User login
What is your diagnosis?
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.

There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.

There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.
There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.
There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
Epidermal nevi are a subset of cutaneous hamartomas resulting from somatic mutations of epidermal cells, presenting as keratinocyte or epidermal appendage overgrowths. The most common type appear in a linear distribution and are termed linear epidermal nevi or linear verrucous epidermal nevi.
There are variations of epidermal nevi (EN) that can be composed of superficial epidermal keratinocytes, sebaceous glands, apocrine or eccrine glands, hair follicles, or smooth muscle. For example, many consider a nevus sebaceous to be a type of epidermal nevus as well. The incidence of EN is approximately 1 in 1,000 newborns. Postzygotic cell mutations result in a mosaic distribution that follows embryonic migration patterns, appearing in a Blaschkoid distribution.
EN present most frequently as unilateral linear or whorled hyperpigmented coalescing papules. The lesions can be present at birth or during childhood, and after appearing, grow with the patient. Typically the lesions become more raised and verrucous around puberty. The differential diagnosis of linear EN include lichen striatus, warts, and incontinentia pigmenti. Lichen striatus can be differentiated because it presents later in life and self-resolves. Verrucae are the most commonly mistaken diagnosis for EN; warts do not usually persist in the same pattern over time with proportionate growth and typically respond to locally destructive treatments such as liquid nitrogen, unlike EN. Incontinentia pigmenti presents as vesicles initially and shows a quick evolution, differentiating it from EN. Inflammatory linear verrucous epidermal nevus (ILVEN) is a variant of linear EN that has associated chronic and intermittent erythema, scale, and pruritus. Lichen nitidus often has a pruritic presentation; however, it is flat topped and skin colored, helping differentiate it from linear EN.
There has been recent research advancing gene associations for linear EN displaying many lesions associated with mosaic mutations in oncogenes. Multiple genes have been identified with EN including RAS, FGFR3, and PIK3CA1. FGFR3 and PIK3CA mutations are associated with 50% of keratinocytic nevi. Of the RAS family, the HRAS pathway has been most closely associated with nevus sebaceous. While KRAS and NRAS genes have been associated with EN, it is to a lesser degree. However, there are multiple recent case reports demonstrating a potential association of G12D mosaicism of the KRAS gene in EN with rhabdomyosarcoma and bladder cancers2.
The diagnosis of epidermal nevus syndrome should be considered when there is a nevus with associated developmental abnormality of the central nervous system, eyes, or musculoskeletal systems. The most common systemic symptoms include delays in developmental milestones, seizure disorders, coloboma, strabismus, muscle weakness, and hemihypertrophy. To date, there are six specific epidermal nevus syndromes identified: sebaceous nevus syndrome, nevus comedonicus syndrome, Becker nevus syndrome, phakomatosis pigmentokeratotica, Proteus syndrome, congenital hemidysplasia with ichthyosiform nevus and limb defects, and cutaneous-skeletal hypophosphatemia syndrome3. In addition to the syndromes described, there are reports of associations between keratinocytic nevi and ILVEN with hypophosphatemic rickets and precocious puberty.
Linear EN are rarely associated with malignant transformation to basal cell carcinoma or squamous cell carcinoma, depending on the cell type involved. Given the low risk of malignancy, the lesions do not need to be removed routinely. For small lesions, monitoring often is the preferred management. However, lesions with functional significance, or causing strangulation or deformity, can be treated with surgical excision, curettage, or laser destruction
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Pediatr Dermatol. 2004 Jul-Aug;21(4):432-9.
2. J Med Genet. 2010 Dec;47(12):859-62.
3. Pediatr Dermatol. 2018 Jan;35(1):21-9.
A 6-year-old, otherwise-healthy male is brought into clinic for evaluation of papules on his neck. The rash has been present since 1 year of age and has been growing in size proportionately. He claims there is occasional itching but no pain or redness. He does not seem to be disturbed by his rash. He has two siblings, aged 2 and 4 years, without lesions.
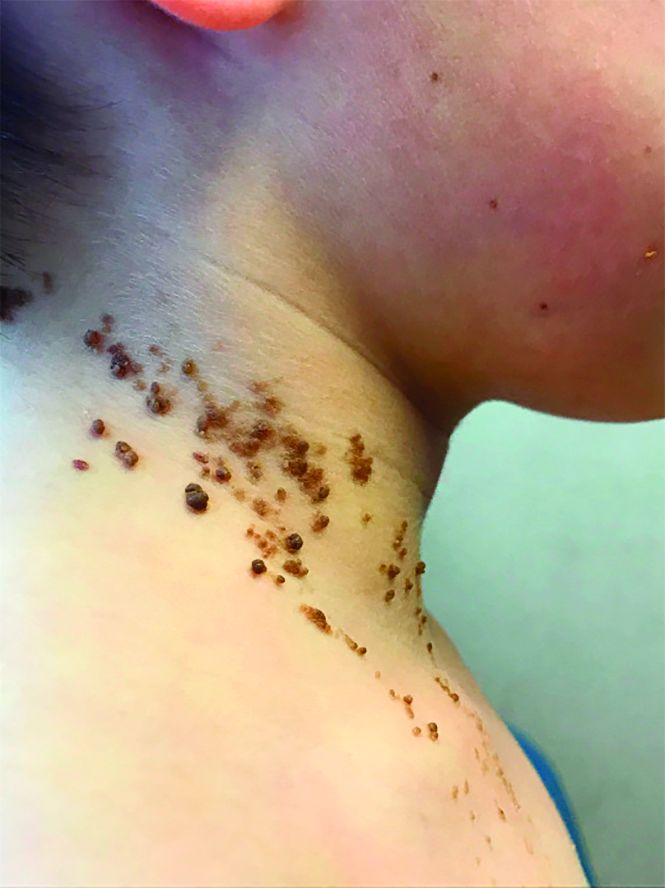
On physical exam, he is noted to have a linear plaque of hyperpigmented verrucous papules on his neck.
Antipsychotic use in young people tied to 80% increased risk of death
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
This study by Wayne A. Ray, PhD, and his colleagues addresses the risks of antipsychotic use in childhood while highlighting the contradictions in how psychiatrically ill children are treated and medicated, according to Barbara Geller, MD, of the department of psychiatry at Washington University in St. Louis.
Before commenting on the study itself, Dr. Geller noted that child psychiatry is not a subspecialty that deals with “little patients and little problems,” despite that lingering perception among some. “Fifty percent of psychiatry disorders begin by age 14 years,” she wrote, “and childhood age at onset is a risk factor for a more severe longitudinal course in mood and other disorders.”
In addition, though it seems instinctually that antipsychotic medications would have lesser side effects on healthy children, that is not always the case. “The opposite is true for certain metabolic and endocrine effects,” she explained, “such as relatively greater weight gain and prolactin level elevation than adults and the onset of type 2 diabetes within the first year of treatment.”
When it came to the study, Dr. Geller posed questions about the findings, including whether an increase in unexpected deaths among the higher-dose group could be attributed to suicide. She also recommended that future investigations “examine outcomes within child, adolescent, and young adult age subgroups, as opposed to combining all youth 6 to 24 years old.”
That said, this research does probe depths that require continued exploration. “Results in the study by Ray et al. heighten the already increased caution about prescribing antipsychotics to children and adolescents,” she wrote, “and emphasize the need to consider situational triggers of psychopathology to avoid medicating the environment.”
These comments are adapted from an accompanying editorial (JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3409). No conflicts of interest were reported.
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
Children and young people who received antipsychotic doses higher than 50-mg chlorpromazine equivalents had an 80% increased risk of death at follow-up, compared with a control group, according to a study of young Medicaid enrollees who recently had begun medication.
“The study findings seem to reinforce existing guidelines for improving the outcomes of antipsychotic therapy in children and youths,” wrote lead author Wayne A. Ray, PhD, of the department of health policy at the Vanderbilt University in Nashville, Tenn., and his coauthors. Those guidelines include using “psychosocial interventions when possible, cardiometabolic assessment before treatment and monitoring after treatment, and limiting therapy to the lowest dose and shortest duration possible,” they wrote.
The study, published online in JAMA Psychiatry, analyzed children and young adults from Tennessee, aged 5-24 years, who were new medication users, and had been enrolled in Medicaid between 1999 and 2014.
They were split into three groups: a control group (189,361) with users primarily taking attention-deficit/hyperactivity disorder medications and antidepressants; a group (28,377) with users who received antipsychotic doses of 50 mg or less chlorpromazine equivalents; and a group (30,120) with users who received doses higher than 50-mg chlorpromazine equivalents.
At follow-up, the incidence of death in the higher-dose group was 146.2 per 100,000 person-years (95% confidence interval, 107.3-199.4 per 100,000 person-years), compared with 49.5 in the lower-dose group (95% CI, 24.8-99.0) and 54.5 in the control group (95% CI, 42.9-69.2). This difference was attributed to unexpected deaths, which accounted for 52.5% of deaths in the higher-dose group. No increased risk of death was noted for injuries or suicides. “The elevated risk persisted for unexpected deaths not due to overdose, with a 4.3-fold increased risk of death from cardiovascular or metabolic causes,” Dr. Ray and his coauthors wrote.
The authors shared potential limitations of their study, including a relatively small number of deaths during follow-up and subsequent statistical adjustment during analysis. They also recognized that their data did not factor in important characteristics such as body mass index and family history, and that a “single-state Medicaid cohort may limit the study’s generalizability.”
Nonetheless, they emphasized Medicaid’s relevance as coverage provider for an estimated 39% of U.S. children, along with noting that
“Further studies are needed that compare antipsychotic users and controls within more narrow comorbidity ranges or in analyses that include richer clinical data,” they wrote.
The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
SOURCE: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
FROM JAMA PSYCHIATRY
Key clinical point: Children and youths who received higher doses of antipsychotic medication had an 80% increased risk of death, compared with those in a control group.
Major finding: The incidence of unexpected death was 76.8 per 100,000 person-years in the higher-dose group, compared with 17.9 per 100,000 person-years in the control group.
Study details: A retrospective cohort study of Medicaid-enrolled children and young adults from Tennessee, aged 5-24 years, who were new users of antipsychotic or control medications.
Disclosures: The study was supported by grants from the National Heart, Lung, and Blood Institute, and the National Institute for Child Health and Human Development. No conflicts of interest were reported.
Source: Ray WA et al. JAMA Psychiatry. 2018 Dec 12. doi: 10.1001/jamapsychiatry.2018.3421.
Preliminary data suggest UCART19 is safe, effective

SAN DIEGO—Preliminary data on UCART19—the first off-the-shelf, anti-CD19, allogeneic chimeric antigen receptor (CAR) T-cell therapy—suggest it can produce complete responses (CRs) and minimal residual disease (MRD) negativity, and side effects are manageable.
Investigators pooled data from the phase 1 pediatric (PALL) and adult (CALM) trials of UCART19 in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) and observed a 67% CR rate in the overall population and an 82% CR rate in patients who received a three-drug lymphodepleting regimen.
Additionally, investigators reported no instance of moderate or severe acute graft-versus-host disease (GVHD) with UCART19.
“We’ve been blessed with the new treatments that have emerged in recent years,” said Reuben Benjamin, MD, PhD, “that include BiTEs, antibody-drug conjugates, and most excitingly, the autologous CAR T-cell therapies.”
Nevertheless, some logistical issues with the autologous CAR T cells leave an unmet need in this group of patients, he noted.
“So an off-the-shelf approach using a product like UCART19 may potentially overcome some of these hurdles that we see in the autologous CAR T-cell therapy field,” he said.
Dr. Benjamin, of King’s College Hospital in London, U.K., presented the analysis of PALL and CALM data at the 2018 ASH Annual Meeting as abstract 896.*
UCART19 product
UCART19 is an allogeneic, genetically modified, CAR T-cell product (anti-CD19 scFv- 41BB-CD3ζ) manufactured from healthy donor T cells.
It has a safety switch—RQR8, which is a CD20 mimotope—that allows the CAR T cells to be targeted by rituximab.
“And importantly,” Dr. Benjamin explained, “the T-cell alpha gene has been knocked out using TALEN® gene-editing technology to prevent T-cell receptor-mediated graft-versus-host disease.”
The CD52 gene is also knocked out, which permits an anti-CD52 monoclonal antibody, such as alemtuzumab, to be used in lymphodepletion.
Study design
The primary objective of both the adult (NCT02746952) and pediatric (NCT02808442) studies was to determine the safety and tolerability of UCART19. Also, the adult study was to determine the maximum tolerated dose of UCART19 and the optimal lymphodepleting regimen.
A secondary objective of both studies was to determine the remission rate at day 28.
Eligible patients received a lymphodepleting regimen for 7 days, followed by a single infusion of UCART19.
Lymphodepletion in the pediatric trial consisted of fludarabine (F) at 150 mg/m2 and cyclophosphamide (C) at 120 mg/kg, with or without alemtuzumab (A) at 1 mg/kg capped at 40 mg.
Adults received lower doses of each agent—90 mg/m2, 1,500 mg/m2, and (optionally) 1 mg/kg or 40 mg, respectively.
Investigators included alemtuzumab in the regimen to minimize viral infections.
The UCART19 dose was weight-banded in the pediatric trial and ranged from 1.1 to 2.3 x 106 cells/kg.
The adult trial included three UCART19 dose levels:
- 6 x 106 cells (≈1 x 105 cells/kg)
- 6 or 8 x 107 cells (≈1 x 106 cells/kg)
- 8 or 2.4 x 108 cells (≈3 x 106 cells/kg).
Patients were assessed for safety and response at day 28 and regularly thereafter for up to 12 months. Patients had the option during the follow-up period to receive a second dose if they did not respond or lost their response.
Patient characteristics/status
Twenty-one patients were enrolled in the trials—seven children and 14 adults. Median ages were 2.7 years (PALL; range, 0.8–16.4) and 29.5 years (CALM; range, 18–62).
Both studies included high-risk, heavily pretreated populations, Dr. Benjamin noted.
The pooled population had a median of 4 prior lines of therapy (range, 1–6), and nine patients had a high-risk cytogenetics, including complex karyotypes, MLL rearrangements, and Ph+ disease.
Thirteen patients had prior allogeneic stem cell transplants.
Nine patients had a bone marrow tumor burden of more than 25% blasts prior to lymphodepletion.
As of the cutoff date of October 23, all patients had been treated with UCART19.
