User login
California study indicates increased melanoma incidence is real
A new analysis in non-Hispanic whites suggests that rising melanoma rates are real, not attributable to increased levels of detection, and that the burden of the disease could rise significantly in the coming years.
The incidence of melanoma in light-skinned individuals has been rising worldwide in recent years, but it remains unclear whether that trend is due to an increase in the disease, or better screening and diagnosis. The new results are drawn from California, and track incidence and stage at diagnosis of melanoma across different socioeconomic status (SES) groups. Across all groups, the researchers found increases not only in incidence, but also in advanced disease.
“Our findings support a true real rise in incidence of melanoma across all thicknesses and stages, and not just thinner, more indolent tumors that may be due to increased screening or diagnosis,” lead researcher Susan Swetter, MD, said in an interview. The study was published online in the Journal of Investigative Dermatology (J Invest Dermatol. 2017 Jul 20. pii: S0022-202X(17)31867-5. doi: 10.1016/j.jid.2017.06.024).

Overall, the incidence rose 25% in men from 1998-2002 to 2008-2012 (an average annual age-adjusted incidence of 34.7 to 43.5 per 100,000 person-years), and by 21% in women between those two time periods (from 21.7 to 26.2 per 100,000). Melanoma incidence rate ratios (IRR) increased across all SES classes: by 27% among men in the highest SES neighborhoods, and by 12% among men in the lowest SES neighborhoods. For women, the rates increased by 28% and 13% respectively.
The highest increases in the incidence of regional and distant disease occurred in the lowest SES neighborhoods, nearly doubling in men (distant disease IRR, 1.87; 95% CI, 1.39-2.53; regional disease IRR, 1.93; 95% CI, 1.51-2.47). Women in these neighborhoods also experienced a significant increase in regional disease (IRR, 1.44; 95% CI, 1.00-2.08), but not distant disease.
Incidence of diagnosis with the thickest tumors (greater than 4 mm) rose significantly in most neighborhood SES quartiles, with the exception of the men in the lowest SES quartiles, who had a lower increase that was of borderline significance.
The results solidify the evidence that melanoma incidence is truly increasing, but they also have public health implications. The rising incidence of more advanced disease suggests a heightening health care burden from melanoma in the coming years, but also points to strategies for prevention, according to Dr. Swetter. “It’s important that we focus not only on primary prevention. We need methods to enhance early detection, especially in areas where there is lower access to dermatologists and even primary care providers, who can assist in this effort,” she said.
The Stanford Cancer Institute funded the study. Dr. Swetter reported having no financial disclosures.
A new analysis in non-Hispanic whites suggests that rising melanoma rates are real, not attributable to increased levels of detection, and that the burden of the disease could rise significantly in the coming years.
The incidence of melanoma in light-skinned individuals has been rising worldwide in recent years, but it remains unclear whether that trend is due to an increase in the disease, or better screening and diagnosis. The new results are drawn from California, and track incidence and stage at diagnosis of melanoma across different socioeconomic status (SES) groups. Across all groups, the researchers found increases not only in incidence, but also in advanced disease.
“Our findings support a true real rise in incidence of melanoma across all thicknesses and stages, and not just thinner, more indolent tumors that may be due to increased screening or diagnosis,” lead researcher Susan Swetter, MD, said in an interview. The study was published online in the Journal of Investigative Dermatology (J Invest Dermatol. 2017 Jul 20. pii: S0022-202X(17)31867-5. doi: 10.1016/j.jid.2017.06.024).
Overall, the incidence rose 25% in men from 1998-2002 to 2008-2012 (an average annual age-adjusted incidence of 34.7 to 43.5 per 100,000 person-years), and by 21% in women between those two time periods (from 21.7 to 26.2 per 100,000). Melanoma incidence rate ratios (IRR) increased across all SES classes: by 27% among men in the highest SES neighborhoods, and by 12% among men in the lowest SES neighborhoods. For women, the rates increased by 28% and 13% respectively.
The highest increases in the incidence of regional and distant disease occurred in the lowest SES neighborhoods, nearly doubling in men (distant disease IRR, 1.87; 95% CI, 1.39-2.53; regional disease IRR, 1.93; 95% CI, 1.51-2.47). Women in these neighborhoods also experienced a significant increase in regional disease (IRR, 1.44; 95% CI, 1.00-2.08), but not distant disease.
Incidence of diagnosis with the thickest tumors (greater than 4 mm) rose significantly in most neighborhood SES quartiles, with the exception of the men in the lowest SES quartiles, who had a lower increase that was of borderline significance.
The results solidify the evidence that melanoma incidence is truly increasing, but they also have public health implications. The rising incidence of more advanced disease suggests a heightening health care burden from melanoma in the coming years, but also points to strategies for prevention, according to Dr. Swetter. “It’s important that we focus not only on primary prevention. We need methods to enhance early detection, especially in areas where there is lower access to dermatologists and even primary care providers, who can assist in this effort,” she said.
The Stanford Cancer Institute funded the study. Dr. Swetter reported having no financial disclosures.
A new analysis in non-Hispanic whites suggests that rising melanoma rates are real, not attributable to increased levels of detection, and that the burden of the disease could rise significantly in the coming years.
The incidence of melanoma in light-skinned individuals has been rising worldwide in recent years, but it remains unclear whether that trend is due to an increase in the disease, or better screening and diagnosis. The new results are drawn from California, and track incidence and stage at diagnosis of melanoma across different socioeconomic status (SES) groups. Across all groups, the researchers found increases not only in incidence, but also in advanced disease.
“Our findings support a true real rise in incidence of melanoma across all thicknesses and stages, and not just thinner, more indolent tumors that may be due to increased screening or diagnosis,” lead researcher Susan Swetter, MD, said in an interview. The study was published online in the Journal of Investigative Dermatology (J Invest Dermatol. 2017 Jul 20. pii: S0022-202X(17)31867-5. doi: 10.1016/j.jid.2017.06.024).
