User login
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.

Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.

The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.

While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.
The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.
While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
To the Editor:
Psoriasis is a chronic inflammatory skin condition that commonly affects the nail matrix and/or nail bed.1 Nail involvement is present in up to 50% of patients with cutaneous psoriasis and 80% of patients with psoriatic arthritis.1 Approximately 5% to 10% of patients with psoriasis demonstrate isolated nail involvement with no skin or joint manifestations.1 Nail psoriasis can cause severe pain and psychological distress, and extreme cases may cause considerable morbidity and functional impairment.2,3 Treatment often requires a long duration and may not result in complete recovery due to the slow rate of nail growth. Patients can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.1,2 Despite the availability of various treatment options, many cases remain refractory to standard interventions, which underscores the need for novel therapeutic approaches. Herein, we present a severe case of refractory isolated nail psoriasis that was successfully treated with deucravacitinib, an oral tyrosine kinase 2 (TYK2) inhibitor.
A 59-year-old woman presented with a progressive, yellow, hyperkeratotic lesion on the left thumbnail of 2 years’ duration. The patient noted initial discoloration and peeling at the distal end of the nail. Over time, the discoloration progressed to encompass the entire nail. Previous treatments performed by outside physicians including topical corticosteroids, calcineurin inhibitors, and 2 surgeries to remove the nail plate and nail bed all were unsuccessful. The patient also reported severe left thumbnail pain and pruritus that considerably impaired her ability to work. The rest of the nails were unaffected, and she had no personal or family history of psoriasis. Her medical history was notable for hypertension, gastroesophageal reflux disease, and osteomyelitis of the right thumb without nail involvement. Drug allergies included penicillin G benzathine, sulfonamides, amoxicillin, and ciprofloxacin.
Physical examination of the left thumbnail revealed severe yellow, hyperkeratotic, dystrophic changes with a large, yellow, crumbling hyperkeratotic plaque that extended from approximately 1 cm beyond the nail plate to the proximal end of the distal interphalangeal joint, to and along the lateral nail folds, with extensive distal onycholysis. The proximal and lateral nail folds demonstrated erythema as well as maceration that was extremely tender to minimal palpation (Figure 1). No cutaneous lesions were noted elsewhere on the body. The patient had no tenderness, swelling, or stiffness in any of the joints. The differential diagnosis at the time included squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
Radiography of the left thumb revealed irregular swelling and nonspecific soft tissue enlargement at the tip of the digit. A nail clipping from the left thumbnail and 3-mm punch biopsies of the lateral and proximal nail folds as well as the horn of the proximal nail fold (Figure 2) were negative for fungus and confirmed psoriasiform dermatitis of the nail.
The patient was started on vinegar soaks (1:1 ratio of vinegar to water) every other day as well as urea cream 10%, ammonium lactate 15%, and petrolatum twice daily for 2 months without considerable improvement. Due to lack of improvement during this 2-month period, the patient subsequently was started on oral deucravacitinib 6 mg/d along with continued use of petrolatum twice daily and vinegar soaks every other day. We selected a trial of deucravacitinib for our patient because of its convenient daily oral dosing and promising clinical evidence.4,5 After 2 months of treatment with deucravacitinib, the patient reported substantial improvement and satisfaction with the treatment results. Physical examination of the left thumbnail after 2 months of deucravacitinib treatment revealed mildly hyperkeratotic, yellow, dystrophic changes of the nail with notable improvement of the yellow hyperkeratotic plaque on the distal thumbnail. Normal-appearing nail growth was noted at the proximal nail fold, demonstrating considerable improvement from the initial presentation (Figure 3). However, the patient had developed multiple oral ulcers, generalized pruritus, and an annular urticarial plaque on the left arm. As such, deucravacitinib was discontinued after 2 months of treatment. These symptoms resolved within a week of discontinuing deucravacitinib.
While the etiology of nail psoriasis remains unclear, it is believed to be due to a combination of immunologic, genetic, and environmental factors.3 Classical clinical features include nail pitting, leukonychia, onycholysis, nail bed hyperkeratosis, and splinter hemorrhages.1,3 Our patient exhibited a severe form of nail psoriasis, encompassing the entire nail matrix and bed and extending to the distal interphalangeal joint and lateral nail folds. Previous surgical interventions may have triggered the Koebner phenomenon—which commonly is associated with psoriasis—and resulted in new skin lesions as a secondary response to the surgical trauma.6 The severity of the condition profoundly impacted her quality of life and considerably hindered her ability to work.
Treatment for nail psoriasis includes topical or systemic therapies such as corticosteroids, vitamin D analogs, tacrolimus, and tumor necrosis factor α inhibitors.1,3 Topical treatment is challenging because it is difficult to deliver medication effectively to the nail bed and nail matrix, and patient adherence may be poor.2 Although it has been shown to be effective, intralesional triamcinolone can be associated with pain as the most common adverse effect.7 Systemic medications such as oral methotrexate also may be effective but are contraindicated in pregnant patients and are associated with potential adverse events (AEs), including hepatotoxicity and acute kidney injury.8 The use of biologics may be challenging due to potential AEs and patient reluctance toward injection-based treatments.9
Deucravacitinib is a TYK2 inhibitor approved for treatment of plaque psoriasis.10 Tyrosine kinase 2 is an intracellular kinase that mediates the signaling of IL-23 and other cytokines involved in psoriasis pathogenesis.10 Deucravacitinib selectively binds to the regulatory domain of TYK2, leading to targeted allosteric inhibition of TYK2-mediated IL-23 and type I interferon signaling.4,5,10 Compared with biologics, deucravacitinib is advantageous because it can be administered as a daily oral pill, encouraging high patient compliance.
In the POETYK PSO-1 and PSO-2 phase 3 randomized controlled trials, 20.9% (n=332) and 20.3% (n=510) of deucravacitinib-treated patients with moderate to severe nail involvement achieved a Physician’s Global Assessment of Fingernail score of 0/1 compared with 8.8% (n=165) and 7.9% (n=254) of patients in the placebo group, respectively. All patients in these trials had a diagnosis of plaque psoriasis with at least 10% body surface area involvement; none of the patients had isolated nail psoriasis.4,5
The phase 3 POETYK PSO-1 and PSO-2 trials demonstrated deucravacitinib to be safe and well tolerated with minimal AEs.4,5 However, the development of AEs in our patient, including oral ulcers and generalized pruritus, underscores the need for close monitoring and consideration of potential risks of treatment. Common AEs associated with deucravacitinib include upper respiratory infections (19.2% [n=840]), increased blood creatine phosphokinase levels (2.7% [n=840]), herpes simplex virus (2.0% [n=840]), and mouth ulcers (1.9% [n=840]).11
Patient education also is a crucial component in the treatment of nail psoriasis. Physicians should emphasize the slow growth of nails and need for prolonged treatment. Clear communication and realistic expectations are essential for ensuring patient adherence to treatment.
Our case highlights the potential efficacy and safety of deucravacitinib for treatment of nail psoriasis, potentially laying the groundwork for future clinical studies. Our patient had a severe case of nail psoriasis that involved the entire nail bed and nail plate, resulting in extreme pain, pruritus, and functional impairment. Her case was unique because involvement was isolated to the nail without any accompanying skin or joint manifestations. She showed a favorable response to deucravacitinib within only 2 months of treatment and exhibited considerable improvement of nail psoriasis, with a reported high level of satisfaction with the treatment. We plan to continue to monitor the patient for long-term results. Future randomized clinical trials with longer follow-up periods are crucial to further establish the efficacy and safety of deucravacitinib for treatment of nail psoriasis.
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Ji C, Wang H, Bao C, et al. Challenge of nail psoriasis: an update review. Clin Rev Allergy Immunol. 2021;61:377-402. doi:10.1007/s12016-021-08896-9
- Muneer H, Sathe NC, Masood S. Nail psoriasis. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated March 1, 2024. Accessed October 24, 2024. https://www.ncbi.nlm.nih.gov/books/NBK559260/
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88:29-39. doi:10.1016/j.jaad.2022.07.002
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J Am Acad Dermatol. 2023;88:40-51. doi:10.1016/j.jaad.2022.08.061
- Sanchez DP, Sonthalia S. Koebner phenomenon. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated November 14, 2022. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK553108/
- Grover C, Kharghoria G, Bansal S. Triamcinolone acetonide injections in nail psoriasis: a pragmatic analysis. Skin Appendage Disord. 2024;10:50-59. doi:10.1159/000534699
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. StatPearls Publishing; 2024 Jan-. Updated August 16, 2023. Accessed April 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;2011:Cd008794. doi:10.1002/14651858.CD008794.pub2
- Thaçi D, Strober B, Gordon KB, et al. Deucravacitinib in moderate to severe psoriasis: clinical and quality-of-life outcomes in a phase 2 trial. Dermatol Ther (Heidelb). 2022;12:495-510. doi:10.1007/s13555-021-00649-y
- Week 0-16: demonstrated safety profile. Bristol-Myers Squibb. 2024. Accessed October 24, 2024. https://www.sotyktuhcp.com/safety-profile?cid=sem_2465603&gclid=CjwKCAiA9ourBhAVEiwA3L5RFnyYqmxbqkz1_zBNPz3dcyHKCSFf1XQ-7acznV0XbR5DDJHYkZcKJxoCWN0QAvD_BwE&gclsrc=aw.ds
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
Successful Treatment of Severe Dystrophic Nail Psoriasis With Deucravacitinib
PRACTICE POINTS
- Nail psoriasis can masquerade as other dermatologic conditions, including squamous cell carcinoma of the nail bed and acrodermatitis continua of Hallopeau.
- Nail psoriasis can progress to permanent nail loss if not treated properly, making early recognition and treatment crucial.
- Deucravacitinib, an oral tyrosine kinase 2 inhibitor approved for the treatment of plaque psoriasis, has shown promise as an effective treatment for nail psoriasis in cases that are refractory to standard therapies.

Adalimumab for Psoriasis: Study Compares Biosimilars Vs. Originator
TOPLINE:
rate than those who remained on Humira.
METHODOLOGY:
- Researchers conducted a cohort study using data on patients with psoriasis who were treated with adalimumab, a tumor necrosis factor alpha inhibitor used to treat moderate to severe psoriasis, from the French National Health Data System, British Association of Dermatologists Biologics and Immunomodulators Register, and Spanish Registry of Systemic Therapy in Psoriasis.
- The analysis included 7387 adalimumab-naive patients who were new users of an adalimumab biosimilar and 3654 patients (switchers) who switched from Humira to a biosimilar. Patients were matched and compared with patients receiving Humira.
- Co-primary outcomes of the study were drug discontinuation and serious adverse events.
- Researchers assessed the following adalimumab biosimilar brands: Amgevita, Imraldi, Hyrimoz, Idacio, and Hulio.
TAKEAWAY:
- All-cause drug discontinuation rates were similar between new users of biosimilars and Humira new users (hazard ratio [HR], 0.99; 95% CI, 0.94-1.04).
- Discontinuation rates were higher among those who switched from Humira to a biosimilar (HR, 1.35; 95% CI, 1.19-1.52) than among those who stayed on Humira. Switching to Amgevita (HR, 1.25; 95% CI, 1.13-1.27), Imraldi (HR, 1.53; 95% CI, 1.33-1.76), and Hyrimoz (HR, 1.80; 95% CI, 1.29-2.52) was associated with higher discontinuation rates.
- Serious adverse events were not significantly different between new users of Humira and biosimilar new users (incidence rate ratio [IRR], 0.91; 95% CI, 0.80-1.05), and between patients who switched from a biosimilar to Humira and those who stayed on Humira (IRR, 0.92; 95% CI, 0.83-1.01).
- No significant differences in discontinuation because of ineffectiveness were found between biosimilar and Humira new users (HR, 0.97; 95% CI, 0.88-1.08). Discontinuation because of adverse events was also comparable for all biosimilars among new users, except for Hyrimoz (HR, 0.54; 95% CI, 0.35-0.85), which showed fewer discontinuations than Humira.
IN PRACTICE:
“This study found comparable drug survival and safety between adalimumab biosimilars and Humira in adalimumab-naive patients, supporting the use of biosimilars as viable alternatives for new patients,” the authors wrote. However, noting that discontinuation was more likely among those who switched from Humira to a biosimilar, they added: “Changes in treatment response, skin or injection site reactions, and nocebo effects may contribute to treatment discontinuation post-switch. Thus, patients who switch from Humira to biosimilars may require closer monitoring and support to alleviate these challenges.”
SOURCE:
The study was led by Duc Binh Phan, Dermatology Centre, Northern Care Alliance NHS Foundation Trust in Manchester, England. It was published online in The British Journal of Dermatology.
LIMITATIONS:
Unmeasured factors including psychological perceptions, regional policies, and drug availability could influence drug survival, making the results not fully reflective of treatment effectiveness or safety. Most Humira users in registries were enrolled before biosimilars became available, making it impractical to match new users on the basis of treatment initiation years. Additionally, reasons for discontinuation were not available in the French National Health Data System.
DISCLOSURES:
In the United Kingdom, the research was funded by the Psoriasis Association PhD studentship and supported by the NIHR Manchester Biomedical Research Centre. In France, the authors are employees of the French National Health Insurance, the French National Agency for the Safety of Medicines and Health Products, and the Assistance Publique — Hôpitaux de Paris and received no funding. The authors reported receiving consulting and speaker fees and clinical trial sponsorship from various pharmaceutical companies. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
rate than those who remained on Humira.
METHODOLOGY:
- Researchers conducted a cohort study using data on patients with psoriasis who were treated with adalimumab, a tumor necrosis factor alpha inhibitor used to treat moderate to severe psoriasis, from the French National Health Data System, British Association of Dermatologists Biologics and Immunomodulators Register, and Spanish Registry of Systemic Therapy in Psoriasis.
- The analysis included 7387 adalimumab-naive patients who were new users of an adalimumab biosimilar and 3654 patients (switchers) who switched from Humira to a biosimilar. Patients were matched and compared with patients receiving Humira.
- Co-primary outcomes of the study were drug discontinuation and serious adverse events.
- Researchers assessed the following adalimumab biosimilar brands: Amgevita, Imraldi, Hyrimoz, Idacio, and Hulio.
TAKEAWAY:
- All-cause drug discontinuation rates were similar between new users of biosimilars and Humira new users (hazard ratio [HR], 0.99; 95% CI, 0.94-1.04).
- Discontinuation rates were higher among those who switched from Humira to a biosimilar (HR, 1.35; 95% CI, 1.19-1.52) than among those who stayed on Humira. Switching to Amgevita (HR, 1.25; 95% CI, 1.13-1.27), Imraldi (HR, 1.53; 95% CI, 1.33-1.76), and Hyrimoz (HR, 1.80; 95% CI, 1.29-2.52) was associated with higher discontinuation rates.
- Serious adverse events were not significantly different between new users of Humira and biosimilar new users (incidence rate ratio [IRR], 0.91; 95% CI, 0.80-1.05), and between patients who switched from a biosimilar to Humira and those who stayed on Humira (IRR, 0.92; 95% CI, 0.83-1.01).
- No significant differences in discontinuation because of ineffectiveness were found between biosimilar and Humira new users (HR, 0.97; 95% CI, 0.88-1.08). Discontinuation because of adverse events was also comparable for all biosimilars among new users, except for Hyrimoz (HR, 0.54; 95% CI, 0.35-0.85), which showed fewer discontinuations than Humira.
IN PRACTICE:
“This study found comparable drug survival and safety between adalimumab biosimilars and Humira in adalimumab-naive patients, supporting the use of biosimilars as viable alternatives for new patients,” the authors wrote. However, noting that discontinuation was more likely among those who switched from Humira to a biosimilar, they added: “Changes in treatment response, skin or injection site reactions, and nocebo effects may contribute to treatment discontinuation post-switch. Thus, patients who switch from Humira to biosimilars may require closer monitoring and support to alleviate these challenges.”
SOURCE:
The study was led by Duc Binh Phan, Dermatology Centre, Northern Care Alliance NHS Foundation Trust in Manchester, England. It was published online in The British Journal of Dermatology.
LIMITATIONS:
Unmeasured factors including psychological perceptions, regional policies, and drug availability could influence drug survival, making the results not fully reflective of treatment effectiveness or safety. Most Humira users in registries were enrolled before biosimilars became available, making it impractical to match new users on the basis of treatment initiation years. Additionally, reasons for discontinuation were not available in the French National Health Data System.
DISCLOSURES:
In the United Kingdom, the research was funded by the Psoriasis Association PhD studentship and supported by the NIHR Manchester Biomedical Research Centre. In France, the authors are employees of the French National Health Insurance, the French National Agency for the Safety of Medicines and Health Products, and the Assistance Publique — Hôpitaux de Paris and received no funding. The authors reported receiving consulting and speaker fees and clinical trial sponsorship from various pharmaceutical companies. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
rate than those who remained on Humira.
METHODOLOGY:
- Researchers conducted a cohort study using data on patients with psoriasis who were treated with adalimumab, a tumor necrosis factor alpha inhibitor used to treat moderate to severe psoriasis, from the French National Health Data System, British Association of Dermatologists Biologics and Immunomodulators Register, and Spanish Registry of Systemic Therapy in Psoriasis.
- The analysis included 7387 adalimumab-naive patients who were new users of an adalimumab biosimilar and 3654 patients (switchers) who switched from Humira to a biosimilar. Patients were matched and compared with patients receiving Humira.
- Co-primary outcomes of the study were drug discontinuation and serious adverse events.
- Researchers assessed the following adalimumab biosimilar brands: Amgevita, Imraldi, Hyrimoz, Idacio, and Hulio.
TAKEAWAY:
- All-cause drug discontinuation rates were similar between new users of biosimilars and Humira new users (hazard ratio [HR], 0.99; 95% CI, 0.94-1.04).
- Discontinuation rates were higher among those who switched from Humira to a biosimilar (HR, 1.35; 95% CI, 1.19-1.52) than among those who stayed on Humira. Switching to Amgevita (HR, 1.25; 95% CI, 1.13-1.27), Imraldi (HR, 1.53; 95% CI, 1.33-1.76), and Hyrimoz (HR, 1.80; 95% CI, 1.29-2.52) was associated with higher discontinuation rates.
- Serious adverse events were not significantly different between new users of Humira and biosimilar new users (incidence rate ratio [IRR], 0.91; 95% CI, 0.80-1.05), and between patients who switched from a biosimilar to Humira and those who stayed on Humira (IRR, 0.92; 95% CI, 0.83-1.01).
- No significant differences in discontinuation because of ineffectiveness were found between biosimilar and Humira new users (HR, 0.97; 95% CI, 0.88-1.08). Discontinuation because of adverse events was also comparable for all biosimilars among new users, except for Hyrimoz (HR, 0.54; 95% CI, 0.35-0.85), which showed fewer discontinuations than Humira.
IN PRACTICE:
“This study found comparable drug survival and safety between adalimumab biosimilars and Humira in adalimumab-naive patients, supporting the use of biosimilars as viable alternatives for new patients,” the authors wrote. However, noting that discontinuation was more likely among those who switched from Humira to a biosimilar, they added: “Changes in treatment response, skin or injection site reactions, and nocebo effects may contribute to treatment discontinuation post-switch. Thus, patients who switch from Humira to biosimilars may require closer monitoring and support to alleviate these challenges.”
SOURCE:
The study was led by Duc Binh Phan, Dermatology Centre, Northern Care Alliance NHS Foundation Trust in Manchester, England. It was published online in The British Journal of Dermatology.
LIMITATIONS:
Unmeasured factors including psychological perceptions, regional policies, and drug availability could influence drug survival, making the results not fully reflective of treatment effectiveness or safety. Most Humira users in registries were enrolled before biosimilars became available, making it impractical to match new users on the basis of treatment initiation years. Additionally, reasons for discontinuation were not available in the French National Health Data System.
DISCLOSURES:
In the United Kingdom, the research was funded by the Psoriasis Association PhD studentship and supported by the NIHR Manchester Biomedical Research Centre. In France, the authors are employees of the French National Health Insurance, the French National Agency for the Safety of Medicines and Health Products, and the Assistance Publique — Hôpitaux de Paris and received no funding. The authors reported receiving consulting and speaker fees and clinical trial sponsorship from various pharmaceutical companies. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Post COVID-19, Long-term Risk for Autoimmune, Autoinflammatory Skin Disorders Increased, Study Finds
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Updated Guidance for Psoriatic Arthritis Ultrasound Comes at Time of Growing Use, New Technology
WASHINGTON — New draft guidance on the use of musculoskeletal ultrasound (MSUS) for diagnosis, monitoring, and prognosis of psoriatic arthritis was presented at the American College of Rheumatology (ACR) 2024 Annual Meeting. The new recommendations, intended to update 2012 guidance on rheumatologic use of MSUS, will go through another round of expert committee voting before being finalized and published.
“Even in the last 12 years, we’ve seen substantive advances, and there’s been significant improvements in musculoskeletal ultrasound technology,” Veena K. Ranganath, MD, professor of clinical medicine at the University of California, Los Angeles, and director of their Rheumatology Fellowship Musculoskeletal Ultrasound Training Program, told attendees. She noted that more than 30,000 articles on MSUS and arthritis have been published since the 2012 guidance. “We’ve seen mastery in teaching and really a wide distribution of this education to the next generation of rheumatologists, and this has led to significant increases in the use of musculoskeletal ultrasound in clinical practices.”
She also noted there have been significant improvements in therapeutic agents and strategies in psoriatic arthritis medications and that differences in today’s patients compared with those of a decade ago have influenced clinical questions related to the use of MSUS in rheumatology.
To develop the guidelines, a committee identified key domains and relevant clinical questions for ultrasonography using the PICO model (patient/population, intervention, comparison, and outcomes). A review of the literature published since 1993 in PubMed, Embase, and the Cochrane Database provided the evidence base, and a committee of 11 experts voted on the strength of the evidence for 22 statements. They rejected two that lacked consensus, and another round of voting will occur before the guidance is published.
Michael Stein, MD, assistant professor of medicine in rheumatology at McGill University in Montreal, Quebec, Canada, who was not involved in the guidance development, said he hopes and expects this new guidance will help persuade more clinicians to recognize the value of using MSUS in their practice.
“Number one, it’ll highlight the huge amount of data that exist that support using this technology for managing these groups of patients, among others, and I think it’ll also highlight the enormous number of questions that still exist that will hopefully be answered in the future, promoting new research,” Stein told this news organization.
“I do think it does allow people who are not comfortable with technology to adopt technology in a very gradual way and make it less threatening,” Stein added.
“Ultrasound is becoming part of the landscape, and so increasingly, we’re trying to promote it as being part of the standard of care, or at least an adjunct to care. I commend the committee for doing all this amazing work.”
Predicting and Diagnosing Early Psoriatic Arthritis
Catherine J. Bakewell, MD, a rheumatologist at Intermountain Health in Salt Lake City, Utah, reviewed the committee’s statements, starting with strong consensus that MSUS can help with diagnosing early psoriatic arthritis. Evidence has shown that patients with psoriasis who have subclinical synovitis, enthesitis, and other features have gone on to develop psoriatic arthritis, and researchers have documented the transition with ultrasonography.
“We can use it to enhance our CASPAR classification criteria” by using ultrasound to change how clinicians apply the classification criteria, Bakewell said. “For example, in order to go through those classification criteria, a patient has to have confirmed inflammatory articular disease, either the joint synthesis or spine, and ultrasound can help clarify that state for us.”
