User login
Drug granted breakthrough designation for ALL

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
Study provides clearer picture of JMML

Photo by Petr Kratochvil
Whole-exome sequencing has provided new insights that may lead to better treatment of juvenile myelomonocytic leukemia (JMML), according to researchers.
The group identified new mutations that appear to drive JMML and could be targeted with drugs that are currently available, such as JAK inhibitors.
The study also suggests it is the number of mutations a patient has—and not the type of mutations—that will influence the patient’s outcome.
Researchers reported these discoveries in Nature Genetics.
“We’ve created the most comprehensive portrait yet of how this cancer evolves from first diagnosis through remission or relapse,” said study author Mignon Loh, MD, of Benioff Children’s Hospital at University of California, San Francisco (USCF). “What we found helps make sense of why patients’ outcomes have been so wildly different.”
“We have personally treated patients with JMML at UCSF with identical driver mutations, some of whom survived, while others died,” added Elliot Stieglitz, MD. “Our frustration was the main impetus that led us to carry out this study.”
So the researchers performed whole-exome sequencing on samples collected at diagnosis and relapse in 27 JMML patients who were 1 month to 3 years of age. The team then performed targeted sequencing of suspected mutation hot spots in another 71 patients.
Previously, just 5 defects in the Ras pathway had been associated with JMML. The new analysis added 10 mutations of known oncogenes and tumor suppressors to the list, including 2 additional Ras pathway genes.
These newly identified mutations occur in genes coding for proteins that function as signaling molecules, transcription factors, epigenetic regulators, and elements of the spliceosome complex.
Several of these mutations raise the possibility of targeting subpopulations of JMML cases with existing drugs.
For instance, JAK inhibitors might inhibit signaling through a hyperactive JAK-STAT pathway identified in some patients. And 5-azacytidine could be used to reduce excessive epigenetic DNA methylation seen in others.
The researchers also performed a 10-year survival study with the same participants and found that patients’ prognosis depended more on the number of mutations they had than on the specific mutations involved.
Patients with more than 1 mutation at the time of diagnosis had a significantly worse long-term prognosis. Of the 34 patients who had at least 2 mutations, only 29% survived for 10 years, compared to a 65% survival rate for patients who had 1 or fewer detectable mutations.
“We have now shown that while driver mutations in the Ras pathway likely cause the leukemia to develop in the first place, it is the presence of these additional mutations that contribute to poor outcome,” Dr Loh said, noting that therapies will likely require targeting multiple pathways at once.
“Precisely how these secondary mutations will interact with the Ras pathway is the focus of our ongoing work.” ![]()
Photo by Petr Kratochvil
Whole-exome sequencing has provided new insights that may lead to better treatment of juvenile myelomonocytic leukemia (JMML), according to researchers.
The group identified new mutations that appear to drive JMML and could be targeted with drugs that are currently available, such as JAK inhibitors.
The study also suggests it is the number of mutations a patient has—and not the type of mutations—that will influence the patient’s outcome.
Researchers reported these discoveries in Nature Genetics.
“We’ve created the most comprehensive portrait yet of how this cancer evolves from first diagnosis through remission or relapse,” said study author Mignon Loh, MD, of Benioff Children’s Hospital at University of California, San Francisco (USCF). “What we found helps make sense of why patients’ outcomes have been so wildly different.”
“We have personally treated patients with JMML at UCSF with identical driver mutations, some of whom survived, while others died,” added Elliot Stieglitz, MD. “Our frustration was the main impetus that led us to carry out this study.”
So the researchers performed whole-exome sequencing on samples collected at diagnosis and relapse in 27 JMML patients who were 1 month to 3 years of age. The team then performed targeted sequencing of suspected mutation hot spots in another 71 patients.
Previously, just 5 defects in the Ras pathway had been associated with JMML. The new analysis added 10 mutations of known oncogenes and tumor suppressors to the list, including 2 additional Ras pathway genes.
These newly identified mutations occur in genes coding for proteins that function as signaling molecules, transcription factors, epigenetic regulators, and elements of the spliceosome complex.