Overall, the incidence rose 25% in men from 1998-2002 to 2008-2012 (an average annual age-adjusted incidence of 34.7 to 43.5 per 100,000 person-years), and by 21% in women between those two time periods (from 21.7 to 26.2 per 100,000). Melanoma incidence rate ratios (IRR) increased across all SES classes: by 27% among men in the highest SES neighborhoods, and by 12% among men in the lowest SES neighborhoods. For women, the rates increased by 28% and 13% respectively.
The highest increases in the incidence of regional and distant disease occurred in the lowest SES neighborhoods, nearly doubling in men (distant disease IRR, 1.87; 95% CI, 1.39-2.53; regional disease IRR, 1.93; 95% CI, 1.51-2.47). Women in these neighborhoods also experienced a significant increase in regional disease (IRR, 1.44; 95% CI, 1.00-2.08), but not distant disease.
Incidence of diagnosis with the thickest tumors (greater than 4 mm) rose significantly in most neighborhood SES quartiles, with the exception of the men in the lowest SES quartiles, who had a lower increase that was of borderline significance.
The results solidify the evidence that melanoma incidence is truly increasing, but they also have public health implications. The rising incidence of more advanced disease suggests a heightening health care burden from melanoma in the coming years, but also points to strategies for prevention, according to Dr. Swetter. “It’s important that we focus not only on primary prevention. We need methods to enhance early detection, especially in areas where there is lower access to dermatologists and even primary care providers, who can assist in this effort,” she said.
The Stanford Cancer Institute funded the study. Dr. Swetter reported having no financial disclosures.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Key clinical point: Increased incidences of more advanced disease suggest a rising health care burden.
Major finding: Between 1998-2002 and 2008-2012, incidence rate ratios rose by 25% in men and 21% in women.
Data source: A retrospective study of over 58,000 melanoma cases.
Disclosures: The Stanford Cancer Institute funded the study. Dr. Swetter reported having no financial disclosures.
Nivolumab Linked to Nephritis in Melanoma
Nivolumab and ipilimumab, new immunotherapies for metastatic melanoma, have both been linked to nephritis. Now, researchers from Centre Hospitalier Lyon-Sud and Université Claude Bernard Lyon in France, report on a patient with melanoma who developed acute interstitial immune nephritis after being treated with nivolumab—not once, but twice.
The patient, a 76-year-old woman with pulmonary metastatic melanoma, was given 4 intravenous cycles of ipilimumab as a first-line treatment. After 16 weeks, the disease was progressing; the ipilimumab was discontinued, and 8 weeks later she was started on second-line treatment with nivolumab. After 3 cycles of nivolumab, she developed acute kidney injury. The patient’s creatinine went from 69 µmol/L before nivolumab to 142 µmol/L before the fourth cycle. Immunotherapy was discontinued.
Related: Getting a Better Picture of Skin Cancer
The patient had not received any other drug that could explain the increased creatinine level, the researchers say, and she was otherwise asymptomatic. The renal failure persisted despite an adequate fluid intake over 3 days. Biopsy revealed interstitial edema.
The clinicians treated the patient with oral prednisolone, and her renal function rapidly improved, although her creatinine level remained higher than before the nivolumab.
A follow-up CT scan found a partial response to the nivolumab. Based on that reponse, the multidisciplinary staff elected to continue the treatment at the same dose. The fourth cycle was administered while the patient was still receiving daily corticosteroids.
The infusion did not cause kidney failure relapse. However, after the corticosteroids were stopped, the patient’s creatinine level increased gradually, to 158 µmol/L, and again she was hospitalized with relapse of immune interstitial nephritis. The clinicians reinstituted prednisolone, and the acute interstitial nephritis improved. Nivolumab was discontinued.
Related: Immunotherapy in Melanoma
Drug-induced acute interstitial nephritis often has been more described with nonsteroidal anti-inflammatory drugs and beta-lactams, among others, the researchers say. Immune interstitial nephritis had been reported in a patient treated with nivolumab and ipilimumab concomitantly, and 3 cases of granulomatous interstitial nephritis have been reported with ipilimumab monotherapy. To the authors’ knowledge, this is the first case of immune interstitial nephritis reported with nivolumab monotherapy in metastatic melanoma. It is important to consider, they add, that the patient had also received ipilimumab, and that due to the drug’s elimination half-life (15.4 days), they can’t exclude an “overlap” between the 2 drugs that might have increased the risk of acute interstitial nephritis.
Source:
Bottlaender L, Breton AL, de Laforcade L, Dijoud F, Thomas L, Dalle S. J Immunother Cancer. 2017;5:56.
doi: 10.1186/s40425-017-0261-2
Nivolumab and ipilimumab, new immunotherapies for metastatic melanoma, have both been linked to nephritis. Now, researchers from Centre Hospitalier Lyon-Sud and Université Claude Bernard Lyon in France, report on a patient with melanoma who developed acute interstitial immune nephritis after being treated with nivolumab—not once, but twice.
The patient, a 76-year-old woman with pulmonary metastatic melanoma, was given 4 intravenous cycles of ipilimumab as a first-line treatment. After 16 weeks, the disease was progressing; the ipilimumab was discontinued, and 8 weeks later she was started on second-line treatment with nivolumab. After 3 cycles of nivolumab, she developed acute kidney injury. The patient’s creatinine went from 69 µmol/L before nivolumab to 142 µmol/L before the fourth cycle. Immunotherapy was discontinued.
Related: Getting a Better Picture of Skin Cancer
The patient had not received any other drug that could explain the increased creatinine level, the researchers say, and she was otherwise asymptomatic. The renal failure persisted despite an adequate fluid intake over 3 days. Biopsy revealed interstitial edema.
The clinicians treated the patient with oral prednisolone, and her renal function rapidly improved, although her creatinine level remained higher than before the nivolumab.