Four of the pediatric patients are still on the trial. Two are in remission, one has relapsed, and one is refractory.
Eight adult patients are still on trial. Three are in remission, three are relapsed, and two are refractory.
Safety
“UCART19 appears to show an acceptable safety profile based on the adverse events reported so far,” Dr. Benjamin said.
Nineteen patients experienced cytokine release syndrome (CRS), primarily grades 1 and 2. Eight patients had grade 1 and 2 neurotoxicity events, and two patients had grade 1 acute skin GVHD.
“In keeping with what is seen in some of the autologous CAR T-cell trials,” Dr. Benjamin explained, “prolonged cytopenias were seen, which we defined in these studies as grade 4 neutropenia or thrombocytopenia occurring at 42 days post-UCART infusion.”
Six of 21 patients developed prolonged cytopenia.
There was also an increased incidence of viral infections occurring in eight patients, including cytomegalovirus, adenovirus, BK virus, and metapneumovirus.
“Most of these infections, however, were manageable,” Dr. Benjamin said.
Two patients developed neutropenic sepsis, one grade 5, which was one of the treatment-related deaths in the CALM trial.
No treatment-related deaths occurred in the PALL study, but there were two in the CALM study—one from pulmonary hemorrhage and the other from neutropenic sepsis and grade 4 CRS.
Twelve patients are still alive, five of whom are in CR.
Efficacy
Of the patients who received FCA lymphodepletion, 82% (14/17) achieved CR/CR with incomplete hematologic recovery (CRi), and 71% (10/14) achieved MRD negativity.
An additional patient gained MRD-negative status after the second dose of UCART19.
Of the 14 patients who achieved a CR/CRi, 78% (n=11) went on to receive an allogeneic transplant.
In the entire pooled population, 67% (14/21) achieved CR/CRi.
Three patients received a second UCART19 dose, and five patients remain in CR/CRi.
UCART19 expansion
UCART19 expansion, as measured by quantitative polymerase chain reaction in PALL and flow-based methods in CALM, occurred primarily in the first 28 days in the FCA-treated population.
Investigators observed expansion in 15 of 17 patients treated with FCA. None of the patients who received FC alone (n=4) had expansion detectable in blood or bone marrow, Dr. Benjamin noted.
“The response we’ve seen in the study so far,” Dr. Benjamin clarified, “is linked to the expansion observed within the first 28-day period.”
UCART cells persisted in three patients beyond day 42. In one patient, they persisted up to day 120.
“Of interest is the T-cell recovery seen in the study,” Dr. Benjamin elaborated. “We only have data from the adult study here—14 patients. And you’ll see that, in the FCA-treated arm (n=11), you have a deeper and more sustained lymphodepletion compared to the FC-treated patients (n=3). And this may play a role in the subsequent UCART19 expansion and disease response.”
Re-dosing
Of the three patients who were re-dosed, two achieved MRD negativity.
One patient achieved MRD-negative status at day 28 but relapsed and received a second infusion 3 months after the first dose. The second expansion was not as deep as the first, but the patient nevertheless achieved MRD negativity after the second dose.
The second patient received FC lymphodepletion and was refractory at day 28.
“The second time around, he received FCA, had a slightly better expansion, and achieved molecular remission,” Dr. Benjamin said.
And the third patient had FCA lymphodepletion but was refractory at day 28.
“We elected to give a second dose at 2.4 months later, but unfortunately, there wasn’t very much expansion, even the second time around, and the patient progressed,” Dr. Benjamin said.
FCA lymphodepletion appears to be required for UCART19 expansion. There was no UCART19 expansion and no response in all four patients lymphodepleted with FC.
The evaluation of UCART19 is ongoing in pediatric and adult B-cell ALL, and “there is a plan for moving into the lymphoma space as well,” Dr. Benjamin added.
Dr. Benjamin disclosed honoraria from Amgen, Takeda, Novartis, Gilead, and Celgene, and research funding from Servier and Pfizer.
Servier and Allogene are supporting the UCART19 trials.
*Data in the abstract differ from the presentation.
SAN DIEGO—Preliminary data on UCART19—the first off-the-shelf, anti-CD19, allogeneic chimeric antigen receptor (CAR) T-cell therapy—suggest it can produce complete responses (CRs) and minimal residual disease (MRD) negativity, and side effects are manageable.
Investigators pooled data from the phase 1 pediatric (PALL) and adult (CALM) trials of UCART19 in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) and observed a 67% CR rate in the overall population and an 82% CR rate in patients who received a three-drug lymphodepleting regimen.
Additionally, investigators reported no instance of moderate or severe acute graft-versus-host disease (GVHD) with UCART19.
“We’ve been blessed with the new treatments that have emerged in recent years,” said Reuben Benjamin, MD, PhD, “that include BiTEs, antibody-drug conjugates, and most excitingly, the autologous CAR T-cell therapies.”
Nevertheless, some logistical issues with the autologous CAR T cells leave an unmet need in this group of patients, he noted.
“So an off-the-shelf approach using a product like UCART19 may potentially overcome some of these hurdles that we see in the autologous CAR T-cell therapy field,” he said.
Dr. Benjamin, of King’s College Hospital in London, U.K., presented the analysis of PALL and CALM data at the 2018 ASH Annual Meeting as abstract 896.*
UCART19 product
UCART19 is an allogeneic, genetically modified, CAR T-cell product (anti-CD19 scFv- 41BB-CD3ζ) manufactured from healthy donor T cells.
It has a safety switch—RQR8, which is a CD20 mimotope—that allows the CAR T cells to be targeted by rituximab.
“And importantly,” Dr. Benjamin explained, “the T-cell alpha gene has been knocked out using TALEN® gene-editing technology to prevent T-cell receptor-mediated graft-versus-host disease.”
The CD52 gene is also knocked out, which permits an anti-CD52 monoclonal antibody, such as alemtuzumab, to be used in lymphodepletion.
Study design
The primary objective of both the adult (NCT02746952) and pediatric (NCT02808442) studies was to determine the safety and tolerability of UCART19. Also, the adult study was to determine the maximum tolerated dose of UCART19 and the optimal lymphodepleting regimen.
A secondary objective of both studies was to determine the remission rate at day 28.
Eligible patients received a lymphodepleting regimen for 7 days, followed by a single infusion of UCART19.
Lymphodepletion in the pediatric trial consisted of fludarabine (F) at 150 mg/m2 and cyclophosphamide (C) at 120 mg/kg, with or without alemtuzumab (A) at 1 mg/kg capped at 40 mg.
Adults received lower doses of each agent—90 mg/m2, 1,500 mg/m2, and (optionally) 1 mg/kg or 40 mg, respectively.
Investigators included alemtuzumab in the regimen to minimize viral infections.
The UCART19 dose was weight-banded in the pediatric trial and ranged from 1.1 to 2.3 x 106 cells/kg.
The adult trial included three UCART19 dose levels:
- 6 x 106 cells (≈1 x 105 cells/kg)
- 6 or 8 x 107 cells (≈1 x 106 cells/kg)
- 8 or 2.4 x 108 cells (≈3 x 106 cells/kg).
Patients were assessed for safety and response at day 28 and regularly thereafter for up to 12 months. Patients had the option during the follow-up period to receive a second dose if they did not respond or lost their response.
Patient characteristics/status
Twenty-one patients were enrolled in the trials—seven children and 14 adults. Median ages were 2.7 years (PALL; range, 0.8–16.4) and 29.5 years (CALM; range, 18–62).
Both studies included high-risk, heavily pretreated populations, Dr. Benjamin noted.
The pooled population had a median of 4 prior lines of therapy (range, 1–6), and nine patients had a high-risk cytogenetics, including complex karyotypes, MLL rearrangements, and Ph+ disease.
Thirteen patients had prior allogeneic stem cell transplants.
Nine patients had a bone marrow tumor burden of more than 25% blasts prior to lymphodepletion.
As of the cutoff date of October 23, all patients had been treated with UCART19.
Four of the pediatric patients are still on the trial. Two are in remission, one has relapsed, and one is refractory.
Eight adult patients are still on trial. Three are in remission, three are relapsed, and two are refractory.
Safety
“UCART19 appears to show an acceptable safety profile based on the adverse events reported so far,” Dr. Benjamin said.
Nineteen patients experienced cytokine release syndrome (CRS), primarily grades 1 and 2. Eight patients had grade 1 and 2 neurotoxicity events, and two patients had grade 1 acute skin GVHD.
“In keeping with what is seen in some of the autologous CAR T-cell trials,” Dr. Benjamin explained, “prolonged cytopenias were seen, which we defined in these studies as grade 4 neutropenia or thrombocytopenia occurring at 42 days post-UCART infusion.”
Six of 21 patients developed prolonged cytopenia.
There was also an increased incidence of viral infections occurring in eight patients, including cytomegalovirus, adenovirus, BK virus, and metapneumovirus.
“Most of these infections, however, were manageable,” Dr. Benjamin said.
Two patients developed neutropenic sepsis, one grade 5, which was one of the treatment-related deaths in the CALM trial.
No treatment-related deaths occurred in the PALL study, but there were two in the CALM study—one from pulmonary hemorrhage and the other from neutropenic sepsis and grade 4 CRS.
Twelve patients are still alive, five of whom are in CR.
Efficacy
Of the patients who received FCA lymphodepletion, 82% (14/17) achieved CR/CR with incomplete hematologic recovery (CRi), and 71% (10/14) achieved MRD negativity.
An additional patient gained MRD-negative status after the second dose of UCART19.
Of the 14 patients who achieved a CR/CRi, 78% (n=11) went on to receive an allogeneic transplant.
In the entire pooled population, 67% (14/21) achieved CR/CRi.
Three patients received a second UCART19 dose, and five patients remain in CR/CRi.
UCART19 expansion
UCART19 expansion, as measured by quantitative polymerase chain reaction in PALL and flow-based methods in CALM, occurred primarily in the first 28 days in the FCA-treated population.
Investigators observed expansion in 15 of 17 patients treated with FCA. None of the patients who received FC alone (n=4) had expansion detectable in blood or bone marrow, Dr. Benjamin noted.
“The response we’ve seen in the study so far,” Dr. Benjamin clarified, “is linked to the expansion observed within the first 28-day period.”
UCART cells persisted in three patients beyond day 42. In one patient, they persisted up to day 120.
“Of interest is the T-cell recovery seen in the study,” Dr. Benjamin elaborated. “We only have data from the adult study here—14 patients. And you’ll see that, in the FCA-treated arm (n=11), you have a deeper and more sustained lymphodepletion compared to the FC-treated patients (n=3). And this may play a role in the subsequent UCART19 expansion and disease response.”
Re-dosing
Of the three patients who were re-dosed, two achieved MRD negativity.
One patient achieved MRD-negative status at day 28 but relapsed and received a second infusion 3 months after the first dose. The second expansion was not as deep as the first, but the patient nevertheless achieved MRD negativity after the second dose.
The second patient received FC lymphodepletion and was refractory at day 28.
“The second time around, he received FCA, had a slightly better expansion, and achieved molecular remission,” Dr. Benjamin said.
And the third patient had FCA lymphodepletion but was refractory at day 28.
“We elected to give a second dose at 2.4 months later, but unfortunately, there wasn’t very much expansion, even the second time around, and the patient progressed,” Dr. Benjamin said.
FCA lymphodepletion appears to be required for UCART19 expansion. There was no UCART19 expansion and no response in all four patients lymphodepleted with FC.
The evaluation of UCART19 is ongoing in pediatric and adult B-cell ALL, and “there is a plan for moving into the lymphoma space as well,” Dr. Benjamin added.
Dr. Benjamin disclosed honoraria from Amgen, Takeda, Novartis, Gilead, and Celgene, and research funding from Servier and Pfizer.
Servier and Allogene are supporting the UCART19 trials.
*Data in the abstract differ from the presentation.
SAN DIEGO—Preliminary data on UCART19—the first off-the-shelf, anti-CD19, allogeneic chimeric antigen receptor (CAR) T-cell therapy—suggest it can produce complete responses (CRs) and minimal residual disease (MRD) negativity, and side effects are manageable.
Investigators pooled data from the phase 1 pediatric (PALL) and adult (CALM) trials of UCART19 in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) and observed a 67% CR rate in the overall population and an 82% CR rate in patients who received a three-drug lymphodepleting regimen.
Additionally, investigators reported no instance of moderate or severe acute graft-versus-host disease (GVHD) with UCART19.
“We’ve been blessed with the new treatments that have emerged in recent years,” said Reuben Benjamin, MD, PhD, “that include BiTEs, antibody-drug conjugates, and most excitingly, the autologous CAR T-cell therapies.”
Nevertheless, some logistical issues with the autologous CAR T cells leave an unmet need in this group of patients, he noted.
“So an off-the-shelf approach using a product like UCART19 may potentially overcome some of these hurdles that we see in the autologous CAR T-cell therapy field,” he said.
Dr. Benjamin, of King’s College Hospital in London, U.K., presented the analysis of PALL and CALM data at the 2018 ASH Annual Meeting as abstract 896.*
UCART19 product
UCART19 is an allogeneic, genetically modified, CAR T-cell product (anti-CD19 scFv- 41BB-CD3ζ) manufactured from healthy donor T cells.
It has a safety switch—RQR8, which is a CD20 mimotope—that allows the CAR T cells to be targeted by rituximab.
“And importantly,” Dr. Benjamin explained, “the T-cell alpha gene has been knocked out using TALEN® gene-editing technology to prevent T-cell receptor-mediated graft-versus-host disease.”
The CD52 gene is also knocked out, which permits an anti-CD52 monoclonal antibody, such as alemtuzumab, to be used in lymphodepletion.
Study design
The primary objective of both the adult (NCT02746952) and pediatric (NCT02808442) studies was to determine the safety and tolerability of UCART19. Also, the adult study was to determine the maximum tolerated dose of UCART19 and the optimal lymphodepleting regimen.
A secondary objective of both studies was to determine the remission rate at day 28.
Eligible patients received a lymphodepleting regimen for 7 days, followed by a single infusion of UCART19.
Lymphodepletion in the pediatric trial consisted of fludarabine (F) at 150 mg/m2 and cyclophosphamide (C) at 120 mg/kg, with or without alemtuzumab (A) at 1 mg/kg capped at 40 mg.