Several of these mutations raise the possibility of targeting subpopulations of JMML cases with existing drugs.
For instance, JAK inhibitors might inhibit signaling through a hyperactive JAK-STAT pathway identified in some patients. And 5-azacytidine could be used to reduce excessive epigenetic DNA methylation seen in others.
The researchers also performed a 10-year survival study with the same participants and found that patients’ prognosis depended more on the number of mutations they had than on the specific mutations involved.
Patients with more than 1 mutation at the time of diagnosis had a significantly worse long-term prognosis. Of the 34 patients who had at least 2 mutations, only 29% survived for 10 years, compared to a 65% survival rate for patients who had 1 or fewer detectable mutations.
“We have now shown that while driver mutations in the Ras pathway likely cause the leukemia to develop in the first place, it is the presence of these additional mutations that contribute to poor outcome,” Dr Loh said, noting that therapies will likely require targeting multiple pathways at once.
“Precisely how these secondary mutations will interact with the Ras pathway is the focus of our ongoing work.” ![]()
Photo by Petr Kratochvil
Whole-exome sequencing has provided new insights that may lead to better treatment of juvenile myelomonocytic leukemia (JMML), according to researchers.
The group identified new mutations that appear to drive JMML and could be targeted with drugs that are currently available, such as JAK inhibitors.
The study also suggests it is the number of mutations a patient has—and not the type of mutations—that will influence the patient’s outcome.
Researchers reported these discoveries in Nature Genetics.
“We’ve created the most comprehensive portrait yet of how this cancer evolves from first diagnosis through remission or relapse,” said study author Mignon Loh, MD, of Benioff Children’s Hospital at University of California, San Francisco (USCF). “What we found helps make sense of why patients’ outcomes have been so wildly different.”
“We have personally treated patients with JMML at UCSF with identical driver mutations, some of whom survived, while others died,” added Elliot Stieglitz, MD. “Our frustration was the main impetus that led us to carry out this study.”
So the researchers performed whole-exome sequencing on samples collected at diagnosis and relapse in 27 JMML patients who were 1 month to 3 years of age. The team then performed targeted sequencing of suspected mutation hot spots in another 71 patients.
Previously, just 5 defects in the Ras pathway had been associated with JMML. The new analysis added 10 mutations of known oncogenes and tumor suppressors to the list, including 2 additional Ras pathway genes.
These newly identified mutations occur in genes coding for proteins that function as signaling molecules, transcription factors, epigenetic regulators, and elements of the spliceosome complex.
Several of these mutations raise the possibility of targeting subpopulations of JMML cases with existing drugs.
For instance, JAK inhibitors might inhibit signaling through a hyperactive JAK-STAT pathway identified in some patients. And 5-azacytidine could be used to reduce excessive epigenetic DNA methylation seen in others.
The researchers also performed a 10-year survival study with the same participants and found that patients’ prognosis depended more on the number of mutations they had than on the specific mutations involved.
Patients with more than 1 mutation at the time of diagnosis had a significantly worse long-term prognosis. Of the 34 patients who had at least 2 mutations, only 29% survived for 10 years, compared to a 65% survival rate for patients who had 1 or fewer detectable mutations.
“We have now shown that while driver mutations in the Ras pathway likely cause the leukemia to develop in the first place, it is the presence of these additional mutations that contribute to poor outcome,” Dr Loh said, noting that therapies will likely require targeting multiple pathways at once.
“Precisely how these secondary mutations will interact with the Ras pathway is the focus of our ongoing work.” ![]()
Model recapitulates cancer susceptibility in DBA

Researchers say they’ve created the first animal model that recapitulates the predisposition to cancer observed in patients with Diamond-Blackfan anemia (DBA).
DBA is caused by mutations in ribosomal genes such as RPL11, so the researchers set out to determine the effects of manipulating RPL11 in mice.
The team found that RPL11-deficient mice
developed anemia, but they also had impaired p53 responses, elevated cMYC levels, and increased susceptibility to radiation-induced lymphomagenesis.
Manuel Serrano, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues described these findings in Cell Reports.