A follow-up CT scan found a partial response to the nivolumab. Based on that reponse, the multidisciplinary staff elected to continue the treatment at the same dose. The fourth cycle was administered while the patient was still receiving daily corticosteroids.
The infusion did not cause kidney failure relapse. However, after the corticosteroids were stopped, the patient’s creatinine level increased gradually, to 158 µmol/L, and again she was hospitalized with relapse of immune interstitial nephritis. The clinicians reinstituted prednisolone, and the acute interstitial nephritis improved. Nivolumab was discontinued.
Related: Immunotherapy in Melanoma
Drug-induced acute interstitial nephritis often has been more described with nonsteroidal anti-inflammatory drugs and beta-lactams, among others, the researchers say. Immune interstitial nephritis had been reported in a patient treated with nivolumab and ipilimumab concomitantly, and 3 cases of granulomatous interstitial nephritis have been reported with ipilimumab monotherapy. To the authors’ knowledge, this is the first case of immune interstitial nephritis reported with nivolumab monotherapy in metastatic melanoma. It is important to consider, they add, that the patient had also received ipilimumab, and that due to the drug’s elimination half-life (15.4 days), they can’t exclude an “overlap” between the 2 drugs that might have increased the risk of acute interstitial nephritis.
Source:
Bottlaender L, Breton AL, de Laforcade L, Dijoud F, Thomas L, Dalle S. J Immunother Cancer. 2017;5:56.
doi: 10.1186/s40425-017-0261-2
Nivolumab and ipilimumab, new immunotherapies for metastatic melanoma, have both been linked to nephritis. Now, researchers from Centre Hospitalier Lyon-Sud and Université Claude Bernard Lyon in France, report on a patient with melanoma who developed acute interstitial immune nephritis after being treated with nivolumab—not once, but twice.
The patient, a 76-year-old woman with pulmonary metastatic melanoma, was given 4 intravenous cycles of ipilimumab as a first-line treatment. After 16 weeks, the disease was progressing; the ipilimumab was discontinued, and 8 weeks later she was started on second-line treatment with nivolumab. After 3 cycles of nivolumab, she developed acute kidney injury. The patient’s creatinine went from 69 µmol/L before nivolumab to 142 µmol/L before the fourth cycle. Immunotherapy was discontinued.
Related: Getting a Better Picture of Skin Cancer
The patient had not received any other drug that could explain the increased creatinine level, the researchers say, and she was otherwise asymptomatic. The renal failure persisted despite an adequate fluid intake over 3 days. Biopsy revealed interstitial edema.
The clinicians treated the patient with oral prednisolone, and her renal function rapidly improved, although her creatinine level remained higher than before the nivolumab.
A follow-up CT scan found a partial response to the nivolumab. Based on that reponse, the multidisciplinary staff elected to continue the treatment at the same dose. The fourth cycle was administered while the patient was still receiving daily corticosteroids.
The infusion did not cause kidney failure relapse. However, after the corticosteroids were stopped, the patient’s creatinine level increased gradually, to 158 µmol/L, and again she was hospitalized with relapse of immune interstitial nephritis. The clinicians reinstituted prednisolone, and the acute interstitial nephritis improved. Nivolumab was discontinued.
Related: Immunotherapy in Melanoma
Drug-induced acute interstitial nephritis often has been more described with nonsteroidal anti-inflammatory drugs and beta-lactams, among others, the researchers say. Immune interstitial nephritis had been reported in a patient treated with nivolumab and ipilimumab concomitantly, and 3 cases of granulomatous interstitial nephritis have been reported with ipilimumab monotherapy. To the authors’ knowledge, this is the first case of immune interstitial nephritis reported with nivolumab monotherapy in metastatic melanoma. It is important to consider, they add, that the patient had also received ipilimumab, and that due to the drug’s elimination half-life (15.4 days), they can’t exclude an “overlap” between the 2 drugs that might have increased the risk of acute interstitial nephritis.
Source:
Bottlaender L, Breton AL, de Laforcade L, Dijoud F, Thomas L, Dalle S. J Immunother Cancer. 2017;5:56.