Adults received lower doses of each agent—90 mg/m2, 1,500 mg/m2, and (optionally) 1 mg/kg or 40 mg, respectively.
Investigators included alemtuzumab in the regimen to minimize viral infections.
The UCART19 dose was weight-banded in the pediatric trial and ranged from 1.1 to 2.3 x 106 cells/kg.
The adult trial included three UCART19 dose levels:
- 6 x 106 cells (≈1 x 105 cells/kg)
- 6 or 8 x 107 cells (≈1 x 106 cells/kg)
- 8 or 2.4 x 108 cells (≈3 x 106 cells/kg).
Patients were assessed for safety and response at day 28 and regularly thereafter for up to 12 months. Patients had the option during the follow-up period to receive a second dose if they did not respond or lost their response.
Patient characteristics/status
Twenty-one patients were enrolled in the trials—seven children and 14 adults. Median ages were 2.7 years (PALL; range, 0.8–16.4) and 29.5 years (CALM; range, 18–62).
Both studies included high-risk, heavily pretreated populations, Dr. Benjamin noted.
The pooled population had a median of 4 prior lines of therapy (range, 1–6), and nine patients had a high-risk cytogenetics, including complex karyotypes, MLL rearrangements, and Ph+ disease.
Thirteen patients had prior allogeneic stem cell transplants.
Nine patients had a bone marrow tumor burden of more than 25% blasts prior to lymphodepletion.
As of the cutoff date of October 23, all patients had been treated with UCART19.
Four of the pediatric patients are still on the trial. Two are in remission, one has relapsed, and one is refractory.
Eight adult patients are still on trial. Three are in remission, three are relapsed, and two are refractory.
Safety
“UCART19 appears to show an acceptable safety profile based on the adverse events reported so far,” Dr. Benjamin said.
Nineteen patients experienced cytokine release syndrome (CRS), primarily grades 1 and 2. Eight patients had grade 1 and 2 neurotoxicity events, and two patients had grade 1 acute skin GVHD.
“In keeping with what is seen in some of the autologous CAR T-cell trials,” Dr. Benjamin explained, “prolonged cytopenias were seen, which we defined in these studies as grade 4 neutropenia or thrombocytopenia occurring at 42 days post-UCART infusion.”
Six of 21 patients developed prolonged cytopenia.
There was also an increased incidence of viral infections occurring in eight patients, including cytomegalovirus, adenovirus, BK virus, and metapneumovirus.
“Most of these infections, however, were manageable,” Dr. Benjamin said.
Two patients developed neutropenic sepsis, one grade 5, which was one of the treatment-related deaths in the CALM trial.
No treatment-related deaths occurred in the PALL study, but there were two in the CALM study—one from pulmonary hemorrhage and the other from neutropenic sepsis and grade 4 CRS.
Twelve patients are still alive, five of whom are in CR.
Efficacy
Of the patients who received FCA lymphodepletion, 82% (14/17) achieved CR/CR with incomplete hematologic recovery (CRi), and 71% (10/14) achieved MRD negativity.
An additional patient gained MRD-negative status after the second dose of UCART19.
Of the 14 patients who achieved a CR/CRi, 78% (n=11) went on to receive an allogeneic transplant.
In the entire pooled population, 67% (14/21) achieved CR/CRi.
Three patients received a second UCART19 dose, and five patients remain in CR/CRi.
UCART19 expansion
UCART19 expansion, as measured by quantitative polymerase chain reaction in PALL and flow-based methods in CALM, occurred primarily in the first 28 days in the FCA-treated population.
Investigators observed expansion in 15 of 17 patients treated with FCA. None of the patients who received FC alone (n=4) had expansion detectable in blood or bone marrow, Dr. Benjamin noted.
“The response we’ve seen in the study so far,” Dr. Benjamin clarified, “is linked to the expansion observed within the first 28-day period.”
UCART cells persisted in three patients beyond day 42. In one patient, they persisted up to day 120.
“Of interest is the T-cell recovery seen in the study,” Dr. Benjamin elaborated. “We only have data from the adult study here—14 patients. And you’ll see that, in the FCA-treated arm (n=11), you have a deeper and more sustained lymphodepletion compared to the FC-treated patients (n=3). And this may play a role in the subsequent UCART19 expansion and disease response.”
Re-dosing
Of the three patients who were re-dosed, two achieved MRD negativity.
One patient achieved MRD-negative status at day 28 but relapsed and received a second infusion 3 months after the first dose. The second expansion was not as deep as the first, but the patient nevertheless achieved MRD negativity after the second dose.
The second patient received FC lymphodepletion and was refractory at day 28.
“The second time around, he received FCA, had a slightly better expansion, and achieved molecular remission,” Dr. Benjamin said.
And the third patient had FCA lymphodepletion but was refractory at day 28.
“We elected to give a second dose at 2.4 months later, but unfortunately, there wasn’t very much expansion, even the second time around, and the patient progressed,” Dr. Benjamin said.
FCA lymphodepletion appears to be required for UCART19 expansion. There was no UCART19 expansion and no response in all four patients lymphodepleted with FC.
The evaluation of UCART19 is ongoing in pediatric and adult B-cell ALL, and “there is a plan for moving into the lymphoma space as well,” Dr. Benjamin added.
Dr. Benjamin disclosed honoraria from Amgen, Takeda, Novartis, Gilead, and Celgene, and research funding from Servier and Pfizer.
Servier and Allogene are supporting the UCART19 trials.
*Data in the abstract differ from the presentation.
Obesity meds used by just over half of pediatric obesity programs
NASHVILLE, TENN. –
Programs that didn’t offer pharmacotherapy for children and adolescents with obesity cited a variety of reasons in responses to a survey of 33 multicomponent pediatric weight management programs (PWMPs).
Simply not being in favor of using pharmacotherapy for obesity treatment was the most frequently cited reason, named by seven PWMPs that didn’t prescribe obesity medications.
The second most common response to the survey, cited by six programs, was a lack of knowledge about prescribing medications for obesity, and concerns about insurance coverage were noted by five programs, said Claudia Fox, MD, and her colleagues in a poster presentation at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery. “Despite recommendations, few youth with severe obesity are treated with medications.”
Of the programs that did offer pharmacotherapy, 14 prescribed topiramate, and 13 prescribed phentermine. Metformin was used by 11 programs, and orlistat by eight. Six programs prescribed the fixed-dose combination of topiramate and phentermine.
Lorcaserin, naltrexone/bupropion, liraglutide, phendimetrazine, and naltrexone alone all were used by fewer than five programs each.
The national Pediatric Obesity Weight Evaluation Registry (POWER) “was established in 2013 to identify and promote effective intervention strategies for pediatric obesity,” wrote Dr. Fox and her colleagues
Of the 33 POWER PWMPs who were invited to participate, 30 completed a program profile survey. Of these, 16 programs (53%) offered pharmacotherapy, wrote Dr. Fox, the codirector of the University of Minnesota’s Center for Pediatric Obesity Medicine, Minneapolis, and her colleagues in the POWER work group.
In addition to not being in favor of prescribing obesity medication for pediatric patients, lack of knowledge, and insurance concerns, one program cited limited outcome studies for pediatric obesity pharmacotherapy. One other program’s response noted that patients couldn’t be seen frequently enough to assess the safety of obesity medications.
Taken together, the POWER sites had 7,880 patients. Just 5% were aged 2- 5 years, 48% were aged 6-11 years, and 47% were aged 12-18 years. Just over half (53%) were female.
At baseline, about a quarter of patients (26.4%) had class 1 obesity, defined as a body mass index of at least the 95th age- and sex-adjusted percentile. Children and adolescents with class 2 obesity (BMI of at least 1.2-1.4 times the 95th percentile) made up 35.3% of patients; 38.3% had class 3 obesity, with BMIs greater than 1.4 times the 95th percentile.
In 2017, the Endocrine Society published updated clinical practice guidelines for the assessment, treatment, and prevention of pediatric obesity (J Clin Endocrin Metab. 2017 Mar;102:3;709-57). The guidelines for pediatric obesity treatment recommend intensive lifestyle modifications including dietary, physical activity, and behavioral interventions. Pharmacotherapy is suggested “only after a formal program of intensive lifestyle modification has failed to limit weight gain or to ameliorate comorbidities.” Additionally, say the guidelines, Food and Drug Administration–approved pharmacotherapy should be used only “with a concomitant lifestyle modification program of the highest intensity available and only by clinicians who are experienced in the use of anti-obesity agents and are aware of the potential for adverse reactions.”
“Most commonly prescribed medications are not FDA approved for indication of obesity in pediatrics,” noted Dr. Fox and her coauthors. “Further research is needed to evaluate efficacy of pharmacotherapy in the pediatric population and to understand factors impacting prescribing practices.”
Dr. Fox reported no outside sources of funding and had no relevant financial disclosures.
NASHVILLE, TENN. –
Programs that didn’t offer pharmacotherapy for children and adolescents with obesity cited a variety of reasons in responses to a survey of 33 multicomponent pediatric weight management programs (PWMPs).
Simply not being in favor of using pharmacotherapy for obesity treatment was the most frequently cited reason, named by seven PWMPs that didn’t prescribe obesity medications.
The second most common response to the survey, cited by six programs, was a lack of knowledge about prescribing medications for obesity, and concerns about insurance coverage were noted by five programs, said Claudia Fox, MD, and her colleagues in a poster presentation at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery. “Despite recommendations, few youth with severe obesity are treated with medications.”
Of the programs that did offer pharmacotherapy, 14 prescribed topiramate, and 13 prescribed phentermine. Metformin was used by 11 programs, and orlistat by eight. Six programs prescribed the fixed-dose combination of topiramate and phentermine.
Lorcaserin, naltrexone/bupropion, liraglutide, phendimetrazine, and naltrexone alone all were used by fewer than five programs each.
The national Pediatric Obesity Weight Evaluation Registry (POWER) “was established in 2013 to identify and promote effective intervention strategies for pediatric obesity,” wrote Dr. Fox and her colleagues
Of the 33 POWER PWMPs who were invited to participate, 30 completed a program profile survey. Of these, 16 programs (53%) offered pharmacotherapy, wrote Dr. Fox, the codirector of the University of Minnesota’s Center for Pediatric Obesity Medicine, Minneapolis, and her colleagues in the POWER work group.
In addition to not being in favor of prescribing obesity medication for pediatric patients, lack of knowledge, and insurance concerns, one program cited limited outcome studies for pediatric obesity pharmacotherapy. One other program’s response noted that patients couldn’t be seen frequently enough to assess the safety of obesity medications.
Taken together, the POWER sites had 7,880 patients. Just 5% were aged 2- 5 years, 48% were aged 6-11 years, and 47% were aged 12-18 years. Just over half (53%) were female.
At baseline, about a quarter of patients (26.4%) had class 1 obesity, defined as a body mass index of at least the 95th age- and sex-adjusted percentile. Children and adolescents with class 2 obesity (BMI of at least 1.2-1.4 times the 95th percentile) made up 35.3% of patients; 38.3% had class 3 obesity, with BMIs greater than 1.4 times the 95th percentile.
In 2017, the Endocrine Society published updated clinical practice guidelines for the assessment, treatment, and prevention of pediatric obesity (J Clin Endocrin Metab. 2017 Mar;102:3;709-57). The guidelines for pediatric obesity treatment recommend intensive lifestyle modifications including dietary, physical activity, and behavioral interventions. Pharmacotherapy is suggested “only after a formal program of intensive lifestyle modification has failed to limit weight gain or to ameliorate comorbidities.” Additionally, say the guidelines, Food and Drug Administration–approved pharmacotherapy should be used only “with a concomitant lifestyle modification program of the highest intensity available and only by clinicians who are experienced in the use of anti-obesity agents and are aware of the potential for adverse reactions.”
“Most commonly prescribed medications are not FDA approved for indication of obesity in pediatrics,” noted Dr. Fox and her coauthors. “Further research is needed to evaluate efficacy of pharmacotherapy in the pediatric population and to understand factors impacting prescribing practices.”
Dr. Fox reported no outside sources of funding and had no relevant financial disclosures.
NASHVILLE, TENN. –
Programs that didn’t offer pharmacotherapy for children and adolescents with obesity cited a variety of reasons in responses to a survey of 33 multicomponent pediatric weight management programs (PWMPs).
Simply not being in favor of using pharmacotherapy for obesity treatment was the most frequently cited reason, named by seven PWMPs that didn’t prescribe obesity medications.
The second most common response to the survey, cited by six programs, was a lack of knowledge about prescribing medications for obesity, and concerns about insurance coverage were noted by five programs, said Claudia Fox, MD, and her colleagues in a poster presentation at a meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery. “Despite recommendations, few youth with severe obesity are treated with medications.”
Of the programs that did offer pharmacotherapy, 14 prescribed topiramate, and 13 prescribed phentermine. Metformin was used by 11 programs, and orlistat by eight. Six programs prescribed the fixed-dose combination of topiramate and phentermine.
Lorcaserin, naltrexone/bupropion, liraglutide, phendimetrazine, and naltrexone alone all were used by fewer than five programs each.
The national Pediatric Obesity Weight Evaluation Registry (POWER) “was established in 2013 to identify and promote effective intervention strategies for pediatric obesity,” wrote Dr. Fox and her colleagues
Of the 33 POWER PWMPs who were invited to participate, 30 completed a program profile survey. Of these, 16 programs (53%) offered pharmacotherapy, wrote Dr. Fox, the codirector of the University of Minnesota’s Center for Pediatric Obesity Medicine, Minneapolis, and her colleagues in the POWER work group.
In addition to not being in favor of prescribing obesity medication for pediatric patients, lack of knowledge, and insurance concerns, one program cited limited outcome studies for pediatric obesity pharmacotherapy. One other program’s response noted that patients couldn’t be seen frequently enough to assess the safety of obesity medications.
Taken together, the POWER sites had 7,880 patients. Just 5% were aged 2- 5 years, 48% were aged 6-11 years, and 47% were aged 12-18 years. Just over half (53%) were female.
At baseline, about a quarter of patients (26.4%) had class 1 obesity, defined as a body mass index of at least the 95th age- and sex-adjusted percentile. Children and adolescents with class 2 obesity (BMI of at least 1.2-1.4 times the 95th percentile) made up 35.3% of patients; 38.3% had class 3 obesity, with BMIs greater than 1.4 times the 95th percentile.
