antibodies against MUM-1 and BCL-2. Although flow cytometry revealed no definitive immunophenotypic lymphoma population, polymerase chain reaction analysis revealed a monoclonal immunoglobulin heavy chain gene rearrangement. Computed tomography (CT) scans of the chest, abdomen, and pelvis were unremarkable. A preliminary diagnosis of primary CBCL (PCBCL) was formulated. Diffuse large B-cell lymphoma (DLBCL) and follicle center lymphoma subtypes were each considered, which triggered further workup to rule out systemic involvement.
|
|
|
|
A bone marrow biopsy from the posterior iliac crest revealed normocellular bone marrow with normal trilineage hematopoiesis. However, whole-body staging with positron emission tomography (PET)–CT scanning revealed osseous disease in the left proximal humerus (Figure 4) as well as a slightly hypermetabolic right axillary lymph node. Magnetic resonance imaging of the brain showed no evidence of intracranial disease. Because of the apparent systemic involvement, stage IV non-Hodgkin lymphoma (DLBCL) became the new suspected diagnosis. The patient was started on the first of 6 cycles of chemotherapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), and the skin lesions quickly dissipated and flattened. A faint pink discoloration remained over a slightly indented area. A repeat PET-CT scan following 4 cycles of R-CHOP chemotherapy also confirmed a complete response to therapy.
In general, CBCL tends to affect adults and presents as relatively firm and plum-colored papules, nodules, tumors, or plaques, which can be either fast or slow growing. Cutaneous B-cell lymphoma may be primary or secondary to systemic involvement. Primary CBCL refers to a group of non-Hodgkin lymphomas that initially present in the skin with no evidence of extracutaneous involvement at the time of diagnosis.1,2 Secondary CBCL (SCBCL) refers to cutaneous disease that occurs secondary to systemic B-cell lymphoma. Detecting systemic involvement and distinguishing between PCBCL and SCBCL is valuable in determining prognosis and therapeutic options, as subtypes of PCBCL often have an improved prognosis and may be treated with local irradiation.
The initial staging techniques that are preferred for cutaneous lymphomas have been debated.3-5 For cutaneous lymphomas, except mycosis fungoides and Sézary syndrome, the International Society for Cutaneous Lymphomas and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer recommends obtaining a complete blood cell count with differential; complete metabolic studies including lactate dehydrogenase; and imaging studies of the chest, abdomen, and pelvis. Bone marrow biopsies and imaging studies of the neck or whole-body PET-CT scanning also may be useful depending on the clinical scenario.3 Although a more limited workup may be sufficient for PCBCLs such as primary cutaneous marginal zone lymphoma,5 a bone marrow biopsy is recommended for cases of primary cutaneous DLBCL (leg type).3 Senff et al5 supported the use of a bone marrow biopsy in the evaluation of follicle center lymphomas first presenting in the skin, though this method is controversial. In our patient, the laboratory results; bone marrow biopsy; and CT scan of the chest, abdomen, and pelvis failed to suggest extracutaneous disease, while the PET-CT scan revealed systemic involvement.
The differential diagnosis of CBCL includes cutaneous lymphoid hyperplasia (pseudolymphoma), which may be the result of insults such as arthropod bites, stings, vaccinations, or trauma. The clinical presentation, histology, and results of molecular studies and immunohistochemistry are essential in differentiating benign versus malignant processes.6 Lymphomas are expected to be larger and more persistent than benign processes, demonstrating an atypical lymphocytic infiltrate and monoclonality; immunohistochemistry will aid in the distinction between B-cell and T-cell processes and can delineate the type of B-cell lymphoma. Histology for CBCL typically reveals an atypical lymphocytic infiltrate showing a CD20+ and CD79a+ immunophenotype. Staining for antibodies against BCL-2, BCL-6, CD10, and MUM-1 also plays an important role in the diagnosis of cutaneous lymphoma and determining where the lesion(s) falls within the classification schemes. For example, to differentiate between primary cutaneous lymphoma subtypes, BCL-2 negativity and BCL-6 positivity in the context of a CD20+ and CD79a+ immunophenotype supports a follicle center lymphoma or a DLBCL (non–leg type). By contrast, CD20, CD79a, BCL-2, and MUM-1 positivity would favor a DLBCL (leg type).7
The natural history and therapeutic options differ greatly between subtypes of CBCL. For example, the prognosis of primary cutaneous follicle center lymphoma is generally favorable with a 5-year disease-specific survival rate of roughly 95%, and radiation therapy is recommended as a first-line therapy for localized disease.2,8 Conversely, primary cutaneous DLBCL (leg type) frequently spreads to extracutaneous sites8 and carries a much lower estimated 5-year disease-specific survival rate of 55%.2 Chemotherapy with R-CHOP is typically included in initial therapy for primary cutaneous DLBCL (leg type).8 The prognosis of systemic B-cell lymphomas also is highly variable and may depend on the type of B-cell lymphoma, the stage of disease at diagnosis, histologic and immunologic characteristics, and the therapy received. Wright et al9 reported that patients with systemic germinal center B cell–like DLBCL had a 5-year survival rate of 62%, whereas patients with activated B cell–like variants of DLBCL had a 5-year survival rate of 26%. Expression of CD40 may be a favorable prognostic factor following treatment with systemic chemotherapy in patients with DLBCL,10 whereas FOXP1 protein overexpression is correlated with poor disease-specific survival in certain DLBCL phenotypes.11
Although it is uncertain whether the cutaneous lesions preceded systemic disease in our patient, the cutaneous lesions could be arbitrarily classified as secondary because extracutaneous disease was discovered within 6 months of the initial diagnosis.1 However, classifying the CBCL as primary or secondary did not alter the course of treatment in our patient, as the presumed systemic disease necessitated treatment with systemic chemotherapy; both PCBCLs that develop systemic involvement and SCBCLs (primary extracutaneous disease) usually are treated with systemic chemotherapy. Our case highlights the importance of whole-body staging, as PET-CT scanning changed the course of care by detecting osseous involvement, necessitating systemic therapy as opposed to local radiation therapy alone. A multidisciplinary team with a focus on the diagnosis and management of cutaneous lymphomas helped streamline our patient’s laboratory testing and imaging studies, diagnostic and therapeutic decision making, and treatment implementation. Open channels and frequent opportunities for communication among dermatologists, dermatopathologists, medical oncologists, hematopathologists, radiologists, and radiation oncologists are needed to optimize and coordinate care for patients with cutaneous lymphoma who require transdisciplinary care.
Acknowledgement—The authors would like to thank Henry Koon, MD (hematology/oncology), for his input and expertise.
A 59-year-old white man presented with 2 large erythematous lesions on the right side of the chest wall that had gradually progressed over the last 1.5 years. The patient denied any fever, night sweats, fatigue, unintentional weight loss, or loss of appetite. Physical examination revealed 2 large, well-circumscribed, nearly contiguous, firm, erythematous tumors. One tumor measured 7.5×4.5 cm and the other measured 4×3.5 cm.
The Diagnosis: Cutaneous B-cell Lymphoma
Biopsies from the right side of the chest wall (Figure 1) revealed an atypical dense and diffuse lymphocytic infiltrate throughout the dermis. There was extensive crush artifact throughout the specimen. However, the findings were consistent with cutaneous B-cell lymphoma (CBCL), and the diffuse large B-cell type was favored (Figure 2). Atypical lymphocytes stained positively for antibodies against CD20 (Figure 3), CD79a, and BCL-6, and stained negatively for antibodies against MUM-1 and BCL-2. Although flow cytometry revealed no definitive immunophenotypic lymphoma population, polymerase chain reaction analysis revealed a monoclonal immunoglobulin heavy chain gene rearrangement. Computed tomography (CT) scans of the chest, abdomen, and pelvis were unremarkable. A preliminary diagnosis of primary CBCL (PCBCL) was formulated. Diffuse large B-cell lymphoma (DLBCL) and follicle center lymphoma subtypes were each considered, which triggered further workup to rule out systemic involvement.
|
|
|
|
A bone marrow biopsy from the posterior iliac crest revealed normocellular bone marrow with normal trilineage hematopoiesis. However, whole-body staging with positron emission tomography (PET)–CT scanning revealed osseous disease in the left proximal humerus (Figure 4) as well as a slightly hypermetabolic right axillary lymph node. Magnetic resonance imaging of the brain showed no evidence of intracranial disease. Because of the apparent systemic involvement, stage IV non-Hodgkin lymphoma (DLBCL) became the new suspected diagnosis. The patient was started on the first of 6 cycles of chemotherapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), and the skin lesions quickly dissipated and flattened. A faint pink discoloration remained over a slightly indented area. A repeat PET-CT scan following 4 cycles of R-CHOP chemotherapy also confirmed a complete response to therapy.