She also noted the potential for ultrasonography to help as a screening tool because studies have suggested that dermatologists’ use of handheld ultrasound transducers can help in screening appropriate patients to refer to rheumatologists.
Patients with psoriasis being evaluated for a potential early psoriatic arthritis diagnosis should undergo MSUS of the bilateral quadriceps tendon, patellar ligament, Achilles tendon, and plantar fascia entheses at a minimum, per moderate consensus.
“This truly is just designed to be the highest bang for your buck. This is designed for clinicians in practice,” Bakewell said. She noted criticism about the exclusion of upper extremities — something that will be discussed in the future published paper — but one reason that was excluded is because common findings have occurred in healthy individuals in some areas.
Moderate consensus also supported reliance on entheseal features — including hypoechogenicity, thickening, Doppler signal, bone erosions, enthesophytes/calcifications, and bursal enlargement — to support a diagnosis. Interpretation of entheseal changes in patients with psoriasis should take into account characteristics such as age, body mass index (BMI), and biomechanical stress.
“There are numerous articles already existing pointing out that people who are over the age of 50 with a BMI over 30 kg/m2 or who have higher levels of biomechanical stress will score more highly on endocytoscoring systems, even in the absence of an underlying disorder,” Bakewell said. Among the mitigating strategies proposed in the literature are to have at least three positive sites to qualify for an indication or to look at the specificity of each elementary lesion. “Whatever mitigating strategy the clinician chooses to use, they need to bear in mind some of these features are not exclusive to spondyloarthritis,” she said. “It has to be taken in the clinical context.”
Scanning the hand, wrist, foot, and relevant symptomatic joints with MSUS to diagnose early psoriatic arthritis in patients with psoriasis received strong consensus. Intracapsular findings of synovitis and erosions may help support an early diagnosis in patients with psoriasis. “These are not obviously specific to psoriatic arthritis but support the diagnosis” with moderate consensus, Bakewell said. “The more specific findings are these extracapsular findings — which did attain a strong level of consensus — which are enthesitis, tenosynovitis, and dactylitis, all supporting that diagnosis of early psoriatic arthritis.”
For patients with psoriatic arthritis, the cutoff for defining a positive joint received moderate consensus for grayscale (GS) of at least 2 or at least 1 with power Doppler (PD) of at least 1.
Strong consensus supported confirming the presence of dactylitis in patients with psoriasis or psoriatic arthritis through a combination of features including tenosynovitis, subcutaneous edema, soft tissue thickening, synovitis, paratenonitis, and pulley thickening.
“I will also note that enthesitis is missing from this definition of dactylitis,” Bakewell said. “It is, however, a feature that is detectable with those higher-frequency transducers, but this is a relatively early area of research and did not make it into this guidance statement.”
Moderate consensus supported determination of an increased risk of radiographic erosions in patients with a dactylitis PD score of at least 1.
“We know as far back as 2005, Brockbank et al taught us that the dactylitic digit is associated with radiographic erosion in that particular digit,” Bakewell said. “Flash forward all the way to 2021: Dubash et al published the paper, ‘Dactylitis is an indicator of a more severe phenotype independently associated with greater swollen joint counts, C-reactive protein, ultrasound synovitis, and erosive damage,’ showing us that this is more than just that particular digit. It is a more severe phenotype, and very minimal Doppler signal, just 1+, is associated with erosive damage.”
Progression of Psoriatic Arthritis and Shared Decision-Making
Strong consensus existed for all statements related to progression of psoriatic arthritis and the role of MSUS in shared decision-making. The first is that synovitis and enthesitis in MSUS can predict radiographic progression and worsening of patient-related outcomes. Second, sonographic features — including increased Doppler signal in synovitis, enthesitis, and tenosynovitis — and presence of bone erosions and dactylitis can help inform decisions regarding therapy escalation.
“This is the first treatment management–specific statement we have made, but we feel this to be justified because each of these ultrasonographic features is associated with overall inflammatory burden and worse outcomes, be it health assessment questionnaires, disability index, or patient-reported outcomes to harder endpoints, such as radiographic erosions or relapse of clinical remission,” Bakewell said.
Finally, MSUS can help inform patients of their disease activity to assist in shared decision-making regarding escalation or de-escalation of therapy.
“We’ve all had this in our practices. You’ve had the patient in front of you who is very inflamed, and they say, ‘Doctor, can’t I please use doTERRA oils? Do I really need to go on one of these toxic drugs? I’ve read the package insert,’” Bakewell said. “Aside from having that conversation about the relative risk–benefit of any individual medication that you recommend, it’s helpful to put the ultrasound transducer on the patient, show them the fire of the Doppler, show them the erosion, show them the damage that is being done. It comes to life for them, especially if they’re not suffering that much with pain or stiffness.”
Bakewell also addressed patients at the other end of the pain spectrum who are suffering more. “You’ve also probably had the patient with psoriatic arthritis and fibromyalgia who comes in and tells you, ‘Doctor, my psoriatic arthritis has been terrible. I’m flaring. I need more immune-suppressing medication,’” she said. “Their exam looks pretty good, and it’s helpful to put that transducer on them and show them the absence of Doppler signal, show them that you’re taking them very seriously. You didn’t just squeeze them and say they’re fine, but you looked more deeply. You looked underneath the skin, and that helps with that patient–provider understanding and communication. I use this every day.”
Clarifying Disease State and Defining Remission
As with patients with psoriasis undergoing evaluation, there was strong consensus for interpreting entheseal changes in psoriatic arthritis in the context of patient characteristics such as age, BMI, and biomechanical stress.
There was moderate consensus for confirming psoriatic arthritis flare with MSUS. Bakewell noted that many have seen in their practices how physical exams can be misleading, such as when a patient appears clinically normal but has ongoing synovitis, or on the flip side, the patient has a swollen joint but nothing is lighting up with Doppler on the ultrasound.
All of the statements on MSUS for remission received moderate consensus. These included defining MSUS remission as a PD score of 0 in entheses and synovial tissues and defining ultrasonographic remission as a total PD ultrasound score of 0, summing all analyzed joints and entheses, at a single given time point.
When using MSUS to evaluate for remission, it’s reasonable to screen the lower-extremity entheses, wrists, metacarpophalangeal joints, interphalangeal hand joints, metatarsophalangeal joints, and relevant symptomatic joints. The inflammatory features to evaluate to confirm ultrasound-defined remission include PD enthesitis, GS and PD synovitis, tenosynovitis, and dactylitis. Finally, for those in remission, subclinical inflammation detected by MSUS likely predicts a higher rate of flare.
During the discussion, Bakewell reiterated that MSUS should be regarded as a tool for patient subsets who can benefit from its use, rather than being used routinely across large patient groups without a clear purpose. “It’s used to answer a question,” she said. “If you’re going to demonstrate the efficacy of a tool, you have to use it appropriately, aka when there’s a question. We don’t need to ultrasound every patient every visit.”
No external funding for the development of the guidance was noted. Ranganath has reported receiving research support from Bristol Myers Squibb and Mallinckrodt. Bakewell has reported receiving speaking/consulting fees from AbbVie, UCB, Lilly, Janssen, Novartis, Sanofi/Regeneron/Genzyme, and Pfizer. Stein had no disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON — New draft guidance on the use of musculoskeletal ultrasound (MSUS) for diagnosis, monitoring, and prognosis of psoriatic arthritis was presented at the American College of Rheumatology (ACR) 2024 Annual Meeting. The new recommendations, intended to update 2012 guidance on rheumatologic use of MSUS, will go through another round of expert committee voting before being finalized and published.
“Even in the last 12 years, we’ve seen substantive advances, and there’s been significant improvements in musculoskeletal ultrasound technology,” Veena K. Ranganath, MD, professor of clinical medicine at the University of California, Los Angeles, and director of their Rheumatology Fellowship Musculoskeletal Ultrasound Training Program, told attendees. She noted that more than 30,000 articles on MSUS and arthritis have been published since the 2012 guidance. “We’ve seen mastery in teaching and really a wide distribution of this education to the next generation of rheumatologists, and this has led to significant increases in the use of musculoskeletal ultrasound in clinical practices.”
She also noted there have been significant improvements in therapeutic agents and strategies in psoriatic arthritis medications and that differences in today’s patients compared with those of a decade ago have influenced clinical questions related to the use of MSUS in rheumatology.
To develop the guidelines, a committee identified key domains and relevant clinical questions for ultrasonography using the PICO model (patient/population, intervention, comparison, and outcomes). A review of the literature published since 1993 in PubMed, Embase, and the Cochrane Database provided the evidence base, and a committee of 11 experts voted on the strength of the evidence for 22 statements. They rejected two that lacked consensus, and another round of voting will occur before the guidance is published.
Michael Stein, MD, assistant professor of medicine in rheumatology at McGill University in Montreal, Quebec, Canada, who was not involved in the guidance development, said he hopes and expects this new guidance will help persuade more clinicians to recognize the value of using MSUS in their practice.
“Number one, it’ll highlight the huge amount of data that exist that support using this technology for managing these groups of patients, among others, and I think it’ll also highlight the enormous number of questions that still exist that will hopefully be answered in the future, promoting new research,” Stein told this news organization.
“I do think it does allow people who are not comfortable with technology to adopt technology in a very gradual way and make it less threatening,” Stein added.
“Ultrasound is becoming part of the landscape, and so increasingly, we’re trying to promote it as being part of the standard of care, or at least an adjunct to care. I commend the committee for doing all this amazing work.”
Predicting and Diagnosing Early Psoriatic Arthritis
Catherine J. Bakewell, MD, a rheumatologist at Intermountain Health in Salt Lake City, Utah, reviewed the committee’s statements, starting with strong consensus that MSUS can help with diagnosing early psoriatic arthritis. Evidence has shown that patients with psoriasis who have subclinical synovitis, enthesitis, and other features have gone on to develop psoriatic arthritis, and researchers have documented the transition with ultrasonography.
“We can use it to enhance our CASPAR classification criteria” by using ultrasound to change how clinicians apply the classification criteria, Bakewell said. “For example, in order to go through those classification criteria, a patient has to have confirmed inflammatory articular disease, either the joint synthesis or spine, and ultrasound can help clarify that state for us.”
She also noted the potential for ultrasonography to help as a screening tool because studies have suggested that dermatologists’ use of handheld ultrasound transducers can help in screening appropriate patients to refer to rheumatologists.
Patients with psoriasis being evaluated for a potential early psoriatic arthritis diagnosis should undergo MSUS of the bilateral quadriceps tendon, patellar ligament, Achilles tendon, and plantar fascia entheses at a minimum, per moderate consensus.
“This truly is just designed to be the highest bang for your buck. This is designed for clinicians in practice,” Bakewell said. She noted criticism about the exclusion of upper extremities — something that will be discussed in the future published paper — but one reason that was excluded is because common findings have occurred in healthy individuals in some areas.
Moderate consensus also supported reliance on entheseal features — including hypoechogenicity, thickening, Doppler signal, bone erosions, enthesophytes/calcifications, and bursal enlargement — to support a diagnosis. Interpretation of entheseal changes in patients with psoriasis should take into account characteristics such as age, body mass index (BMI), and biomechanical stress.
“There are numerous articles already existing pointing out that people who are over the age of 50 with a BMI over 30 kg/m2 or who have higher levels of biomechanical stress will score more highly on endocytoscoring systems, even in the absence of an underlying disorder,” Bakewell said. Among the mitigating strategies proposed in the literature are to have at least three positive sites to qualify for an indication or to look at the specificity of each elementary lesion. “Whatever mitigating strategy the clinician chooses to use, they need to bear in mind some of these features are not exclusive to spondyloarthritis,” she said. “It has to be taken in the clinical context.”
Scanning the hand, wrist, foot, and relevant symptomatic joints with MSUS to diagnose early psoriatic arthritis in patients with psoriasis received strong consensus. Intracapsular findings of synovitis and erosions may help support an early diagnosis in patients with psoriasis. “These are not obviously specific to psoriatic arthritis but support the diagnosis” with moderate consensus, Bakewell said. “The more specific findings are these extracapsular findings — which did attain a strong level of consensus — which are enthesitis, tenosynovitis, and dactylitis, all supporting that diagnosis of early psoriatic arthritis.”
For patients with psoriatic arthritis, the cutoff for defining a positive joint received moderate consensus for grayscale (GS) of at least 2 or at least 1 with power Doppler (PD) of at least 1.
Strong consensus supported confirming the presence of dactylitis in patients with psoriasis or psoriatic arthritis through a combination of features including tenosynovitis, subcutaneous edema, soft tissue thickening, synovitis, paratenonitis, and pulley thickening.
“I will also note that enthesitis is missing from this definition of dactylitis,” Bakewell said. “It is, however, a feature that is detectable with those higher-frequency transducers, but this is a relatively early area of research and did not make it into this guidance statement.”
Moderate consensus supported determination of an increased risk of radiographic erosions in patients with a dactylitis PD score of at least 1.
“We know as far back as 2005, Brockbank et al taught us that the dactylitic digit is associated with radiographic erosion in that particular digit,” Bakewell said. “Flash forward all the way to 2021: Dubash et al published the paper, ‘Dactylitis is an indicator of a more severe phenotype independently associated with greater swollen joint counts, C-reactive protein, ultrasound synovitis, and erosive damage,’ showing us that this is more than just that particular digit. It is a more severe phenotype, and very minimal Doppler signal, just 1+, is associated with erosive damage.”
Progression of Psoriatic Arthritis and Shared Decision-Making
Strong consensus existed for all statements related to progression of psoriatic arthritis and the role of MSUS in shared decision-making. The first is that synovitis and enthesitis in MSUS can predict radiographic progression and worsening of patient-related outcomes. Second, sonographic features — including increased Doppler signal in synovitis, enthesitis, and tenosynovitis — and presence of bone erosions and dactylitis can help inform decisions regarding therapy escalation.
“This is the first treatment management–specific statement we have made, but we feel this to be justified because each of these ultrasonographic features is associated with overall inflammatory burden and worse outcomes, be it health assessment questionnaires, disability index, or patient-reported outcomes to harder endpoints, such as radiographic erosions or relapse of clinical remission,” Bakewell said.
Finally, MSUS can help inform patients of their disease activity to assist in shared decision-making regarding escalation or de-escalation of therapy.
“We’ve all had this in our practices. You’ve had the patient in front of you who is very inflamed, and they say, ‘Doctor, can’t I please use doTERRA oils? Do I really need to go on one of these toxic drugs? I’ve read the package insert,’” Bakewell said. “Aside from having that conversation about the relative risk–benefit of any individual medication that you recommend, it’s helpful to put the ultrasound transducer on the patient, show them the fire of the Doppler, show them the erosion, show them the damage that is being done. It comes to life for them, especially if they’re not suffering that much with pain or stiffness.”
Bakewell also addressed patients at the other end of the pain spectrum who are suffering more. “You’ve also probably had the patient with psoriatic arthritis and fibromyalgia who comes in and tells you, ‘Doctor, my psoriatic arthritis has been terrible. I’m flaring. I need more immune-suppressing medication,’” she said. “Their exam looks pretty good, and it’s helpful to put that transducer on them and show them the absence of Doppler signal, show them that you’re taking them very seriously. You didn’t just squeeze them and say they’re fine, but you looked more deeply. You looked underneath the skin, and that helps with that patient–provider understanding and communication. I use this every day.”
Clarifying Disease State and Defining Remission
As with patients with psoriasis undergoing evaluation, there was strong consensus for interpreting entheseal changes in psoriatic arthritis in the context of patient characteristics such as age, BMI, and biomechanical stress.
There was moderate consensus for confirming psoriatic arthritis flare with MSUS. Bakewell noted that many have seen in their practices how physical exams can be misleading, such as when a patient appears clinically normal but has ongoing synovitis, or on the flip side, the patient has a swollen joint but nothing is lighting up with Doppler on the ultrasound.
All of the statements on MSUS for remission received moderate consensus. These included defining MSUS remission as a PD score of 0 in entheses and synovial tissues and defining ultrasonographic remission as a total PD ultrasound score of 0, summing all analyzed joints and entheses, at a single given time point.
When using MSUS to evaluate for remission, it’s reasonable to screen the lower-extremity entheses, wrists, metacarpophalangeal joints, interphalangeal hand joints, metatarsophalangeal joints, and relevant symptomatic joints. The inflammatory features to evaluate to confirm ultrasound-defined remission include PD enthesitis, GS and PD synovitis, tenosynovitis, and dactylitis. Finally, for those in remission, subclinical inflammation detected by MSUS likely predicts a higher rate of flare.
During the discussion, Bakewell reiterated that MSUS should be regarded as a tool for patient subsets who can benefit from its use, rather than being used routinely across large patient groups without a clear purpose. “It’s used to answer a question,” she said. “If you’re going to demonstrate the efficacy of a tool, you have to use it appropriately, aka when there’s a question. We don’t need to ultrasound every patient every visit.”
No external funding for the development of the guidance was noted. Ranganath has reported receiving research support from Bristol Myers Squibb and Mallinckrodt. Bakewell has reported receiving speaking/consulting fees from AbbVie, UCB, Lilly, Janssen, Novartis, Sanofi/Regeneron/Genzyme, and Pfizer. Stein had no disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON — New draft guidance on the use of musculoskeletal ultrasound (MSUS) for diagnosis, monitoring, and prognosis of psoriatic arthritis was presented at the American College of Rheumatology (ACR) 2024 Annual Meeting. The new recommendations, intended to update 2012 guidance on rheumatologic use of MSUS, will go through another round of expert committee voting before being finalized and published.
“Even in the last 12 years, we’ve seen substantive advances, and there’s been significant improvements in musculoskeletal ultrasound technology,” Veena K. Ranganath, MD, professor of clinical medicine at the University of California, Los Angeles, and director of their Rheumatology Fellowship Musculoskeletal Ultrasound Training Program, told attendees. She noted that more than 30,000 articles on MSUS and arthritis have been published since the 2012 guidance. “We’ve seen mastery in teaching and really a wide distribution of this education to the next generation of rheumatologists, and this has led to significant increases in the use of musculoskeletal ultrasound in clinical practices.”
She also noted there have been significant improvements in therapeutic agents and strategies in psoriatic arthritis medications and that differences in today’s patients compared with those of a decade ago have influenced clinical questions related to the use of MSUS in rheumatology.
To develop the guidelines, a committee identified key domains and relevant clinical questions for ultrasonography using the PICO model (patient/population, intervention, comparison, and outcomes). A review of the literature published since 1993 in PubMed, Embase, and the Cochrane Database provided the evidence base, and a committee of 11 experts voted on the strength of the evidence for 22 statements. They rejected two that lacked consensus, and another round of voting will occur before the guidance is published.
Michael Stein, MD, assistant professor of medicine in rheumatology at McGill University in Montreal, Quebec, Canada, who was not involved in the guidance development, said he hopes and expects this new guidance will help persuade more clinicians to recognize the value of using MSUS in their practice.
“Number one, it’ll highlight the huge amount of data that exist that support using this technology for managing these groups of patients, among others, and I think it’ll also highlight the enormous number of questions that still exist that will hopefully be answered in the future, promoting new research,” Stein told this news organization.
“I do think it does allow people who are not comfortable with technology to adopt technology in a very gradual way and make it less threatening,” Stein added.
“Ultrasound is becoming part of the landscape, and so increasingly, we’re trying to promote it as being part of the standard of care, or at least an adjunct to care. I commend the committee for doing all this amazing work.”
Predicting and Diagnosing Early Psoriatic Arthritis
Catherine J. Bakewell, MD, a rheumatologist at Intermountain Health in Salt Lake City, Utah, reviewed the committee’s statements, starting with strong consensus that MSUS can help with diagnosing early psoriatic arthritis. Evidence has shown that patients with psoriasis who have subclinical synovitis, enthesitis, and other features have gone on to develop psoriatic arthritis, and researchers have documented the transition with ultrasonography.
“We can use it to enhance our CASPAR classification criteria” by using ultrasound to change how clinicians apply the classification criteria, Bakewell said. “For example, in order to go through those classification criteria, a patient has to have confirmed inflammatory articular disease, either the joint synthesis or spine, and ultrasound can help clarify that state for us.”
She also noted the potential for ultrasonography to help as a screening tool because studies have suggested that dermatologists’ use of handheld ultrasound transducers can help in screening appropriate patients to refer to rheumatologists.
Patients with psoriasis being evaluated for a potential early psoriatic arthritis diagnosis should undergo MSUS of the bilateral quadriceps tendon, patellar ligament, Achilles tendon, and plantar fascia entheses at a minimum, per moderate consensus.
“This truly is just designed to be the highest bang for your buck. This is designed for clinicians in practice,” Bakewell said. She noted criticism about the exclusion of upper extremities — something that will be discussed in the future published paper — but one reason that was excluded is because common findings have occurred in healthy individuals in some areas.
Moderate consensus also supported reliance on entheseal features — including hypoechogenicity, thickening, Doppler signal, bone erosions, enthesophytes/calcifications, and bursal enlargement — to support a diagnosis. Interpretation of entheseal changes in patients with psoriasis should take into account characteristics such as age, body mass index (BMI), and biomechanical stress.
“There are numerous articles already existing pointing out that people who are over the age of 50 with a BMI over 30 kg/m2 or who have higher levels of biomechanical stress will score more highly on endocytoscoring systems, even in the absence of an underlying disorder,” Bakewell said. Among the mitigating strategies proposed in the literature are to have at least three positive sites to qualify for an indication or to look at the specificity of each elementary lesion. “Whatever mitigating strategy the clinician chooses to use, they need to bear in mind some of these features are not exclusive to spondyloarthritis,” she said. “It has to be taken in the clinical context.”
Scanning the hand, wrist, foot, and relevant symptomatic joints with MSUS to diagnose early psoriatic arthritis in patients with psoriasis received strong consensus. Intracapsular findings of synovitis and erosions may help support an early diagnosis in patients with psoriasis. “These are not obviously specific to psoriatic arthritis but support the diagnosis” with moderate consensus, Bakewell said. “The more specific findings are these extracapsular findings — which did attain a strong level of consensus — which are enthesitis, tenosynovitis, and dactylitis, all supporting that diagnosis of early psoriatic arthritis.”
For patients with psoriatic arthritis, the cutoff for defining a positive joint received moderate consensus for grayscale (GS) of at least 2 or at least 1 with power Doppler (PD) of at least 1.
Strong consensus supported confirming the presence of dactylitis in patients with psoriasis or psoriatic arthritis through a combination of features including tenosynovitis, subcutaneous edema, soft tissue thickening, synovitis, paratenonitis, and pulley thickening.
“I will also note that enthesitis is missing from this definition of dactylitis,” Bakewell said. “It is, however, a feature that is detectable with those higher-frequency transducers, but this is a relatively early area of research and did not make it into this guidance statement.”
Moderate consensus supported determination of an increased risk of radiographic erosions in patients with a dactylitis PD score of at least 1.
“We know as far back as 2005, Brockbank et al taught us that the dactylitic digit is associated with radiographic erosion in that particular digit,” Bakewell said. “Flash forward all the way to 2021: Dubash et al published the paper, ‘Dactylitis is an indicator of a more severe phenotype independently associated with greater swollen joint counts, C-reactive protein, ultrasound synovitis, and erosive damage,’ showing us that this is more than just that particular digit. It is a more severe phenotype, and very minimal Doppler signal, just 1+, is associated with erosive damage.”