In 2017, the Endocrine Society published updated clinical practice guidelines for the assessment, treatment, and prevention of pediatric obesity (J Clin Endocrin Metab. 2017 Mar;102:3;709-57). The guidelines for pediatric obesity treatment recommend intensive lifestyle modifications including dietary, physical activity, and behavioral interventions. Pharmacotherapy is suggested “only after a formal program of intensive lifestyle modification has failed to limit weight gain or to ameliorate comorbidities.” Additionally, say the guidelines, Food and Drug Administration–approved pharmacotherapy should be used only “with a concomitant lifestyle modification program of the highest intensity available and only by clinicians who are experienced in the use of anti-obesity agents and are aware of the potential for adverse reactions.”
“Most commonly prescribed medications are not FDA approved for indication of obesity in pediatrics,” noted Dr. Fox and her coauthors. “Further research is needed to evaluate efficacy of pharmacotherapy in the pediatric population and to understand factors impacting prescribing practices.”
Dr. Fox reported no outside sources of funding and had no relevant financial disclosures.
REPORTING FROM OBESITY WEEK 2018
Key clinical point: Just over half of pediatric weight management programs prescribed obesity medications.
Major finding: Of 30 programs responding, 16 (53%) prescribed obesity medication.
Study details: Survey of 33 programs in the Pediatric Obesity Weight Evaluation Registry (POWER).
Disclosures: Dr. Fox reported no outside sources of funding and no conflicts of interest.
Ghost busting in pediatric primary care
As clinicians trained in the care of children, we have struggled in recent years with how much care is appropriate to provide to the parents of our young charges.

Gradual progression has occurred from recognizing postpartum depression as affecting infants, to recommending screening, to creation of a billing code for screening as “for the benefit of” the child, and increasingly even being paid for that code. We now see referral of depressed parents as within our scope of practice with the goal of protecting the child’s emotional development from the caregiver’s altered mental condition, as well as relieving the parent’s suffering. Some of us even provide treatment ourselves.
While the family history has been our standard way of assessing “transgenerational transmission” of risk for physical and mental health conditions, parenting practices are a more direct transmission threat, and one more amenable to our intervention.
Aversive parenting acts happen to many people growing up, but how the parent thinks about these seems to make the difference between consciously protecting the child from similar experiences or unconsciously playing them out in the child’s life. With 64% of U.S. adults reporting at least one adverse childhood experience (ACE), many of which were acts or omissions by their parents, we need to be vigilant to track their translation of past events, “the ghosts,” into present parenting.
Just ask
“I barely have time to talk about the child,” you may be saying, “how can I have time to dig into the parent’s issues, much less know what to do?” Exploring for connections to the parent’s past in primary care is most crucial when the parent-child relationship is strained, or the parent’s handling of typical or problematic child behaviors is abnormal, clinically symptomatic, or dangerous. Nonetheless, helping all parents make these connections enriches life and meaning for families, and dramatically strengthens the doctor-family relationship. Then all of our care is more effective.
In my experience, this valuable connection is not difficult to make – it lives just below the surface for most parents. We may want to ask permission first, noting that “our ideas about how to parent tend to be shaped by how we were parented.” By simply asking, “May I ask how your parents would have handled this [behavior or situation]?” we may hear a description of a reasonable approach (sent to my room), denial that this ever came up (I was never as hardheaded as this kid!), blanking out (Things were tough. I have tried to block it all out), or clues to a pattern better not repeated (Oh, my father would have beat me ...). This question also may be useful in elucidating cultural or generational differences between what was done to them and their own intentions that can be hard to bridge. All of these are opportunities for promoting positive parenting by creating empathy for that child of the past to carry forward to the own child in the present.
While we may be lucky to have even one parent at the visit, we should ask the one present the equivalent question of the partner’s past. Even if one parent had a model that he or she wanted to emulate or a ghost to bust, the other may not agree. Conflict between partners undermines management and can create harmful tension. If the parent does not know, this is an important homework assignment to being collaborative coparents.
Empathize
After hearing about the past experiences, we should empathize with the parent regarding pain experienced as a child in the past (“That would be very scary for any child”) and ask “How much is this a burden for you now?” to see if help is needed. But this is a key educational moment for us as child development experts to suggest how children of the age they were then might process the events. For example, one might explain reaction to abandonment by a father by saying, “Any 6-year-old whose father left would feel sad and mad, but also might think he had done something wrong or wasn’t worth staying around for.” One might react to a story of abusive discipline by saying, “Children need to feel safe and protected at home. Not knowing when your parent is going to hurt you could produce lifelong anxiety and trouble trusting your closest relationships.” Watch to see if this connects for them.
Selma Fraiberg, in the classic article “Ghosts in the Nursery,”1 noted that if parents have come to empathize with their past hurting selves, they will work to prevent similar pain for their own children. If they have dealt with these experiences by identifying with the aggressive or neglectful adult or blanking the memory, they are more likely to act out similar practices with their children.
For some, being able to tolerate reviewing these painful times enough to experience empathy for the child may require years of work with a trusted therapist. We should be prepared to refer if the parents are in distress. But for many, getting our help to understand how a child might feel and later act after these experiences may be enough to interrupt the transmission. We can try to elicit current impact of the past (“How are those experiences affecting your parenting now?”). This question, expecting impact, often causes parents to stop short and think. While at first denying impact, if I have been compassionate and nonjudgmental in asking, they often return with more insight.
Help with parenting issues
After eliciting perceptions of the past, I find it useful to ask, “So, what have (the two of) you decided” about how to manage [the problematic parenting situation]?” The implication is that parenting actions are decisions. Making this decision process overt may reveal that they are having blank out moments of impulsive action, or ambivalence with thoughts and feelings in conflict, or arguments resulting in standoffs. A common reaction to hurts in the past is for parents to strongly avoid doing as their own parents did, but then have no plan at all, get increasingly emotional, and finally blow up and scream or hit or storm off ineffectually. We can help them pick out one or two stressful situations, often perceived disrespect or defiance by the child, and plan steps for when it comes up again – as hot-button issues always do. It is important to let them know that their “emotion brain” is likely to speak up first under stress and the “thinking brain” takes longer. We, and they, need to be patient and congratulate them for little bits of progress in having rationality win.

Don’t forget that children adapt to the parenting they receive and develop reactions that may interfere with seeing their parents in a new mode of trust and kindness. A child may have defended him/herself from the emotional pain of not feeling safe or protected by the parent who is acting out a ghost and may react by laughing, running, spitting, hitting, shutting down, pushing the parent away, or saying “I don’t care.” The child’s reaction, too, takes time and consistent responsiveness to change to accept new parenting patterns. It can be painful to the newly-aware parents to recognize these behaviors are caused, at least in part, by their own actions, especially when it is a repetition of their own childhood experiences. We can be the patient, empathic coach – believing in their good intentions as they develop as parents – just as they would have wanted from their parents when they were growing up.
Dr. Howard is assistant professor of pediatrics at The Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert for MDedge News. E-mail her at pdnews@mdedge.com.
Reference
1. “Ghosts in the Nursery: A Psychoanalytic Approach to the Problems of Impaired Infant-Mother Relationships,” J Am Acad Child Psychiatry. 1975 Summer;14(3);387-421.
As clinicians trained in the care of children, we have struggled in recent years with how much care is appropriate to provide to the parents of our young charges.
Gradual progression has occurred from recognizing postpartum depression as affecting infants, to recommending screening, to creation of a billing code for screening as “for the benefit of” the child, and increasingly even being paid for that code. We now see referral of depressed parents as within our scope of practice with the goal of protecting the child’s emotional development from the caregiver’s altered mental condition, as well as relieving the parent’s suffering. Some of us even provide treatment ourselves.
While the family history has been our standard way of assessing “transgenerational transmission” of risk for physical and mental health conditions, parenting practices are a more direct transmission threat, and one more amenable to our intervention.
Aversive parenting acts happen to many people growing up, but how the parent thinks about these seems to make the difference between consciously protecting the child from similar experiences or unconsciously playing them out in the child’s life. With 64% of U.S. adults reporting at least one adverse childhood experience (ACE), many of which were acts or omissions by their parents, we need to be vigilant to track their translation of past events, “the ghosts,” into present parenting.
Just ask
“I barely have time to talk about the child,” you may be saying, “how can I have time to dig into the parent’s issues, much less know what to do?” Exploring for connections to the parent’s past in primary care is most crucial when the parent-child relationship is strained, or the parent’s handling of typical or problematic child behaviors is abnormal, clinically symptomatic, or dangerous. Nonetheless, helping all parents make these connections enriches life and meaning for families, and dramatically strengthens the doctor-family relationship. Then all of our care is more effective.
In my experience, this valuable connection is not difficult to make – it lives just below the surface for most parents. We may want to ask permission first, noting that “our ideas about how to parent tend to be shaped by how we were parented.” By simply asking, “May I ask how your parents would have handled this [behavior or situation]?” we may hear a description of a reasonable approach (sent to my room), denial that this ever came up (I was never as hardheaded as this kid!), blanking out (Things were tough. I have tried to block it all out), or clues to a pattern better not repeated (Oh, my father would have beat me ...). This question also may be useful in elucidating cultural or generational differences between what was done to them and their own intentions that can be hard to bridge. All of these are opportunities for promoting positive parenting by creating empathy for that child of the past to carry forward to the own child in the present.
While we may be lucky to have even one parent at the visit, we should ask the one present the equivalent question of the partner’s past. Even if one parent had a model that he or she wanted to emulate or a ghost to bust, the other may not agree. Conflict between partners undermines management and can create harmful tension. If the parent does not know, this is an important homework assignment to being collaborative coparents.
Empathize
After hearing about the past experiences, we should empathize with the parent regarding pain experienced as a child in the past (“That would be very scary for any child”) and ask “How much is this a burden for you now?” to see if help is needed. But this is a key educational moment for us as child development experts to suggest how children of the age they were then might process the events. For example, one might explain reaction to abandonment by a father by saying, “Any 6-year-old whose father left would feel sad and mad, but also might think he had done something wrong or wasn’t worth staying around for.” One might react to a story of abusive discipline by saying, “Children need to feel safe and protected at home. Not knowing when your parent is going to hurt you could produce lifelong anxiety and trouble trusting your closest relationships.” Watch to see if this connects for them.
Selma Fraiberg, in the classic article “Ghosts in the Nursery,”1 noted that if parents have come to empathize with their past hurting selves, they will work to prevent similar pain for their own children. If they have dealt with these experiences by identifying with the aggressive or neglectful adult or blanking the memory, they are more likely to act out similar practices with their children.
For some, being able to tolerate reviewing these painful times enough to experience empathy for the child may require years of work with a trusted therapist. We should be prepared to refer if the parents are in distress. But for many, getting our help to understand how a child might feel and later act after these experiences may be enough to interrupt the transmission. We can try to elicit current impact of the past (“How are those experiences affecting your parenting now?”). This question, expecting impact, often causes parents to stop short and think. While at first denying impact, if I have been compassionate and nonjudgmental in asking, they often return with more insight.
Help with parenting issues
After eliciting perceptions of the past, I find it useful to ask, “So, what have (the two of) you decided” about how to manage [the problematic parenting situation]?” The implication is that parenting actions are decisions. Making this decision process overt may reveal that they are having blank out moments of impulsive action, or ambivalence with thoughts and feelings in conflict, or arguments resulting in standoffs. A common reaction to hurts in the past is for parents to strongly avoid doing as their own parents did, but then have no plan at all, get increasingly emotional, and finally blow up and scream or hit or storm off ineffectually. We can help them pick out one or two stressful situations, often perceived disrespect or defiance by the child, and plan steps for when it comes up again – as hot-button issues always do. It is important to let them know that their “emotion brain” is likely to speak up first under stress and the “thinking brain” takes longer. We, and they, need to be patient and congratulate them for little bits of progress in having rationality win.
Don’t forget that children adapt to the parenting they receive and develop reactions that may interfere with seeing their parents in a new mode of trust and kindness. A child may have defended him/herself from the emotional pain of not feeling safe or protected by the parent who is acting out a ghost and may react by laughing, running, spitting, hitting, shutting down, pushing the parent away, or saying “I don’t care.” The child’s reaction, too, takes time and consistent responsiveness to change to accept new parenting patterns. It can be painful to the newly-aware parents to recognize these behaviors are caused, at least in part, by their own actions, especially when it is a repetition of their own childhood experiences. We can be the patient, empathic coach – believing in their good intentions as they develop as parents – just as they would have wanted from their parents when they were growing up.
Dr. Howard is assistant professor of pediatrics at The Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert for MDedge News. E-mail her at pdnews@mdedge.com.
Reference
1. “Ghosts in the Nursery: A Psychoanalytic Approach to the Problems of Impaired Infant-Mother Relationships,” J Am Acad Child Psychiatry. 1975 Summer;14(3);387-421.
As clinicians trained in the care of children, we have struggled in recent years with how much care is appropriate to provide to the parents of our young charges.
Gradual progression has occurred from recognizing postpartum depression as affecting infants, to recommending screening, to creation of a billing code for screening as “for the benefit of” the child, and increasingly even being paid for that code. We now see referral of depressed parents as within our scope of practice with the goal of protecting the child’s emotional development from the caregiver’s altered mental condition, as well as relieving the parent’s suffering. Some of us even provide treatment ourselves.
While the family history has been our standard way of assessing “transgenerational transmission” of risk for physical and mental health conditions, parenting practices are a more direct transmission threat, and one more amenable to our intervention.
Aversive parenting acts happen to many people growing up, but how the parent thinks about these seems to make the difference between consciously protecting the child from similar experiences or unconsciously playing them out in the child’s life. With 64% of U.S. adults reporting at least one adverse childhood experience (ACE), many of which were acts or omissions by their parents, we need to be vigilant to track their translation of past events, “the ghosts,” into present parenting.
Just ask
“I barely have time to talk about the child,” you may be saying, “how can I have time to dig into the parent’s issues, much less know what to do?” Exploring for connections to the parent’s past in primary care is most crucial when the parent-child relationship is strained, or the parent’s handling of typical or problematic child behaviors is abnormal, clinically symptomatic, or dangerous. Nonetheless, helping all parents make these connections enriches life and meaning for families, and dramatically strengthens the doctor-family relationship. Then all of our care is more effective.