In general, CBCL tends to affect adults and presents as relatively firm and plum-colored papules, nodules, tumors, or plaques, which can be either fast or slow growing. Cutaneous B-cell lymphoma may be primary or secondary to systemic involvement. Primary CBCL refers to a group of non-Hodgkin lymphomas that initially present in the skin with no evidence of extracutaneous involvement at the time of diagnosis.1,2 Secondary CBCL (SCBCL) refers to cutaneous disease that occurs secondary to systemic B-cell lymphoma. Detecting systemic involvement and distinguishing between PCBCL and SCBCL is valuable in determining prognosis and therapeutic options, as subtypes of PCBCL often have an improved prognosis and may be treated with local irradiation.
The initial staging techniques that are preferred for cutaneous lymphomas have been debated.3-5 For cutaneous lymphomas, except mycosis fungoides and Sézary syndrome, the International Society for Cutaneous Lymphomas and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer recommends obtaining a complete blood cell count with differential; complete metabolic studies including lactate dehydrogenase; and imaging studies of the chest, abdomen, and pelvis. Bone marrow biopsies and imaging studies of the neck or whole-body PET-CT scanning also may be useful depending on the clinical scenario.3 Although a more limited workup may be sufficient for PCBCLs such as primary cutaneous marginal zone lymphoma,5 a bone marrow biopsy is recommended for cases of primary cutaneous DLBCL (leg type).3 Senff et al5 supported the use of a bone marrow biopsy in the evaluation of follicle center lymphomas first presenting in the skin, though this method is controversial. In our patient, the laboratory results; bone marrow biopsy; and CT scan of the chest, abdomen, and pelvis failed to suggest extracutaneous disease, while the PET-CT scan revealed systemic involvement.
The differential diagnosis of CBCL includes cutaneous lymphoid hyperplasia (pseudolymphoma), which may be the result of insults such as arthropod bites, stings, vaccinations, or trauma. The clinical presentation, histology, and results of molecular studies and immunohistochemistry are essential in differentiating benign versus malignant processes.6 Lymphomas are expected to be larger and more persistent than benign processes, demonstrating an atypical lymphocytic infiltrate and monoclonality; immunohistochemistry will aid in the distinction between B-cell and T-cell processes and can delineate the type of B-cell lymphoma. Histology for CBCL typically reveals an atypical lymphocytic infiltrate showing a CD20+ and CD79a+ immunophenotype. Staining for antibodies against BCL-2, BCL-6, CD10, and MUM-1 also plays an important role in the diagnosis of cutaneous lymphoma and determining where the lesion(s) falls within the classification schemes. For example, to differentiate between primary cutaneous lymphoma subtypes, BCL-2 negativity and BCL-6 positivity in the context of a CD20+ and CD79a+ immunophenotype supports a follicle center lymphoma or a DLBCL (non–leg type). By contrast, CD20, CD79a, BCL-2, and MUM-1 positivity would favor a DLBCL (leg type).7
The natural history and therapeutic options differ greatly between subtypes of CBCL. For example, the prognosis of primary cutaneous follicle center lymphoma is generally favorable with a 5-year disease-specific survival rate of roughly 95%, and radiation therapy is recommended as a first-line therapy for localized disease.2,8 Conversely, primary cutaneous DLBCL (leg type) frequently spreads to extracutaneous sites8 and carries a much lower estimated 5-year disease-specific survival rate of 55%.2 Chemotherapy with R-CHOP is typically included in initial therapy for primary cutaneous DLBCL (leg type).8 The prognosis of systemic B-cell lymphomas also is highly variable and may depend on the type of B-cell lymphoma, the stage of disease at diagnosis, histologic and immunologic characteristics, and the therapy received. Wright et al9 reported that patients with systemic germinal center B cell–like DLBCL had a 5-year survival rate of 62%, whereas patients with activated B cell–like variants of DLBCL had a 5-year survival rate of 26%. Expression of CD40 may be a favorable prognostic factor following treatment with systemic chemotherapy in patients with DLBCL,10 whereas FOXP1 protein overexpression is correlated with poor disease-specific survival in certain DLBCL phenotypes.11
Although it is uncertain whether the cutaneous lesions preceded systemic disease in our patient, the cutaneous lesions could be arbitrarily classified as secondary because extracutaneous disease was discovered within 6 months of the initial diagnosis.1 However, classifying the CBCL as primary or secondary did not alter the course of treatment in our patient, as the presumed systemic disease necessitated treatment with systemic chemotherapy; both PCBCLs that develop systemic involvement and SCBCLs (primary extracutaneous disease) usually are treated with systemic chemotherapy. Our case highlights the importance of whole-body staging, as PET-CT scanning changed the course of care by detecting osseous involvement, necessitating systemic therapy as opposed to local radiation therapy alone. A multidisciplinary team with a focus on the diagnosis and management of cutaneous lymphomas helped streamline our patient’s laboratory testing and imaging studies, diagnostic and therapeutic decision making, and treatment implementation. Open channels and frequent opportunities for communication among dermatologists, dermatopathologists, medical oncologists, hematopathologists, radiologists, and radiation oncologists are needed to optimize and coordinate care for patients with cutaneous lymphoma who require transdisciplinary care.
Acknowledgement—The authors would like to thank Henry Koon, MD (hematology/oncology), for his input and expertise.
1. Willemze R, Kerl H, Sterry W, et al. EORTC classification for primary cutaneous lymphomas: a proposal from the cutaneous lymphoma study group of the European organization for research and treatment of cancer. Blood. 1997;90:354-371.
2. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
3. Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
4. Quereux G, Frot AS, Brocard A, et al. Routine bone marrow biopsy in the initial evaluation of primary cutaneous B-cell lymphoma does not appear justified. Eur J Dermatol. 2009;19:216-220.
5. Senff NJ, Kluin-Nelemans HC, Willemze R. Results of bone marrow examination in 275 patients with histological features that suggest an indolent type of cutaneous B-cell lymphoma. Br J Haematol. 2008;142:52-56.
6. Gilliam AC, Wood GS. Cutaneous lymphoid hyperplasias. Semin Cutan Med Surg. 2000;19:133-141.
7. Burg G, Kempf W, Cozzio A, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32:647-674.
8. Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112:1600-1609.
9. Wright G, Tan B, Rosenwald A, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9991-9996.
10. Rydström K, Linderoth J, Nyman H, et al. CD40 is a potential marker of favorable prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Leuk Lymphoma. 2010;51:1643-1648.
11. Hoeller S, Schneider A, Haralambieva E, et al. FOXP1 protein overexpression is associated with inferior outcome in nodal diffuse large B-cell lymphomas with non-gerzminal centre phenotype, independent of gains and structural aberrations at 3p14.1. Histopathology. 2010;57:73-80.
1. Willemze R, Kerl H, Sterry W, et al. EORTC classification for primary cutaneous lymphomas: a proposal from the cutaneous lymphoma study group of the European organization for research and treatment of cancer. Blood. 1997;90:354-371.
2. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
3. Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
4. Quereux G, Frot AS, Brocard A, et al. Routine bone marrow biopsy in the initial evaluation of primary cutaneous B-cell lymphoma does not appear justified. Eur J Dermatol. 2009;19:216-220.
5. Senff NJ, Kluin-Nelemans HC, Willemze R. Results of bone marrow examination in 275 patients with histological features that suggest an indolent type of cutaneous B-cell lymphoma. Br J Haematol. 2008;142:52-56.
6. Gilliam AC, Wood GS. Cutaneous lymphoid hyperplasias. Semin Cutan Med Surg. 2000;19:133-141.
7. Burg G, Kempf W, Cozzio A, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32:647-674.
8. Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112:1600-1609.
9. Wright G, Tan B, Rosenwald A, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9991-9996.
10. Rydström K, Linderoth J, Nyman H, et al. CD40 is a potential marker of favorable prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Leuk Lymphoma. 2010;51:1643-1648.