doi: 10.1186/s40425-017-0261-2
Ex Vivo Confocal Microscopy: A Diagnostic Tool for Skin Malignancies
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
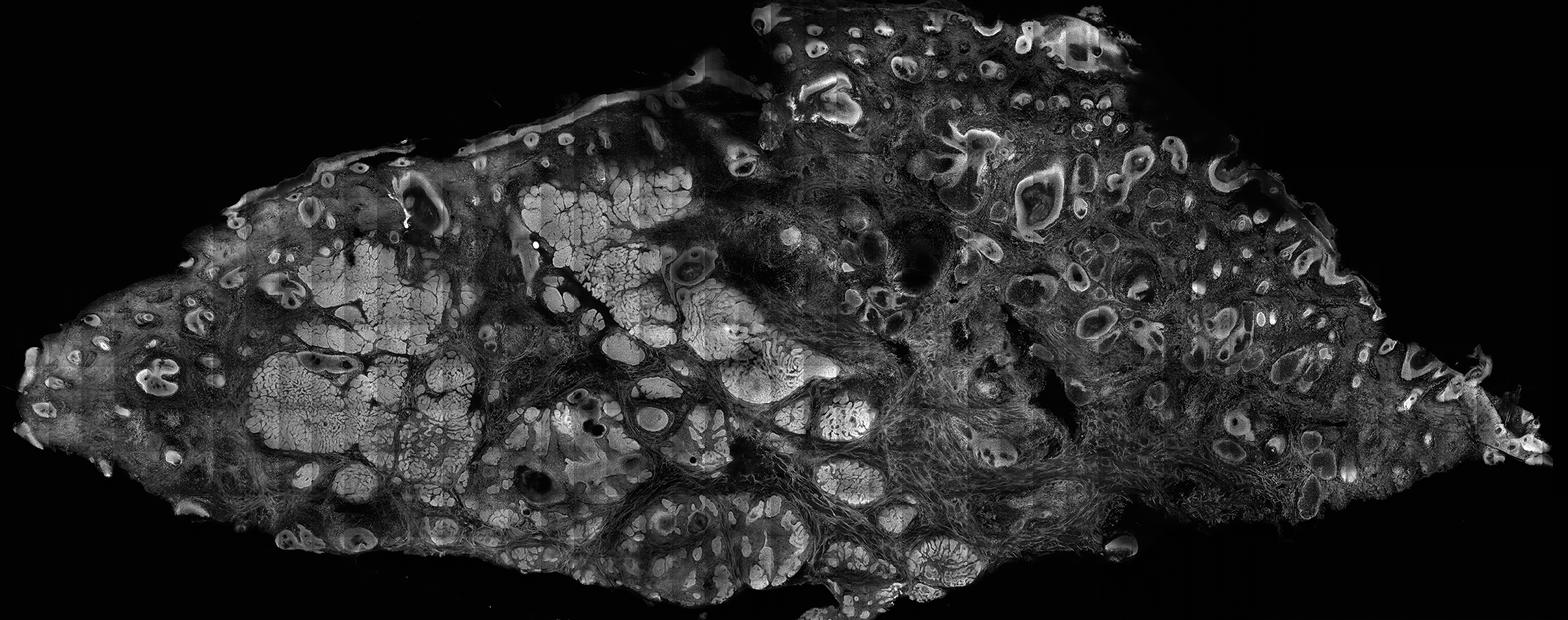
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Practice Points
- Confocal microscopy is an imaging tool that can be used both in vivo and ex vivo to aid in the diagnosis and management of cutaneous neoplasms, including melanoma, basal cell carcinoma, and squamous cell carcinoma, as well as inflammatory dermatoses.
- Ex vivo confocal microscopy can be used in both reflectance and fluorescent modes to render diagnosis in excised tissue or check surgical margins.
- Both in vivo and ex vivo confocal microscopy produces images with cellular resolution with a main limitation being depth of imaging.
What’s on the dermatopathologist’s wish list
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
Ex Vivo Confocal Microscopy in Clinical Practice: Report From the AAD Meeting

Comorbidity and Survival With Receipt of Adjuvant Immunotherapy in Stage III Melanoma: An Analysis of the National Cancer Database
Background: Within melanoma, comorbidity is associated with delayed diagnosis, advanced stage, and less aggressive treatment. Using the National Cancer Database (NCDB), this is the largest study to determine the influence of comorbidity (Charlson-Deyo) on receipt of adjuvant immunotherapy and survival in stage III melanoma.
Methods: We identified 10,739 patients with stage III melanoma between 2006-2012, for which high dose interferon was the standard adjuvant treatment. The probability of receipt was estimated using multivariable marginal logistic regression model, whereas survival was estimated using both the Kaplan-Meier method and multivariable marginal Cox regression model. Multivariable models adjusted for patient and facility-level characteristics.
Results: Greater receipt of adjuvant immunotherapy was observed in patients with fewer comorbidities (28.2%, 23.6%, and 13.8%, respectively, for 0, 1, and 2 or more comorbidities; P < .001). Patients with two or more comorbid conditions had a 44.0% lower adjusted odds of receiving adjuvant immunotherapy relative to patients with none (P < .001), and 45.5% lower adjusted odds relative to 1 comorbidity (P < .001). Regarding survival estimates, patients receiving adjuvant immunotherapy had significantly longer survival with fewer than 2 comorbid conditions (both P < .001); no difference was observed in patients with 2 or more (P = .077). In patients with no comorbidities, at 5-years postdiagnosis, 60.4% of those receiving adjuvant immunotherapy were alive compared to 49.8% of those who did not. In patients with 1 comorbid condition, 51.3% of those receiving adjuvant immunotherapy were alive compared to 37.7% of those who did not. The adjusted risk of death in patients who received adjuvant immunotherapy was not moderated by the number of comorbidities (χ2 = 0.51, P = .775). As such, the 20.4% lower risk of death favoring patients who received adjuvant immunotherapy (P < .001) was constant across different numbers of comorbidities. Lower risk of death and receipt of adjuvant immunotherapy were observed in younger patients with private insurance.
Conclusions: Risk of death findings suggest that adjuvant immunotherapy works equally well across numbers of comorbidities, despite a decrease in receipt with greater comorbidity. Additionally, overall survival findings support this in patients with 1 comorbidity.
Background: Within melanoma, comorbidity is associated with delayed diagnosis, advanced stage, and less aggressive treatment. Using the National Cancer Database (NCDB), this is the largest study to determine the influence of comorbidity (Charlson-Deyo) on receipt of adjuvant immunotherapy and survival in stage III melanoma.
Methods: We identified 10,739 patients with stage III melanoma between 2006-2012, for which high dose interferon was the standard adjuvant treatment. The probability of receipt was estimated using multivariable marginal logistic regression model, whereas survival was estimated using both the Kaplan-Meier method and multivariable marginal Cox regression model. Multivariable models adjusted for patient and facility-level characteristics.