Progression of Psoriatic Arthritis and Shared Decision-Making
Strong consensus existed for all statements related to progression of psoriatic arthritis and the role of MSUS in shared decision-making. The first is that synovitis and enthesitis in MSUS can predict radiographic progression and worsening of patient-related outcomes. Second, sonographic features — including increased Doppler signal in synovitis, enthesitis, and tenosynovitis — and presence of bone erosions and dactylitis can help inform decisions regarding therapy escalation.
“This is the first treatment management–specific statement we have made, but we feel this to be justified because each of these ultrasonographic features is associated with overall inflammatory burden and worse outcomes, be it health assessment questionnaires, disability index, or patient-reported outcomes to harder endpoints, such as radiographic erosions or relapse of clinical remission,” Bakewell said.
Finally, MSUS can help inform patients of their disease activity to assist in shared decision-making regarding escalation or de-escalation of therapy.
“We’ve all had this in our practices. You’ve had the patient in front of you who is very inflamed, and they say, ‘Doctor, can’t I please use doTERRA oils? Do I really need to go on one of these toxic drugs? I’ve read the package insert,’” Bakewell said. “Aside from having that conversation about the relative risk–benefit of any individual medication that you recommend, it’s helpful to put the ultrasound transducer on the patient, show them the fire of the Doppler, show them the erosion, show them the damage that is being done. It comes to life for them, especially if they’re not suffering that much with pain or stiffness.”
Bakewell also addressed patients at the other end of the pain spectrum who are suffering more. “You’ve also probably had the patient with psoriatic arthritis and fibromyalgia who comes in and tells you, ‘Doctor, my psoriatic arthritis has been terrible. I’m flaring. I need more immune-suppressing medication,’” she said. “Their exam looks pretty good, and it’s helpful to put that transducer on them and show them the absence of Doppler signal, show them that you’re taking them very seriously. You didn’t just squeeze them and say they’re fine, but you looked more deeply. You looked underneath the skin, and that helps with that patient–provider understanding and communication. I use this every day.”
Clarifying Disease State and Defining Remission
As with patients with psoriasis undergoing evaluation, there was strong consensus for interpreting entheseal changes in psoriatic arthritis in the context of patient characteristics such as age, BMI, and biomechanical stress.
There was moderate consensus for confirming psoriatic arthritis flare with MSUS. Bakewell noted that many have seen in their practices how physical exams can be misleading, such as when a patient appears clinically normal but has ongoing synovitis, or on the flip side, the patient has a swollen joint but nothing is lighting up with Doppler on the ultrasound.
All of the statements on MSUS for remission received moderate consensus. These included defining MSUS remission as a PD score of 0 in entheses and synovial tissues and defining ultrasonographic remission as a total PD ultrasound score of 0, summing all analyzed joints and entheses, at a single given time point.
When using MSUS to evaluate for remission, it’s reasonable to screen the lower-extremity entheses, wrists, metacarpophalangeal joints, interphalangeal hand joints, metatarsophalangeal joints, and relevant symptomatic joints. The inflammatory features to evaluate to confirm ultrasound-defined remission include PD enthesitis, GS and PD synovitis, tenosynovitis, and dactylitis. Finally, for those in remission, subclinical inflammation detected by MSUS likely predicts a higher rate of flare.
During the discussion, Bakewell reiterated that MSUS should be regarded as a tool for patient subsets who can benefit from its use, rather than being used routinely across large patient groups without a clear purpose. “It’s used to answer a question,” she said. “If you’re going to demonstrate the efficacy of a tool, you have to use it appropriately, aka when there’s a question. We don’t need to ultrasound every patient every visit.”
No external funding for the development of the guidance was noted. Ranganath has reported receiving research support from Bristol Myers Squibb and Mallinckrodt. Bakewell has reported receiving speaking/consulting fees from AbbVie, UCB, Lilly, Janssen, Novartis, Sanofi/Regeneron/Genzyme, and Pfizer. Stein had no disclosures.
A version of this article first appeared on Medscape.com.
FROM ACR 2024
Eating Disorder Risk Factors and the Impact of Obesity in Patients With Psoriasis
Psoriasis is a chronic multisystemic inflammatory skin disease with a worldwide prevalence of 2% to 3%.1 Psoriasis can be accompanied by other conditions such as psoriatic arthritis, obesity, metabolic syndrome, diabetes mellitus, hypertension, dyslipidemia, atherosclerotic disease, inflammatory bowel disease, and anxiety/depression. It is important to manage comorbidities of psoriasis in addition to treating the cutaneous manifestations of the disease.1
Obesity is a major public health concern worldwide. Numerous observational and epidemiologic studies have reported a high prevalence of obesity among patients with psoriasis.2 Current evidence indicates that obesity may initiate or worsen psoriasis; furthermore, it is important to note that obesity may negatively impact the effectiveness of psoriasis-specific treatments or increase the incidence of adverse effects. Therefore, managing obesity is crucial in the treatment of psoriasis.3 Numerous studies have investigated the association between psoriasis and obesity, and they commonly conclude that both conditions share the same genetic metabolic pathways.2-4 However, it is important to consider environmental factors such as dietary habits, smoking, alcohol consumption, and a sedentary lifestyle—all of which are associated with psoriasis and also can contribute to the development of obesity.5 Because of the effects of obesity in psoriasis patients, factors that impact the development of obesity have become a popular research topic.
Eating disorders (EDs) are a crucial risk factor for both developing and maintaining obesity. In particular, two EDs that are associated with obesity include binge eating disorder and bulimia nervosa.6 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,7 binge eating disorder can be diagnosed when a patient has at least 1 episode of binge eating per week over a 3-month period. Bulimia nervosa can be diagnosed when a patient is excessively concerned with their body weight and shape and engages in behaviors to prevent weight gain (eg, forced vomiting, excessive use of laxatives).7 Psychiatrists who specialize in EDs make diagnoses based on these criteria. In daily practice, there are several quick and simple questionnaires available to screen for EDs that can be used by nonpsychiatrist physicians, including the commonly used 26-item Eating Attitudes Test (EAT-26).8 The EAT-26 has been used to screen for EDs in patients with inflammatory disorders.9
The aim of this study was to screen for EDs in patients with psoriasis to identify potential risk factors for development of obesity.
Materials and Methods
This study included patients with psoriasis who were screened for EDs at a tertiary dermatology clinic in Turkey between January 2021 and December 2023. This study was approved by the local ethics committee and was in accordance with the Declaration of Helsinki (decision number E-93471371-514.99-225000079).
Study Design and Patient Inclusion Criteria—This quantitative cross-sectional study utilized EAT-26, Dermatology Life Quality Index (DLQI), Attitude Scale for Healthy Nutrition (ASHN), and Depression Anxiety Stress Scale-21 (DASS-21) scores. All the questionnaire scales used in the study were adapted and validated in Turkey.8,10-12 The inclusion criteria consisted of being older than 18 years of age, being literate, having psoriasis for at least 1 year that was not treated topically or systemically, and having no psychiatric diseases outside an ED. The questionnaires were presented in written format following the clinical examination. Literacy was an inclusion criterion in this study due to the absence of auxiliary health personnel.
Study Variables—The study variables included age, sex, marital status (single/divorced or married), education status (primary/secondary school or high school/university), employment status (employed or unemployed/retired), body mass index (BMI), smoking status, alcohol-consumption status, Psoriasis Area Severity Index score, presence of nail psoriasis and psoriatic arthritis, duration of psoriasis, family history of psoriasis, EAT-26 score, ASHN score, DLQI score, and DASS-21 score. Body mass index was calculated by taking a participant’s weight in kilograms and dividing it by their height in meters squared. The BMI values were classified into 3 categories: normal (18.5–24.9 kg/m2), overweight (25.0–29.9 kg/m2), and obese (≥30 kg/m2).13
Questionnaires—The EAT-26 questionnaire includes 26 questions that are used to detect EDs. Responses to each question include Likert-type answer options (ie, “always,” “usually,” “often,” “sometimes,” “rarely,” and “never.”) Patients with scores of 20 points or higher (range, 0–78) are classified as high risk for EDs.8 In our study, EAT-26 scores were grouped into 2 categories: patients scoring less than 20 points and those scoring 20 points or higher.
The DLQI questionnaire includes 10 questions to measure dermatologic symptoms and qualiy of life. Responses to each question include Likert-type answer options (ie, “not at all,” “a little,” “a lot,” or “very much.”) On the DLQI scale, the higher the score, the lower the quality of life (score range, 0–30).10
The ASHN questionnaire includes 21 questions that measure attitudes toward healthy nutrition with 5 possible answer options (“strongly disagree,” “disagree,” “undecided,” “agree,” and “strongly agree”). On this scale, higher scores indicate the participant is more knowledgeable about healthy nutrition (score range, 0–78).11
The DASS-21 questionnaire includes 21 questions that measure the severity of a range of symptoms common to depression, anxiety, and stress. Responses include Likert-type answer options (eg, “never,” “sometimes,” “often,” and “almost always.”) On this scale, a higher score (range of 0–21 for each) indicates higher levels of depression, anxiety, and stress.12
Statistical Analysis—Descriptive statistics were analyzed using SPSS software version 22.0 (IBM). The Shapiro-Wilk test was applied to determine whether the data were normally distributed. For categorical variables, frequency differences among groups were compared using the Pearson χ2 test. A t test was used to compare the means of 2 independent groups with a normal distribution. One-way analysis of variance and Tukey Honest Significant Difference post hoc analysis were used to test whether there was a statistically significant difference among the normally distributed means of independent groups. Pearson correlation analysis was used to determine whether there was a linear relationship between 2 numeric measurements and, if so, to determine the direction and severity of this relationship. P<.05 indicated statistical significance in this study.
Results
Study Participant Demographics—This study included 82 participants with a mean age of 44.3 years; 52.4% (43/82) were female, and 85.4% (70/82) were married. The questionnaire took an average of 4.2 minutes for participants to complete. A total of 57.3% (47/82) of patients had completed primary/secondary education and 59.8% (49/82) were employed. The mean BMI was 28.1 kg/m2. According to the BMI classification, 26.8% (22/82) participants had a normal weight, 36.6% (30/82) were overweight, and 43.9% (36/82) were obese. A total of 48.8% (40/82) of participants smoked, and 4.9% (4/82) consumed alcohol. The mean Psoriasis Area and Severity Index score was 5.4. A total of 54.9% (45/82) of participants had nail psoriasis, and 24.4% (20/82) had psoriatic arthritis. The mean duration of psoriasis was 153 months. A total of 29.3% (24/82) of participants had a positive family history of psoriasis. The mean EAT-26 score was 11.1. A total of 12.2% (10/82) of participants had an EAT-26 score of 20 points or higher and were considered at high risk for an ED. The mean ASHN score was 72.9; the mean DLQI score was 5.5; and on the DASS-21 scale, mean scores for depression, anxiety, and stress were 6.3, 8.7, and 10.0, respectively (Table).
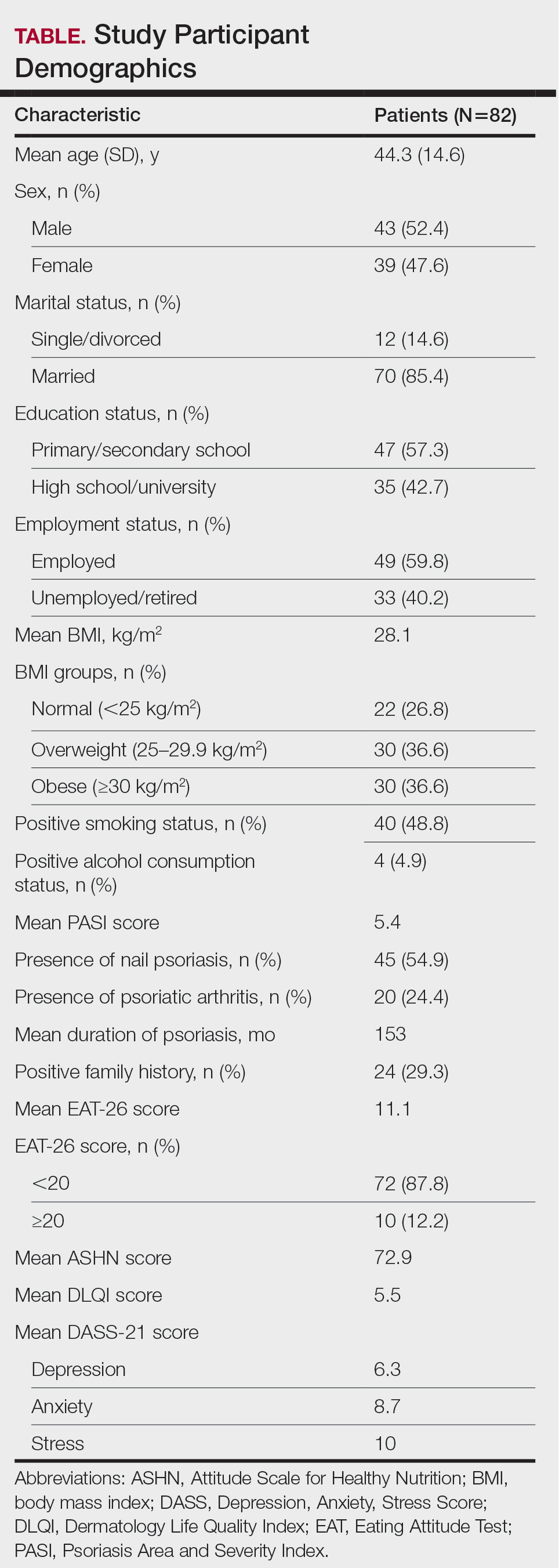
Comparative Evaluation of the BMI Groups—The only statistically significant differences among the 3 BMI groups were related to marital status, EAT-26 score, and anxiety and stress scores (P=.02, <.01, <.01, and <.01, respectively)(eTable 1). The number of single/divorced participants in the overweight group was significantly (P=.02) greater than in the normal weight group. The mean EAT-26 score for the normal weight group was significantly (P<.01) lower than for the overweight and obese groups; there was no significant difference in mean EAT-26 scores between the overweight and obese groups. The mean anxiety score was significantly (P<.01) lower in the normal weight group compared with the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean depression score. The mean stress and anxiety scores were significantly (P<.01) lower in the normal weight group than in the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean anxiety score.
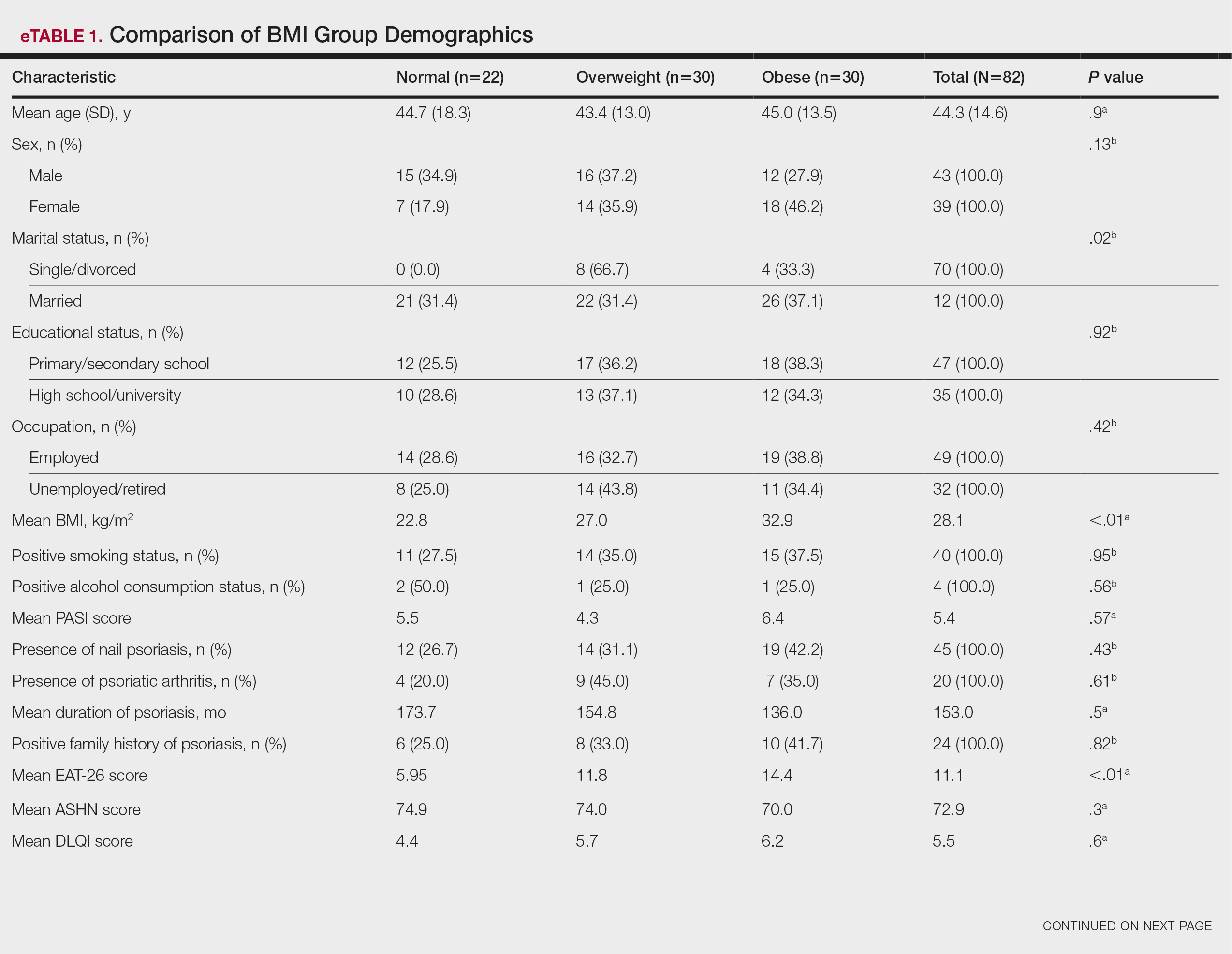
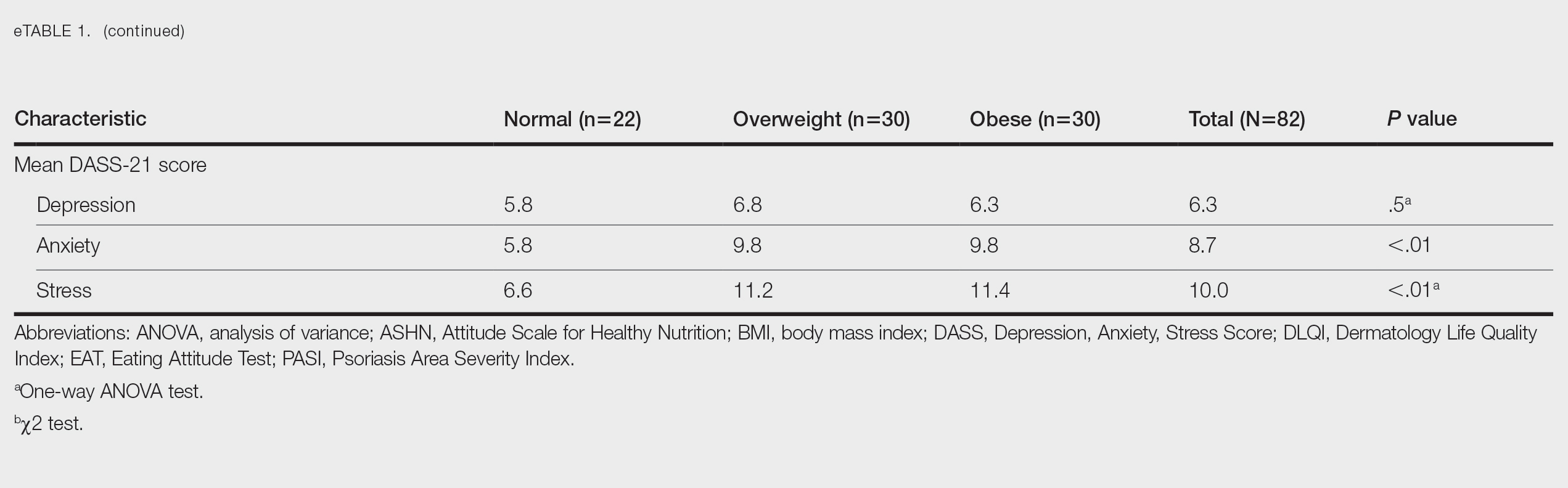
Comparative Evaluation of the EAT-26 Scores—There were statistically significant differences among the EAT-26 scores related to sex; BMI; and depression, anxiety, and stress scores (P=.04, .02, <.01, <.01, and <.01, respectively). The number of females in the group with a score of 20 points or higher was significantly (P=.04) less than that in the group scoring less than 20 points. The mean BMI in the group with a score of 20 points or higher was significantly (P=.02) greater than in group scoring less than 20 points. The mean depression, anxiety, and stress scores of the group scoring 20 points or higher were significantly (P<.01 for all) greater than in the group scoring less than 20 points (eTable 2).