Previous observational studies suggested that around 20% of patients with DBA develop cancers, particularly lymphomas. Other research groups have developed animal models that recapitulate certain characteristics of DBA but not the predisposition to cancer.
In an attempt to change that, Dr Serrano and his colleagues focused their work on RPL11.
“Cells need the ribosomes to function properly in order to proliferate and grow,” Dr Serrano explained. “We knew that when something goes wrong in these organelles, RPL11 operates as a switch that activates the p53 gene to stop the cells from proliferating and forming tumors. This mechanism is called ribosomal stress.”
“P53 is one of the main tumor suppressor genes identified to date, to the extent that its relevance in preventing cancer has led to it being named the ‘guardian of the genome.’ This important function made us think that the protein could play a crucial role in the cancer predisposition observed in patients with DBA. If RPL11 is mutated, it loses the ability to activate p53 to prevent tumors caused by cellular damage.”
In fact, the researchers found that total or partial deletion of RPL11 impairs the normal function of p53 and increases levels of cMYC, which can promote tumor development.
“We believe that, in DBA, both factors combined contribute to induce the development of cancer,” said Lucía Morgado-Palacín, also of CNIO.
The researchers’ experiments supported this idea, as mice with heterozygous RPL11 deletion exhibited increased susceptibility to radiation-induced lymphomagenesis.
Mice with heterozygous RPL11 deletion also developed anemia that was associated with decreased erythroid
progenitors and defective erythroid maturation.
Homozygous deletion of RPL11, on the other hand, led to bone marrow aplasia
and intestinal atrophy in adult mice. And these mice died within a few weeks. ![]()
Researchers say they’ve created the first animal model that recapitulates the predisposition to cancer observed in patients with Diamond-Blackfan anemia (DBA).
DBA is caused by mutations in ribosomal genes such as RPL11, so the researchers set out to determine the effects of manipulating RPL11 in mice.
The team found that RPL11-deficient mice
developed anemia, but they also had impaired p53 responses, elevated cMYC levels, and increased susceptibility to radiation-induced lymphomagenesis.
Manuel Serrano, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues described these findings in Cell Reports.
Previous observational studies suggested that around 20% of patients with DBA develop cancers, particularly lymphomas. Other research groups have developed animal models that recapitulate certain characteristics of DBA but not the predisposition to cancer.
In an attempt to change that, Dr Serrano and his colleagues focused their work on RPL11.
“Cells need the ribosomes to function properly in order to proliferate and grow,” Dr Serrano explained. “We knew that when something goes wrong in these organelles, RPL11 operates as a switch that activates the p53 gene to stop the cells from proliferating and forming tumors. This mechanism is called ribosomal stress.”
“P53 is one of the main tumor suppressor genes identified to date, to the extent that its relevance in preventing cancer has led to it being named the ‘guardian of the genome.’ This important function made us think that the protein could play a crucial role in the cancer predisposition observed in patients with DBA. If RPL11 is mutated, it loses the ability to activate p53 to prevent tumors caused by cellular damage.”
In fact, the researchers found that total or partial deletion of RPL11 impairs the normal function of p53 and increases levels of cMYC, which can promote tumor development.
“We believe that, in DBA, both factors combined contribute to induce the development of cancer,” said Lucía Morgado-Palacín, also of CNIO.
The researchers’ experiments supported this idea, as mice with heterozygous RPL11 deletion exhibited increased susceptibility to radiation-induced lymphomagenesis.
Mice with heterozygous RPL11 deletion also developed anemia that was associated with decreased erythroid
progenitors and defective erythroid maturation.
Homozygous deletion of RPL11, on the other hand, led to bone marrow aplasia
and intestinal atrophy in adult mice. And these mice died within a few weeks. ![]()
Researchers say they’ve created the first animal model that recapitulates the predisposition to cancer observed in patients with Diamond-Blackfan anemia (DBA).
DBA is caused by mutations in ribosomal genes such as RPL11, so the researchers set out to determine the effects of manipulating RPL11 in mice.
The team found that RPL11-deficient mice
developed anemia, but they also had impaired p53 responses, elevated cMYC levels, and increased susceptibility to radiation-induced lymphomagenesis.