11. Hoeller S, Schneider A, Haralambieva E, et al. FOXP1 protein overexpression is associated with inferior outcome in nodal diffuse large B-cell lymphomas with non-gerzminal centre phenotype, independent of gains and structural aberrations at 3p14.1. Histopathology. 2010;57:73-80.
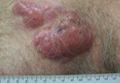
Atrophic Plaques on the Back
The Diagnosis: Atrophic Pityriasis Versicolor
Pityriasis versicolor lesions accompanied by skin atrophy were first reported by De Graciansky and Mery1 in 1971. Since then, few reports have been described and it remains a rare condition.2-5 It manifests with oval to round, ivory-colored lesions with a typically depressed and sometimes finely pleated surface.3 The pathogenesis of the skin atrophy remains controversial. In some of the cases described, the onset of atrophy was related to long-term use of topical steroids.1 This fact as well as impaired barrier function due to fungal infection may explain the atrophy occurring only in the pityriasis versicolor lesions.2,6,7 Some authors call this disease “pityriasis versicolor pseudoatrophicans.”7 However, case reports have been described without use of topical corticosteroids. Crowson and Magro8 maintained that skin atrophy in these cases may occur due to mechanisms of delayed-type hypersensitivity and coined the term atrophying pityriasis versicolor as a variant of this disease. Our patient did not report prior use of topical corticosteroids.
The differential diagnosis consists of other diseases that cause skin atrophy, such as collagen vascular diseases including anetoderma, morphea or atrophoderma, lupus erythematosus, dermatomyositis, and poikilodermatous T-cell dyscrasia; parapsoriasis or mycosis fungoides; sarcoidosis; cutis laxa; acrodermatitis chronica atrophicans; necrobiosis lipoidica; and atrophy due to intralesional steroid therapy.2,3,6-8 Histologic examination helps to achieve proper diagnosis. In our patient, cutaneous biopsy showed the presence of multiple short hyphae and spores in the horny layer with hematoxylin and eosin as well as periodic acid–Schiff stains, with typical “spaghetti and meatballs” appearance. Partial atrophy of the epidermis was observed with flattening of the epidermic ridges. A comparison with the normal areas could not be made because the biopsy was taken from cutaneous lesions without areas of uninvolved skin (Figure).
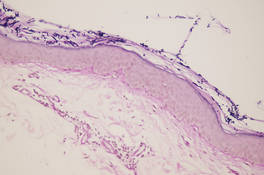
Treatment of this variant does not differ from conventional therapies for pityriasis versicolor, except that a longer treatment period might be required. Atrophy usually disappears, showing that atrophic pityriasis versicolor has a relatively good prognosis compared with other diseases that cause skin atrophy.2
Our patient was treated with ketoconazole gel 2% once daily for 3 weeks with complete resolution of the lesions and no evidence of atrophy.
- De Graciansky P, Mery F. Atrophie sur pityriasis verscolor après corticotherapie locales prolongee. Bull Soc Fr Dermatol Syphiligr. 1971;78:295.
- Yang YS, Shin MK, Haw CR. Atrophying pityriasis versicolor: is this a new variant of pityriasis versicolor? Ann Dermatol. 2010;22:456-459.
- Romano C, Maritati E, Ghilardi A, et al. A case of pityriasis versicolor atrophicans. Mycoses. 2005;48:439-441.
- Park JS, Chae IS, Kim IY, et al. Achromatic atrophic macules and patches of upper extremities. Indian J Dermatol Venereol Leprol. 2013;79:270.
- Tellechea O, Cravo M, Brinca A, et al. Pityriasis versicolor atrophicans. Eur J Dermatol. 2012;22:287-288.
- Mazuecos Blanca J, García-Bravo B, Moreno Giménez JC, et al. Pseudoatrophic pityriasis versicolor. Med Cutan Ibero Lat Am. 1990;18:101-103.
- Tatnall FM, Rycroft RJ. Pityriasis versicolor with cutaneous atrophy induced by topical steroid application. Clin Exp Dermatol. 1985;10:258-261.
- Crowson AN, Magro CM. Atrophying tinea versicolor: a clinical and histological study of 12 patients. Int J Dermatol. 2003;42:928-932.
The Diagnosis: Atrophic Pityriasis Versicolor
Pityriasis versicolor lesions accompanied by skin atrophy were first reported by De Graciansky and Mery1 in 1971. Since then, few reports have been described and it remains a rare condition.2-5 It manifests with oval to round, ivory-colored lesions with a typically depressed and sometimes finely pleated surface.3 The pathogenesis of the skin atrophy remains controversial. In some of the cases described, the onset of atrophy was related to long-term use of topical steroids.1 This fact as well as impaired barrier function due to fungal infection may explain the atrophy occurring only in the pityriasis versicolor lesions.2,6,7 Some authors call this disease “pityriasis versicolor pseudoatrophicans.”7 However, case reports have been described without use of topical corticosteroids. Crowson and Magro8 maintained that skin atrophy in these cases may occur due to mechanisms of delayed-type hypersensitivity and coined the term atrophying pityriasis versicolor as a variant of this disease. Our patient did not report prior use of topical corticosteroids.
The differential diagnosis consists of other diseases that cause skin atrophy, such as collagen vascular diseases including anetoderma, morphea or atrophoderma, lupus erythematosus, dermatomyositis, and poikilodermatous T-cell dyscrasia; parapsoriasis or mycosis fungoides; sarcoidosis; cutis laxa; acrodermatitis chronica atrophicans; necrobiosis lipoidica; and atrophy due to intralesional steroid therapy.2,3,6-8 Histologic examination helps to achieve proper diagnosis. In our patient, cutaneous biopsy showed the presence of multiple short hyphae and spores in the horny layer with hematoxylin and eosin as well as periodic acid–Schiff stains, with typical “spaghetti and meatballs” appearance. Partial atrophy of the epidermis was observed with flattening of the epidermic ridges. A comparison with the normal areas could not be made because the biopsy was taken from cutaneous lesions without areas of uninvolved skin (Figure).
Treatment of this variant does not differ from conventional therapies for pityriasis versicolor, except that a longer treatment period might be required. Atrophy usually disappears, showing that atrophic pityriasis versicolor has a relatively good prognosis compared with other diseases that cause skin atrophy.2
Our patient was treated with ketoconazole gel 2% once daily for 3 weeks with complete resolution of the lesions and no evidence of atrophy.
The Diagnosis: Atrophic Pityriasis Versicolor
Pityriasis versicolor lesions accompanied by skin atrophy were first reported by De Graciansky and Mery1 in 1971. Since then, few reports have been described and it remains a rare condition.2-5 It manifests with oval to round, ivory-colored lesions with a typically depressed and sometimes finely pleated surface.3 The pathogenesis of the skin atrophy remains controversial. In some of the cases described, the onset of atrophy was related to long-term use of topical steroids.1 This fact as well as impaired barrier function due to fungal infection may explain the atrophy occurring only in the pityriasis versicolor lesions.2,6,7 Some authors call this disease “pityriasis versicolor pseudoatrophicans.”7 However, case reports have been described without use of topical corticosteroids. Crowson and Magro8 maintained that skin atrophy in these cases may occur due to mechanisms of delayed-type hypersensitivity and coined the term atrophying pityriasis versicolor as a variant of this disease. Our patient did not report prior use of topical corticosteroids.
The differential diagnosis consists of other diseases that cause skin atrophy, such as collagen vascular diseases including anetoderma, morphea or atrophoderma, lupus erythematosus, dermatomyositis, and poikilodermatous T-cell dyscrasia; parapsoriasis or mycosis fungoides; sarcoidosis; cutis laxa; acrodermatitis chronica atrophicans; necrobiosis lipoidica; and atrophy due to intralesional steroid therapy.2,3,6-8 Histologic examination helps to achieve proper diagnosis. In our patient, cutaneous biopsy showed the presence of multiple short hyphae and spores in the horny layer with hematoxylin and eosin as well as periodic acid–Schiff stains, with typical “spaghetti and meatballs” appearance. Partial atrophy of the epidermis was observed with flattening of the epidermic ridges. A comparison with the normal areas could not be made because the biopsy was taken from cutaneous lesions without areas of uninvolved skin (Figure).
