User login
Suicide rates up significantly among adolescents, young adults
Suicide rates in young people aged 10-24 years increased significantly in 42 states from 2007-2009 to 2016-2018, according to a recent analysis from the National Center for Health Statistics.
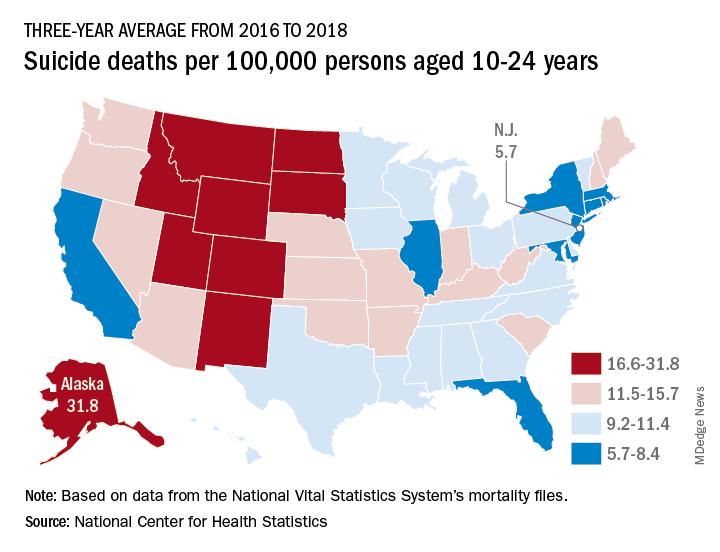
Nationally, the suicide rate jumped 47%, based on the averages for the two 3-year periods, rising from 7.0 per 100,000 persons aged 10-24 years to 10.3 per 100,000. For all ages, the corresponding increase was 47%, Sally C. Curtin, MA, of the NCHS, said in a National Vital Statistics Report.
There was no state with a decrease in suicide rates for adolescents and young adults, as the other eight all had nonsignificant increases, the smallest being 14% in South Dakota. Three-year averages were used to increase statistical power for states with relatively small numbers of deaths but were still not enough to show significance for some large increases, such as the 48% rise in Delaware, Ms. Curtin noted.
In 2016-2018, Alaska’s suicide rate of 31.8 per 100,000 persons aged 10-24 years was the highest in the country, followed by South Dakota (23.6), Montana (23.2), and Wyoming (20.5). New Jersey had the lowest rate at 5.7 per 100,000, with New York and Rhode Island both slightly higher at 5.9 and Connecticut at 6.3, based on data from the National Vital Statistics System.
Even the low numbers, however, hide some large changes, as New Jersey (up by 39%) and New York (up by 44%) were among the 42 states with statistically significant increases, which ranged from 21.7% in Maryland to 110% in New Hampshire, Ms. Curtin said in the report. The increases seen in this analysis contrast with data from the preceding time period, as “the suicide rate among persons aged 10-24 was statistically stable from 2000 to 2007.”
SOURCE: Curtin SC. National Vital Statistics Reports. 2020;69(11)1-9.
Suicide rates in young people aged 10-24 years increased significantly in 42 states from 2007-2009 to 2016-2018, according to a recent analysis from the National Center for Health Statistics.
Nationally, the suicide rate jumped 47%, based on the averages for the two 3-year periods, rising from 7.0 per 100,000 persons aged 10-24 years to 10.3 per 100,000. For all ages, the corresponding increase was 47%, Sally C. Curtin, MA, of the NCHS, said in a National Vital Statistics Report.
There was no state with a decrease in suicide rates for adolescents and young adults, as the other eight all had nonsignificant increases, the smallest being 14% in South Dakota. Three-year averages were used to increase statistical power for states with relatively small numbers of deaths but were still not enough to show significance for some large increases, such as the 48% rise in Delaware, Ms. Curtin noted.
In 2016-2018, Alaska’s suicide rate of 31.8 per 100,000 persons aged 10-24 years was the highest in the country, followed by South Dakota (23.6), Montana (23.2), and Wyoming (20.5). New Jersey had the lowest rate at 5.7 per 100,000, with New York and Rhode Island both slightly higher at 5.9 and Connecticut at 6.3, based on data from the National Vital Statistics System.
Even the low numbers, however, hide some large changes, as New Jersey (up by 39%) and New York (up by 44%) were among the 42 states with statistically significant increases, which ranged from 21.7% in Maryland to 110% in New Hampshire, Ms. Curtin said in the report. The increases seen in this analysis contrast with data from the preceding time period, as “the suicide rate among persons aged 10-24 was statistically stable from 2000 to 2007.”
SOURCE: Curtin SC. National Vital Statistics Reports. 2020;69(11)1-9.
Suicide rates in young people aged 10-24 years increased significantly in 42 states from 2007-2009 to 2016-2018, according to a recent analysis from the National Center for Health Statistics.
Nationally, the suicide rate jumped 47%, based on the averages for the two 3-year periods, rising from 7.0 per 100,000 persons aged 10-24 years to 10.3 per 100,000. For all ages, the corresponding increase was 47%, Sally C. Curtin, MA, of the NCHS, said in a National Vital Statistics Report.
There was no state with a decrease in suicide rates for adolescents and young adults, as the other eight all had nonsignificant increases, the smallest being 14% in South Dakota. Three-year averages were used to increase statistical power for states with relatively small numbers of deaths but were still not enough to show significance for some large increases, such as the 48% rise in Delaware, Ms. Curtin noted.
In 2016-2018, Alaska’s suicide rate of 31.8 per 100,000 persons aged 10-24 years was the highest in the country, followed by South Dakota (23.6), Montana (23.2), and Wyoming (20.5). New Jersey had the lowest rate at 5.7 per 100,000, with New York and Rhode Island both slightly higher at 5.9 and Connecticut at 6.3, based on data from the National Vital Statistics System.
Even the low numbers, however, hide some large changes, as New Jersey (up by 39%) and New York (up by 44%) were among the 42 states with statistically significant increases, which ranged from 21.7% in Maryland to 110% in New Hampshire, Ms. Curtin said in the report. The increases seen in this analysis contrast with data from the preceding time period, as “the suicide rate among persons aged 10-24 was statistically stable from 2000 to 2007.”
SOURCE: Curtin SC. National Vital Statistics Reports. 2020;69(11)1-9.
Health Care Disparities Among Adolescents and Adults With Sickle Cell Disease: A Community-Based Needs Assessment to Inform Intervention Strategies
From the University of California San Francisco (Dr. Treadwell, Dr. Hessler, Yumei Chen, Swapandeep Mushiana, Dr. Potter, and Dr. Vichinsky), the University of California Los Angeles (Dr. Jacob), and the University of California Berkeley (Alex Chen).
Abstract
- Objective: Adolescents and adults with sickle cell disease (SCD) face pervasive disparities in health resources and outcomes. We explored barriers to and facilitators of care to identify opportunities to support implementation of evidence-based interventions aimed at improving care quality for patients with SCD.
- Methods: We engaged a representative sample of adolescents and adults with SCD (n = 58), health care providers (n = 51), and community stakeholders (health care administrators and community-based organization leads (n = 5) in Northern California in a community-based needs assessment. We conducted group interviews separately with participant groups to obtain in-depth perspectives. Adolescents and adults with SCD completed validated measures of pain interference, quality of care, self-efficacy, and barriers to care. Providers and community stakeholders completed surveys about barriers to SCD care.
- Results: We triangulated qualitative and quantitative data and found that participants with SCD (mean age, 31 ± 8.6 years), providers, and community stakeholders emphasized the social and emotional burden of SCD as barriers. Concrete barriers agreed upon included insurance and lack of resources for addressing pain impact. Adolescents and adults with SCD identified provider issues (lack of knowledge, implicit bias), transportation, and limited social support as barriers. Negative encounters with the health care system contributed to 84% of adolescents and adults with SCD reporting they chose to manage severe pain at home. Providers focused on structural barriers: lack of access to care guidelines, comfort level with and knowledge of SCD management, and poor care coordination.
- Conclusion: Strategies for improving access to compassionate, evidence-based quality care, as well as strategies for minimizing the burden of having SCD, are warranted for this medically complex population.
Keywords: barriers to care; quality of care; care access; care coordination.
Sickle cell disease (SCD), an inherited chronic medical condition, affects about 100,000 individuals in the United States, a population that is predominantly African American.1 These individuals experience multiple serious and life-threatening complications, most frequently recurrent vaso-occlusive pain episodes,2 and they require interactions with multidisciplinary specialists from childhood. Because of advances in treatments, the majority are reaching adulthood; however, there is a dearth of adult health care providers with the training and expertise to manage their complex medical needs.3 Other concrete barriers to adequate SCD care include insurance and distance to comprehensive SCD centers.4,5
Social, behavioral, and emotional factors may also contribute to challenges with SCD management. SCD may limit daily functional abilities and lead to diminished overall quality of life.6,7 Some adolescents and adults may require high doses of opioids, which contributes to health care providers’ perceptions that there is a high prevalence of drug addiction in the population.8,9 These providers express negative attitudes towards adults with SCD, and, consequently, delay medication administration when it is acutely needed and provide otherwise suboptimal treatment.8,10,11 Adult care providers may also be uncomfortable with prescribing and managing disease-modifying therapies (blood transfusion, hydroxyurea) that have established efficacy.12-17
As 1 of 8 programs funded by the National Heart, Lung, and Blood Institute’s (NHLBI) Sickle Cell Disease Implementation Consortium (SCDIC), we are using implementation science to reduce barriers to care and improve quality of care and health care outcomes in SCD.18,19 Given that adolescents and adults with SCD experience high mortality, severe pain, and progressive decline in their ability to function day to day, and also face lack of access to knowledgeable, compassionate providers in primary and emergency settings, the SCDIC focuses on individuals aged 15 to 45 years.6,8,9,11,12
Our regional SCDIC program, the Sickle Cell Care Coordination Initiative (SCCCI), brings together researchers, clinicians, adolescents, and adults with SCD and their families, dedicated community members, policy makers, and administrators to identify and address barriers to health care within 5 counties in Northern California. One of our first steps was to conduct a community-based needs assessment, designed to inform implementation of evidence-based interventions, accounting for unique contextual factors in our region.
Conceptual Framework for Improving Medical Practice
Our needs assessment is guided by Solberg’s Conceptual Framework for Improving Medical Practice (Figure 1).20 Consistent with the overarching principles of the SCDIC, this conceptual framework focuses on the inadequate implementation of evidence-based guidelines, and on the need to first understand multifactorial facilitators and barriers to guideline implementation in order to effect change. The framework identifies 3 main elements that must be present to ensure improvements in quality-of-care processes and patient outcomes: priority, change process capability, and care process content. Priority refers to ample resource allocation for the specific change, as well as freedom from competing priorities for those implementing the change. Change process capability includes strong, effective leadership, adequate infrastructure for managing change (including resources and time), change management skills at all levels, and an established clinical information system. Care process content refers to context and systems-level changes, such as delivery system redesign as needed, support for self-management to lessen the impact of the disease, and decision support.21-23

The purpose of our community-based needs assessment was to evaluate barriers to care and quality of care in SCD, within Solberg’s conceptual model for improving medical practice. The specific aims were to evaluate access and barriers to care (eg, lack of provider expertise and training, health care system barriers such as poor care coordination and provider communication); evaluate quality of care; and assess patient needs related to pain, pain interference, self-efficacy, and self-management for adolescents and adults with SCD. We gathered the perspectives of a representative community of adolescents and adults with SCD, their providers, and community stakeholders in order to examine barriers, quality of life and care, and patient experiences in our region.
Methods
Design
In this cross-sectional study, adolescents and adults with SCD, their providers, and community stakeholders participated in group or individual qualitative interviews and completed surveys between October 2017 and March 2018.
Setting and Sample
Recruitment flyers were posted on a regional SCD-focused website, and clinical providers or a study coordinator introduced information about the needs assessment to potential participants with SCD during clinic visits at the participating centers. Participants with SCD were eligible if they had any diagnosis of SCD, were aged 15 to 48 years, and received health services within 5 Northern California counties (Alameda, Contra Costa, Sacramento, San Francisco, and Solano). They were excluded if they did not have a SCD diagnosis or had not received health services within the catchment area. As the project proceeded, participants were asked to refer other adolescents and adults with SCD for the interviews and surveys (snowball sampling). Our goal was to recruit 50 adolescents and adults with SCD into the study, aiming for 10 representatives from each county.
Providers and community stakeholders were recruited via emails, letters and informational flyers. We engaged our partner, the Sickle Cell Data Collection Program,2 to generate a list of providers and institutions that had seen patients with SCD in primary, emergency, or inpatient settings in the region. We contacted these institutions to describe the SCCCI and invite participation in the needs assessment. We also invited community-based organization leads and health care administrators who worked with SCD to participate. Providers accessed confidential surveys via a secure link on the study website or completed paper versions. Common data collected across providers included demographics and descriptions of practice settings.
Participants were eligible to be part of the study if they were health care providers (physicians and nurses) representing hematology, primary care, family medicine, internal medicine, or emergency medicine; ancillary staff (social work, psychology, child life); or leaders or administrators of clinical or sickle cell community-based organizations in Northern California (recruitment goal of n = 50). Providers were excluded if they practiced in specialties other than those noted or did not practice within the region.
Data Collection Procedures
After providing assent/consent, participating adolescents and adults with SCD took part in individual and group interviews and completed survey questionnaires. All procedures were conducted in a private space in the sickle cell center or community. Adolescents and adults with SCD completed the survey questionnaire on a tablet, with responses recorded directly in a REDCap (Research Electronic Data Capture) database,24 or on a paper version. Interviews lasted 60 (individual) to 90 (group) minutes, while survey completion time was 20 to 25 minutes. Each participant received a gift card upon completion as an expression of appreciation. All procedures were approved by the institutional review boards of the participating health care facilities.
Group and Individual Interviews
Participants with SCD and providers were invited to participate in a semi-structured qualitative interview prior to being presented with the surveys. Adolescents and adults with SCD were interviewed about barriers to care, quality of care, and pain-related experiences. Providers were asked about barriers to care and treatments. Interview guides were modified for community-based organization leaders and health care administrators who did not provide clinical services. Interview guides can be found in the Appendix. Interviews were conducted by research coordinators trained in qualitative research methods by the first author (MT). As appropriate with semi-structured interviews, the interviewers could word questions spontaneously, change the order of questions for ease of flow of conversation, and inform simultaneous coding of interviews with new themes as those might arise, as long as they touched on all topics within the interview guide.25 The interview guides were written, per qualitative research standards, based on the aims and purpose of the research,26 and were informed by existing literature on access and barriers to care in SCD, quality of care, and the needs of individuals with SCD, including in relation to impact of the disease, self-efficacy, and self-management.
Interviewees participated in either individual or group interviews, but not both. The decision for which type of interview an individual participated in was based on 2 factors: if there were not comparable participants for group interviews (eg, health care administrator and community-based organization lead), these interviews were done individually; and given that we were drawing participants from a 5-county area in Northern California, scheduling was challenging for individuals with SCD with regard to aligning schedules and traveling to a central location where the group interviews were conducted. Provider group interviews were easier to arrange because we could schedule them at the same time as regularly scheduled meetings at the participants’ health care institutions.
Interview Data Gathering and Analysis
Digital recordings of the interviews were cleaned of any participant identifying data and sent for transcription to an outside service. Transcripts were reviewed for completeness and imported into NVivo (www.qsrinternational.com), a qualitative data management program.
A thematic content analysis and deductive and inductive approaches were used to analyze the verbatim transcripts generated from the interviews. The research team was trained in the use of NVivo software to facilitate the coding process. A deductive coding scheme was initially used based on existing concepts in the literature regarding challenges to optimal SCD care, with new codes added as the thematic content analyses progressed. The initial coding, pattern coding, and use of displays to examine the relationships between different categories were conducted simultaneously.27,28 Using the constant comparative method, new concepts from participants with SCD and providers could be incorporated into subsequent interviews with other participants. For this study, the only additional concepts added were in relation to participant recruitment and retention in the SCDIC Registry. Research team members coded transcripts separately and came together weekly, constantly comparing codes and developing the consensus coding scheme. Where differences between coders existed, code meanings were discussed and clarified until consensus was reached.29
Quantitative data were analyzed using SPSS (v. 25, Chicago, IL). Descriptive statistics (means, standard deviations, frequencies, percentages) were used to summarize demographics (eg, age, gender, and race), economic status, and type of SCD. No systematic differences were detected from cases with missing values. Scale reliabilities (ie, Cronbach α) were evaluated for self-report measures.
Measurement
Adolescents and adults with SCD completed items from the PhenX Toolkit (consensus measures for Phenotypes and eXposures), assessing sociodemographics (age, sex, race, ethnicity, educational attainment, occupation, marital status, annual income, insurance), and clinical characteristics (sickle cell diagnosis and emergency department [ED] and hospital utilization for pain).30
Pain Interference Short Form (Patient-Reported Outcomes Measurement Information System [PROMIS]). The Pain Interference Form consists of 8 items that assess the degree to which pain interfered with day-to-day activities in the previous 7 days at home, including impacts on social, cognitive, emotional, and physical functioning; household chores and recreational activities; sleep; and enjoyment in life. Reliability and validity of the PROMIS Pain Interference Scale has been demonstrated, with strong negative correlations with Physical Function Scales (r = 0.717, P < 0.01), indicating that higher scores are associated with lower function (β = 0.707, P < 0.001).31 The Cronbach α estimate for the other items on the pain interference scale was 0.99. Validity analysis indicated strong correlations with pain-related domains: BPI Interference Subscale (rho = 0.90), SF-36 Bodily Pain Subscale (rho = –0.84), and 0–10 Numerical Rating of Pain Intensity (rho = 0.48).32
Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) Quality of Care (QOC). ASCQ-Me QOC consists of 27 items that measure the quality of care that adults with SCD have received from health care providers.33 There are 3 composites: provider communication (quality of patient and provider communication), ED care (quality of care in the ED), and access (to routine and emergency care). Internal consistency reliability for all 3 composites is greater than 0.70. Strong correlations of the provider communication composite with overall ratings of routine care (r = 0.65) and overall provider ratings (r = 0.83) provided evidence of construct validity. Similarly, the ED care composite was strongly correlated with overall ratings of QOC in the ED, and the access composite was highly correlated with overall evaluations of ED care (r = 0.70). Access, provider interaction, and ED care composites were reliable (Cronbach α, 0.70–0.83) and correlated with ratings of global care (r = 0.32–0.83), further indicating construct validity.33
Sickle Cell Self-Efficacy Scale (SCSES). The SCSES is a 9-item, self-administered questionnaire measuring perceptions of the ability to manage day-to-day issues resulting from SCD. SCSES items are scored on a 5-point scale ranging from Not sure at all (1) to Very sure (5). Individual item responses are summed to give an overall score, with higher scores indicating greater self-efficacy. The SCSES has acceptable reliability (r = 0.45, P < 0.001) and validity (α = 0.89).34,35
Sickle Cell Disease Barriers Checklist. This checklist consists of 53 items organized into 8 categories: insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers (fear, anger), and disease-related barriers. Participants check applicable barriers, with a total score range of 0 to 53 and higher scores indicating more barriers to care. The SCD Barriers Checklist has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).5
ED Provider Checklist. The ED provider survey is a checklist of 14 statements pertaining to issues regarding patient care, with which the provider rates level of agreement. Items representing the attitudes and beliefs of providers towards patients with SCD are rated on a Likert-type scale, with level of agreement indicated as 1 (strongly disagree) to 6 (strongly agree). The positive attitudes subscale consists of 4 items (Cronbach α= 0.85), and the negative attitudes subscale consists of 6 items (Cronbach α = 0.89). The Red-Flag Behaviors subscale includes 4 items that indicate behavior concerns about drug-seeking, such as requesting specific narcotics and changing behavior when the provider walks in.8,36,37
Sickle cell and primary care providers also completed a survey consisting of sets of items compiled from existing provider surveys; this survey consisted of a list of 16 barriers to using opioids, which the providers rated on a 5-point Likert-type scale (1, not a barrier; 5, complete barrier).13,16,38 Providers indicated their level of experience with caring for patients with SCD; care provided, such as routine health screenings; and comfort level with providing preventive care, managing comorbidities, and managing acute and chronic pain. Providers were asked what potential facilitators might improve care for patients with SCD, including higher reimbursement, case management services, access to pain management specialists, and access to clinical decision-support tools. Providers responded to specific questions about management with hydroxyurea (eg, criteria for, barriers to, and comfort level with prescribing).39 The surveys are included in the Appendix.
Triangulation
Data from the interviews and surveys were triangulated to enhance understanding of results generated from the different data sources.40 Convergence of findings, different facets of the same phenomenon, or new perspectives were examined.
Results
Qualitative Data
Adolescents and adults with SCD (n = 55) and health care providers and community stakeholders (n = 56) participated in group or individual interviews to help us gain an in-depth understanding of the needs and barriers related to SCD care in our 5-county region. Participants with SCD described their experiences, which included stigma, racism, labeling, and, consequently, stress. They also identified barriers such as lack of transportation, challenges with insurance, and lack of access to providers who were competent with pain management. They reported that having SCD in a health care system that was unable to meet their needs was burdensome.
Barriers to Care and Treatments. Adolescents and adults indicated that SCD and its sequelae posed significant barriers to health care. Feelings of tiredness and pain make it more difficult for them to seek care. The emotional burden of SCD (fear and anger) was a frequently cited barrier, which was fueled by previous negative encounters with the health care system. All adolescents and adults with SCD reported that they knew of stigma in relation to seeking pain management that was pervasive and long-standing, and the majority reported they had directly experienced stigma. They reported that being labeled as “drug-seekers” was typical when in the ED for pain management. Participants articulated unconscious bias or overt racism among providers: “people with sickle cell are Black ... and Black pain is never as valuable as White pain” (25-year-old male). Respondents with SCD described challenges to the credibility of their pain reports in the ED. They reported that ED providers expressed doubts regarding the existence and/or severity of their pain, consequently creating a feeling of disrespect for patients seeking pain relief. The issue of stigma was mentioned by only 2 of 56 providers during their interviews.
Lack of Access to Knowledgeable, Compassionate Providers. Lack of access to knowledgeable care providers was another prevalent theme expressed by adolescents and adults with SCD. Frustration occurred when providers did not have knowledge of SCD and its management, particularly pain assessment. Adolescents and adults with SCD noted the lack of compassion among providers: “I’ve been kicked out of the hospital because they felt like okay, well we gave you enough medication, you should be all right” (29-year-old female). Providers specifically mentioned lack of compassion and knowledge as barriers to SCD care much less often during their interviews compared with the adolescents and adults with SCD.
Health Care System Barriers. Patient participants often expressed concerns about concrete and structural aspects of care. Getting to their appointments was a challenge for half of the interviewees, as they either did not have access to a vehicle or could not afford to travel the needed distance to obtain quality care. Even when hospitals were accessible by public transportation, those with excruciating pain understandably preferred a more comfortable and private way to travel: “I would like to change that, something that will be much easier, convenient for sickle cell patients that do suffer with pain, that they don’t have to travel always to see the doctor” (30-year-old male).
Insurance and other financial barriers also played an important role in influencing decisions to seek health care services. Medical expenses were not covered, or co-pays were too high. The Medicaid managed care system could prevent access to knowledgeable providers who were not within network. Such a lack of access discouraged some adolescents and adults with SCD from seeking acute and preventive care.
Transition From Pediatric to Adult Care. Interviewees with SCD expressed distress about the gap between pediatric and adult care. They described how they had a long-standing relationship with their medical providers, who were familiar with their medical background and history from childhood. Adolescent interviewees reported an understanding of their own pain management as well as adherence to and satisfaction with their individualized pain plans. However, adults noted that satisfaction plummeted with increasing age due to the limited number of experienced adult SCD providers, which was compounded by negative experiences (stigma, racism, drug-seeking label).
One interviewee emphasized the difficulty of finding knowledgeable providers after transition: “When you’re a pediatric sickle cell [patient], you have the doctors there every step of the way, but not with adult sickle cell… I know when I first transitioned I never felt more alone in my life… you look at that ER doctor kind of with the same mindset as you would your hematologist who just hand walked you through everything. And adult care providers were a lot more blunt and cold and they’re like… ‘I don’t know; I’m not really educated in sickle cell.’” A sickle cell provider shared his insight about the problem of transitioning: “I think it’s particularly challenging because we, as a community, don’t really set them up for success. It’s different from other chronic conditions [in that] it’s much harder to find an adult sickle cell provider. There’s not a lot of adult hematologists that will take care of our adult patients, and so I know statistically, there’s like a drop-down in the overall outcomes of our kids after they age out of our pediatric program.”
Self-Management, Supporting Hydroxyurea Use. Interview participants with SCD reported using a variety of methods to manage pain at home and chose to go to the ED only when the pain became intolerable. Patients and providers expressed awareness of different resources for managing pain at home, yet they also indicated that these resources have not been consolidated in an accessible way for patients and families. Some resources cited included heat therapy, acupuncture, meditation, medical marijuana, virtual reality devices, and pain medications other than opioids.
Patients and providers expressed the need for increasing awareness and education about hydroxyurea. Many interview participants with SCD were concerned about side effects, multiple visits with a provider during dose titration, and ongoing laboratory monitoring. They also expressed difficulties with scheduling multiple appointments, depending on access to transportation and limited provider clinic hours. They were aware of strategies for improving adherence with hydroxyurea, including setting phone alarms, educating family members about hydroxyurea, and eliciting family support, but expressed needing help to consistently implement these strategies.
Safe Opioid Prescribing. Adult care providers expressed concerns about safe opioid prescribing for patients with SCD. They were reluctant to prescribe opioid doses needed to adequately control SCD pain. Providers expressed uncertainty and fear or concern about medical/legal liability or about their judgment about what’s safe and not safe for patients with chronic use/very high doses of opioids. “I know we’re in like this opiate epidemic here in this country but I feel like these patients don’t really fit under that umbrella that the problem is coming from so [I am] just trying to learn more about how to take care of them.”
Care Coordination and Provider Communication. Adolescents and adults with SCD reported having positive experiences—good communication, established trust, and compassionate care—with their usual providers. However, they perceived that ED physicians and nurses did not really care about them. Both interviewees with SCD and providers recognized the importance of good communication in all settings as the key to overcoming barriers to receiving quality care. All agreed on the importance of using individual pain plans so that all providers, especially ED providers, can be more at ease with treating adolescents and adults with SCD.
Quantitative Data: Adolescents and Adults With SCD
Fifty-eight adolescents and adults with SCD (aged 15 to 48 years) completed the survey. Three additional individuals who did not complete the interview completed the survey. Reasons for not completing the interview included scheduling challenges (n = 2) or a sickle cell pain episode (n = 1). The average age of participants was 31 years ± 8.6, more than half (57%) were female, and the majority (93%) were African American (Table 1). Most (71%) had never been married. Half (50%) had some college or an associate degree, and 40% were employed and reported an annual household income of less than $30,000. Insurance coverage was predominantly Medi-Cal (Medicaid, 69%). The majority of participants resided in Alameda (34.5%) or Contra Costa (21%) counties. The majority of sickle cell care was received in Alameda County, whether outpatient (52%), inpatient (40%), or ED care (41%). The majority (71%) had a diagnosis of SCD hemoglobin SS.

Pain. More than one-third of individuals with SCD reported 1 or 2 ED visits for pain in the previous 6 months (34%), and more than 3 hospitalizations (36%) related to pain in the previous year (Table 2). The majority (85%) reported having severe pain at home in the previous 6 months that they did not seek health care for, consistent with their reports in the qualitative interviews. More than half (59%) reported 4 or more of these severe pain episodes that led to inability to perform daily activities for 1 week or more. While pain interference on the PROMIS Pain Interference Short Form on average (T-score, 59.6 ± 8.6) was similar to that of the general population (T-score, 50 ± 10), a higher proportion of patients with SCD reported pain interference compared with the general population. The mean self-efficacy (confidence in ability to manage complications of SCD) score on the SCSES of 30.0 ± 7.3 (range, 9–45) was similar to that of other adults with SCD (mean, 32.2 ± 7.0). Twenty-five percent of the present sample had a low self-efficacy score (< 25).

Barriers to Care and Treatments. Consistent with the qualitative data, SCD-related symptoms such as tiredness (64%) and pain (62%) were reported most often as barriers to care (Table 3). Emotions (> 25%) such as worry/fear, frustration/anger, and lack of confidence were other important barriers to care. Provider knowledge and attitudes were cited next most often, with 38% of the sample indicating “Providers accuse me of drug-seeking” and “It is hard for me to find a provider who has enough experiences with or knowledge about SCD.” Participants expressed that they were not believed when in pain and “I am treated differently from other patients.” Almost half of respondents cited “I am not seen quickly enough when I am in pain” as a barrier to their care.