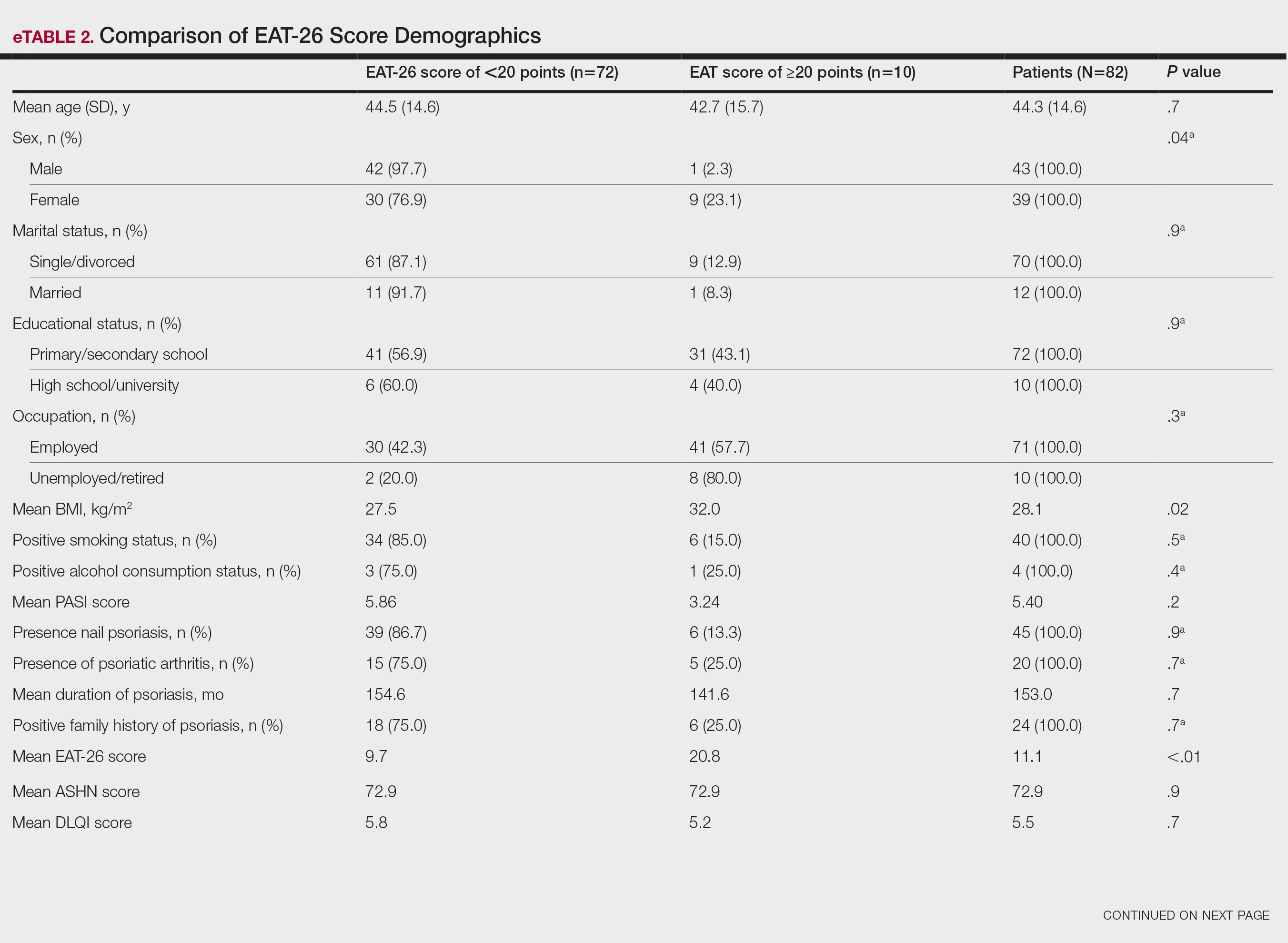
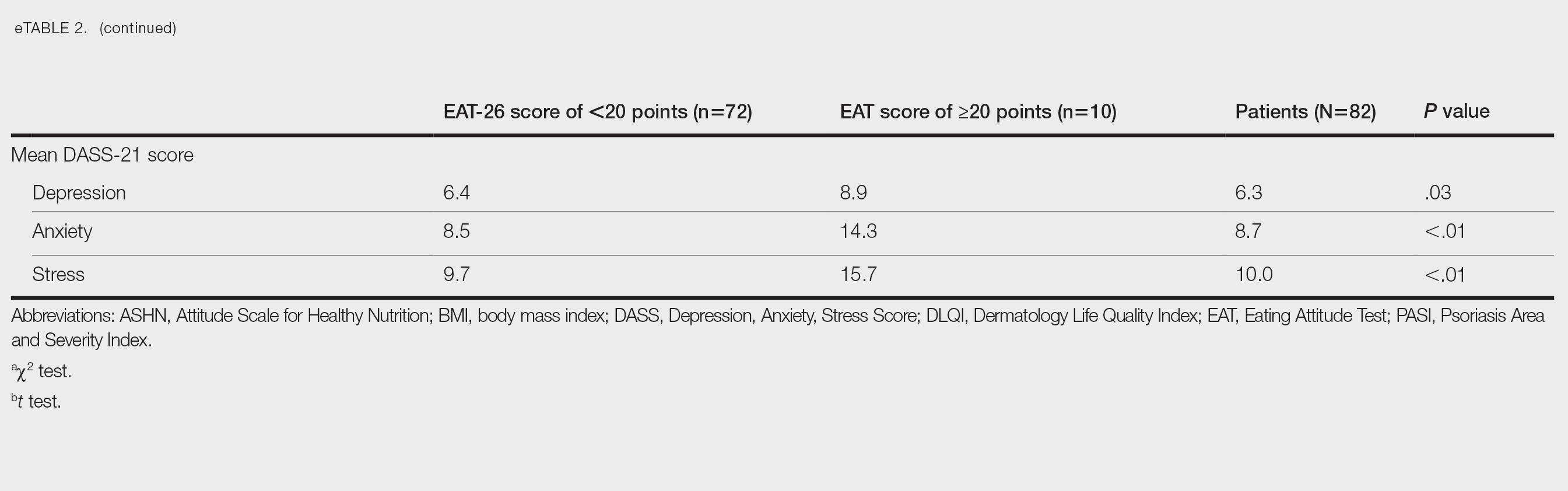
Correlation Analysis of the Study Variables—The EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress (P<.01 for all)(eTable 3).
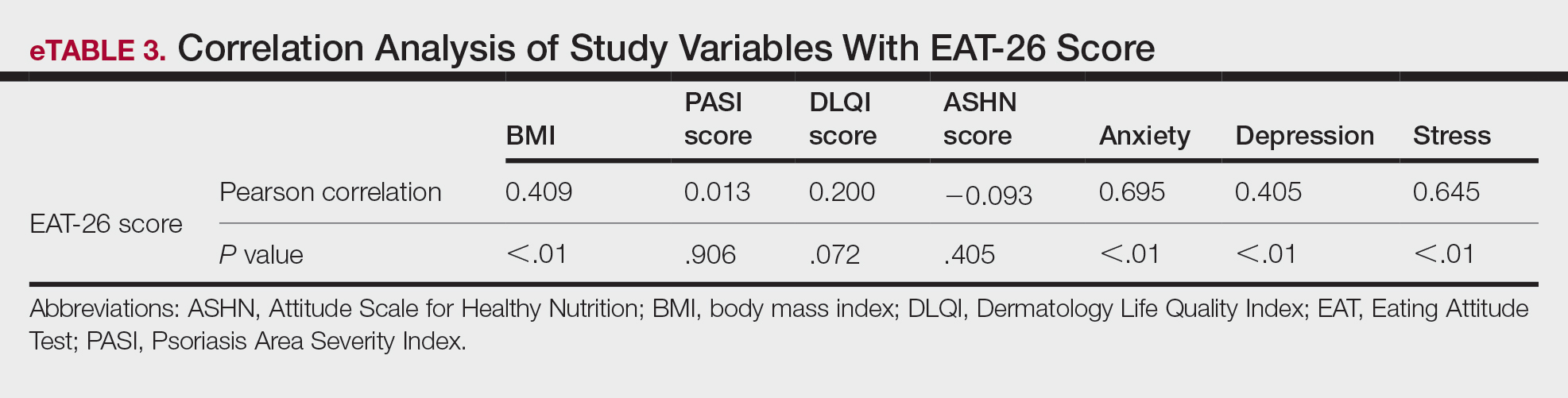
Comment
Eating disorders are psychiatric conditions that require a multidisciplinary approach. Nonpsychiatric medical departments may be involved due to the severe consequences (eg, various skin changes14) of these disorders. Psoriasis is not known to be directly affected by the presence of an ED; however, it is possible that EDs could indirectly affect patients with psoriasis by influencing obesity. Therefore, this study aimed to examine the relationship between ED risk factors and obesity in this population.
The relationship between psoriasis and obesity has been a popular research topic in dermatology since the 1990s.15 Epidemiologic and observational studies have reported that patients with psoriasis are more likely to be overweight or have obesity, which is an independent risk factor for psoriasis.3,16 However, the causal relationship between psoriasis and obesity remains unclear. In a comprehensive review, Barros et al17 emphasized the causal relationship between obesity and psoriasis under several headings. Firstly, a higher BMI increases the risk for psoriasis by promoting cytokine release and immune system dysregulation. Secondly, a Western diet (eg, processed foods and fast food) triggers obesity and psoriasis by increasing adipose tissue. Thirdly, the alteration of the skin and gut microbiota triggers chronic inflammation as a result of bacterial translocation in patients with obesity. Fourthly, a high-fat diet and palmitic acid disrupt the intestinal integrity of the gut and increase the risk for psoriasis and obesity by triggering chronic inflammation of bacterial fragments that pass into the blood. Finally, the decrease in the amount of adiponectin and the increase in the amount of leptin in patients with obesity may cause psoriasis by increasing proinflammatory cytokines, which are similar to those involved in the pathogenesis of psoriasis.17 Additionally, psoriatic inflammation can cause insulin resistance and metabolic dysfunction, leading to obesity.18 The relationship between psoriasis and obesity cannot be solely explained by metabolic pathways. Smoking, alcohol consumption, and a sedentary lifestyle all are associated with psoriasis and also can contribute to obesity.5 Our study revealed no significant difference in smoking or alcohol consumption between the normal weight and overweight/obesity groups. Based on our data, we determined that smoking and alcohol consumption did not affect obesity in our patients with psoriasis.
Observational and epidemiologic studies have shown that patients with psoriasis experience increased rates of depression, anxiety, and stress.19 In studies of pathogenesis, a connection between depression and psoriatic inflammation has been established.20 It is known that inflammatory cytokines similar to those in psoriasis are involved in the development of obesity.18 In addition, depression and anxiety can lead to binge eating, unhealthy food choices, and a more sedentary lifestyle.5 All of these variables may contribute to the associations between depression and anxiety with psoriasis and obesity. Zafiriou et al21 conducted a study to investigate the relationship between psoriasis, obesity, and depression through inflammatory pathways with a focus on the importance of IL-17. Data showing that IL-17–producing Th17-cell subgroups play a considerable role in the development of obesity and depression prompted the authors to suggest that psoriasis, obesity, and anxiety/depression may be interconnected manifestations of immune dysregulation, potentially linked to IL-17 and its associated cells.21 Mrowietz et al22 also suggested that metabolic inflammation may contribute to obesity and depression in patients with psoriasis and highlighted the importance of several cytokines, including tumor necrosis factor α, IL-6, IL-8, IL-17, and IL-23. Our study revealed no significant differences in depression scores between BMI groups. Another meta-analysis reported conflicting findings on the incidence of depression in obese patients with psoriasis.23 Some of the studies had a small number of participants. Compared to depression, anxiety has received less attention in studies of patients with obesity with psoriasis. However, these studies have shown a positive correlation between anxiety scores and BMI in patients with psoriasis.24,25 In our study, similar to the findings of previous studies, overweight patients and those with obesitywho have psoriasis had significantly (P<.01) greater anxiety and stress scores than did normal weight patients with psoriasis.
Obesity should be assessed in patients with psoriasis via a biopsychosocial approach that takes into account genetic, behavioral, and environmental factors.26 Eating disorders are considered to be one of the factors contributing to obesity. Numerous studies in the literature have demonstrated a greater incidence of EDs in patients with obesity vs those without obesity.5,6,27 Obesity and EDs have a bidirectional relationship: individuals with obesity are at risk for EDs due to body dissatisfaction, dieting habits, and depressive states. Conversely, poor eating behaviors in individuals with a normal weight can lead to obesity.28
There are few studies in the literature exploring the relationship between psoriasis and EDs. Crosta et al29 demonstrated that patients with psoriasis had impaired results on ED screening tests and that these scores deteriorated further as BMI increased. Moreover, Altunay et al30 demonstrated that patients with psoriasis and metabolic syndrome had higher scores on the ED screening test. In this study, patients with higher scores also exhibited high levels of anxiety.30 In our study, similar to the findings of previous studies, patients with psoriasis who were overweight or had obesity had significantly (P<.01) greater EAT-26 scores than those in the normal weight group. Patients with high EAT-26 scores also exhibited elevated levels of depression, anxiety, and stress. Additionally, EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress scores. Our study as well as other studies in the literature indicate that additional research is needed to determine the associations between EDs and obesity in psoriasis.
Conclusion
Managing obesity is crucial for patients with psoriasis. This study showed that EAT-26 scores were higher in patients with psoriasis who were overweight or had obesity than in those who were normal weight. Participants with high EAT-26 scores (≥20 points) were more likely to be female and have higher anxiety and stress scores. In addition, EAT-26 scores were positively correlated with BMI as well as depression, anxiety, and stress scores. Eating disorders may contribute to the development of obesity in patients with psoriasis. Although our study was limited by a small sample size, the results suggest that there is a need for large-scale multicenter studies to investigate the relationship between psoriasis and EDs.
- Kalkan G. Comorbidities in psoriasis: the recognition of psoriasis as a systemic disease and current management. Turkderm-Turk Arch Dermatol Venereol. 2017;51:71-77.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Mirghani H, Altemani AT, Altemani ST, et al. The cross talk between psoriasis, obesity, and dyslipidemia: a meta-analysis. Cureus. 2023;15:e49253.
- Roehring M, Mashep MR, White MA, et al. The metabolic syndrome and behavioral correlates in obese patients with binge disorders. Obesity. 2009;17:481-486.
- da Luz FQ, Hay P, Touyz S, et al. Obesity with comorbid eating disorders: associated health risks and treatment approaches. Nutrients. 2018;10:829.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. American Psychiatric Association; 2013.
- Ergüney Okumus¸ FE, Sertel Berk HÖ. The psychometric properties of the Eating Attitudes Test short form (EAT-26) in a college sample. Stud Psychol. 2020;40:57-78.
- Stoleru G, Leopold A, Auerbach A, et al. Female gender, dissatisfaction with weight, and number of IBD related surgeries as independent risk factors for eating disorders among patients with inflammatory bowel diseases. BMC Gastroenterol. 2022;22:438.
- Öztürkcan S, Ermertcan AT, Eser E, et al. Cross validation of the Turkish version of dermatology life quality index. Int J Dermatol. 2006;45:1300-1307.
- Demir GT, Ciciog˘lu HI˙. Attitude scale for healthy nutrition (ASHN): validity and reliability study. Gaziantep Univ J Sport Sci. 2019;4:256-274.
- Yılmaz O, Boz H, Arslan A. The validity and reliability of depression stress and anxiety scale (DASS 21) Turkish short form. Res Financial Econ Soc Stud. 2017;2:78-91.
- Nuttall FQ. Body mass index: obesity, BMI, and health: a critical review. Nutr Today. 2015;50:117-128.
- Strumia R, Manzata E, Gualandi M. Is there a role for dermatologists in eating disorders? Expert Rev Dermatol. 2017; 2:109-112.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Naldi L, Addis A, Chimenti S, et al. Impact of body mass index and obesity on clinical response to systemic treatment for psoriasis. evidence from the Psocare project. Dermatology. 2008;217:365-373.
- Barros G, Duran P, Vera I, et al. Exploring the links between obesity and psoriasis: a comprehensive review. Int J Mol Sci. 2022;23:7499.
- Hao Y, Zhu YJ, Zou S, et al. Metabolic syndrome and psoriasis: mechanisms and future directions. Front Immunol. 2021;12:711060.
- Jing D, Xiao H, Shen M, et al. Association of psoriasis with anxiety and depression: a case–control study in Chinese patients. Front Med (Lausanne). 2021;8:771645.
- Sahi FM, Masood A, Danawar NA, et al. Association between psoriasis and depression: a traditional review. Cureus. 2020;12:E9708.
- Zafiriou E, Daponte AI, Siokas V, et al. Depression and obesity in patients with psoriasis and psoriatic arthritis: is IL-17–mediated immune dysregulation the connecting link? Front Immunol. 2021;12:699848.
- Mrowietz U, Sümbül M, Gerdes S. Depression, a major comorbidity of psoriatic disease, is caused by metabolic inflammation. J Eur Acad Dermatol Venereol. 2023;37:1731-1738.
- Pavlova NT, Kioskli K, Smith C, et al. Psychosocial aspects of obesity in adults with psoriasis: a systematic review. Skin Health Dis. 2021;1:E33.
- Innamorati M, Quinto RM, Imperatori C, et al. Health-related quality of life and its association with alexithymia and difficulties in emotion regulation in patients with psoriasis. Compr Psychiatry. 2016;70:200-208.
- Tabolli S, Naldi L, Pagliarello C, et al. Evaluation of the impact of writing exercises interventions on quality of life in patients with psoriasis undergoing systemic treatments. Br J Dermatol. 2012;167:1254‐1264.
- Albuquerque D, Nóbrega C, Manco L, et al. The contribution of genetics and environment to obesity. Br Med Bull. 2017;123:159‐173.
- Balantekin KN, Grammer AC, Fitzsimmons-Craft EE, et al. Overweight and obesity are associated with increased eating disorder correlates and general psychopathology in university women with eating disorders. Eat Behav. 2021;41:101482.
- Jebeile H, Lister NB, Baur LA, et al. Eating disorder risk in adolescents with obesity. Obes Rev. 2021;22:E13173.
- Crosta ML, Caldarola G, Fraietta S, et al. Psychopathology and eating disorders in patients with psoriasis. G Ital Dermatol Venereol. 2014;149:355-361.
- Altunay I, Demirci GT, Ates B, et al. Do eating disorders accompany metabolic syndrome in psoriasis patients? results of a preliminary study. Clin Cosmet Investig Dermatol. 2011;4:139-143.
Psoriasis is a chronic multisystemic inflammatory skin disease with a worldwide prevalence of 2% to 3%.1 Psoriasis can be accompanied by other conditions such as psoriatic arthritis, obesity, metabolic syndrome, diabetes mellitus, hypertension, dyslipidemia, atherosclerotic disease, inflammatory bowel disease, and anxiety/depression. It is important to manage comorbidities of psoriasis in addition to treating the cutaneous manifestations of the disease.1
Obesity is a major public health concern worldwide. Numerous observational and epidemiologic studies have reported a high prevalence of obesity among patients with psoriasis.2 Current evidence indicates that obesity may initiate or worsen psoriasis; furthermore, it is important to note that obesity may negatively impact the effectiveness of psoriasis-specific treatments or increase the incidence of adverse effects. Therefore, managing obesity is crucial in the treatment of psoriasis.3 Numerous studies have investigated the association between psoriasis and obesity, and they commonly conclude that both conditions share the same genetic metabolic pathways.2-4 However, it is important to consider environmental factors such as dietary habits, smoking, alcohol consumption, and a sedentary lifestyle—all of which are associated with psoriasis and also can contribute to the development of obesity.5 Because of the effects of obesity in psoriasis patients, factors that impact the development of obesity have become a popular research topic.
Eating disorders (EDs) are a crucial risk factor for both developing and maintaining obesity. In particular, two EDs that are associated with obesity include binge eating disorder and bulimia nervosa.6 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,7 binge eating disorder can be diagnosed when a patient has at least 1 episode of binge eating per week over a 3-month period. Bulimia nervosa can be diagnosed when a patient is excessively concerned with their body weight and shape and engages in behaviors to prevent weight gain (eg, forced vomiting, excessive use of laxatives).7 Psychiatrists who specialize in EDs make diagnoses based on these criteria. In daily practice, there are several quick and simple questionnaires available to screen for EDs that can be used by nonpsychiatrist physicians, including the commonly used 26-item Eating Attitudes Test (EAT-26).8 The EAT-26 has been used to screen for EDs in patients with inflammatory disorders.9
The aim of this study was to screen for EDs in patients with psoriasis to identify potential risk factors for development of obesity.
Materials and Methods
This study included patients with psoriasis who were screened for EDs at a tertiary dermatology clinic in Turkey between January 2021 and December 2023. This study was approved by the local ethics committee and was in accordance with the Declaration of Helsinki (decision number E-93471371-514.99-225000079).
Study Design and Patient Inclusion Criteria—This quantitative cross-sectional study utilized EAT-26, Dermatology Life Quality Index (DLQI), Attitude Scale for Healthy Nutrition (ASHN), and Depression Anxiety Stress Scale-21 (DASS-21) scores. All the questionnaire scales used in the study were adapted and validated in Turkey.8,10-12 The inclusion criteria consisted of being older than 18 years of age, being literate, having psoriasis for at least 1 year that was not treated topically or systemically, and having no psychiatric diseases outside an ED. The questionnaires were presented in written format following the clinical examination. Literacy was an inclusion criterion in this study due to the absence of auxiliary health personnel.
Study Variables—The study variables included age, sex, marital status (single/divorced or married), education status (primary/secondary school or high school/university), employment status (employed or unemployed/retired), body mass index (BMI), smoking status, alcohol-consumption status, Psoriasis Area Severity Index score, presence of nail psoriasis and psoriatic arthritis, duration of psoriasis, family history of psoriasis, EAT-26 score, ASHN score, DLQI score, and DASS-21 score. Body mass index was calculated by taking a participant’s weight in kilograms and dividing it by their height in meters squared. The BMI values were classified into 3 categories: normal (18.5–24.9 kg/m2), overweight (25.0–29.9 kg/m2), and obese (≥30 kg/m2).13
Questionnaires—The EAT-26 questionnaire includes 26 questions that are used to detect EDs. Responses to each question include Likert-type answer options (ie, “always,” “usually,” “often,” “sometimes,” “rarely,” and “never.”) Patients with scores of 20 points or higher (range, 0–78) are classified as high risk for EDs.8 In our study, EAT-26 scores were grouped into 2 categories: patients scoring less than 20 points and those scoring 20 points or higher.
The DLQI questionnaire includes 10 questions to measure dermatologic symptoms and qualiy of life. Responses to each question include Likert-type answer options (ie, “not at all,” “a little,” “a lot,” or “very much.”) On the DLQI scale, the higher the score, the lower the quality of life (score range, 0–30).10
The ASHN questionnaire includes 21 questions that measure attitudes toward healthy nutrition with 5 possible answer options (“strongly disagree,” “disagree,” “undecided,” “agree,” and “strongly agree”). On this scale, higher scores indicate the participant is more knowledgeable about healthy nutrition (score range, 0–78).11
The DASS-21 questionnaire includes 21 questions that measure the severity of a range of symptoms common to depression, anxiety, and stress. Responses include Likert-type answer options (eg, “never,” “sometimes,” “often,” and “almost always.”) On this scale, a higher score (range of 0–21 for each) indicates higher levels of depression, anxiety, and stress.12
Statistical Analysis—Descriptive statistics were analyzed using SPSS software version 22.0 (IBM). The Shapiro-Wilk test was applied to determine whether the data were normally distributed. For categorical variables, frequency differences among groups were compared using the Pearson χ2 test. A t test was used to compare the means of 2 independent groups with a normal distribution. One-way analysis of variance and Tukey Honest Significant Difference post hoc analysis were used to test whether there was a statistically significant difference among the normally distributed means of independent groups. Pearson correlation analysis was used to determine whether there was a linear relationship between 2 numeric measurements and, if so, to determine the direction and severity of this relationship. P<.05 indicated statistical significance in this study.
Results
Study Participant Demographics—This study included 82 participants with a mean age of 44.3 years; 52.4% (43/82) were female, and 85.4% (70/82) were married. The questionnaire took an average of 4.2 minutes for participants to complete. A total of 57.3% (47/82) of patients had completed primary/secondary education and 59.8% (49/82) were employed. The mean BMI was 28.1 kg/m2. According to the BMI classification, 26.8% (22/82) participants had a normal weight, 36.6% (30/82) were overweight, and 43.9% (36/82) were obese. A total of 48.8% (40/82) of participants smoked, and 4.9% (4/82) consumed alcohol. The mean Psoriasis Area and Severity Index score was 5.4. A total of 54.9% (45/82) of participants had nail psoriasis, and 24.4% (20/82) had psoriatic arthritis. The mean duration of psoriasis was 153 months. A total of 29.3% (24/82) of participants had a positive family history of psoriasis. The mean EAT-26 score was 11.1. A total of 12.2% (10/82) of participants had an EAT-26 score of 20 points or higher and were considered at high risk for an ED. The mean ASHN score was 72.9; the mean DLQI score was 5.5; and on the DASS-21 scale, mean scores for depression, anxiety, and stress were 6.3, 8.7, and 10.0, respectively (Table).
Comparative Evaluation of the BMI Groups—The only statistically significant differences among the 3 BMI groups were related to marital status, EAT-26 score, and anxiety and stress scores (P=.02, <.01, <.01, and <.01, respectively)(eTable 1). The number of single/divorced participants in the overweight group was significantly (P=.02) greater than in the normal weight group. The mean EAT-26 score for the normal weight group was significantly (P<.01) lower than for the overweight and obese groups; there was no significant difference in mean EAT-26 scores between the overweight and obese groups. The mean anxiety score was significantly (P<.01) lower in the normal weight group compared with the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean depression score. The mean stress and anxiety scores were significantly (P<.01) lower in the normal weight group than in the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean anxiety score.