Results: Greater receipt of adjuvant immunotherapy was observed in patients with fewer comorbidities (28.2%, 23.6%, and 13.8%, respectively, for 0, 1, and 2 or more comorbidities; P < .001). Patients with two or more comorbid conditions had a 44.0% lower adjusted odds of receiving adjuvant immunotherapy relative to patients with none (P < .001), and 45.5% lower adjusted odds relative to 1 comorbidity (P < .001). Regarding survival estimates, patients receiving adjuvant immunotherapy had significantly longer survival with fewer than 2 comorbid conditions (both P < .001); no difference was observed in patients with 2 or more (P = .077). In patients with no comorbidities, at 5-years postdiagnosis, 60.4% of those receiving adjuvant immunotherapy were alive compared to 49.8% of those who did not. In patients with 1 comorbid condition, 51.3% of those receiving adjuvant immunotherapy were alive compared to 37.7% of those who did not. The adjusted risk of death in patients who received adjuvant immunotherapy was not moderated by the number of comorbidities (χ2 = 0.51, P = .775). As such, the 20.4% lower risk of death favoring patients who received adjuvant immunotherapy (P < .001) was constant across different numbers of comorbidities. Lower risk of death and receipt of adjuvant immunotherapy were observed in younger patients with private insurance.
Conclusions: Risk of death findings suggest that adjuvant immunotherapy works equally well across numbers of comorbidities, despite a decrease in receipt with greater comorbidity. Additionally, overall survival findings support this in patients with 1 comorbidity.
Background: Within melanoma, comorbidity is associated with delayed diagnosis, advanced stage, and less aggressive treatment. Using the National Cancer Database (NCDB), this is the largest study to determine the influence of comorbidity (Charlson-Deyo) on receipt of adjuvant immunotherapy and survival in stage III melanoma.
Methods: We identified 10,739 patients with stage III melanoma between 2006-2012, for which high dose interferon was the standard adjuvant treatment. The probability of receipt was estimated using multivariable marginal logistic regression model, whereas survival was estimated using both the Kaplan-Meier method and multivariable marginal Cox regression model. Multivariable models adjusted for patient and facility-level characteristics.
Results: Greater receipt of adjuvant immunotherapy was observed in patients with fewer comorbidities (28.2%, 23.6%, and 13.8%, respectively, for 0, 1, and 2 or more comorbidities; P < .001). Patients with two or more comorbid conditions had a 44.0% lower adjusted odds of receiving adjuvant immunotherapy relative to patients with none (P < .001), and 45.5% lower adjusted odds relative to 1 comorbidity (P < .001). Regarding survival estimates, patients receiving adjuvant immunotherapy had significantly longer survival with fewer than 2 comorbid conditions (both P < .001); no difference was observed in patients with 2 or more (P = .077). In patients with no comorbidities, at 5-years postdiagnosis, 60.4% of those receiving adjuvant immunotherapy were alive compared to 49.8% of those who did not. In patients with 1 comorbid condition, 51.3% of those receiving adjuvant immunotherapy were alive compared to 37.7% of those who did not. The adjusted risk of death in patients who received adjuvant immunotherapy was not moderated by the number of comorbidities (χ2 = 0.51, P = .775). As such, the 20.4% lower risk of death favoring patients who received adjuvant immunotherapy (P < .001) was constant across different numbers of comorbidities. Lower risk of death and receipt of adjuvant immunotherapy were observed in younger patients with private insurance.
Conclusions: Risk of death findings suggest that adjuvant immunotherapy works equally well across numbers of comorbidities, despite a decrease in receipt with greater comorbidity. Additionally, overall survival findings support this in patients with 1 comorbidity.
Metabolic Reprogramming in BRAF Inhibitor-Resistant Melanoma Cells Leads to Hypersensitvity to Arginine Depletion and Upregulation of PD-L1 Expression
Purpose: The time to response to immunotherapy is long and not suitable for rapidly growing BRAF resistant (BR) or BMR ( BRAF and MEK) resistant melanoma. We exploit a new approach to treat these tumors.
Background: We have previously shown that BR/BMR cells do not express argininosuccinate synthetase (ASS) and arginine deprivation induced apoptosis instead of autophagy (Oncotarget 7:14). We plan to exploited this alteration to treat BR/BMR.
Methods: Five BR cells A375, MEL-1220, A2058, UACC-62, and SK-MEL28 were established (Oncotarget). Arginine deprivation was achieved using arginine degrading enzyme (ADI-PEG20, Polaris) which degrade arginine to citrulline.
Results: BR cells are hypersensitive to ADI-PEG20. Treatment resulted in 10-30% increase in apoptosis compared to their parental cells. The mechanisms involved are as follows: All BR cells do not express ASS, and have attenuated glucose uptake. They acquire exogenous arginine by expressing high levels of arginine transporter CAT2. The mechanism for low levels of ASS is due to diminished c-Myc, a positive regulator of ASS. Additionally, AMPK-α1 (govern autophagy and glucose uptake) was attenuated in BR cells. This is proved by knockdown and overexpress AMPK-α1. Immunohistochemical staining further confirmed lower levels of AMPK-α1 in tumor tissues (average H-scores of ASS and AMPK in parental tissues vs BR (BMR) tissues are 58.2 vs 7.8, and 146 vs 78.3, respectively, P < .03). Importantly, these findings also apply to tumor from BMR patients. Interestingly, treatment with ADI-PEG20 leads to robust expression of immune checkpoint PD-L1 in both parental and BR cells and PBMCs from BR patients. Importantly, macrophage polarization may involve in metabolic reprogramming.
Conclusions: Attenuation of AMPK-α1-in BR results in diminished autophagy and metabolic alteration. These BR cells depend less on glucose but more on arginine, and hence vulnerable to arginine deprivation. Additionally, arginine deprivation upregulates PD-L1 expression and leads to sensitivity to anti PDL-1 antibody. Combination of ADI-PEG20 with checkpoint inhibitors can lead to robust antitumor effect in BR and BMR patients. Supported by VA Merit Review (BLR&D BX00333280-01) and R01CA152197.
Purpose: The time to response to immunotherapy is long and not suitable for rapidly growing BRAF resistant (BR) or BMR ( BRAF and MEK) resistant melanoma. We exploit a new approach to treat these tumors.
Background: We have previously shown that BR/BMR cells do not express argininosuccinate synthetase (ASS) and arginine deprivation induced apoptosis instead of autophagy (Oncotarget 7:14). We plan to exploited this alteration to treat BR/BMR.