Treatment of this variant does not differ from conventional therapies for pityriasis versicolor, except that a longer treatment period might be required. Atrophy usually disappears, showing that atrophic pityriasis versicolor has a relatively good prognosis compared with other diseases that cause skin atrophy.2
Our patient was treated with ketoconazole gel 2% once daily for 3 weeks with complete resolution of the lesions and no evidence of atrophy.
- De Graciansky P, Mery F. Atrophie sur pityriasis verscolor après corticotherapie locales prolongee. Bull Soc Fr Dermatol Syphiligr. 1971;78:295.
- Yang YS, Shin MK, Haw CR. Atrophying pityriasis versicolor: is this a new variant of pityriasis versicolor? Ann Dermatol. 2010;22:456-459.
- Romano C, Maritati E, Ghilardi A, et al. A case of pityriasis versicolor atrophicans. Mycoses. 2005;48:439-441.
- Park JS, Chae IS, Kim IY, et al. Achromatic atrophic macules and patches of upper extremities. Indian J Dermatol Venereol Leprol. 2013;79:270.
- Tellechea O, Cravo M, Brinca A, et al. Pityriasis versicolor atrophicans. Eur J Dermatol. 2012;22:287-288.
- Mazuecos Blanca J, García-Bravo B, Moreno Giménez JC, et al. Pseudoatrophic pityriasis versicolor. Med Cutan Ibero Lat Am. 1990;18:101-103.
- Tatnall FM, Rycroft RJ. Pityriasis versicolor with cutaneous atrophy induced by topical steroid application. Clin Exp Dermatol. 1985;10:258-261.
- Crowson AN, Magro CM. Atrophying tinea versicolor: a clinical and histological study of 12 patients. Int J Dermatol. 2003;42:928-932.
- De Graciansky P, Mery F. Atrophie sur pityriasis verscolor après corticotherapie locales prolongee. Bull Soc Fr Dermatol Syphiligr. 1971;78:295.
- Yang YS, Shin MK, Haw CR. Atrophying pityriasis versicolor: is this a new variant of pityriasis versicolor? Ann Dermatol. 2010;22:456-459.
- Romano C, Maritati E, Ghilardi A, et al. A case of pityriasis versicolor atrophicans. Mycoses. 2005;48:439-441.
- Park JS, Chae IS, Kim IY, et al. Achromatic atrophic macules and patches of upper extremities. Indian J Dermatol Venereol Leprol. 2013;79:270.
- Tellechea O, Cravo M, Brinca A, et al. Pityriasis versicolor atrophicans. Eur J Dermatol. 2012;22:287-288.
- Mazuecos Blanca J, García-Bravo B, Moreno Giménez JC, et al. Pseudoatrophic pityriasis versicolor. Med Cutan Ibero Lat Am. 1990;18:101-103.
- Tatnall FM, Rycroft RJ. Pityriasis versicolor with cutaneous atrophy induced by topical steroid application. Clin Exp Dermatol. 1985;10:258-261.
- Crowson AN, Magro CM. Atrophying tinea versicolor: a clinical and histological study of 12 patients. Int J Dermatol. 2003;42:928-932.
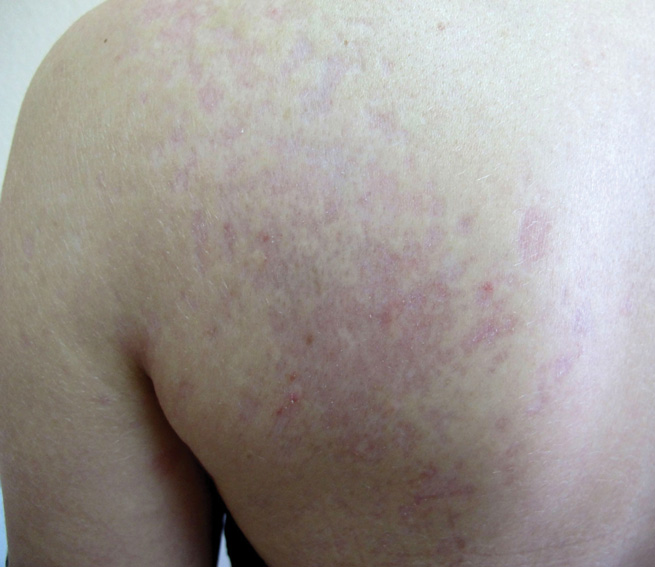
A 41-year-old woman presented with recurrent skin color and violaceous atrophic plaques that were slightly depressed and symmetrically distributed on the back and upper extremities. She had been given oral azithromycin for 3 days without improvement. Laboratory tests, including IgE levels, were within reference range. Her medical history was unremarkable.
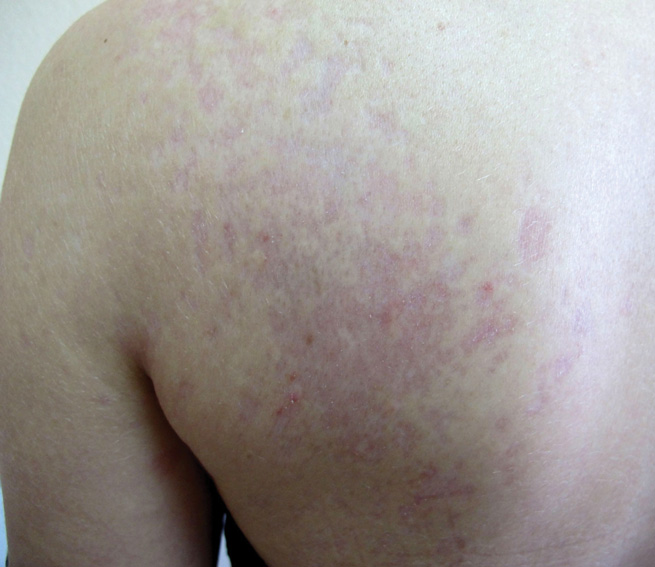
Erythematous Seropurulent Ulcerations
The Diagnosis: Cutaneous Leishmaniasis
On examination, the patient had multiple punched-out ulcers with indurated borders and surrounding erythema arranged in a sporotrichoid pattern from the left forearm to the left lateral chest (Figure 1).
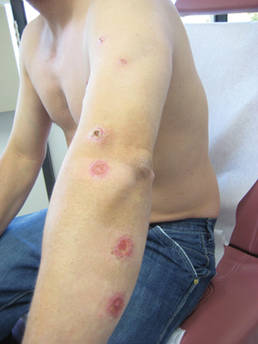
Bacterial culture of a tissue specimen was negative, and tissue fungal culture failed to grow any organisms. Serological studies included a complete blood cell count with differential, a chemistry panel, and liver function tests, which were all unremarkable. Coccidioidomycosis and human immunodefi-ciency virus antibodies were negative. A 4-mm punch biopsy was obtained and sent to the Armed Forces Institute of Pathology for review. Histopathologic examination revealed marked inflammation with ill-formed noncaseating granulomas and focal ulceration, necrosis in the deep dermis, and both intra-cellular and extracellular amastigotes within areas of necrosis (Figures 2 and 3).
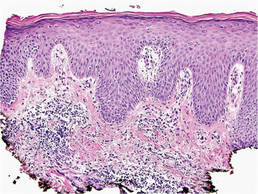
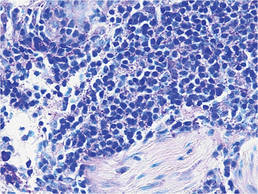
The rise in the number of cases of cutaneous leishmaniasis in the United States, particularly in the veteran population, can be attributed to the recent conflicts in the Middle East and Afghanistan. Infection with Leishmania species can result in a variety of clinical presentations, ranging from localized, self-limited cutaneous lesions to a life-threatening infection with visceral involvement.1 Additionally, the host immune response is variable. This variation in clinical presentation and disease progression explains why there is no single best treatment identified for leishmaniasis to date.
The clinical pattern of spread along the lymphatics in this patient is unique. The differential diagnosis of lesions with sporotrichoid spread includes Mycobacterium marinum and other atypical mycobacterial infections, Sporothrix schenckii, nocardiosis, leishmaniasis, coccidioidomycosis, tularemia, cat scratch disease, anthrax, chromoblastomycosis, pyogenic bacteria, and other fungal or bacterial infections. With such a broad differential diagnosis, histologic confirmation is paramount.
