User login
Diffuse Annular Plaques in an Infant
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
A 5-week-old infant boy presented with a rash at birth (left). The pregnancy was full term without complications, and he was otherwise healthy. A family history revealed that his older brother developed a similar rash 2 weeks after birth (right). Physical examination revealed polycyclic annular patches with an erythematous border and central clearing diffusely located on the trunk, extremities, scalp, and face with periorbital edema.


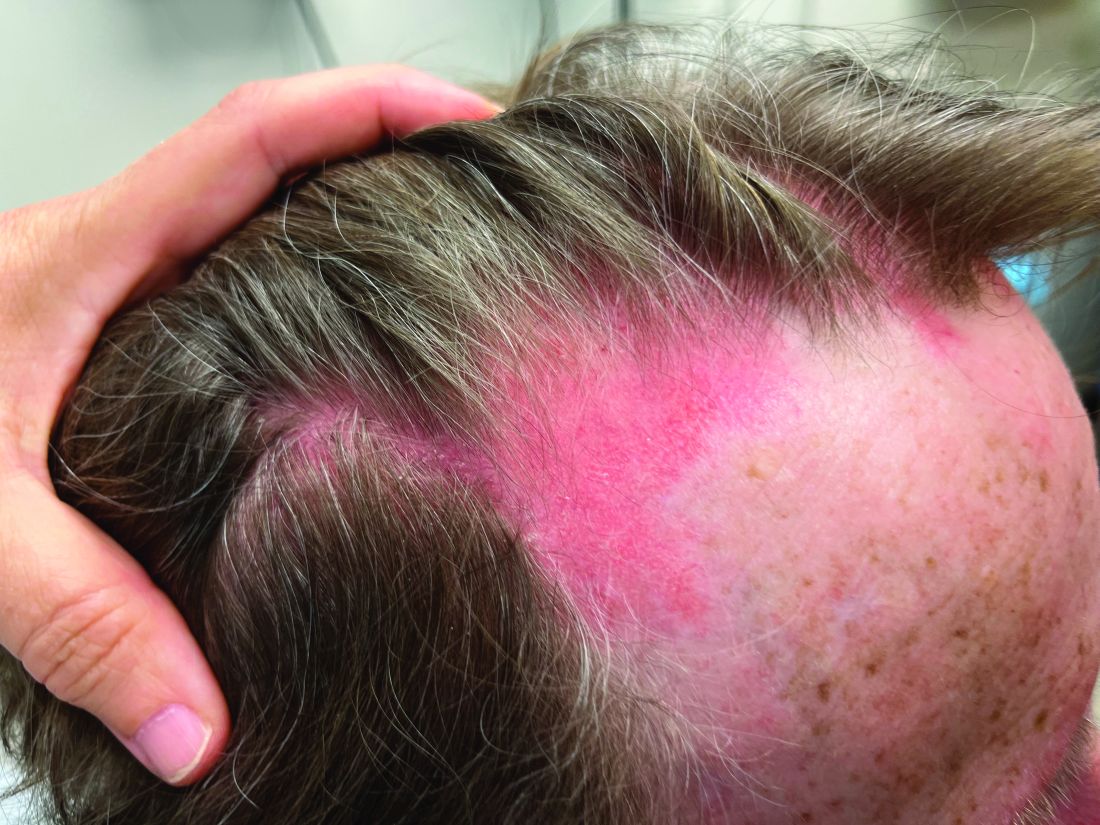
A 75-year-old White woman presented with diffuse erythema, scale, and pruritus on her scalp
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.

Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
Shiny Indurated Plaques on the Legs
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
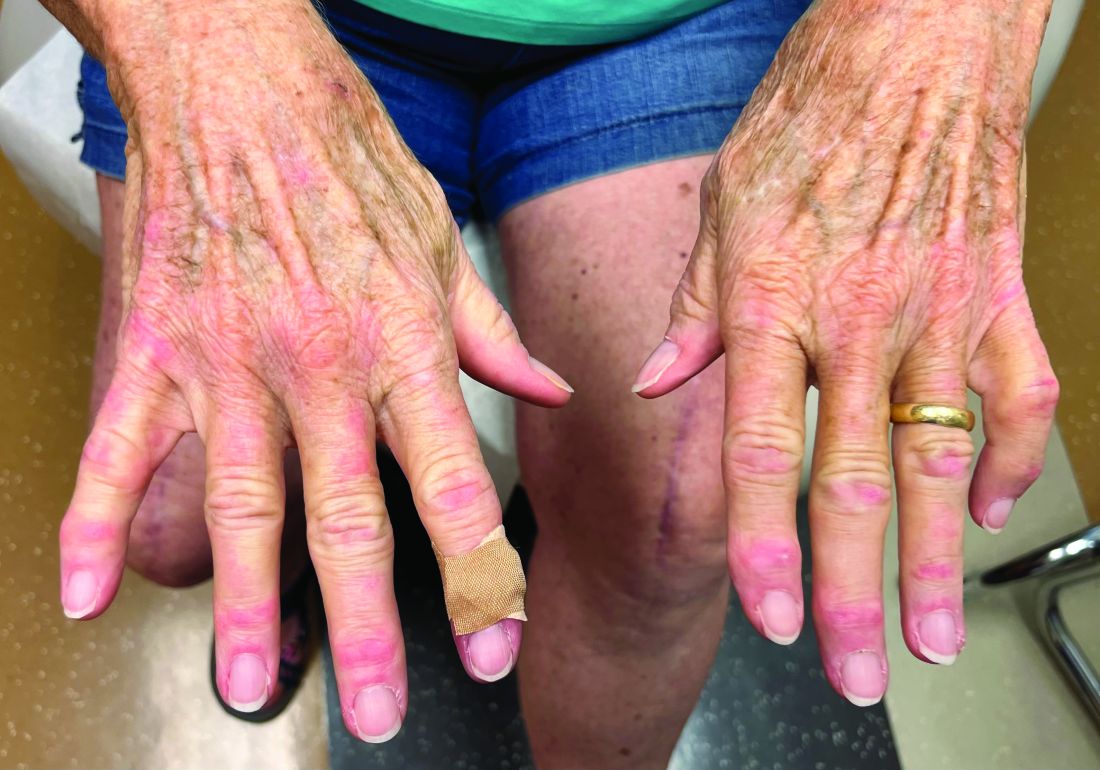
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
The Diagnosis: Pretibial Myxedema
Histopathology showed superficial and deep mucin deposition with proliferation of fibroblasts and thin wiry collagen bundles that were consistent with a diagnosis of pretibial myxedema. The patient was treated with clobetasol ointment 0.05% twice daily for 3 months, followed by a trial of pentoxifylline 400 mg 3 times daily for 3 months. After this treatment failed, she was started on rituximab infusions of 1 g biweekly for 1 month, followed by 500 mg at 6 months, with marked improvement after the first 2 doses of 1 g.