In my experience, this valuable connection is not difficult to make – it lives just below the surface for most parents. We may want to ask permission first, noting that “our ideas about how to parent tend to be shaped by how we were parented.” By simply asking, “May I ask how your parents would have handled this [behavior or situation]?” we may hear a description of a reasonable approach (sent to my room), denial that this ever came up (I was never as hardheaded as this kid!), blanking out (Things were tough. I have tried to block it all out), or clues to a pattern better not repeated (Oh, my father would have beat me ...). This question also may be useful in elucidating cultural or generational differences between what was done to them and their own intentions that can be hard to bridge. All of these are opportunities for promoting positive parenting by creating empathy for that child of the past to carry forward to the own child in the present.
While we may be lucky to have even one parent at the visit, we should ask the one present the equivalent question of the partner’s past. Even if one parent had a model that he or she wanted to emulate or a ghost to bust, the other may not agree. Conflict between partners undermines management and can create harmful tension. If the parent does not know, this is an important homework assignment to being collaborative coparents.
Empathize
After hearing about the past experiences, we should empathize with the parent regarding pain experienced as a child in the past (“That would be very scary for any child”) and ask “How much is this a burden for you now?” to see if help is needed. But this is a key educational moment for us as child development experts to suggest how children of the age they were then might process the events. For example, one might explain reaction to abandonment by a father by saying, “Any 6-year-old whose father left would feel sad and mad, but also might think he had done something wrong or wasn’t worth staying around for.” One might react to a story of abusive discipline by saying, “Children need to feel safe and protected at home. Not knowing when your parent is going to hurt you could produce lifelong anxiety and trouble trusting your closest relationships.” Watch to see if this connects for them.
Selma Fraiberg, in the classic article “Ghosts in the Nursery,”1 noted that if parents have come to empathize with their past hurting selves, they will work to prevent similar pain for their own children. If they have dealt with these experiences by identifying with the aggressive or neglectful adult or blanking the memory, they are more likely to act out similar practices with their children.
For some, being able to tolerate reviewing these painful times enough to experience empathy for the child may require years of work with a trusted therapist. We should be prepared to refer if the parents are in distress. But for many, getting our help to understand how a child might feel and later act after these experiences may be enough to interrupt the transmission. We can try to elicit current impact of the past (“How are those experiences affecting your parenting now?”). This question, expecting impact, often causes parents to stop short and think. While at first denying impact, if I have been compassionate and nonjudgmental in asking, they often return with more insight.
Help with parenting issues
After eliciting perceptions of the past, I find it useful to ask, “So, what have (the two of) you decided” about how to manage [the problematic parenting situation]?” The implication is that parenting actions are decisions. Making this decision process overt may reveal that they are having blank out moments of impulsive action, or ambivalence with thoughts and feelings in conflict, or arguments resulting in standoffs. A common reaction to hurts in the past is for parents to strongly avoid doing as their own parents did, but then have no plan at all, get increasingly emotional, and finally blow up and scream or hit or storm off ineffectually. We can help them pick out one or two stressful situations, often perceived disrespect or defiance by the child, and plan steps for when it comes up again – as hot-button issues always do. It is important to let them know that their “emotion brain” is likely to speak up first under stress and the “thinking brain” takes longer. We, and they, need to be patient and congratulate them for little bits of progress in having rationality win.
Don’t forget that children adapt to the parenting they receive and develop reactions that may interfere with seeing their parents in a new mode of trust and kindness. A child may have defended him/herself from the emotional pain of not feeling safe or protected by the parent who is acting out a ghost and may react by laughing, running, spitting, hitting, shutting down, pushing the parent away, or saying “I don’t care.” The child’s reaction, too, takes time and consistent responsiveness to change to accept new parenting patterns. It can be painful to the newly-aware parents to recognize these behaviors are caused, at least in part, by their own actions, especially when it is a repetition of their own childhood experiences. We can be the patient, empathic coach – believing in their good intentions as they develop as parents – just as they would have wanted from their parents when they were growing up.
Dr. Howard is assistant professor of pediatrics at The Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert for MDedge News. E-mail her at pdnews@mdedge.com.
Reference
1. “Ghosts in the Nursery: A Psychoanalytic Approach to the Problems of Impaired Infant-Mother Relationships,” J Am Acad Child Psychiatry. 1975 Summer;14(3);387-421.
Emapalumab found safe, effective in primary HLH

SAN DIEGO—Emapalumab, an interferon gamma-blocking antibody, controls disease activity and has a favorable safety profile in pediatric patients with primary hemophagocytic lymphohistiocytosis (HLH), according to research presented at the 2018 ASH Annual Meeting.
Investigators believe emapalumab, which was recently approved to treat HLH in the United States, should be considered a new therapeutic option for this rare and life-threatening syndrome because of the drug’s targeted mode of action.
Multiple lines of evidence have pointed to interferon gamma as a “rational target” in HLH, and elevated levels of interferon gamma are consistently observed in patients with HLH, said Franco Locatelli, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Emapalumab binds to its target with high affinity, recognizing both free and receptor-bound interferon gamma, he added.
Dr. Locatelli described trial results with emapalumab in children with HLH during the late-breaking abstracts session at ASH (abstract LBA-6).
This phase 2/3 study (NCT01818492) included 34 children with primary HLH—7 who were treatment-naïve and 27 who had failed conventional HLH therapy.
The patients received emapalumab intravenously with concomitant dexamethasone for up to 8 weeks or extended to the point of allogeneic hematopoietic stem cell transplant if needed.
The study met its primary endpoint of overall response rate higher than 40%, Dr. Locatelli reported.
The overall response rate was 64.7% for all 34 treated patients (95% confidence interval [CI], 46% to 80%; P=0.0031) and 63% for the 27 patients who had failed prior therapy (95% CI, 42% to 81%; P=0.0134).
Response was rapid, occurring at a median of 8 days after starting emapalumab, and patients were in response for a median of 75% of days during treatment, Dr. Locatelli said.
Ninety-four percent of patients had at least one adverse event (AE), and 63% had serious AEs. Common AEs included infections (56%), hypertension (35%), infusion-related reactions (27%), and pyrexia (24%).
One patient had disseminated histoplasmosis that led to discontinuation of emapalumab but resolved with appropriate treatment.
This study was sponsored by Novimmune. Investigators provided disclosures related to Sobi, Novimmune, Rocket Pharmaceuticals, Inc., AB2Bio, Novartis, Eli Lilly, Sanofi, UCB, Pfizer, and AbbVie. Two investigators reported employment with Novimmune.
SAN DIEGO—Emapalumab, an interferon gamma-blocking antibody, controls disease activity and has a favorable safety profile in pediatric patients with primary hemophagocytic lymphohistiocytosis (HLH), according to research presented at the 2018 ASH Annual Meeting.
Investigators believe emapalumab, which was recently approved to treat HLH in the United States, should be considered a new therapeutic option for this rare and life-threatening syndrome because of the drug’s targeted mode of action.
Multiple lines of evidence have pointed to interferon gamma as a “rational target” in HLH, and elevated levels of interferon gamma are consistently observed in patients with HLH, said Franco Locatelli, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Emapalumab binds to its target with high affinity, recognizing both free and receptor-bound interferon gamma, he added.
Dr. Locatelli described trial results with emapalumab in children with HLH during the late-breaking abstracts session at ASH (abstract LBA-6).
This phase 2/3 study (NCT01818492) included 34 children with primary HLH—7 who were treatment-naïve and 27 who had failed conventional HLH therapy.
The patients received emapalumab intravenously with concomitant dexamethasone for up to 8 weeks or extended to the point of allogeneic hematopoietic stem cell transplant if needed.
The study met its primary endpoint of overall response rate higher than 40%, Dr. Locatelli reported.
The overall response rate was 64.7% for all 34 treated patients (95% confidence interval [CI], 46% to 80%; P=0.0031) and 63% for the 27 patients who had failed prior therapy (95% CI, 42% to 81%; P=0.0134).
Response was rapid, occurring at a median of 8 days after starting emapalumab, and patients were in response for a median of 75% of days during treatment, Dr. Locatelli said.
Ninety-four percent of patients had at least one adverse event (AE), and 63% had serious AEs. Common AEs included infections (56%), hypertension (35%), infusion-related reactions (27%), and pyrexia (24%).
One patient had disseminated histoplasmosis that led to discontinuation of emapalumab but resolved with appropriate treatment.
This study was sponsored by Novimmune. Investigators provided disclosures related to Sobi, Novimmune, Rocket Pharmaceuticals, Inc., AB2Bio, Novartis, Eli Lilly, Sanofi, UCB, Pfizer, and AbbVie. Two investigators reported employment with Novimmune.
SAN DIEGO—Emapalumab, an interferon gamma-blocking antibody, controls disease activity and has a favorable safety profile in pediatric patients with primary hemophagocytic lymphohistiocytosis (HLH), according to research presented at the 2018 ASH Annual Meeting.
Investigators believe emapalumab, which was recently approved to treat HLH in the United States, should be considered a new therapeutic option for this rare and life-threatening syndrome because of the drug’s targeted mode of action.
Multiple lines of evidence have pointed to interferon gamma as a “rational target” in HLH, and elevated levels of interferon gamma are consistently observed in patients with HLH, said Franco Locatelli, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Emapalumab binds to its target with high affinity, recognizing both free and receptor-bound interferon gamma, he added.
Dr. Locatelli described trial results with emapalumab in children with HLH during the late-breaking abstracts session at ASH (abstract LBA-6).
This phase 2/3 study (NCT01818492) included 34 children with primary HLH—7 who were treatment-naïve and 27 who had failed conventional HLH therapy.
The patients received emapalumab intravenously with concomitant dexamethasone for up to 8 weeks or extended to the point of allogeneic hematopoietic stem cell transplant if needed.
The study met its primary endpoint of overall response rate higher than 40%, Dr. Locatelli reported.
The overall response rate was 64.7% for all 34 treated patients (95% confidence interval [CI], 46% to 80%; P=0.0031) and 63% for the 27 patients who had failed prior therapy (95% CI, 42% to 81%; P=0.0134).
Response was rapid, occurring at a median of 8 days after starting emapalumab, and patients were in response for a median of 75% of days during treatment, Dr. Locatelli said.
Ninety-four percent of patients had at least one adverse event (AE), and 63% had serious AEs. Common AEs included infections (56%), hypertension (35%), infusion-related reactions (27%), and pyrexia (24%).
One patient had disseminated histoplasmosis that led to discontinuation of emapalumab but resolved with appropriate treatment.
This study was sponsored by Novimmune. Investigators provided disclosures related to Sobi, Novimmune, Rocket Pharmaceuticals, Inc., AB2Bio, Novartis, Eli Lilly, Sanofi, UCB, Pfizer, and AbbVie. Two investigators reported employment with Novimmune.
HU could save millions of lives in Africa, speaker says

SAN DIEGO—Daily hydroxyurea (HU) treatment is feasible, safe, and effective for children with sickle cell disease (SCD) in sub-Saharan Africa, according to a phase 1/2 trial.
During HU treatment, children experienced less vaso-occlusive pain, fewer cases of malaria and other infections, and lower rates of transfusions and death, compared to rates observed in the pretreatment screening phase of the trial.
“Based on that data, we believe that wider access to hydroxyurea for sickle cell anemia has the potential to save millions of lives in Africa,” said Léon Tshilolo, MD, PhD, of Centre Hospitalier Monkole in Kinshasa, Democratic Republic of the Congo.
Dr. Tshilolo reported the data, from the REACH trial (NCT01966731), during the plenary session at the 2018 ASH Annual Meeting (abstract 3*). Data were simultaneously published in The New England Journal of Medicine.
Use of HU has been limited in Africa because of cost, access issues, and challenges associated with laboratory monitoring, according to researchers.
Moreover, most of the efficacy data on HU come from studies conducted in the United States, Europe, and other high-income settings, said senior study author Russell E. Ware, MD, PhD, of Cincinnati Children’s Hospital Center in Ohio.
“Now that there’s data in an African setting, I think this will go a long way to advancing [HU therapy] and encouraging governments, organizations, and pharmaceutical companies to bring it in,” Dr. Ware said.
To collect the data, Drs. Ware and Tshilolo and their colleagues evaluated SCD patients, ages 1 to 10, living in four sub-Saharan African countries—Angola, Democratic Republic of the Congo, Kenya, and Uganda.
The children completed a 2-month pretreatment screening phase designed to capture baseline clinical and laboratory data.
The children were started at 15 mg/kg to 20 mg/kg of HU for 6 months, followed by escalation to the maximum-tolerated dose.
A total of 606 children were treated, 600 of them for 3 months. Treatment is ongoing, but the mean treatment duration at the time of analysis was 29 months.
Results
The average maximum tolerated dose was 22.5 mg/kg/day. Dose-limiting toxicities occurred in 5.1% of the children, which was below the 20% protocol-specified threshold for safety, Dr. Tshilolo said.
Dose-limiting toxicities included severe anemia, reticulocytopenia, neutropenia, and thrombocytopenia. However, there were similar rates of these events during the screening period and the treatment period.
The rate of vaso-occlusive pain during HU treatment was 44.6 events per 100 patient-years, compared with 98.3 events per 100 patient-years in the pretreatment period (incidence rate ratio [IRR], 0.45; 95% confidence interval [CI], 0.37-0.56).
The rate of malaria infection was 22.9 events per 100 patient-years in the HU treatment period, compared to 46.9 events in the pretreatment period (IRR, 0.49; 95% CI, 0.37-0.66).
The rate of nonmalaria infections was 90.0 events per 100 patient-years in the HU treatment period, compared to 142.5 events per 100 patient-years in the pretreatment period (IRR, 0.62; 95% CI, 0.53-0.72).
Dr. Tshilolo said the researchers were “encouraged” by the reduced infection rates, particularly in light of previous concerns that HU could suppress the immune system and put children at risk for malaria.
The rate of transfusion during HU treatment was 14.2 events per 100 patient-years, compared to 43.3 events per 100 patient-years (IRR, 0.33; 95% CI, 0.23 to 0.47).
Death rates were 1.1 per 100 patient-years in the HU treatment period and 3.6 per 100 patient-years in the pretreatment period (IRR, 0.30; 95% CI, 0.10-0.88).
Dr. Tshilolo reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Cincinnati Children’s Research Foundation, along with nonfinancial support from Bristol-Myers Squibb. Dr. Ware reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Bristol-Myers Squibb.