The most widely used pharmacotherapy for leishmaniasis is with pentavalent antimony compounds, which have been studied in randomized controlled trials for leishmaniasis more than any other drug.2 These antimony compounds are associated with a large spectrum of clinical adverse events, and there is increasing evidence for emerging parasite resistance to the antimonies.3-5 Historically, amphotericin B was considered a second-line treatment of leishmaniasis due to its systemic toxicity.6 However, this treatment has come back into favor due to its newer, more tolerable, lipid-associated formulation.
Our patient was treated with intravenous liposomal amphotericin B at a dosage of 3 mg/kg daily for days 1 to 5, then again on days 14 and 21. He tolerated the therapeutic regimen without difficulty or adverse effects. The ulcers eventually became smaller and ceased to weep, fully healing over a course of several months.
1. Martin-Ezquerra G, Fisa R, Riera C, et al. Role of Leishmania spp. infestation in nondiagnostic cutaneous granulomatous lesions: report of a series of patients from a Western Mediterranean area. Br J Dermatol. 2009;161:320-325.
2. Khatami A, Firooz A, Gorouhi F, et al. Treatment of acute old world cutaneous leishmaniasis: a systemic review of the randomized controlled trials. J Am Acad Dermatol. 2007;57:335.e1-335.e29.
3. Rojas R, Valderrama L, Valderrama M, et al. Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J Infect Dis. 2006;193:1375-1383.
4. Hadighi R, Mohebali M, Boucher P, et al. Unresponsiveness to glucantime treatment in Iranian cutaneous leishmaniasis due to drug-resistant Leishmania tropica parasites. PLoS Med. 2006;3:e162.
5. Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111-126.
6. Croft S, Seifert K, Yardley V. Current scenario of drug development for leishmaniasis. Indian J Med Res. 2006;123:399-410.
The Diagnosis: Cutaneous Leishmaniasis
On examination, the patient had multiple punched-out ulcers with indurated borders and surrounding erythema arranged in a sporotrichoid pattern from the left forearm to the left lateral chest (Figure 1).
Bacterial culture of a tissue specimen was negative, and tissue fungal culture failed to grow any organisms. Serological studies included a complete blood cell count with differential, a chemistry panel, and liver function tests, which were all unremarkable. Coccidioidomycosis and human immunodefi-ciency virus antibodies were negative. A 4-mm punch biopsy was obtained and sent to the Armed Forces Institute of Pathology for review. Histopathologic examination revealed marked inflammation with ill-formed noncaseating granulomas and focal ulceration, necrosis in the deep dermis, and both intra-cellular and extracellular amastigotes within areas of necrosis (Figures 2 and 3).
The rise in the number of cases of cutaneous leishmaniasis in the United States, particularly in the veteran population, can be attributed to the recent conflicts in the Middle East and Afghanistan. Infection with Leishmania species can result in a variety of clinical presentations, ranging from localized, self-limited cutaneous lesions to a life-threatening infection with visceral involvement.1 Additionally, the host immune response is variable. This variation in clinical presentation and disease progression explains why there is no single best treatment identified for leishmaniasis to date.
The clinical pattern of spread along the lymphatics in this patient is unique. The differential diagnosis of lesions with sporotrichoid spread includes Mycobacterium marinum and other atypical mycobacterial infections, Sporothrix schenckii, nocardiosis, leishmaniasis, coccidioidomycosis, tularemia, cat scratch disease, anthrax, chromoblastomycosis, pyogenic bacteria, and other fungal or bacterial infections. With such a broad differential diagnosis, histologic confirmation is paramount.
The most widely used pharmacotherapy for leishmaniasis is with pentavalent antimony compounds, which have been studied in randomized controlled trials for leishmaniasis more than any other drug.2 These antimony compounds are associated with a large spectrum of clinical adverse events, and there is increasing evidence for emerging parasite resistance to the antimonies.3-5 Historically, amphotericin B was considered a second-line treatment of leishmaniasis due to its systemic toxicity.6 However, this treatment has come back into favor due to its newer, more tolerable, lipid-associated formulation.
Our patient was treated with intravenous liposomal amphotericin B at a dosage of 3 mg/kg daily for days 1 to 5, then again on days 14 and 21. He tolerated the therapeutic regimen without difficulty or adverse effects. The ulcers eventually became smaller and ceased to weep, fully healing over a course of several months.
The Diagnosis: Cutaneous Leishmaniasis
On examination, the patient had multiple punched-out ulcers with indurated borders and surrounding erythema arranged in a sporotrichoid pattern from the left forearm to the left lateral chest (Figure 1).
Bacterial culture of a tissue specimen was negative, and tissue fungal culture failed to grow any organisms. Serological studies included a complete blood cell count with differential, a chemistry panel, and liver function tests, which were all unremarkable. Coccidioidomycosis and human immunodefi-ciency virus antibodies were negative. A 4-mm punch biopsy was obtained and sent to the Armed Forces Institute of Pathology for review. Histopathologic examination revealed marked inflammation with ill-formed noncaseating granulomas and focal ulceration, necrosis in the deep dermis, and both intra-cellular and extracellular amastigotes within areas of necrosis (Figures 2 and 3).
The rise in the number of cases of cutaneous leishmaniasis in the United States, particularly in the veteran population, can be attributed to the recent conflicts in the Middle East and Afghanistan. Infection with Leishmania species can result in a variety of clinical presentations, ranging from localized, self-limited cutaneous lesions to a life-threatening infection with visceral involvement.1 Additionally, the host immune response is variable. This variation in clinical presentation and disease progression explains why there is no single best treatment identified for leishmaniasis to date.
The clinical pattern of spread along the lymphatics in this patient is unique. The differential diagnosis of lesions with sporotrichoid spread includes Mycobacterium marinum and other atypical mycobacterial infections, Sporothrix schenckii, nocardiosis, leishmaniasis, coccidioidomycosis, tularemia, cat scratch disease, anthrax, chromoblastomycosis, pyogenic bacteria, and other fungal or bacterial infections. With such a broad differential diagnosis, histologic confirmation is paramount.
The most widely used pharmacotherapy for leishmaniasis is with pentavalent antimony compounds, which have been studied in randomized controlled trials for leishmaniasis more than any other drug.2 These antimony compounds are associated with a large spectrum of clinical adverse events, and there is increasing evidence for emerging parasite resistance to the antimonies.3-5 Historically, amphotericin B was considered a second-line treatment of leishmaniasis due to its systemic toxicity.6 However, this treatment has come back into favor due to its newer, more tolerable, lipid-associated formulation.
Our patient was treated with intravenous liposomal amphotericin B at a dosage of 3 mg/kg daily for days 1 to 5, then again on days 14 and 21. He tolerated the therapeutic regimen without difficulty or adverse effects. The ulcers eventually became smaller and ceased to weep, fully healing over a course of several months.
1. Martin-Ezquerra G, Fisa R, Riera C, et al. Role of Leishmania spp. infestation in nondiagnostic cutaneous granulomatous lesions: report of a series of patients from a Western Mediterranean area. Br J Dermatol. 2009;161:320-325.
2. Khatami A, Firooz A, Gorouhi F, et al. Treatment of acute old world cutaneous leishmaniasis: a systemic review of the randomized controlled trials. J Am Acad Dermatol. 2007;57:335.e1-335.e29.
3. Rojas R, Valderrama L, Valderrama M, et al. Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J Infect Dis. 2006;193:1375-1383.
4. Hadighi R, Mohebali M, Boucher P, et al. Unresponsiveness to glucantime treatment in Iranian cutaneous leishmaniasis due to drug-resistant Leishmania tropica parasites. PLoS Med. 2006;3:e162.
5. Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111-126.
6. Croft S, Seifert K, Yardley V. Current scenario of drug development for leishmaniasis. Indian J Med Res. 2006;123:399-410.
1. Martin-Ezquerra G, Fisa R, Riera C, et al. Role of Leishmania spp. infestation in nondiagnostic cutaneous granulomatous lesions: report of a series of patients from a Western Mediterranean area. Br J Dermatol. 2009;161:320-325.
2. Khatami A, Firooz A, Gorouhi F, et al. Treatment of acute old world cutaneous leishmaniasis: a systemic review of the randomized controlled trials. J Am Acad Dermatol. 2007;57:335.e1-335.e29.
3. Rojas R, Valderrama L, Valderrama M, et al. Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J Infect Dis. 2006;193:1375-1383.