Pretibial myxedema is an uncommon cutaneous manifestation of autoimmune thyroid disease, occurring in 1% to 5% of patients with Graves disease. It usually occurs in older adult women on the pretibial regions and less commonly on the upper extremities, face, and areas of prior trauma.1-3 Although typically asymptomatic, it can be painful and ulcerate.3 The clinical presentation consists of bilateral nonpitting edema with overlying indurated skin as well as flesh-colored, yellow-brown, violaceous, or peau d’orange papules and plaques.2,3 Lesions develop over months and often have been associated with hyperhidrosis and hypertrichosis.2 Many variants have been identified including nodular, plaquelike, diffuse swelling (ie, nonpitting edema), tumor, mixture, polypoid, and elephantiasis; severe cases with acral involvement are termed thyroid acropachy.1-3 Pathogenesis likely involves the activation of thyrotropin receptors on fibroblasts by the circulating thyrotropin autoantibodies found in Graves disease. Activated fibroblasts upregulate glycosaminoglycan production, which osmotically drives the accumulation of dermal and subdermal fluid.1,3
This diagnosis should be considered in any patient with pretibial edema or edema in areas of trauma. Graves disease most commonly is diagnosed 1 to 2 years prior to the development of pretibial myxedema; other extrathyroidal manifestations, most commonly ophthalmopathies, almost always are found in patients with pretibial myxedema. If a diagnosis of Graves disease has not been established, thyroid studies, including thyrotropin receptor antibody serum levels, should be obtained. Histopathology showing increased mucin in the dermis and increased fibroblasts can aid in diagnosis.2,3
The differential diagnosis includes inflammatory dermatoses, such as stasis dermatitis and lipodermatosclerosis. Stasis dermatitis is characterized by lichenified yellowbrown plaques that present on the lower extremities; lipodermatosclerosis then can develop and present as atrophic sclerotic plaques with a champagne bottle–like appearance. Necrobiosis lipoidica demonstrates atrophic, shiny, yellow plaques with telangiectases and ulcerations. Hypertrophic lichen planus presents with hyperkeratotic hyperpigmented plaques on the shins.1,2 Other diseases of cutaneous mucin deposition, namely scleromyxedema, demonstrate similar physical findings but more commonly are located on the trunk, face, and dorsal hands rather than the lower extremities.1-3
Treatment of pretibial myxedema is difficult; normalization of thyroid function, weight reduction, and compression stockings can help reduce edema. Medical therapies aim to decrease glycosaminoglycan production by fibroblasts. First-line treatment includes topical steroids under occlusion, and second-line therapies include intralesional steroids, systemic corticosteroids, pentoxifylline, and octreotide.2,3 Therapies for refractory disease include plasmapheresis, surgical excision, radiotherapy, and intravenous immunoglobulin; more recent studies also endorse the use of isotretinoin, intralesional hyaluronidase, and rituximab.2,4 Success also has been observed with the insulin growth factor 1 receptor inhibitor teprotumumab in active thyroid eye disease, in which insulin growth factor 1 receptor is overexpressed by fibroblasts. Given the similar pathogenesis of thyroid ophthalmopathy with other extrathyroidal manifestations, teprotumumab is a promising option for refractory cases of pretibial myxedema and has led to disease resolution in several patients.4
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). review of 150 cases. Medicine (Baltimore). 1994;73:1-7. doi:10.1097/00005792-199401000-00001
- Ai J, Leonhardt JM, Heymann WR. Autoimmune thyroid diseases: etiology, pathogenesis, and dermatologic manifestations. J Am Acad Dermatol. 2003;48:641-662. doi:10.1067/mjd.2003.257
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446. doi:10.1210/jcem.87.2.8220
- Varma A, Rheeman C, Levitt J. Resolution of pretibial myxedema with teprotumumab in a patient with Graves disease. JAAD Case Reports. 2020;6:1281-1282. doi:10.1016/j.jdcr.2020.09.003
A 70-year-old woman presented with pain and swelling in both legs of many years’ duration. She had no history of skin disease. Physical examination revealed shiny indurated plaques on the legs, ankles, and toes with limited range of motion in the ankles (top). Marked thickening of the hands and index fingers also was noted (bottom). A punch biopsy of the distal pretibial region was performed.


Autoantibody against enteric nervous system protein linked to GI dysfunction in systemic sclerosis
(SSc), new research suggests. Researchers also found that gephyrin is expressed in the patient’s enteric nervous system (ENS), which regulates gut motility.

“While there are many antibodies that are helpful in identifying patients at risk for extraintestinal complications of this disease, markers that identify patients at higher risk for gastrointestinal complications are limited. Furthermore, the biological mechanisms that cause and perpetuate the progression of gastrointestinal disease in scleroderma are not well understood, making it challenging to distinguish between patients whose gastrointestinal disease will progress from those whose GI disease will remain stable/mild,” Zsuzsanna H. McMahan, MD, MHS, told this news organization in an email. Dr. McMahan is co–first author on the study along with Subhash Kulkarni, PhD. They conducted the research with colleagues when they both worked at Johns Hopkins University in Baltimore, Md.

When asked for comment, Kimberly Lakin, MD, MS, assistant professor of medicine at Weill Cornell Medicine and a rheumatologist at Hospital for Special Surgery, New York, called the study “interesting and novel.”
“Not only did [antigephyrin antibodies] correlate with the presence of lower GI symptoms, but also higher levels of antibodies correlated with worse lower GI symptoms. This suggests that not only could this antibody be used to predict who may have constipation and potentially need more aggressive GI interventions, but it may also be useful in quantifying GI severity in systemic sclerosis, although more research is still needed,” said Dr. Lakin, who was not involved with the research.
The study was published online in Arthritis & Rheumatology.
In the cross-sectional study, researchers identified gephyrin as an autoantigen in sera from a single patient with SSc by isolating it from immunoprecipitations performed with murine myenteric plexus neuron lysates, and then characterizing it by mass spectrometry and validating it in further assays. That patient had GI dysfunction but no defined SSc-associated autoantibodies.
Dr. McMahan and colleagues then investigated the prevalence of the autoantibody by screening the sera of 188 patients with SSc who presented consecutively to the Johns Hopkins Scleroderma Center between April 2016 and August 2017, as well as 40 controls, and compared GI symptom severity between antibody-positive and antibody-negative patients with SSc.
A total of 16 (8.5%) of the 188 patients with SSc had antigephyrin antibodies, compared with none of the controls. Of these 16 patients, 4 had no other defined SSc antibodies. In the SSc cohort, severe constipation was more common in antigephyrin antibody–positive patients, compared with antibody-negative patients (46% vs. 15%). Antibody-positive patients also had higher constipation scores, and severe distension and bloating occurred in the antibody-positive group more than twice as often (54% vs. 25%).
Patients with severe constipation, distention, and bloating had higher antigephyrin antibody levels. After adjusting for confounders such as disease duration, patients with severe constipation were nearly five times as likely (odds ratio, 4.74; P = .010) to be antigephyrin antibody–positive, and patients with severe distention and bloating were nearly four times as likely (OR, 3.71; P = .027) to be antibody-positive.
Last, the authors showed via immunohistochemistry that gephyrin is expressed in the myenteric ganglia of human GI tissue.
“Gastrointestinal function is highly regulated by the ENS, so it is interesting that antibodies that target a protein expressed by ENS cells (gephyrin) were identified in patients with scleroderma who have severe lower bowel dysfunction,” said Dr. McMahan, who is associate professor in the division of rheumatology and codirector of the scleroderma program at the University of Texas Health Science Center at Houston. “Gephyrin is a key mediator of normal communications between nerves in the gut, so it is tantalizing to speculate that autoimmune-mediated disruption (e.g., an inhibitory or blocking antibody) in neural (ENS) communications in the gut might lead to impaired bowel transit and prominent constipation.”
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and other NIH grants, as well as the Scleroderma Research Foundation, Rheumatology Research Foundation, Jerome L. Greene Foundation, Martha McCrory Professorship, and Chresanthe Stauraluakis Memorial Discovery Fund. The study authors and Dr. Lakin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(SSc), new research suggests. Researchers also found that gephyrin is expressed in the patient’s enteric nervous system (ENS), which regulates gut motility.