*Data in the abstract differ from the presentation and the article.
SAN DIEGO—Daily hydroxyurea (HU) treatment is feasible, safe, and effective for children with sickle cell disease (SCD) in sub-Saharan Africa, according to a phase 1/2 trial.
During HU treatment, children experienced less vaso-occlusive pain, fewer cases of malaria and other infections, and lower rates of transfusions and death, compared to rates observed in the pretreatment screening phase of the trial.
“Based on that data, we believe that wider access to hydroxyurea for sickle cell anemia has the potential to save millions of lives in Africa,” said Léon Tshilolo, MD, PhD, of Centre Hospitalier Monkole in Kinshasa, Democratic Republic of the Congo.
Dr. Tshilolo reported the data, from the REACH trial (NCT01966731), during the plenary session at the 2018 ASH Annual Meeting (abstract 3*). Data were simultaneously published in The New England Journal of Medicine.
Use of HU has been limited in Africa because of cost, access issues, and challenges associated with laboratory monitoring, according to researchers.
Moreover, most of the efficacy data on HU come from studies conducted in the United States, Europe, and other high-income settings, said senior study author Russell E. Ware, MD, PhD, of Cincinnati Children’s Hospital Center in Ohio.
“Now that there’s data in an African setting, I think this will go a long way to advancing [HU therapy] and encouraging governments, organizations, and pharmaceutical companies to bring it in,” Dr. Ware said.
To collect the data, Drs. Ware and Tshilolo and their colleagues evaluated SCD patients, ages 1 to 10, living in four sub-Saharan African countries—Angola, Democratic Republic of the Congo, Kenya, and Uganda.
The children completed a 2-month pretreatment screening phase designed to capture baseline clinical and laboratory data.
The children were started at 15 mg/kg to 20 mg/kg of HU for 6 months, followed by escalation to the maximum-tolerated dose.
A total of 606 children were treated, 600 of them for 3 months. Treatment is ongoing, but the mean treatment duration at the time of analysis was 29 months.
Results
The average maximum tolerated dose was 22.5 mg/kg/day. Dose-limiting toxicities occurred in 5.1% of the children, which was below the 20% protocol-specified threshold for safety, Dr. Tshilolo said.
Dose-limiting toxicities included severe anemia, reticulocytopenia, neutropenia, and thrombocytopenia. However, there were similar rates of these events during the screening period and the treatment period.
The rate of vaso-occlusive pain during HU treatment was 44.6 events per 100 patient-years, compared with 98.3 events per 100 patient-years in the pretreatment period (incidence rate ratio [IRR], 0.45; 95% confidence interval [CI], 0.37-0.56).
The rate of malaria infection was 22.9 events per 100 patient-years in the HU treatment period, compared to 46.9 events in the pretreatment period (IRR, 0.49; 95% CI, 0.37-0.66).
The rate of nonmalaria infections was 90.0 events per 100 patient-years in the HU treatment period, compared to 142.5 events per 100 patient-years in the pretreatment period (IRR, 0.62; 95% CI, 0.53-0.72).
Dr. Tshilolo said the researchers were “encouraged” by the reduced infection rates, particularly in light of previous concerns that HU could suppress the immune system and put children at risk for malaria.
The rate of transfusion during HU treatment was 14.2 events per 100 patient-years, compared to 43.3 events per 100 patient-years (IRR, 0.33; 95% CI, 0.23 to 0.47).
Death rates were 1.1 per 100 patient-years in the HU treatment period and 3.6 per 100 patient-years in the pretreatment period (IRR, 0.30; 95% CI, 0.10-0.88).
Dr. Tshilolo reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Cincinnati Children’s Research Foundation, along with nonfinancial support from Bristol-Myers Squibb. Dr. Ware reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Bristol-Myers Squibb.
*Data in the abstract differ from the presentation and the article.
SAN DIEGO—Daily hydroxyurea (HU) treatment is feasible, safe, and effective for children with sickle cell disease (SCD) in sub-Saharan Africa, according to a phase 1/2 trial.
During HU treatment, children experienced less vaso-occlusive pain, fewer cases of malaria and other infections, and lower rates of transfusions and death, compared to rates observed in the pretreatment screening phase of the trial.
“Based on that data, we believe that wider access to hydroxyurea for sickle cell anemia has the potential to save millions of lives in Africa,” said Léon Tshilolo, MD, PhD, of Centre Hospitalier Monkole in Kinshasa, Democratic Republic of the Congo.
Dr. Tshilolo reported the data, from the REACH trial (NCT01966731), during the plenary session at the 2018 ASH Annual Meeting (abstract 3*). Data were simultaneously published in The New England Journal of Medicine.
Use of HU has been limited in Africa because of cost, access issues, and challenges associated with laboratory monitoring, according to researchers.
Moreover, most of the efficacy data on HU come from studies conducted in the United States, Europe, and other high-income settings, said senior study author Russell E. Ware, MD, PhD, of Cincinnati Children’s Hospital Center in Ohio.
“Now that there’s data in an African setting, I think this will go a long way to advancing [HU therapy] and encouraging governments, organizations, and pharmaceutical companies to bring it in,” Dr. Ware said.
To collect the data, Drs. Ware and Tshilolo and their colleagues evaluated SCD patients, ages 1 to 10, living in four sub-Saharan African countries—Angola, Democratic Republic of the Congo, Kenya, and Uganda.
The children completed a 2-month pretreatment screening phase designed to capture baseline clinical and laboratory data.
The children were started at 15 mg/kg to 20 mg/kg of HU for 6 months, followed by escalation to the maximum-tolerated dose.
A total of 606 children were treated, 600 of them for 3 months. Treatment is ongoing, but the mean treatment duration at the time of analysis was 29 months.
Results
The average maximum tolerated dose was 22.5 mg/kg/day. Dose-limiting toxicities occurred in 5.1% of the children, which was below the 20% protocol-specified threshold for safety, Dr. Tshilolo said.
Dose-limiting toxicities included severe anemia, reticulocytopenia, neutropenia, and thrombocytopenia. However, there were similar rates of these events during the screening period and the treatment period.
The rate of vaso-occlusive pain during HU treatment was 44.6 events per 100 patient-years, compared with 98.3 events per 100 patient-years in the pretreatment period (incidence rate ratio [IRR], 0.45; 95% confidence interval [CI], 0.37-0.56).
The rate of malaria infection was 22.9 events per 100 patient-years in the HU treatment period, compared to 46.9 events in the pretreatment period (IRR, 0.49; 95% CI, 0.37-0.66).
The rate of nonmalaria infections was 90.0 events per 100 patient-years in the HU treatment period, compared to 142.5 events per 100 patient-years in the pretreatment period (IRR, 0.62; 95% CI, 0.53-0.72).
Dr. Tshilolo said the researchers were “encouraged” by the reduced infection rates, particularly in light of previous concerns that HU could suppress the immune system and put children at risk for malaria.
The rate of transfusion during HU treatment was 14.2 events per 100 patient-years, compared to 43.3 events per 100 patient-years (IRR, 0.33; 95% CI, 0.23 to 0.47).
Death rates were 1.1 per 100 patient-years in the HU treatment period and 3.6 per 100 patient-years in the pretreatment period (IRR, 0.30; 95% CI, 0.10-0.88).
Dr. Tshilolo reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Cincinnati Children’s Research Foundation, along with nonfinancial support from Bristol-Myers Squibb. Dr. Ware reported grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and Bristol-Myers Squibb.
*Data in the abstract differ from the presentation and the article.
Biologics options for pediatric asthma continue to grow
ORLANDO – The goal of treatment is the same for all asthma cases, regardless of severity: “to enable a patient to achieve and maintain control over their asthma,” according to Stanley J. Szefler, MD, a professor of pediatrics at the University of Colorado at Denver, Aurora.

That goal includes “reducing the risk of exacerbations, emergency department visits, hospitalizations, and progression as well as reducing impairments, including symptoms, functional limitations, poor quality of life, and other manifestations of asthma,” Dr. Szefler, also director of the Children’s Hospital of Colorado pediatric asthma research program, told colleagues at the annual meeting of the American Academy of Pediatrics.
Severe asthma challenges
These aims are more difficult with severe asthma, defined by the World Health Organization as “the current level of clinical control and risks which can result in frequent severe exacerbations and/or adverse reactions to medications and/or chronic morbidity,” Dr. Szefler explained. Severe asthma includes untreated severe asthma, difficult-to-treat asthma, and treatment-resistant severe asthma, whether controlled on high-dose medication or not.
Allergen sensitization, viral respiratory infections, and respiratory irritants (such as air pollution and smoking) are common features of severe asthma in children. Also common are challenges specific to management: poor medication adherence, poor technique for inhaled medications, and undertreatment. Poor management can lead to repeated exacerbations, adverse effects from drugs, disease progression, possible development of chronic obstructive pulmonary disease (COPD), and early mortality.
The National Heart, Lung, and Blood Institute EPR-3 guidelines for treatment of pediatric asthma recommend a stepwise approach to therapy, starting with short-acting beta2-agonists as needed (SABA p.r.n.). The clinician then assesses the patient’s symptoms, exacerbations, side effects, quality of life, and lung function to determine whether the asthma is well managed or requires inhaled corticosteroids, or another therapy in moving through the steps. Each step also involves patient education, environmental control, and management of the child’s comorbidities.
It is not until steps 5 and 6 that the guidelines advise considering the biologic omalizumab for patients who have allergies. But other biologic options exist as well. Four biologics currently approved for treating asthma include omalizumab, mepolizumab, benralizumab, and reslizumab, but reslizumab is approved only for patients at least 18 years old.
Biologics for pediatric asthma
Omalizumab, which targets IgE, is appropriate for patients at least 6 years old in whom inhaled corticosteroids could not adequately control the symptoms of moderate to-severe persistent asthma. Dosing of omalizumab is a subcutaneous injection every 2-4 weeks based on pretreatment serum IgE and body weight using a dosing table that starts at 0.016 mg/kg/IgE (IU/mL). Maximum dose is 375 mg every 2 weeks in the United States and 600 mg every 2 weeks in the European Union.
The advantages of an anti-IgE drug are its use only once a month and its substantial effect on reducing exacerbations in a clearly identified population. However, these drugs are costly and require supervised administration, Dr. Szefler noted. They also carry a risk of anaphylaxis in less than 0.2% of patients, requiring the patient to be monitored after first administration and to carry an injectable epinephrine after omalizumab administration as a precaution for late-occurring anaphylaxis.
Mepolizumab is an anti–interleukin (IL)–5 drug used in patients at least 12 years old with severe persistent asthma that’s inadequately controlled with inhaled corticosteroids. Peripheral blood counts of eosinophilia determine if a patient has an eosinophilic phenotype, which has the best response to mepolizumab. People with at least 150 cells per microliter at baseline or at least 300 cells per microliter within the past year have shown a good response to mepolizumab. Dosing is 100 mg subcutaneously every 4 weeks.
For patients with atopic asthma, mepolizumab is effective in reducing the daily oral corticosteroid dose and the number of both annual exacerbations and exacerbations requiring hospitalization or an emergency visit. Other benefits of mepolizumab include increasing the time to a first exacerbation, the pre- and postbronchodilator forced expiratory volume in one second (FEV1) and overall quality of life.
Patient reductions in exacerbations while taking mepolizumab were associated with eosinophil count but not IgE, atopic status, FEV1 or bronchodilator response in the DREAM study (Lancet. 2012 Aug 18;380[9842]:651-9.).
Two safety considerations with mepolizumab include an increased risk of shingles and the risk of a preexisting helminth infection getting worse. Providers should screen for helminth infection and might consider a herpes zoster vaccination prior to starting therapy, Dr. Szefler said.
Benralizumab is an anti-IL5Ra for use in people at least 12 years old with severe persistent asthma and an eosinophilic phenotype (at least 300 cells per microliter). Dosing begins with three subcutaneous injections of 30 mg every 4 weeks, followed by administration every 8 weeks thereafter.
Benralizumab’s clinical effects include reduced exacerbations and oral corticosteroid use, and improved asthma symptom scores and prebronchodilator FEV1. Higher serum eosinophils and a history of more frequent exacerbations are both biomarkers for reduced exacerbations with benralizumab treatment.
Dupilumab: New kid on the block
The newest biologic for asthma is dupilumab, approved Oct. 19, 2018, by the Food and Drug Administration as the only asthma biologic that patients can administer at home. Dupilumab is an anti–IL-4 and anti–IL-13 biologic whose most recent study results showed a severe exacerbations rate 50% lower than placebo (N Engl J Med. 2018 Jun 28;378[26]:2486-96.). Patients with higher baseline levels of eosinophils had the best response, although some patients showed hypereosinophilia following dupilumab therapy.
The study had a low number of adolescents enrolled, however, and more data on predictive biomarkers are needed. Dupilumab also requires a twice-monthly administration.
“It could be potentially better than those currently available due to additional effect on FEV1,” Dr. Szefler said, but cost and safety may determine how dupilumab is recommended and used, including possible use for early intervention.
As development in biologics for pediatric asthma continues to grow, questions about best practices for management remain, such as what age is best for starting biologics, what strategies are most safe and effective, and what risks and benefits exist for each strategy. Questions also remain regarding the risk factors for asthma and what early intervention strategies might change the disease’s natural history.
“Look at asthma in children as a chronic disease that can result in potentially preventable adverse respiratory outcomes in adulthood,” Dr. Szefler said. He recommended monitoring children’s lung function over time and using “measures of clinical outcomes, lung function, and biomarkers to assess potential benefits of biologic therapy.”
Dr. Szefler has served on the advisory board for Regeneron and Sanofi, and he has consulted for AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, GlaxoSmithKline, Novartis, and Propeller Health.
ORLANDO – The goal of treatment is the same for all asthma cases, regardless of severity: “to enable a patient to achieve and maintain control over their asthma,” according to Stanley J. Szefler, MD, a professor of pediatrics at the University of Colorado at Denver, Aurora.
That goal includes “reducing the risk of exacerbations, emergency department visits, hospitalizations, and progression as well as reducing impairments, including symptoms, functional limitations, poor quality of life, and other manifestations of asthma,” Dr. Szefler, also director of the Children’s Hospital of Colorado pediatric asthma research program, told colleagues at the annual meeting of the American Academy of Pediatrics.