Consistent with the qualitative data, transportation barriers (not having a vehicle, costs of transportation, public transit not easy to get to) were cited by 55% of participants. About half of participants reported that insurance was an important barrier, with high co-pays and medications and other services not covered. In addition, gathering approvals was a long and fragmented process, particularly for consultations among providers (hematology, primary care provider, pain specialist). Furthermore, insurance provided limited choices about location for services.
Participants reported social support system burnout (22%), help needed with daily activities (21%), and social isolation or generally not having enough support (33%) as ongoing barriers. Difficulties were encountered with self-management (eg, taking medications on time or making follow-up appointments, 19%), with 22% of participants finding the health care system confusing or hard to understand. Thirty percent reported “Places for me to go to learn how to stay well are not close by or easy to get to.” ”Worry about side effects” (33%) was a common barrier to hydroxyurea use. Participants described “forgetting to take the medicine,” “tried before but it did not work,” “heard scary things” about hydroxyurea, and “not interested in taking another medicine” as barriers.
Quality of Care. More than half (51%) of the 53 participants who had accessed health care in the previous year rated their overall health care as poor on the ASCQ-Me QOC measure. This was significantly higher compared to the reports from more than 47,000 adults with Medicaid in 2017 (16%),41 and to the 2008-2009 report from 556 adults with SCD from across the United States (37%, Figure 2).33 The major contributor to these poor ratings for participants in our sample was low satisfaction with ED care.

Sixty percent of the 42 participants who had accessed ED care in the past year indicated “never” or “sometimes” to the question “When you went to the ED for care, how often did you get it as soon as you wanted?” compared with only 16% of the 2017 adult Medicaid population responding (n = 25,789) (Figure 3). Forty-seven percent of those with an ED visit indicated that, in the previous 12 months, they had been made to wait “more than 2 hours before receiving treatment for acute pain in the ED.” However, in the previous 12 months, 39% reported that their wait time in the ED had been only “between five minutes and one hour.”

On the ASCQ-Me QOC Access to Care composite measure, 33% of 42 participants responding reported they were seen at a routine appointment as soon as they would have liked. This is significantly lower compared to 56% of the adult Medicaid population responding to the same question. Reports of provider communication (Provider Communication composite) for adolescents and adults with SCD were comparable to reports of adults with SCD from the ASCQ-Me field test,33 but adults with Medicaid reported higher ratings of quality communication behaviors (Figure 4).33,41 Nearly 60% of both groups with SCD reported that providers “always” performed quality communication behaviors—listened carefully, spent enough time, treated them with respect, and explained things well—compared with more than 70% of adults with Medicaid.

Participants from all counties reported the same number of barriers to care on average (3.3 ± 2.1). Adolescents and adults who reported more barriers to care also reported lower satisfaction with care (r = –0.47, P < 0.01) and less confidence in their ability to manage their SCD (self-efficacy, r = – 0.36, P < 0.05). Female participants reported more barriers to care on average compared with male participants (2.6 ± 2.4 vs 1.4 ± 2.0, P = 0.05). Participants with higher self-efficacy reported lower pain ratings (r = –0.47, P < 0.001).
Quantitative Data: Health Care Providers
Providers (n = 56) and community stakeholders (2 leaders of community-based organizations and 3 health care administrators) were interviewed, with 29 also completing the survey. The reason for not completing (n = 22) was not having the time once the interview was complete. A link to the survey was sent to any provider not completing at the time of the interview, with 2 follow-up reminders. The majority of providers were between the ages of 31 and 50 years (46.4%), female (71.4%), and white (66.1%) (Table 4). None were of Hispanic, Latinx, or Spanish origin. Thirty-six were physicians (64.3%), and 16 were allied health professionals (28.6%). Of the 56 providers, 32 indicated they had expertise caring for patients with SCD (57.1%), 14 were ED providers (25%), and 5 were primary care providers. Most of the providers practiced in an urban setting (91.1%).