Methods: Five BR cells A375, MEL-1220, A2058, UACC-62, and SK-MEL28 were established (Oncotarget). Arginine deprivation was achieved using arginine degrading enzyme (ADI-PEG20, Polaris) which degrade arginine to citrulline.
Results: BR cells are hypersensitive to ADI-PEG20. Treatment resulted in 10-30% increase in apoptosis compared to their parental cells. The mechanisms involved are as follows: All BR cells do not express ASS, and have attenuated glucose uptake. They acquire exogenous arginine by expressing high levels of arginine transporter CAT2. The mechanism for low levels of ASS is due to diminished c-Myc, a positive regulator of ASS. Additionally, AMPK-α1 (govern autophagy and glucose uptake) was attenuated in BR cells. This is proved by knockdown and overexpress AMPK-α1. Immunohistochemical staining further confirmed lower levels of AMPK-α1 in tumor tissues (average H-scores of ASS and AMPK in parental tissues vs BR (BMR) tissues are 58.2 vs 7.8, and 146 vs 78.3, respectively, P < .03). Importantly, these findings also apply to tumor from BMR patients. Interestingly, treatment with ADI-PEG20 leads to robust expression of immune checkpoint PD-L1 in both parental and BR cells and PBMCs from BR patients. Importantly, macrophage polarization may involve in metabolic reprogramming.
Conclusions: Attenuation of AMPK-α1-in BR results in diminished autophagy and metabolic alteration. These BR cells depend less on glucose but more on arginine, and hence vulnerable to arginine deprivation. Additionally, arginine deprivation upregulates PD-L1 expression and leads to sensitivity to anti PDL-1 antibody. Combination of ADI-PEG20 with checkpoint inhibitors can lead to robust antitumor effect in BR and BMR patients. Supported by VA Merit Review (BLR&D BX00333280-01) and R01CA152197.
Purpose: The time to response to immunotherapy is long and not suitable for rapidly growing BRAF resistant (BR) or BMR ( BRAF and MEK) resistant melanoma. We exploit a new approach to treat these tumors.
Background: We have previously shown that BR/BMR cells do not express argininosuccinate synthetase (ASS) and arginine deprivation induced apoptosis instead of autophagy (Oncotarget 7:14). We plan to exploited this alteration to treat BR/BMR.
Methods: Five BR cells A375, MEL-1220, A2058, UACC-62, and SK-MEL28 were established (Oncotarget). Arginine deprivation was achieved using arginine degrading enzyme (ADI-PEG20, Polaris) which degrade arginine to citrulline.
Results: BR cells are hypersensitive to ADI-PEG20. Treatment resulted in 10-30% increase in apoptosis compared to their parental cells. The mechanisms involved are as follows: All BR cells do not express ASS, and have attenuated glucose uptake. They acquire exogenous arginine by expressing high levels of arginine transporter CAT2. The mechanism for low levels of ASS is due to diminished c-Myc, a positive regulator of ASS. Additionally, AMPK-α1 (govern autophagy and glucose uptake) was attenuated in BR cells. This is proved by knockdown and overexpress AMPK-α1. Immunohistochemical staining further confirmed lower levels of AMPK-α1 in tumor tissues (average H-scores of ASS and AMPK in parental tissues vs BR (BMR) tissues are 58.2 vs 7.8, and 146 vs 78.3, respectively, P < .03). Importantly, these findings also apply to tumor from BMR patients. Interestingly, treatment with ADI-PEG20 leads to robust expression of immune checkpoint PD-L1 in both parental and BR cells and PBMCs from BR patients. Importantly, macrophage polarization may involve in metabolic reprogramming.
Conclusions: Attenuation of AMPK-α1-in BR results in diminished autophagy and metabolic alteration. These BR cells depend less on glucose but more on arginine, and hence vulnerable to arginine deprivation. Additionally, arginine deprivation upregulates PD-L1 expression and leads to sensitivity to anti PDL-1 antibody. Combination of ADI-PEG20 with checkpoint inhibitors can lead to robust antitumor effect in BR and BMR patients. Supported by VA Merit Review (BLR&D BX00333280-01) and R01CA152197.
Successful 3-Year Melanoma Treatment After Nivolumab Failure
Background: Since approval of kinase inhibitors and programmed death receptor-1 (PD-1) and CTLA-4 blocking antibody, therapeutic approach to treat metastatic melanoma have demonstrated better efficacy then traditional chemotherapy. While kinase inhibitors target specific mutations and have demonstrated response rate around 50%, resistance to treatment ultimately develops. Treatment with check point inhibitor can produce durable response which translated into long-term survival, but the key question is that the marker(s) to predict response is not
clear yet. Other questions are: whether both drugs have equal efficacy, target similar domain on PD-1, and possible cross resistance. Here, we report a melanoma patient who clearly progressed after 10 doses of nivolumab. The treatment was changed to pembrolizumab with good response.
Case: We report a case of a 76-year-old man with aggressive metastatic melanoma and a 3-year history of treatment. After lymph node dissection in Dec 2013 for a T4bN3 disease of neck (BRAF V600E positive), he was planned for postoperative radiation. Unfortunately, his malignancy recurred very quickly in Jan 2014 with dermal infiltration and subcutaneous nodule. Initial treatment with vemurafenib was not tolerated. Subsequently, he was given trametinib plus dabrafenib with some response, then melanoma recurred after 10 months. Ipilimumab was started, patient tolerated it well, but disease progressed. Nivolumab was started, but disease progressed with lymph nodes, liver, lung and spleen metastasis. While there are no data, we decided to try pembrolizumab and the patient showed a significant response, with disappearing of the metastasis. The only metastatic site that remained was in spleen. Interestingly, biopsy from metastatic lesion in the spleen showed 40% of tumor cells were positive for PD-L1 in the membrane but immune cells were negative. Patient has received a total of 29 cycles of pembrolizumab and is doing well.