Comparative Evaluation of the EAT-26 Scores—There were statistically significant differences among the EAT-26 scores related to sex; BMI; and depression, anxiety, and stress scores (P=.04, .02, <.01, <.01, and <.01, respectively). The number of females in the group with a score of 20 points or higher was significantly (P=.04) less than that in the group scoring less than 20 points. The mean BMI in the group with a score of 20 points or higher was significantly (P=.02) greater than in group scoring less than 20 points. The mean depression, anxiety, and stress scores of the group scoring 20 points or higher were significantly (P<.01 for all) greater than in the group scoring less than 20 points (eTable 2).
Correlation Analysis of the Study Variables—The EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress (P<.01 for all)(eTable 3).
Comment
Eating disorders are psychiatric conditions that require a multidisciplinary approach. Nonpsychiatric medical departments may be involved due to the severe consequences (eg, various skin changes14) of these disorders. Psoriasis is not known to be directly affected by the presence of an ED; however, it is possible that EDs could indirectly affect patients with psoriasis by influencing obesity. Therefore, this study aimed to examine the relationship between ED risk factors and obesity in this population.
The relationship between psoriasis and obesity has been a popular research topic in dermatology since the 1990s.15 Epidemiologic and observational studies have reported that patients with psoriasis are more likely to be overweight or have obesity, which is an independent risk factor for psoriasis.3,16 However, the causal relationship between psoriasis and obesity remains unclear. In a comprehensive review, Barros et al17 emphasized the causal relationship between obesity and psoriasis under several headings. Firstly, a higher BMI increases the risk for psoriasis by promoting cytokine release and immune system dysregulation. Secondly, a Western diet (eg, processed foods and fast food) triggers obesity and psoriasis by increasing adipose tissue. Thirdly, the alteration of the skin and gut microbiota triggers chronic inflammation as a result of bacterial translocation in patients with obesity. Fourthly, a high-fat diet and palmitic acid disrupt the intestinal integrity of the gut and increase the risk for psoriasis and obesity by triggering chronic inflammation of bacterial fragments that pass into the blood. Finally, the decrease in the amount of adiponectin and the increase in the amount of leptin in patients with obesity may cause psoriasis by increasing proinflammatory cytokines, which are similar to those involved in the pathogenesis of psoriasis.17 Additionally, psoriatic inflammation can cause insulin resistance and metabolic dysfunction, leading to obesity.18 The relationship between psoriasis and obesity cannot be solely explained by metabolic pathways. Smoking, alcohol consumption, and a sedentary lifestyle all are associated with psoriasis and also can contribute to obesity.5 Our study revealed no significant difference in smoking or alcohol consumption between the normal weight and overweight/obesity groups. Based on our data, we determined that smoking and alcohol consumption did not affect obesity in our patients with psoriasis.
Observational and epidemiologic studies have shown that patients with psoriasis experience increased rates of depression, anxiety, and stress.19 In studies of pathogenesis, a connection between depression and psoriatic inflammation has been established.20 It is known that inflammatory cytokines similar to those in psoriasis are involved in the development of obesity.18 In addition, depression and anxiety can lead to binge eating, unhealthy food choices, and a more sedentary lifestyle.5 All of these variables may contribute to the associations between depression and anxiety with psoriasis and obesity. Zafiriou et al21 conducted a study to investigate the relationship between psoriasis, obesity, and depression through inflammatory pathways with a focus on the importance of IL-17. Data showing that IL-17–producing Th17-cell subgroups play a considerable role in the development of obesity and depression prompted the authors to suggest that psoriasis, obesity, and anxiety/depression may be interconnected manifestations of immune dysregulation, potentially linked to IL-17 and its associated cells.21 Mrowietz et al22 also suggested that metabolic inflammation may contribute to obesity and depression in patients with psoriasis and highlighted the importance of several cytokines, including tumor necrosis factor α, IL-6, IL-8, IL-17, and IL-23. Our study revealed no significant differences in depression scores between BMI groups. Another meta-analysis reported conflicting findings on the incidence of depression in obese patients with psoriasis.23 Some of the studies had a small number of participants. Compared to depression, anxiety has received less attention in studies of patients with obesity with psoriasis. However, these studies have shown a positive correlation between anxiety scores and BMI in patients with psoriasis.24,25 In our study, similar to the findings of previous studies, overweight patients and those with obesitywho have psoriasis had significantly (P<.01) greater anxiety and stress scores than did normal weight patients with psoriasis.
Obesity should be assessed in patients with psoriasis via a biopsychosocial approach that takes into account genetic, behavioral, and environmental factors.26 Eating disorders are considered to be one of the factors contributing to obesity. Numerous studies in the literature have demonstrated a greater incidence of EDs in patients with obesity vs those without obesity.5,6,27 Obesity and EDs have a bidirectional relationship: individuals with obesity are at risk for EDs due to body dissatisfaction, dieting habits, and depressive states. Conversely, poor eating behaviors in individuals with a normal weight can lead to obesity.28
There are few studies in the literature exploring the relationship between psoriasis and EDs. Crosta et al29 demonstrated that patients with psoriasis had impaired results on ED screening tests and that these scores deteriorated further as BMI increased. Moreover, Altunay et al30 demonstrated that patients with psoriasis and metabolic syndrome had higher scores on the ED screening test. In this study, patients with higher scores also exhibited high levels of anxiety.30 In our study, similar to the findings of previous studies, patients with psoriasis who were overweight or had obesity had significantly (P<.01) greater EAT-26 scores than those in the normal weight group. Patients with high EAT-26 scores also exhibited elevated levels of depression, anxiety, and stress. Additionally, EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress scores. Our study as well as other studies in the literature indicate that additional research is needed to determine the associations between EDs and obesity in psoriasis.
Conclusion
Managing obesity is crucial for patients with psoriasis. This study showed that EAT-26 scores were higher in patients with psoriasis who were overweight or had obesity than in those who were normal weight. Participants with high EAT-26 scores (≥20 points) were more likely to be female and have higher anxiety and stress scores. In addition, EAT-26 scores were positively correlated with BMI as well as depression, anxiety, and stress scores. Eating disorders may contribute to the development of obesity in patients with psoriasis. Although our study was limited by a small sample size, the results suggest that there is a need for large-scale multicenter studies to investigate the relationship between psoriasis and EDs.
Psoriasis is a chronic multisystemic inflammatory skin disease with a worldwide prevalence of 2% to 3%.1 Psoriasis can be accompanied by other conditions such as psoriatic arthritis, obesity, metabolic syndrome, diabetes mellitus, hypertension, dyslipidemia, atherosclerotic disease, inflammatory bowel disease, and anxiety/depression. It is important to manage comorbidities of psoriasis in addition to treating the cutaneous manifestations of the disease.1
Obesity is a major public health concern worldwide. Numerous observational and epidemiologic studies have reported a high prevalence of obesity among patients with psoriasis.2 Current evidence indicates that obesity may initiate or worsen psoriasis; furthermore, it is important to note that obesity may negatively impact the effectiveness of psoriasis-specific treatments or increase the incidence of adverse effects. Therefore, managing obesity is crucial in the treatment of psoriasis.3 Numerous studies have investigated the association between psoriasis and obesity, and they commonly conclude that both conditions share the same genetic metabolic pathways.2-4 However, it is important to consider environmental factors such as dietary habits, smoking, alcohol consumption, and a sedentary lifestyle—all of which are associated with psoriasis and also can contribute to the development of obesity.5 Because of the effects of obesity in psoriasis patients, factors that impact the development of obesity have become a popular research topic.
Eating disorders (EDs) are a crucial risk factor for both developing and maintaining obesity. In particular, two EDs that are associated with obesity include binge eating disorder and bulimia nervosa.6 According to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,7 binge eating disorder can be diagnosed when a patient has at least 1 episode of binge eating per week over a 3-month period. Bulimia nervosa can be diagnosed when a patient is excessively concerned with their body weight and shape and engages in behaviors to prevent weight gain (eg, forced vomiting, excessive use of laxatives).7 Psychiatrists who specialize in EDs make diagnoses based on these criteria. In daily practice, there are several quick and simple questionnaires available to screen for EDs that can be used by nonpsychiatrist physicians, including the commonly used 26-item Eating Attitudes Test (EAT-26).8 The EAT-26 has been used to screen for EDs in patients with inflammatory disorders.9
The aim of this study was to screen for EDs in patients with psoriasis to identify potential risk factors for development of obesity.
Materials and Methods
This study included patients with psoriasis who were screened for EDs at a tertiary dermatology clinic in Turkey between January 2021 and December 2023. This study was approved by the local ethics committee and was in accordance with the Declaration of Helsinki (decision number E-93471371-514.99-225000079).
Study Design and Patient Inclusion Criteria—This quantitative cross-sectional study utilized EAT-26, Dermatology Life Quality Index (DLQI), Attitude Scale for Healthy Nutrition (ASHN), and Depression Anxiety Stress Scale-21 (DASS-21) scores. All the questionnaire scales used in the study were adapted and validated in Turkey.8,10-12 The inclusion criteria consisted of being older than 18 years of age, being literate, having psoriasis for at least 1 year that was not treated topically or systemically, and having no psychiatric diseases outside an ED. The questionnaires were presented in written format following the clinical examination. Literacy was an inclusion criterion in this study due to the absence of auxiliary health personnel.
Study Variables—The study variables included age, sex, marital status (single/divorced or married), education status (primary/secondary school or high school/university), employment status (employed or unemployed/retired), body mass index (BMI), smoking status, alcohol-consumption status, Psoriasis Area Severity Index score, presence of nail psoriasis and psoriatic arthritis, duration of psoriasis, family history of psoriasis, EAT-26 score, ASHN score, DLQI score, and DASS-21 score. Body mass index was calculated by taking a participant’s weight in kilograms and dividing it by their height in meters squared. The BMI values were classified into 3 categories: normal (18.5–24.9 kg/m2), overweight (25.0–29.9 kg/m2), and obese (≥30 kg/m2).13
Questionnaires—The EAT-26 questionnaire includes 26 questions that are used to detect EDs. Responses to each question include Likert-type answer options (ie, “always,” “usually,” “often,” “sometimes,” “rarely,” and “never.”) Patients with scores of 20 points or higher (range, 0–78) are classified as high risk for EDs.8 In our study, EAT-26 scores were grouped into 2 categories: patients scoring less than 20 points and those scoring 20 points or higher.
The DLQI questionnaire includes 10 questions to measure dermatologic symptoms and qualiy of life. Responses to each question include Likert-type answer options (ie, “not at all,” “a little,” “a lot,” or “very much.”) On the DLQI scale, the higher the score, the lower the quality of life (score range, 0–30).10
The ASHN questionnaire includes 21 questions that measure attitudes toward healthy nutrition with 5 possible answer options (“strongly disagree,” “disagree,” “undecided,” “agree,” and “strongly agree”). On this scale, higher scores indicate the participant is more knowledgeable about healthy nutrition (score range, 0–78).11
The DASS-21 questionnaire includes 21 questions that measure the severity of a range of symptoms common to depression, anxiety, and stress. Responses include Likert-type answer options (eg, “never,” “sometimes,” “often,” and “almost always.”) On this scale, a higher score (range of 0–21 for each) indicates higher levels of depression, anxiety, and stress.12
Statistical Analysis—Descriptive statistics were analyzed using SPSS software version 22.0 (IBM). The Shapiro-Wilk test was applied to determine whether the data were normally distributed. For categorical variables, frequency differences among groups were compared using the Pearson χ2 test. A t test was used to compare the means of 2 independent groups with a normal distribution. One-way analysis of variance and Tukey Honest Significant Difference post hoc analysis were used to test whether there was a statistically significant difference among the normally distributed means of independent groups. Pearson correlation analysis was used to determine whether there was a linear relationship between 2 numeric measurements and, if so, to determine the direction and severity of this relationship. P<.05 indicated statistical significance in this study.
Results
Study Participant Demographics—This study included 82 participants with a mean age of 44.3 years; 52.4% (43/82) were female, and 85.4% (70/82) were married. The questionnaire took an average of 4.2 minutes for participants to complete. A total of 57.3% (47/82) of patients had completed primary/secondary education and 59.8% (49/82) were employed. The mean BMI was 28.1 kg/m2. According to the BMI classification, 26.8% (22/82) participants had a normal weight, 36.6% (30/82) were overweight, and 43.9% (36/82) were obese. A total of 48.8% (40/82) of participants smoked, and 4.9% (4/82) consumed alcohol. The mean Psoriasis Area and Severity Index score was 5.4. A total of 54.9% (45/82) of participants had nail psoriasis, and 24.4% (20/82) had psoriatic arthritis. The mean duration of psoriasis was 153 months. A total of 29.3% (24/82) of participants had a positive family history of psoriasis. The mean EAT-26 score was 11.1. A total of 12.2% (10/82) of participants had an EAT-26 score of 20 points or higher and were considered at high risk for an ED. The mean ASHN score was 72.9; the mean DLQI score was 5.5; and on the DASS-21 scale, mean scores for depression, anxiety, and stress were 6.3, 8.7, and 10.0, respectively (Table).
Comparative Evaluation of the BMI Groups—The only statistically significant differences among the 3 BMI groups were related to marital status, EAT-26 score, and anxiety and stress scores (P=.02, <.01, <.01, and <.01, respectively)(eTable 1). The number of single/divorced participants in the overweight group was significantly (P=.02) greater than in the normal weight group. The mean EAT-26 score for the normal weight group was significantly (P<.01) lower than for the overweight and obese groups; there was no significant difference in mean EAT-26 scores between the overweight and obese groups. The mean anxiety score was significantly (P<.01) lower in the normal weight group compared with the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean depression score. The mean stress and anxiety scores were significantly (P<.01) lower in the normal weight group than in the overweight and obese groups. There was no significant difference between the overweight and obese groups according to the mean anxiety score.
Comparative Evaluation of the EAT-26 Scores—There were statistically significant differences among the EAT-26 scores related to sex; BMI; and depression, anxiety, and stress scores (P=.04, .02, <.01, <.01, and <.01, respectively). The number of females in the group with a score of 20 points or higher was significantly (P=.04) less than that in the group scoring less than 20 points. The mean BMI in the group with a score of 20 points or higher was significantly (P=.02) greater than in group scoring less than 20 points. The mean depression, anxiety, and stress scores of the group scoring 20 points or higher were significantly (P<.01 for all) greater than in the group scoring less than 20 points (eTable 2).
Correlation Analysis of the Study Variables—The EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress (P<.01 for all)(eTable 3).
Comment
Eating disorders are psychiatric conditions that require a multidisciplinary approach. Nonpsychiatric medical departments may be involved due to the severe consequences (eg, various skin changes14) of these disorders. Psoriasis is not known to be directly affected by the presence of an ED; however, it is possible that EDs could indirectly affect patients with psoriasis by influencing obesity. Therefore, this study aimed to examine the relationship between ED risk factors and obesity in this population.
The relationship between psoriasis and obesity has been a popular research topic in dermatology since the 1990s.15 Epidemiologic and observational studies have reported that patients with psoriasis are more likely to be overweight or have obesity, which is an independent risk factor for psoriasis.3,16 However, the causal relationship between psoriasis and obesity remains unclear. In a comprehensive review, Barros et al17 emphasized the causal relationship between obesity and psoriasis under several headings. Firstly, a higher BMI increases the risk for psoriasis by promoting cytokine release and immune system dysregulation. Secondly, a Western diet (eg, processed foods and fast food) triggers obesity and psoriasis by increasing adipose tissue. Thirdly, the alteration of the skin and gut microbiota triggers chronic inflammation as a result of bacterial translocation in patients with obesity. Fourthly, a high-fat diet and palmitic acid disrupt the intestinal integrity of the gut and increase the risk for psoriasis and obesity by triggering chronic inflammation of bacterial fragments that pass into the blood. Finally, the decrease in the amount of adiponectin and the increase in the amount of leptin in patients with obesity may cause psoriasis by increasing proinflammatory cytokines, which are similar to those involved in the pathogenesis of psoriasis.17 Additionally, psoriatic inflammation can cause insulin resistance and metabolic dysfunction, leading to obesity.18 The relationship between psoriasis and obesity cannot be solely explained by metabolic pathways. Smoking, alcohol consumption, and a sedentary lifestyle all are associated with psoriasis and also can contribute to obesity.5 Our study revealed no significant difference in smoking or alcohol consumption between the normal weight and overweight/obesity groups. Based on our data, we determined that smoking and alcohol consumption did not affect obesity in our patients with psoriasis.
Observational and epidemiologic studies have shown that patients with psoriasis experience increased rates of depression, anxiety, and stress.19 In studies of pathogenesis, a connection between depression and psoriatic inflammation has been established.20 It is known that inflammatory cytokines similar to those in psoriasis are involved in the development of obesity.18 In addition, depression and anxiety can lead to binge eating, unhealthy food choices, and a more sedentary lifestyle.5 All of these variables may contribute to the associations between depression and anxiety with psoriasis and obesity. Zafiriou et al21 conducted a study to investigate the relationship between psoriasis, obesity, and depression through inflammatory pathways with a focus on the importance of IL-17. Data showing that IL-17–producing Th17-cell subgroups play a considerable role in the development of obesity and depression prompted the authors to suggest that psoriasis, obesity, and anxiety/depression may be interconnected manifestations of immune dysregulation, potentially linked to IL-17 and its associated cells.21 Mrowietz et al22 also suggested that metabolic inflammation may contribute to obesity and depression in patients with psoriasis and highlighted the importance of several cytokines, including tumor necrosis factor α, IL-6, IL-8, IL-17, and IL-23. Our study revealed no significant differences in depression scores between BMI groups. Another meta-analysis reported conflicting findings on the incidence of depression in obese patients with psoriasis.23 Some of the studies had a small number of participants. Compared to depression, anxiety has received less attention in studies of patients with obesity with psoriasis. However, these studies have shown a positive correlation between anxiety scores and BMI in patients with psoriasis.24,25 In our study, similar to the findings of previous studies, overweight patients and those with obesitywho have psoriasis had significantly (P<.01) greater anxiety and stress scores than did normal weight patients with psoriasis.
Obesity should be assessed in patients with psoriasis via a biopsychosocial approach that takes into account genetic, behavioral, and environmental factors.26 Eating disorders are considered to be one of the factors contributing to obesity. Numerous studies in the literature have demonstrated a greater incidence of EDs in patients with obesity vs those without obesity.5,6,27 Obesity and EDs have a bidirectional relationship: individuals with obesity are at risk for EDs due to body dissatisfaction, dieting habits, and depressive states. Conversely, poor eating behaviors in individuals with a normal weight can lead to obesity.28
There are few studies in the literature exploring the relationship between psoriasis and EDs. Crosta et al29 demonstrated that patients with psoriasis had impaired results on ED screening tests and that these scores deteriorated further as BMI increased. Moreover, Altunay et al30 demonstrated that patients with psoriasis and metabolic syndrome had higher scores on the ED screening test. In this study, patients with higher scores also exhibited high levels of anxiety.30 In our study, similar to the findings of previous studies, patients with psoriasis who were overweight or had obesity had significantly (P<.01) greater EAT-26 scores than those in the normal weight group. Patients with high EAT-26 scores also exhibited elevated levels of depression, anxiety, and stress. Additionally, EAT-26 scores were positively correlated with BMI, anxiety, depression, and stress scores. Our study as well as other studies in the literature indicate that additional research is needed to determine the associations between EDs and obesity in psoriasis.
Conclusion
Managing obesity is crucial for patients with psoriasis. This study showed that EAT-26 scores were higher in patients with psoriasis who were overweight or had obesity than in those who were normal weight. Participants with high EAT-26 scores (≥20 points) were more likely to be female and have higher anxiety and stress scores. In addition, EAT-26 scores were positively correlated with BMI as well as depression, anxiety, and stress scores. Eating disorders may contribute to the development of obesity in patients with psoriasis. Although our study was limited by a small sample size, the results suggest that there is a need for large-scale multicenter studies to investigate the relationship between psoriasis and EDs.
- Kalkan G. Comorbidities in psoriasis: the recognition of psoriasis as a systemic disease and current management. Turkderm-Turk Arch Dermatol Venereol. 2017;51:71-77.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Mirghani H, Altemani AT, Altemani ST, et al. The cross talk between psoriasis, obesity, and dyslipidemia: a meta-analysis. Cureus. 2023;15:e49253.
- Roehring M, Mashep MR, White MA, et al. The metabolic syndrome and behavioral correlates in obese patients with binge disorders. Obesity. 2009;17:481-486.
- da Luz FQ, Hay P, Touyz S, et al. Obesity with comorbid eating disorders: associated health risks and treatment approaches. Nutrients. 2018;10:829.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. American Psychiatric Association; 2013.
- Ergüney Okumus¸ FE, Sertel Berk HÖ. The psychometric properties of the Eating Attitudes Test short form (EAT-26) in a college sample. Stud Psychol. 2020;40:57-78.
- Stoleru G, Leopold A, Auerbach A, et al. Female gender, dissatisfaction with weight, and number of IBD related surgeries as independent risk factors for eating disorders among patients with inflammatory bowel diseases. BMC Gastroenterol. 2022;22:438.
- Öztürkcan S, Ermertcan AT, Eser E, et al. Cross validation of the Turkish version of dermatology life quality index. Int J Dermatol. 2006;45:1300-1307.
- Demir GT, Ciciog˘lu HI˙. Attitude scale for healthy nutrition (ASHN): validity and reliability study. Gaziantep Univ J Sport Sci. 2019;4:256-274.
- Yılmaz O, Boz H, Arslan A. The validity and reliability of depression stress and anxiety scale (DASS 21) Turkish short form. Res Financial Econ Soc Stud. 2017;2:78-91.
- Nuttall FQ. Body mass index: obesity, BMI, and health: a critical review. Nutr Today. 2015;50:117-128.
- Strumia R, Manzata E, Gualandi M. Is there a role for dermatologists in eating disorders? Expert Rev Dermatol. 2017; 2:109-112.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Naldi L, Addis A, Chimenti S, et al. Impact of body mass index and obesity on clinical response to systemic treatment for psoriasis. evidence from the Psocare project. Dermatology. 2008;217:365-373.
- Barros G, Duran P, Vera I, et al. Exploring the links between obesity and psoriasis: a comprehensive review. Int J Mol Sci. 2022;23:7499.
- Hao Y, Zhu YJ, Zou S, et al. Metabolic syndrome and psoriasis: mechanisms and future directions. Front Immunol. 2021;12:711060.
- Jing D, Xiao H, Shen M, et al. Association of psoriasis with anxiety and depression: a case–control study in Chinese patients. Front Med (Lausanne). 2021;8:771645.
- Sahi FM, Masood A, Danawar NA, et al. Association between psoriasis and depression: a traditional review. Cureus. 2020;12:E9708.
- Zafiriou E, Daponte AI, Siokas V, et al. Depression and obesity in patients with psoriasis and psoriatic arthritis: is IL-17–mediated immune dysregulation the connecting link? Front Immunol. 2021;12:699848.
- Mrowietz U, Sümbül M, Gerdes S. Depression, a major comorbidity of psoriatic disease, is caused by metabolic inflammation. J Eur Acad Dermatol Venereol. 2023;37:1731-1738.
- Pavlova NT, Kioskli K, Smith C, et al. Psychosocial aspects of obesity in adults with psoriasis: a systematic review. Skin Health Dis. 2021;1:E33.
- Innamorati M, Quinto RM, Imperatori C, et al. Health-related quality of life and its association with alexithymia and difficulties in emotion regulation in patients with psoriasis. Compr Psychiatry. 2016;70:200-208.