Barriers to Care: ED Provider Perspectives. Nine of 14 ED providers interviewed completed the survey on their perspectives regarding barriers to care in the ED, difficulty with follow-ups, ED training resources, and pain control for patients with SCD. ED providers (n = 8) indicated that “provider attitudes” were a barrier to care delivery in the ED for patients with SCD. Some providers (n = 7) indicated that “implicit bias,” “opioid epidemic,” “concern about addiction,” and “patient behavior” were barriers. Respondents indicated that “overcrowding” (n = 6) and “lack of care pathway/protocol” (n = 5) were barriers. When asked to express their level of agreement with statements about SCD care in the ED, respondents disagreed/strongly disagreed (n = 5) that they were “able to make a follow-up appointment” with a sickle cell specialist or primary care provider upon discharge from the ED, and others disagreed/strongly disagreed (n = 4) that they were able to make a “referral to a case management program.”
ED training and resources. Providers agreed/strongly agreed (n = 8) that they had the knowledge and training to care for patients with SCD, that they had access to needed medications, and that they had access to knowledgeable nursing staff with expertise in SCD care. All 9 ED providers indicated that they had sufficient physician/provider staffing to provide good pain management to persons with SCD in the ED.
Pain control in the ED. Seven ED providers indicated that their ED used individualized dosing protocols to treat sickle cell pain, and 5 respondents indicated their ED had a protocol for treating sickle cell pain. Surprisingly, only 3 indicated that they were aware of the NHLBI recommendations for the treatment of vaso-occlusive pain.
Barriers to Care: Primary Care Provider Perspectives. Twenty providers completed the SCD provider section of the survey, including 17 multidisciplinary SCD providers from 4 sickle cell special care centers and 3 community primary care providers. Of the 20, 12 were primary care providers for patients with SCD (Table 4).
Patient needs. Six primary care providers indicated that the medical needs of patients with SCD were being met, but none indicated that the behavioral health or mental health needs were being met.
Managing SCD comorbidities. Five primary care providers indicated they were very comfortable providing preventive ambulatory care to patients with SCD. Six indicated they were very comfortable managing acute pain episodes, but none were very comfortable managing comorbidities such as pulmonary hypertension, diabetes, or chronic pain.
Barriers to opioid use. Only 3 of 12 providers reviewing a list of 15 potential barriers to the use of opioids for SCD pain management indicated a perceived lack of efficacy of opioids, development of tolerance and dependence, and concerns about community perceptions as barriers. Two providers selected potential for diversion as a moderate barrier to opioid use.
Barriers to hydroxyurea use. Eight of 12 providers indicated that the common reasons that patients/families refuse hydroxyurea were “worry about side effects”; 7 chose “don’t want to take another medicine,” and 6 chose “worry about carcinogenic potential.” Others (n = 10) indicated that “patient/family adherence with hydroxyurea” and “patient/family adherence with required blood tests” were important barriers to hydroxyurea use. Eight of the 12 providers indicated that they were comfortable with managing hydroxyurea in patients with SCD.
Care redesign. Twenty SCD and primary care providers completed the Care Redesign section of the survey. Respondents (n = 11) indicated that they would see more patients with SCD if they had accessible case management services available without charge or if patient access to transportation to clinic was also available. Ten indicated that they would see more patients with SCD if they had an accessible community health worker (who understands patient’s/family’s social situation) and access to a pain management specialist on call to answer questions and who would manage chronic pain. All (n = 20) were willing to see more patients with SCD in their practices. Most reported that a clinical decision-support tool for SCD treatment (n = 13) and avoidance of complications (n = 12) would be useful.
Discussion
We evaluated access and barriers to care, quality of care, care coordination, and provider communication from the perspectives of adolescents and adults with SCD, their care providers, and community stakeholders, within the Solberg conceptual model for quality improvement. We found that barriers within the care process content domain (context and systems) were most salient for this population of adolescents and adults with SCD, with lack of provider knowledge and poor attitudes toward adolescents and adults with SCD, particularly in the ED, cited consistently by participant groups. Stigmatization and lack of provider compassion that affected the quality of care were particularly problematic. These findings are consistent with previous reports.42,43 Adult health care (particularly ED) provider biases and negative attitudes have been recognized as major barriers to optimal pain management in SCD.8,11,44,45 Interestingly, ED providers in our needs assessment indicated that they felt they had the training and resources to manage patients with SCD. However, only a few actually reported knowing about the NHLBI recommendations for the treatment of vaso-occlusive pain.
Within the care process content domain, we also found that SCD-related complications and associated emotions (fear, worry, anxiety), compounded by lack of access to knowledgeable and compassionate providers, pose a significant burden. Negative encounters with the health care system contributed to a striking 84% of patient participants choosing to manage severe pain at home, with pain seriously interfering with their ability to function on a daily basis. ED providers agreed that provider attitudes and implicit bias pose important barriers to care for adolescents and adults with SCD. Adolescents and adults with SCD wanted, and understood the need, to enhance self-management skills. Both they and their providers agreed that barriers to hydroxyurea uptake included worries about potential side effects, challenges with adherence to repeated laboratory testing, and support with remembering to take the medicine. However, providers uniformly expressed that access to behavioral and mental health services were, if not nonexistent, impossible to access.
Participants with SCD and their providers reported infrastructural challenges (change process capability), as manifested in limitations with accessing acute and preventive care due to transportation- and insurance- related issues. There were health system barriers that were particularly encountered during the transition from pediatric to adult care. These findings are consistent with previous reports that have found fewer interdisciplinary services available in the adult care settings compared with pediatrics.46,47 Furthermore, adult care providers were less willing to accept adults with SCD because of the complexity of their management, for which the providers did not have the necessary expertise.3,48-50 In addition, both adolescents and adults with SCD and primary care providers highlighted the inadequacies of the current system in addressing the chronic pain needs of this population. Linking back to the Solberg conceptual framework, our needs assessment results confirm the important role of establishing SCD care as a priority within a health care system—this requires leadership and vision. The vision and priorities must be implemented by effective health care teams. Multilevel approaches or interventions, when implemented, will lead to the desired outcomes.
Findings from our needs assessment within our 5-county region mirror needs assessment results from the broader consortium.51 The SCDIC has prioritized developing an intervention that addresses the challenges identified within the care process domain by directly enhancing provider access to patient individualized care plans in the electronic health record in the ED. Importantly, ED providers will be asked to view a short video that directly challenges bias and stigma in the ED. Previous studies have indeed found that attitudes can be improved by providers viewing short video segments of adults with SCD discussing their experiences.36,52 This ED protocol will be one of the interventions that we will roll out in Northern California, given the significance of negative ED encounters reported by needs assessment participants. An additional feature of the intervention is a script for adults with SCD that guides them through introducing their individualized pain plan to their ED providers, thereby enhancing their self-efficacy in a situation that has been so overwhelmingly challenging.
We will implement a second SCDIC intervention that utilizes a mobile app to support self-management on the part of the patient, by supporting motivation and adherence with hydroxyurea.53 A companion app supports hydroxyurea guideline adherence on the part of the provider, in keeping with one of our findings that providers are in need of decision-support tools. Elements of the intervention also align with our findings related to the importance of a support system in managing SCD, in that participants will identify a supportive partner who will play a specific role in supporting their adherence with hydroxyurea.
On our local level, we have, by necessity, partnered with leaders and community stakeholders throughout the region to ensure that these interventions to improve SCD care are prioritized. Grant funds provide initial resources for the SCDIC interventions, but our partnering health care administrators and medical directors must ensure that participating ED and hematology providers are free from competing priorities in order to implement the changes. We have partnered with a SCD community-based organization that is designing additional educational presentations for local emergency medicine providers, with the goal to bring to life very personal stories of bias and stigma within the EDs that directly contribute to decisions to avoid ED care despite severe symptoms.
Although we attempted to obtain samples of adolescents and adults with SCD and their providers that were representative across the 5-county region, the larger proportion of respondents were from 1 county. We did not assess concerns of age- and race-matched adults in our catchment area, so we cannot definitively say that our findings are unique to SCD. However, our results are consistent with findings from the national sample of adults with SCD who participated in the ASCQ-Me field test, and with results from the SCDIC needs assessment.33,51 Interviews and surveys are subject to self-report bias and, therefore, may or may not reflect the actual behaviors or thoughts of participants. Confidence is increased in our results given the triangulation of expressed concerns across participant groups and across data collection strategies. The majority of adolescents and adults with SCD (95%) completed both the interview and survey, while 64% of ED providers interviewed completed the survey, compared with 54% of SCD specialists and primary care providers. These response rates are more than acceptable within the realm of survey response rates.54,55
Although we encourage examining issues with care delivery within the conceptual framework for quality improvement presented, we recognize that grant funding allowed us to conduct an in-depth needs assessment that might not be feasible in other settings. Still, we would like readers to understand the importance of gathering data for improvement in a systematic manner across a range of participant groups, to ultimately inform the development of interventions and provide for evaluation of outcomes as a result of the interventions. This is particularly important for a disease, such as SCD, that is both medically and sociopolitically complex.
Conclusion
Our needs assessment brought into focus the multiple factors contributing to the disparities in health care experienced by adolescents and adults with SCD on our local level, and within the context of inequities in health resources and outcomes on the national level. We propose solutions that include specific interventions developed by a consortium of SCD and implementation science experts. We utilize a quality improvement framework to ensure that the elements of the interventions also address the barriers identified by our local providers and patients that are unique to our community. The pervasive challenges in SCD care, coupled with its medical complexities, may seem insurmountable, but our survey and qualitative results provide us with a road map for the way forward.
Acknowledgments: The authors thank the adolescents and adults with sickle cell disease, the providers, and the community stakeholders who completed the interviews and surveys. The authors also acknowledge the SCCCI co-investigators for their contributions to this project, including Michael Bell, MD, Ward Hagar, MD, Christine Hoehner, FNP, Kimberly Major, MSW, Anne Marsh, MD, Lynne Neumayr, MD, and Ted Wun, MD. We also thank Kamilah Bailey, Jameelah Hodge, Jennifer Kim, Michael Rowland, Adria Stauber, Amber Fearon, and Shanda Robertson, and the Sickle Cell Data Collection Program for their contributions.
Corresponding author: Marsha J. Treadwell, PhD, University of California San Francisco Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609; marsha.treadwell@ucsf.edu.
Financial disclosures: None.
Funding/support: This work was supported by grant # 1U01HL134007 from the National Heart, Lung, and Blood Institute to the University of California San Francisco Benioff Children’s Hospital Oakland.
1. Hassell KL. Population Estimates of sickle cell disease in the U.S. Am J Prev Med. 2010; 38:S512-S521.
2. Data & Statistics on Sickle Cell Disease. Centers for Disease Control and Prevention website. www.cdc.gov/ncbddd/sicklecell/data.html. Accessed March 25, 2020.
3. Inusa BPD, Stewart CE, Mathurin-Charles S, et al. Paediatric to adult transition care for patients with sickle cell disease: a global perspective. Lancet Haematol. 2020;7:e329-e341.
4. Smith SK, Johnston J, Rutherford C, et al. Identifying social-behavioral health needs of adults with sickle cell disease in the emergency department. J Emerg Nurs. 2017;43:444-450.
5. Treadwell MJ, Barreda F, Kaur K, et al. Emotional distress, barriers to care, and health-related quality of life in sickle cell disease. J Clin Outcomes Manag. 2015;22:8-17.
6. Treadwell MJ, Hassell K, Levine R, et al. Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-Me): conceptual model based on review of the literature and formative research. Clin J Pain. 2014;30:902-914.
7. Rizio AA, Bhor M, Lin X, et al. The relationship between frequency and severity of vaso-occlusive crises and health-related quality of life and work productivity in adults with sickle cell disease. Qual Life Res. 2020;29:1533-1547.
8. Freiermuth CE, Haywood C, Silva S, et al. Attitudes toward patients with sickle cell disease in a multicenter sample of emergency department providers. Adv Emerg Nurs J. 2014;36:335-347.
9. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Natl Med Assoc. 2010;102:1050-1055.
10. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain. 2010;26:199-205.
11. Haywood C, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med. 2013;31:651-656.
12. Haywood C, Beach MC, Lanzkron S, et al. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101:1022-1033.
13. Mainous AG, Tanner RJ, Harle CA, et al. Attitudes toward management of sickle cell disease and its complications: a national survey of academic family physicians. Anemia. 2015;2015:1-6.
14. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033.
15. Lunyera J, Jonassaint C, Jonassaint J, et al. Attitudes of primary care physicians toward sickle cell disease care, guidelines, and comanaging hydroxyurea with a specialist. J Prim Care Community Health. 2017;8:37-40.
16. Whiteman LN, Haywood C, Lanzkron S, et al. Primary care providers’ comfort levels in caring for patients with sickle cell disease. South Med J. 2015;108:531-536.
17. Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood. 2014;124:3850-4004.
18. DiMartino LD, Baumann AA, Hsu LL, et al. The sickle cell disease implementation consortium: Translating evidence-based guidelines into practice for sickle cell disease. Am J Hematol. 2018;93:E391-E395.
19. King AA, Baumann AA. Sickle cell disease and implementation science: A partnership to accelerate advances. Pediatr Blood Cancer. 2017;64:e26649.
20. Solberg LI. Improving medical practice: a conceptual framework. Ann Fam Med. 2007;5:251-256.
21. Bodenheimer T, Wagner EH, Grumbach K. Improving primary care for patients with chronic illness. J Am Med Assoc. 2002;288:5.
22. Bodenheimer T. Interventions to improve chronic illness care: evaluating their effectiveness. Dis Manag. 2003;6:63-71.
23. Tsai AC, Morton SC, Mangione CM, Keeler EB. A meta-analysis of interventions to improve care for chronic illnesses. Am J Manag Care. 2005;11:478-488.
24. Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
25. Kallio H, Pietilä A-M, Johnson M, et al. Systematic methodological review: developing a framework for a qualitative semi-structured interview guide. J Adv Nurs. 2016;72:2954-2965.
26. Clarke V, Braun V. Successful Qualitative Research: A Practical Guide for Beginners. First. Thousand Oaks, CA: Sage; 2013.
27. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15:1277-1288.
28. Creswell JW, Hanson WE, Clark Plano VL, et al. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35:236-264.
29. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis A Methods Sourcebook. 4th ed. Thousand Oaks, CA: Sage; 2019.
30. Eckman JR, Hassell KL, Huggins W, et al. Standard measures for sickle cell disease research: the PhenX Toolkit sickle cell disease collections. Blood Adv. 2017; 1: 2703-2711.
31. Kendall R, Wagner B, Brodke D, et al. The relationship of PROMIS pain interference and physical function scales. Pain Med. 2018;19:1720-1724.
32. Amtmann D, Cook KF, Jensen MP, et al. Development of a PROMIS item bank to measure pain interference. Pain. 2010;150:173-182.
33. Evensen CT, Treadwell MJ, Keller S, et al. Quality of care in sickle cell disease: Cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine (Baltimore). 2016;95:e4528.
34. Edwards R, Telfair J, Cecil H, et al. Reliability and validity of a self-efficacy instrument specific to sickle cell disease. Behav Res Ther. 2000;38:951-963.
35. Edwards R, Telfair J, Cecil H, et al. Self-efficacy as a predictor of adult adjustment to sickle cell disease: one-year outcomes. Psychosom Med. 2001;63:850-858.
36. Puri Singh A, Haywood C, Beach MC, et al. Improving emergency providers’ attitudes toward sickle cell patients in pain. J Pain Symptom Manage. 2016;51:628-632.e3.
37. Glassberg JA, Tanabe P, Chow A, et al. Emergency provider analgesic practices and attitudes towards patients with sickle cell disease. Ann Emerg Med. 2013;62:293-302.e10.
38. Grahmann PH, Jackson KC 2nd, Lipman AG. Clinician beliefs about opioid use and barriers in chronic nonmalignant pain [published correction appears in J Pain Palliat Care Pharmacother. 2004;18:145-6]. J Pain Palliat Care Pharmacother. 2004;18:7-28.
39. Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010;3:255-260.
40. Fielding N. Triangulation and mixed methods designs: data integration with new research technologies. J Mixed Meth Res. 2012;6:124-136.
41. 2017 CAHPS Health Plan Survey Chartbook. Agency for Healthcare Research and Quality website. www.ahrq.gov/cahps/cahps-database/comparative-data/2017-health-plan-chartbook/results-enrollee-population.html. Accessed September 8, 2020.
42. Bulgin D, Tanabe P, Jenerette C. Stigma of sickle cell disease: a systematic review. Issues Ment Health Nurs. 2018;1-11.
43. Wakefield EO, Zempsky WT, Puhl RM, et al. Conceptualizing pain-related stigma in adolescent chronic pain: a literature review and preliminary focus group findings. PAIN Rep. 2018;3:e679.
44. Nelson SC, Hackman HW. Race matters: Perceptions of race and racism in a sickle cell center. Pediatr Blood Cancer. 2013;60:451-454.
45. Dyal BW, Abudawood K, Schoppee TM, et al. Reflections of healthcare experiences of african americans with sickle cell disease or cancer: a qualitative study. Cancer Nurs. 2019;10.1097/NCC.0000000000000750.
46. Renedo A. Not being heard: barriers to high quality unplanned hospital care during young people’s transition to adult services - evidence from ‘this sickle cell life’ research. BMC Health Serv Res. 2019;19:876.
47. Ballas S, Vichinsky E. Is the medical home for adult patients with sickle cell disease a reality or an illusion? Hemoglobin. 2015;39:130-133.
48. Hankins JS, Osarogiagbon R, Adams-Graves P, et al. A transition pilot program for adolescents with sickle cell disease. J Pediatr Health Care. 2012;26 e45-e49.
49. Smith WR, Sisler IY, Johnson S, et al. Lessons learned from building a pediatric-to-adult sickle cell transition program. South Med J. 2019;112:190-197.
50. Lanzkron S, Sawicki GS, Hassell KL, et al. Transition to adulthood and adult health care for patients with sickle cell disease or cystic fibrosis: Current practices and research priorities. J Clin Transl Sci. 2018;2:334-342.
51. Kanter J, Gibson R, Lawrence RH, et al. Perceptions of US adolescents and adults with sickle cell disease on their quality of care. JAMA Netw Open. 2020;3:e206016.
52. Haywood C, Lanzkron S, Hughes MT, et al. A video-intervention to improve clinician attitudes toward patients with sickle cell disease: the results of a randomized experiment. J Gen Intern Med. 2011;26:518-523.
53. Hankins JS, Shah N, DiMartino L, et al. Integration of mobile health into sickle cell disease care to increase hydroxyurea utilization: protocol for an efficacy and implementation study. JMIR Res Protoc. 2020;9:e16319.
54. Fan W, Yan Z. Factors affecting response rates of the web survey: A systematic review. Comput Hum Behav. 2010;26:132-139.
55. Millar MM, Dillman DA. Improving response to web and mixed-mode surveys. Public Opin Q. 2011;75:249-269.
From the University of California San Francisco (Dr. Treadwell, Dr. Hessler, Yumei Chen, Swapandeep Mushiana, Dr. Potter, and Dr. Vichinsky), the University of California Los Angeles (Dr. Jacob), and the University of California Berkeley (Alex Chen).
Abstract
- Objective: Adolescents and adults with sickle cell disease (SCD) face pervasive disparities in health resources and outcomes. We explored barriers to and facilitators of care to identify opportunities to support implementation of evidence-based interventions aimed at improving care quality for patients with SCD.
- Methods: We engaged a representative sample of adolescents and adults with SCD (n = 58), health care providers (n = 51), and community stakeholders (health care administrators and community-based organization leads (n = 5) in Northern California in a community-based needs assessment. We conducted group interviews separately with participant groups to obtain in-depth perspectives. Adolescents and adults with SCD completed validated measures of pain interference, quality of care, self-efficacy, and barriers to care. Providers and community stakeholders completed surveys about barriers to SCD care.
- Results: We triangulated qualitative and quantitative data and found that participants with SCD (mean age, 31 ± 8.6 years), providers, and community stakeholders emphasized the social and emotional burden of SCD as barriers. Concrete barriers agreed upon included insurance and lack of resources for addressing pain impact. Adolescents and adults with SCD identified provider issues (lack of knowledge, implicit bias), transportation, and limited social support as barriers. Negative encounters with the health care system contributed to 84% of adolescents and adults with SCD reporting they chose to manage severe pain at home. Providers focused on structural barriers: lack of access to care guidelines, comfort level with and knowledge of SCD management, and poor care coordination.
- Conclusion: Strategies for improving access to compassionate, evidence-based quality care, as well as strategies for minimizing the burden of having SCD, are warranted for this medically complex population.
Keywords: barriers to care; quality of care; care access; care coordination.
Sickle cell disease (SCD), an inherited chronic medical condition, affects about 100,000 individuals in the United States, a population that is predominantly African American.1 These individuals experience multiple serious and life-threatening complications, most frequently recurrent vaso-occlusive pain episodes,2 and they require interactions with multidisciplinary specialists from childhood. Because of advances in treatments, the majority are reaching adulthood; however, there is a dearth of adult health care providers with the training and expertise to manage their complex medical needs.3 Other concrete barriers to adequate SCD care include insurance and distance to comprehensive SCD centers.4,5
Social, behavioral, and emotional factors may also contribute to challenges with SCD management. SCD may limit daily functional abilities and lead to diminished overall quality of life.6,7 Some adolescents and adults may require high doses of opioids, which contributes to health care providers’ perceptions that there is a high prevalence of drug addiction in the population.8,9 These providers express negative attitudes towards adults with SCD, and, consequently, delay medication administration when it is acutely needed and provide otherwise suboptimal treatment.8,10,11 Adult care providers may also be uncomfortable with prescribing and managing disease-modifying therapies (blood transfusion, hydroxyurea) that have established efficacy.12-17
As 1 of 8 programs funded by the National Heart, Lung, and Blood Institute’s (NHLBI) Sickle Cell Disease Implementation Consortium (SCDIC), we are using implementation science to reduce barriers to care and improve quality of care and health care outcomes in SCD.18,19 Given that adolescents and adults with SCD experience high mortality, severe pain, and progressive decline in their ability to function day to day, and also face lack of access to knowledgeable, compassionate providers in primary and emergency settings, the SCDIC focuses on individuals aged 15 to 45 years.6,8,9,11,12
Our regional SCDIC program, the Sickle Cell Care Coordination Initiative (SCCCI), brings together researchers, clinicians, adolescents, and adults with SCD and their families, dedicated community members, policy makers, and administrators to identify and address barriers to health care within 5 counties in Northern California. One of our first steps was to conduct a community-based needs assessment, designed to inform implementation of evidence-based interventions, accounting for unique contextual factors in our region.
Conceptual Framework for Improving Medical Practice
Our needs assessment is guided by Solberg’s Conceptual Framework for Improving Medical Practice (Figure 1).20 Consistent with the overarching principles of the SCDIC, this conceptual framework focuses on the inadequate implementation of evidence-based guidelines, and on the need to first understand multifactorial facilitators and barriers to guideline implementation in order to effect change. The framework identifies 3 main elements that must be present to ensure improvements in quality-of-care processes and patient outcomes: priority, change process capability, and care process content. Priority refers to ample resource allocation for the specific change, as well as freedom from competing priorities for those implementing the change. Change process capability includes strong, effective leadership, adequate infrastructure for managing change (including resources and time), change management skills at all levels, and an established clinical information system. Care process content refers to context and systems-level changes, such as delivery system redesign as needed, support for self-management to lessen the impact of the disease, and decision support.21-23
The purpose of our community-based needs assessment was to evaluate barriers to care and quality of care in SCD, within Solberg’s conceptual model for improving medical practice. The specific aims were to evaluate access and barriers to care (eg, lack of provider expertise and training, health care system barriers such as poor care coordination and provider communication); evaluate quality of care; and assess patient needs related to pain, pain interference, self-efficacy, and self-management for adolescents and adults with SCD. We gathered the perspectives of a representative community of adolescents and adults with SCD, their providers, and community stakeholders in order to examine barriers, quality of life and care, and patient experiences in our region.
Methods
Design
In this cross-sectional study, adolescents and adults with SCD, their providers, and community stakeholders participated in group or individual qualitative interviews and completed surveys between October 2017 and March 2018.
Setting and Sample
Recruitment flyers were posted on a regional SCD-focused website, and clinical providers or a study coordinator introduced information about the needs assessment to potential participants with SCD during clinic visits at the participating centers. Participants with SCD were eligible if they had any diagnosis of SCD, were aged 15 to 48 years, and received health services within 5 Northern California counties (Alameda, Contra Costa, Sacramento, San Francisco, and Solano). They were excluded if they did not have a SCD diagnosis or had not received health services within the catchment area. As the project proceeded, participants were asked to refer other adolescents and adults with SCD for the interviews and surveys (snowball sampling). Our goal was to recruit 50 adolescents and adults with SCD into the study, aiming for 10 representatives from each county.
Providers and community stakeholders were recruited via emails, letters and informational flyers. We engaged our partner, the Sickle Cell Data Collection Program,2 to generate a list of providers and institutions that had seen patients with SCD in primary, emergency, or inpatient settings in the region. We contacted these institutions to describe the SCCCI and invite participation in the needs assessment. We also invited community-based organization leads and health care administrators who worked with SCD to participate. Providers accessed confidential surveys via a secure link on the study website or completed paper versions. Common data collected across providers included demographics and descriptions of practice settings.
Participants were eligible to be part of the study if they were health care providers (physicians and nurses) representing hematology, primary care, family medicine, internal medicine, or emergency medicine; ancillary staff (social work, psychology, child life); or leaders or administrators of clinical or sickle cell community-based organizations in Northern California (recruitment goal of n = 50). Providers were excluded if they practiced in specialties other than those noted or did not practice within the region.
Data Collection Procedures
After providing assent/consent, participating adolescents and adults with SCD took part in individual and group interviews and completed survey questionnaires. All procedures were conducted in a private space in the sickle cell center or community. Adolescents and adults with SCD completed the survey questionnaire on a tablet, with responses recorded directly in a REDCap (Research Electronic Data Capture) database,24 or on a paper version. Interviews lasted 60 (individual) to 90 (group) minutes, while survey completion time was 20 to 25 minutes. Each participant received a gift card upon completion as an expression of appreciation. All procedures were approved by the institutional review boards of the participating health care facilities.
Group and Individual Interviews
Participants with SCD and providers were invited to participate in a semi-structured qualitative interview prior to being presented with the surveys. Adolescents and adults with SCD were interviewed about barriers to care, quality of care, and pain-related experiences. Providers were asked about barriers to care and treatments. Interview guides were modified for community-based organization leaders and health care administrators who did not provide clinical services. Interview guides can be found in the Appendix. Interviews were conducted by research coordinators trained in qualitative research methods by the first author (MT). As appropriate with semi-structured interviews, the interviewers could word questions spontaneously, change the order of questions for ease of flow of conversation, and inform simultaneous coding of interviews with new themes as those might arise, as long as they touched on all topics within the interview guide.25 The interview guides were written, per qualitative research standards, based on the aims and purpose of the research,26 and were informed by existing literature on access and barriers to care in SCD, quality of care, and the needs of individuals with SCD, including in relation to impact of the disease, self-efficacy, and self-management.
Interviewees participated in either individual or group interviews, but not both. The decision for which type of interview an individual participated in was based on 2 factors: if there were not comparable participants for group interviews (eg, health care administrator and community-based organization lead), these interviews were done individually; and given that we were drawing participants from a 5-county area in Northern California, scheduling was challenging for individuals with SCD with regard to aligning schedules and traveling to a central location where the group interviews were conducted. Provider group interviews were easier to arrange because we could schedule them at the same time as regularly scheduled meetings at the participants’ health care institutions.
Interview Data Gathering and Analysis
Digital recordings of the interviews were cleaned of any participant identifying data and sent for transcription to an outside service. Transcripts were reviewed for completeness and imported into NVivo (www.qsrinternational.com), a qualitative data management program.
A thematic content analysis and deductive and inductive approaches were used to analyze the verbatim transcripts generated from the interviews. The research team was trained in the use of NVivo software to facilitate the coding process. A deductive coding scheme was initially used based on existing concepts in the literature regarding challenges to optimal SCD care, with new codes added as the thematic content analyses progressed. The initial coding, pattern coding, and use of displays to examine the relationships between different categories were conducted simultaneously.27,28 Using the constant comparative method, new concepts from participants with SCD and providers could be incorporated into subsequent interviews with other participants. For this study, the only additional concepts added were in relation to participant recruitment and retention in the SCDIC Registry. Research team members coded transcripts separately and came together weekly, constantly comparing codes and developing the consensus coding scheme. Where differences between coders existed, code meanings were discussed and clarified until consensus was reached.29
Quantitative data were analyzed using SPSS (v. 25, Chicago, IL). Descriptive statistics (means, standard deviations, frequencies, percentages) were used to summarize demographics (eg, age, gender, and race), economic status, and type of SCD. No systematic differences were detected from cases with missing values. Scale reliabilities (ie, Cronbach α) were evaluated for self-report measures.
Measurement
Adolescents and adults with SCD completed items from the PhenX Toolkit (consensus measures for Phenotypes and eXposures), assessing sociodemographics (age, sex, race, ethnicity, educational attainment, occupation, marital status, annual income, insurance), and clinical characteristics (sickle cell diagnosis and emergency department [ED] and hospital utilization for pain).30
Pain Interference Short Form (Patient-Reported Outcomes Measurement Information System [PROMIS]). The Pain Interference Form consists of 8 items that assess the degree to which pain interfered with day-to-day activities in the previous 7 days at home, including impacts on social, cognitive, emotional, and physical functioning; household chores and recreational activities; sleep; and enjoyment in life. Reliability and validity of the PROMIS Pain Interference Scale has been demonstrated, with strong negative correlations with Physical Function Scales (r = 0.717, P < 0.01), indicating that higher scores are associated with lower function (β = 0.707, P < 0.001).31 The Cronbach α estimate for the other items on the pain interference scale was 0.99. Validity analysis indicated strong correlations with pain-related domains: BPI Interference Subscale (rho = 0.90), SF-36 Bodily Pain Subscale (rho = –0.84), and 0–10 Numerical Rating of Pain Intensity (rho = 0.48).32
Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) Quality of Care (QOC). ASCQ-Me QOC consists of 27 items that measure the quality of care that adults with SCD have received from health care providers.33 There are 3 composites: provider communication (quality of patient and provider communication), ED care (quality of care in the ED), and access (to routine and emergency care). Internal consistency reliability for all 3 composites is greater than 0.70. Strong correlations of the provider communication composite with overall ratings of routine care (r = 0.65) and overall provider ratings (r = 0.83) provided evidence of construct validity. Similarly, the ED care composite was strongly correlated with overall ratings of QOC in the ED, and the access composite was highly correlated with overall evaluations of ED care (r = 0.70). Access, provider interaction, and ED care composites were reliable (Cronbach α, 0.70–0.83) and correlated with ratings of global care (r = 0.32–0.83), further indicating construct validity.33
Sickle Cell Self-Efficacy Scale (SCSES). The SCSES is a 9-item, self-administered questionnaire measuring perceptions of the ability to manage day-to-day issues resulting from SCD. SCSES items are scored on a 5-point scale ranging from Not sure at all (1) to Very sure (5). Individual item responses are summed to give an overall score, with higher scores indicating greater self-efficacy. The SCSES has acceptable reliability (r = 0.45, P < 0.001) and validity (α = 0.89).34,35
Sickle Cell Disease Barriers Checklist. This checklist consists of 53 items organized into 8 categories: insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers (fear, anger), and disease-related barriers. Participants check applicable barriers, with a total score range of 0 to 53 and higher scores indicating more barriers to care. The SCD Barriers Checklist has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).5
ED Provider Checklist. The ED provider survey is a checklist of 14 statements pertaining to issues regarding patient care, with which the provider rates level of agreement. Items representing the attitudes and beliefs of providers towards patients with SCD are rated on a Likert-type scale, with level of agreement indicated as 1 (strongly disagree) to 6 (strongly agree). The positive attitudes subscale consists of 4 items (Cronbach α= 0.85), and the negative attitudes subscale consists of 6 items (Cronbach α = 0.89). The Red-Flag Behaviors subscale includes 4 items that indicate behavior concerns about drug-seeking, such as requesting specific narcotics and changing behavior when the provider walks in.8,36,37
Sickle cell and primary care providers also completed a survey consisting of sets of items compiled from existing provider surveys; this survey consisted of a list of 16 barriers to using opioids, which the providers rated on a 5-point Likert-type scale (1, not a barrier; 5, complete barrier).13,16,38 Providers indicated their level of experience with caring for patients with SCD; care provided, such as routine health screenings; and comfort level with providing preventive care, managing comorbidities, and managing acute and chronic pain. Providers were asked what potential facilitators might improve care for patients with SCD, including higher reimbursement, case management services, access to pain management specialists, and access to clinical decision-support tools. Providers responded to specific questions about management with hydroxyurea (eg, criteria for, barriers to, and comfort level with prescribing).39 The surveys are included in the Appendix.
Triangulation
Data from the interviews and surveys were triangulated to enhance understanding of results generated from the different data sources.40 Convergence of findings, different facets of the same phenomenon, or new perspectives were examined.
Results
Qualitative Data
Adolescents and adults with SCD (n = 55) and health care providers and community stakeholders (n = 56) participated in group or individual interviews to help us gain an in-depth understanding of the needs and barriers related to SCD care in our 5-county region. Participants with SCD described their experiences, which included stigma, racism, labeling, and, consequently, stress. They also identified barriers such as lack of transportation, challenges with insurance, and lack of access to providers who were competent with pain management. They reported that having SCD in a health care system that was unable to meet their needs was burdensome.
Barriers to Care and Treatments. Adolescents and adults indicated that SCD and its sequelae posed significant barriers to health care. Feelings of tiredness and pain make it more difficult for them to seek care. The emotional burden of SCD (fear and anger) was a frequently cited barrier, which was fueled by previous negative encounters with the health care system. All adolescents and adults with SCD reported that they knew of stigma in relation to seeking pain management that was pervasive and long-standing, and the majority reported they had directly experienced stigma. They reported that being labeled as “drug-seekers” was typical when in the ED for pain management. Participants articulated unconscious bias or overt racism among providers: “people with sickle cell are Black ... and Black pain is never as valuable as White pain” (25-year-old male). Respondents with SCD described challenges to the credibility of their pain reports in the ED. They reported that ED providers expressed doubts regarding the existence and/or severity of their pain, consequently creating a feeling of disrespect for patients seeking pain relief. The issue of stigma was mentioned by only 2 of 56 providers during their interviews.
Lack of Access to Knowledgeable, Compassionate Providers. Lack of access to knowledgeable care providers was another prevalent theme expressed by adolescents and adults with SCD. Frustration occurred when providers did not have knowledge of SCD and its management, particularly pain assessment. Adolescents and adults with SCD noted the lack of compassion among providers: “I’ve been kicked out of the hospital because they felt like okay, well we gave you enough medication, you should be all right” (29-year-old female). Providers specifically mentioned lack of compassion and knowledge as barriers to SCD care much less often during their interviews compared with the adolescents and adults with SCD.
Health Care System Barriers. Patient participants often expressed concerns about concrete and structural aspects of care. Getting to their appointments was a challenge for half of the interviewees, as they either did not have access to a vehicle or could not afford to travel the needed distance to obtain quality care. Even when hospitals were accessible by public transportation, those with excruciating pain understandably preferred a more comfortable and private way to travel: “I would like to change that, something that will be much easier, convenient for sickle cell patients that do suffer with pain, that they don’t have to travel always to see the doctor” (30-year-old male).
Insurance and other financial barriers also played an important role in influencing decisions to seek health care services. Medical expenses were not covered, or co-pays were too high. The Medicaid managed care system could prevent access to knowledgeable providers who were not within network. Such a lack of access discouraged some adolescents and adults with SCD from seeking acute and preventive care.
Transition From Pediatric to Adult Care. Interviewees with SCD expressed distress about the gap between pediatric and adult care. They described how they had a long-standing relationship with their medical providers, who were familiar with their medical background and history from childhood. Adolescent interviewees reported an understanding of their own pain management as well as adherence to and satisfaction with their individualized pain plans. However, adults noted that satisfaction plummeted with increasing age due to the limited number of experienced adult SCD providers, which was compounded by negative experiences (stigma, racism, drug-seeking label).
One interviewee emphasized the difficulty of finding knowledgeable providers after transition: “When you’re a pediatric sickle cell [patient], you have the doctors there every step of the way, but not with adult sickle cell… I know when I first transitioned I never felt more alone in my life… you look at that ER doctor kind of with the same mindset as you would your hematologist who just hand walked you through everything. And adult care providers were a lot more blunt and cold and they’re like… ‘I don’t know; I’m not really educated in sickle cell.’” A sickle cell provider shared his insight about the problem of transitioning: “I think it’s particularly challenging because we, as a community, don’t really set them up for success. It’s different from other chronic conditions [in that] it’s much harder to find an adult sickle cell provider. There’s not a lot of adult hematologists that will take care of our adult patients, and so I know statistically, there’s like a drop-down in the overall outcomes of our kids after they age out of our pediatric program.”
Self-Management, Supporting Hydroxyurea Use. Interview participants with SCD reported using a variety of methods to manage pain at home and chose to go to the ED only when the pain became intolerable. Patients and providers expressed awareness of different resources for managing pain at home, yet they also indicated that these resources have not been consolidated in an accessible way for patients and families. Some resources cited included heat therapy, acupuncture, meditation, medical marijuana, virtual reality devices, and pain medications other than opioids.
Patients and providers expressed the need for increasing awareness and education about hydroxyurea. Many interview participants with SCD were concerned about side effects, multiple visits with a provider during dose titration, and ongoing laboratory monitoring. They also expressed difficulties with scheduling multiple appointments, depending on access to transportation and limited provider clinic hours. They were aware of strategies for improving adherence with hydroxyurea, including setting phone alarms, educating family members about hydroxyurea, and eliciting family support, but expressed needing help to consistently implement these strategies.
Safe Opioid Prescribing. Adult care providers expressed concerns about safe opioid prescribing for patients with SCD. They were reluctant to prescribe opioid doses needed to adequately control SCD pain. Providers expressed uncertainty and fear or concern about medical/legal liability or about their judgment about what’s safe and not safe for patients with chronic use/very high doses of opioids. “I know we’re in like this opiate epidemic here in this country but I feel like these patients don’t really fit under that umbrella that the problem is coming from so [I am] just trying to learn more about how to take care of them.”
Care Coordination and Provider Communication. Adolescents and adults with SCD reported having positive experiences—good communication, established trust, and compassionate care—with their usual providers. However, they perceived that ED physicians and nurses did not really care about them. Both interviewees with SCD and providers recognized the importance of good communication in all settings as the key to overcoming barriers to receiving quality care. All agreed on the importance of using individual pain plans so that all providers, especially ED providers, can be more at ease with treating adolescents and adults with SCD.
Quantitative Data: Adolescents and Adults With SCD
Fifty-eight adolescents and adults with SCD (aged 15 to 48 years) completed the survey. Three additional individuals who did not complete the interview completed the survey. Reasons for not completing the interview included scheduling challenges (n = 2) or a sickle cell pain episode (n = 1). The average age of participants was 31 years ± 8.6, more than half (57%) were female, and the majority (93%) were African American (Table 1). Most (71%) had never been married. Half (50%) had some college or an associate degree, and 40% were employed and reported an annual household income of less than $30,000. Insurance coverage was predominantly Medi-Cal (Medicaid, 69%). The majority of participants resided in Alameda (34.5%) or Contra Costa (21%) counties. The majority of sickle cell care was received in Alameda County, whether outpatient (52%), inpatient (40%), or ED care (41%). The majority (71%) had a diagnosis of SCD hemoglobin SS.
Pain. More than one-third of individuals with SCD reported 1 or 2 ED visits for pain in the previous 6 months (34%), and more than 3 hospitalizations (36%) related to pain in the previous year (Table 2). The majority (85%) reported having severe pain at home in the previous 6 months that they did not seek health care for, consistent with their reports in the qualitative interviews. More than half (59%) reported 4 or more of these severe pain episodes that led to inability to perform daily activities for 1 week or more. While pain interference on the PROMIS Pain Interference Short Form on average (T-score, 59.6 ± 8.6) was similar to that of the general population (T-score, 50 ± 10), a higher proportion of patients with SCD reported pain interference compared with the general population. The mean self-efficacy (confidence in ability to manage complications of SCD) score on the SCSES of 30.0 ± 7.3 (range, 9–45) was similar to that of other adults with SCD (mean, 32.2 ± 7.0). Twenty-five percent of the present sample had a low self-efficacy score (< 25).
Barriers to Care and Treatments. Consistent with the qualitative data, SCD-related symptoms such as tiredness (64%) and pain (62%) were reported most often as barriers to care (Table 3). Emotions (> 25%) such as worry/fear, frustration/anger, and lack of confidence were other important barriers to care. Provider knowledge and attitudes were cited next most often, with 38% of the sample indicating “Providers accuse me of drug-seeking” and “It is hard for me to find a provider who has enough experiences with or knowledge about SCD.” Participants expressed that they were not believed when in pain and “I am treated differently from other patients.” Almost half of respondents cited “I am not seen quickly enough when I am in pain” as a barrier to their care.
Consistent with the qualitative data, transportation barriers (not having a vehicle, costs of transportation, public transit not easy to get to) were cited by 55% of participants. About half of participants reported that insurance was an important barrier, with high co-pays and medications and other services not covered. In addition, gathering approvals was a long and fragmented process, particularly for consultations among providers (hematology, primary care provider, pain specialist). Furthermore, insurance provided limited choices about location for services.
Participants reported social support system burnout (22%), help needed with daily activities (21%), and social isolation or generally not having enough support (33%) as ongoing barriers. Difficulties were encountered with self-management (eg, taking medications on time or making follow-up appointments, 19%), with 22% of participants finding the health care system confusing or hard to understand. Thirty percent reported “Places for me to go to learn how to stay well are not close by or easy to get to.” ”Worry about side effects” (33%) was a common barrier to hydroxyurea use. Participants described “forgetting to take the medicine,” “tried before but it did not work,” “heard scary things” about hydroxyurea, and “not interested in taking another medicine” as barriers.
Quality of Care. More than half (51%) of the 53 participants who had accessed health care in the previous year rated their overall health care as poor on the ASCQ-Me QOC measure. This was significantly higher compared to the reports from more than 47,000 adults with Medicaid in 2017 (16%),41 and to the 2008-2009 report from 556 adults with SCD from across the United States (37%, Figure 2).33 The major contributor to these poor ratings for participants in our sample was low satisfaction with ED care.
Sixty percent of the 42 participants who had accessed ED care in the past year indicated “never” or “sometimes” to the question “When you went to the ED for care, how often did you get it as soon as you wanted?” compared with only 16% of the 2017 adult Medicaid population responding (n = 25,789) (Figure 3). Forty-seven percent of those with an ED visit indicated that, in the previous 12 months, they had been made to wait “more than 2 hours before receiving treatment for acute pain in the ED.” However, in the previous 12 months, 39% reported that their wait time in the ED had been only “between five minutes and one hour.”
On the ASCQ-Me QOC Access to Care composite measure, 33% of 42 participants responding reported they were seen at a routine appointment as soon as they would have liked. This is significantly lower compared to 56% of the adult Medicaid population responding to the same question. Reports of provider communication (Provider Communication composite) for adolescents and adults with SCD were comparable to reports of adults with SCD from the ASCQ-Me field test,33 but adults with Medicaid reported higher ratings of quality communication behaviors (Figure 4).33,41 Nearly 60% of both groups with SCD reported that providers “always” performed quality communication behaviors—listened carefully, spent enough time, treated them with respect, and explained things well—compared with more than 70% of adults with Medicaid.
Participants from all counties reported the same number of barriers to care on average (3.3 ± 2.1). Adolescents and adults who reported more barriers to care also reported lower satisfaction with care (r = –0.47, P < 0.01) and less confidence in their ability to manage their SCD (self-efficacy, r = – 0.36, P < 0.05). Female participants reported more barriers to care on average compared with male participants (2.6 ± 2.4 vs 1.4 ± 2.0, P = 0.05). Participants with higher self-efficacy reported lower pain ratings (r = –0.47, P < 0.001).
Quantitative Data: Health Care Providers
Providers (n = 56) and community stakeholders (2 leaders of community-based organizations and 3 health care administrators) were interviewed, with 29 also completing the survey. The reason for not completing (n = 22) was not having the time once the interview was complete. A link to the survey was sent to any provider not completing at the time of the interview, with 2 follow-up reminders. The majority of providers were between the ages of 31 and 50 years (46.4%), female (71.4%), and white (66.1%) (Table 4). None were of Hispanic, Latinx, or Spanish origin. Thirty-six were physicians (64.3%), and 16 were allied health professionals (28.6%). Of the 56 providers, 32 indicated they had expertise caring for patients with SCD (57.1%), 14 were ED providers (25%), and 5 were primary care providers. Most of the providers practiced in an urban setting (91.1%).
Barriers to Care: ED Provider Perspectives. Nine of 14 ED providers interviewed completed the survey on their perspectives regarding barriers to care in the ED, difficulty with follow-ups, ED training resources, and pain control for patients with SCD. ED providers (n = 8) indicated that “provider attitudes” were a barrier to care delivery in the ED for patients with SCD. Some providers (n = 7) indicated that “implicit bias,” “opioid epidemic,” “concern about addiction,” and “patient behavior” were barriers. Respondents indicated that “overcrowding” (n = 6) and “lack of care pathway/protocol” (n = 5) were barriers. When asked to express their level of agreement with statements about SCD care in the ED, respondents disagreed/strongly disagreed (n = 5) that they were “able to make a follow-up appointment” with a sickle cell specialist or primary care provider upon discharge from the ED, and others disagreed/strongly disagreed (n = 4) that they were able to make a “referral to a case management program.”
ED training and resources. Providers agreed/strongly agreed (n = 8) that they had the knowledge and training to care for patients with SCD, that they had access to needed medications, and that they had access to knowledgeable nursing staff with expertise in SCD care. All 9 ED providers indicated that they had sufficient physician/provider staffing to provide good pain management to persons with SCD in the ED.
Pain control in the ED. Seven ED providers indicated that their ED used individualized dosing protocols to treat sickle cell pain, and 5 respondents indicated their ED had a protocol for treating sickle cell pain. Surprisingly, only 3 indicated that they were aware of the NHLBI recommendations for the treatment of vaso-occlusive pain.
Barriers to Care: Primary Care Provider Perspectives. Twenty providers completed the SCD provider section of the survey, including 17 multidisciplinary SCD providers from 4 sickle cell special care centers and 3 community primary care providers. Of the 20, 12 were primary care providers for patients with SCD (Table 4).
Patient needs. Six primary care providers indicated that the medical needs of patients with SCD were being met, but none indicated that the behavioral health or mental health needs were being met.
Managing SCD comorbidities. Five primary care providers indicated they were very comfortable providing preventive ambulatory care to patients with SCD. Six indicated they were very comfortable managing acute pain episodes, but none were very comfortable managing comorbidities such as pulmonary hypertension, diabetes, or chronic pain.
Barriers to opioid use. Only 3 of 12 providers reviewing a list of 15 potential barriers to the use of opioids for SCD pain management indicated a perceived lack of efficacy of opioids, development of tolerance and dependence, and concerns about community perceptions as barriers. Two providers selected potential for diversion as a moderate barrier to opioid use.
Barriers to hydroxyurea use. Eight of 12 providers indicated that the common reasons that patients/families refuse hydroxyurea were “worry about side effects”; 7 chose “don’t want to take another medicine,” and 6 chose “worry about carcinogenic potential.” Others (n = 10) indicated that “patient/family adherence with hydroxyurea” and “patient/family adherence with required blood tests” were important barriers to hydroxyurea use. Eight of the 12 providers indicated that they were comfortable with managing hydroxyurea in patients with SCD.
Care redesign. Twenty SCD and primary care providers completed the Care Redesign section of the survey. Respondents (n = 11) indicated that they would see more patients with SCD if they had accessible case management services available without charge or if patient access to transportation to clinic was also available. Ten indicated that they would see more patients with SCD if they had an accessible community health worker (who understands patient’s/family’s social situation) and access to a pain management specialist on call to answer questions and who would manage chronic pain. All (n = 20) were willing to see more patients with SCD in their practices. Most reported that a clinical decision-support tool for SCD treatment (n = 13) and avoidance of complications (n = 12) would be useful.
Discussion
We evaluated access and barriers to care, quality of care, care coordination, and provider communication from the perspectives of adolescents and adults with SCD, their care providers, and community stakeholders, within the Solberg conceptual model for quality improvement. We found that barriers within the care process content domain (context and systems) were most salient for this population of adolescents and adults with SCD, with lack of provider knowledge and poor attitudes toward adolescents and adults with SCD, particularly in the ED, cited consistently by participant groups. Stigmatization and lack of provider compassion that affected the quality of care were particularly problematic. These findings are consistent with previous reports.42,43 Adult health care (particularly ED) provider biases and negative attitudes have been recognized as major barriers to optimal pain management in SCD.8,11,44,45 Interestingly, ED providers in our needs assessment indicated that they felt they had the training and resources to manage patients with SCD. However, only a few actually reported knowing about the NHLBI recommendations for the treatment of vaso-occlusive pain.
Within the care process content domain, we also found that SCD-related complications and associated emotions (fear, worry, anxiety), compounded by lack of access to knowledgeable and compassionate providers, pose a significant burden. Negative encounters with the health care system contributed to a striking 84% of patient participants choosing to manage severe pain at home, with pain seriously interfering with their ability to function on a daily basis. ED providers agreed that provider attitudes and implicit bias pose important barriers to care for adolescents and adults with SCD. Adolescents and adults with SCD wanted, and understood the need, to enhance self-management skills. Both they and their providers agreed that barriers to hydroxyurea uptake included worries about potential side effects, challenges with adherence to repeated laboratory testing, and support with remembering to take the medicine. However, providers uniformly expressed that access to behavioral and mental health services were, if not nonexistent, impossible to access.
Participants with SCD and their providers reported infrastructural challenges (change process capability), as manifested in limitations with accessing acute and preventive care due to transportation- and insurance- related issues. There were health system barriers that were particularly encountered during the transition from pediatric to adult care. These findings are consistent with previous reports that have found fewer interdisciplinary services available in the adult care settings compared with pediatrics.46,47 Furthermore, adult care providers were less willing to accept adults with SCD because of the complexity of their management, for which the providers did not have the necessary expertise.3,48-50 In addition, both adolescents and adults with SCD and primary care providers highlighted the inadequacies of the current system in addressing the chronic pain needs of this population. Linking back to the Solberg conceptual framework, our needs assessment results confirm the important role of establishing SCD care as a priority within a health care system—this requires leadership and vision. The vision and priorities must be implemented by effective health care teams. Multilevel approaches or interventions, when implemented, will lead to the desired outcomes.
Findings from our needs assessment within our 5-county region mirror needs assessment results from the broader consortium.51 The SCDIC has prioritized developing an intervention that addresses the challenges identified within the care process domain by directly enhancing provider access to patient individualized care plans in the electronic health record in the ED. Importantly, ED providers will be asked to view a short video that directly challenges bias and stigma in the ED. Previous studies have indeed found that attitudes can be improved by providers viewing short video segments of adults with SCD discussing their experiences.36,52 This ED protocol will be one of the interventions that we will roll out in Northern California, given the significance of negative ED encounters reported by needs assessment participants. An additional feature of the intervention is a script for adults with SCD that guides them through introducing their individualized pain plan to their ED providers, thereby enhancing their self-efficacy in a situation that has been so overwhelmingly challenging.
We will implement a second SCDIC intervention that utilizes a mobile app to support self-management on the part of the patient, by supporting motivation and adherence with hydroxyurea.53 A companion app supports hydroxyurea guideline adherence on the part of the provider, in keeping with one of our findings that providers are in need of decision-support tools. Elements of the intervention also align with our findings related to the importance of a support system in managing SCD, in that participants will identify a supportive partner who will play a specific role in supporting their adherence with hydroxyurea.
On our local level, we have, by necessity, partnered with leaders and community stakeholders throughout the region to ensure that these interventions to improve SCD care are prioritized. Grant funds provide initial resources for the SCDIC interventions, but our partnering health care administrators and medical directors must ensure that participating ED and hematology providers are free from competing priorities in order to implement the changes. We have partnered with a SCD community-based organization that is designing additional educational presentations for local emergency medicine providers, with the goal to bring to life very personal stories of bias and stigma within the EDs that directly contribute to decisions to avoid ED care despite severe symptoms.
Although we attempted to obtain samples of adolescents and adults with SCD and their providers that were representative across the 5-county region, the larger proportion of respondents were from 1 county. We did not assess concerns of age- and race-matched adults in our catchment area, so we cannot definitively say that our findings are unique to SCD. However, our results are consistent with findings from the national sample of adults with SCD who participated in the ASCQ-Me field test, and with results from the SCDIC needs assessment.33,51 Interviews and surveys are subject to self-report bias and, therefore, may or may not reflect the actual behaviors or thoughts of participants. Confidence is increased in our results given the triangulation of expressed concerns across participant groups and across data collection strategies. The majority of adolescents and adults with SCD (95%) completed both the interview and survey, while 64% of ED providers interviewed completed the survey, compared with 54% of SCD specialists and primary care providers. These response rates are more than acceptable within the realm of survey response rates.54,55