“While there are many antibodies that are helpful in identifying patients at risk for extraintestinal complications of this disease, markers that identify patients at higher risk for gastrointestinal complications are limited. Furthermore, the biological mechanisms that cause and perpetuate the progression of gastrointestinal disease in scleroderma are not well understood, making it challenging to distinguish between patients whose gastrointestinal disease will progress from those whose GI disease will remain stable/mild,” Zsuzsanna H. McMahan, MD, MHS, told this news organization in an email. Dr. McMahan is co–first author on the study along with Subhash Kulkarni, PhD. They conducted the research with colleagues when they both worked at Johns Hopkins University in Baltimore, Md.
When asked for comment, Kimberly Lakin, MD, MS, assistant professor of medicine at Weill Cornell Medicine and a rheumatologist at Hospital for Special Surgery, New York, called the study “interesting and novel.”
“Not only did [antigephyrin antibodies] correlate with the presence of lower GI symptoms, but also higher levels of antibodies correlated with worse lower GI symptoms. This suggests that not only could this antibody be used to predict who may have constipation and potentially need more aggressive GI interventions, but it may also be useful in quantifying GI severity in systemic sclerosis, although more research is still needed,” said Dr. Lakin, who was not involved with the research.
The study was published online in Arthritis & Rheumatology.
In the cross-sectional study, researchers identified gephyrin as an autoantigen in sera from a single patient with SSc by isolating it from immunoprecipitations performed with murine myenteric plexus neuron lysates, and then characterizing it by mass spectrometry and validating it in further assays. That patient had GI dysfunction but no defined SSc-associated autoantibodies.
Dr. McMahan and colleagues then investigated the prevalence of the autoantibody by screening the sera of 188 patients with SSc who presented consecutively to the Johns Hopkins Scleroderma Center between April 2016 and August 2017, as well as 40 controls, and compared GI symptom severity between antibody-positive and antibody-negative patients with SSc.
A total of 16 (8.5%) of the 188 patients with SSc had antigephyrin antibodies, compared with none of the controls. Of these 16 patients, 4 had no other defined SSc antibodies. In the SSc cohort, severe constipation was more common in antigephyrin antibody–positive patients, compared with antibody-negative patients (46% vs. 15%). Antibody-positive patients also had higher constipation scores, and severe distension and bloating occurred in the antibody-positive group more than twice as often (54% vs. 25%).
Patients with severe constipation, distention, and bloating had higher antigephyrin antibody levels. After adjusting for confounders such as disease duration, patients with severe constipation were nearly five times as likely (odds ratio, 4.74; P = .010) to be antigephyrin antibody–positive, and patients with severe distention and bloating were nearly four times as likely (OR, 3.71; P = .027) to be antibody-positive.
Last, the authors showed via immunohistochemistry that gephyrin is expressed in the myenteric ganglia of human GI tissue.
“Gastrointestinal function is highly regulated by the ENS, so it is interesting that antibodies that target a protein expressed by ENS cells (gephyrin) were identified in patients with scleroderma who have severe lower bowel dysfunction,” said Dr. McMahan, who is associate professor in the division of rheumatology and codirector of the scleroderma program at the University of Texas Health Science Center at Houston. “Gephyrin is a key mediator of normal communications between nerves in the gut, so it is tantalizing to speculate that autoimmune-mediated disruption (e.g., an inhibitory or blocking antibody) in neural (ENS) communications in the gut might lead to impaired bowel transit and prominent constipation.”
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and other NIH grants, as well as the Scleroderma Research Foundation, Rheumatology Research Foundation, Jerome L. Greene Foundation, Martha McCrory Professorship, and Chresanthe Stauraluakis Memorial Discovery Fund. The study authors and Dr. Lakin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(SSc), new research suggests. Researchers also found that gephyrin is expressed in the patient’s enteric nervous system (ENS), which regulates gut motility.
“While there are many antibodies that are helpful in identifying patients at risk for extraintestinal complications of this disease, markers that identify patients at higher risk for gastrointestinal complications are limited. Furthermore, the biological mechanisms that cause and perpetuate the progression of gastrointestinal disease in scleroderma are not well understood, making it challenging to distinguish between patients whose gastrointestinal disease will progress from those whose GI disease will remain stable/mild,” Zsuzsanna H. McMahan, MD, MHS, told this news organization in an email. Dr. McMahan is co–first author on the study along with Subhash Kulkarni, PhD. They conducted the research with colleagues when they both worked at Johns Hopkins University in Baltimore, Md.
When asked for comment, Kimberly Lakin, MD, MS, assistant professor of medicine at Weill Cornell Medicine and a rheumatologist at Hospital for Special Surgery, New York, called the study “interesting and novel.”
“Not only did [antigephyrin antibodies] correlate with the presence of lower GI symptoms, but also higher levels of antibodies correlated with worse lower GI symptoms. This suggests that not only could this antibody be used to predict who may have constipation and potentially need more aggressive GI interventions, but it may also be useful in quantifying GI severity in systemic sclerosis, although more research is still needed,” said Dr. Lakin, who was not involved with the research.
The study was published online in Arthritis & Rheumatology.
In the cross-sectional study, researchers identified gephyrin as an autoantigen in sera from a single patient with SSc by isolating it from immunoprecipitations performed with murine myenteric plexus neuron lysates, and then characterizing it by mass spectrometry and validating it in further assays. That patient had GI dysfunction but no defined SSc-associated autoantibodies.
Dr. McMahan and colleagues then investigated the prevalence of the autoantibody by screening the sera of 188 patients with SSc who presented consecutively to the Johns Hopkins Scleroderma Center between April 2016 and August 2017, as well as 40 controls, and compared GI symptom severity between antibody-positive and antibody-negative patients with SSc.
A total of 16 (8.5%) of the 188 patients with SSc had antigephyrin antibodies, compared with none of the controls. Of these 16 patients, 4 had no other defined SSc antibodies. In the SSc cohort, severe constipation was more common in antigephyrin antibody–positive patients, compared with antibody-negative patients (46% vs. 15%). Antibody-positive patients also had higher constipation scores, and severe distension and bloating occurred in the antibody-positive group more than twice as often (54% vs. 25%).
Patients with severe constipation, distention, and bloating had higher antigephyrin antibody levels. After adjusting for confounders such as disease duration, patients with severe constipation were nearly five times as likely (odds ratio, 4.74; P = .010) to be antigephyrin antibody–positive, and patients with severe distention and bloating were nearly four times as likely (OR, 3.71; P = .027) to be antibody-positive.
Last, the authors showed via immunohistochemistry that gephyrin is expressed in the myenteric ganglia of human GI tissue.
“Gastrointestinal function is highly regulated by the ENS, so it is interesting that antibodies that target a protein expressed by ENS cells (gephyrin) were identified in patients with scleroderma who have severe lower bowel dysfunction,” said Dr. McMahan, who is associate professor in the division of rheumatology and codirector of the scleroderma program at the University of Texas Health Science Center at Houston. “Gephyrin is a key mediator of normal communications between nerves in the gut, so it is tantalizing to speculate that autoimmune-mediated disruption (e.g., an inhibitory or blocking antibody) in neural (ENS) communications in the gut might lead to impaired bowel transit and prominent constipation.”