- Tabolli S, Naldi L, Pagliarello C, et al. Evaluation of the impact of writing exercises interventions on quality of life in patients with psoriasis undergoing systemic treatments. Br J Dermatol. 2012;167:1254‐1264.
- Albuquerque D, Nóbrega C, Manco L, et al. The contribution of genetics and environment to obesity. Br Med Bull. 2017;123:159‐173.
- Balantekin KN, Grammer AC, Fitzsimmons-Craft EE, et al. Overweight and obesity are associated with increased eating disorder correlates and general psychopathology in university women with eating disorders. Eat Behav. 2021;41:101482.
- Jebeile H, Lister NB, Baur LA, et al. Eating disorder risk in adolescents with obesity. Obes Rev. 2021;22:E13173.
- Crosta ML, Caldarola G, Fraietta S, et al. Psychopathology and eating disorders in patients with psoriasis. G Ital Dermatol Venereol. 2014;149:355-361.
- Altunay I, Demirci GT, Ates B, et al. Do eating disorders accompany metabolic syndrome in psoriasis patients? results of a preliminary study. Clin Cosmet Investig Dermatol. 2011;4:139-143.
- Kalkan G. Comorbidities in psoriasis: the recognition of psoriasis as a systemic disease and current management. Turkderm-Turk Arch Dermatol Venereol. 2017;51:71-77.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Mirghani H, Altemani AT, Altemani ST, et al. The cross talk between psoriasis, obesity, and dyslipidemia: a meta-analysis. Cureus. 2023;15:e49253.
- Roehring M, Mashep MR, White MA, et al. The metabolic syndrome and behavioral correlates in obese patients with binge disorders. Obesity. 2009;17:481-486.
- da Luz FQ, Hay P, Touyz S, et al. Obesity with comorbid eating disorders: associated health risks and treatment approaches. Nutrients. 2018;10:829.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. American Psychiatric Association; 2013.
- Ergüney Okumus¸ FE, Sertel Berk HÖ. The psychometric properties of the Eating Attitudes Test short form (EAT-26) in a college sample. Stud Psychol. 2020;40:57-78.
- Stoleru G, Leopold A, Auerbach A, et al. Female gender, dissatisfaction with weight, and number of IBD related surgeries as independent risk factors for eating disorders among patients with inflammatory bowel diseases. BMC Gastroenterol. 2022;22:438.
- Öztürkcan S, Ermertcan AT, Eser E, et al. Cross validation of the Turkish version of dermatology life quality index. Int J Dermatol. 2006;45:1300-1307.
- Demir GT, Ciciog˘lu HI˙. Attitude scale for healthy nutrition (ASHN): validity and reliability study. Gaziantep Univ J Sport Sci. 2019;4:256-274.
- Yılmaz O, Boz H, Arslan A. The validity and reliability of depression stress and anxiety scale (DASS 21) Turkish short form. Res Financial Econ Soc Stud. 2017;2:78-91.
- Nuttall FQ. Body mass index: obesity, BMI, and health: a critical review. Nutr Today. 2015;50:117-128.
- Strumia R, Manzata E, Gualandi M. Is there a role for dermatologists in eating disorders? Expert Rev Dermatol. 2017; 2:109-112.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Naldi L, Addis A, Chimenti S, et al. Impact of body mass index and obesity on clinical response to systemic treatment for psoriasis. evidence from the Psocare project. Dermatology. 2008;217:365-373.
- Barros G, Duran P, Vera I, et al. Exploring the links between obesity and psoriasis: a comprehensive review. Int J Mol Sci. 2022;23:7499.
- Hao Y, Zhu YJ, Zou S, et al. Metabolic syndrome and psoriasis: mechanisms and future directions. Front Immunol. 2021;12:711060.
- Jing D, Xiao H, Shen M, et al. Association of psoriasis with anxiety and depression: a case–control study in Chinese patients. Front Med (Lausanne). 2021;8:771645.
- Sahi FM, Masood A, Danawar NA, et al. Association between psoriasis and depression: a traditional review. Cureus. 2020;12:E9708.
- Zafiriou E, Daponte AI, Siokas V, et al. Depression and obesity in patients with psoriasis and psoriatic arthritis: is IL-17–mediated immune dysregulation the connecting link? Front Immunol. 2021;12:699848.
- Mrowietz U, Sümbül M, Gerdes S. Depression, a major comorbidity of psoriatic disease, is caused by metabolic inflammation. J Eur Acad Dermatol Venereol. 2023;37:1731-1738.
- Pavlova NT, Kioskli K, Smith C, et al. Psychosocial aspects of obesity in adults with psoriasis: a systematic review. Skin Health Dis. 2021;1:E33.
- Innamorati M, Quinto RM, Imperatori C, et al. Health-related quality of life and its association with alexithymia and difficulties in emotion regulation in patients with psoriasis. Compr Psychiatry. 2016;70:200-208.
- Tabolli S, Naldi L, Pagliarello C, et al. Evaluation of the impact of writing exercises interventions on quality of life in patients with psoriasis undergoing systemic treatments. Br J Dermatol. 2012;167:1254‐1264.
- Albuquerque D, Nóbrega C, Manco L, et al. The contribution of genetics and environment to obesity. Br Med Bull. 2017;123:159‐173.
- Balantekin KN, Grammer AC, Fitzsimmons-Craft EE, et al. Overweight and obesity are associated with increased eating disorder correlates and general psychopathology in university women with eating disorders. Eat Behav. 2021;41:101482.
- Jebeile H, Lister NB, Baur LA, et al. Eating disorder risk in adolescents with obesity. Obes Rev. 2021;22:E13173.
- Crosta ML, Caldarola G, Fraietta S, et al. Psychopathology and eating disorders in patients with psoriasis. G Ital Dermatol Venereol. 2014;149:355-361.
- Altunay I, Demirci GT, Ates B, et al. Do eating disorders accompany metabolic syndrome in psoriasis patients? results of a preliminary study. Clin Cosmet Investig Dermatol. 2011;4:139-143.
Practice Points
- Eating disorders are considered a contributing factor in obesity.
- Obesity is prevalent in patients with psoriasis, and current evidence indicates that obesity may initiate psoriasis or worsen existing disease.
- Obesity should be considered as contributory to the development of psoriasis via a biopsychosocial approach that accounts for genetic, behavioral, and environmental factors.
Projected 2023 Cost Reduction From Tumor Necrosis Factor α Inhibitor Biosimilars in Dermatology: A National Medicare Analysis
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).

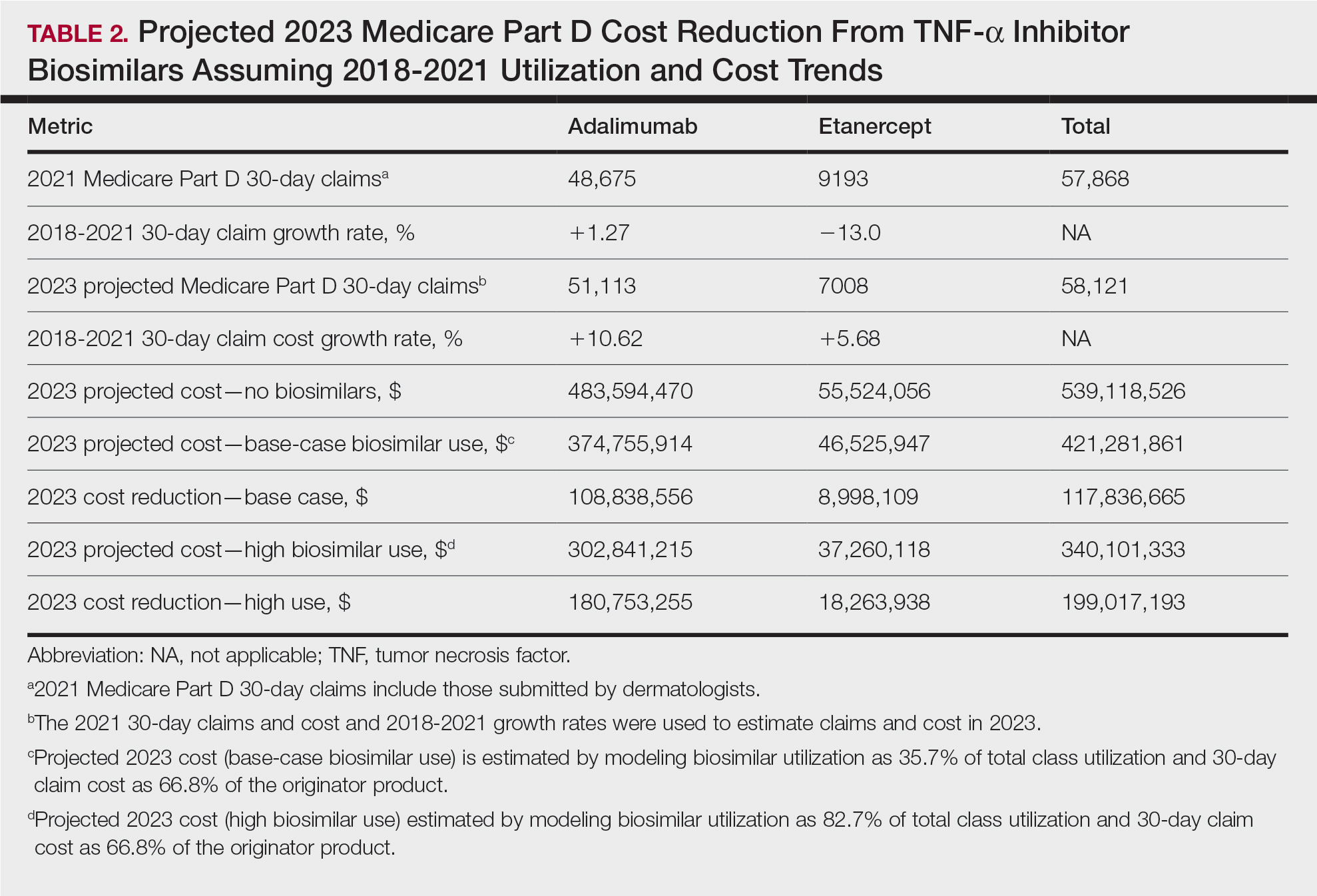
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
Practice Points
- Biosimilars for adalimumab and etanercept are safe and effective alternatives with the potential to reduce health care costs in dermatology by approximately $118 million.
- A high utilization rate of biosimilars by dermatologists would increase cost savings even further.
Considerations for the Use of Biologics in Pregnancy
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
Treat-to-Target Outcomes With Tapinarof Cream 1% in Phase 3 Trials for Plaque Psoriasis
Psoriasis is a chronic inflammatory disease affecting approximately 8 million adults in the United States and 2% of the global population.1,2 Psoriasis causes pain, itching, and disfigurement and is associated with a physical, psychological, and economic burden that substantially affects health-related quality of life.3-5
Setting treatment goals and treating to target are evidence-based approaches that have been successfully applied to several chronic diseases to improve patient outcomes, including diabetes, hypertension, and rheumatoid arthritis.6-9 Treat-to-target strategies generally set low disease activity (or remission) as an overall goal and seek to achieve this using available therapeutic options as necessary. Introduced following the availability of biologics and targeted systemic therapies, treat-to-target strategies generally provide guidance on expectations of treatment but not specific treatments, as personalized treatment decisions depend on an assessment of individual patients and consider clinical and demographic features as well as preferences for available therapeutic options. If targets are not achieved in the assigned time span, adjustments can be made to the treatment approach in close consultation with the patient. If the target is reached, follow-up visits can be scheduled to ensure improvement is maintained or to establish if more aggressive goals could be selected.
Treat-to-target strategies for the management of psoriasis developed by the National Psoriasis Foundation (NPF) Medical Board include reducing the extent of psoriasis to 1% or lower total body surface area (BSA) after 3 months of treatment.10 Treatment targets endorsed by the European Academy of Dermatology and Venereology (EADV) in guidelines on the use of systemic therapies in psoriasis include achieving a 75% or greater reduction in Psoriasis Area and Severity Index (PASI) score within 3 to 4 months of treatment.11
In clinical practice, many patients do not achieve these treatment targets, and topical treatments alone generally are insufficient in achieving treatment goals for psoriasis.12,13 Moreover, conventional topical treatments (eg, topical corticosteroids) used by most patients with psoriasis regardless of disease severity are associated with adverse events that can limit their use. Most topical corticosteroids have US Food and Drug Administration label restrictions relating to sites of application, duration and extent of use, and frequency of administration.14,15
Tapinarof cream 1% (VTAMA [Dermavant Sciences, Inc]) is a first-in-class topical nonsteroidal aryl hydrocarbon receptor agonist that was approved by the US Food and Drug Administration for the treatment of plaque psoriasis in adults16 and is being studied for the treatment of plaque psoriasis in children 2 years and older as well as for atopic dermatitis in adults and children 2 years and older. In PSOARING 1 (ClinicalTrials .gov identifier NCT03956355) and PSOARING 2 (NCT03983980)—identical 12-week pivotal phase 3 trials—monotherapy with tapinarof cream 1% once daily (QD) demonstrated statistically significant efficacy vs vehicle cream and was well tolerated in adults with mild to severe plaque psoriasis (Supplementary Figure S1).17 Lebwohl et al17 reported that significantly higher PASI75 responses were observed at week 12 with tapinarof cream vs vehicle in PSOARING 1 and PSOARING 2 (36% and 48% vs 10% and 7%, respectively; both P<.0001). A significantly higher PASI90 response of 19% and 21% at week 12 also was observed with tapinarof cream vs 2% and 3% with vehicle in PSOARING 1 and PSOARING 2, respectively (P=.0005 and P<.0001).17
In PSOARING 3 (NCT04053387)—the long-term extension trial (Supplementary Figure S1)—efficacy continued to improve or was maintained beyond the two 12-week trials, with improvements in total BSA affected and PASI scores for up to 52 weeks.18 Tapinarof cream 1% QD demonstrated positive, rapid, and durable outcomes in PSOARING 3, including high rates of complete disease clearance (Physician Global Assessment [PGA] score=0 [clear])(40.9% [312/763]), durability of response on treatment with no evidence of tachyphylaxis, and a remittive effect of approximately 4 months when off therapy (defined as maintenance of a PGA score of 0 [clear] or 1 [almost clear] after first achieving a PGA score of 0).18
Herein, we report absolute treatment targets for patients with plaque psoriasis who received tapinarof cream 1% QD in the PSOARING trials that are at least as stringent as the corresponding NPF and EADV targets of achieving a total BSA affected of 1% or lower or a PASI75 response within 3 to 4 months, respectively.
METHODS
Study Design
The pooled efficacy analyses included all patients with a baseline PGA score of 2 or higher (mild or worse) before treatment with tapinarof cream 1% QD in the PSOARING trials. This included patients who received tapinarof cream 1% in PSOARING 1 and PSOARING 2 who may or may not have continued into PSOARING 3, as well as those who received the vehicle in PSOARING 1 and PSOARING 2 who enrolled in PSOARING 3 and had a PGA score of 2 or higher before receiving tapinarof cream 1%.
Trial Participants
Full methods, including inclusion and exclusion criteria, for the PSOARING trials have been previously reported.17,18 Patients were aged 18 to 75 years and had chronic plaque psoriasis that was stable for at least 6 months before randomization; 3% to 20% total BSA affected (excluding the scalp, palms, fingernails, toenails, and soles); and a PGA score of 2 (mild), 3 (moderate), or 4 (severe) at baseline.
The clinical trials were conducted in compliance with the guidelines for Good Clinical Practice and the Declaration of Helsinki. Approval was obtained from local ethics committees or institutional review boards at each center. All patients provided written informed consent.
Trial Treatment
In PSOARING 1 and PSOARING 2, patients were randomized (2:1) to receive tapinarof cream 1% or vehicle QD for 12 weeks. In PSOARING 3 (the long-term extension trial), patients received up to 40 weeks of open-label tapinarof, followed by 4 weeks of follow-up off treatment. Patients received intermittent or continuous treatment with tapinarof cream 1% in PSOARING 3 based on PGA score: those entering the trial with a PGA score of 1 or higher received tapinarof cream 1% until complete disease clearance was achieved (defined as a PGA score of 0 [clear]). Those entering PSOARING 3 with or achieving a PGA score of 0 (clear) discontinued treatment and were observed for the duration of maintenance of a PGA score of 0 (clear) or 1 (almost clear) while off therapy (the protocol-defined “duration of remittive effect”). If disease worsening (defined as a PGA score 2 or higher) occurred, tapinarof cream 1% was restarted and continued until a PGA score of 0 (clear) was achieved. This pattern of treatment, discontinuation on achieving a PGA score of 0 (clear), and retreatment on disease worsening continued until the end of the trial. As a result, patients in PSOARING 3 could receive tapinarof cream 1% continuously or intermittently for 40 weeks.
Outcome Measures and Statistical Analyses
The assessment of total BSA affected by plaque psoriasis is an estimate of the total extent of disease as a percentage of total skin area. In the PSOARING trials, the skin surface of one hand (palm and digits) was assumed to be approximately equivalent to 1% BSA. The total BSA affected by psoriasis was evaluated from 0% to 100%, with greater total BSA affected being an indication of more extensive disease. The BSA efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved a 1% or lower or 0.5% or lower total BSA affected.
Psoriasis Area and Severity Index scores assess both the severity and extent of psoriasis. A PASI score lower than 5 often is considered indicative of mild psoriasis, a score of 5 to 10 indicates moderate disease, and a score higher than 10 indicates severe disease.19 The maximum PASI score is 72. The PASI efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved an absolute total PASI score of 3 or lower, 2 or lower, and 1 or lower.
Efficacy analyses were based on pooled data for all patients in the PSOARING trials who had a PGA score of 2 to 4 (mild to severe) before treatment with tapinarof cream 1% in the intention-to-treat population using observed cases. Time-to-target analyses were based on Kaplan-Meier (KM) estimates using observed cases.
Safety analyses included the incidence and frequency of adverse events and were based on all patients who received tapinarof cream 1% in the PSOARING trials.
RESULTS
Baseline Patient Demographics and Disease Characteristics
The pooled efficacy analyses included 915 eligible patients (Table). At baseline, the mean (SD) age was 50.2 (13.25) years, 58.7% were male, the mean (SD) weight was 92.2 (23.67) kg, and the mean (SD) body mass index was 31.6 (7.53) kg/m2. The percentage of patients with a PGA score of 2 (mild), 3 (moderate), or 4 (severe) was 13.9%, 78.1%, and 8.0%, respectively. The mean (SD) PASI score was 8.7 (4.23) and mean (SD) total BSA affected was 7.8% (4.98).
Efficacy
Achievement of BSA-Affected Targets—
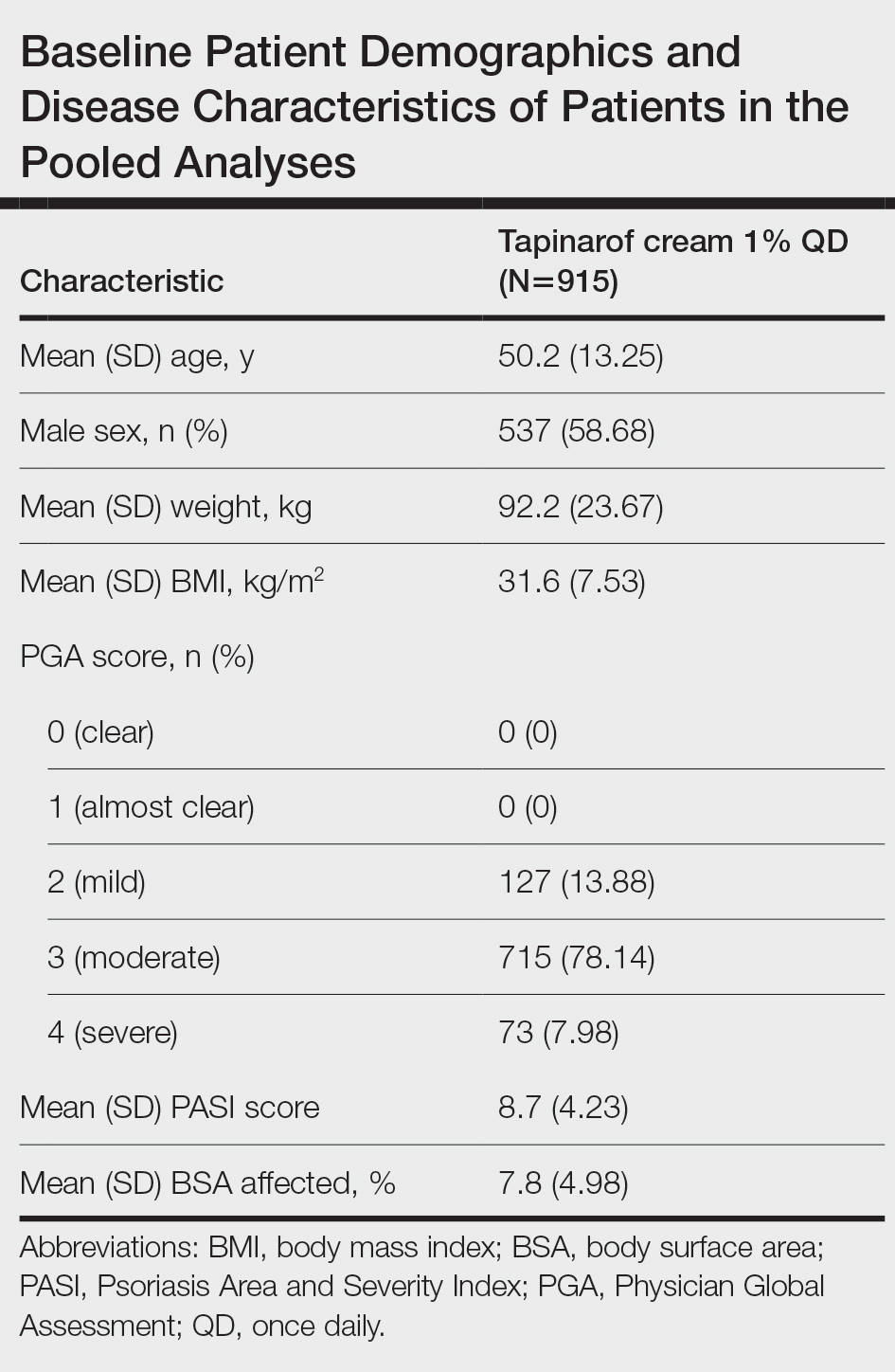
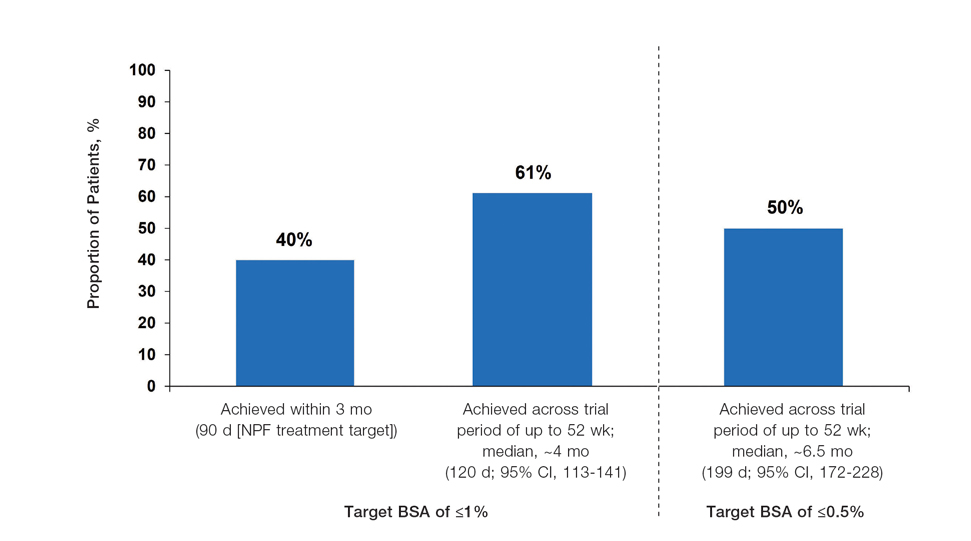
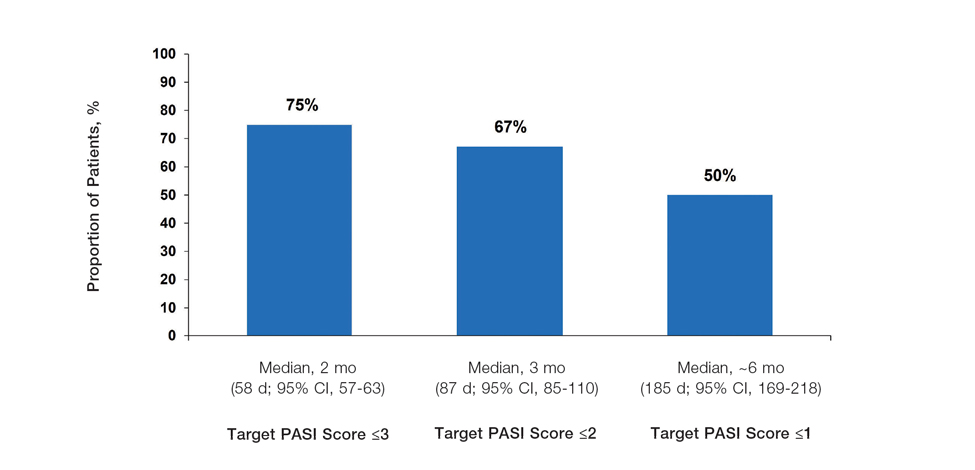
Achievement of Absolute PASI Targets—Across the total trial period (up to 52 weeks), an absolute total PASI score of 3 or lower was achieved by 75% of patients (686/915), with a median time to achieve this of 2 months (KM estimate: 58 days [95% CI, 57-63]); approximately 67% of patients (612/915) achieved a total PASI score of 2 or lower, with a median time to achieve of 3 months (KM estimate: 87 days [95% CI, 85-110])(Figure 2; Supplementary Figures S3a and S3b). A PASI score of 1 or lower was achieved by approximately 50% of patients (460/915), with a median time to achieve of approximately 6 months (KM estimate: 185 days [95% CI, 169-218])(Figure 2, Supplementary Figure S3c).