Although we encourage examining issues with care delivery within the conceptual framework for quality improvement presented, we recognize that grant funding allowed us to conduct an in-depth needs assessment that might not be feasible in other settings. Still, we would like readers to understand the importance of gathering data for improvement in a systematic manner across a range of participant groups, to ultimately inform the development of interventions and provide for evaluation of outcomes as a result of the interventions. This is particularly important for a disease, such as SCD, that is both medically and sociopolitically complex.
Conclusion
Our needs assessment brought into focus the multiple factors contributing to the disparities in health care experienced by adolescents and adults with SCD on our local level, and within the context of inequities in health resources and outcomes on the national level. We propose solutions that include specific interventions developed by a consortium of SCD and implementation science experts. We utilize a quality improvement framework to ensure that the elements of the interventions also address the barriers identified by our local providers and patients that are unique to our community. The pervasive challenges in SCD care, coupled with its medical complexities, may seem insurmountable, but our survey and qualitative results provide us with a road map for the way forward.
Acknowledgments: The authors thank the adolescents and adults with sickle cell disease, the providers, and the community stakeholders who completed the interviews and surveys. The authors also acknowledge the SCCCI co-investigators for their contributions to this project, including Michael Bell, MD, Ward Hagar, MD, Christine Hoehner, FNP, Kimberly Major, MSW, Anne Marsh, MD, Lynne Neumayr, MD, and Ted Wun, MD. We also thank Kamilah Bailey, Jameelah Hodge, Jennifer Kim, Michael Rowland, Adria Stauber, Amber Fearon, and Shanda Robertson, and the Sickle Cell Data Collection Program for their contributions.
Corresponding author: Marsha J. Treadwell, PhD, University of California San Francisco Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609; marsha.treadwell@ucsf.edu.
Financial disclosures: None.
Funding/support: This work was supported by grant # 1U01HL134007 from the National Heart, Lung, and Blood Institute to the University of California San Francisco Benioff Children’s Hospital Oakland.
From the University of California San Francisco (Dr. Treadwell, Dr. Hessler, Yumei Chen, Swapandeep Mushiana, Dr. Potter, and Dr. Vichinsky), the University of California Los Angeles (Dr. Jacob), and the University of California Berkeley (Alex Chen).
Abstract
- Objective: Adolescents and adults with sickle cell disease (SCD) face pervasive disparities in health resources and outcomes. We explored barriers to and facilitators of care to identify opportunities to support implementation of evidence-based interventions aimed at improving care quality for patients with SCD.
- Methods: We engaged a representative sample of adolescents and adults with SCD (n = 58), health care providers (n = 51), and community stakeholders (health care administrators and community-based organization leads (n = 5) in Northern California in a community-based needs assessment. We conducted group interviews separately with participant groups to obtain in-depth perspectives. Adolescents and adults with SCD completed validated measures of pain interference, quality of care, self-efficacy, and barriers to care. Providers and community stakeholders completed surveys about barriers to SCD care.
- Results: We triangulated qualitative and quantitative data and found that participants with SCD (mean age, 31 ± 8.6 years), providers, and community stakeholders emphasized the social and emotional burden of SCD as barriers. Concrete barriers agreed upon included insurance and lack of resources for addressing pain impact. Adolescents and adults with SCD identified provider issues (lack of knowledge, implicit bias), transportation, and limited social support as barriers. Negative encounters with the health care system contributed to 84% of adolescents and adults with SCD reporting they chose to manage severe pain at home. Providers focused on structural barriers: lack of access to care guidelines, comfort level with and knowledge of SCD management, and poor care coordination.
- Conclusion: Strategies for improving access to compassionate, evidence-based quality care, as well as strategies for minimizing the burden of having SCD, are warranted for this medically complex population.
Keywords: barriers to care; quality of care; care access; care coordination.
Sickle cell disease (SCD), an inherited chronic medical condition, affects about 100,000 individuals in the United States, a population that is predominantly African American.1 These individuals experience multiple serious and life-threatening complications, most frequently recurrent vaso-occlusive pain episodes,2 and they require interactions with multidisciplinary specialists from childhood. Because of advances in treatments, the majority are reaching adulthood; however, there is a dearth of adult health care providers with the training and expertise to manage their complex medical needs.3 Other concrete barriers to adequate SCD care include insurance and distance to comprehensive SCD centers.4,5
Social, behavioral, and emotional factors may also contribute to challenges with SCD management. SCD may limit daily functional abilities and lead to diminished overall quality of life.6,7 Some adolescents and adults may require high doses of opioids, which contributes to health care providers’ perceptions that there is a high prevalence of drug addiction in the population.8,9 These providers express negative attitudes towards adults with SCD, and, consequently, delay medication administration when it is acutely needed and provide otherwise suboptimal treatment.8,10,11 Adult care providers may also be uncomfortable with prescribing and managing disease-modifying therapies (blood transfusion, hydroxyurea) that have established efficacy.12-17
As 1 of 8 programs funded by the National Heart, Lung, and Blood Institute’s (NHLBI) Sickle Cell Disease Implementation Consortium (SCDIC), we are using implementation science to reduce barriers to care and improve quality of care and health care outcomes in SCD.18,19 Given that adolescents and adults with SCD experience high mortality, severe pain, and progressive decline in their ability to function day to day, and also face lack of access to knowledgeable, compassionate providers in primary and emergency settings, the SCDIC focuses on individuals aged 15 to 45 years.6,8,9,11,12
Our regional SCDIC program, the Sickle Cell Care Coordination Initiative (SCCCI), brings together researchers, clinicians, adolescents, and adults with SCD and their families, dedicated community members, policy makers, and administrators to identify and address barriers to health care within 5 counties in Northern California. One of our first steps was to conduct a community-based needs assessment, designed to inform implementation of evidence-based interventions, accounting for unique contextual factors in our region.
Conceptual Framework for Improving Medical Practice
Our needs assessment is guided by Solberg’s Conceptual Framework for Improving Medical Practice (Figure 1).20 Consistent with the overarching principles of the SCDIC, this conceptual framework focuses on the inadequate implementation of evidence-based guidelines, and on the need to first understand multifactorial facilitators and barriers to guideline implementation in order to effect change. The framework identifies 3 main elements that must be present to ensure improvements in quality-of-care processes and patient outcomes: priority, change process capability, and care process content. Priority refers to ample resource allocation for the specific change, as well as freedom from competing priorities for those implementing the change. Change process capability includes strong, effective leadership, adequate infrastructure for managing change (including resources and time), change management skills at all levels, and an established clinical information system. Care process content refers to context and systems-level changes, such as delivery system redesign as needed, support for self-management to lessen the impact of the disease, and decision support.21-23
The purpose of our community-based needs assessment was to evaluate barriers to care and quality of care in SCD, within Solberg’s conceptual model for improving medical practice. The specific aims were to evaluate access and barriers to care (eg, lack of provider expertise and training, health care system barriers such as poor care coordination and provider communication); evaluate quality of care; and assess patient needs related to pain, pain interference, self-efficacy, and self-management for adolescents and adults with SCD. We gathered the perspectives of a representative community of adolescents and adults with SCD, their providers, and community stakeholders in order to examine barriers, quality of life and care, and patient experiences in our region.
Methods
Design
In this cross-sectional study, adolescents and adults with SCD, their providers, and community stakeholders participated in group or individual qualitative interviews and completed surveys between October 2017 and March 2018.
Setting and Sample
Recruitment flyers were posted on a regional SCD-focused website, and clinical providers or a study coordinator introduced information about the needs assessment to potential participants with SCD during clinic visits at the participating centers. Participants with SCD were eligible if they had any diagnosis of SCD, were aged 15 to 48 years, and received health services within 5 Northern California counties (Alameda, Contra Costa, Sacramento, San Francisco, and Solano). They were excluded if they did not have a SCD diagnosis or had not received health services within the catchment area. As the project proceeded, participants were asked to refer other adolescents and adults with SCD for the interviews and surveys (snowball sampling). Our goal was to recruit 50 adolescents and adults with SCD into the study, aiming for 10 representatives from each county.
Providers and community stakeholders were recruited via emails, letters and informational flyers. We engaged our partner, the Sickle Cell Data Collection Program,2 to generate a list of providers and institutions that had seen patients with SCD in primary, emergency, or inpatient settings in the region. We contacted these institutions to describe the SCCCI and invite participation in the needs assessment. We also invited community-based organization leads and health care administrators who worked with SCD to participate. Providers accessed confidential surveys via a secure link on the study website or completed paper versions. Common data collected across providers included demographics and descriptions of practice settings.
Participants were eligible to be part of the study if they were health care providers (physicians and nurses) representing hematology, primary care, family medicine, internal medicine, or emergency medicine; ancillary staff (social work, psychology, child life); or leaders or administrators of clinical or sickle cell community-based organizations in Northern California (recruitment goal of n = 50). Providers were excluded if they practiced in specialties other than those noted or did not practice within the region.
Data Collection Procedures
After providing assent/consent, participating adolescents and adults with SCD took part in individual and group interviews and completed survey questionnaires. All procedures were conducted in a private space in the sickle cell center or community. Adolescents and adults with SCD completed the survey questionnaire on a tablet, with responses recorded directly in a REDCap (Research Electronic Data Capture) database,24 or on a paper version. Interviews lasted 60 (individual) to 90 (group) minutes, while survey completion time was 20 to 25 minutes. Each participant received a gift card upon completion as an expression of appreciation. All procedures were approved by the institutional review boards of the participating health care facilities.
Group and Individual Interviews
Participants with SCD and providers were invited to participate in a semi-structured qualitative interview prior to being presented with the surveys. Adolescents and adults with SCD were interviewed about barriers to care, quality of care, and pain-related experiences. Providers were asked about barriers to care and treatments. Interview guides were modified for community-based organization leaders and health care administrators who did not provide clinical services. Interview guides can be found in the Appendix. Interviews were conducted by research coordinators trained in qualitative research methods by the first author (MT). As appropriate with semi-structured interviews, the interviewers could word questions spontaneously, change the order of questions for ease of flow of conversation, and inform simultaneous coding of interviews with new themes as those might arise, as long as they touched on all topics within the interview guide.25 The interview guides were written, per qualitative research standards, based on the aims and purpose of the research,26 and were informed by existing literature on access and barriers to care in SCD, quality of care, and the needs of individuals with SCD, including in relation to impact of the disease, self-efficacy, and self-management.
Interviewees participated in either individual or group interviews, but not both. The decision for which type of interview an individual participated in was based on 2 factors: if there were not comparable participants for group interviews (eg, health care administrator and community-based organization lead), these interviews were done individually; and given that we were drawing participants from a 5-county area in Northern California, scheduling was challenging for individuals with SCD with regard to aligning schedules and traveling to a central location where the group interviews were conducted. Provider group interviews were easier to arrange because we could schedule them at the same time as regularly scheduled meetings at the participants’ health care institutions.
Interview Data Gathering and Analysis
Digital recordings of the interviews were cleaned of any participant identifying data and sent for transcription to an outside service. Transcripts were reviewed for completeness and imported into NVivo (www.qsrinternational.com), a qualitative data management program.
A thematic content analysis and deductive and inductive approaches were used to analyze the verbatim transcripts generated from the interviews. The research team was trained in the use of NVivo software to facilitate the coding process. A deductive coding scheme was initially used based on existing concepts in the literature regarding challenges to optimal SCD care, with new codes added as the thematic content analyses progressed. The initial coding, pattern coding, and use of displays to examine the relationships between different categories were conducted simultaneously.27,28 Using the constant comparative method, new concepts from participants with SCD and providers could be incorporated into subsequent interviews with other participants. For this study, the only additional concepts added were in relation to participant recruitment and retention in the SCDIC Registry. Research team members coded transcripts separately and came together weekly, constantly comparing codes and developing the consensus coding scheme. Where differences between coders existed, code meanings were discussed and clarified until consensus was reached.29
Quantitative data were analyzed using SPSS (v. 25, Chicago, IL). Descriptive statistics (means, standard deviations, frequencies, percentages) were used to summarize demographics (eg, age, gender, and race), economic status, and type of SCD. No systematic differences were detected from cases with missing values. Scale reliabilities (ie, Cronbach α) were evaluated for self-report measures.
Measurement
Adolescents and adults with SCD completed items from the PhenX Toolkit (consensus measures for Phenotypes and eXposures), assessing sociodemographics (age, sex, race, ethnicity, educational attainment, occupation, marital status, annual income, insurance), and clinical characteristics (sickle cell diagnosis and emergency department [ED] and hospital utilization for pain).30
Pain Interference Short Form (Patient-Reported Outcomes Measurement Information System [PROMIS]). The Pain Interference Form consists of 8 items that assess the degree to which pain interfered with day-to-day activities in the previous 7 days at home, including impacts on social, cognitive, emotional, and physical functioning; household chores and recreational activities; sleep; and enjoyment in life. Reliability and validity of the PROMIS Pain Interference Scale has been demonstrated, with strong negative correlations with Physical Function Scales (r = 0.717, P < 0.01), indicating that higher scores are associated with lower function (β = 0.707, P < 0.001).31 The Cronbach α estimate for the other items on the pain interference scale was 0.99. Validity analysis indicated strong correlations with pain-related domains: BPI Interference Subscale (rho = 0.90), SF-36 Bodily Pain Subscale (rho = –0.84), and 0–10 Numerical Rating of Pain Intensity (rho = 0.48).32
Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) Quality of Care (QOC). ASCQ-Me QOC consists of 27 items that measure the quality of care that adults with SCD have received from health care providers.33 There are 3 composites: provider communication (quality of patient and provider communication), ED care (quality of care in the ED), and access (to routine and emergency care). Internal consistency reliability for all 3 composites is greater than 0.70. Strong correlations of the provider communication composite with overall ratings of routine care (r = 0.65) and overall provider ratings (r = 0.83) provided evidence of construct validity. Similarly, the ED care composite was strongly correlated with overall ratings of QOC in the ED, and the access composite was highly correlated with overall evaluations of ED care (r = 0.70). Access, provider interaction, and ED care composites were reliable (Cronbach α, 0.70–0.83) and correlated with ratings of global care (r = 0.32–0.83), further indicating construct validity.33
Sickle Cell Self-Efficacy Scale (SCSES). The SCSES is a 9-item, self-administered questionnaire measuring perceptions of the ability to manage day-to-day issues resulting from SCD. SCSES items are scored on a 5-point scale ranging from Not sure at all (1) to Very sure (5). Individual item responses are summed to give an overall score, with higher scores indicating greater self-efficacy. The SCSES has acceptable reliability (r = 0.45, P < 0.001) and validity (α = 0.89).34,35
Sickle Cell Disease Barriers Checklist. This checklist consists of 53 items organized into 8 categories: insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers (fear, anger), and disease-related barriers. Participants check applicable barriers, with a total score range of 0 to 53 and higher scores indicating more barriers to care. The SCD Barriers Checklist has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).5
ED Provider Checklist. The ED provider survey is a checklist of 14 statements pertaining to issues regarding patient care, with which the provider rates level of agreement. Items representing the attitudes and beliefs of providers towards patients with SCD are rated on a Likert-type scale, with level of agreement indicated as 1 (strongly disagree) to 6 (strongly agree). The positive attitudes subscale consists of 4 items (Cronbach α= 0.85), and the negative attitudes subscale consists of 6 items (Cronbach α = 0.89). The Red-Flag Behaviors subscale includes 4 items that indicate behavior concerns about drug-seeking, such as requesting specific narcotics and changing behavior when the provider walks in.8,36,37
Sickle cell and primary care providers also completed a survey consisting of sets of items compiled from existing provider surveys; this survey consisted of a list of 16 barriers to using opioids, which the providers rated on a 5-point Likert-type scale (1, not a barrier; 5, complete barrier).13,16,38 Providers indicated their level of experience with caring for patients with SCD; care provided, such as routine health screenings; and comfort level with providing preventive care, managing comorbidities, and managing acute and chronic pain. Providers were asked what potential facilitators might improve care for patients with SCD, including higher reimbursement, case management services, access to pain management specialists, and access to clinical decision-support tools. Providers responded to specific questions about management with hydroxyurea (eg, criteria for, barriers to, and comfort level with prescribing).39 The surveys are included in the Appendix.
Triangulation
Data from the interviews and surveys were triangulated to enhance understanding of results generated from the different data sources.40 Convergence of findings, different facets of the same phenomenon, or new perspectives were examined.
Results
Qualitative Data
Adolescents and adults with SCD (n = 55) and health care providers and community stakeholders (n = 56) participated in group or individual interviews to help us gain an in-depth understanding of the needs and barriers related to SCD care in our 5-county region. Participants with SCD described their experiences, which included stigma, racism, labeling, and, consequently, stress. They also identified barriers such as lack of transportation, challenges with insurance, and lack of access to providers who were competent with pain management. They reported that having SCD in a health care system that was unable to meet their needs was burdensome.
Barriers to Care and Treatments. Adolescents and adults indicated that SCD and its sequelae posed significant barriers to health care. Feelings of tiredness and pain make it more difficult for them to seek care. The emotional burden of SCD (fear and anger) was a frequently cited barrier, which was fueled by previous negative encounters with the health care system. All adolescents and adults with SCD reported that they knew of stigma in relation to seeking pain management that was pervasive and long-standing, and the majority reported they had directly experienced stigma. They reported that being labeled as “drug-seekers” was typical when in the ED for pain management. Participants articulated unconscious bias or overt racism among providers: “people with sickle cell are Black ... and Black pain is never as valuable as White pain” (25-year-old male). Respondents with SCD described challenges to the credibility of their pain reports in the ED. They reported that ED providers expressed doubts regarding the existence and/or severity of their pain, consequently creating a feeling of disrespect for patients seeking pain relief. The issue of stigma was mentioned by only 2 of 56 providers during their interviews.
Lack of Access to Knowledgeable, Compassionate Providers. Lack of access to knowledgeable care providers was another prevalent theme expressed by adolescents and adults with SCD. Frustration occurred when providers did not have knowledge of SCD and its management, particularly pain assessment. Adolescents and adults with SCD noted the lack of compassion among providers: “I’ve been kicked out of the hospital because they felt like okay, well we gave you enough medication, you should be all right” (29-year-old female). Providers specifically mentioned lack of compassion and knowledge as barriers to SCD care much less often during their interviews compared with the adolescents and adults with SCD.
Health Care System Barriers. Patient participants often expressed concerns about concrete and structural aspects of care. Getting to their appointments was a challenge for half of the interviewees, as they either did not have access to a vehicle or could not afford to travel the needed distance to obtain quality care. Even when hospitals were accessible by public transportation, those with excruciating pain understandably preferred a more comfortable and private way to travel: “I would like to change that, something that will be much easier, convenient for sickle cell patients that do suffer with pain, that they don’t have to travel always to see the doctor” (30-year-old male).
Insurance and other financial barriers also played an important role in influencing decisions to seek health care services. Medical expenses were not covered, or co-pays were too high. The Medicaid managed care system could prevent access to knowledgeable providers who were not within network. Such a lack of access discouraged some adolescents and adults with SCD from seeking acute and preventive care.
Transition From Pediatric to Adult Care. Interviewees with SCD expressed distress about the gap between pediatric and adult care. They described how they had a long-standing relationship with their medical providers, who were familiar with their medical background and history from childhood. Adolescent interviewees reported an understanding of their own pain management as well as adherence to and satisfaction with their individualized pain plans. However, adults noted that satisfaction plummeted with increasing age due to the limited number of experienced adult SCD providers, which was compounded by negative experiences (stigma, racism, drug-seeking label).
One interviewee emphasized the difficulty of finding knowledgeable providers after transition: “When you’re a pediatric sickle cell [patient], you have the doctors there every step of the way, but not with adult sickle cell… I know when I first transitioned I never felt more alone in my life… you look at that ER doctor kind of with the same mindset as you would your hematologist who just hand walked you through everything. And adult care providers were a lot more blunt and cold and they’re like… ‘I don’t know; I’m not really educated in sickle cell.’” A sickle cell provider shared his insight about the problem of transitioning: “I think it’s particularly challenging because we, as a community, don’t really set them up for success. It’s different from other chronic conditions [in that] it’s much harder to find an adult sickle cell provider. There’s not a lot of adult hematologists that will take care of our adult patients, and so I know statistically, there’s like a drop-down in the overall outcomes of our kids after they age out of our pediatric program.”
Self-Management, Supporting Hydroxyurea Use. Interview participants with SCD reported using a variety of methods to manage pain at home and chose to go to the ED only when the pain became intolerable. Patients and providers expressed awareness of different resources for managing pain at home, yet they also indicated that these resources have not been consolidated in an accessible way for patients and families. Some resources cited included heat therapy, acupuncture, meditation, medical marijuana, virtual reality devices, and pain medications other than opioids.
Patients and providers expressed the need for increasing awareness and education about hydroxyurea. Many interview participants with SCD were concerned about side effects, multiple visits with a provider during dose titration, and ongoing laboratory monitoring. They also expressed difficulties with scheduling multiple appointments, depending on access to transportation and limited provider clinic hours. They were aware of strategies for improving adherence with hydroxyurea, including setting phone alarms, educating family members about hydroxyurea, and eliciting family support, but expressed needing help to consistently implement these strategies.
Safe Opioid Prescribing. Adult care providers expressed concerns about safe opioid prescribing for patients with SCD. They were reluctant to prescribe opioid doses needed to adequately control SCD pain. Providers expressed uncertainty and fear or concern about medical/legal liability or about their judgment about what’s safe and not safe for patients with chronic use/very high doses of opioids. “I know we’re in like this opiate epidemic here in this country but I feel like these patients don’t really fit under that umbrella that the problem is coming from so [I am] just trying to learn more about how to take care of them.”
Care Coordination and Provider Communication. Adolescents and adults with SCD reported having positive experiences—good communication, established trust, and compassionate care—with their usual providers. However, they perceived that ED physicians and nurses did not really care about them. Both interviewees with SCD and providers recognized the importance of good communication in all settings as the key to overcoming barriers to receiving quality care. All agreed on the importance of using individual pain plans so that all providers, especially ED providers, can be more at ease with treating adolescents and adults with SCD.
Quantitative Data: Adolescents and Adults With SCD
Fifty-eight adolescents and adults with SCD (aged 15 to 48 years) completed the survey. Three additional individuals who did not complete the interview completed the survey. Reasons for not completing the interview included scheduling challenges (n = 2) or a sickle cell pain episode (n = 1). The average age of participants was 31 years ± 8.6, more than half (57%) were female, and the majority (93%) were African American (Table 1). Most (71%) had never been married. Half (50%) had some college or an associate degree, and 40% were employed and reported an annual household income of less than $30,000. Insurance coverage was predominantly Medi-Cal (Medicaid, 69%). The majority of participants resided in Alameda (34.5%) or Contra Costa (21%) counties. The majority of sickle cell care was received in Alameda County, whether outpatient (52%), inpatient (40%), or ED care (41%). The majority (71%) had a diagnosis of SCD hemoglobin SS.
Pain. More than one-third of individuals with SCD reported 1 or 2 ED visits for pain in the previous 6 months (34%), and more than 3 hospitalizations (36%) related to pain in the previous year (Table 2). The majority (85%) reported having severe pain at home in the previous 6 months that they did not seek health care for, consistent with their reports in the qualitative interviews. More than half (59%) reported 4 or more of these severe pain episodes that led to inability to perform daily activities for 1 week or more. While pain interference on the PROMIS Pain Interference Short Form on average (T-score, 59.6 ± 8.6) was similar to that of the general population (T-score, 50 ± 10), a higher proportion of patients with SCD reported pain interference compared with the general population. The mean self-efficacy (confidence in ability to manage complications of SCD) score on the SCSES of 30.0 ± 7.3 (range, 9–45) was similar to that of other adults with SCD (mean, 32.2 ± 7.0). Twenty-five percent of the present sample had a low self-efficacy score (< 25).
Barriers to Care and Treatments. Consistent with the qualitative data, SCD-related symptoms such as tiredness (64%) and pain (62%) were reported most often as barriers to care (Table 3). Emotions (> 25%) such as worry/fear, frustration/anger, and lack of confidence were other important barriers to care. Provider knowledge and attitudes were cited next most often, with 38% of the sample indicating “Providers accuse me of drug-seeking” and “It is hard for me to find a provider who has enough experiences with or knowledge about SCD.” Participants expressed that they were not believed when in pain and “I am treated differently from other patients.” Almost half of respondents cited “I am not seen quickly enough when I am in pain” as a barrier to their care.
Consistent with the qualitative data, transportation barriers (not having a vehicle, costs of transportation, public transit not easy to get to) were cited by 55% of participants. About half of participants reported that insurance was an important barrier, with high co-pays and medications and other services not covered. In addition, gathering approvals was a long and fragmented process, particularly for consultations among providers (hematology, primary care provider, pain specialist). Furthermore, insurance provided limited choices about location for services.
Participants reported social support system burnout (22%), help needed with daily activities (21%), and social isolation or generally not having enough support (33%) as ongoing barriers. Difficulties were encountered with self-management (eg, taking medications on time or making follow-up appointments, 19%), with 22% of participants finding the health care system confusing or hard to understand. Thirty percent reported “Places for me to go to learn how to stay well are not close by or easy to get to.” ”Worry about side effects” (33%) was a common barrier to hydroxyurea use. Participants described “forgetting to take the medicine,” “tried before but it did not work,” “heard scary things” about hydroxyurea, and “not interested in taking another medicine” as barriers.
Quality of Care. More than half (51%) of the 53 participants who had accessed health care in the previous year rated their overall health care as poor on the ASCQ-Me QOC measure. This was significantly higher compared to the reports from more than 47,000 adults with Medicaid in 2017 (16%),41 and to the 2008-2009 report from 556 adults with SCD from across the United States (37%, Figure 2).33 The major contributor to these poor ratings for participants in our sample was low satisfaction with ED care.
Sixty percent of the 42 participants who had accessed ED care in the past year indicated “never” or “sometimes” to the question “When you went to the ED for care, how often did you get it as soon as you wanted?” compared with only 16% of the 2017 adult Medicaid population responding (n = 25,789) (Figure 3). Forty-seven percent of those with an ED visit indicated that, in the previous 12 months, they had been made to wait “more than 2 hours before receiving treatment for acute pain in the ED.” However, in the previous 12 months, 39% reported that their wait time in the ED had been only “between five minutes and one hour.”
On the ASCQ-Me QOC Access to Care composite measure, 33% of 42 participants responding reported they were seen at a routine appointment as soon as they would have liked. This is significantly lower compared to 56% of the adult Medicaid population responding to the same question. Reports of provider communication (Provider Communication composite) for adolescents and adults with SCD were comparable to reports of adults with SCD from the ASCQ-Me field test,33 but adults with Medicaid reported higher ratings of quality communication behaviors (Figure 4).33,41 Nearly 60% of both groups with SCD reported that providers “always” performed quality communication behaviors—listened carefully, spent enough time, treated them with respect, and explained things well—compared with more than 70% of adults with Medicaid.
Participants from all counties reported the same number of barriers to care on average (3.3 ± 2.1). Adolescents and adults who reported more barriers to care also reported lower satisfaction with care (r = –0.47, P < 0.01) and less confidence in their ability to manage their SCD (self-efficacy, r = – 0.36, P < 0.05). Female participants reported more barriers to care on average compared with male participants (2.6 ± 2.4 vs 1.4 ± 2.0, P = 0.05). Participants with higher self-efficacy reported lower pain ratings (r = –0.47, P < 0.001).
Quantitative Data: Health Care Providers
Providers (n = 56) and community stakeholders (2 leaders of community-based organizations and 3 health care administrators) were interviewed, with 29 also completing the survey. The reason for not completing (n = 22) was not having the time once the interview was complete. A link to the survey was sent to any provider not completing at the time of the interview, with 2 follow-up reminders. The majority of providers were between the ages of 31 and 50 years (46.4%), female (71.4%), and white (66.1%) (Table 4). None were of Hispanic, Latinx, or Spanish origin. Thirty-six were physicians (64.3%), and 16 were allied health professionals (28.6%). Of the 56 providers, 32 indicated they had expertise caring for patients with SCD (57.1%), 14 were ED providers (25%), and 5 were primary care providers. Most of the providers practiced in an urban setting (91.1%).
Barriers to Care: ED Provider Perspectives. Nine of 14 ED providers interviewed completed the survey on their perspectives regarding barriers to care in the ED, difficulty with follow-ups, ED training resources, and pain control for patients with SCD. ED providers (n = 8) indicated that “provider attitudes” were a barrier to care delivery in the ED for patients with SCD. Some providers (n = 7) indicated that “implicit bias,” “opioid epidemic,” “concern about addiction,” and “patient behavior” were barriers. Respondents indicated that “overcrowding” (n = 6) and “lack of care pathway/protocol” (n = 5) were barriers. When asked to express their level of agreement with statements about SCD care in the ED, respondents disagreed/strongly disagreed (n = 5) that they were “able to make a follow-up appointment” with a sickle cell specialist or primary care provider upon discharge from the ED, and others disagreed/strongly disagreed (n = 4) that they were able to make a “referral to a case management program.”
ED training and resources. Providers agreed/strongly agreed (n = 8) that they had the knowledge and training to care for patients with SCD, that they had access to needed medications, and that they had access to knowledgeable nursing staff with expertise in SCD care. All 9 ED providers indicated that they had sufficient physician/provider staffing to provide good pain management to persons with SCD in the ED.
Pain control in the ED. Seven ED providers indicated that their ED used individualized dosing protocols to treat sickle cell pain, and 5 respondents indicated their ED had a protocol for treating sickle cell pain. Surprisingly, only 3 indicated that they were aware of the NHLBI recommendations for the treatment of vaso-occlusive pain.
Barriers to Care: Primary Care Provider Perspectives. Twenty providers completed the SCD provider section of the survey, including 17 multidisciplinary SCD providers from 4 sickle cell special care centers and 3 community primary care providers. Of the 20, 12 were primary care providers for patients with SCD (Table 4).
Patient needs. Six primary care providers indicated that the medical needs of patients with SCD were being met, but none indicated that the behavioral health or mental health needs were being met.
Managing SCD comorbidities. Five primary care providers indicated they were very comfortable providing preventive ambulatory care to patients with SCD. Six indicated they were very comfortable managing acute pain episodes, but none were very comfortable managing comorbidities such as pulmonary hypertension, diabetes, or chronic pain.
Barriers to opioid use. Only 3 of 12 providers reviewing a list of 15 potential barriers to the use of opioids for SCD pain management indicated a perceived lack of efficacy of opioids, development of tolerance and dependence, and concerns about community perceptions as barriers. Two providers selected potential for diversion as a moderate barrier to opioid use.
Barriers to hydroxyurea use. Eight of 12 providers indicated that the common reasons that patients/families refuse hydroxyurea were “worry about side effects”; 7 chose “don’t want to take another medicine,” and 6 chose “worry about carcinogenic potential.” Others (n = 10) indicated that “patient/family adherence with hydroxyurea” and “patient/family adherence with required blood tests” were important barriers to hydroxyurea use. Eight of the 12 providers indicated that they were comfortable with managing hydroxyurea in patients with SCD.
Care redesign. Twenty SCD and primary care providers completed the Care Redesign section of the survey. Respondents (n = 11) indicated that they would see more patients with SCD if they had accessible case management services available without charge or if patient access to transportation to clinic was also available. Ten indicated that they would see more patients with SCD if they had an accessible community health worker (who understands patient’s/family’s social situation) and access to a pain management specialist on call to answer questions and who would manage chronic pain. All (n = 20) were willing to see more patients with SCD in their practices. Most reported that a clinical decision-support tool for SCD treatment (n = 13) and avoidance of complications (n = 12) would be useful.
Discussion
We evaluated access and barriers to care, quality of care, care coordination, and provider communication from the perspectives of adolescents and adults with SCD, their care providers, and community stakeholders, within the Solberg conceptual model for quality improvement. We found that barriers within the care process content domain (context and systems) were most salient for this population of adolescents and adults with SCD, with lack of provider knowledge and poor attitudes toward adolescents and adults with SCD, particularly in the ED, cited consistently by participant groups. Stigmatization and lack of provider compassion that affected the quality of care were particularly problematic. These findings are consistent with previous reports.42,43 Adult health care (particularly ED) provider biases and negative attitudes have been recognized as major barriers to optimal pain management in SCD.8,11,44,45 Interestingly, ED providers in our needs assessment indicated that they felt they had the training and resources to manage patients with SCD. However, only a few actually reported knowing about the NHLBI recommendations for the treatment of vaso-occlusive pain.
Within the care process content domain, we also found that SCD-related complications and associated emotions (fear, worry, anxiety), compounded by lack of access to knowledgeable and compassionate providers, pose a significant burden. Negative encounters with the health care system contributed to a striking 84% of patient participants choosing to manage severe pain at home, with pain seriously interfering with their ability to function on a daily basis. ED providers agreed that provider attitudes and implicit bias pose important barriers to care for adolescents and adults with SCD. Adolescents and adults with SCD wanted, and understood the need, to enhance self-management skills. Both they and their providers agreed that barriers to hydroxyurea uptake included worries about potential side effects, challenges with adherence to repeated laboratory testing, and support with remembering to take the medicine. However, providers uniformly expressed that access to behavioral and mental health services were, if not nonexistent, impossible to access.
Participants with SCD and their providers reported infrastructural challenges (change process capability), as manifested in limitations with accessing acute and preventive care due to transportation- and insurance- related issues. There were health system barriers that were particularly encountered during the transition from pediatric to adult care. These findings are consistent with previous reports that have found fewer interdisciplinary services available in the adult care settings compared with pediatrics.46,47 Furthermore, adult care providers were less willing to accept adults with SCD because of the complexity of their management, for which the providers did not have the necessary expertise.3,48-50 In addition, both adolescents and adults with SCD and primary care providers highlighted the inadequacies of the current system in addressing the chronic pain needs of this population. Linking back to the Solberg conceptual framework, our needs assessment results confirm the important role of establishing SCD care as a priority within a health care system—this requires leadership and vision. The vision and priorities must be implemented by effective health care teams. Multilevel approaches or interventions, when implemented, will lead to the desired outcomes.
Findings from our needs assessment within our 5-county region mirror needs assessment results from the broader consortium.51 The SCDIC has prioritized developing an intervention that addresses the challenges identified within the care process domain by directly enhancing provider access to patient individualized care plans in the electronic health record in the ED. Importantly, ED providers will be asked to view a short video that directly challenges bias and stigma in the ED. Previous studies have indeed found that attitudes can be improved by providers viewing short video segments of adults with SCD discussing their experiences.36,52 This ED protocol will be one of the interventions that we will roll out in Northern California, given the significance of negative ED encounters reported by needs assessment participants. An additional feature of the intervention is a script for adults with SCD that guides them through introducing their individualized pain plan to their ED providers, thereby enhancing their self-efficacy in a situation that has been so overwhelmingly challenging.
We will implement a second SCDIC intervention that utilizes a mobile app to support self-management on the part of the patient, by supporting motivation and adherence with hydroxyurea.53 A companion app supports hydroxyurea guideline adherence on the part of the provider, in keeping with one of our findings that providers are in need of decision-support tools. Elements of the intervention also align with our findings related to the importance of a support system in managing SCD, in that participants will identify a supportive partner who will play a specific role in supporting their adherence with hydroxyurea.
On our local level, we have, by necessity, partnered with leaders and community stakeholders throughout the region to ensure that these interventions to improve SCD care are prioritized. Grant funds provide initial resources for the SCDIC interventions, but our partnering health care administrators and medical directors must ensure that participating ED and hematology providers are free from competing priorities in order to implement the changes. We have partnered with a SCD community-based organization that is designing additional educational presentations for local emergency medicine providers, with the goal to bring to life very personal stories of bias and stigma within the EDs that directly contribute to decisions to avoid ED care despite severe symptoms.
Although we attempted to obtain samples of adolescents and adults with SCD and their providers that were representative across the 5-county region, the larger proportion of respondents were from 1 county. We did not assess concerns of age- and race-matched adults in our catchment area, so we cannot definitively say that our findings are unique to SCD. However, our results are consistent with findings from the national sample of adults with SCD who participated in the ASCQ-Me field test, and with results from the SCDIC needs assessment.33,51 Interviews and surveys are subject to self-report bias and, therefore, may or may not reflect the actual behaviors or thoughts of participants. Confidence is increased in our results given the triangulation of expressed concerns across participant groups and across data collection strategies. The majority of adolescents and adults with SCD (95%) completed both the interview and survey, while 64% of ED providers interviewed completed the survey, compared with 54% of SCD specialists and primary care providers. These response rates are more than acceptable within the realm of survey response rates.54,55
Although we encourage examining issues with care delivery within the conceptual framework for quality improvement presented, we recognize that grant funding allowed us to conduct an in-depth needs assessment that might not be feasible in other settings. Still, we would like readers to understand the importance of gathering data for improvement in a systematic manner across a range of participant groups, to ultimately inform the development of interventions and provide for evaluation of outcomes as a result of the interventions. This is particularly important for a disease, such as SCD, that is both medically and sociopolitically complex.
Conclusion
Our needs assessment brought into focus the multiple factors contributing to the disparities in health care experienced by adolescents and adults with SCD on our local level, and within the context of inequities in health resources and outcomes on the national level. We propose solutions that include specific interventions developed by a consortium of SCD and implementation science experts. We utilize a quality improvement framework to ensure that the elements of the interventions also address the barriers identified by our local providers and patients that are unique to our community. The pervasive challenges in SCD care, coupled with its medical complexities, may seem insurmountable, but our survey and qualitative results provide us with a road map for the way forward.
Acknowledgments: The authors thank the adolescents and adults with sickle cell disease, the providers, and the community stakeholders who completed the interviews and surveys. The authors also acknowledge the SCCCI co-investigators for their contributions to this project, including Michael Bell, MD, Ward Hagar, MD, Christine Hoehner, FNP, Kimberly Major, MSW, Anne Marsh, MD, Lynne Neumayr, MD, and Ted Wun, MD. We also thank Kamilah Bailey, Jameelah Hodge, Jennifer Kim, Michael Rowland, Adria Stauber, Amber Fearon, and Shanda Robertson, and the Sickle Cell Data Collection Program for their contributions.
Corresponding author: Marsha J. Treadwell, PhD, University of California San Francisco Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609; marsha.treadwell@ucsf.edu.
Financial disclosures: None.
Funding/support: This work was supported by grant # 1U01HL134007 from the National Heart, Lung, and Blood Institute to the University of California San Francisco Benioff Children’s Hospital Oakland.
1. Hassell KL. Population Estimates of sickle cell disease in the U.S. Am J Prev Med. 2010; 38:S512-S521.
2. Data & Statistics on Sickle Cell Disease. Centers for Disease Control and Prevention website. www.cdc.gov/ncbddd/sicklecell/data.html. Accessed March 25, 2020.
3. Inusa BPD, Stewart CE, Mathurin-Charles S, et al. Paediatric to adult transition care for patients with sickle cell disease: a global perspective. Lancet Haematol. 2020;7:e329-e341.
4. Smith SK, Johnston J, Rutherford C, et al. Identifying social-behavioral health needs of adults with sickle cell disease in the emergency department. J Emerg Nurs. 2017;43:444-450.
5. Treadwell MJ, Barreda F, Kaur K, et al. Emotional distress, barriers to care, and health-related quality of life in sickle cell disease. J Clin Outcomes Manag. 2015;22:8-17.
6. Treadwell MJ, Hassell K, Levine R, et al. Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-Me): conceptual model based on review of the literature and formative research. Clin J Pain. 2014;30:902-914.
7. Rizio AA, Bhor M, Lin X, et al. The relationship between frequency and severity of vaso-occlusive crises and health-related quality of life and work productivity in adults with sickle cell disease. Qual Life Res. 2020;29:1533-1547.
8. Freiermuth CE, Haywood C, Silva S, et al. Attitudes toward patients with sickle cell disease in a multicenter sample of emergency department providers. Adv Emerg Nurs J. 2014;36:335-347.
9. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Natl Med Assoc. 2010;102:1050-1055.
10. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain. 2010;26:199-205.
11. Haywood C, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med. 2013;31:651-656.
12. Haywood C, Beach MC, Lanzkron S, et al. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101:1022-1033.
13. Mainous AG, Tanner RJ, Harle CA, et al. Attitudes toward management of sickle cell disease and its complications: a national survey of academic family physicians. Anemia. 2015;2015:1-6.
14. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033.
15. Lunyera J, Jonassaint C, Jonassaint J, et al. Attitudes of primary care physicians toward sickle cell disease care, guidelines, and comanaging hydroxyurea with a specialist. J Prim Care Community Health. 2017;8:37-40.
16. Whiteman LN, Haywood C, Lanzkron S, et al. Primary care providers’ comfort levels in caring for patients with sickle cell disease. South Med J. 2015;108:531-536.
17. Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood. 2014;124:3850-4004.
18. DiMartino LD, Baumann AA, Hsu LL, et al. The sickle cell disease implementation consortium: Translating evidence-based guidelines into practice for sickle cell disease. Am J Hematol. 2018;93:E391-E395.
19. King AA, Baumann AA. Sickle cell disease and implementation science: A partnership to accelerate advances. Pediatr Blood Cancer. 2017;64:e26649.
20. Solberg LI. Improving medical practice: a conceptual framework. Ann Fam Med. 2007;5:251-256.
21. Bodenheimer T, Wagner EH, Grumbach K. Improving primary care for patients with chronic illness. J Am Med Assoc. 2002;288:5.
22. Bodenheimer T. Interventions to improve chronic illness care: evaluating their effectiveness. Dis Manag. 2003;6:63-71.
23. Tsai AC, Morton SC, Mangione CM, Keeler EB. A meta-analysis of interventions to improve care for chronic illnesses. Am J Manag Care. 2005;11:478-488.
24. Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
25. Kallio H, Pietilä A-M, Johnson M, et al. Systematic methodological review: developing a framework for a qualitative semi-structured interview guide. J Adv Nurs. 2016;72:2954-2965.
26. Clarke V, Braun V. Successful Qualitative Research: A Practical Guide for Beginners. First. Thousand Oaks, CA: Sage; 2013.
27. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15:1277-1288.
28. Creswell JW, Hanson WE, Clark Plano VL, et al. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35:236-264.
29. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis A Methods Sourcebook. 4th ed. Thousand Oaks, CA: Sage; 2019.
30. Eckman JR, Hassell KL, Huggins W, et al. Standard measures for sickle cell disease research: the PhenX Toolkit sickle cell disease collections. Blood Adv. 2017; 1: 2703-2711.
31. Kendall R, Wagner B, Brodke D, et al. The relationship of PROMIS pain interference and physical function scales. Pain Med. 2018;19:1720-1724.
32. Amtmann D, Cook KF, Jensen MP, et al. Development of a PROMIS item bank to measure pain interference. Pain. 2010;150:173-182.
33. Evensen CT, Treadwell MJ, Keller S, et al. Quality of care in sickle cell disease: Cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine (Baltimore). 2016;95:e4528.
34. Edwards R, Telfair J, Cecil H, et al. Reliability and validity of a self-efficacy instrument specific to sickle cell disease. Behav Res Ther. 2000;38:951-963.
35. Edwards R, Telfair J, Cecil H, et al. Self-efficacy as a predictor of adult adjustment to sickle cell disease: one-year outcomes. Psychosom Med. 2001;63:850-858.
36. Puri Singh A, Haywood C, Beach MC, et al. Improving emergency providers’ attitudes toward sickle cell patients in pain. J Pain Symptom Manage. 2016;51:628-632.e3.
37. Glassberg JA, Tanabe P, Chow A, et al. Emergency provider analgesic practices and attitudes towards patients with sickle cell disease. Ann Emerg Med. 2013;62:293-302.e10.
38. Grahmann PH, Jackson KC 2nd, Lipman AG. Clinician beliefs about opioid use and barriers in chronic nonmalignant pain [published correction appears in J Pain Palliat Care Pharmacother. 2004;18:145-6]. J Pain Palliat Care Pharmacother. 2004;18:7-28.
39. Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010;3:255-260.
40. Fielding N. Triangulation and mixed methods designs: data integration with new research technologies. J Mixed Meth Res. 2012;6:124-136.
41. 2017 CAHPS Health Plan Survey Chartbook. Agency for Healthcare Research and Quality website. www.ahrq.gov/cahps/cahps-database/comparative-data/2017-health-plan-chartbook/results-enrollee-population.html. Accessed September 8, 2020.
42. Bulgin D, Tanabe P, Jenerette C. Stigma of sickle cell disease: a systematic review. Issues Ment Health Nurs. 2018;1-11.
43. Wakefield EO, Zempsky WT, Puhl RM, et al. Conceptualizing pain-related stigma in adolescent chronic pain: a literature review and preliminary focus group findings. PAIN Rep. 2018;3:e679.
44. Nelson SC, Hackman HW. Race matters: Perceptions of race and racism in a sickle cell center. Pediatr Blood Cancer. 2013;60:451-454.
45. Dyal BW, Abudawood K, Schoppee TM, et al. Reflections of healthcare experiences of african americans with sickle cell disease or cancer: a qualitative study. Cancer Nurs. 2019;10.1097/NCC.0000000000000750.
46. Renedo A. Not being heard: barriers to high quality unplanned hospital care during young people’s transition to adult services - evidence from ‘this sickle cell life’ research. BMC Health Serv Res. 2019;19:876.
47. Ballas S, Vichinsky E. Is the medical home for adult patients with sickle cell disease a reality or an illusion? Hemoglobin. 2015;39:130-133.
48. Hankins JS, Osarogiagbon R, Adams-Graves P, et al. A transition pilot program for adolescents with sickle cell disease. J Pediatr Health Care. 2012;26 e45-e49.
49. Smith WR, Sisler IY, Johnson S, et al. Lessons learned from building a pediatric-to-adult sickle cell transition program. South Med J. 2019;112:190-197.
50. Lanzkron S, Sawicki GS, Hassell KL, et al. Transition to adulthood and adult health care for patients with sickle cell disease or cystic fibrosis: Current practices and research priorities. J Clin Transl Sci. 2018;2:334-342.
51. Kanter J, Gibson R, Lawrence RH, et al. Perceptions of US adolescents and adults with sickle cell disease on their quality of care. JAMA Netw Open. 2020;3:e206016.
52. Haywood C, Lanzkron S, Hughes MT, et al. A video-intervention to improve clinician attitudes toward patients with sickle cell disease: the results of a randomized experiment. J Gen Intern Med. 2011;26:518-523.
53. Hankins JS, Shah N, DiMartino L, et al. Integration of mobile health into sickle cell disease care to increase hydroxyurea utilization: protocol for an efficacy and implementation study. JMIR Res Protoc. 2020;9:e16319.
54. Fan W, Yan Z. Factors affecting response rates of the web survey: A systematic review. Comput Hum Behav. 2010;26:132-139.
55. Millar MM, Dillman DA. Improving response to web and mixed-mode surveys. Public Opin Q. 2011;75:249-269.
1. Hassell KL. Population Estimates of sickle cell disease in the U.S. Am J Prev Med. 2010; 38:S512-S521.
2. Data & Statistics on Sickle Cell Disease. Centers for Disease Control and Prevention website. www.cdc.gov/ncbddd/sicklecell/data.html. Accessed March 25, 2020.
3. Inusa BPD, Stewart CE, Mathurin-Charles S, et al. Paediatric to adult transition care for patients with sickle cell disease: a global perspective. Lancet Haematol. 2020;7:e329-e341.
4. Smith SK, Johnston J, Rutherford C, et al. Identifying social-behavioral health needs of adults with sickle cell disease in the emergency department. J Emerg Nurs. 2017;43:444-450.
5. Treadwell MJ, Barreda F, Kaur K, et al. Emotional distress, barriers to care, and health-related quality of life in sickle cell disease. J Clin Outcomes Manag. 2015;22:8-17.
6. Treadwell MJ, Hassell K, Levine R, et al. Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-Me): conceptual model based on review of the literature and formative research. Clin J Pain. 2014;30:902-914.
7. Rizio AA, Bhor M, Lin X, et al. The relationship between frequency and severity of vaso-occlusive crises and health-related quality of life and work productivity in adults with sickle cell disease. Qual Life Res. 2020;29:1533-1547.
8. Freiermuth CE, Haywood C, Silva S, et al. Attitudes toward patients with sickle cell disease in a multicenter sample of emergency department providers. Adv Emerg Nurs J. 2014;36:335-347.
9. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Natl Med Assoc. 2010;102:1050-1055.
10. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain. 2010;26:199-205.
11. Haywood C, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med. 2013;31:651-656.
12. Haywood C, Beach MC, Lanzkron S, et al. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101:1022-1033.
13. Mainous AG, Tanner RJ, Harle CA, et al. Attitudes toward management of sickle cell disease and its complications: a national survey of academic family physicians. Anemia. 2015;2015:1-6.
14. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312:1033.
15. Lunyera J, Jonassaint C, Jonassaint J, et al. Attitudes of primary care physicians toward sickle cell disease care, guidelines, and comanaging hydroxyurea with a specialist. J Prim Care Community Health. 2017;8:37-40.
16. Whiteman LN, Haywood C, Lanzkron S, et al. Primary care providers’ comfort levels in caring for patients with sickle cell disease. South Med J. 2015;108:531-536.
17. Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood. 2014;124:3850-4004.
18. DiMartino LD, Baumann AA, Hsu LL, et al. The sickle cell disease implementation consortium: Translating evidence-based guidelines into practice for sickle cell disease. Am J Hematol. 2018;93:E391-E395.
19. King AA, Baumann AA. Sickle cell disease and implementation science: A partnership to accelerate advances. Pediatr Blood Cancer. 2017;64:e26649.
20. Solberg LI. Improving medical practice: a conceptual framework. Ann Fam Med. 2007;5:251-256.
21. Bodenheimer T, Wagner EH, Grumbach K. Improving primary care for patients with chronic illness. J Am Med Assoc. 2002;288:5.
22. Bodenheimer T. Interventions to improve chronic illness care: evaluating their effectiveness. Dis Manag. 2003;6:63-71.
23. Tsai AC, Morton SC, Mangione CM, Keeler EB. A meta-analysis of interventions to improve care for chronic illnesses. Am J Manag Care. 2005;11:478-488.
24. Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
25. Kallio H, Pietilä A-M, Johnson M, et al. Systematic methodological review: developing a framework for a qualitative semi-structured interview guide. J Adv Nurs. 2016;72:2954-2965.
26. Clarke V, Braun V. Successful Qualitative Research: A Practical Guide for Beginners. First. Thousand Oaks, CA: Sage; 2013.
27. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15:1277-1288.
28. Creswell JW, Hanson WE, Clark Plano VL, et al. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35:236-264.
29. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis A Methods Sourcebook. 4th ed. Thousand Oaks, CA: Sage; 2019.
30. Eckman JR, Hassell KL, Huggins W, et al. Standard measures for sickle cell disease research: the PhenX Toolkit sickle cell disease collections. Blood Adv. 2017; 1: 2703-2711.
31. Kendall R, Wagner B, Brodke D, et al. The relationship of PROMIS pain interference and physical function scales. Pain Med. 2018;19:1720-1724.
32. Amtmann D, Cook KF, Jensen MP, et al. Development of a PROMIS item bank to measure pain interference. Pain. 2010;150:173-182.
33. Evensen CT, Treadwell MJ, Keller S, et al. Quality of care in sickle cell disease: Cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine (Baltimore). 2016;95:e4528.
34. Edwards R, Telfair J, Cecil H, et al. Reliability and validity of a self-efficacy instrument specific to sickle cell disease. Behav Res Ther. 2000;38:951-963.
35. Edwards R, Telfair J, Cecil H, et al. Self-efficacy as a predictor of adult adjustment to sickle cell disease: one-year outcomes. Psychosom Med. 2001;63:850-858.
36. Puri Singh A, Haywood C, Beach MC, et al. Improving emergency providers’ attitudes toward sickle cell patients in pain. J Pain Symptom Manage. 2016;51:628-632.e3.
37. Glassberg JA, Tanabe P, Chow A, et al. Emergency provider analgesic practices and attitudes towards patients with sickle cell disease. Ann Emerg Med. 2013;62:293-302.e10.
38. Grahmann PH, Jackson KC 2nd, Lipman AG. Clinician beliefs about opioid use and barriers in chronic nonmalignant pain [published correction appears in J Pain Palliat Care Pharmacother. 2004;18:145-6]. J Pain Palliat Care Pharmacother. 2004;18:7-28.
39. Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010;3:255-260.
40. Fielding N. Triangulation and mixed methods designs: data integration with new research technologies. J Mixed Meth Res. 2012;6:124-136.
41. 2017 CAHPS Health Plan Survey Chartbook. Agency for Healthcare Research and Quality website. www.ahrq.gov/cahps/cahps-database/comparative-data/2017-health-plan-chartbook/results-enrollee-population.html. Accessed September 8, 2020.
42. Bulgin D, Tanabe P, Jenerette C. Stigma of sickle cell disease: a systematic review. Issues Ment Health Nurs. 2018;1-11.
43. Wakefield EO, Zempsky WT, Puhl RM, et al. Conceptualizing pain-related stigma in adolescent chronic pain: a literature review and preliminary focus group findings. PAIN Rep. 2018;3:e679.
44. Nelson SC, Hackman HW. Race matters: Perceptions of race and racism in a sickle cell center. Pediatr Blood Cancer. 2013;60:451-454.
45. Dyal BW, Abudawood K, Schoppee TM, et al. Reflections of healthcare experiences of african americans with sickle cell disease or cancer: a qualitative study. Cancer Nurs. 2019;10.1097/NCC.0000000000000750.
46. Renedo A. Not being heard: barriers to high quality unplanned hospital care during young people’s transition to adult services - evidence from ‘this sickle cell life’ research. BMC Health Serv Res. 2019;19:876.
47. Ballas S, Vichinsky E. Is the medical home for adult patients with sickle cell disease a reality or an illusion? Hemoglobin. 2015;39:130-133.
48. Hankins JS, Osarogiagbon R, Adams-Graves P, et al. A transition pilot program for adolescents with sickle cell disease. J Pediatr Health Care. 2012;26 e45-e49.
49. Smith WR, Sisler IY, Johnson S, et al. Lessons learned from building a pediatric-to-adult sickle cell transition program. South Med J. 2019;112:190-197.
50. Lanzkron S, Sawicki GS, Hassell KL, et al. Transition to adulthood and adult health care for patients with sickle cell disease or cystic fibrosis: Current practices and research priorities. J Clin Transl Sci. 2018;2:334-342.
51. Kanter J, Gibson R, Lawrence RH, et al. Perceptions of US adolescents and adults with sickle cell disease on their quality of care. JAMA Netw Open. 2020;3:e206016.
52. Haywood C, Lanzkron S, Hughes MT, et al. A video-intervention to improve clinician attitudes toward patients with sickle cell disease: the results of a randomized experiment. J Gen Intern Med. 2011;26:518-523.
53. Hankins JS, Shah N, DiMartino L, et al. Integration of mobile health into sickle cell disease care to increase hydroxyurea utilization: protocol for an efficacy and implementation study. JMIR Res Protoc. 2020;9:e16319.
54. Fan W, Yan Z. Factors affecting response rates of the web survey: A systematic review. Comput Hum Behav. 2010;26:132-139.
55. Millar MM, Dillman DA. Improving response to web and mixed-mode surveys. Public Opin Q. 2011;75:249-269.