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and other NIH grants, as well as the Scleroderma Research Foundation, Rheumatology Research Foundation, Jerome L. Greene Foundation, Martha McCrory Professorship, and Chresanthe Stauraluakis Memorial Discovery Fund. The study authors and Dr. Lakin report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ARTHRITIS & RHEUMATOLOGY
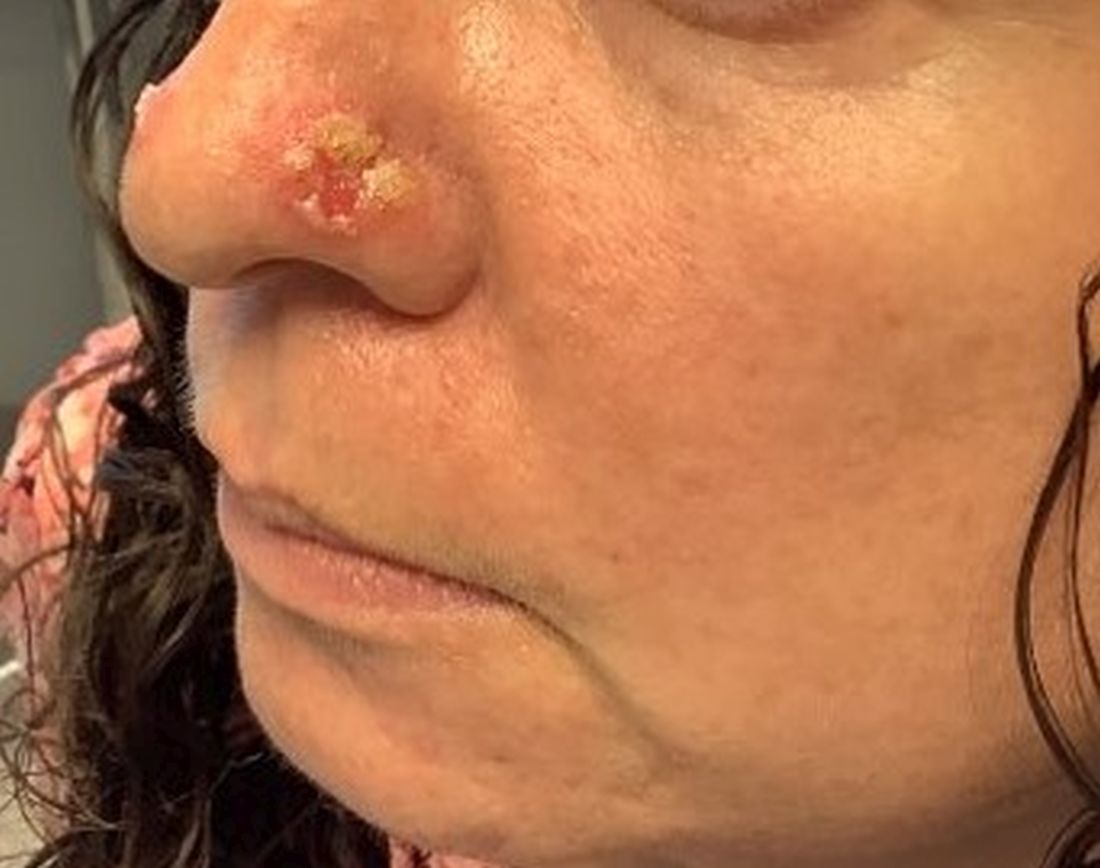
A 45-year-old White woman with no significant medical history presented with a 1-month history of lesions on the nose and right cheek
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Autoantibodies could help predict cancer risk in scleroderma
TOPLINE:
METHODOLOGY:
- Included patients from the Johns Hopkins Scleroderma Center Research Registry and the University of Pittsburgh Scleroderma Center, Pittsburgh.
- A total of 676 patients with scleroderma and a history of cancer were compared with 687 control patients with scleroderma but without a history of cancer.
- Serum tested via line blot and enzyme-linked immunosorbent assay for an array of scleroderma autoantibodies.
- Examined association between autoantibodies and overall cancer risk.
TAKEAWAYS:
- Anti-POLR3 and monospecific anti-Ro52 were associated with significantly increased overall cancer risk.
- Anti-centromere and anti-U1RNP were associated with a decreased cancer risk.
- These associations remained when looking specifically at cancer-associated scleroderma.
- Patients positive for anti-Ro52 in combination with either anti-U1RNP or anti-Th/To had a decreased risk of cancer, compared with those who had anti-Ro52 alone.
IN PRACTICE:
This study is too preliminary to have practice application.
SOURCE:
Ji Soo Kim, PhD, of John Hopkins University, Baltimore, was the first author of the study, published in Arthritis & Rheumatology on July 24, 2023. Fellow Johns Hopkins researchers Livia Casciola-Rosen, PhD, and Ami A. Shah, MD, were joint senior authors.
DISCLOSURES:
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the Donald B. and Dorothy L. Stabler Foundation, the Jerome L. Greene Foundation, the Chresanthe Staurulakis Memorial Discovery Fund, the Martha McCrory Professorship, and the Johns Hopkins inHealth initiative. The authors disclosed the following patents or patent applications: Autoimmune Antigens and Cancer, Materials and Methods for Assessing Cancer Risk and Treating Cancer.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Included patients from the Johns Hopkins Scleroderma Center Research Registry and the University of Pittsburgh Scleroderma Center, Pittsburgh.
- A total of 676 patients with scleroderma and a history of cancer were compared with 687 control patients with scleroderma but without a history of cancer.
- Serum tested via line blot and enzyme-linked immunosorbent assay for an array of scleroderma autoantibodies.
- Examined association between autoantibodies and overall cancer risk.
TAKEAWAYS:
- Anti-POLR3 and monospecific anti-Ro52 were associated with significantly increased overall cancer risk.
- Anti-centromere and anti-U1RNP were associated with a decreased cancer risk.
- These associations remained when looking specifically at cancer-associated scleroderma.
- Patients positive for anti-Ro52 in combination with either anti-U1RNP or anti-Th/To had a decreased risk of cancer, compared with those who had anti-Ro52 alone.
IN PRACTICE:
This study is too preliminary to have practice application.
SOURCE:
Ji Soo Kim, PhD, of John Hopkins University, Baltimore, was the first author of the study, published in Arthritis & Rheumatology on July 24, 2023. Fellow Johns Hopkins researchers Livia Casciola-Rosen, PhD, and Ami A. Shah, MD, were joint senior authors.
DISCLOSURES:
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the Donald B. and Dorothy L. Stabler Foundation, the Jerome L. Greene Foundation, the Chresanthe Staurulakis Memorial Discovery Fund, the Martha McCrory Professorship, and the Johns Hopkins inHealth initiative. The authors disclosed the following patents or patent applications: Autoimmune Antigens and Cancer, Materials and Methods for Assessing Cancer Risk and Treating Cancer.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Included patients from the Johns Hopkins Scleroderma Center Research Registry and the University of Pittsburgh Scleroderma Center, Pittsburgh.
- A total of 676 patients with scleroderma and a history of cancer were compared with 687 control patients with scleroderma but without a history of cancer.
- Serum tested via line blot and enzyme-linked immunosorbent assay for an array of scleroderma autoantibodies.
- Examined association between autoantibodies and overall cancer risk.
TAKEAWAYS:
- Anti-POLR3 and monospecific anti-Ro52 were associated with significantly increased overall cancer risk.
- Anti-centromere and anti-U1RNP were associated with a decreased cancer risk.
- These associations remained when looking specifically at cancer-associated scleroderma.
- Patients positive for anti-Ro52 in combination with either anti-U1RNP or anti-Th/To had a decreased risk of cancer, compared with those who had anti-Ro52 alone.
IN PRACTICE:
This study is too preliminary to have practice application.
SOURCE:
Ji Soo Kim, PhD, of John Hopkins University, Baltimore, was the first author of the study, published in Arthritis & Rheumatology on July 24, 2023. Fellow Johns Hopkins researchers Livia Casciola-Rosen, PhD, and Ami A. Shah, MD, were joint senior authors.