Severe asthma challenges
These aims are more difficult with severe asthma, defined by the World Health Organization as “the current level of clinical control and risks which can result in frequent severe exacerbations and/or adverse reactions to medications and/or chronic morbidity,” Dr. Szefler explained. Severe asthma includes untreated severe asthma, difficult-to-treat asthma, and treatment-resistant severe asthma, whether controlled on high-dose medication or not.
Allergen sensitization, viral respiratory infections, and respiratory irritants (such as air pollution and smoking) are common features of severe asthma in children. Also common are challenges specific to management: poor medication adherence, poor technique for inhaled medications, and undertreatment. Poor management can lead to repeated exacerbations, adverse effects from drugs, disease progression, possible development of chronic obstructive pulmonary disease (COPD), and early mortality.
The National Heart, Lung, and Blood Institute EPR-3 guidelines for treatment of pediatric asthma recommend a stepwise approach to therapy, starting with short-acting beta2-agonists as needed (SABA p.r.n.). The clinician then assesses the patient’s symptoms, exacerbations, side effects, quality of life, and lung function to determine whether the asthma is well managed or requires inhaled corticosteroids, or another therapy in moving through the steps. Each step also involves patient education, environmental control, and management of the child’s comorbidities.
It is not until steps 5 and 6 that the guidelines advise considering the biologic omalizumab for patients who have allergies. But other biologic options exist as well. Four biologics currently approved for treating asthma include omalizumab, mepolizumab, benralizumab, and reslizumab, but reslizumab is approved only for patients at least 18 years old.
Biologics for pediatric asthma
Omalizumab, which targets IgE, is appropriate for patients at least 6 years old in whom inhaled corticosteroids could not adequately control the symptoms of moderate to-severe persistent asthma. Dosing of omalizumab is a subcutaneous injection every 2-4 weeks based on pretreatment serum IgE and body weight using a dosing table that starts at 0.016 mg/kg/IgE (IU/mL). Maximum dose is 375 mg every 2 weeks in the United States and 600 mg every 2 weeks in the European Union.
The advantages of an anti-IgE drug are its use only once a month and its substantial effect on reducing exacerbations in a clearly identified population. However, these drugs are costly and require supervised administration, Dr. Szefler noted. They also carry a risk of anaphylaxis in less than 0.2% of patients, requiring the patient to be monitored after first administration and to carry an injectable epinephrine after omalizumab administration as a precaution for late-occurring anaphylaxis.
Mepolizumab is an anti–interleukin (IL)–5 drug used in patients at least 12 years old with severe persistent asthma that’s inadequately controlled with inhaled corticosteroids. Peripheral blood counts of eosinophilia determine if a patient has an eosinophilic phenotype, which has the best response to mepolizumab. People with at least 150 cells per microliter at baseline or at least 300 cells per microliter within the past year have shown a good response to mepolizumab. Dosing is 100 mg subcutaneously every 4 weeks.
For patients with atopic asthma, mepolizumab is effective in reducing the daily oral corticosteroid dose and the number of both annual exacerbations and exacerbations requiring hospitalization or an emergency visit. Other benefits of mepolizumab include increasing the time to a first exacerbation, the pre- and postbronchodilator forced expiratory volume in one second (FEV1) and overall quality of life.
Patient reductions in exacerbations while taking mepolizumab were associated with eosinophil count but not IgE, atopic status, FEV1 or bronchodilator response in the DREAM study (Lancet. 2012 Aug 18;380[9842]:651-9.).
Two safety considerations with mepolizumab include an increased risk of shingles and the risk of a preexisting helminth infection getting worse. Providers should screen for helminth infection and might consider a herpes zoster vaccination prior to starting therapy, Dr. Szefler said.
Benralizumab is an anti-IL5Ra for use in people at least 12 years old with severe persistent asthma and an eosinophilic phenotype (at least 300 cells per microliter). Dosing begins with three subcutaneous injections of 30 mg every 4 weeks, followed by administration every 8 weeks thereafter.
Benralizumab’s clinical effects include reduced exacerbations and oral corticosteroid use, and improved asthma symptom scores and prebronchodilator FEV1. Higher serum eosinophils and a history of more frequent exacerbations are both biomarkers for reduced exacerbations with benralizumab treatment.
Dupilumab: New kid on the block
The newest biologic for asthma is dupilumab, approved Oct. 19, 2018, by the Food and Drug Administration as the only asthma biologic that patients can administer at home. Dupilumab is an anti–IL-4 and anti–IL-13 biologic whose most recent study results showed a severe exacerbations rate 50% lower than placebo (N Engl J Med. 2018 Jun 28;378[26]:2486-96.). Patients with higher baseline levels of eosinophils had the best response, although some patients showed hypereosinophilia following dupilumab therapy.
The study had a low number of adolescents enrolled, however, and more data on predictive biomarkers are needed. Dupilumab also requires a twice-monthly administration.
“It could be potentially better than those currently available due to additional effect on FEV1,” Dr. Szefler said, but cost and safety may determine how dupilumab is recommended and used, including possible use for early intervention.
As development in biologics for pediatric asthma continues to grow, questions about best practices for management remain, such as what age is best for starting biologics, what strategies are most safe and effective, and what risks and benefits exist for each strategy. Questions also remain regarding the risk factors for asthma and what early intervention strategies might change the disease’s natural history.
“Look at asthma in children as a chronic disease that can result in potentially preventable adverse respiratory outcomes in adulthood,” Dr. Szefler said. He recommended monitoring children’s lung function over time and using “measures of clinical outcomes, lung function, and biomarkers to assess potential benefits of biologic therapy.”
Dr. Szefler has served on the advisory board for Regeneron and Sanofi, and he has consulted for AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, GlaxoSmithKline, Novartis, and Propeller Health.
ORLANDO – The goal of treatment is the same for all asthma cases, regardless of severity: “to enable a patient to achieve and maintain control over their asthma,” according to Stanley J. Szefler, MD, a professor of pediatrics at the University of Colorado at Denver, Aurora.
That goal includes “reducing the risk of exacerbations, emergency department visits, hospitalizations, and progression as well as reducing impairments, including symptoms, functional limitations, poor quality of life, and other manifestations of asthma,” Dr. Szefler, also director of the Children’s Hospital of Colorado pediatric asthma research program, told colleagues at the annual meeting of the American Academy of Pediatrics.
Severe asthma challenges
These aims are more difficult with severe asthma, defined by the World Health Organization as “the current level of clinical control and risks which can result in frequent severe exacerbations and/or adverse reactions to medications and/or chronic morbidity,” Dr. Szefler explained. Severe asthma includes untreated severe asthma, difficult-to-treat asthma, and treatment-resistant severe asthma, whether controlled on high-dose medication or not.
Allergen sensitization, viral respiratory infections, and respiratory irritants (such as air pollution and smoking) are common features of severe asthma in children. Also common are challenges specific to management: poor medication adherence, poor technique for inhaled medications, and undertreatment. Poor management can lead to repeated exacerbations, adverse effects from drugs, disease progression, possible development of chronic obstructive pulmonary disease (COPD), and early mortality.
The National Heart, Lung, and Blood Institute EPR-3 guidelines for treatment of pediatric asthma recommend a stepwise approach to therapy, starting with short-acting beta2-agonists as needed (SABA p.r.n.). The clinician then assesses the patient’s symptoms, exacerbations, side effects, quality of life, and lung function to determine whether the asthma is well managed or requires inhaled corticosteroids, or another therapy in moving through the steps. Each step also involves patient education, environmental control, and management of the child’s comorbidities.
It is not until steps 5 and 6 that the guidelines advise considering the biologic omalizumab for patients who have allergies. But other biologic options exist as well. Four biologics currently approved for treating asthma include omalizumab, mepolizumab, benralizumab, and reslizumab, but reslizumab is approved only for patients at least 18 years old.
Biologics for pediatric asthma
Omalizumab, which targets IgE, is appropriate for patients at least 6 years old in whom inhaled corticosteroids could not adequately control the symptoms of moderate to-severe persistent asthma. Dosing of omalizumab is a subcutaneous injection every 2-4 weeks based on pretreatment serum IgE and body weight using a dosing table that starts at 0.016 mg/kg/IgE (IU/mL). Maximum dose is 375 mg every 2 weeks in the United States and 600 mg every 2 weeks in the European Union.
The advantages of an anti-IgE drug are its use only once a month and its substantial effect on reducing exacerbations in a clearly identified population. However, these drugs are costly and require supervised administration, Dr. Szefler noted. They also carry a risk of anaphylaxis in less than 0.2% of patients, requiring the patient to be monitored after first administration and to carry an injectable epinephrine after omalizumab administration as a precaution for late-occurring anaphylaxis.
Mepolizumab is an anti–interleukin (IL)–5 drug used in patients at least 12 years old with severe persistent asthma that’s inadequately controlled with inhaled corticosteroids. Peripheral blood counts of eosinophilia determine if a patient has an eosinophilic phenotype, which has the best response to mepolizumab. People with at least 150 cells per microliter at baseline or at least 300 cells per microliter within the past year have shown a good response to mepolizumab. Dosing is 100 mg subcutaneously every 4 weeks.
For patients with atopic asthma, mepolizumab is effective in reducing the daily oral corticosteroid dose and the number of both annual exacerbations and exacerbations requiring hospitalization or an emergency visit. Other benefits of mepolizumab include increasing the time to a first exacerbation, the pre- and postbronchodilator forced expiratory volume in one second (FEV1) and overall quality of life.
Patient reductions in exacerbations while taking mepolizumab were associated with eosinophil count but not IgE, atopic status, FEV1 or bronchodilator response in the DREAM study (Lancet. 2012 Aug 18;380[9842]:651-9.).
Two safety considerations with mepolizumab include an increased risk of shingles and the risk of a preexisting helminth infection getting worse. Providers should screen for helminth infection and might consider a herpes zoster vaccination prior to starting therapy, Dr. Szefler said.
Benralizumab is an anti-IL5Ra for use in people at least 12 years old with severe persistent asthma and an eosinophilic phenotype (at least 300 cells per microliter). Dosing begins with three subcutaneous injections of 30 mg every 4 weeks, followed by administration every 8 weeks thereafter.
Benralizumab’s clinical effects include reduced exacerbations and oral corticosteroid use, and improved asthma symptom scores and prebronchodilator FEV1. Higher serum eosinophils and a history of more frequent exacerbations are both biomarkers for reduced exacerbations with benralizumab treatment.
Dupilumab: New kid on the block
The newest biologic for asthma is dupilumab, approved Oct. 19, 2018, by the Food and Drug Administration as the only asthma biologic that patients can administer at home. Dupilumab is an anti–IL-4 and anti–IL-13 biologic whose most recent study results showed a severe exacerbations rate 50% lower than placebo (N Engl J Med. 2018 Jun 28;378[26]:2486-96.). Patients with higher baseline levels of eosinophils had the best response, although some patients showed hypereosinophilia following dupilumab therapy.
The study had a low number of adolescents enrolled, however, and more data on predictive biomarkers are needed. Dupilumab also requires a twice-monthly administration.
“It could be potentially better than those currently available due to additional effect on FEV1,” Dr. Szefler said, but cost and safety may determine how dupilumab is recommended and used, including possible use for early intervention.
As development in biologics for pediatric asthma continues to grow, questions about best practices for management remain, such as what age is best for starting biologics, what strategies are most safe and effective, and what risks and benefits exist for each strategy. Questions also remain regarding the risk factors for asthma and what early intervention strategies might change the disease’s natural history.
“Look at asthma in children as a chronic disease that can result in potentially preventable adverse respiratory outcomes in adulthood,” Dr. Szefler said. He recommended monitoring children’s lung function over time and using “measures of clinical outcomes, lung function, and biomarkers to assess potential benefits of biologic therapy.”
Dr. Szefler has served on the advisory board for Regeneron and Sanofi, and he has consulted for AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, GlaxoSmithKline, Novartis, and Propeller Health.
EXPERT ANALYSIS FROM AAP 18
ALL regimens clear disease in kids with MPAL
SAN DIEGO—Pediatric patients with mixed phenotype acute leukemia (MPAL) can achieve minimal residual disease (MRD) negativity with acute lymphoblastic leukemia (ALL)-directed chemotherapy, according to new research.
In a retrospective study, most pediatric MPAL patients who received ALL-directed chemotherapy achieved an MRD-negative complete response (CR).
Ninety-three percent of patients achieved a CR at the end of induction with an ALL regimen, 70% were MRD-negative at the end of induction, and 86% were MRD-negative at the end of induction or consolidation.
Etan Orgel, MD, of the University of Southern California, Los Angeles, presented these findings at the ASH 2018 Annual Meeting (abstract 558*).
The study included 94 patients aged 1-21 years who met World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions.
Most patients had B/Myeloid phenotype (89%, n=84), 10% (n=9) had T/Myeloid, and 1% (n=1) had B/T phenotype.
Eighty-seven patients (93%) received ALL induction, and 83 (89%) continued on ALL therapy after induction.
Ninety-three percent (81/87) of patients treated with an ALL induction regimen had a CR at the end of induction. One patient died during induction, and six had induction failures, defined as either disease progression (n=2) or MRD of 5% or greater (n=4).
The MRD-negative rates, defined as MRD less than 0.01%, were 70% (59/84) at the end of induction and 86% (68/79) at the end of induction or consolidation.
Twelve of 14 patients (86%) who were MRD-positive at the end of induction and continued on ALL therapy achieved MRD negativity at the end of consolidation.
Survival
The researchers assessed 5-year survival in patients who received an ALL regimen but did not go on to transplant.
In these patients, the 5-year event-free survival (EFS) was 75%, and the 5-year overall survival (OS) was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said.
“[T]his is very different from the approach used at many adult centers and many of the adult recommendations,” he added.
The 5-year EFS rate was 80% in patients who were MRD-negative at the end of induction and 52% in patients who were MRD-positive at the end of induction. Five-year OS rates were 91% and 84%, respectively.
The 5-year EFS rate was 77% in patients who were MRD-negative at the end of consolidation and was unavailable in the three patients who were MRD-positive. The 5-year OS rates were 89% and not available, respectively.
In a multivariable analysis, MRD was the strongest predictor of EFS (hazard ratio [HR]=3.5) and OS (HR=4.6).
There was a trend toward earlier failure and worse OS (HR=4.49, P=0.074) for T-lineage-containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” Dr. Orgel said.