Children’s share of COVID-19 burden continues to increase
Children continue to represent an increasing proportion of reported COVID-19 cases in the United States, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The previous week, children represented 10.0% of all cases, and that proportion has continued to rise throughout the pandemic, the AAP and CHA report shows.
Looking at just new cases for the latest week, the 38,000+ pediatric cases made up almost 17% of the 228,396 cases reported for all ages, compared with 16% and 15% the two previous weeks. For the weeks ending Aug. 13 and Aug. 6, the corresponding figures were 8% and 13%, based on the data in the AAP/CHA report, which cover 49 states (New York City but not New York state), the District of Columbia, Puerto Rico, and Guam.
The state with the highest proportion of child COVID-19 cases as of Sept. 17 was Wyoming, with 20.6%, followed by North Dakota at 18.3% and Tennessee at 17.9%. New York City has a cumulative rate of just 3.4%, but New Jersey is the state with the lowest rate at 3.6%. Florida comes in at 5.9% but is using an age range of 0-14 years for children, and Texas has a rate of 6.0% but has reported ages for only 8% of confirmed cases, the AAP and CHA noted.
Severe illness, however, continues to be rare in children. The overall hospitalization rate for children was down to 1.7% among the 26 jurisdictions providing ages as Sept. 17 – down from 1.8% the week before and 2.3% on Aug. 20. The death rate is just 0.02% among 43 jurisdictions, the report said.
Children continue to represent an increasing proportion of reported COVID-19 cases in the United States, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The previous week, children represented 10.0% of all cases, and that proportion has continued to rise throughout the pandemic, the AAP and CHA report shows.
Looking at just new cases for the latest week, the 38,000+ pediatric cases made up almost 17% of the 228,396 cases reported for all ages, compared with 16% and 15% the two previous weeks. For the weeks ending Aug. 13 and Aug. 6, the corresponding figures were 8% and 13%, based on the data in the AAP/CHA report, which cover 49 states (New York City but not New York state), the District of Columbia, Puerto Rico, and Guam.
The state with the highest proportion of child COVID-19 cases as of Sept. 17 was Wyoming, with 20.6%, followed by North Dakota at 18.3% and Tennessee at 17.9%. New York City has a cumulative rate of just 3.4%, but New Jersey is the state with the lowest rate at 3.6%. Florida comes in at 5.9% but is using an age range of 0-14 years for children, and Texas has a rate of 6.0% but has reported ages for only 8% of confirmed cases, the AAP and CHA noted.
Severe illness, however, continues to be rare in children. The overall hospitalization rate for children was down to 1.7% among the 26 jurisdictions providing ages as Sept. 17 – down from 1.8% the week before and 2.3% on Aug. 20. The death rate is just 0.02% among 43 jurisdictions, the report said.
Children continue to represent an increasing proportion of reported COVID-19 cases in the United States, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The previous week, children represented 10.0% of all cases, and that proportion has continued to rise throughout the pandemic, the AAP and CHA report shows.
Looking at just new cases for the latest week, the 38,000+ pediatric cases made up almost 17% of the 228,396 cases reported for all ages, compared with 16% and 15% the two previous weeks. For the weeks ending Aug. 13 and Aug. 6, the corresponding figures were 8% and 13%, based on the data in the AAP/CHA report, which cover 49 states (New York City but not New York state), the District of Columbia, Puerto Rico, and Guam.
The state with the highest proportion of child COVID-19 cases as of Sept. 17 was Wyoming, with 20.6%, followed by North Dakota at 18.3% and Tennessee at 17.9%. New York City has a cumulative rate of just 3.4%, but New Jersey is the state with the lowest rate at 3.6%. Florida comes in at 5.9% but is using an age range of 0-14 years for children, and Texas has a rate of 6.0% but has reported ages for only 8% of confirmed cases, the AAP and CHA noted.
Severe illness, however, continues to be rare in children. The overall hospitalization rate for children was down to 1.7% among the 26 jurisdictions providing ages as Sept. 17 – down from 1.8% the week before and 2.3% on Aug. 20. The death rate is just 0.02% among 43 jurisdictions, the report said.
More female specialists, but gender gap persists in pay, survey finds
More female physicians are becoming specialists, a Medscape survey finds, and five specialties have seen particularly large increases during the last 5 years.