Conclusions: This case provides an interesting observation of long duration of response of pembrolizumab after failing nivolumab. This could be due to some pharmacological differences between these inhibitors as well as variations of immune cells in the tumor microenvironment. Further study is needed to clarify this issue.
Background: Since approval of kinase inhibitors and programmed death receptor-1 (PD-1) and CTLA-4 blocking antibody, therapeutic approach to treat metastatic melanoma have demonstrated better efficacy then traditional chemotherapy. While kinase inhibitors target specific mutations and have demonstrated response rate around 50%, resistance to treatment ultimately develops. Treatment with check point inhibitor can produce durable response which translated into long-term survival, but the key question is that the marker(s) to predict response is not
clear yet. Other questions are: whether both drugs have equal efficacy, target similar domain on PD-1, and possible cross resistance. Here, we report a melanoma patient who clearly progressed after 10 doses of nivolumab. The treatment was changed to pembrolizumab with good response.
Case: We report a case of a 76-year-old man with aggressive metastatic melanoma and a 3-year history of treatment. After lymph node dissection in Dec 2013 for a T4bN3 disease of neck (BRAF V600E positive), he was planned for postoperative radiation. Unfortunately, his malignancy recurred very quickly in Jan 2014 with dermal infiltration and subcutaneous nodule. Initial treatment with vemurafenib was not tolerated. Subsequently, he was given trametinib plus dabrafenib with some response, then melanoma recurred after 10 months. Ipilimumab was started, patient tolerated it well, but disease progressed. Nivolumab was started, but disease progressed with lymph nodes, liver, lung and spleen metastasis. While there are no data, we decided to try pembrolizumab and the patient showed a significant response, with disappearing of the metastasis. The only metastatic site that remained was in spleen. Interestingly, biopsy from metastatic lesion in the spleen showed 40% of tumor cells were positive for PD-L1 in the membrane but immune cells were negative. Patient has received a total of 29 cycles of pembrolizumab and is doing well.
Conclusions: This case provides an interesting observation of long duration of response of pembrolizumab after failing nivolumab. This could be due to some pharmacological differences between these inhibitors as well as variations of immune cells in the tumor microenvironment. Further study is needed to clarify this issue.
Background: Since approval of kinase inhibitors and programmed death receptor-1 (PD-1) and CTLA-4 blocking antibody, therapeutic approach to treat metastatic melanoma have demonstrated better efficacy then traditional chemotherapy. While kinase inhibitors target specific mutations and have demonstrated response rate around 50%, resistance to treatment ultimately develops. Treatment with check point inhibitor can produce durable response which translated into long-term survival, but the key question is that the marker(s) to predict response is not
clear yet. Other questions are: whether both drugs have equal efficacy, target similar domain on PD-1, and possible cross resistance. Here, we report a melanoma patient who clearly progressed after 10 doses of nivolumab. The treatment was changed to pembrolizumab with good response.
Case: We report a case of a 76-year-old man with aggressive metastatic melanoma and a 3-year history of treatment. After lymph node dissection in Dec 2013 for a T4bN3 disease of neck (BRAF V600E positive), he was planned for postoperative radiation. Unfortunately, his malignancy recurred very quickly in Jan 2014 with dermal infiltration and subcutaneous nodule. Initial treatment with vemurafenib was not tolerated. Subsequently, he was given trametinib plus dabrafenib with some response, then melanoma recurred after 10 months. Ipilimumab was started, patient tolerated it well, but disease progressed. Nivolumab was started, but disease progressed with lymph nodes, liver, lung and spleen metastasis. While there are no data, we decided to try pembrolizumab and the patient showed a significant response, with disappearing of the metastasis. The only metastatic site that remained was in spleen. Interestingly, biopsy from metastatic lesion in the spleen showed 40% of tumor cells were positive for PD-L1 in the membrane but immune cells were negative. Patient has received a total of 29 cycles of pembrolizumab and is doing well.
Conclusions: This case provides an interesting observation of long duration of response of pembrolizumab after failing nivolumab. This could be due to some pharmacological differences between these inhibitors as well as variations of immune cells in the tumor microenvironment. Further study is needed to clarify this issue.
Getting a Better Picture of Skin Cancer
A handheld detector that offers noninvasive real-time imaging can help dermatologic surgeons get a better idea of skin cancer dimensions before committing to surgery, according to researchers from National Skin Centre and Singapore Bioimaging Consortium, both in Singapore, and Technical University of Munich and iThera Medical GmbH, both in Germany.
Current imaging technologies can lead to excessive or incomplete removal of the cancer, the researchers say. The multispectral optoacoustic tomography (MSOT) allows the user to differentiate tissue chromophores (the chromophore is the part of the molecule responsible for its color) and exogenous contrast agents based on their spectral signatures.
The researchers performed MSOT imaging with 2- and 3-dimensional handheld scanners on 21 patients with nonmelanoma skin cancers. All the skin lesions had recognizable images on MSOT with both detectors, visualizing the shape and thickness of the lesions. The 2D and 3D detectors also offered images with well-resolved tissue chromophores. But the volumetric probe gave more accurate tumor dimensions compared with those from histology analysis.
Aggressive types of skin cancers can involve deeper structures, such as predominant deep blood vessels, the researchers note—another reason the MSOT detector could be useful. In one case, the depth of the basal cell carcinoma, which included its underlying vasculature, reached beyond 3 mm, which might have gone undetected by other imaging modalities, they say.
The fact that the device is also noninvasive and offers real-time imaging, the researchers suggest, makes volumetric MSOT “an ideal modality for longitudinal monitoring of skin diseases and treatment responses.”
Source:
Attia ABE, Chuah SY, Razansky D, et al. Photoacoustics. 2017;7:20-26.
doi: 10.1016/j.pacs.2017.05.003.