Manuel Serrano, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues described these findings in Cell Reports.
Previous observational studies suggested that around 20% of patients with DBA develop cancers, particularly lymphomas. Other research groups have developed animal models that recapitulate certain characteristics of DBA but not the predisposition to cancer.
In an attempt to change that, Dr Serrano and his colleagues focused their work on RPL11.
“Cells need the ribosomes to function properly in order to proliferate and grow,” Dr Serrano explained. “We knew that when something goes wrong in these organelles, RPL11 operates as a switch that activates the p53 gene to stop the cells from proliferating and forming tumors. This mechanism is called ribosomal stress.”
“P53 is one of the main tumor suppressor genes identified to date, to the extent that its relevance in preventing cancer has led to it being named the ‘guardian of the genome.’ This important function made us think that the protein could play a crucial role in the cancer predisposition observed in patients with DBA. If RPL11 is mutated, it loses the ability to activate p53 to prevent tumors caused by cellular damage.”
In fact, the researchers found that total or partial deletion of RPL11 impairs the normal function of p53 and increases levels of cMYC, which can promote tumor development.
“We believe that, in DBA, both factors combined contribute to induce the development of cancer,” said Lucía Morgado-Palacín, also of CNIO.
The researchers’ experiments supported this idea, as mice with heterozygous RPL11 deletion exhibited increased susceptibility to radiation-induced lymphomagenesis.
Mice with heterozygous RPL11 deletion also developed anemia that was associated with decreased erythroid
progenitors and defective erythroid maturation.
Homozygous deletion of RPL11, on the other hand, led to bone marrow aplasia
and intestinal atrophy in adult mice. And these mice died within a few weeks. ![]()
Study reveals potential drivers of CLL

Photo courtesy of Dana-
Farber Cancer Institute
Researchers say they have identified dozens of genetic abnormalities that may drive chronic lymphocytic leukemia (CLL), including some that were never before linked to human cancer.
The team began to trace how some of these abnormalities affect the course of the disease and its susceptibility to treatment.
And they started tracking the evolutionary path of CLL as its genome spawns new groups and subgroups of tumor cells in a single patient.
“Sequencing the DNA of CLL has taught us a great deal about the genetic basis of the disease,” said Catherine Wu, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“Previous studies, however, were limited by the relatively small number of tumor tissue samples analyzed and by the fact that those samples were taken at different stages of the treatment process from patients treated with different drug agents.”
“In our new study, we wanted to determine if analyzing tissue samples from a large, similarly treated group of patients provides the statistical power necessary to study the disease in all its genetic diversity, to draw connections between certain mutations and the aggressiveness of the disease, and to chart the emergence of new mutations and their role in helping the disease advance. Our results demonstrate the range of insights to be gained by this approach.”
Dr Wu and her colleagues reported these results in Nature.
The researchers collected tumor and normal tissue samples from 538 patients with CLL and performed whole-exome sequencing on each sample.
In this way, the team identified 44 putative CLL driver genes, including 18 CLL mutated drivers that were previously identified and 26 additional putative CLL genes. About 34% of the CLL samples harbored a mutation in at least 1 of these 26 genes.
Nearly 9% of the patients had mutations in CLL genes in the MAPK-ERK pathway. The researchers therefore believe that further exploration of MAPK-ERK pathway inhibitors may be warranted.
The team found that mutations differed between IGHV-mutated and unmutated CLL. IGHV-unmutated CLL tended to have a higher proportion of most driving mutations, while only 3 driver genes were enriched in IGHV-mutated CLL—del(13q), MYD88, and CHD2.
The researchers also discovered that certain mutations were common in patients who had already undergone treatment.
Previous treatment was associated with enrichment in TP53 and BIRC3 mutations del(17p) and del(11q), as well as in mutated DDX3X and MAP2K1. The team therefore believes these mutations may help CLL rebound after initial therapy.
Another key finding was that therapy tended to produce shorter remissions in patients with mutations in TP53 or SF3B1.