In closing, he said this research suggests that ALL therapy without transplant may be sufficient to treat most patients with pediatric MPAL. However, he noted that clinical trials are necessary to prospectively validate MRD thresholds at end of induction and consolidation and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” Dr. Orgel said.
He disclosed no conflicts of interest.
* Data in the presentation differ from the abstract.
SAN DIEGO—Pediatric patients with mixed phenotype acute leukemia (MPAL) can achieve minimal residual disease (MRD) negativity with acute lymphoblastic leukemia (ALL)-directed chemotherapy, according to new research.
In a retrospective study, most pediatric MPAL patients who received ALL-directed chemotherapy achieved an MRD-negative complete response (CR).
Ninety-three percent of patients achieved a CR at the end of induction with an ALL regimen, 70% were MRD-negative at the end of induction, and 86% were MRD-negative at the end of induction or consolidation.
Etan Orgel, MD, of the University of Southern California, Los Angeles, presented these findings at the ASH 2018 Annual Meeting (abstract 558*).
The study included 94 patients aged 1-21 years who met World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions.
Most patients had B/Myeloid phenotype (89%, n=84), 10% (n=9) had T/Myeloid, and 1% (n=1) had B/T phenotype.
Eighty-seven patients (93%) received ALL induction, and 83 (89%) continued on ALL therapy after induction.
Ninety-three percent (81/87) of patients treated with an ALL induction regimen had a CR at the end of induction. One patient died during induction, and six had induction failures, defined as either disease progression (n=2) or MRD of 5% or greater (n=4).
The MRD-negative rates, defined as MRD less than 0.01%, were 70% (59/84) at the end of induction and 86% (68/79) at the end of induction or consolidation.
Twelve of 14 patients (86%) who were MRD-positive at the end of induction and continued on ALL therapy achieved MRD negativity at the end of consolidation.
Survival
The researchers assessed 5-year survival in patients who received an ALL regimen but did not go on to transplant.
In these patients, the 5-year event-free survival (EFS) was 75%, and the 5-year overall survival (OS) was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said.
“[T]his is very different from the approach used at many adult centers and many of the adult recommendations,” he added.
The 5-year EFS rate was 80% in patients who were MRD-negative at the end of induction and 52% in patients who were MRD-positive at the end of induction. Five-year OS rates were 91% and 84%, respectively.
The 5-year EFS rate was 77% in patients who were MRD-negative at the end of consolidation and was unavailable in the three patients who were MRD-positive. The 5-year OS rates were 89% and not available, respectively.
In a multivariable analysis, MRD was the strongest predictor of EFS (hazard ratio [HR]=3.5) and OS (HR=4.6).
There was a trend toward earlier failure and worse OS (HR=4.49, P=0.074) for T-lineage-containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” Dr. Orgel said.
In closing, he said this research suggests that ALL therapy without transplant may be sufficient to treat most patients with pediatric MPAL. However, he noted that clinical trials are necessary to prospectively validate MRD thresholds at end of induction and consolidation and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” Dr. Orgel said.
He disclosed no conflicts of interest.
* Data in the presentation differ from the abstract.
SAN DIEGO—Pediatric patients with mixed phenotype acute leukemia (MPAL) can achieve minimal residual disease (MRD) negativity with acute lymphoblastic leukemia (ALL)-directed chemotherapy, according to new research.
In a retrospective study, most pediatric MPAL patients who received ALL-directed chemotherapy achieved an MRD-negative complete response (CR).
Ninety-three percent of patients achieved a CR at the end of induction with an ALL regimen, 70% were MRD-negative at the end of induction, and 86% were MRD-negative at the end of induction or consolidation.
Etan Orgel, MD, of the University of Southern California, Los Angeles, presented these findings at the ASH 2018 Annual Meeting (abstract 558*).
The study included 94 patients aged 1-21 years who met World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions.
Most patients had B/Myeloid phenotype (89%, n=84), 10% (n=9) had T/Myeloid, and 1% (n=1) had B/T phenotype.
Eighty-seven patients (93%) received ALL induction, and 83 (89%) continued on ALL therapy after induction.
Ninety-three percent (81/87) of patients treated with an ALL induction regimen had a CR at the end of induction. One patient died during induction, and six had induction failures, defined as either disease progression (n=2) or MRD of 5% or greater (n=4).
The MRD-negative rates, defined as MRD less than 0.01%, were 70% (59/84) at the end of induction and 86% (68/79) at the end of induction or consolidation.
Twelve of 14 patients (86%) who were MRD-positive at the end of induction and continued on ALL therapy achieved MRD negativity at the end of consolidation.
Survival
The researchers assessed 5-year survival in patients who received an ALL regimen but did not go on to transplant.
In these patients, the 5-year event-free survival (EFS) was 75%, and the 5-year overall survival (OS) was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said.
“[T]his is very different from the approach used at many adult centers and many of the adult recommendations,” he added.
The 5-year EFS rate was 80% in patients who were MRD-negative at the end of induction and 52% in patients who were MRD-positive at the end of induction. Five-year OS rates were 91% and 84%, respectively.
The 5-year EFS rate was 77% in patients who were MRD-negative at the end of consolidation and was unavailable in the three patients who were MRD-positive. The 5-year OS rates were 89% and not available, respectively.
In a multivariable analysis, MRD was the strongest predictor of EFS (hazard ratio [HR]=3.5) and OS (HR=4.6).
There was a trend toward earlier failure and worse OS (HR=4.49, P=0.074) for T-lineage-containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” Dr. Orgel said.
In closing, he said this research suggests that ALL therapy without transplant may be sufficient to treat most patients with pediatric MPAL. However, he noted that clinical trials are necessary to prospectively validate MRD thresholds at end of induction and consolidation and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” Dr. Orgel said.
He disclosed no conflicts of interest.
* Data in the presentation differ from the abstract.
Early caffeine therapy linked to improved neurologic outcomes in premature babies
Premature babies may benefit more if caffeine therapy is given within 2 days of birth, based on a retrospective observational cohort study of more than 2,000 newborns.
When caffeine was given within the first 2 days of birth, neonates had an adjusted odds ratio of significant neurodevelopmental impairment of 0.68, compared with neonates who received caffeine after 2 or more days. Further, the early-caffeine group had a 0.67 adjusted odds ratio for having cognitive scores of less than 85 on the Bayley Scales of Infant and Toddler Development, Third Edition, compared with the late-caffeine group. After researchers corrected for small-for-gestational-age status and other risk factors, however, early-caffeine therapy was associated with lower odds of cerebral palsy and hearing impairment only, according to the study published online in Pediatrics.
Caffeine administration should be a priority once extremely preterm neonates are stabilized, Abhay Lodha, MD, of the University of Calgary (Alta.), and his coauthors wrote. “It is rather easy to organize the administration of caffeine as early as possible for Level 3 nurseries, and many units have already accomplished this. However, certain Level 2 nurseries may not have facilities available for such early administration. We do not have data that indicate the earliest that caffeine would have to be given to get maximum benefit, and thus, it should not be counted as an emergency medication yet,” they wrote.
The study examined data from 2,108 neonates born before 29 weeks of gestational age and given caffeine to treat or prevent apnea; 1,545 received the caffeine within 2 days of birth and the remaining 563 were treated with caffeine after 2 days. Data were adjusted for gestational age, sex, antenatal steroids, and SNAP-II (Score of Neonatal Acute Physiology-II) score.
The early-caffeine group had a significantly reduced odds of hearing impairment and cerebral palsy, bronchopulmonary dysplasia, patent ductus arteriosus, and severe neurologic injury. When the data were further analyzed using propensity-matched groups – which also accounted for small-for-gestational-age status – the difference in outcomes was a nonsignificant trend in favor of early caffeine.
The authors noted that the late-caffeine group contained a higher proportion of infants born at or before 24 weeks’ gestational age, and a lower proportion of infants born at 25-28 weeks’ gestational age, compared with the early-caffeine group. The infants in the early-caffeine group also had higher Apgar scores, higher median birth weight, and lower SNAP-II scores, and received a longer median duration of caffeine treatment.
Dr. Lodha and his coauthors said the reason for the differences between the early- and late-caffeine groups was unclear. “However, it could be attributable to an increased growth of dendrites and spines in neurons that is initiated by the especially prolonged use of caffeine in the early-caffeine group,” they wrote. “The other speculation is that caffeine improves cardiac output and blood pressure in infants who are relatively stable.”
No funding or conflicts of interest were declared.
SOURCE: Lodha A et al. Pediatrics. 2018 Dec. 5. doi. org/10.1542/peds.2018-1348.
Premature babies may benefit more if caffeine therapy is given within 2 days of birth, based on a retrospective observational cohort study of more than 2,000 newborns.
When caffeine was given within the first 2 days of birth, neonates had an adjusted odds ratio of significant neurodevelopmental impairment of 0.68, compared with neonates who received caffeine after 2 or more days. Further, the early-caffeine group had a 0.67 adjusted odds ratio for having cognitive scores of less than 85 on the Bayley Scales of Infant and Toddler Development, Third Edition, compared with the late-caffeine group. After researchers corrected for small-for-gestational-age status and other risk factors, however, early-caffeine therapy was associated with lower odds of cerebral palsy and hearing impairment only, according to the study published online in Pediatrics.
Caffeine administration should be a priority once extremely preterm neonates are stabilized, Abhay Lodha, MD, of the University of Calgary (Alta.), and his coauthors wrote. “It is rather easy to organize the administration of caffeine as early as possible for Level 3 nurseries, and many units have already accomplished this. However, certain Level 2 nurseries may not have facilities available for such early administration. We do not have data that indicate the earliest that caffeine would have to be given to get maximum benefit, and thus, it should not be counted as an emergency medication yet,” they wrote.
The study examined data from 2,108 neonates born before 29 weeks of gestational age and given caffeine to treat or prevent apnea; 1,545 received the caffeine within 2 days of birth and the remaining 563 were treated with caffeine after 2 days. Data were adjusted for gestational age, sex, antenatal steroids, and SNAP-II (Score of Neonatal Acute Physiology-II) score.
The early-caffeine group had a significantly reduced odds of hearing impairment and cerebral palsy, bronchopulmonary dysplasia, patent ductus arteriosus, and severe neurologic injury. When the data were further analyzed using propensity-matched groups – which also accounted for small-for-gestational-age status – the difference in outcomes was a nonsignificant trend in favor of early caffeine.
The authors noted that the late-caffeine group contained a higher proportion of infants born at or before 24 weeks’ gestational age, and a lower proportion of infants born at 25-28 weeks’ gestational age, compared with the early-caffeine group. The infants in the early-caffeine group also had higher Apgar scores, higher median birth weight, and lower SNAP-II scores, and received a longer median duration of caffeine treatment.
Dr. Lodha and his coauthors said the reason for the differences between the early- and late-caffeine groups was unclear. “However, it could be attributable to an increased growth of dendrites and spines in neurons that is initiated by the especially prolonged use of caffeine in the early-caffeine group,” they wrote. “The other speculation is that caffeine improves cardiac output and blood pressure in infants who are relatively stable.”
No funding or conflicts of interest were declared.
SOURCE: Lodha A et al. Pediatrics. 2018 Dec. 5. doi. org/10.1542/peds.2018-1348.
Premature babies may benefit more if caffeine therapy is given within 2 days of birth, based on a retrospective observational cohort study of more than 2,000 newborns.
When caffeine was given within the first 2 days of birth, neonates had an adjusted odds ratio of significant neurodevelopmental impairment of 0.68, compared with neonates who received caffeine after 2 or more days. Further, the early-caffeine group had a 0.67 adjusted odds ratio for having cognitive scores of less than 85 on the Bayley Scales of Infant and Toddler Development, Third Edition, compared with the late-caffeine group. After researchers corrected for small-for-gestational-age status and other risk factors, however, early-caffeine therapy was associated with lower odds of cerebral palsy and hearing impairment only, according to the study published online in Pediatrics.
Caffeine administration should be a priority once extremely preterm neonates are stabilized, Abhay Lodha, MD, of the University of Calgary (Alta.), and his coauthors wrote. “It is rather easy to organize the administration of caffeine as early as possible for Level 3 nurseries, and many units have already accomplished this. However, certain Level 2 nurseries may not have facilities available for such early administration. We do not have data that indicate the earliest that caffeine would have to be given to get maximum benefit, and thus, it should not be counted as an emergency medication yet,” they wrote.
The study examined data from 2,108 neonates born before 29 weeks of gestational age and given caffeine to treat or prevent apnea; 1,545 received the caffeine within 2 days of birth and the remaining 563 were treated with caffeine after 2 days. Data were adjusted for gestational age, sex, antenatal steroids, and SNAP-II (Score of Neonatal Acute Physiology-II) score.
The early-caffeine group had a significantly reduced odds of hearing impairment and cerebral palsy, bronchopulmonary dysplasia, patent ductus arteriosus, and severe neurologic injury. When the data were further analyzed using propensity-matched groups – which also accounted for small-for-gestational-age status – the difference in outcomes was a nonsignificant trend in favor of early caffeine.
The authors noted that the late-caffeine group contained a higher proportion of infants born at or before 24 weeks’ gestational age, and a lower proportion of infants born at 25-28 weeks’ gestational age, compared with the early-caffeine group. The infants in the early-caffeine group also had higher Apgar scores, higher median birth weight, and lower SNAP-II scores, and received a longer median duration of caffeine treatment.
Dr. Lodha and his coauthors said the reason for the differences between the early- and late-caffeine groups was unclear. “However, it could be attributable to an increased growth of dendrites and spines in neurons that is initiated by the especially prolonged use of caffeine in the early-caffeine group,” they wrote. “The other speculation is that caffeine improves cardiac output and blood pressure in infants who are relatively stable.”
No funding or conflicts of interest were declared.
SOURCE: Lodha A et al. Pediatrics. 2018 Dec. 5. doi. org/10.1542/peds.2018-1348.
FROM PEDIATRICS
Key clinical point: Earlier caffeine treatment for premature infants may improve neurologic outcomes.
Major finding: Preterm neonates treated with caffeine within 2 days of birth have a significantly lower risk of hearing impairment and cerebral palsy.
Study details: A retrospective observational cohort study in 2,108 preterm neonates.
Disclosures: No funding or conflicts of interest were declared.
Source: Lodha A et al. Pediatrics. 2018 Dec. 5. doi. org/10.1542/peds.2018-1348.