DISCLOSURES:
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the Donald B. and Dorothy L. Stabler Foundation, the Jerome L. Greene Foundation, the Chresanthe Staurulakis Memorial Discovery Fund, the Martha McCrory Professorship, and the Johns Hopkins inHealth initiative. The authors disclosed the following patents or patent applications: Autoimmune Antigens and Cancer, Materials and Methods for Assessing Cancer Risk and Treating Cancer.
A version of this article appeared on Medscape.com.
FROM ARTHRITIS & RHEUMATOLOGY
Rheumatology summit tackles racial disparities in lupus trials
Clinical research in lupus has a mammoth diversity problem: Black individuals are most likely to develop the disease, but they’re the least likely to take part in studies. By the numbers, a 2018 analysis of randomized controlled trials in systemic lupus erythematosus from the years 1997 to 2017 found that 51% of trial participants were White and 14% were Black, even though an estimated 33% of patients with lupus were White and 43% were Black.
Are there ways to fix this disparity? The topic is getting plenty of attention, and speakers at a July 21 online conference touted research projects that aim to boost the numbers of non-White participants in lupus trials.
So far there doesn’t seem to be anything like a magic bullet. Still, the stakes are high. “While race is a social construct, genetic polymorphisms as well as environmental and social differences may influence drugs, safety, and efficacy,” Joy Buie, PhD, MSCR, research director for the Lupus Foundation of America, said at the “Engaging Diverse Participants in Lupus Clinical Trials: The Path Forward” summit held by the American College of Rheumatology (ACR).
As African American patients explained, minority populations often don’t trust the medical system and feel burned by their lengthy struggles to get diagnosed. In some cases, they don’t have full faith in their clinicians and feel unheard.
In a video presentation developed as part of a federal education campaign, Shanelle Gabriel, a poet and musician diagnosed with lupus, described her first reaction when her physician suggested she join a clinical trial. “My first reaction was no. I know my history,” she said, apparently referring to the infamous Tuskegee study that withheld proper treatment from Black men with syphilis for decades. “As an African American woman, I was scared. I didn’t want to be a guinea pig.”
Stacey Kennedy-Conner, a Chicago-area patient and advocate, told the summit audience about how patients can feel that clinical trial information can add “an extra layer of confusion” to their experience. “There’s also the mentality of, ‘If it’s not broke, don’t fix it’: If this medication regimen is working, I don’t want anybody to touch me.”
Monique Gore-Massy, a New York City patient and advocate, added that there can be a perception that patients with lupus “are stuck at home in bed.” In reality, she said, “we have jobs, we have families. Think about that, and consider everything that you’re asking from us: Is this taking me away from my family? Am I going to have to take off work? There may be incentives, but is that worth me taking time off work that I may not get paid for? These are some of the realities that we have to look at in terms of the whole entire clinical trial process.”
It’s also important to keep patients informed of progress being made in trials, she said. “You don’t want to say you just felt like a number and then not get any kind of follow-up.”
In the big picture, “there has to be something that builds up the confidence of individuals so that they are more mindful to participate in these clinical trials,” said Aleta McLean, an Atlanta patient who was diagnosed with lupus 14 years ago.
Several researchers highlighted ongoing projects at the summit. The ACR, for example, has launched a $500,000 initiative called Training to Increase Minority Enrollment in Lupus Clinical Trials with Community Engagement (TIMELY). The federally funded project aims to evaluate whether training of health care professionals can boost clinical trial participation among Black and Hispanic patients.
“We hope to disseminate the results of our project to the scientific community through abstracts, manuscripts, presentations at national meetings,” said rheumatologist Saira Z. Sheikh, MD, of the University of North Carolina at Chapel Hill. “Overall, our goal is to establish new partnerships to support the TIMELY model and advance the education and engagement of providers and community health workers.”
Pamela Payne-Foster, MD, MPH, preventive medicine/public health physician at the University of Alabama College of Community Health Sciences, Tuscaloosa, spoke about the federally funded Deep South Health Equity Project, which is paying patients to take part in an online education program and attend an online regional conference.
Other efforts are underway. The Lupus Research Alliance and its clinical affiliate Lupus Therapeutics have launched two initiatives. One is a program called Project Change (Community-based Health Action Network to Generate Trial Participation and Eliminate Disparities), and the Diversity in Lupus Research Program aims to fund scientists’ work.
Will any of this work boost diversity in clinical trials? As one audience member noted in a Q&A session, health care disparities – and knowledge about them – are nothing new: “Why are we not able to narrow the gap?”
Rear Admiral Richardae Araojo, PharmD, MS, director of the FDA’s Office of Minority Health and Health Equity and associate commissioner for minority health, replied that waves of interest in disparities come and go. “That contributes to why we may not see solutions. But ultimately, there are a lot of people doing a lot of work trying to solve the issues.”
The summit was sponsored by Bristol-Myers Squibb, Genentech, and RemeGen.
A version of this article appeared on Medscape.com.
Clinical research in lupus has a mammoth diversity problem: Black individuals are most likely to develop the disease, but they’re the least likely to take part in studies. By the numbers, a 2018 analysis of randomized controlled trials in systemic lupus erythematosus from the years 1997 to 2017 found that 51% of trial participants were White and 14% were Black, even though an estimated 33% of patients with lupus were White and 43% were Black.
Are there ways to fix this disparity? The topic is getting plenty of attention, and speakers at a July 21 online conference touted research projects that aim to boost the numbers of non-White participants in lupus trials.
So far there doesn’t seem to be anything like a magic bullet. Still, the stakes are high. “While race is a social construct, genetic polymorphisms as well as environmental and social differences may influence drugs, safety, and efficacy,” Joy Buie, PhD, MSCR, research director for the Lupus Foundation of America, said at the “Engaging Diverse Participants in Lupus Clinical Trials: The Path Forward” summit held by the American College of Rheumatology (ACR).
As African American patients explained, minority populations often don’t trust the medical system and feel burned by their lengthy struggles to get diagnosed. In some cases, they don’t have full faith in their clinicians and feel unheard.
In a video presentation developed as part of a federal education campaign, Shanelle Gabriel, a poet and musician diagnosed with lupus, described her first reaction when her physician suggested she join a clinical trial. “My first reaction was no. I know my history,” she said, apparently referring to the infamous Tuskegee study that withheld proper treatment from Black men with syphilis for decades. “As an African American woman, I was scared. I didn’t want to be a guinea pig.”
Stacey Kennedy-Conner, a Chicago-area patient and advocate, told the summit audience about how patients can feel that clinical trial information can add “an extra layer of confusion” to their experience. “There’s also the mentality of, ‘If it’s not broke, don’t fix it’: If this medication regimen is working, I don’t want anybody to touch me.”
Monique Gore-Massy, a New York City patient and advocate, added that there can be a perception that patients with lupus “are stuck at home in bed.” In reality, she said, “we have jobs, we have families. Think about that, and consider everything that you’re asking from us: Is this taking me away from my family? Am I going to have to take off work? There may be incentives, but is that worth me taking time off work that I may not get paid for? These are some of the realities that we have to look at in terms of the whole entire clinical trial process.”
It’s also important to keep patients informed of progress being made in trials, she said. “You don’t want to say you just felt like a number and then not get any kind of follow-up.”
In the big picture, “there has to be something that builds up the confidence of individuals so that they are more mindful to participate in these clinical trials,” said Aleta McLean, an Atlanta patient who was diagnosed with lupus 14 years ago.