4. Hadighi R, Mohebali M, Boucher P, et al. Unresponsiveness to glucantime treatment in Iranian cutaneous leishmaniasis due to drug-resistant Leishmania tropica parasites. PLoS Med. 2006;3:e162.
5. Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111-126.
6. Croft S, Seifert K, Yardley V. Current scenario of drug development for leishmaniasis. Indian J Med Res. 2006;123:399-410.
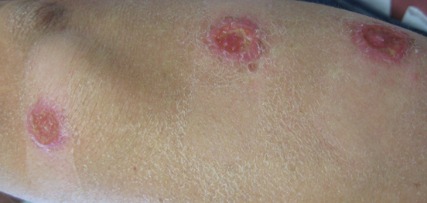
A 34-year-old male veteran who was otherwise healthy presented with multiple ulcerated skin lesions on the left arm and forearm as well as the chest. After returning to the United States from being stationed in Qatar and Saudi Arabia, he noticed multiple “bug bites” on the left arm that eventually progressed to larger crusted ulcerations. He denied fever, chills, nausea, vomiting, abdominal pain, tenderness, or any other symptoms. He had been given doxycycline for a possible bacterial infection, but the lesions did not improve.
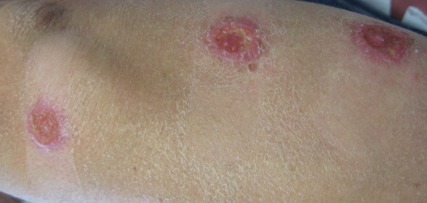
What Is Your Diagnosis? Rhino-orbital-cerebral Mucormycosis
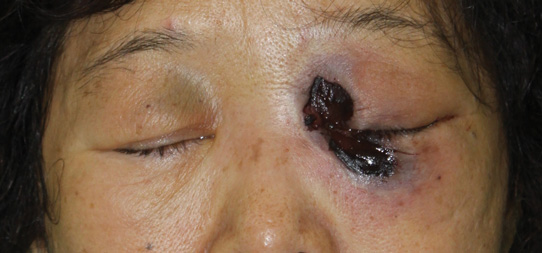
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
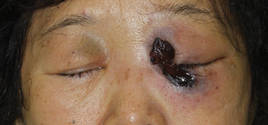 |
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
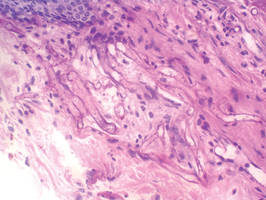 |
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
|
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
|
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
A 56-year-old woman presented with painful, erythematous to violaceous patches with necrosis of the left eye and periorbital area of 1 day’s duration. She reported headaches and periorbital pain in the 3 weeks prior to presentation. She was being treated for hypertension, type 2 diabetes mellitus, and end-stage renal disease. The patient denied prior trauma to the area.
The Diagnosis: Rhino-orbital-cerebral Mucormycosis
Cutaneous examination revealed a dusky, erythematous to violaceous patch with a necrotic center involving the left eye and periorbital area (Figure 1). The differential diagnosis included herpes zoster, cellulitis, and fungal infection. We obtained patient consent for a punch biopsy. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (Figure 2). Magnetic resonance imaging of the orbit and head showed involvement of the periorbital soft tissues; the ethmoidal, sphenoidal, and maxillary sinuses; and the left medial temporal lobe. The patient was started on an empirical antifungal treatment of amphotericin B deoxycholate 50 mg daily but died 4 days later due to multiorgan failure.
|
| Figure 1. A dusky, erythematous to violaceous patch with a necrotic center on the left eye and periorbital area. |
|
| Figure 2. Histopathologic examination revealed irregularly shaped, broad, nonseptate hyphae with right-angle branching (periodic acid–Schiff, original magnification ×400). |
Mucormycosis is a rare but fatal infection that may rapidly progress.1 Risk factors include defects in host defense such as malignancy, immunodeficiency from bone marrow or solid organ transplantation, diabetes mellitus, malnutrition, abnormal metabolic states, and deferoxamine use.1,2 Rhino-orbital-cerebral mucormycosis usually starts with eye or facial pain and unilateral facial swelling.3,4 Visual impairment, fever, and mental status changes may follow.1,3,4 Skin findings may progress from erythema to violaceous color changes and lastly to a black necrotic eschar resulting from tissue infarction.5
Radiologic imaging may be helpful but rarely is diagnostic in mucormycosis, and reliable serologic tests are lacking.1 Therefore, suspicion of mucormycosis based on clinical and histopathologic factors followed by immediate initiation of empirical antifungal treatment is critical. The key factors in treating mucormycosis include early diagnosis, correction of underlying risk factors, prompt antifungal therapy, and surgical debridement.1 Amphotericin B deoxycholate and its lipid derivatives (eg, amphotericin B lipid complex, liposomal amphotericin B) are the standard antifungal agents used in the treatment of mucormycosis.6,7 Posaconazole is an extended-spectrum triazole with in vitro activity against Mucorales. Posaconazole may be useful as salvage therapy; however, strong clinical evidence to support its role as a primary therapeutic agent is lacking in the literature.6,7 Blood vessel thrombosis and tissue necrosis can result in poor penetration of antifungal agents to the infection site; therefore, surgical debridement also may be critical for complete eradication of the disease.6 Confirmative diagnosis of mucormycosis can be made based on histopathologic findings.
Our case highlights the importance of clinician awareness of the typical presentation of rhino-orbital-cerebral mucormycosis to ensure prompt diagnosis and initiation of immediate treatment of this possibly fatal infection.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
1. Spellberg B, Edwards J Jr, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation, and management. Clin Microbiol Rev. 2005;18:556-569.
2. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope. 1982;92:1140-1143.
3. Peterson KL, Wang M, Canalis RF, et al. Rhinocerebral mucormycosis: evolution of the disease and treatment options. Laryngoscope. 1997;107:855-862.
4. Khor BS, Lee MH, Leu HS, et al. Rhinocerebral mucormycosis in Taiwan. J Microbiol Immunol Infect. 2003;36:266-269.
5. Petrikkos G, Skiada A, Sambatakou H, et al. Mucormycosis: ten-year experience at a tertiary-care center in Greece. Eur J Clin Microbiol Infect Dis. 2003;22:753-756.
6. Spellberg B, Ibrahim AS. Recent advances in the treatment of mucormycosis. Curr Infect Dis Rep. 2010;12:423-429.
7. Enoch DA, Aliyu SH, Sule O, et al. Posaconazole for the treatment of mucormycosis. Int J Antimicrob Agents. 2011;38:465-473.
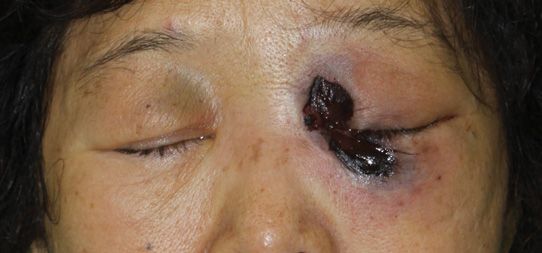
Asymptomatic Annular Plaques on the Neck
The Diagnosis: D-Penicillamine–Induced Elastosis Perforans Serpiginosa
A 27-year-old woman was referred to our clinic by her rheumatologist with a 2×5-cm annular plaque on the right side of the lateral neck along with nummular plaques on the left side of 1 year’s duration (Figure 1). Her medical history was notable for systemic sclerosis that had been treated with oral D-penicillamine (750 mg daily) for the last 10 years. Histopathologic examination of the skin biopsy specimen revealed an epidermis with perforating channels of elastic fibers admixed with collagen (Figure 2). Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (Figure 3), which confirmed the diagnosis of D-penicillamine–induced elastosis perforans serpiginosa (EPS).