A handheld detector that offers noninvasive real-time imaging can help dermatologic surgeons get a better idea of skin cancer dimensions before committing to surgery, according to researchers from National Skin Centre and Singapore Bioimaging Consortium, both in Singapore, and Technical University of Munich and iThera Medical GmbH, both in Germany.
Current imaging technologies can lead to excessive or incomplete removal of the cancer, the researchers say. The multispectral optoacoustic tomography (MSOT) allows the user to differentiate tissue chromophores (the chromophore is the part of the molecule responsible for its color) and exogenous contrast agents based on their spectral signatures.
The researchers performed MSOT imaging with 2- and 3-dimensional handheld scanners on 21 patients with nonmelanoma skin cancers. All the skin lesions had recognizable images on MSOT with both detectors, visualizing the shape and thickness of the lesions. The 2D and 3D detectors also offered images with well-resolved tissue chromophores. But the volumetric probe gave more accurate tumor dimensions compared with those from histology analysis.
Aggressive types of skin cancers can involve deeper structures, such as predominant deep blood vessels, the researchers note—another reason the MSOT detector could be useful. In one case, the depth of the basal cell carcinoma, which included its underlying vasculature, reached beyond 3 mm, which might have gone undetected by other imaging modalities, they say.
The fact that the device is also noninvasive and offers real-time imaging, the researchers suggest, makes volumetric MSOT “an ideal modality for longitudinal monitoring of skin diseases and treatment responses.”
Source:
Attia ABE, Chuah SY, Razansky D, et al. Photoacoustics. 2017;7:20-26.
doi: 10.1016/j.pacs.2017.05.003.
A handheld detector that offers noninvasive real-time imaging can help dermatologic surgeons get a better idea of skin cancer dimensions before committing to surgery, according to researchers from National Skin Centre and Singapore Bioimaging Consortium, both in Singapore, and Technical University of Munich and iThera Medical GmbH, both in Germany.
Current imaging technologies can lead to excessive or incomplete removal of the cancer, the researchers say. The multispectral optoacoustic tomography (MSOT) allows the user to differentiate tissue chromophores (the chromophore is the part of the molecule responsible for its color) and exogenous contrast agents based on their spectral signatures.
The researchers performed MSOT imaging with 2- and 3-dimensional handheld scanners on 21 patients with nonmelanoma skin cancers. All the skin lesions had recognizable images on MSOT with both detectors, visualizing the shape and thickness of the lesions. The 2D and 3D detectors also offered images with well-resolved tissue chromophores. But the volumetric probe gave more accurate tumor dimensions compared with those from histology analysis.
Aggressive types of skin cancers can involve deeper structures, such as predominant deep blood vessels, the researchers note—another reason the MSOT detector could be useful. In one case, the depth of the basal cell carcinoma, which included its underlying vasculature, reached beyond 3 mm, which might have gone undetected by other imaging modalities, they say.
The fact that the device is also noninvasive and offers real-time imaging, the researchers suggest, makes volumetric MSOT “an ideal modality for longitudinal monitoring of skin diseases and treatment responses.”
Source:
Attia ABE, Chuah SY, Razansky D, et al. Photoacoustics. 2017;7:20-26.
doi: 10.1016/j.pacs.2017.05.003.
Targeted Therapy and Immunotherapy in the Treatment of Metastatic Cutaneous Melanoma
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.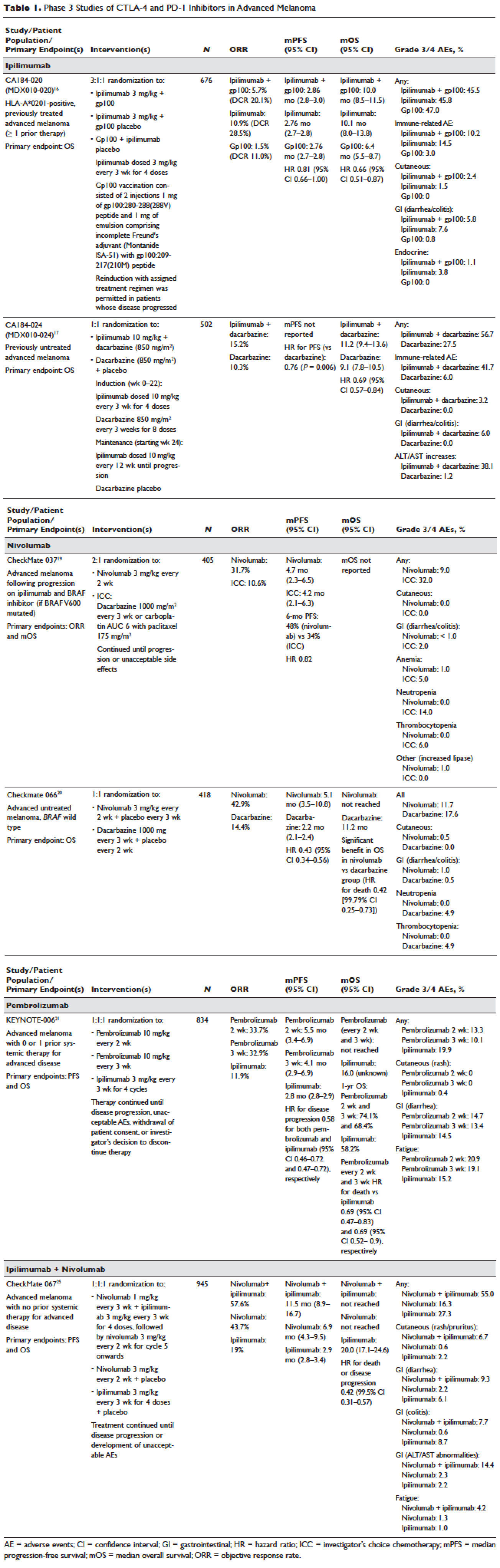
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.