“We found that genomic evolution after therapy is the rule rather than the exception,” Dr Wu noted. “Certain mutations were present in a greater number of leukemia cells within a sample after relapse, showing that these mutations, presumably, allow the tumor to persevere.” ![]()
Photo courtesy of Dana-
Farber Cancer Institute
Researchers say they have identified dozens of genetic abnormalities that may drive chronic lymphocytic leukemia (CLL), including some that were never before linked to human cancer.
The team began to trace how some of these abnormalities affect the course of the disease and its susceptibility to treatment.
And they started tracking the evolutionary path of CLL as its genome spawns new groups and subgroups of tumor cells in a single patient.
“Sequencing the DNA of CLL has taught us a great deal about the genetic basis of the disease,” said Catherine Wu, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“Previous studies, however, were limited by the relatively small number of tumor tissue samples analyzed and by the fact that those samples were taken at different stages of the treatment process from patients treated with different drug agents.”
“In our new study, we wanted to determine if analyzing tissue samples from a large, similarly treated group of patients provides the statistical power necessary to study the disease in all its genetic diversity, to draw connections between certain mutations and the aggressiveness of the disease, and to chart the emergence of new mutations and their role in helping the disease advance. Our results demonstrate the range of insights to be gained by this approach.”
Dr Wu and her colleagues reported these results in Nature.
The researchers collected tumor and normal tissue samples from 538 patients with CLL and performed whole-exome sequencing on each sample.
In this way, the team identified 44 putative CLL driver genes, including 18 CLL mutated drivers that were previously identified and 26 additional putative CLL genes. About 34% of the CLL samples harbored a mutation in at least 1 of these 26 genes.
Nearly 9% of the patients had mutations in CLL genes in the MAPK-ERK pathway. The researchers therefore believe that further exploration of MAPK-ERK pathway inhibitors may be warranted.
The team found that mutations differed between IGHV-mutated and unmutated CLL. IGHV-unmutated CLL tended to have a higher proportion of most driving mutations, while only 3 driver genes were enriched in IGHV-mutated CLL—del(13q), MYD88, and CHD2.
The researchers also discovered that certain mutations were common in patients who had already undergone treatment.
Previous treatment was associated with enrichment in TP53 and BIRC3 mutations del(17p) and del(11q), as well as in mutated DDX3X and MAP2K1. The team therefore believes these mutations may help CLL rebound after initial therapy.
Another key finding was that therapy tended to produce shorter remissions in patients with mutations in TP53 or SF3B1.
“We found that genomic evolution after therapy is the rule rather than the exception,” Dr Wu noted. “Certain mutations were present in a greater number of leukemia cells within a sample after relapse, showing that these mutations, presumably, allow the tumor to persevere.” ![]()
Photo courtesy of Dana-
Farber Cancer Institute
Researchers say they have identified dozens of genetic abnormalities that may drive chronic lymphocytic leukemia (CLL), including some that were never before linked to human cancer.
The team began to trace how some of these abnormalities affect the course of the disease and its susceptibility to treatment.
And they started tracking the evolutionary path of CLL as its genome spawns new groups and subgroups of tumor cells in a single patient.
“Sequencing the DNA of CLL has taught us a great deal about the genetic basis of the disease,” said Catherine Wu, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“Previous studies, however, were limited by the relatively small number of tumor tissue samples analyzed and by the fact that those samples were taken at different stages of the treatment process from patients treated with different drug agents.”
“In our new study, we wanted to determine if analyzing tissue samples from a large, similarly treated group of patients provides the statistical power necessary to study the disease in all its genetic diversity, to draw connections between certain mutations and the aggressiveness of the disease, and to chart the emergence of new mutations and their role in helping the disease advance. Our results demonstrate the range of insights to be gained by this approach.”
Dr Wu and her colleagues reported these results in Nature.
The researchers collected tumor and normal tissue samples from 538 patients with CLL and performed whole-exome sequencing on each sample.
In this way, the team identified 44 putative CLL driver genes, including 18 CLL mutated drivers that were previously identified and 26 additional putative CLL genes. About 34% of the CLL samples harbored a mutation in at least 1 of these 26 genes.