Several researchers highlighted ongoing projects at the summit. The ACR, for example, has launched a $500,000 initiative called Training to Increase Minority Enrollment in Lupus Clinical Trials with Community Engagement (TIMELY). The federally funded project aims to evaluate whether training of health care professionals can boost clinical trial participation among Black and Hispanic patients.
“We hope to disseminate the results of our project to the scientific community through abstracts, manuscripts, presentations at national meetings,” said rheumatologist Saira Z. Sheikh, MD, of the University of North Carolina at Chapel Hill. “Overall, our goal is to establish new partnerships to support the TIMELY model and advance the education and engagement of providers and community health workers.”
Pamela Payne-Foster, MD, MPH, preventive medicine/public health physician at the University of Alabama College of Community Health Sciences, Tuscaloosa, spoke about the federally funded Deep South Health Equity Project, which is paying patients to take part in an online education program and attend an online regional conference.
Other efforts are underway. The Lupus Research Alliance and its clinical affiliate Lupus Therapeutics have launched two initiatives. One is a program called Project Change (Community-based Health Action Network to Generate Trial Participation and Eliminate Disparities), and the Diversity in Lupus Research Program aims to fund scientists’ work.
Will any of this work boost diversity in clinical trials? As one audience member noted in a Q&A session, health care disparities – and knowledge about them – are nothing new: “Why are we not able to narrow the gap?”
Rear Admiral Richardae Araojo, PharmD, MS, director of the FDA’s Office of Minority Health and Health Equity and associate commissioner for minority health, replied that waves of interest in disparities come and go. “That contributes to why we may not see solutions. But ultimately, there are a lot of people doing a lot of work trying to solve the issues.”
The summit was sponsored by Bristol-Myers Squibb, Genentech, and RemeGen.
A version of this article appeared on Medscape.com.
Clinical research in lupus has a mammoth diversity problem: Black individuals are most likely to develop the disease, but they’re the least likely to take part in studies. By the numbers, a 2018 analysis of randomized controlled trials in systemic lupus erythematosus from the years 1997 to 2017 found that 51% of trial participants were White and 14% were Black, even though an estimated 33% of patients with lupus were White and 43% were Black.
Are there ways to fix this disparity? The topic is getting plenty of attention, and speakers at a July 21 online conference touted research projects that aim to boost the numbers of non-White participants in lupus trials.
So far there doesn’t seem to be anything like a magic bullet. Still, the stakes are high. “While race is a social construct, genetic polymorphisms as well as environmental and social differences may influence drugs, safety, and efficacy,” Joy Buie, PhD, MSCR, research director for the Lupus Foundation of America, said at the “Engaging Diverse Participants in Lupus Clinical Trials: The Path Forward” summit held by the American College of Rheumatology (ACR).
As African American patients explained, minority populations often don’t trust the medical system and feel burned by their lengthy struggles to get diagnosed. In some cases, they don’t have full faith in their clinicians and feel unheard.
In a video presentation developed as part of a federal education campaign, Shanelle Gabriel, a poet and musician diagnosed with lupus, described her first reaction when her physician suggested she join a clinical trial. “My first reaction was no. I know my history,” she said, apparently referring to the infamous Tuskegee study that withheld proper treatment from Black men with syphilis for decades. “As an African American woman, I was scared. I didn’t want to be a guinea pig.”
Stacey Kennedy-Conner, a Chicago-area patient and advocate, told the summit audience about how patients can feel that clinical trial information can add “an extra layer of confusion” to their experience. “There’s also the mentality of, ‘If it’s not broke, don’t fix it’: If this medication regimen is working, I don’t want anybody to touch me.”
Monique Gore-Massy, a New York City patient and advocate, added that there can be a perception that patients with lupus “are stuck at home in bed.” In reality, she said, “we have jobs, we have families. Think about that, and consider everything that you’re asking from us: Is this taking me away from my family? Am I going to have to take off work? There may be incentives, but is that worth me taking time off work that I may not get paid for? These are some of the realities that we have to look at in terms of the whole entire clinical trial process.”
It’s also important to keep patients informed of progress being made in trials, she said. “You don’t want to say you just felt like a number and then not get any kind of follow-up.”
In the big picture, “there has to be something that builds up the confidence of individuals so that they are more mindful to participate in these clinical trials,” said Aleta McLean, an Atlanta patient who was diagnosed with lupus 14 years ago.
Several researchers highlighted ongoing projects at the summit. The ACR, for example, has launched a $500,000 initiative called Training to Increase Minority Enrollment in Lupus Clinical Trials with Community Engagement (TIMELY). The federally funded project aims to evaluate whether training of health care professionals can boost clinical trial participation among Black and Hispanic patients.
“We hope to disseminate the results of our project to the scientific community through abstracts, manuscripts, presentations at national meetings,” said rheumatologist Saira Z. Sheikh, MD, of the University of North Carolina at Chapel Hill. “Overall, our goal is to establish new partnerships to support the TIMELY model and advance the education and engagement of providers and community health workers.”
Pamela Payne-Foster, MD, MPH, preventive medicine/public health physician at the University of Alabama College of Community Health Sciences, Tuscaloosa, spoke about the federally funded Deep South Health Equity Project, which is paying patients to take part in an online education program and attend an online regional conference.
Other efforts are underway. The Lupus Research Alliance and its clinical affiliate Lupus Therapeutics have launched two initiatives. One is a program called Project Change (Community-based Health Action Network to Generate Trial Participation and Eliminate Disparities), and the Diversity in Lupus Research Program aims to fund scientists’ work.
Will any of this work boost diversity in clinical trials? As one audience member noted in a Q&A session, health care disparities – and knowledge about them – are nothing new: “Why are we not able to narrow the gap?”
Rear Admiral Richardae Araojo, PharmD, MS, director of the FDA’s Office of Minority Health and Health Equity and associate commissioner for minority health, replied that waves of interest in disparities come and go. “That contributes to why we may not see solutions. But ultimately, there are a lot of people doing a lot of work trying to solve the issues.”
The summit was sponsored by Bristol-Myers Squibb, Genentech, and RemeGen.
A version of this article appeared on Medscape.com.
FROM AN ACR CLINICAL TRIAL SUMMIT
Porcelain White, Crinkled, Violaceous Patches on the Inner Thighs
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).

Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.

Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
A 50-year-old woman presented with multiple pruritic lesions on the right inner thigh of 2 years’ duration. Physical examination revealed porcelain white, crinkled, violaceous patches extending from the right inner thigh to the inguinal fold (top). Dermoscopic examination revealed follicular plugs, white structureless areas, white lines, and a rainbow pattern arranged over white polygonal clods on polarized mode (bottom).


Lupus flares linked to gut bacteria overgrowth
Flares of systemic lupus erythematosus (SLE), particularly those involving severe kidney disease, were associated with growth spikes of the gut bacteria Ruminococcus blautia gnavus in a small, 4-year observational study that also demonstrated an underlying, inherent instability in the gut microbiome of patients with SLE.
Of 16 patients with SLE studied during the provision of routine care and monitoring, 5 had R. gnavus blooms that were “strikingly concordant with periods of raised disease activity,” Gregg J. Silverman, MD, of NYU Grossman School of Medicine, New York, and coinvestigators reported in Annals of the Rheumatic Diseases.

Four of the five patients with flare-associated R. gnavus blooms had lupus nephritis (LN); the other had a flare involving inflammation in multiple joints. The four patients with concurrent LN and spikes in R. gnavus also represented almost half of patients who had LN disease flares (four of nine) during the study period. The nine patients in the study with renal involvement, and the four with concurrent R. gnavus spikes and flares, represented different races and ethnicities.