Illustrative Case—Case photography showing the clinical response in a 63-year-old man with moderate plaque psoriasis in PSOARING 2 is shown in Figure 3. After 12 weeks of treatment with tapinarof cream 1% QD, the patient achieved all primary and secondary efficacy end points. In addition to achieving the regulatory end point of a PGA score of 0 (clear) or 1 (almost clear) and a decrease from baseline of at least 2 points, achievement of 0% total BSA affected and a total PASI score of 0 at week 12 exceeded the NPF and EADV consensus treatment targets.10,11 Targets were achieved as early as week 4, with a total BSA affected of 0.5% or lower and a total PASI score of 1 or lower, illustrated by marked skin clearing and only faint residual erythema that completely resolved at week 12, with the absence of postinflammatory hyperpigmentation.
Safety
Safety data for the PSOARING trials have been previously reported.17,18 The most common treatment-emergent adverse events were folliculitis, contact dermatitis, upper respiratory tract infection, and nasopharyngitis. Treatment-emergent adverse events generally were mild or moderate in severity and did not lead to trial discontinuation.17,18
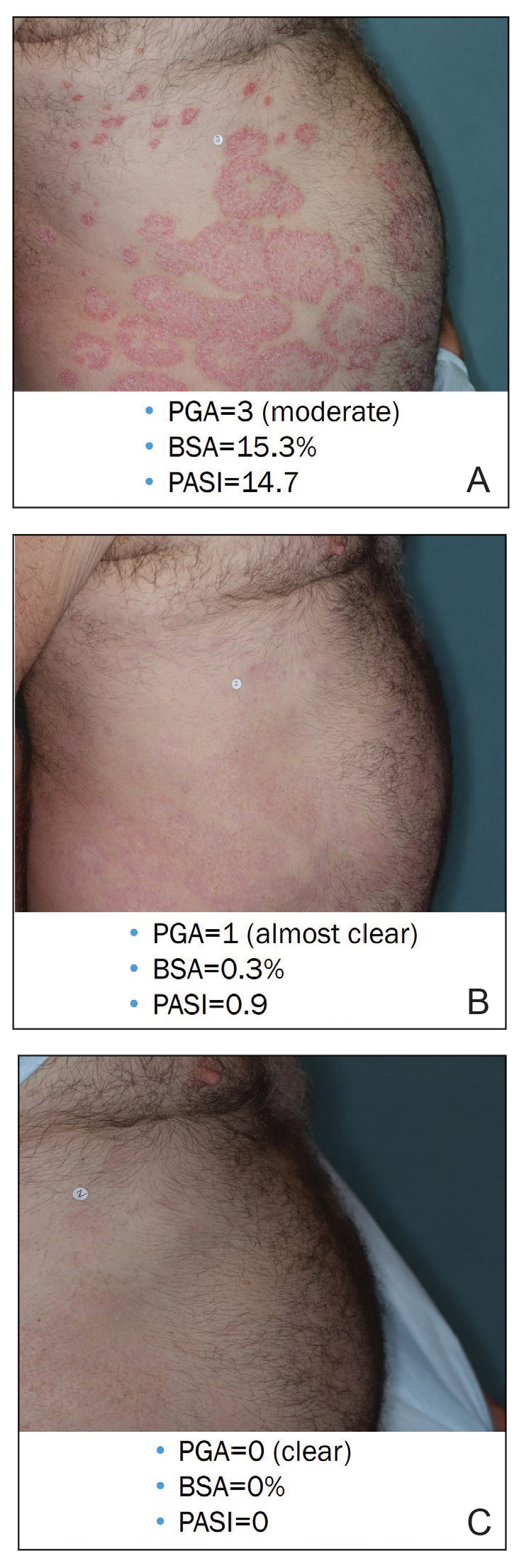
COMMENT
Treat-to-target management approaches aim to improve patient outcomes by striving to achieve optimal goals. The treat-to-target approach supports shared decision-making between clinicians and patients based on common expectations of what constitutes treatment success.
The findings of this analysis based on pooled data from a large cohort of patients demonstrate that a high proportion of patients can achieve or exceed recommended treatment targets with tapinarof cream 1% QD and maintain improvements long-term. The NPF-recommended treatment target of 1% or lower BSA affected within approximately 3 months (90 days) of treatment was achieved by 40% of tapinarof-treated patients. In addition, 1% or lower BSA affected at any time during the trials was achieved by 61% of patients (median, approximately 4 months). The analyses also indicated that PASI total scores of 3 or lower and 2 or lower were achieved by 75% and 67% of tapinarof-treated patients, respectively, within 2 to 3 months.
These findings support the previously reported efficacy of tapinarof cream, including high rates of complete disease clearance (40.9% [312/763]), durable response following treatment interruption, an off-therapy remittive effect of approximately 4 months, and good disease control on therapy with no evidence of tachyphylaxis.17,18
CONCLUSION
Taken together with previously reported tapinarof efficacy and safety results, our findings demonstrate that a high proportion of patients treated with tapinarof cream as monotherapy can achieve aggressive treatment targets set by both US and European guidelines developed for systemic and biologic therapies. Tapinarof cream 1% QD is an effective topical treatment option for patients with plaque psoriasis that has been approved without restrictions relating to severity or extent of disease treated, duration of use, or application sites, including application to sensitive and intertriginous skin.
Acknowledgments—Editorial and medical writing support under the guidance of the authors was provided by Melanie Govender, MSc (Med), ApotheCom (United Kingdom), and was funded by Dermavant Sciences, Inc, in accordance with Good Publication Practice (GPP) guidelines.
- Armstrong AW, Mehta MD, Schupp CW, et al. Psoriasis prevalence in adults in the United States. JAMA Dermatol. 2021;157:940-946.
- Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369:m1590.
- Pilon D, Teeple A, Zhdanava M, et al. The economic burden of psoriasis with high comorbidity among privately insured patients in the United States. J Med Econ. 2019;22:196-203.
- Singh S, Taylor C, Kornmehl H, et al. Psoriasis and suicidality: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:425-440.e2.
- Feldman SR, Goffe B, Rice G, et al. The challenge of managing psoriasis: unmet medical needs and stakeholder perspectives. Am Health Drug Benefits. 2016;9:504-513.
- Ford JA, Solomon DH. Challenges in implementing treat-to-target strategies in rheumatology. Rheum Dis Clin North Am. 2019;45:101-112.
- Sitbon O, Galiè N. Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur Respir Rev. 2010;19:272-278.
- Smolen JS, Aletaha D, Bijlsma JW, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69:631-637.
- Wangnoo SK, Sethi B, Sahay RK, et al. Treat-to-target trials in diabetes. Indian J Endocrinol Metab. 2014;18:166-174.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Pathirana D, Ormerod AD, Saiag P, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris. J Eur Acad Dermatol Venereol. 2009;23(Suppl 2):1-70.
- Strober BE, van der Walt JM, Armstrong AW, et al. Clinical goals and barriers to effective psoriasis care. Dermatol Ther (Heidelb). 2019; 9:5-18.
- Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
- Elmets CA, Korman NJ, Prater EF, et al. Joint AAD-NPF Guidelines of care for the management and treatment of psoriasis with topical therapy and alternative medicine modalities for psoriasis severity measures. J Am Acad Dermatol. 2021;84:432-470.
- Stein Gold LF. Topical therapies for psoriasis: improving management strategies and patient adherence. Semin Cutan Med Surg. 2016;35 (2 Suppl 2):S36-S44; quiz S45.
- VTAMA® (tapinarof) cream. Prescribing information. Dermavant Sciences; 2022. Accessed September 13, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215272s000lbl.pdf
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229 and supplementary appendix.
- Strober B, Stein Gold L, Bissonnette R, et al. One-year safety and efficacy of tapinarof cream for the treatment of plaque psoriasis: results from the PSOARING 3 trial. J Am Acad Dermatol. 2022;87:800-806.
- Clinical Review Report: Guselkumab (Tremfya) [Internet]. Canadian Agency for Drugs and Technologies in Health; 2018. Accessed September 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534047/pdf/Bookshelf_NBK534047.pdf
Psoriasis is a chronic inflammatory disease affecting approximately 8 million adults in the United States and 2% of the global population.1,2 Psoriasis causes pain, itching, and disfigurement and is associated with a physical, psychological, and economic burden that substantially affects health-related quality of life.3-5
Setting treatment goals and treating to target are evidence-based approaches that have been successfully applied to several chronic diseases to improve patient outcomes, including diabetes, hypertension, and rheumatoid arthritis.6-9 Treat-to-target strategies generally set low disease activity (or remission) as an overall goal and seek to achieve this using available therapeutic options as necessary. Introduced following the availability of biologics and targeted systemic therapies, treat-to-target strategies generally provide guidance on expectations of treatment but not specific treatments, as personalized treatment decisions depend on an assessment of individual patients and consider clinical and demographic features as well as preferences for available therapeutic options. If targets are not achieved in the assigned time span, adjustments can be made to the treatment approach in close consultation with the patient. If the target is reached, follow-up visits can be scheduled to ensure improvement is maintained or to establish if more aggressive goals could be selected.
Treat-to-target strategies for the management of psoriasis developed by the National Psoriasis Foundation (NPF) Medical Board include reducing the extent of psoriasis to 1% or lower total body surface area (BSA) after 3 months of treatment.10 Treatment targets endorsed by the European Academy of Dermatology and Venereology (EADV) in guidelines on the use of systemic therapies in psoriasis include achieving a 75% or greater reduction in Psoriasis Area and Severity Index (PASI) score within 3 to 4 months of treatment.11
In clinical practice, many patients do not achieve these treatment targets, and topical treatments alone generally are insufficient in achieving treatment goals for psoriasis.12,13 Moreover, conventional topical treatments (eg, topical corticosteroids) used by most patients with psoriasis regardless of disease severity are associated with adverse events that can limit their use. Most topical corticosteroids have US Food and Drug Administration label restrictions relating to sites of application, duration and extent of use, and frequency of administration.14,15
Tapinarof cream 1% (VTAMA [Dermavant Sciences, Inc]) is a first-in-class topical nonsteroidal aryl hydrocarbon receptor agonist that was approved by the US Food and Drug Administration for the treatment of plaque psoriasis in adults16 and is being studied for the treatment of plaque psoriasis in children 2 years and older as well as for atopic dermatitis in adults and children 2 years and older. In PSOARING 1 (ClinicalTrials .gov identifier NCT03956355) and PSOARING 2 (NCT03983980)—identical 12-week pivotal phase 3 trials—monotherapy with tapinarof cream 1% once daily (QD) demonstrated statistically significant efficacy vs vehicle cream and was well tolerated in adults with mild to severe plaque psoriasis (Supplementary Figure S1).17 Lebwohl et al17 reported that significantly higher PASI75 responses were observed at week 12 with tapinarof cream vs vehicle in PSOARING 1 and PSOARING 2 (36% and 48% vs 10% and 7%, respectively; both P<.0001). A significantly higher PASI90 response of 19% and 21% at week 12 also was observed with tapinarof cream vs 2% and 3% with vehicle in PSOARING 1 and PSOARING 2, respectively (P=.0005 and P<.0001).17
In PSOARING 3 (NCT04053387)—the long-term extension trial (Supplementary Figure S1)—efficacy continued to improve or was maintained beyond the two 12-week trials, with improvements in total BSA affected and PASI scores for up to 52 weeks.18 Tapinarof cream 1% QD demonstrated positive, rapid, and durable outcomes in PSOARING 3, including high rates of complete disease clearance (Physician Global Assessment [PGA] score=0 [clear])(40.9% [312/763]), durability of response on treatment with no evidence of tachyphylaxis, and a remittive effect of approximately 4 months when off therapy (defined as maintenance of a PGA score of 0 [clear] or 1 [almost clear] after first achieving a PGA score of 0).18
Herein, we report absolute treatment targets for patients with plaque psoriasis who received tapinarof cream 1% QD in the PSOARING trials that are at least as stringent as the corresponding NPF and EADV targets of achieving a total BSA affected of 1% or lower or a PASI75 response within 3 to 4 months, respectively.
METHODS
Study Design
The pooled efficacy analyses included all patients with a baseline PGA score of 2 or higher (mild or worse) before treatment with tapinarof cream 1% QD in the PSOARING trials. This included patients who received tapinarof cream 1% in PSOARING 1 and PSOARING 2 who may or may not have continued into PSOARING 3, as well as those who received the vehicle in PSOARING 1 and PSOARING 2 who enrolled in PSOARING 3 and had a PGA score of 2 or higher before receiving tapinarof cream 1%.
Trial Participants
Full methods, including inclusion and exclusion criteria, for the PSOARING trials have been previously reported.17,18 Patients were aged 18 to 75 years and had chronic plaque psoriasis that was stable for at least 6 months before randomization; 3% to 20% total BSA affected (excluding the scalp, palms, fingernails, toenails, and soles); and a PGA score of 2 (mild), 3 (moderate), or 4 (severe) at baseline.
The clinical trials were conducted in compliance with the guidelines for Good Clinical Practice and the Declaration of Helsinki. Approval was obtained from local ethics committees or institutional review boards at each center. All patients provided written informed consent.
Trial Treatment
In PSOARING 1 and PSOARING 2, patients were randomized (2:1) to receive tapinarof cream 1% or vehicle QD for 12 weeks. In PSOARING 3 (the long-term extension trial), patients received up to 40 weeks of open-label tapinarof, followed by 4 weeks of follow-up off treatment. Patients received intermittent or continuous treatment with tapinarof cream 1% in PSOARING 3 based on PGA score: those entering the trial with a PGA score of 1 or higher received tapinarof cream 1% until complete disease clearance was achieved (defined as a PGA score of 0 [clear]). Those entering PSOARING 3 with or achieving a PGA score of 0 (clear) discontinued treatment and were observed for the duration of maintenance of a PGA score of 0 (clear) or 1 (almost clear) while off therapy (the protocol-defined “duration of remittive effect”). If disease worsening (defined as a PGA score 2 or higher) occurred, tapinarof cream 1% was restarted and continued until a PGA score of 0 (clear) was achieved. This pattern of treatment, discontinuation on achieving a PGA score of 0 (clear), and retreatment on disease worsening continued until the end of the trial. As a result, patients in PSOARING 3 could receive tapinarof cream 1% continuously or intermittently for 40 weeks.
Outcome Measures and Statistical Analyses
The assessment of total BSA affected by plaque psoriasis is an estimate of the total extent of disease as a percentage of total skin area. In the PSOARING trials, the skin surface of one hand (palm and digits) was assumed to be approximately equivalent to 1% BSA. The total BSA affected by psoriasis was evaluated from 0% to 100%, with greater total BSA affected being an indication of more extensive disease. The BSA efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved a 1% or lower or 0.5% or lower total BSA affected.
Psoriasis Area and Severity Index scores assess both the severity and extent of psoriasis. A PASI score lower than 5 often is considered indicative of mild psoriasis, a score of 5 to 10 indicates moderate disease, and a score higher than 10 indicates severe disease.19 The maximum PASI score is 72. The PASI efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved an absolute total PASI score of 3 or lower, 2 or lower, and 1 or lower.
Efficacy analyses were based on pooled data for all patients in the PSOARING trials who had a PGA score of 2 to 4 (mild to severe) before treatment with tapinarof cream 1% in the intention-to-treat population using observed cases. Time-to-target analyses were based on Kaplan-Meier (KM) estimates using observed cases.
Safety analyses included the incidence and frequency of adverse events and were based on all patients who received tapinarof cream 1% in the PSOARING trials.
RESULTS
Baseline Patient Demographics and Disease Characteristics
The pooled efficacy analyses included 915 eligible patients (Table). At baseline, the mean (SD) age was 50.2 (13.25) years, 58.7% were male, the mean (SD) weight was 92.2 (23.67) kg, and the mean (SD) body mass index was 31.6 (7.53) kg/m2. The percentage of patients with a PGA score of 2 (mild), 3 (moderate), or 4 (severe) was 13.9%, 78.1%, and 8.0%, respectively. The mean (SD) PASI score was 8.7 (4.23) and mean (SD) total BSA affected was 7.8% (4.98).
Efficacy
Achievement of BSA-Affected Targets—
Achievement of Absolute PASI Targets—Across the total trial period (up to 52 weeks), an absolute total PASI score of 3 or lower was achieved by 75% of patients (686/915), with a median time to achieve this of 2 months (KM estimate: 58 days [95% CI, 57-63]); approximately 67% of patients (612/915) achieved a total PASI score of 2 or lower, with a median time to achieve of 3 months (KM estimate: 87 days [95% CI, 85-110])(Figure 2; Supplementary Figures S3a and S3b). A PASI score of 1 or lower was achieved by approximately 50% of patients (460/915), with a median time to achieve of approximately 6 months (KM estimate: 185 days [95% CI, 169-218])(Figure 2, Supplementary Figure S3c).
Illustrative Case—Case photography showing the clinical response in a 63-year-old man with moderate plaque psoriasis in PSOARING 2 is shown in Figure 3. After 12 weeks of treatment with tapinarof cream 1% QD, the patient achieved all primary and secondary efficacy end points. In addition to achieving the regulatory end point of a PGA score of 0 (clear) or 1 (almost clear) and a decrease from baseline of at least 2 points, achievement of 0% total BSA affected and a total PASI score of 0 at week 12 exceeded the NPF and EADV consensus treatment targets.10,11 Targets were achieved as early as week 4, with a total BSA affected of 0.5% or lower and a total PASI score of 1 or lower, illustrated by marked skin clearing and only faint residual erythema that completely resolved at week 12, with the absence of postinflammatory hyperpigmentation.
Safety
Safety data for the PSOARING trials have been previously reported.17,18 The most common treatment-emergent adverse events were folliculitis, contact dermatitis, upper respiratory tract infection, and nasopharyngitis. Treatment-emergent adverse events generally were mild or moderate in severity and did not lead to trial discontinuation.17,18
COMMENT
Treat-to-target management approaches aim to improve patient outcomes by striving to achieve optimal goals. The treat-to-target approach supports shared decision-making between clinicians and patients based on common expectations of what constitutes treatment success.
The findings of this analysis based on pooled data from a large cohort of patients demonstrate that a high proportion of patients can achieve or exceed recommended treatment targets with tapinarof cream 1% QD and maintain improvements long-term. The NPF-recommended treatment target of 1% or lower BSA affected within approximately 3 months (90 days) of treatment was achieved by 40% of tapinarof-treated patients. In addition, 1% or lower BSA affected at any time during the trials was achieved by 61% of patients (median, approximately 4 months). The analyses also indicated that PASI total scores of 3 or lower and 2 or lower were achieved by 75% and 67% of tapinarof-treated patients, respectively, within 2 to 3 months.
These findings support the previously reported efficacy of tapinarof cream, including high rates of complete disease clearance (40.9% [312/763]), durable response following treatment interruption, an off-therapy remittive effect of approximately 4 months, and good disease control on therapy with no evidence of tachyphylaxis.17,18
CONCLUSION
Taken together with previously reported tapinarof efficacy and safety results, our findings demonstrate that a high proportion of patients treated with tapinarof cream as monotherapy can achieve aggressive treatment targets set by both US and European guidelines developed for systemic and biologic therapies. Tapinarof cream 1% QD is an effective topical treatment option for patients with plaque psoriasis that has been approved without restrictions relating to severity or extent of disease treated, duration of use, or application sites, including application to sensitive and intertriginous skin.
Acknowledgments—Editorial and medical writing support under the guidance of the authors was provided by Melanie Govender, MSc (Med), ApotheCom (United Kingdom), and was funded by Dermavant Sciences, Inc, in accordance with Good Publication Practice (GPP) guidelines.
Psoriasis is a chronic inflammatory disease affecting approximately 8 million adults in the United States and 2% of the global population.1,2 Psoriasis causes pain, itching, and disfigurement and is associated with a physical, psychological, and economic burden that substantially affects health-related quality of life.3-5
Setting treatment goals and treating to target are evidence-based approaches that have been successfully applied to several chronic diseases to improve patient outcomes, including diabetes, hypertension, and rheumatoid arthritis.6-9 Treat-to-target strategies generally set low disease activity (or remission) as an overall goal and seek to achieve this using available therapeutic options as necessary. Introduced following the availability of biologics and targeted systemic therapies, treat-to-target strategies generally provide guidance on expectations of treatment but not specific treatments, as personalized treatment decisions depend on an assessment of individual patients and consider clinical and demographic features as well as preferences for available therapeutic options. If targets are not achieved in the assigned time span, adjustments can be made to the treatment approach in close consultation with the patient. If the target is reached, follow-up visits can be scheduled to ensure improvement is maintained or to establish if more aggressive goals could be selected.