Obstetrician/gynecologists and pediatricians had the largest female representation at 58% and those percentages were both up from 50% in 2015, according to the Medscape Female Physician Compensation Report 2020.
Rheumatology saw a dramatic jump in numbers of women from 29% in 2015 to 54% now. Dermatology increased from 32% to 49%, and family medicine rose from 35% to 43% during that time.
Specialist pay gap narrows slightly
The gender gap was the same this year in primary care — women made 25% less ($212,000 vs. $264,000).
The gap in specialists narrowed slightly. Women made 31% less this year ($286,000 vs $375,000) instead of the 33% less reported in last year’s survey, a difference of $89,000 this year.
The gender pay gap was consistent across all race and age groups and was consistent in responses about net worth. Whereas 57% of male physicians had a net worth of $1 million or more, only 40% of female physicians did. Twice as many male physicians as female physicians had a net worth of more than $5 million (10% vs. 5%).
“Many physicians expect the gender pay gap to narrow in the coming years,” John Prescott, MD, chief academic officer of the Association of American Medical Colleges, said in an interview.
“Yet, it is a challenging task, requiring an institutional commitment to transparency, cross-campus collaboration, ongoing communication, dedicated resources, and enlightened leadership,” he said.
Female physicians working in office-based, solo practices made the most overall at $290,000; women in outpatient settings made the least at $223,000.
The survey included more than 4,500 responses. The responses were collected during the early part of the year and do not reflect changes in income expected from the COVID-19 pandemic.
An analysis in Health Affairs, for instance, predicted that primary care practices would lose $67,774 in gross revenue per full-time-equivalent physician in calendar year 2020 because of the pandemic.
Most physicians did not experience a significant financial loss in 2019, but COVID-19 may, at least temporarily, change those answers in next year’s report, physicians predicted.
Women more likely than men to live above their means
More women this year (39%) said they live below their means than answered that way last year (31%). Female physicians were more likely to say they lived above their means than were their male counterparts (8% vs. 6%).
Greenwald Wealth Management in St. Louis Park, Minn., says aiming for putting away 20% of total gross salary is a good financial goal.
Women in this year’s survey spent about 7% less time seeing patients than did their male counterparts (35.9 hours a week vs. 38.8). The average for all physicians was 37.8 hours a week. Add the 15.6 average hours per week physicians spend on paperwork, and they are putting in 53-hour workweeks on average overall.
Asked what parts of their job they found most rewarding, women were more likely than were men to say “gratitude/relationships with patients” (31% vs. 25%). They were less likely than were men to answer that the most rewarding part was “being very good at what I do/finding answers/diagnoses” (22% vs. 25%) or “making good money at a job I like” (9% vs. 13%).
Most female physicians — and physicians overall — said they would choose medicine again. But two specialties saw a substantial increase in that answer.
This year, 79% of those in physical medicine and rehabilitation said they would choose medicine again (compared with 66% last year) and 84% of gastroenterologists answered that way (compared with 76% in 2019).
Psychiatrists, however, were in the group least likely to say they would choose their specialty again along with those in internal medicine, family medicine, and diabetes and endocrinology.
Female physicians in orthopedics, radiology, and dermatology were most likely to choose their specialties again (91% - 92%).
Female physicians were less likely to use physician assistants in their practices than were their male colleagues (31% vs. 38%) but more likely to use NPs (52% vs. 50%). More than a third (38%) of male and female physicians reported they use neither.
A version of this article originally appeared on Medscape.com.
More female physicians are becoming specialists, a Medscape survey finds, and five specialties have seen particularly large increases during the last 5 years.
Obstetrician/gynecologists and pediatricians had the largest female representation at 58% and those percentages were both up from 50% in 2015, according to the Medscape Female Physician Compensation Report 2020.
Rheumatology saw a dramatic jump in numbers of women from 29% in 2015 to 54% now. Dermatology increased from 32% to 49%, and family medicine rose from 35% to 43% during that time.
Specialist pay gap narrows slightly
The gender gap was the same this year in primary care — women made 25% less ($212,000 vs. $264,000).
The gap in specialists narrowed slightly. Women made 31% less this year ($286,000 vs $375,000) instead of the 33% less reported in last year’s survey, a difference of $89,000 this year.
The gender pay gap was consistent across all race and age groups and was consistent in responses about net worth. Whereas 57% of male physicians had a net worth of $1 million or more, only 40% of female physicians did. Twice as many male physicians as female physicians had a net worth of more than $5 million (10% vs. 5%).
“Many physicians expect the gender pay gap to narrow in the coming years,” John Prescott, MD, chief academic officer of the Association of American Medical Colleges, said in an interview.
“Yet, it is a challenging task, requiring an institutional commitment to transparency, cross-campus collaboration, ongoing communication, dedicated resources, and enlightened leadership,” he said.
Female physicians working in office-based, solo practices made the most overall at $290,000; women in outpatient settings made the least at $223,000.
The survey included more than 4,500 responses. The responses were collected during the early part of the year and do not reflect changes in income expected from the COVID-19 pandemic.
An analysis in Health Affairs, for instance, predicted that primary care practices would lose $67,774 in gross revenue per full-time-equivalent physician in calendar year 2020 because of the pandemic.
Most physicians did not experience a significant financial loss in 2019, but COVID-19 may, at least temporarily, change those answers in next year’s report, physicians predicted.
Women more likely than men to live above their means
More women this year (39%) said they live below their means than answered that way last year (31%). Female physicians were more likely to say they lived above their means than were their male counterparts (8% vs. 6%).
Greenwald Wealth Management in St. Louis Park, Minn., says aiming for putting away 20% of total gross salary is a good financial goal.
Women in this year’s survey spent about 7% less time seeing patients than did their male counterparts (35.9 hours a week vs. 38.8). The average for all physicians was 37.8 hours a week. Add the 15.6 average hours per week physicians spend on paperwork, and they are putting in 53-hour workweeks on average overall.
Asked what parts of their job they found most rewarding, women were more likely than were men to say “gratitude/relationships with patients” (31% vs. 25%). They were less likely than were men to answer that the most rewarding part was “being very good at what I do/finding answers/diagnoses” (22% vs. 25%) or “making good money at a job I like” (9% vs. 13%).
Most female physicians — and physicians overall — said they would choose medicine again. But two specialties saw a substantial increase in that answer.
This year, 79% of those in physical medicine and rehabilitation said they would choose medicine again (compared with 66% last year) and 84% of gastroenterologists answered that way (compared with 76% in 2019).
Psychiatrists, however, were in the group least likely to say they would choose their specialty again along with those in internal medicine, family medicine, and diabetes and endocrinology.
Female physicians in orthopedics, radiology, and dermatology were most likely to choose their specialties again (91% - 92%).
Female physicians were less likely to use physician assistants in their practices than were their male colleagues (31% vs. 38%) but more likely to use NPs (52% vs. 50%). More than a third (38%) of male and female physicians reported they use neither.
A version of this article originally appeared on Medscape.com.
More female physicians are becoming specialists, a Medscape survey finds, and five specialties have seen particularly large increases during the last 5 years.
Obstetrician/gynecologists and pediatricians had the largest female representation at 58% and those percentages were both up from 50% in 2015, according to the Medscape Female Physician Compensation Report 2020.
Rheumatology saw a dramatic jump in numbers of women from 29% in 2015 to 54% now. Dermatology increased from 32% to 49%, and family medicine rose from 35% to 43% during that time.
Specialist pay gap narrows slightly
The gender gap was the same this year in primary care — women made 25% less ($212,000 vs. $264,000).
The gap in specialists narrowed slightly. Women made 31% less this year ($286,000 vs $375,000) instead of the 33% less reported in last year’s survey, a difference of $89,000 this year.
The gender pay gap was consistent across all race and age groups and was consistent in responses about net worth. Whereas 57% of male physicians had a net worth of $1 million or more, only 40% of female physicians did. Twice as many male physicians as female physicians had a net worth of more than $5 million (10% vs. 5%).
“Many physicians expect the gender pay gap to narrow in the coming years,” John Prescott, MD, chief academic officer of the Association of American Medical Colleges, said in an interview.
“Yet, it is a challenging task, requiring an institutional commitment to transparency, cross-campus collaboration, ongoing communication, dedicated resources, and enlightened leadership,” he said.
Female physicians working in office-based, solo practices made the most overall at $290,000; women in outpatient settings made the least at $223,000.
The survey included more than 4,500 responses. The responses were collected during the early part of the year and do not reflect changes in income expected from the COVID-19 pandemic.
An analysis in Health Affairs, for instance, predicted that primary care practices would lose $67,774 in gross revenue per full-time-equivalent physician in calendar year 2020 because of the pandemic.
Most physicians did not experience a significant financial loss in 2019, but COVID-19 may, at least temporarily, change those answers in next year’s report, physicians predicted.
Women more likely than men to live above their means
More women this year (39%) said they live below their means than answered that way last year (31%). Female physicians were more likely to say they lived above their means than were their male counterparts (8% vs. 6%).
Greenwald Wealth Management in St. Louis Park, Minn., says aiming for putting away 20% of total gross salary is a good financial goal.
Women in this year’s survey spent about 7% less time seeing patients than did their male counterparts (35.9 hours a week vs. 38.8). The average for all physicians was 37.8 hours a week. Add the 15.6 average hours per week physicians spend on paperwork, and they are putting in 53-hour workweeks on average overall.
Asked what parts of their job they found most rewarding, women were more likely than were men to say “gratitude/relationships with patients” (31% vs. 25%). They were less likely than were men to answer that the most rewarding part was “being very good at what I do/finding answers/diagnoses” (22% vs. 25%) or “making good money at a job I like” (9% vs. 13%).
Most female physicians — and physicians overall — said they would choose medicine again. But two specialties saw a substantial increase in that answer.
This year, 79% of those in physical medicine and rehabilitation said they would choose medicine again (compared with 66% last year) and 84% of gastroenterologists answered that way (compared with 76% in 2019).
Psychiatrists, however, were in the group least likely to say they would choose their specialty again along with those in internal medicine, family medicine, and diabetes and endocrinology.
Female physicians in orthopedics, radiology, and dermatology were most likely to choose their specialties again (91% - 92%).
Female physicians were less likely to use physician assistants in their practices than were their male colleagues (31% vs. 38%) but more likely to use NPs (52% vs. 50%). More than a third (38%) of male and female physicians reported they use neither.
A version of this article originally appeared on Medscape.com.
“I Really Didn’t Want To Come In”: The Unseen Effects of COVID-19 on Children
The Children’s Hospital of Philadelphia, Philadelphia, PA.
The effects of COVID-19 on children’s health are multifaceted. In comparison to adults, children typically experience far milder physical consequences when infected with the virus. A notable exception is the newly described multisystem inflammatory syndrome associated with COVID-19 (MIS-C), which has proven to be a source of significant morbidity among the children it affects.1 Nevertheless, even those children not infected with COVID-19 have suffered due to the disease. School closures have deprived children of opportunities for social and academic growth and, in some cases, the provision of food, social services, medication administration, and many different therapies. Social distancing rules have limited play among children, which is crucial to their development and mental health. The impact on children who have lost family members, including parents, is monumental. Amidst all of this observable suffering, however, the pandemic poses a less visible threat to the health of children.
It is well documented that concern about exposure to COVID-19 has led many adults to avoid emergency departments (EDs) around the world. We believe parents may be avoiding ED visits for their children for the same reason. In the United States, ED volumes dropped approximately 50% during spring 2020.2 While EDs saw increasing, and at times overwhelming, numbers of patients with COVID-19, the number of patients presenting with other life-threatening medical issues, including heart attacks and strokes, declined.3,4 Data from the National Center for Health Statistics this past spring revealed nationwide increases in deaths due to nonrespiratory causes such as diabetes, heart disease, and stroke.5 ED avoidance and unprecedented lack of access to outpatient care, though with the intent to reduce overall risk, are likely significant contributors to these deaths.
Pediatric patients, especially the most vulnerable, are similarly at risk for deleterious health-related consequences from ED avoidance and from limited access to primary and outpatient specialty care. Data from Europe indicate dramatic drops in pediatric ED (PED) volumes, as well as an increase in the proportion of ED visits leading to hospitalization.6,7 These studies suggest that when patients do ultimately present to the PED, they may be more seriously ill.
At our institution, we have seen many COVID-19-negative patients whose medical care has been negatively influenced by the pandemic. A few months ago, a 1-month-old infant with an underlying health condition presented to the PED in extremis after weeks of progressively worsening feeding issues. The infant had been closely followed by the primary care provider (PCP) and subspecialty team via phone calls, televisits, and some office visits. Both physicians and parents had tried to resolve the feeding issues within the outpatient context, explicitly hoping to avoid potential exposure of this fragile patient to COVID-19 in the hospital. On eventual presentation to the PED, the infant was profoundly dehydrated, with significant electrolyte derangement and an acute abdomen, requiring admission to the intensive care unit. Ultimately, a new diagnosis of Hirschsprung disease was made, and the infant was hospitalized for several weeks for weight gain.
Later this summer, a school-aged child with a history of poorly controlled type 1 diabetes presented to an affiliated community hospital comatose and with Kussmaul respirations. Prior to the pandemic, a school nurse administered the child’s morning insulin. Since school closed, the patient had been responsible for administering this dose of insulin while the parents worked outside the home. Despite close and frequent communication between the patient’s endocrinology team and the family, the patient’s glucose and ketone levels began to rise. The parent administered repeated boluses of insulin at home in an attempt to avoid the perceived exposure risk associated with an ED visit. On presentation to the PED, the patient was profoundly altered, with a pH of 7.0. When transfer to a tertiary care center was recommended, the patient’s parent expressed persistent concerns about COVID-19 exposure in the larger hospital, although ultimately consent to transfer was given.
A third case from this summer provides an example of a different type of patient affected by COVID-19: the neonate whose birth circumstances were altered due to the virus. A 3-day-old, full-term infant presented to the ED with hypothermia after PCP referral. The parents had considered both home birth and hospital delivery earlier in the pregnancy, ultimately opting for home birth due to concerns about COVID-19 exposure in the hospital. The pregnancy and delivery were uncomplicated. The neonate did not receive the first hepatitis B vaccine, erythromycin eye ointment, or vitamin K after delivery. In the first 3 days of life, the patient had voided once and stooled once per day. The patient’s mother, inexperienced with breastfeeding and without access to a lactation consultant, was unsure about latch or emptying of her breasts. At the first pediatrician visit, the infant was noted to be hypothermic to 35°C, intermittently bradycardic to the 80s, and with diminished arousal. In the PED, a full sepsis work-up was initiated. Though multiple attempts were made by different providers, only a minimal amount of blood could be drawn, presumably due to dehydration. Of note, the neonate received vitamin K subcutaneously prior to lumbar puncture.
Pediatricians across the country have gone to great lengths to protect their patients and to provide high-quality care both inside and outside the office during this unprecedented time. Nevertheless, these 3 cases illustrate the detrimental effects of COVID-19 on the delivery of pediatric health care. The first 2 cases in particular demonstrate the limitations of even close and consistent phone and televisit follow-up. Telehealth has provided a lifeline for patients and families during the pandemic, and, in most cases, has provided an excellent temporary substitution for office visits. There are, however, limitations to care without physical evaluation. Had the children in the first 2 cases been evaluated in person sooner, they may have been referred to a higher level of care more expediently. Likewise, in all 3 cases, parental reservations about exposing their children to COVID-19 through a trip to the hospital, however well-intentioned, likely played a role in the eventual severity of illness with which each child presented to the hospital.
If we are encountering children in the PED with severe illness due to delayed presentation to care, what about the children we aren’t seeing? As COVID-19 cases rise daily in the United States, we must be aware of the possibility of ED avoidance. We propose a multimodal approach to combat this dangerous phenomenon. Inpatient and ED-based pediatricians must maintain clear and open lines of communication with outpatient colleagues so that we can partner in considering which cases warrant prompt ED evaluation, even in the midst of a pandemic. All pediatricians must remind families that our hospitals remain open and ready to treat children safely. We must promote community awareness of the numerous safety precautions we take every day so that patients and families can feel comfortable seeking care at the hospital; the message of ED and hospital safety must be even more robust for caregivers of our particularly vulnerable children. As always, how we communicate with patients and their families matters. Validating and addressing concerns about COVID-19 exposure, while providing reassurance about the safety of our hospitals, could save children’s lives.
Acknowledgment: Thank you to Dr. Cynthia Mollen and Dr. Kathy Shaw for their reviews of the manuscript.
Corresponding author: Regina L. Toto, MD, Department of Pediatrics, The Children’s Hospital of Philadelphia, 3401 Civic Center Blvd., Philadelphia, PA 19104; totor@email.chop.edu.
Financial disclosures: None.
Keywords: coronavirus; pediatric; children; access to care; emergency department.
1. Riphagen S, Gomez X, Gonzalez-Martinez C, et al. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395:1607-1608.
2. Wong LE, Hawkins JE, Langness S, et al. Where are all the patients? addressing COVID-19 fear to encourage sick patients to seek emergency care. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0193
3. Moroni F, Gramegna M, Ajello S, et al. Collateral damage: medical care avoidance behavior among patients with acute coronary syndrome during the COVID-19 pandemic. JACC. 2020. doi:10.1016/j.jaccas.2020.04.010
4. Deerberg-Wittram J, Knothe C. Do not stay home: we are ready for you. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0146
5. Woolf SH, Chapman DA, Sabo RT, et al. Excess deaths From COVID-19 and other causes, March-April 2020. JAMA. 2020. doi:10.1001.jama.2020.11787
6. Lazzerini M, Barbi E, Apicella A, et al. Delayed access or provision of care in Italy resulting from fear of COVID-19. Lancet Child Adolesc Health. 2020;4:E10-1.
7. Happle C, Dopfer C, Wetzke M, et al. Covid-19 related reduction in paediatric emergency healthcare utilization--a concerning trend. BMC Pediatrics. [under review]. 2020. doi:10.21203/rs.3.rs-2
The Children’s Hospital of Philadelphia, Philadelphia, PA.
The effects of COVID-19 on children’s health are multifaceted. In comparison to adults, children typically experience far milder physical consequences when infected with the virus. A notable exception is the newly described multisystem inflammatory syndrome associated with COVID-19 (MIS-C), which has proven to be a source of significant morbidity among the children it affects.1 Nevertheless, even those children not infected with COVID-19 have suffered due to the disease. School closures have deprived children of opportunities for social and academic growth and, in some cases, the provision of food, social services, medication administration, and many different therapies. Social distancing rules have limited play among children, which is crucial to their development and mental health. The impact on children who have lost family members, including parents, is monumental. Amidst all of this observable suffering, however, the pandemic poses a less visible threat to the health of children.
It is well documented that concern about exposure to COVID-19 has led many adults to avoid emergency departments (EDs) around the world. We believe parents may be avoiding ED visits for their children for the same reason. In the United States, ED volumes dropped approximately 50% during spring 2020.2 While EDs saw increasing, and at times overwhelming, numbers of patients with COVID-19, the number of patients presenting with other life-threatening medical issues, including heart attacks and strokes, declined.3,4 Data from the National Center for Health Statistics this past spring revealed nationwide increases in deaths due to nonrespiratory causes such as diabetes, heart disease, and stroke.5 ED avoidance and unprecedented lack of access to outpatient care, though with the intent to reduce overall risk, are likely significant contributors to these deaths.
Pediatric patients, especially the most vulnerable, are similarly at risk for deleterious health-related consequences from ED avoidance and from limited access to primary and outpatient specialty care. Data from Europe indicate dramatic drops in pediatric ED (PED) volumes, as well as an increase in the proportion of ED visits leading to hospitalization.6,7 These studies suggest that when patients do ultimately present to the PED, they may be more seriously ill.
At our institution, we have seen many COVID-19-negative patients whose medical care has been negatively influenced by the pandemic. A few months ago, a 1-month-old infant with an underlying health condition presented to the PED in extremis after weeks of progressively worsening feeding issues. The infant had been closely followed by the primary care provider (PCP) and subspecialty team via phone calls, televisits, and some office visits. Both physicians and parents had tried to resolve the feeding issues within the outpatient context, explicitly hoping to avoid potential exposure of this fragile patient to COVID-19 in the hospital. On eventual presentation to the PED, the infant was profoundly dehydrated, with significant electrolyte derangement and an acute abdomen, requiring admission to the intensive care unit. Ultimately, a new diagnosis of Hirschsprung disease was made, and the infant was hospitalized for several weeks for weight gain.
Later this summer, a school-aged child with a history of poorly controlled type 1 diabetes presented to an affiliated community hospital comatose and with Kussmaul respirations. Prior to the pandemic, a school nurse administered the child’s morning insulin. Since school closed, the patient had been responsible for administering this dose of insulin while the parents worked outside the home. Despite close and frequent communication between the patient’s endocrinology team and the family, the patient’s glucose and ketone levels began to rise. The parent administered repeated boluses of insulin at home in an attempt to avoid the perceived exposure risk associated with an ED visit. On presentation to the PED, the patient was profoundly altered, with a pH of 7.0. When transfer to a tertiary care center was recommended, the patient’s parent expressed persistent concerns about COVID-19 exposure in the larger hospital, although ultimately consent to transfer was given.
A third case from this summer provides an example of a different type of patient affected by COVID-19: the neonate whose birth circumstances were altered due to the virus. A 3-day-old, full-term infant presented to the ED with hypothermia after PCP referral. The parents had considered both home birth and hospital delivery earlier in the pregnancy, ultimately opting for home birth due to concerns about COVID-19 exposure in the hospital. The pregnancy and delivery were uncomplicated. The neonate did not receive the first hepatitis B vaccine, erythromycin eye ointment, or vitamin K after delivery. In the first 3 days of life, the patient had voided once and stooled once per day. The patient’s mother, inexperienced with breastfeeding and without access to a lactation consultant, was unsure about latch or emptying of her breasts. At the first pediatrician visit, the infant was noted to be hypothermic to 35°C, intermittently bradycardic to the 80s, and with diminished arousal. In the PED, a full sepsis work-up was initiated. Though multiple attempts were made by different providers, only a minimal amount of blood could be drawn, presumably due to dehydration. Of note, the neonate received vitamin K subcutaneously prior to lumbar puncture.
Pediatricians across the country have gone to great lengths to protect their patients and to provide high-quality care both inside and outside the office during this unprecedented time. Nevertheless, these 3 cases illustrate the detrimental effects of COVID-19 on the delivery of pediatric health care. The first 2 cases in particular demonstrate the limitations of even close and consistent phone and televisit follow-up. Telehealth has provided a lifeline for patients and families during the pandemic, and, in most cases, has provided an excellent temporary substitution for office visits. There are, however, limitations to care without physical evaluation. Had the children in the first 2 cases been evaluated in person sooner, they may have been referred to a higher level of care more expediently. Likewise, in all 3 cases, parental reservations about exposing their children to COVID-19 through a trip to the hospital, however well-intentioned, likely played a role in the eventual severity of illness with which each child presented to the hospital.
If we are encountering children in the PED with severe illness due to delayed presentation to care, what about the children we aren’t seeing? As COVID-19 cases rise daily in the United States, we must be aware of the possibility of ED avoidance. We propose a multimodal approach to combat this dangerous phenomenon. Inpatient and ED-based pediatricians must maintain clear and open lines of communication with outpatient colleagues so that we can partner in considering which cases warrant prompt ED evaluation, even in the midst of a pandemic. All pediatricians must remind families that our hospitals remain open and ready to treat children safely. We must promote community awareness of the numerous safety precautions we take every day so that patients and families can feel comfortable seeking care at the hospital; the message of ED and hospital safety must be even more robust for caregivers of our particularly vulnerable children. As always, how we communicate with patients and their families matters. Validating and addressing concerns about COVID-19 exposure, while providing reassurance about the safety of our hospitals, could save children’s lives.
Acknowledgment: Thank you to Dr. Cynthia Mollen and Dr. Kathy Shaw for their reviews of the manuscript.
Corresponding author: Regina L. Toto, MD, Department of Pediatrics, The Children’s Hospital of Philadelphia, 3401 Civic Center Blvd., Philadelphia, PA 19104; totor@email.chop.edu.
Financial disclosures: None.
Keywords: coronavirus; pediatric; children; access to care; emergency department.
The Children’s Hospital of Philadelphia, Philadelphia, PA.
The effects of COVID-19 on children’s health are multifaceted. In comparison to adults, children typically experience far milder physical consequences when infected with the virus. A notable exception is the newly described multisystem inflammatory syndrome associated with COVID-19 (MIS-C), which has proven to be a source of significant morbidity among the children it affects.1 Nevertheless, even those children not infected with COVID-19 have suffered due to the disease. School closures have deprived children of opportunities for social and academic growth and, in some cases, the provision of food, social services, medication administration, and many different therapies. Social distancing rules have limited play among children, which is crucial to their development and mental health. The impact on children who have lost family members, including parents, is monumental. Amidst all of this observable suffering, however, the pandemic poses a less visible threat to the health of children.
It is well documented that concern about exposure to COVID-19 has led many adults to avoid emergency departments (EDs) around the world. We believe parents may be avoiding ED visits for their children for the same reason. In the United States, ED volumes dropped approximately 50% during spring 2020.2 While EDs saw increasing, and at times overwhelming, numbers of patients with COVID-19, the number of patients presenting with other life-threatening medical issues, including heart attacks and strokes, declined.3,4 Data from the National Center for Health Statistics this past spring revealed nationwide increases in deaths due to nonrespiratory causes such as diabetes, heart disease, and stroke.5 ED avoidance and unprecedented lack of access to outpatient care, though with the intent to reduce overall risk, are likely significant contributors to these deaths.
Pediatric patients, especially the most vulnerable, are similarly at risk for deleterious health-related consequences from ED avoidance and from limited access to primary and outpatient specialty care. Data from Europe indicate dramatic drops in pediatric ED (PED) volumes, as well as an increase in the proportion of ED visits leading to hospitalization.6,7 These studies suggest that when patients do ultimately present to the PED, they may be more seriously ill.
At our institution, we have seen many COVID-19-negative patients whose medical care has been negatively influenced by the pandemic. A few months ago, a 1-month-old infant with an underlying health condition presented to the PED in extremis after weeks of progressively worsening feeding issues. The infant had been closely followed by the primary care provider (PCP) and subspecialty team via phone calls, televisits, and some office visits. Both physicians and parents had tried to resolve the feeding issues within the outpatient context, explicitly hoping to avoid potential exposure of this fragile patient to COVID-19 in the hospital. On eventual presentation to the PED, the infant was profoundly dehydrated, with significant electrolyte derangement and an acute abdomen, requiring admission to the intensive care unit. Ultimately, a new diagnosis of Hirschsprung disease was made, and the infant was hospitalized for several weeks for weight gain.
Later this summer, a school-aged child with a history of poorly controlled type 1 diabetes presented to an affiliated community hospital comatose and with Kussmaul respirations. Prior to the pandemic, a school nurse administered the child’s morning insulin. Since school closed, the patient had been responsible for administering this dose of insulin while the parents worked outside the home. Despite close and frequent communication between the patient’s endocrinology team and the family, the patient’s glucose and ketone levels began to rise. The parent administered repeated boluses of insulin at home in an attempt to avoid the perceived exposure risk associated with an ED visit. On presentation to the PED, the patient was profoundly altered, with a pH of 7.0. When transfer to a tertiary care center was recommended, the patient’s parent expressed persistent concerns about COVID-19 exposure in the larger hospital, although ultimately consent to transfer was given.
A third case from this summer provides an example of a different type of patient affected by COVID-19: the neonate whose birth circumstances were altered due to the virus. A 3-day-old, full-term infant presented to the ED with hypothermia after PCP referral. The parents had considered both home birth and hospital delivery earlier in the pregnancy, ultimately opting for home birth due to concerns about COVID-19 exposure in the hospital. The pregnancy and delivery were uncomplicated. The neonate did not receive the first hepatitis B vaccine, erythromycin eye ointment, or vitamin K after delivery. In the first 3 days of life, the patient had voided once and stooled once per day. The patient’s mother, inexperienced with breastfeeding and without access to a lactation consultant, was unsure about latch or emptying of her breasts. At the first pediatrician visit, the infant was noted to be hypothermic to 35°C, intermittently bradycardic to the 80s, and with diminished arousal. In the PED, a full sepsis work-up was initiated. Though multiple attempts were made by different providers, only a minimal amount of blood could be drawn, presumably due to dehydration. Of note, the neonate received vitamin K subcutaneously prior to lumbar puncture.
Pediatricians across the country have gone to great lengths to protect their patients and to provide high-quality care both inside and outside the office during this unprecedented time. Nevertheless, these 3 cases illustrate the detrimental effects of COVID-19 on the delivery of pediatric health care. The first 2 cases in particular demonstrate the limitations of even close and consistent phone and televisit follow-up. Telehealth has provided a lifeline for patients and families during the pandemic, and, in most cases, has provided an excellent temporary substitution for office visits. There are, however, limitations to care without physical evaluation. Had the children in the first 2 cases been evaluated in person sooner, they may have been referred to a higher level of care more expediently. Likewise, in all 3 cases, parental reservations about exposing their children to COVID-19 through a trip to the hospital, however well-intentioned, likely played a role in the eventual severity of illness with which each child presented to the hospital.
If we are encountering children in the PED with severe illness due to delayed presentation to care, what about the children we aren’t seeing? As COVID-19 cases rise daily in the United States, we must be aware of the possibility of ED avoidance. We propose a multimodal approach to combat this dangerous phenomenon. Inpatient and ED-based pediatricians must maintain clear and open lines of communication with outpatient colleagues so that we can partner in considering which cases warrant prompt ED evaluation, even in the midst of a pandemic. All pediatricians must remind families that our hospitals remain open and ready to treat children safely. We must promote community awareness of the numerous safety precautions we take every day so that patients and families can feel comfortable seeking care at the hospital; the message of ED and hospital safety must be even more robust for caregivers of our particularly vulnerable children. As always, how we communicate with patients and their families matters. Validating and addressing concerns about COVID-19 exposure, while providing reassurance about the safety of our hospitals, could save children’s lives.
Acknowledgment: Thank you to Dr. Cynthia Mollen and Dr. Kathy Shaw for their reviews of the manuscript.
Corresponding author: Regina L. Toto, MD, Department of Pediatrics, The Children’s Hospital of Philadelphia, 3401 Civic Center Blvd., Philadelphia, PA 19104; totor@email.chop.edu.
Financial disclosures: None.
Keywords: coronavirus; pediatric; children; access to care; emergency department.
1. Riphagen S, Gomez X, Gonzalez-Martinez C, et al. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395:1607-1608.
2. Wong LE, Hawkins JE, Langness S, et al. Where are all the patients? addressing COVID-19 fear to encourage sick patients to seek emergency care. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0193
3. Moroni F, Gramegna M, Ajello S, et al. Collateral damage: medical care avoidance behavior among patients with acute coronary syndrome during the COVID-19 pandemic. JACC. 2020. doi:10.1016/j.jaccas.2020.04.010
4. Deerberg-Wittram J, Knothe C. Do not stay home: we are ready for you. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0146
5. Woolf SH, Chapman DA, Sabo RT, et al. Excess deaths From COVID-19 and other causes, March-April 2020. JAMA. 2020. doi:10.1001.jama.2020.11787
6. Lazzerini M, Barbi E, Apicella A, et al. Delayed access or provision of care in Italy resulting from fear of COVID-19. Lancet Child Adolesc Health. 2020;4:E10-1.
7. Happle C, Dopfer C, Wetzke M, et al. Covid-19 related reduction in paediatric emergency healthcare utilization--a concerning trend. BMC Pediatrics. [under review]. 2020. doi:10.21203/rs.3.rs-2
1. Riphagen S, Gomez X, Gonzalez-Martinez C, et al. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395:1607-1608.
2. Wong LE, Hawkins JE, Langness S, et al. Where are all the patients? addressing COVID-19 fear to encourage sick patients to seek emergency care. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0193
3. Moroni F, Gramegna M, Ajello S, et al. Collateral damage: medical care avoidance behavior among patients with acute coronary syndrome during the COVID-19 pandemic. JACC. 2020. doi:10.1016/j.jaccas.2020.04.010
4. Deerberg-Wittram J, Knothe C. Do not stay home: we are ready for you. NEJM Catalyst. 2020. doi:10.1056/CAT.20.0146
5. Woolf SH, Chapman DA, Sabo RT, et al. Excess deaths From COVID-19 and other causes, March-April 2020. JAMA. 2020. doi:10.1001.jama.2020.11787
6. Lazzerini M, Barbi E, Apicella A, et al. Delayed access or provision of care in Italy resulting from fear of COVID-19. Lancet Child Adolesc Health. 2020;4:E10-1.
7. Happle C, Dopfer C, Wetzke M, et al. Covid-19 related reduction in paediatric emergency healthcare utilization--a concerning trend. BMC Pediatrics. [under review]. 2020. doi:10.21203/rs.3.rs-2
Excessive screen time can harm a child’s eyesight
As an ophthalmologist who’s been performing vision correction surgery in the San Francisco Bay Area for more than 20 years, I want to address a rising concern I’ve seen among parents I’ve recently treated who currently have kids at home attending virtual classroom instruction.