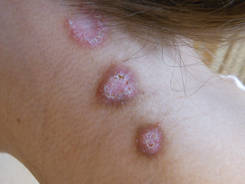 |
Figure 1. Nummular plaques on the left side of the |
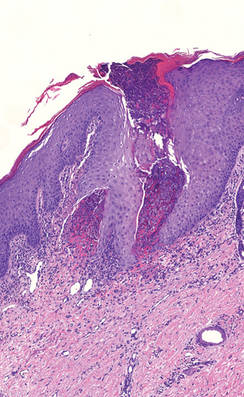 |
Figure 2. A punch biopsy specimen showed an epidermis with perforating channels of elastic fibers admixed with collagen (H&E, original magnification ×10). |
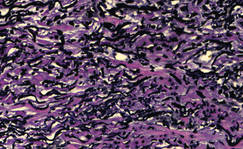 |
Figure 3. Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (original magnification ×40). |
Elastosis perforans serpiginosa is a rare entity that may present in many different settings. Some associated genetic conditions include Down syndrome, pseudoxanthoma elasticum, Marfan syndrome, Ehlers-Danlos syndrome, acrogeria, osteogenesis imperfecta, Rothmund-Thomson syndrome, and moya-moya disease. Elastosis perforans serpiginosa also may be inherited in rare cases in an autosomal-dominant pattern.1 There are solitary reports of EPS in the setting of renal disease, morphea, and systemic sclerosis. Most cases of EPS are iatrogenically acquired. As first reported in 1973, long-term D-penicillamine therapy for Wilson disease has been classically associated with the rare development of EPS.2 Putative mechanisms include copper chelation by D-penicillamine in the setting of altered copper homeostasis in Wilson disease and subsequent inhibition of elastic fiber cross-linking by copper-dependent lysyl oxidase. Another proposed mechanism is the direct inhibition of collagen cross-linking by D-penicillamine resulting in abnormal elastic fiber maturation.3 Outside of the context of Wilson disease, D-penicillamine–induced EPS also has developed during the treatment of juvenile rheumatoid arthritis and cystinuria.4 Our patient withsystemic sclerosis also exemplifies the possibility of developing EPS from long-term D-penicillamine therapy even in the absence of coexisting Wilson disease.
Elastosis perforans serpiginosa lesions classically present as asymptomatic, serpiginously arranged, hyperkeratotic papules, nodules, and annular plaques in young adults and children. Lesions usually present on the neck, though other locations have been described. Histologically, transepidermal elimination of elastic fibers, degenerated keratinocytes, and collagen is seen in the background of a foreign-body reaction with hematoxylin and eosin stain. Elastin stains show increased thickened elastic fibers in the dermis underlying the perforation. The histology of D-penicillamine–induced EPS is distinctive in that the elastic fibers are arranged in a bramble bush pattern with lateral buds.
The clinical course of D-penicillamine–induced EPS is variable, ranging from slow to no resolution after drug discontinuation, with residual scarring, atrophy, and concern for systemic elastosis. Adjunctive therapies include oral and topical retinoids, cryotherapy, imiquimod, and CO2 laser.5 For our patient, tazarotene gel 0.1% was recommended, but the patient became pregnant soon after the diagnosis was made. Despite being untreated, her lesions have remarkably improved during her pregnancy.
1. Langeveld-Wildschut EG, Toonstra J, van Vloten WA, et al. Familial elastosis perforans serpiginosa. Arch Dermatol. 1993;129:205-207.
2. Pass F, Goldfischer S, Sternlieb I, et al. Elastosis perforans serpiginosa during penicillamine therapy for Wilson disease. Arch Dermatol. 1973;108:713-715.
3. Deguti MM, Mucenic M, Cancado EL, et al. Elastosis perforans serpiginosa secondary to D-penicillamine treatment in a Wilson’s disease patient. Am J Gastroenterol. 2002;97:2153-2154.
4. Sahn EE, Maize JC, Garen PD, et al. D-penicillamine–induced elastosis perforans serpiginosa in a child with juvenile rheumatoid arthritis. report of a case and review of the literature. J Am Acad Dermatol. 1989;20:979-988.
5. Atzori L, Pinna AL, Pau M, et al. D-penicillamine elastosis perforans serpiginosa: description of two cases and review of the literature. Dermatol Online J. 2011;17:3.
The Diagnosis: D-Penicillamine–Induced Elastosis Perforans Serpiginosa
A 27-year-old woman was referred to our clinic by her rheumatologist with a 2×5-cm annular plaque on the right side of the lateral neck along with nummular plaques on the left side of 1 year’s duration (Figure 1). Her medical history was notable for systemic sclerosis that had been treated with oral D-penicillamine (750 mg daily) for the last 10 years. Histopathologic examination of the skin biopsy specimen revealed an epidermis with perforating channels of elastic fibers admixed with collagen (Figure 2). Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (Figure 3), which confirmed the diagnosis of D-penicillamine–induced elastosis perforans serpiginosa (EPS).
|
Figure 1. Nummular plaques on the left side of the |
|
Figure 2. A punch biopsy specimen showed an epidermis with perforating channels of elastic fibers admixed with collagen (H&E, original magnification ×10). |
|
Figure 3. Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (original magnification ×40). |
Elastosis perforans serpiginosa is a rare entity that may present in many different settings. Some associated genetic conditions include Down syndrome, pseudoxanthoma elasticum, Marfan syndrome, Ehlers-Danlos syndrome, acrogeria, osteogenesis imperfecta, Rothmund-Thomson syndrome, and moya-moya disease. Elastosis perforans serpiginosa also may be inherited in rare cases in an autosomal-dominant pattern.1 There are solitary reports of EPS in the setting of renal disease, morphea, and systemic sclerosis. Most cases of EPS are iatrogenically acquired. As first reported in 1973, long-term D-penicillamine therapy for Wilson disease has been classically associated with the rare development of EPS.2 Putative mechanisms include copper chelation by D-penicillamine in the setting of altered copper homeostasis in Wilson disease and subsequent inhibition of elastic fiber cross-linking by copper-dependent lysyl oxidase. Another proposed mechanism is the direct inhibition of collagen cross-linking by D-penicillamine resulting in abnormal elastic fiber maturation.3 Outside of the context of Wilson disease, D-penicillamine–induced EPS also has developed during the treatment of juvenile rheumatoid arthritis and cystinuria.4 Our patient withsystemic sclerosis also exemplifies the possibility of developing EPS from long-term D-penicillamine therapy even in the absence of coexisting Wilson disease.
Elastosis perforans serpiginosa lesions classically present as asymptomatic, serpiginously arranged, hyperkeratotic papules, nodules, and annular plaques in young adults and children. Lesions usually present on the neck, though other locations have been described. Histologically, transepidermal elimination of elastic fibers, degenerated keratinocytes, and collagen is seen in the background of a foreign-body reaction with hematoxylin and eosin stain. Elastin stains show increased thickened elastic fibers in the dermis underlying the perforation. The histology of D-penicillamine–induced EPS is distinctive in that the elastic fibers are arranged in a bramble bush pattern with lateral buds.
The clinical course of D-penicillamine–induced EPS is variable, ranging from slow to no resolution after drug discontinuation, with residual scarring, atrophy, and concern for systemic elastosis. Adjunctive therapies include oral and topical retinoids, cryotherapy, imiquimod, and CO2 laser.5 For our patient, tazarotene gel 0.1% was recommended, but the patient became pregnant soon after the diagnosis was made. Despite being untreated, her lesions have remarkably improved during her pregnancy.
The Diagnosis: D-Penicillamine–Induced Elastosis Perforans Serpiginosa
A 27-year-old woman was referred to our clinic by her rheumatologist with a 2×5-cm annular plaque on the right side of the lateral neck along with nummular plaques on the left side of 1 year’s duration (Figure 1). Her medical history was notable for systemic sclerosis that had been treated with oral D-penicillamine (750 mg daily) for the last 10 years. Histopathologic examination of the skin biopsy specimen revealed an epidermis with perforating channels of elastic fibers admixed with collagen (Figure 2). Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (Figure 3), which confirmed the diagnosis of D-penicillamine–induced elastosis perforans serpiginosa (EPS).
|
Figure 1. Nummular plaques on the left side of the |
|
Figure 2. A punch biopsy specimen showed an epidermis with perforating channels of elastic fibers admixed with collagen (H&E, original magnification ×10). |
|
Figure 3. Verhoeff–van Gieson elastin stain highlighted “bramble bush or lumpy-bumpy” appearance of elastic fibers in the dermis (original magnification ×40). |
Elastosis perforans serpiginosa is a rare entity that may present in many different settings. Some associated genetic conditions include Down syndrome, pseudoxanthoma elasticum, Marfan syndrome, Ehlers-Danlos syndrome, acrogeria, osteogenesis imperfecta, Rothmund-Thomson syndrome, and moya-moya disease. Elastosis perforans serpiginosa also may be inherited in rare cases in an autosomal-dominant pattern.1 There are solitary reports of EPS in the setting of renal disease, morphea, and systemic sclerosis. Most cases of EPS are iatrogenically acquired. As first reported in 1973, long-term D-penicillamine therapy for Wilson disease has been classically associated with the rare development of EPS.2 Putative mechanisms include copper chelation by D-penicillamine in the setting of altered copper homeostasis in Wilson disease and subsequent inhibition of elastic fiber cross-linking by copper-dependent lysyl oxidase. Another proposed mechanism is the direct inhibition of collagen cross-linking by D-penicillamine resulting in abnormal elastic fiber maturation.3 Outside of the context of Wilson disease, D-penicillamine–induced EPS also has developed during the treatment of juvenile rheumatoid arthritis and cystinuria.4 Our patient withsystemic sclerosis also exemplifies the possibility of developing EPS from long-term D-penicillamine therapy even in the absence of coexisting Wilson disease.