The findings build upon research published by the NYU group several years ago showing that patients with SLE had more R. gnavus in the gut than similar patients without the disease, and that flares closely tracked major increases in R. gnavus growth. Evidence of R. gnavus expansions in patients with SLE now comes from several cohorts in the United States as well as cohorts in Europe and China, the researchers noted in their new paper.
An underlying, unstable microbiome
The new study at NYU took a “deeper dive” than previous research, looking at individuals over a longer period of time, Dr. Silverman, the study’s senior investigator and associate director of rheumatology at NYU Langone Health, said in an interview. Blood and a total of 44 stool samples from the 16 patients were analyzed, as were a total of 72 stool samples from 22 healthy control volunteers.
Importantly, he said, the gut microbiome in patients with SLE was found to be inherently unstable over time, compared with the microbiota communities of the controls. “There was an instability, a shifting dynamic composition of the microbiome [in patients with lupus]. ... Healthy individuals had more of a balance, with small changes over time” and a stable, low abundance of R. gnavus, Dr. Silverman said.
Transient expansions of several pathogenic species occurred in some of the patients with lupus (and not in controls), but blooms of R. gnavus were the most common. The researchers said in their paper that they “speculate that susceptibility for specific clinical features during R. gnavus blooms reflect in part differences in genetic susceptibility of the patient.”
Patients on cytotoxic agents or antibiotics were excluded from the study, but the study was not designed to disentangle the potential influence of diet or prior antibiotic exposure, they noted. Larger studies are needed that are better controlled and that include more frequent assessments, Dr. Silverman added.
A sure association and probable cause
“There seems to be a special connection [of R. gnavus] to lupus nephritis, which is an important, major subset of disease,” said Martin Kriegel, MD, PhD, chief or rheumatology and clinical immunology at the University of Munster (Germany). Dr. Kriegel also researches the gut microbiome in lupus and was asked to comment on the new findings from NYU.

The “difficult question is, is the bug driving the flare [as the NYU paper proposes], or is it the lupus nephritis that leads to overgrowth?” he said, noting that it “is well known that kidney disease, whether from lupus or other causes, creates disturbances in the microbiome.”
It’s “likely the case” that the pathobiome – with R. gnavus being an important pathobiont – helps to drive flares, he said. The new research shows only an association, but studies done in mice – including prior research by Dr. Silverman – support a mechanistic link, said Dr. Kriegel, also adjunct associate professor of immunobiology and of medicine at Yale University, New Haven, Conn.
Investigators in the microbiome space are moving toward more strain-level analysis – “not only measuring what organisms are there, but culturing them and sequencing them,” Dr. Kriegel noted, and the new study does just this.
The R. gnavus strains isolated during lupus flares were distinguishable from strains found in healthy people – and from strains found by other researchers in patients with inflammatory bowel disease – by their common expression of a novel type of cell membrane–associated lipoglycan. The lipoglycans were recognized by specific serum IgG2 antibodies that were detected concurrently with R. gnavus blooms and lupus flares, Dr. Silverman and his colleagues reported.
Dr. Silverman and Dr. Kriegel both study the paradigm of a gut-barrier breach, whereby pathogenic bacteria cause intestinal permeability, allowing bacterial leakages that trigger inflammation and immune responses. “We think that in lupus and other rheumatic diseases like rheumatoid arthritis, a leaky gut barrier is an important mechanism, even though these patients don’t have gastrointestinal symptoms,” said Dr. Kriegel, who has studied the role of another potentially pathogenic bacteria, Enterococcus gallinarum, in SLE.
Strengthening the gut barrier may be a plausible, general approach to reducing the severity of diseases like SLE and RA until more personalized approaches targeting individuals’ microbiome are developed, he noted.
Future treatments involving antibacterial agents, probiotics or dietary regimens that prevent imbalances in the gut microbiome would be “benign,” compared with currently utilized immunosuppressive medications, Dr. Silverman said.
The NYU study was funded in part by grants from the National Institutes of Health and the Lupus Research Alliance. Dr. Silverman disclosed that NYU has filed a patent application for an antibody test to detect serum antibodies to the lipoglycan made by some strains of R. gnavus. Dr. Kriegel disclosed that he holds a patent at Yale related to the Enterococcus bacteria he studies, and that he consults for Roche, Enterome, and Eligo Biosciences.
Flares of systemic lupus erythematosus (SLE), particularly those involving severe kidney disease, were associated with growth spikes of the gut bacteria Ruminococcus blautia gnavus in a small, 4-year observational study that also demonstrated an underlying, inherent instability in the gut microbiome of patients with SLE.
Of 16 patients with SLE studied during the provision of routine care and monitoring, 5 had R. gnavus blooms that were “strikingly concordant with periods of raised disease activity,” Gregg J. Silverman, MD, of NYU Grossman School of Medicine, New York, and coinvestigators reported in Annals of the Rheumatic Diseases.
Four of the five patients with flare-associated R. gnavus blooms had lupus nephritis (LN); the other had a flare involving inflammation in multiple joints. The four patients with concurrent LN and spikes in R. gnavus also represented almost half of patients who had LN disease flares (four of nine) during the study period. The nine patients in the study with renal involvement, and the four with concurrent R. gnavus spikes and flares, represented different races and ethnicities.
The findings build upon research published by the NYU group several years ago showing that patients with SLE had more R. gnavus in the gut than similar patients without the disease, and that flares closely tracked major increases in R. gnavus growth. Evidence of R. gnavus expansions in patients with SLE now comes from several cohorts in the United States as well as cohorts in Europe and China, the researchers noted in their new paper.
An underlying, unstable microbiome
The new study at NYU took a “deeper dive” than previous research, looking at individuals over a longer period of time, Dr. Silverman, the study’s senior investigator and associate director of rheumatology at NYU Langone Health, said in an interview. Blood and a total of 44 stool samples from the 16 patients were analyzed, as were a total of 72 stool samples from 22 healthy control volunteers.
Importantly, he said, the gut microbiome in patients with SLE was found to be inherently unstable over time, compared with the microbiota communities of the controls. “There was an instability, a shifting dynamic composition of the microbiome [in patients with lupus]. ... Healthy individuals had more of a balance, with small changes over time” and a stable, low abundance of R. gnavus, Dr. Silverman said.
Transient expansions of several pathogenic species occurred in some of the patients with lupus (and not in controls), but blooms of R. gnavus were the most common. The researchers said in their paper that they “speculate that susceptibility for specific clinical features during R. gnavus blooms reflect in part differences in genetic susceptibility of the patient.”
Patients on cytotoxic agents or antibiotics were excluded from the study, but the study was not designed to disentangle the potential influence of diet or prior antibiotic exposure, they noted. Larger studies are needed that are better controlled and that include more frequent assessments, Dr. Silverman added.
A sure association and probable cause
“There seems to be a special connection [of R. gnavus] to lupus nephritis, which is an important, major subset of disease,” said Martin Kriegel, MD, PhD, chief or rheumatology and clinical immunology at the University of Munster (Germany). Dr. Kriegel also researches the gut microbiome in lupus and was asked to comment on the new findings from NYU.
The “difficult question is, is the bug driving the flare [as the NYU paper proposes], or is it the lupus nephritis that leads to overgrowth?” he said, noting that it “is well known that kidney disease, whether from lupus or other causes, creates disturbances in the microbiome.”
It’s “likely the case” that the pathobiome – with R. gnavus being an important pathobiont – helps to drive flares, he said. The new research shows only an association, but studies done in mice – including prior research by Dr. Silverman – support a mechanistic link, said Dr. Kriegel, also adjunct associate professor of immunobiology and of medicine at Yale University, New Haven, Conn.