Many of these parents are asking me if excessive screen time can be harmful to their children’s developing vision, especially if they are potentially genetically predisposed to myopia – also known as nearsightedness. While the short answer is, yes it can,
Eye fatigue, also called digital eye strain, is a physical eye discomfort that is caused by excessive screen use, and is a common condition in both children and adults.
Today, more than 40% of Americans are myopic, and that number is increasing at an alarming rate, especially among school-aged children. One in four parents have a child with myopia, and about three quarters of children with myopia were diagnosed between the ages of 3 and 12 years.
A recent ParentsTogether study found that a majority of parents surveyed said they are concerned about a massive spike in their children’s screen time during the coronavirus pandemic. And nearly half of respondents’ children (48%) are currently spending more than 6 hours per day online – a nearly 500% increase from before the crisis.

While glasses or contact lenses can correct a child’s vision, research in the journal Retina by the Ikuno Eye Center in Osaka, Japan, suggests that having severe myopia puts children at risk for a number of eye problems down the road, including retinal detachment, glaucoma, and macular degeneration. Research published in PLOS ONE found that working up close – such as reading or using a tablet – increased the odds of myopia. And in a report published in Ophthalmology, researchers studying myopia have estimated that, by 2050, about half the world’s population could be myopic.
Watch for warning signs
To determine if excessive screen time is affecting your child, there warning signs to watch out for including the following:
- Eye irritation.
- Watery eyes.
- Headaches.
- Intermittent blurry vision or double vision.
- Sore eyes.
- Difficulty concentrating.
- Sore neck, shoulders, and/or back.
- Increased sensitivity to light.
- Tiredness.
- Poor posture.
Use these approaches to reduce eye fatigue
To reduce eye fatigue associated with excessive screen exposure, parents should set limits on screen time if possible, and at minimum, schedule between 8 and 15 hours of outdoor activity per week. Screens should be at least 20 centimeters away from the child’s eyes, the child’s desk should be placed near a window, and she should be instructed to look outside every hour. Lastly, children should be kept in fully corrected glasses, not under corrected, and those glasses need to be worn full time.
As we plan the future of education in the age of COVID-19, I firmly believe that schools and policymakers must consider children’s vision needs while designing new initiatives. Schools, teachers, and parents must work together to incorporate eye health strategies that protect children as they learn online. By doing so, we can decrease the chance that a child’s vision becomes compromised from unwittingly staring into a screen for too many hours during the day.
Dr. Faktorovich is an ophthalmologist, founder of Pacific Vision Institute in San Francisco, California, and founder of the annual San Francisco Cornea, Cataract, and Refractive Surgery Symposium. She had no relevant financial disclosures. Email her at pdnews@mdedge.com.
As an ophthalmologist who’s been performing vision correction surgery in the San Francisco Bay Area for more than 20 years, I want to address a rising concern I’ve seen among parents I’ve recently treated who currently have kids at home attending virtual classroom instruction.
Many of these parents are asking me if excessive screen time can be harmful to their children’s developing vision, especially if they are potentially genetically predisposed to myopia – also known as nearsightedness. While the short answer is, yes it can,
Eye fatigue, also called digital eye strain, is a physical eye discomfort that is caused by excessive screen use, and is a common condition in both children and adults.
Today, more than 40% of Americans are myopic, and that number is increasing at an alarming rate, especially among school-aged children. One in four parents have a child with myopia, and about three quarters of children with myopia were diagnosed between the ages of 3 and 12 years.
A recent ParentsTogether study found that a majority of parents surveyed said they are concerned about a massive spike in their children’s screen time during the coronavirus pandemic. And nearly half of respondents’ children (48%) are currently spending more than 6 hours per day online – a nearly 500% increase from before the crisis.
While glasses or contact lenses can correct a child’s vision, research in the journal Retina by the Ikuno Eye Center in Osaka, Japan, suggests that having severe myopia puts children at risk for a number of eye problems down the road, including retinal detachment, glaucoma, and macular degeneration. Research published in PLOS ONE found that working up close – such as reading or using a tablet – increased the odds of myopia. And in a report published in Ophthalmology, researchers studying myopia have estimated that, by 2050, about half the world’s population could be myopic.
Watch for warning signs
To determine if excessive screen time is affecting your child, there warning signs to watch out for including the following:
- Eye irritation.
- Watery eyes.
- Headaches.
- Intermittent blurry vision or double vision.
- Sore eyes.
- Difficulty concentrating.
- Sore neck, shoulders, and/or back.
- Increased sensitivity to light.
- Tiredness.
- Poor posture.
Use these approaches to reduce eye fatigue
To reduce eye fatigue associated with excessive screen exposure, parents should set limits on screen time if possible, and at minimum, schedule between 8 and 15 hours of outdoor activity per week. Screens should be at least 20 centimeters away from the child’s eyes, the child’s desk should be placed near a window, and she should be instructed to look outside every hour. Lastly, children should be kept in fully corrected glasses, not under corrected, and those glasses need to be worn full time.
As we plan the future of education in the age of COVID-19, I firmly believe that schools and policymakers must consider children’s vision needs while designing new initiatives. Schools, teachers, and parents must work together to incorporate eye health strategies that protect children as they learn online. By doing so, we can decrease the chance that a child’s vision becomes compromised from unwittingly staring into a screen for too many hours during the day.
Dr. Faktorovich is an ophthalmologist, founder of Pacific Vision Institute in San Francisco, California, and founder of the annual San Francisco Cornea, Cataract, and Refractive Surgery Symposium. She had no relevant financial disclosures. Email her at pdnews@mdedge.com.
As an ophthalmologist who’s been performing vision correction surgery in the San Francisco Bay Area for more than 20 years, I want to address a rising concern I’ve seen among parents I’ve recently treated who currently have kids at home attending virtual classroom instruction.
Many of these parents are asking me if excessive screen time can be harmful to their children’s developing vision, especially if they are potentially genetically predisposed to myopia – also known as nearsightedness. While the short answer is, yes it can,
Eye fatigue, also called digital eye strain, is a physical eye discomfort that is caused by excessive screen use, and is a common condition in both children and adults.
Today, more than 40% of Americans are myopic, and that number is increasing at an alarming rate, especially among school-aged children. One in four parents have a child with myopia, and about three quarters of children with myopia were diagnosed between the ages of 3 and 12 years.
A recent ParentsTogether study found that a majority of parents surveyed said they are concerned about a massive spike in their children’s screen time during the coronavirus pandemic. And nearly half of respondents’ children (48%) are currently spending more than 6 hours per day online – a nearly 500% increase from before the crisis.
While glasses or contact lenses can correct a child’s vision, research in the journal Retina by the Ikuno Eye Center in Osaka, Japan, suggests that having severe myopia puts children at risk for a number of eye problems down the road, including retinal detachment, glaucoma, and macular degeneration. Research published in PLOS ONE found that working up close – such as reading or using a tablet – increased the odds of myopia. And in a report published in Ophthalmology, researchers studying myopia have estimated that, by 2050, about half the world’s population could be myopic.
Watch for warning signs
To determine if excessive screen time is affecting your child, there warning signs to watch out for including the following:
- Eye irritation.
- Watery eyes.
- Headaches.
- Intermittent blurry vision or double vision.
- Sore eyes.
- Difficulty concentrating.
- Sore neck, shoulders, and/or back.
- Increased sensitivity to light.
- Tiredness.
- Poor posture.
Use these approaches to reduce eye fatigue
To reduce eye fatigue associated with excessive screen exposure, parents should set limits on screen time if possible, and at minimum, schedule between 8 and 15 hours of outdoor activity per week. Screens should be at least 20 centimeters away from the child’s eyes, the child’s desk should be placed near a window, and she should be instructed to look outside every hour. Lastly, children should be kept in fully corrected glasses, not under corrected, and those glasses need to be worn full time.
As we plan the future of education in the age of COVID-19, I firmly believe that schools and policymakers must consider children’s vision needs while designing new initiatives. Schools, teachers, and parents must work together to incorporate eye health strategies that protect children as they learn online. By doing so, we can decrease the chance that a child’s vision becomes compromised from unwittingly staring into a screen for too many hours during the day.
Dr. Faktorovich is an ophthalmologist, founder of Pacific Vision Institute in San Francisco, California, and founder of the annual San Francisco Cornea, Cataract, and Refractive Surgery Symposium. She had no relevant financial disclosures. Email her at pdnews@mdedge.com.
A teen girl presents with a pinkish-red bump on her right leg
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6

The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.

Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6
The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.
Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6
The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.
Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
2020-2021 respiratory viral season: Onset, presentations, and testing likely to differ in pandemic
Respiratory virus seasons usually follow a fairly well-known pattern. Enterovirus 68 (EV-D68) is a summer-to-early fall virus with biennial peak years. Rhinovirus (HRv) and adenovirus (Adv) occur nearly year-round but may have small upticks in the first month or so that children return to school. Early in the school year, upper respiratory infections from both HRv and Adv and viral sore throats from Adv are common, with conjunctivitis from Adv outbreaks in some years. October to November is human parainfluenza (HPiV) 1 and 2 season, often presenting as croup. Human metapneumovirus infections span October through April. In late November to December, influenza begins, usually with an A type, later transitioning to a B type in February through April. Also in December, respiratory syncytial virus (RSV) starts, characteristically with bronchiolitis presentations, peaking in February to March and tapering off in May. In late March to April, HPiV 3 also appears for 4-6 weeks.

Will 2020-2021 be different?
Summer was remarkably free of expected enterovirus activity, suggesting that the seasonal parade may differ this year. Remember that the 2019-2020 respiratory season suddenly and nearly completely stopped in March because of social distancing and lockdowns needed to address the SARS-CoV-2 pandemic.
The mild influenza season in the southern hemisphere suggests that our influenza season also could be mild. But perhaps not – most southern hemisphere countries that are surveyed for influenza activities had the most intense SARS-CoV-2 mitigations, making the observed mildness potentially related more to social mitigation than less virulent influenza strains. If so, southern hemisphere influenza data may not apply to the United States, where social distancing and masks are ignored or used inconsistently by almost half the population.