Elastosis perforans serpiginosa lesions classically present as asymptomatic, serpiginously arranged, hyperkeratotic papules, nodules, and annular plaques in young adults and children. Lesions usually present on the neck, though other locations have been described. Histologically, transepidermal elimination of elastic fibers, degenerated keratinocytes, and collagen is seen in the background of a foreign-body reaction with hematoxylin and eosin stain. Elastin stains show increased thickened elastic fibers in the dermis underlying the perforation. The histology of D-penicillamine–induced EPS is distinctive in that the elastic fibers are arranged in a bramble bush pattern with lateral buds.
The clinical course of D-penicillamine–induced EPS is variable, ranging from slow to no resolution after drug discontinuation, with residual scarring, atrophy, and concern for systemic elastosis. Adjunctive therapies include oral and topical retinoids, cryotherapy, imiquimod, and CO2 laser.5 For our patient, tazarotene gel 0.1% was recommended, but the patient became pregnant soon after the diagnosis was made. Despite being untreated, her lesions have remarkably improved during her pregnancy.
1. Langeveld-Wildschut EG, Toonstra J, van Vloten WA, et al. Familial elastosis perforans serpiginosa. Arch Dermatol. 1993;129:205-207.
2. Pass F, Goldfischer S, Sternlieb I, et al. Elastosis perforans serpiginosa during penicillamine therapy for Wilson disease. Arch Dermatol. 1973;108:713-715.
3. Deguti MM, Mucenic M, Cancado EL, et al. Elastosis perforans serpiginosa secondary to D-penicillamine treatment in a Wilson’s disease patient. Am J Gastroenterol. 2002;97:2153-2154.
4. Sahn EE, Maize JC, Garen PD, et al. D-penicillamine–induced elastosis perforans serpiginosa in a child with juvenile rheumatoid arthritis. report of a case and review of the literature. J Am Acad Dermatol. 1989;20:979-988.
5. Atzori L, Pinna AL, Pau M, et al. D-penicillamine elastosis perforans serpiginosa: description of two cases and review of the literature. Dermatol Online J. 2011;17:3.
1. Langeveld-Wildschut EG, Toonstra J, van Vloten WA, et al. Familial elastosis perforans serpiginosa. Arch Dermatol. 1993;129:205-207.
2. Pass F, Goldfischer S, Sternlieb I, et al. Elastosis perforans serpiginosa during penicillamine therapy for Wilson disease. Arch Dermatol. 1973;108:713-715.
3. Deguti MM, Mucenic M, Cancado EL, et al. Elastosis perforans serpiginosa secondary to D-penicillamine treatment in a Wilson’s disease patient. Am J Gastroenterol. 2002;97:2153-2154.
4. Sahn EE, Maize JC, Garen PD, et al. D-penicillamine–induced elastosis perforans serpiginosa in a child with juvenile rheumatoid arthritis. report of a case and review of the literature. J Am Acad Dermatol. 1989;20:979-988.
5. Atzori L, Pinna AL, Pau M, et al. D-penicillamine elastosis perforans serpiginosa: description of two cases and review of the literature. Dermatol Online J. 2011;17:3.
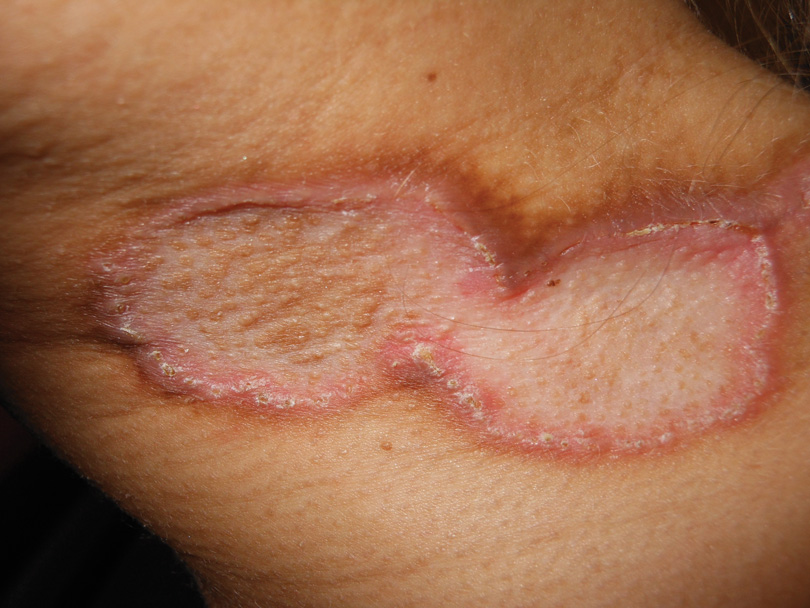
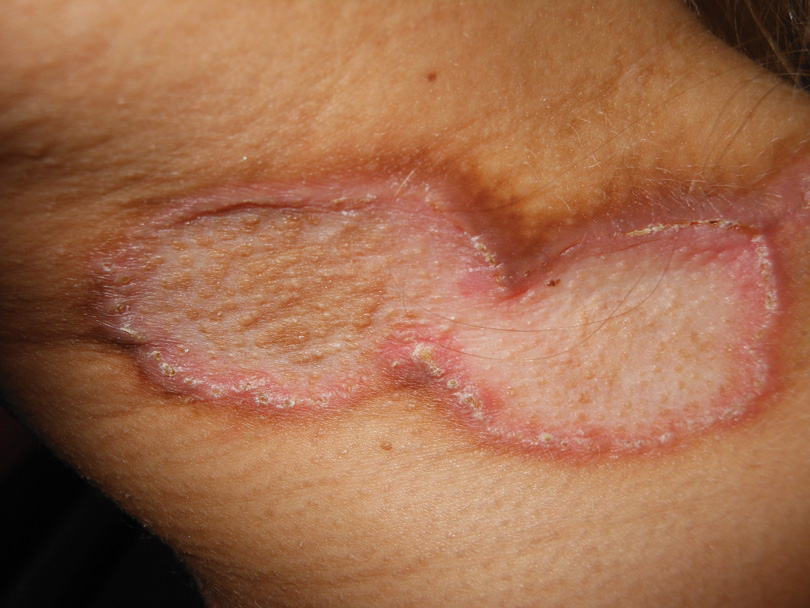
Rapidly Enlarging Noduloulcerative Lesions
The Diagnosis: Lues Maligna
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
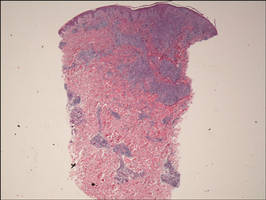 |
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
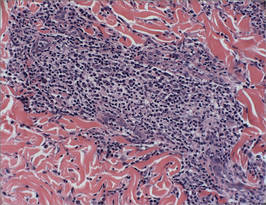 |
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
The Diagnosis: Lues Maligna
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
|
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
|
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
The Diagnosis: Lues Maligna
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
|
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
|
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
A 43-year-old man presented with a rapidly enlarging ulcerated nodule on the right ankle with a necrotic and crusted center. He also had multiple red-brown papules on the trunk and extremities. Some of these lesions had central erosions, while others had surface scale. He was known to be human immunodeficiency virus positive but had no lymphadenopathy. The CD4+ lymphocyte count was 153 cells/mm3 (reference range, 400–1600 cells/mm3) and he was on highly active antiretroviral therapy.