Investigators in the microbiome space are moving toward more strain-level analysis – “not only measuring what organisms are there, but culturing them and sequencing them,” Dr. Kriegel noted, and the new study does just this.
The R. gnavus strains isolated during lupus flares were distinguishable from strains found in healthy people – and from strains found by other researchers in patients with inflammatory bowel disease – by their common expression of a novel type of cell membrane–associated lipoglycan. The lipoglycans were recognized by specific serum IgG2 antibodies that were detected concurrently with R. gnavus blooms and lupus flares, Dr. Silverman and his colleagues reported.
Dr. Silverman and Dr. Kriegel both study the paradigm of a gut-barrier breach, whereby pathogenic bacteria cause intestinal permeability, allowing bacterial leakages that trigger inflammation and immune responses. “We think that in lupus and other rheumatic diseases like rheumatoid arthritis, a leaky gut barrier is an important mechanism, even though these patients don’t have gastrointestinal symptoms,” said Dr. Kriegel, who has studied the role of another potentially pathogenic bacteria, Enterococcus gallinarum, in SLE.
Strengthening the gut barrier may be a plausible, general approach to reducing the severity of diseases like SLE and RA until more personalized approaches targeting individuals’ microbiome are developed, he noted.
Future treatments involving antibacterial agents, probiotics or dietary regimens that prevent imbalances in the gut microbiome would be “benign,” compared with currently utilized immunosuppressive medications, Dr. Silverman said.
The NYU study was funded in part by grants from the National Institutes of Health and the Lupus Research Alliance. Dr. Silverman disclosed that NYU has filed a patent application for an antibody test to detect serum antibodies to the lipoglycan made by some strains of R. gnavus. Dr. Kriegel disclosed that he holds a patent at Yale related to the Enterococcus bacteria he studies, and that he consults for Roche, Enterome, and Eligo Biosciences.
Flares of systemic lupus erythematosus (SLE), particularly those involving severe kidney disease, were associated with growth spikes of the gut bacteria Ruminococcus blautia gnavus in a small, 4-year observational study that also demonstrated an underlying, inherent instability in the gut microbiome of patients with SLE.
Of 16 patients with SLE studied during the provision of routine care and monitoring, 5 had R. gnavus blooms that were “strikingly concordant with periods of raised disease activity,” Gregg J. Silverman, MD, of NYU Grossman School of Medicine, New York, and coinvestigators reported in Annals of the Rheumatic Diseases.
Four of the five patients with flare-associated R. gnavus blooms had lupus nephritis (LN); the other had a flare involving inflammation in multiple joints. The four patients with concurrent LN and spikes in R. gnavus also represented almost half of patients who had LN disease flares (four of nine) during the study period. The nine patients in the study with renal involvement, and the four with concurrent R. gnavus spikes and flares, represented different races and ethnicities.
The findings build upon research published by the NYU group several years ago showing that patients with SLE had more R. gnavus in the gut than similar patients without the disease, and that flares closely tracked major increases in R. gnavus growth. Evidence of R. gnavus expansions in patients with SLE now comes from several cohorts in the United States as well as cohorts in Europe and China, the researchers noted in their new paper.
An underlying, unstable microbiome
The new study at NYU took a “deeper dive” than previous research, looking at individuals over a longer period of time, Dr. Silverman, the study’s senior investigator and associate director of rheumatology at NYU Langone Health, said in an interview. Blood and a total of 44 stool samples from the 16 patients were analyzed, as were a total of 72 stool samples from 22 healthy control volunteers.
Importantly, he said, the gut microbiome in patients with SLE was found to be inherently unstable over time, compared with the microbiota communities of the controls. “There was an instability, a shifting dynamic composition of the microbiome [in patients with lupus]. ... Healthy individuals had more of a balance, with small changes over time” and a stable, low abundance of R. gnavus, Dr. Silverman said.
Transient expansions of several pathogenic species occurred in some of the patients with lupus (and not in controls), but blooms of R. gnavus were the most common. The researchers said in their paper that they “speculate that susceptibility for specific clinical features during R. gnavus blooms reflect in part differences in genetic susceptibility of the patient.”
Patients on cytotoxic agents or antibiotics were excluded from the study, but the study was not designed to disentangle the potential influence of diet or prior antibiotic exposure, they noted. Larger studies are needed that are better controlled and that include more frequent assessments, Dr. Silverman added.
A sure association and probable cause
“There seems to be a special connection [of R. gnavus] to lupus nephritis, which is an important, major subset of disease,” said Martin Kriegel, MD, PhD, chief or rheumatology and clinical immunology at the University of Munster (Germany). Dr. Kriegel also researches the gut microbiome in lupus and was asked to comment on the new findings from NYU.
The “difficult question is, is the bug driving the flare [as the NYU paper proposes], or is it the lupus nephritis that leads to overgrowth?” he said, noting that it “is well known that kidney disease, whether from lupus or other causes, creates disturbances in the microbiome.”
It’s “likely the case” that the pathobiome – with R. gnavus being an important pathobiont – helps to drive flares, he said. The new research shows only an association, but studies done in mice – including prior research by Dr. Silverman – support a mechanistic link, said Dr. Kriegel, also adjunct associate professor of immunobiology and of medicine at Yale University, New Haven, Conn.
Investigators in the microbiome space are moving toward more strain-level analysis – “not only measuring what organisms are there, but culturing them and sequencing them,” Dr. Kriegel noted, and the new study does just this.
The R. gnavus strains isolated during lupus flares were distinguishable from strains found in healthy people – and from strains found by other researchers in patients with inflammatory bowel disease – by their common expression of a novel type of cell membrane–associated lipoglycan. The lipoglycans were recognized by specific serum IgG2 antibodies that were detected concurrently with R. gnavus blooms and lupus flares, Dr. Silverman and his colleagues reported.
Dr. Silverman and Dr. Kriegel both study the paradigm of a gut-barrier breach, whereby pathogenic bacteria cause intestinal permeability, allowing bacterial leakages that trigger inflammation and immune responses. “We think that in lupus and other rheumatic diseases like rheumatoid arthritis, a leaky gut barrier is an important mechanism, even though these patients don’t have gastrointestinal symptoms,” said Dr. Kriegel, who has studied the role of another potentially pathogenic bacteria, Enterococcus gallinarum, in SLE.
Strengthening the gut barrier may be a plausible, general approach to reducing the severity of diseases like SLE and RA until more personalized approaches targeting individuals’ microbiome are developed, he noted.
Future treatments involving antibacterial agents, probiotics or dietary regimens that prevent imbalances in the gut microbiome would be “benign,” compared with currently utilized immunosuppressive medications, Dr. Silverman said.
The NYU study was funded in part by grants from the National Institutes of Health and the Lupus Research Alliance. Dr. Silverman disclosed that NYU has filed a patent application for an antibody test to detect serum antibodies to the lipoglycan made by some strains of R. gnavus. Dr. Kriegel disclosed that he holds a patent at Yale related to the Enterococcus bacteria he studies, and that he consults for Roche, Enterome, and Eligo Biosciences.
FROM ANNALS OF THE RHEUMATIC DISEASES
Racial Disparities in Hidradenitis Suppurativa–Related Pain: A Cross-sectional Analysis
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).


Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
")
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).
Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).
Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
Practice Points
- Racial disparities exist in the management of hidradenitis suppurativa (HS)–related pain.
- Black/African American patients with HS are 4 times more likely to experience very severe pain than patients of other races or ethnicities.
- Lower income levels, higher HS disease severity, and a history of prescription pain medication use are all independent risk factors for very severe pain in patients with HS.