Further, the stop-and-go pattern of in-person school/college attendance adds to uncertainties for the usual orderly virus-specific seasonality. The result may be multiple stop-and-go “pop-up” or “mini” outbreaks for any given virus potentially reflected as exaggerated local or regional differences in circulation of various viruses. The erratic seasonality also would increase coinfections, which could present with more severe or different symptoms.
SARS-CoV-2’s potential interaction
Will the relatively mild presentations for most children with SARS-CoV-2 hold up in the setting of coinfections or sequential respiratory viral infections? Could SARS-CoV-2 cause worse/more prolonged symptoms or more sequelae if paired simultaneously or in tandem with a traditional respiratory virus? To date, data on the frequency and severity of SARS-CoV-2 coinfections are conflicting and sparse, but it appears that non-SARS-CoV-2 viruses can be involved in 15%-50% pediatric acute respiratory infections.1,2
However, it may not be important to know about coinfecting viruses other than influenza (can be treated) or SARS-CoV-2 (needs quarantine and contact tracing), unless symptoms are atypical or more severe than usual. For example, a young child with bronchiolitis is most likely infected with RSV, but HPiV, influenza, metapneumovirus, HRv, and even SARS-CoV-2 can cause bronchiolitis. Even so, testing outpatients for RSV or non-influenza is not routine or even clinically helpful. Supportive treatment and restriction from daycare attendance are sufficient management for outpatient ARIs whether presenting as bronchiolitis or not.
Considerations for SARS-CoV-2 testing: Outpatient bronchiolitis
If a child presents with classic bronchiolitis but has above moderate to severe symptoms, is SARS-CoV-2 a consideration? Perhaps, if SARS-CoV-2 acts similarly to non-SARS-CoV-2s.
A recent report from the 30th Multicenter Airway Research Collaboration (MARC-30) surveillance study (2007-2014) of children hospitalized with clinical bronchiolitis evaluated respiratory viruses, including RSV and the four common non-SARS coronaviruses using molecular testing.3 Among 1,880 subjects, a CoV (alpha CoV: NL63 or 229E, or beta CoV: KKU1 or OC43) was detected in 12%. Yet most had only RSV (n = 1,661); 32 had only CoV (n = 32). But note that 219 had both.
Bronchiolitis subjects with CoV were older – median 3.7 (1.4-5.8) vs. 2.8 (1.9-7.2) years – and more likely male than were RSV subjects (68% vs. 58%). OC43 was most frequent followed by equal numbers of HKU1 and NL63, while 229E was the least frequent. Medical utilization and severity did not differ among the CoVs, or between RSV+CoV vs. RSV alone, unless one considered CoV viral load as a variable. ICU use increased when the polymerase chain reaction cycle threshold result indicated a high CoV viral load.
These data suggest CoVs are not infrequent coinfectors with RSV in bronchiolitis – and that SARS-CoV-2 is the same. Therefore, a bronchiolitis presentation doesn’t necessarily take us off the hook for the need to consider SARS-CoV-2 testing, particularly in the somewhat older bronchiolitis patient with more than mild symptoms.
Considerations for SARS-CoV-2 testing: Outpatient influenza-like illness
In 2020-2021, the Centers for Disease Control and Prevention recommends considering empiric antiviral treatment for ILIs (fever plus either cough or sore throat) based upon our clinical judgement, even in non-high-risk children.4
While pediatric COVID-19 illnesses are predominantly asymptomatic or mild, a febrile ARI is also a SARS-CoV-2 compatible presentation. So, if all we use is our clinical judgment, how do we know if the febrile ARI is due to influenza or SARS-CoV-2 or both? At least one study used a highly sensitive and specific molecular influenza test to show that the accuracy of clinically diagnosing influenza in children is not much better than flipping a coin and would lead to potential antiviral overuse.5
So, it seems ideal to test for influenza when possible. Point-of-care (POC) tests are frequently used for outpatients. Eight POC Clinical Laboratory Improvement Amendments (CLIA)–waived kits, some also detecting RSV, are available but most have modest sensitivity (60%-80%) compared with lab-based molecular tests.6 That said, if supplies and kits for one of the POC tests are available to us during these SARS-CoV-2 stressed times (back orders seem more common this year), a positive influenza test in the first 48 hours of symptoms confirms the option to prescribe an antiviral. Yet how will we have confidence that the febrile ARI is not also partly due to SARS-CoV-2? Currently febrile ARIs usually are considered SARS-CoV-2 and the children are sent for SARS-CoV-2 testing. During influenza season, it seems we will need to continue to send febrile outpatients for SARS-CoV-2 testing, even if POC influenza positive, via whatever mechanisms are available as time goes on.
We expect more rapid pediatric testing modalities for SARS-CoV-2 (maybe even saliva tests) to become available over the next months. Indeed, rapid antigen tests and rapid molecular tests are being evaluated in adults and seem destined for CLIA waivers as POC tests, and even home testing kits. Pediatric approvals hopefully also will occur. So, the pathways for SARS-CoV-2 testing available now will likely change over this winter. But be aware that supplies/kits will be prioritized to locations within high need areas and bulk purchase contracts. So POC kits may remain scarce for practices, meaning a reference laboratory still could be the way to go for SARS-CoV-2 for at least the rest of 2020. Reference labs are becoming creative as well; one combined detection of influenza A, influenza B, RSV, and SARS-CoV-2 into one test, and hopes to get approval for swab collection that can be done by families at home and mailed in.
Summary
Expect variations on the traditional parade of seasonal respiratory viruses, with increased numbers of coinfections. Choosing the outpatient who needs influenza testing is the same as in past years, although we have CDC permissive recommendations to prescribe antivirals for any outpatient ILI within the first 48 hours of symptoms. Still, POC testing for influenza remains potentially valuable in the ILI patient. The choice of whether and how to test for SARS-CoV-2 given its potential to be a primary or coinfecting agent in presentations linked more closely to a traditional virus (e.g. RSV bronchiolitis) will be a test of our clinical judgement until more data and easier testing are available. Further complicating coinfection recognition is the fact that many sick visits occur by telehealth and much testing is done at drive-through SARS-CoV-2 testing facilities with no clinician exam. Unless we are liberal in SARS-CoV-2 testing, detecting SARS-CoV-2 coinfections is easier said than done given its usually mild presentation being overshadowed by any coinfecting virus.
But understanding who has SARS-CoV-2, even as a coinfection, still is essential in controlling the pandemic. We will need to be vigilant for evolving approaches to SARS-CoV-2 testing in the context of symptomatic ARI presentations, knowing this will likely remain a moving target for the foreseeable future.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatrics. 2020;146(1):e20200961.
2. JAMA. 2020 May 26;323(20):2085-6.
3. Pediatrics. 2020. doi: 10.1542/peds.2020-1267.
4. www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm.
5. J. Pediatr. 2020. doi: 10.1016/j.jpeds.2020.08.007.
6. www.cdc.gov/flu/professionals/diagnosis/table-nucleic-acid-detection.html.
Respiratory virus seasons usually follow a fairly well-known pattern. Enterovirus 68 (EV-D68) is a summer-to-early fall virus with biennial peak years. Rhinovirus (HRv) and adenovirus (Adv) occur nearly year-round but may have small upticks in the first month or so that children return to school. Early in the school year, upper respiratory infections from both HRv and Adv and viral sore throats from Adv are common, with conjunctivitis from Adv outbreaks in some years. October to November is human parainfluenza (HPiV) 1 and 2 season, often presenting as croup. Human metapneumovirus infections span October through April. In late November to December, influenza begins, usually with an A type, later transitioning to a B type in February through April. Also in December, respiratory syncytial virus (RSV) starts, characteristically with bronchiolitis presentations, peaking in February to March and tapering off in May. In late March to April, HPiV 3 also appears for 4-6 weeks.
Will 2020-2021 be different?
Summer was remarkably free of expected enterovirus activity, suggesting that the seasonal parade may differ this year. Remember that the 2019-2020 respiratory season suddenly and nearly completely stopped in March because of social distancing and lockdowns needed to address the SARS-CoV-2 pandemic.
The mild influenza season in the southern hemisphere suggests that our influenza season also could be mild. But perhaps not – most southern hemisphere countries that are surveyed for influenza activities had the most intense SARS-CoV-2 mitigations, making the observed mildness potentially related more to social mitigation than less virulent influenza strains. If so, southern hemisphere influenza data may not apply to the United States, where social distancing and masks are ignored or used inconsistently by almost half the population.
Further, the stop-and-go pattern of in-person school/college attendance adds to uncertainties for the usual orderly virus-specific seasonality. The result may be multiple stop-and-go “pop-up” or “mini” outbreaks for any given virus potentially reflected as exaggerated local or regional differences in circulation of various viruses. The erratic seasonality also would increase coinfections, which could present with more severe or different symptoms.
SARS-CoV-2’s potential interaction
Will the relatively mild presentations for most children with SARS-CoV-2 hold up in the setting of coinfections or sequential respiratory viral infections? Could SARS-CoV-2 cause worse/more prolonged symptoms or more sequelae if paired simultaneously or in tandem with a traditional respiratory virus? To date, data on the frequency and severity of SARS-CoV-2 coinfections are conflicting and sparse, but it appears that non-SARS-CoV-2 viruses can be involved in 15%-50% pediatric acute respiratory infections.1,2
However, it may not be important to know about coinfecting viruses other than influenza (can be treated) or SARS-CoV-2 (needs quarantine and contact tracing), unless symptoms are atypical or more severe than usual. For example, a young child with bronchiolitis is most likely infected with RSV, but HPiV, influenza, metapneumovirus, HRv, and even SARS-CoV-2 can cause bronchiolitis. Even so, testing outpatients for RSV or non-influenza is not routine or even clinically helpful. Supportive treatment and restriction from daycare attendance are sufficient management for outpatient ARIs whether presenting as bronchiolitis or not.
Considerations for SARS-CoV-2 testing: Outpatient bronchiolitis
If a child presents with classic bronchiolitis but has above moderate to severe symptoms, is SARS-CoV-2 a consideration? Perhaps, if SARS-CoV-2 acts similarly to non-SARS-CoV-2s.
A recent report from the 30th Multicenter Airway Research Collaboration (MARC-30) surveillance study (2007-2014) of children hospitalized with clinical bronchiolitis evaluated respiratory viruses, including RSV and the four common non-SARS coronaviruses using molecular testing.3 Among 1,880 subjects, a CoV (alpha CoV: NL63 or 229E, or beta CoV: KKU1 or OC43) was detected in 12%. Yet most had only RSV (n = 1,661); 32 had only CoV (n = 32). But note that 219 had both.
Bronchiolitis subjects with CoV were older – median 3.7 (1.4-5.8) vs. 2.8 (1.9-7.2) years – and more likely male than were RSV subjects (68% vs. 58%). OC43 was most frequent followed by equal numbers of HKU1 and NL63, while 229E was the least frequent. Medical utilization and severity did not differ among the CoVs, or between RSV+CoV vs. RSV alone, unless one considered CoV viral load as a variable. ICU use increased when the polymerase chain reaction cycle threshold result indicated a high CoV viral load.
These data suggest CoVs are not infrequent coinfectors with RSV in bronchiolitis – and that SARS-CoV-2 is the same. Therefore, a bronchiolitis presentation doesn’t necessarily take us off the hook for the need to consider SARS-CoV-2 testing, particularly in the somewhat older bronchiolitis patient with more than mild symptoms.
Considerations for SARS-CoV-2 testing: Outpatient influenza-like illness
In 2020-2021, the Centers for Disease Control and Prevention recommends considering empiric antiviral treatment for ILIs (fever plus either cough or sore throat) based upon our clinical judgement, even in non-high-risk children.4
While pediatric COVID-19 illnesses are predominantly asymptomatic or mild, a febrile ARI is also a SARS-CoV-2 compatible presentation. So, if all we use is our clinical judgment, how do we know if the febrile ARI is due to influenza or SARS-CoV-2 or both? At least one study used a highly sensitive and specific molecular influenza test to show that the accuracy of clinically diagnosing influenza in children is not much better than flipping a coin and would lead to potential antiviral overuse.5
So, it seems ideal to test for influenza when possible. Point-of-care (POC) tests are frequently used for outpatients. Eight POC Clinical Laboratory Improvement Amendments (CLIA)–waived kits, some also detecting RSV, are available but most have modest sensitivity (60%-80%) compared with lab-based molecular tests.6 That said, if supplies and kits for one of the POC tests are available to us during these SARS-CoV-2 stressed times (back orders seem more common this year), a positive influenza test in the first 48 hours of symptoms confirms the option to prescribe an antiviral. Yet how will we have confidence that the febrile ARI is not also partly due to SARS-CoV-2? Currently febrile ARIs usually are considered SARS-CoV-2 and the children are sent for SARS-CoV-2 testing. During influenza season, it seems we will need to continue to send febrile outpatients for SARS-CoV-2 testing, even if POC influenza positive, via whatever mechanisms are available as time goes on.
We expect more rapid pediatric testing modalities for SARS-CoV-2 (maybe even saliva tests) to become available over the next months. Indeed, rapid antigen tests and rapid molecular tests are being evaluated in adults and seem destined for CLIA waivers as POC tests, and even home testing kits. Pediatric approvals hopefully also will occur. So, the pathways for SARS-CoV-2 testing available now will likely change over this winter. But be aware that supplies/kits will be prioritized to locations within high need areas and bulk purchase contracts. So POC kits may remain scarce for practices, meaning a reference laboratory still could be the way to go for SARS-CoV-2 for at least the rest of 2020. Reference labs are becoming creative as well; one combined detection of influenza A, influenza B, RSV, and SARS-CoV-2 into one test, and hopes to get approval for swab collection that can be done by families at home and mailed in.
Summary
Expect variations on the traditional parade of seasonal respiratory viruses, with increased numbers of coinfections. Choosing the outpatient who needs influenza testing is the same as in past years, although we have CDC permissive recommendations to prescribe antivirals for any outpatient ILI within the first 48 hours of symptoms. Still, POC testing for influenza remains potentially valuable in the ILI patient. The choice of whether and how to test for SARS-CoV-2 given its potential to be a primary or coinfecting agent in presentations linked more closely to a traditional virus (e.g. RSV bronchiolitis) will be a test of our clinical judgement until more data and easier testing are available. Further complicating coinfection recognition is the fact that many sick visits occur by telehealth and much testing is done at drive-through SARS-CoV-2 testing facilities with no clinician exam. Unless we are liberal in SARS-CoV-2 testing, detecting SARS-CoV-2 coinfections is easier said than done given its usually mild presentation being overshadowed by any coinfecting virus.
But understanding who has SARS-CoV-2, even as a coinfection, still is essential in controlling the pandemic. We will need to be vigilant for evolving approaches to SARS-CoV-2 testing in the context of symptomatic ARI presentations, knowing this will likely remain a moving target for the foreseeable future.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatrics. 2020;146(1):e20200961.
2. JAMA. 2020 May 26;323(20):2085-6.
3. Pediatrics. 2020. doi: 10.1542/peds.2020-1267.
4. www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm.
5. J. Pediatr. 2020. doi: 10.1016/j.jpeds.2020.08.007.
6. www.cdc.gov/flu/professionals/diagnosis/table-nucleic-acid-detection.html.
Respiratory virus seasons usually follow a fairly well-known pattern. Enterovirus 68 (EV-D68) is a summer-to-early fall virus with biennial peak years. Rhinovirus (HRv) and adenovirus (Adv) occur nearly year-round but may have small upticks in the first month or so that children return to school. Early in the school year, upper respiratory infections from both HRv and Adv and viral sore throats from Adv are common, with conjunctivitis from Adv outbreaks in some years. October to November is human parainfluenza (HPiV) 1 and 2 season, often presenting as croup. Human metapneumovirus infections span October through April. In late November to December, influenza begins, usually with an A type, later transitioning to a B type in February through April. Also in December, respiratory syncytial virus (RSV) starts, characteristically with bronchiolitis presentations, peaking in February to March and tapering off in May. In late March to April, HPiV 3 also appears for 4-6 weeks.
Will 2020-2021 be different?
Summer was remarkably free of expected enterovirus activity, suggesting that the seasonal parade may differ this year. Remember that the 2019-2020 respiratory season suddenly and nearly completely stopped in March because of social distancing and lockdowns needed to address the SARS-CoV-2 pandemic.
The mild influenza season in the southern hemisphere suggests that our influenza season also could be mild. But perhaps not – most southern hemisphere countries that are surveyed for influenza activities had the most intense SARS-CoV-2 mitigations, making the observed mildness potentially related more to social mitigation than less virulent influenza strains. If so, southern hemisphere influenza data may not apply to the United States, where social distancing and masks are ignored or used inconsistently by almost half the population.
Further, the stop-and-go pattern of in-person school/college attendance adds to uncertainties for the usual orderly virus-specific seasonality. The result may be multiple stop-and-go “pop-up” or “mini” outbreaks for any given virus potentially reflected as exaggerated local or regional differences in circulation of various viruses. The erratic seasonality also would increase coinfections, which could present with more severe or different symptoms.
SARS-CoV-2’s potential interaction
Will the relatively mild presentations for most children with SARS-CoV-2 hold up in the setting of coinfections or sequential respiratory viral infections? Could SARS-CoV-2 cause worse/more prolonged symptoms or more sequelae if paired simultaneously or in tandem with a traditional respiratory virus? To date, data on the frequency and severity of SARS-CoV-2 coinfections are conflicting and sparse, but it appears that non-SARS-CoV-2 viruses can be involved in 15%-50% pediatric acute respiratory infections.1,2
However, it may not be important to know about coinfecting viruses other than influenza (can be treated) or SARS-CoV-2 (needs quarantine and contact tracing), unless symptoms are atypical or more severe than usual. For example, a young child with bronchiolitis is most likely infected with RSV, but HPiV, influenza, metapneumovirus, HRv, and even SARS-CoV-2 can cause bronchiolitis. Even so, testing outpatients for RSV or non-influenza is not routine or even clinically helpful. Supportive treatment and restriction from daycare attendance are sufficient management for outpatient ARIs whether presenting as bronchiolitis or not.
Considerations for SARS-CoV-2 testing: Outpatient bronchiolitis
If a child presents with classic bronchiolitis but has above moderate to severe symptoms, is SARS-CoV-2 a consideration? Perhaps, if SARS-CoV-2 acts similarly to non-SARS-CoV-2s.
A recent report from the 30th Multicenter Airway Research Collaboration (MARC-30) surveillance study (2007-2014) of children hospitalized with clinical bronchiolitis evaluated respiratory viruses, including RSV and the four common non-SARS coronaviruses using molecular testing.3 Among 1,880 subjects, a CoV (alpha CoV: NL63 or 229E, or beta CoV: KKU1 or OC43) was detected in 12%. Yet most had only RSV (n = 1,661); 32 had only CoV (n = 32). But note that 219 had both.
Bronchiolitis subjects with CoV were older – median 3.7 (1.4-5.8) vs. 2.8 (1.9-7.2) years – and more likely male than were RSV subjects (68% vs. 58%). OC43 was most frequent followed by equal numbers of HKU1 and NL63, while 229E was the least frequent. Medical utilization and severity did not differ among the CoVs, or between RSV+CoV vs. RSV alone, unless one considered CoV viral load as a variable. ICU use increased when the polymerase chain reaction cycle threshold result indicated a high CoV viral load.
These data suggest CoVs are not infrequent coinfectors with RSV in bronchiolitis – and that SARS-CoV-2 is the same. Therefore, a bronchiolitis presentation doesn’t necessarily take us off the hook for the need to consider SARS-CoV-2 testing, particularly in the somewhat older bronchiolitis patient with more than mild symptoms.
Considerations for SARS-CoV-2 testing: Outpatient influenza-like illness
In 2020-2021, the Centers for Disease Control and Prevention recommends considering empiric antiviral treatment for ILIs (fever plus either cough or sore throat) based upon our clinical judgement, even in non-high-risk children.4
While pediatric COVID-19 illnesses are predominantly asymptomatic or mild, a febrile ARI is also a SARS-CoV-2 compatible presentation. So, if all we use is our clinical judgment, how do we know if the febrile ARI is due to influenza or SARS-CoV-2 or both? At least one study used a highly sensitive and specific molecular influenza test to show that the accuracy of clinically diagnosing influenza in children is not much better than flipping a coin and would lead to potential antiviral overuse.5
So, it seems ideal to test for influenza when possible. Point-of-care (POC) tests are frequently used for outpatients. Eight POC Clinical Laboratory Improvement Amendments (CLIA)–waived kits, some also detecting RSV, are available but most have modest sensitivity (60%-80%) compared with lab-based molecular tests.6 That said, if supplies and kits for one of the POC tests are available to us during these SARS-CoV-2 stressed times (back orders seem more common this year), a positive influenza test in the first 48 hours of symptoms confirms the option to prescribe an antiviral. Yet how will we have confidence that the febrile ARI is not also partly due to SARS-CoV-2? Currently febrile ARIs usually are considered SARS-CoV-2 and the children are sent for SARS-CoV-2 testing. During influenza season, it seems we will need to continue to send febrile outpatients for SARS-CoV-2 testing, even if POC influenza positive, via whatever mechanisms are available as time goes on.
We expect more rapid pediatric testing modalities for SARS-CoV-2 (maybe even saliva tests) to become available over the next months. Indeed, rapid antigen tests and rapid molecular tests are being evaluated in adults and seem destined for CLIA waivers as POC tests, and even home testing kits. Pediatric approvals hopefully also will occur. So, the pathways for SARS-CoV-2 testing available now will likely change over this winter. But be aware that supplies/kits will be prioritized to locations within high need areas and bulk purchase contracts. So POC kits may remain scarce for practices, meaning a reference laboratory still could be the way to go for SARS-CoV-2 for at least the rest of 2020. Reference labs are becoming creative as well; one combined detection of influenza A, influenza B, RSV, and SARS-CoV-2 into one test, and hopes to get approval for swab collection that can be done by families at home and mailed in.
Summary
Expect variations on the traditional parade of seasonal respiratory viruses, with increased numbers of coinfections. Choosing the outpatient who needs influenza testing is the same as in past years, although we have CDC permissive recommendations to prescribe antivirals for any outpatient ILI within the first 48 hours of symptoms. Still, POC testing for influenza remains potentially valuable in the ILI patient. The choice of whether and how to test for SARS-CoV-2 given its potential to be a primary or coinfecting agent in presentations linked more closely to a traditional virus (e.g. RSV bronchiolitis) will be a test of our clinical judgement until more data and easier testing are available. Further complicating coinfection recognition is the fact that many sick visits occur by telehealth and much testing is done at drive-through SARS-CoV-2 testing facilities with no clinician exam. Unless we are liberal in SARS-CoV-2 testing, detecting SARS-CoV-2 coinfections is easier said than done given its usually mild presentation being overshadowed by any coinfecting virus.
But understanding who has SARS-CoV-2, even as a coinfection, still is essential in controlling the pandemic. We will need to be vigilant for evolving approaches to SARS-CoV-2 testing in the context of symptomatic ARI presentations, knowing this will likely remain a moving target for the foreseeable future.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatrics. 2020;146(1):e20200961.
2. JAMA. 2020 May 26;323(20):2085-6.
3. Pediatrics. 2020. doi: 10.1542/peds.2020-1267.
4. www.cdc.gov/flu/professionals/antivirals/summary-clinicians.htm.
5. J. Pediatr. 2020. doi: 10.1016/j.jpeds.2020.08.007.
6. www.cdc.gov/flu/professionals/diagnosis/table-nucleic-acid-detection.html.
Reassuring findings on SSRIs and diabetes risk in children
SSRIs are associated with a much lower risk of type 2 diabetes (T2D) in children and adolescents than previously reported, new research shows.
Investigators found publicly insured patients treated with SSRIs had a 13% increased risk for T2D, compared with those not treated with these agents. In addition, those taking SSRIs continuously (defined as receiving one or more prescriptions every 3 months) had a 33% increased risk of T2D.
On the other hand, privately insured youth had a much lower increased risk – a finding that may be attributable to a lower prevalence of risk factors for T2D in this group.
“We cannot exclude that children and adolescents treated with SSRIs may be at a small increased risk of developing T2D, particularly publicly insured patients, but the magnitude of association was weaker than previous thought and much smaller than other known risk factors for T2DM, such as obesity, race, and poverty,” lead investigator Jenny Sun, PhD, said in an interview.
“When weighing the known benefits and risks of SSRI treatment in children and adolescents, our findings provide reassurance that the risk of T2DM is not as substantial as initially reported,” said Dr. Sun, a postdoctoral research fellow in the department of population medicine at Harvard Medical School’s Harvard Pilgrim Health Care Institute, Boston.
The study was published online Sept. 2 in JAMA Psychiatry.
Limited evidence
Previous research suggested that SSRIs increase the risk of T2D by up to 90% in children and adolescents.
However, the investigators noted, the study reporting this finding was too small to draw conclusions about the SSRI class as a whole also did not examine specific SSRIs.
In addition, although “several studies have reported that antidepressant use may be a risk factor for T2D in adults, evidence was limited in children and adolescents,” said Dr. Sun.
“Rapid changes in growth during childhood and adolescents can alter drugs’ pharmacokinetics and pharmacodynamics, so high-quality, age-specific data are needed to inform prescribing decisions,” she said.
For the current study, the researchers analyzed claims data on almost 1.6 million patients aged 10-19 years (58.3% female; mean age, 15.1 years) from two large claims databases.
The analysis focused on those with a diagnosis warranting treatment with an SSRI, including depression, generalized or social anxiety disorder, obsessive compulsive disorder, PTSD, panic disorder, or bulimia nervosa.
The Medicaid Analytic Extract database consisted of 316,178 patients insured through Medicaid or the Children’s Health Insurance Program. The IBM MarketScan database consisted of 211,460 privately insured patients. Patients were followed up for a mean of 2.3 and 2.2 years, respectively.
Patients who initiated SSRI treatment were compared with those with a similar indication but who were not taking an SSRI. Secondary analyses compared new SSRI users with patients who recently initiated treatment with bupropion, which has no metabolic side effects, or with patients who recently initiated psychotherapy.
“In observational data, it is difficult to mimic a placebo group, often used in RCTs [randomized, controlled trials], therefore several comparator groups were explored to broaden our understanding,” said Dr. Sun.
In addition, the researchers compared the individual SSRI medications, using fluoxetine as a comparator.
A wide range of more than 100 potential confounders or “proxies of confounders,” were taken into account, including demographic characteristics, psychiatric diagnoses, metabolic conditions, concomitant medications, and use of health care services.
The researchers conducted two analyses. They included an intention-to-treat (ITT) analysis that was restricted to patients with one or more additional SSRI prescriptions during the 6 months following the index exposure assessment period.
Close monitoring required
An as-treated analysis estimated the association of continuous SSRI treatment (vs. untreated, bupropion treatment, and psychotherapy), with adherence assessed at 3-month intervals.
Initiation and continuation of SSRI treatment in publicly insured patients were both associated with a considerably higher risk of T2D, compared with untreated patients, and a steeper risk, compared with their privately insured counterparts.
For newly treated publicly insured patients initiated on SSRI treatment, the ITT adjusted hazard ratio was 1.13 (95% confidence interval, 1.04-1.22).
There was an even stronger association among continuously treated publicly insured patients, with an as-treated aHR of 1.33 (95% CI, 1.21-1.47). The authors noted that this corresponds to 6.6 additional T2D cases per 10,000 patients continuously treated for at least 2 years.
The association was weaker in privately insured patients (ITT aHR, 1.01; 95% CI, 0.84-1.23; as-treated aHR, 1.10; 95% CI, 0.88-1.36).
The secondary analyses yielded similar findings: When SSRI treatment was compared with psychotherapy, the as-treated aHR for publicly insured patients was 1.44 (95% CI, 1.25-1.65), whereas the aHR for privately insured patients was lower at 1.21 (95% CI, 0.93-1.57)
The investigators found no increased risk when SSRIs were compared with bupropion, and the within-class analysis showed that none of the SSRIs carried an increased hazard of T2D, compared with fluoxetine.
“Publicly insured patients are enrolled in Medicaid and the Children’s Health Insurance Program, whereas privately insured patients are generally covered by their parent’s employer-sponsored insurance,” said Dr. Sun.
“Publicly insured patients are of lower socioeconomic status and represent a population with greater overall medical burden, more comorbidities, and a higher prevalence of risk factors for T2D, such as obesity, at the time of treatment initiation,” she said.
She added that high-risk children and youth should be closely monitored and clinicians should also consider recommending dietary modifications and increased exercise to offset T2D risk.
Useful ‘real-world data’
William Cooper, MD, MPH, professor of pediatrics and health policy at Vanderbilt University Medical Center in Nashville, Tenn., said that the study “provides a fascinating look at risks of SSRI medications in children and adolescents.”
Dr. Cooper, who was not involved with the study, said that the authors “draw from real-world data representing two different populations and carefully consider factors which might confound the associations.”
The results, he said, “provide important benefits for patients, families, and clinicians as they weigh the risks and benefits of using SSRIs for children who need treatment for depression and anxiety disorders.
The study was supported by a training grant from the program in pharmacoepidemiology at the Harvard School of Public Health. Dr. Sun disclosed no relevant financial relationships. Dr. Cooper disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
SSRIs are associated with a much lower risk of type 2 diabetes (T2D) in children and adolescents than previously reported, new research shows.
Investigators found publicly insured patients treated with SSRIs had a 13% increased risk for T2D, compared with those not treated with these agents. In addition, those taking SSRIs continuously (defined as receiving one or more prescriptions every 3 months) had a 33% increased risk of T2D.
On the other hand, privately insured youth had a much lower increased risk – a finding that may be attributable to a lower prevalence of risk factors for T2D in this group.
“We cannot exclude that children and adolescents treated with SSRIs may be at a small increased risk of developing T2D, particularly publicly insured patients, but the magnitude of association was weaker than previous thought and much smaller than other known risk factors for T2DM, such as obesity, race, and poverty,” lead investigator Jenny Sun, PhD, said in an interview.
“When weighing the known benefits and risks of SSRI treatment in children and adolescents, our findings provide reassurance that the risk of T2DM is not as substantial as initially reported,” said Dr. Sun, a postdoctoral research fellow in the department of population medicine at Harvard Medical School’s Harvard Pilgrim Health Care Institute, Boston.
The study was published online Sept. 2 in JAMA Psychiatry.
Limited evidence
Previous research suggested that SSRIs increase the risk of T2D by up to 90% in children and adolescents.
However, the investigators noted, the study reporting this finding was too small to draw conclusions about the SSRI class as a whole also did not examine specific SSRIs.
In addition, although “several studies have reported that antidepressant use may be a risk factor for T2D in adults, evidence was limited in children and adolescents,” said Dr. Sun.
“Rapid changes in growth during childhood and adolescents can alter drugs’ pharmacokinetics and pharmacodynamics, so high-quality, age-specific data are needed to inform prescribing decisions,” she said.
For the current study, the researchers analyzed claims data on almost 1.6 million patients aged 10-19 years (58.3% female; mean age, 15.1 years) from two large claims databases.
The analysis focused on those with a diagnosis warranting treatment with an SSRI, including depression, generalized or social anxiety disorder, obsessive compulsive disorder, PTSD, panic disorder, or bulimia nervosa.
The Medicaid Analytic Extract database consisted of 316,178 patients insured through Medicaid or the Children’s Health Insurance Program. The IBM MarketScan database consisted of 211,460 privately insured patients. Patients were followed up for a mean of 2.3 and 2.2 years, respectively.
Patients who initiated SSRI treatment were compared with those with a similar indication but who were not taking an SSRI. Secondary analyses compared new SSRI users with patients who recently initiated treatment with bupropion, which has no metabolic side effects, or with patients who recently initiated psychotherapy.
“In observational data, it is difficult to mimic a placebo group, often used in RCTs [randomized, controlled trials], therefore several comparator groups were explored to broaden our understanding,” said Dr. Sun.
In addition, the researchers compared the individual SSRI medications, using fluoxetine as a comparator.
A wide range of more than 100 potential confounders or “proxies of confounders,” were taken into account, including demographic characteristics, psychiatric diagnoses, metabolic conditions, concomitant medications, and use of health care services.
The researchers conducted two analyses. They included an intention-to-treat (ITT) analysis that was restricted to patients with one or more additional SSRI prescriptions during the 6 months following the index exposure assessment period.
Close monitoring required
An as-treated analysis estimated the association of continuous SSRI treatment (vs. untreated, bupropion treatment, and psychotherapy), with adherence assessed at 3-month intervals.
Initiation and continuation of SSRI treatment in publicly insured patients were both associated with a considerably higher risk of T2D, compared with untreated patients, and a steeper risk, compared with their privately insured counterparts.
For newly treated publicly insured patients initiated on SSRI treatment, the ITT adjusted hazard ratio was 1.13 (95% confidence interval, 1.04-1.22).
There was an even stronger association among continuously treated publicly insured patients, with an as-treated aHR of 1.33 (95% CI, 1.21-1.47). The authors noted that this corresponds to 6.6 additional T2D cases per 10,000 patients continuously treated for at least 2 years.
The association was weaker in privately insured patients (ITT aHR, 1.01; 95% CI, 0.84-1.23; as-treated aHR, 1.10; 95% CI, 0.88-1.36).
The secondary analyses yielded similar findings: When SSRI treatment was compared with psychotherapy, the as-treated aHR for publicly insured patients was 1.44 (95% CI, 1.25-1.65), whereas the aHR for privately insured patients was lower at 1.21 (95% CI, 0.93-1.57)
The investigators found no increased risk when SSRIs were compared with bupropion, and the within-class analysis showed that none of the SSRIs carried an increased hazard of T2D, compared with fluoxetine.
“Publicly insured patients are enrolled in Medicaid and the Children’s Health Insurance Program, whereas privately insured patients are generally covered by their parent’s employer-sponsored insurance,” said Dr. Sun.
“Publicly insured patients are of lower socioeconomic status and represent a population with greater overall medical burden, more comorbidities, and a higher prevalence of risk factors for T2D, such as obesity, at the time of treatment initiation,” she said.
She added that high-risk children and youth should be closely monitored and clinicians should also consider recommending dietary modifications and increased exercise to offset T2D risk.
Useful ‘real-world data’
William Cooper, MD, MPH, professor of pediatrics and health policy at Vanderbilt University Medical Center in Nashville, Tenn., said that the study “provides a fascinating look at risks of SSRI medications in children and adolescents.”
Dr. Cooper, who was not involved with the study, said that the authors “draw from real-world data representing two different populations and carefully consider factors which might confound the associations.”
The results, he said, “provide important benefits for patients, families, and clinicians as they weigh the risks and benefits of using SSRIs for children who need treatment for depression and anxiety disorders.
The study was supported by a training grant from the program in pharmacoepidemiology at the Harvard School of Public Health. Dr. Sun disclosed no relevant financial relationships. Dr. Cooper disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
SSRIs are associated with a much lower risk of type 2 diabetes (T2D) in children and adolescents than previously reported, new research shows.
Investigators found publicly insured patients treated with SSRIs had a 13% increased risk for T2D, compared with those not treated with these agents. In addition, those taking SSRIs continuously (defined as receiving one or more prescriptions every 3 months) had a 33% increased risk of T2D.
On the other hand, privately insured youth had a much lower increased risk – a finding that may be attributable to a lower prevalence of risk factors for T2D in this group.
“We cannot exclude that children and adolescents treated with SSRIs may be at a small increased risk of developing T2D, particularly publicly insured patients, but the magnitude of association was weaker than previous thought and much smaller than other known risk factors for T2DM, such as obesity, race, and poverty,” lead investigator Jenny Sun, PhD, said in an interview.
“When weighing the known benefits and risks of SSRI treatment in children and adolescents, our findings provide reassurance that the risk of T2DM is not as substantial as initially reported,” said Dr. Sun, a postdoctoral research fellow in the department of population medicine at Harvard Medical School’s Harvard Pilgrim Health Care Institute, Boston.
The study was published online Sept. 2 in JAMA Psychiatry.
Limited evidence
Previous research suggested that SSRIs increase the risk of T2D by up to 90% in children and adolescents.
However, the investigators noted, the study reporting this finding was too small to draw conclusions about the SSRI class as a whole also did not examine specific SSRIs.
In addition, although “several studies have reported that antidepressant use may be a risk factor for T2D in adults, evidence was limited in children and adolescents,” said Dr. Sun.
“Rapid changes in growth during childhood and adolescents can alter drugs’ pharmacokinetics and pharmacodynamics, so high-quality, age-specific data are needed to inform prescribing decisions,” she said.
For the current study, the researchers analyzed claims data on almost 1.6 million patients aged 10-19 years (58.3% female; mean age, 15.1 years) from two large claims databases.
The analysis focused on those with a diagnosis warranting treatment with an SSRI, including depression, generalized or social anxiety disorder, obsessive compulsive disorder, PTSD, panic disorder, or bulimia nervosa.
The Medicaid Analytic Extract database consisted of 316,178 patients insured through Medicaid or the Children’s Health Insurance Program. The IBM MarketScan database consisted of 211,460 privately insured patients. Patients were followed up for a mean of 2.3 and 2.2 years, respectively.
Patients who initiated SSRI treatment were compared with those with a similar indication but who were not taking an SSRI. Secondary analyses compared new SSRI users with patients who recently initiated treatment with bupropion, which has no metabolic side effects, or with patients who recently initiated psychotherapy.
“In observational data, it is difficult to mimic a placebo group, often used in RCTs [randomized, controlled trials], therefore several comparator groups were explored to broaden our understanding,” said Dr. Sun.
In addition, the researchers compared the individual SSRI medications, using fluoxetine as a comparator.
A wide range of more than 100 potential confounders or “proxies of confounders,” were taken into account, including demographic characteristics, psychiatric diagnoses, metabolic conditions, concomitant medications, and use of health care services.
The researchers conducted two analyses. They included an intention-to-treat (ITT) analysis that was restricted to patients with one or more additional SSRI prescriptions during the 6 months following the index exposure assessment period.
Close monitoring required
An as-treated analysis estimated the association of continuous SSRI treatment (vs. untreated, bupropion treatment, and psychotherapy), with adherence assessed at 3-month intervals.
Initiation and continuation of SSRI treatment in publicly insured patients were both associated with a considerably higher risk of T2D, compared with untreated patients, and a steeper risk, compared with their privately insured counterparts.
For newly treated publicly insured patients initiated on SSRI treatment, the ITT adjusted hazard ratio was 1.13 (95% confidence interval, 1.04-1.22).
There was an even stronger association among continuously treated publicly insured patients, with an as-treated aHR of 1.33 (95% CI, 1.21-1.47). The authors noted that this corresponds to 6.6 additional T2D cases per 10,000 patients continuously treated for at least 2 years.
The association was weaker in privately insured patients (ITT aHR, 1.01; 95% CI, 0.84-1.23; as-treated aHR, 1.10; 95% CI, 0.88-1.36).
The secondary analyses yielded similar findings: When SSRI treatment was compared with psychotherapy, the as-treated aHR for publicly insured patients was 1.44 (95% CI, 1.25-1.65), whereas the aHR for privately insured patients was lower at 1.21 (95% CI, 0.93-1.57)
The investigators found no increased risk when SSRIs were compared with bupropion, and the within-class analysis showed that none of the SSRIs carried an increased hazard of T2D, compared with fluoxetine.
“Publicly insured patients are enrolled in Medicaid and the Children’s Health Insurance Program, whereas privately insured patients are generally covered by their parent’s employer-sponsored insurance,” said Dr. Sun.
“Publicly insured patients are of lower socioeconomic status and represent a population with greater overall medical burden, more comorbidities, and a higher prevalence of risk factors for T2D, such as obesity, at the time of treatment initiation,” she said.
She added that high-risk children and youth should be closely monitored and clinicians should also consider recommending dietary modifications and increased exercise to offset T2D risk.
Useful ‘real-world data’
William Cooper, MD, MPH, professor of pediatrics and health policy at Vanderbilt University Medical Center in Nashville, Tenn., said that the study “provides a fascinating look at risks of SSRI medications in children and adolescents.”
Dr. Cooper, who was not involved with the study, said that the authors “draw from real-world data representing two different populations and carefully consider factors which might confound the associations.”
The results, he said, “provide important benefits for patients, families, and clinicians as they weigh the risks and benefits of using SSRIs for children who need treatment for depression and anxiety disorders.
The study was supported by a training grant from the program in pharmacoepidemiology at the Harvard School of Public Health. Dr. Sun disclosed no relevant financial relationships. Dr. Cooper disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Children and COVID-19: New cases may be leveling off
Growth in new pediatric COVID-19 cases has evened out in recent weeks, but children now represent 10% of all COVID-19 cases in the United States, and that measurement has been rising throughout the pandemic, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and the CHA said in the report, based on data from 49 states (New York City is included but not New York state), the District of Columbia, Puerto Rico, and Guam.
The weekly percentage of increase in the number of new cases has not reached double digits since early August and has been no higher than 7.8% over the last 3 weeks. The number of child COVID-19 cases, however, has finally reached 10% of the total for Americans of all ages, which stands at 5.49 million in the jurisdictions included in the report, the AHA and CHA reported.
Measures, however, continue to show low levels of severe illness in children, they noted, including the following:
- Child cases as a proportion of all COVID-19 hospitalizations: 1.7%.
- Hospitalization rate for children: 1.8%.
- Child deaths as a proportion of all deaths: 0.07%.
- Percent of child cases resulting in death: 0.01%.
The number of cumulative cases per 100,000 children is now up to 728.5 nationally, with a range by state that goes from 154.0 in Vermont to 1,670.3 in Tennessee, which is one of only two states reporting cases in those aged 0-20 years as children (the other is South Carolina). The age range for children is 0-17 or 0-19 for most other states, although Florida uses a range of 0-14, the report notes.
Other than Tennessee, there are 10 states with overall rates higher than 1,000 COVID-19 cases per 100,000 children, and there are nine states with cumulative totals over 15,000 cases (California is the highest with just over 75,000), according to the report.
Growth in new pediatric COVID-19 cases has evened out in recent weeks, but children now represent 10% of all COVID-19 cases in the United States, and that measurement has been rising throughout the pandemic, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and the CHA said in the report, based on data from 49 states (New York City is included but not New York state), the District of Columbia, Puerto Rico, and Guam.
The weekly percentage of increase in the number of new cases has not reached double digits since early August and has been no higher than 7.8% over the last 3 weeks. The number of child COVID-19 cases, however, has finally reached 10% of the total for Americans of all ages, which stands at 5.49 million in the jurisdictions included in the report, the AHA and CHA reported.
Measures, however, continue to show low levels of severe illness in children, they noted, including the following:
- Child cases as a proportion of all COVID-19 hospitalizations: 1.7%.
- Hospitalization rate for children: 1.8%.
- Child deaths as a proportion of all deaths: 0.07%.
- Percent of child cases resulting in death: 0.01%.
The number of cumulative cases per 100,000 children is now up to 728.5 nationally, with a range by state that goes from 154.0 in Vermont to 1,670.3 in Tennessee, which is one of only two states reporting cases in those aged 0-20 years as children (the other is South Carolina). The age range for children is 0-17 or 0-19 for most other states, although Florida uses a range of 0-14, the report notes.
Other than Tennessee, there are 10 states with overall rates higher than 1,000 COVID-19 cases per 100,000 children, and there are nine states with cumulative totals over 15,000 cases (California is the highest with just over 75,000), according to the report.
Growth in new pediatric COVID-19 cases has evened out in recent weeks, but children now represent 10% of all COVID-19 cases in the United States, and that measurement has been rising throughout the pandemic, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
the AAP and the CHA said in the report, based on data from 49 states (New York City is included but not New York state), the District of Columbia, Puerto Rico, and Guam.
The weekly percentage of increase in the number of new cases has not reached double digits since early August and has been no higher than 7.8% over the last 3 weeks. The number of child COVID-19 cases, however, has finally reached 10% of the total for Americans of all ages, which stands at 5.49 million in the jurisdictions included in the report, the AHA and CHA reported.
Measures, however, continue to show low levels of severe illness in children, they noted, including the following:
- Child cases as a proportion of all COVID-19 hospitalizations: 1.7%.
- Hospitalization rate for children: 1.8%.
- Child deaths as a proportion of all deaths: 0.07%.
- Percent of child cases resulting in death: 0.01%.
The number of cumulative cases per 100,000 children is now up to 728.5 nationally, with a range by state that goes from 154.0 in Vermont to 1,670.3 in Tennessee, which is one of only two states reporting cases in those aged 0-20 years as children (the other is South Carolina). The age range for children is 0-17 or 0-19 for most other states, although Florida uses a range of 0-14, the report notes.
Other than Tennessee, there are 10 states with overall rates higher than 1,000 COVID-19 cases per 100,000 children, and there are nine states with cumulative totals over 15,000 cases (California is the highest with just over 75,000), according to the report.