Treat-to-target strategies for the management of psoriasis developed by the National Psoriasis Foundation (NPF) Medical Board include reducing the extent of psoriasis to 1% or lower total body surface area (BSA) after 3 months of treatment.10 Treatment targets endorsed by the European Academy of Dermatology and Venereology (EADV) in guidelines on the use of systemic therapies in psoriasis include achieving a 75% or greater reduction in Psoriasis Area and Severity Index (PASI) score within 3 to 4 months of treatment.11
In clinical practice, many patients do not achieve these treatment targets, and topical treatments alone generally are insufficient in achieving treatment goals for psoriasis.12,13 Moreover, conventional topical treatments (eg, topical corticosteroids) used by most patients with psoriasis regardless of disease severity are associated with adverse events that can limit their use. Most topical corticosteroids have US Food and Drug Administration label restrictions relating to sites of application, duration and extent of use, and frequency of administration.14,15
Tapinarof cream 1% (VTAMA [Dermavant Sciences, Inc]) is a first-in-class topical nonsteroidal aryl hydrocarbon receptor agonist that was approved by the US Food and Drug Administration for the treatment of plaque psoriasis in adults16 and is being studied for the treatment of plaque psoriasis in children 2 years and older as well as for atopic dermatitis in adults and children 2 years and older. In PSOARING 1 (ClinicalTrials .gov identifier NCT03956355) and PSOARING 2 (NCT03983980)—identical 12-week pivotal phase 3 trials—monotherapy with tapinarof cream 1% once daily (QD) demonstrated statistically significant efficacy vs vehicle cream and was well tolerated in adults with mild to severe plaque psoriasis (Supplementary Figure S1).17 Lebwohl et al17 reported that significantly higher PASI75 responses were observed at week 12 with tapinarof cream vs vehicle in PSOARING 1 and PSOARING 2 (36% and 48% vs 10% and 7%, respectively; both P<.0001). A significantly higher PASI90 response of 19% and 21% at week 12 also was observed with tapinarof cream vs 2% and 3% with vehicle in PSOARING 1 and PSOARING 2, respectively (P=.0005 and P<.0001).17
In PSOARING 3 (NCT04053387)—the long-term extension trial (Supplementary Figure S1)—efficacy continued to improve or was maintained beyond the two 12-week trials, with improvements in total BSA affected and PASI scores for up to 52 weeks.18 Tapinarof cream 1% QD demonstrated positive, rapid, and durable outcomes in PSOARING 3, including high rates of complete disease clearance (Physician Global Assessment [PGA] score=0 [clear])(40.9% [312/763]), durability of response on treatment with no evidence of tachyphylaxis, and a remittive effect of approximately 4 months when off therapy (defined as maintenance of a PGA score of 0 [clear] or 1 [almost clear] after first achieving a PGA score of 0).18
Herein, we report absolute treatment targets for patients with plaque psoriasis who received tapinarof cream 1% QD in the PSOARING trials that are at least as stringent as the corresponding NPF and EADV targets of achieving a total BSA affected of 1% or lower or a PASI75 response within 3 to 4 months, respectively.
METHODS
Study Design
The pooled efficacy analyses included all patients with a baseline PGA score of 2 or higher (mild or worse) before treatment with tapinarof cream 1% QD in the PSOARING trials. This included patients who received tapinarof cream 1% in PSOARING 1 and PSOARING 2 who may or may not have continued into PSOARING 3, as well as those who received the vehicle in PSOARING 1 and PSOARING 2 who enrolled in PSOARING 3 and had a PGA score of 2 or higher before receiving tapinarof cream 1%.
Trial Participants
Full methods, including inclusion and exclusion criteria, for the PSOARING trials have been previously reported.17,18 Patients were aged 18 to 75 years and had chronic plaque psoriasis that was stable for at least 6 months before randomization; 3% to 20% total BSA affected (excluding the scalp, palms, fingernails, toenails, and soles); and a PGA score of 2 (mild), 3 (moderate), or 4 (severe) at baseline.
The clinical trials were conducted in compliance with the guidelines for Good Clinical Practice and the Declaration of Helsinki. Approval was obtained from local ethics committees or institutional review boards at each center. All patients provided written informed consent.
Trial Treatment
In PSOARING 1 and PSOARING 2, patients were randomized (2:1) to receive tapinarof cream 1% or vehicle QD for 12 weeks. In PSOARING 3 (the long-term extension trial), patients received up to 40 weeks of open-label tapinarof, followed by 4 weeks of follow-up off treatment. Patients received intermittent or continuous treatment with tapinarof cream 1% in PSOARING 3 based on PGA score: those entering the trial with a PGA score of 1 or higher received tapinarof cream 1% until complete disease clearance was achieved (defined as a PGA score of 0 [clear]). Those entering PSOARING 3 with or achieving a PGA score of 0 (clear) discontinued treatment and were observed for the duration of maintenance of a PGA score of 0 (clear) or 1 (almost clear) while off therapy (the protocol-defined “duration of remittive effect”). If disease worsening (defined as a PGA score 2 or higher) occurred, tapinarof cream 1% was restarted and continued until a PGA score of 0 (clear) was achieved. This pattern of treatment, discontinuation on achieving a PGA score of 0 (clear), and retreatment on disease worsening continued until the end of the trial. As a result, patients in PSOARING 3 could receive tapinarof cream 1% continuously or intermittently for 40 weeks.
Outcome Measures and Statistical Analyses
The assessment of total BSA affected by plaque psoriasis is an estimate of the total extent of disease as a percentage of total skin area. In the PSOARING trials, the skin surface of one hand (palm and digits) was assumed to be approximately equivalent to 1% BSA. The total BSA affected by psoriasis was evaluated from 0% to 100%, with greater total BSA affected being an indication of more extensive disease. The BSA efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved a 1% or lower or 0.5% or lower total BSA affected.
Psoriasis Area and Severity Index scores assess both the severity and extent of psoriasis. A PASI score lower than 5 often is considered indicative of mild psoriasis, a score of 5 to 10 indicates moderate disease, and a score higher than 10 indicates severe disease.19 The maximum PASI score is 72. The PASI efficacy outcomes used in these analyses were based post hoc on the proportion of patients who achieved an absolute total PASI score of 3 or lower, 2 or lower, and 1 or lower.
Efficacy analyses were based on pooled data for all patients in the PSOARING trials who had a PGA score of 2 to 4 (mild to severe) before treatment with tapinarof cream 1% in the intention-to-treat population using observed cases. Time-to-target analyses were based on Kaplan-Meier (KM) estimates using observed cases.
Safety analyses included the incidence and frequency of adverse events and were based on all patients who received tapinarof cream 1% in the PSOARING trials.
RESULTS
Baseline Patient Demographics and Disease Characteristics
The pooled efficacy analyses included 915 eligible patients (Table). At baseline, the mean (SD) age was 50.2 (13.25) years, 58.7% were male, the mean (SD) weight was 92.2 (23.67) kg, and the mean (SD) body mass index was 31.6 (7.53) kg/m2. The percentage of patients with a PGA score of 2 (mild), 3 (moderate), or 4 (severe) was 13.9%, 78.1%, and 8.0%, respectively. The mean (SD) PASI score was 8.7 (4.23) and mean (SD) total BSA affected was 7.8% (4.98).
Efficacy
Achievement of BSA-Affected Targets—
Achievement of Absolute PASI Targets—Across the total trial period (up to 52 weeks), an absolute total PASI score of 3 or lower was achieved by 75% of patients (686/915), with a median time to achieve this of 2 months (KM estimate: 58 days [95% CI, 57-63]); approximately 67% of patients (612/915) achieved a total PASI score of 2 or lower, with a median time to achieve of 3 months (KM estimate: 87 days [95% CI, 85-110])(Figure 2; Supplementary Figures S3a and S3b). A PASI score of 1 or lower was achieved by approximately 50% of patients (460/915), with a median time to achieve of approximately 6 months (KM estimate: 185 days [95% CI, 169-218])(Figure 2, Supplementary Figure S3c).
Illustrative Case—Case photography showing the clinical response in a 63-year-old man with moderate plaque psoriasis in PSOARING 2 is shown in Figure 3. After 12 weeks of treatment with tapinarof cream 1% QD, the patient achieved all primary and secondary efficacy end points. In addition to achieving the regulatory end point of a PGA score of 0 (clear) or 1 (almost clear) and a decrease from baseline of at least 2 points, achievement of 0% total BSA affected and a total PASI score of 0 at week 12 exceeded the NPF and EADV consensus treatment targets.10,11 Targets were achieved as early as week 4, with a total BSA affected of 0.5% or lower and a total PASI score of 1 or lower, illustrated by marked skin clearing and only faint residual erythema that completely resolved at week 12, with the absence of postinflammatory hyperpigmentation.
Safety
Safety data for the PSOARING trials have been previously reported.17,18 The most common treatment-emergent adverse events were folliculitis, contact dermatitis, upper respiratory tract infection, and nasopharyngitis. Treatment-emergent adverse events generally were mild or moderate in severity and did not lead to trial discontinuation.17,18
COMMENT
Treat-to-target management approaches aim to improve patient outcomes by striving to achieve optimal goals. The treat-to-target approach supports shared decision-making between clinicians and patients based on common expectations of what constitutes treatment success.
The findings of this analysis based on pooled data from a large cohort of patients demonstrate that a high proportion of patients can achieve or exceed recommended treatment targets with tapinarof cream 1% QD and maintain improvements long-term. The NPF-recommended treatment target of 1% or lower BSA affected within approximately 3 months (90 days) of treatment was achieved by 40% of tapinarof-treated patients. In addition, 1% or lower BSA affected at any time during the trials was achieved by 61% of patients (median, approximately 4 months). The analyses also indicated that PASI total scores of 3 or lower and 2 or lower were achieved by 75% and 67% of tapinarof-treated patients, respectively, within 2 to 3 months.
These findings support the previously reported efficacy of tapinarof cream, including high rates of complete disease clearance (40.9% [312/763]), durable response following treatment interruption, an off-therapy remittive effect of approximately 4 months, and good disease control on therapy with no evidence of tachyphylaxis.17,18
CONCLUSION
Taken together with previously reported tapinarof efficacy and safety results, our findings demonstrate that a high proportion of patients treated with tapinarof cream as monotherapy can achieve aggressive treatment targets set by both US and European guidelines developed for systemic and biologic therapies. Tapinarof cream 1% QD is an effective topical treatment option for patients with plaque psoriasis that has been approved without restrictions relating to severity or extent of disease treated, duration of use, or application sites, including application to sensitive and intertriginous skin.
Acknowledgments—Editorial and medical writing support under the guidance of the authors was provided by Melanie Govender, MSc (Med), ApotheCom (United Kingdom), and was funded by Dermavant Sciences, Inc, in accordance with Good Publication Practice (GPP) guidelines.
- Armstrong AW, Mehta MD, Schupp CW, et al. Psoriasis prevalence in adults in the United States. JAMA Dermatol. 2021;157:940-946.
- Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369:m1590.
- Pilon D, Teeple A, Zhdanava M, et al. The economic burden of psoriasis with high comorbidity among privately insured patients in the United States. J Med Econ. 2019;22:196-203.
- Singh S, Taylor C, Kornmehl H, et al. Psoriasis and suicidality: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:425-440.e2.
- Feldman SR, Goffe B, Rice G, et al. The challenge of managing psoriasis: unmet medical needs and stakeholder perspectives. Am Health Drug Benefits. 2016;9:504-513.
- Ford JA, Solomon DH. Challenges in implementing treat-to-target strategies in rheumatology. Rheum Dis Clin North Am. 2019;45:101-112.
- Sitbon O, Galiè N. Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur Respir Rev. 2010;19:272-278.
- Smolen JS, Aletaha D, Bijlsma JW, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69:631-637.
- Wangnoo SK, Sethi B, Sahay RK, et al. Treat-to-target trials in diabetes. Indian J Endocrinol Metab. 2014;18:166-174.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Pathirana D, Ormerod AD, Saiag P, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris. J Eur Acad Dermatol Venereol. 2009;23(Suppl 2):1-70.
- Strober BE, van der Walt JM, Armstrong AW, et al. Clinical goals and barriers to effective psoriasis care. Dermatol Ther (Heidelb). 2019; 9:5-18.
- Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
- Elmets CA, Korman NJ, Prater EF, et al. Joint AAD-NPF Guidelines of care for the management and treatment of psoriasis with topical therapy and alternative medicine modalities for psoriasis severity measures. J Am Acad Dermatol. 2021;84:432-470.
- Stein Gold LF. Topical therapies for psoriasis: improving management strategies and patient adherence. Semin Cutan Med Surg. 2016;35 (2 Suppl 2):S36-S44; quiz S45.
- VTAMA® (tapinarof) cream. Prescribing information. Dermavant Sciences; 2022. Accessed September 13, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215272s000lbl.pdf
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229 and supplementary appendix.
- Strober B, Stein Gold L, Bissonnette R, et al. One-year safety and efficacy of tapinarof cream for the treatment of plaque psoriasis: results from the PSOARING 3 trial. J Am Acad Dermatol. 2022;87:800-806.
- Clinical Review Report: Guselkumab (Tremfya) [Internet]. Canadian Agency for Drugs and Technologies in Health; 2018. Accessed September 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534047/pdf/Bookshelf_NBK534047.pdf
- Armstrong AW, Mehta MD, Schupp CW, et al. Psoriasis prevalence in adults in the United States. JAMA Dermatol. 2021;157:940-946.
- Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369:m1590.
- Pilon D, Teeple A, Zhdanava M, et al. The economic burden of psoriasis with high comorbidity among privately insured patients in the United States. J Med Econ. 2019;22:196-203.
- Singh S, Taylor C, Kornmehl H, et al. Psoriasis and suicidality: a systematic review and meta-analysis. J Am Acad Dermatol. 2017;77:425-440.e2.
- Feldman SR, Goffe B, Rice G, et al. The challenge of managing psoriasis: unmet medical needs and stakeholder perspectives. Am Health Drug Benefits. 2016;9:504-513.
- Ford JA, Solomon DH. Challenges in implementing treat-to-target strategies in rheumatology. Rheum Dis Clin North Am. 2019;45:101-112.
- Sitbon O, Galiè N. Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur Respir Rev. 2010;19:272-278.
- Smolen JS, Aletaha D, Bijlsma JW, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69:631-637.
- Wangnoo SK, Sethi B, Sahay RK, et al. Treat-to-target trials in diabetes. Indian J Endocrinol Metab. 2014;18:166-174.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Pathirana D, Ormerod AD, Saiag P, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris. J Eur Acad Dermatol Venereol. 2009;23(Suppl 2):1-70.
- Strober BE, van der Walt JM, Armstrong AW, et al. Clinical goals and barriers to effective psoriasis care. Dermatol Ther (Heidelb). 2019; 9:5-18.
- Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16:1209-1222.
- Elmets CA, Korman NJ, Prater EF, et al. Joint AAD-NPF Guidelines of care for the management and treatment of psoriasis with topical therapy and alternative medicine modalities for psoriasis severity measures. J Am Acad Dermatol. 2021;84:432-470.
- Stein Gold LF. Topical therapies for psoriasis: improving management strategies and patient adherence. Semin Cutan Med Surg. 2016;35 (2 Suppl 2):S36-S44; quiz S45.
- VTAMA® (tapinarof) cream. Prescribing information. Dermavant Sciences; 2022. Accessed September 13, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215272s000lbl.pdf
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229 and supplementary appendix.
- Strober B, Stein Gold L, Bissonnette R, et al. One-year safety and efficacy of tapinarof cream for the treatment of plaque psoriasis: results from the PSOARING 3 trial. J Am Acad Dermatol. 2022;87:800-806.
- Clinical Review Report: Guselkumab (Tremfya) [Internet]. Canadian Agency for Drugs and Technologies in Health; 2018. Accessed September 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534047/pdf/Bookshelf_NBK534047.pdf
Practice Points
- In clinical practice, many patients with psoriasis do not achieve treatment targets set forth by the National Psoriasis Foundation and the European Academy of Dermatology and Venereology, and topical treatments alone generally are insufficient in achieving treatment goals for psoriasis.
- Tapinarof cream 1% is a nonsteroidal aryl hydrocarbon receptor agonist approved by the US Food and Drug Administration for the treatment of plaque psoriasis in adults; it also is being studied for the treatment of plaque psoriasis in children 2 years and older.
- Tapinarof cream 1% is an effective topical treatment option for patients with plaque psoriasis of any severity, with no limitations on treatment duration, total extent of use, or application sites, including intertriginous skin and sensitive areas.
Study Supports Efficacy of Home-Based Phototherapy for Psoriasis
TOPLINE:
study.
METHODOLOGY:
- The pragmatic, investigator-initiated, open-label, noninferiority, randomized trial compared the effectiveness of 12 weeks of treatment with narrow-band ultraviolet B phototherapy administered at home (n = 393) vs at the doctor’s office (n = 390).
- Overall, 783 patients with plaque or guttate psoriasis (mean age, 48 years; 48% women) were enrolled at 42 academic and private clinical dermatology practices in the United States from March 1, 2019, to December 4, 2023, and were followed up through June 2024. At baseline, the mean Physician Global Assessment (PGA) and the mean Dermatology Life Quality Index (DLQI) scores were 2.7 and 12.2, respectively.
- The two co-primary endpoints were a PGA score ≤ 1 indicating clear or almost clear skin and a DLQI score ≤ 5.
TAKEAWAY:
- At 12 weeks, a PGA score ≤ 1 was achieved in 32.8% of patients using home-based phototherapy and in 25.6% of those who received office-based phototherapy (P < .001).
- At 12 weeks, a DLQI score ≤ 5 was achieved in 52.4% and 33.6% of home- and office-treated patients, respectively (P < .001).
- Similar benefits were seen across all Fitzpatrick skin types.
- A higher percentage of patients were adherent to home-based (51.4%) vs office-based (15.9%) phototherapy (P < .001).
IN PRACTICE:
“These data support the use of home phototherapy as a first-line treatment option for psoriasis,” and “efforts are needed to make home and office phototherapy more available to patients,” said the study’s lead author.
SOURCE:
Joel M. Gelfand, MD, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania, Philadelphia, presented the findings at the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis meeting during the annual meeting of the European Academy of Dermatology and Venereology, with simultaneous publication in JAMA Dermatology.
LIMITATIONS:
This was an open-label trial and because of its pragmatic design, outcome data were missing. The cost of the home-based phototherapy equipment used in the study was $6040.88, which was mostly covered by Medicare, but direct costs to patients may have varied depending on their insurance plan.
DISCLOSURES:
The Patient-Centered Outcomes Research Institute funded the study. Daavlin provided and shipped machines for home-based phototherapy to patients at no cost. Dr. Gelfand disclosed serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, Boehringer Ingelheim, Celldex, and other companies. The full list of author disclosures can be found in the published study.
A version of this article first appeared on Medscape.com.
TOPLINE:
study.
METHODOLOGY:
- The pragmatic, investigator-initiated, open-label, noninferiority, randomized trial compared the effectiveness of 12 weeks of treatment with narrow-band ultraviolet B phototherapy administered at home (n = 393) vs at the doctor’s office (n = 390).
- Overall, 783 patients with plaque or guttate psoriasis (mean age, 48 years; 48% women) were enrolled at 42 academic and private clinical dermatology practices in the United States from March 1, 2019, to December 4, 2023, and were followed up through June 2024. At baseline, the mean Physician Global Assessment (PGA) and the mean Dermatology Life Quality Index (DLQI) scores were 2.7 and 12.2, respectively.
- The two co-primary endpoints were a PGA score ≤ 1 indicating clear or almost clear skin and a DLQI score ≤ 5.
TAKEAWAY:
- At 12 weeks, a PGA score ≤ 1 was achieved in 32.8% of patients using home-based phototherapy and in 25.6% of those who received office-based phototherapy (P < .001).
- At 12 weeks, a DLQI score ≤ 5 was achieved in 52.4% and 33.6% of home- and office-treated patients, respectively (P < .001).
- Similar benefits were seen across all Fitzpatrick skin types.
- A higher percentage of patients were adherent to home-based (51.4%) vs office-based (15.9%) phototherapy (P < .001).
IN PRACTICE:
“These data support the use of home phototherapy as a first-line treatment option for psoriasis,” and “efforts are needed to make home and office phototherapy more available to patients,” said the study’s lead author.
SOURCE:
Joel M. Gelfand, MD, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania, Philadelphia, presented the findings at the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis meeting during the annual meeting of the European Academy of Dermatology and Venereology, with simultaneous publication in JAMA Dermatology.
LIMITATIONS:
This was an open-label trial and because of its pragmatic design, outcome data were missing. The cost of the home-based phototherapy equipment used in the study was $6040.88, which was mostly covered by Medicare, but direct costs to patients may have varied depending on their insurance plan.
DISCLOSURES:
The Patient-Centered Outcomes Research Institute funded the study. Daavlin provided and shipped machines for home-based phototherapy to patients at no cost. Dr. Gelfand disclosed serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, Boehringer Ingelheim, Celldex, and other companies. The full list of author disclosures can be found in the published study.
A version of this article first appeared on Medscape.com.
TOPLINE:
study.
METHODOLOGY:
- The pragmatic, investigator-initiated, open-label, noninferiority, randomized trial compared the effectiveness of 12 weeks of treatment with narrow-band ultraviolet B phototherapy administered at home (n = 393) vs at the doctor’s office (n = 390).
- Overall, 783 patients with plaque or guttate psoriasis (mean age, 48 years; 48% women) were enrolled at 42 academic and private clinical dermatology practices in the United States from March 1, 2019, to December 4, 2023, and were followed up through June 2024. At baseline, the mean Physician Global Assessment (PGA) and the mean Dermatology Life Quality Index (DLQI) scores were 2.7 and 12.2, respectively.
- The two co-primary endpoints were a PGA score ≤ 1 indicating clear or almost clear skin and a DLQI score ≤ 5.
TAKEAWAY:
- At 12 weeks, a PGA score ≤ 1 was achieved in 32.8% of patients using home-based phototherapy and in 25.6% of those who received office-based phototherapy (P < .001).
- At 12 weeks, a DLQI score ≤ 5 was achieved in 52.4% and 33.6% of home- and office-treated patients, respectively (P < .001).
- Similar benefits were seen across all Fitzpatrick skin types.
- A higher percentage of patients were adherent to home-based (51.4%) vs office-based (15.9%) phototherapy (P < .001).
IN PRACTICE:
“These data support the use of home phototherapy as a first-line treatment option for psoriasis,” and “efforts are needed to make home and office phototherapy more available to patients,” said the study’s lead author.
SOURCE:
Joel M. Gelfand, MD, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania, Philadelphia, presented the findings at the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis meeting during the annual meeting of the European Academy of Dermatology and Venereology, with simultaneous publication in JAMA Dermatology.
LIMITATIONS:
This was an open-label trial and because of its pragmatic design, outcome data were missing. The cost of the home-based phototherapy equipment used in the study was $6040.88, which was mostly covered by Medicare, but direct costs to patients may have varied depending on their insurance plan.
DISCLOSURES:
The Patient-Centered Outcomes Research Institute funded the study. Daavlin provided and shipped machines for home-based phototherapy to patients at no cost. Dr. Gelfand disclosed serving as a consultant for AbbVie, Artax, Bristol-Myers Squibb, Boehringer Ingelheim, Celldex, and other companies. The full list of author disclosures can be found in the published study.
A version of this article first appeared on Medscape.com.
Bimekizumab Gains FDA Approval for Psoriatic Arthritis, Axial Spondyloarthritis
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.