Nearly 9% of the patients had mutations in CLL genes in the MAPK-ERK pathway. The researchers therefore believe that further exploration of MAPK-ERK pathway inhibitors may be warranted.
The team found that mutations differed between IGHV-mutated and unmutated CLL. IGHV-unmutated CLL tended to have a higher proportion of most driving mutations, while only 3 driver genes were enriched in IGHV-mutated CLL—del(13q), MYD88, and CHD2.
The researchers also discovered that certain mutations were common in patients who had already undergone treatment.
Previous treatment was associated with enrichment in TP53 and BIRC3 mutations del(17p) and del(11q), as well as in mutated DDX3X and MAP2K1. The team therefore believes these mutations may help CLL rebound after initial therapy.
Another key finding was that therapy tended to produce shorter remissions in patients with mutations in TP53 or SF3B1.
“We found that genomic evolution after therapy is the rule rather than the exception,” Dr Wu noted. “Certain mutations were present in a greater number of leukemia cells within a sample after relapse, showing that these mutations, presumably, allow the tumor to persevere.” ![]()
Team identifies problems with preclinical research

Poor study design and the tendency to publish positive—but not negative—results threaten the validity of preclinical research, according to an article published in eLife.
“Only a fraction of drugs that show promise in animals end up proving safe and effective in humans,” said study author Jonathan Kimmelman, PhD, of McGill University in Montreal, Quebec, Canada.
“An important reason is because studies in animals are often not well designed and because positive results have a higher chance of being published. They end up skewing what we think we know about the potential of a drug.”
Dr Kimmelman and his colleagues came to this conclusion after evaluating all published animal studies of sunitinib, a drug used to treat advanced kidney cancer, a rare type of stomach cancer, and rare tumors of the neuroendocrine system.
The investigators found evidence to suggest that studies reporting little or no anticancer effect were simply not published, leading anticancer effects of the drug to be overestimated by as much as 45%.
The team noted, however, that these findings do not raise any concerns about the clinical use of sunitinib.
Dr Kimmelman and his colleagues also found that few studies used practices like blinding or randomization. And it was often unclear how many animals had been tested because the sample size was not reported.
The drug was tested against different cancers, and all types tested showed statistically significant anticancer activity, a result that “strains credibility”, according to Dr Kimmelman.
He added that researchers failed to observe the dose-dependent response to the drug that is known to occur in humans.
Finally, the researchers did not test the drug on a range of animal models, focusing instead on juvenile female mice with a compromised immune system. Malignancies tested in a wider range of animal types, such as mice that have spontaneously developed tumors, showed less extreme effect sizes.
“Preclinical research is plagued by poor design and reporting practices, exposing patients to harmful and inactive agents, wasting time in the lab, and driving up the price of drugs,” Dr Kimmelman said.
“Our findings provide compelling reasons for developing and implementing guidelines for the design and reporting of preclinical studies in cancer, similar to those already in use for stroke, epilepsy, and cardiology.” ![]()
Poor study design and the tendency to publish positive—but not negative—results threaten the validity of preclinical research, according to an article published in eLife.
“Only a fraction of drugs that show promise in animals end up proving safe and effective in humans,” said study author Jonathan Kimmelman, PhD, of McGill University in Montreal, Quebec, Canada.
“An important reason is because studies in animals are often not well designed and because positive results have a higher chance of being published. They end up skewing what we think we know about the potential of a drug.”
Dr Kimmelman and his colleagues came to this conclusion after evaluating all published animal studies of sunitinib, a drug used to treat advanced kidney cancer, a rare type of stomach cancer, and rare tumors of the neuroendocrine system.
The investigators found evidence to suggest that studies reporting little or no anticancer effect were simply not published, leading anticancer effects of the drug to be overestimated by as much as 45%.
The team noted, however, that these findings do not raise any concerns about the clinical use of sunitinib.
Dr Kimmelman and his colleagues also found that few studies used practices like blinding or randomization. And it was often unclear how many animals had been tested because the sample size was not reported.
The drug was tested against different cancers, and all types tested showed statistically significant anticancer activity, a result that “strains credibility”, according to Dr Kimmelman.
He added that researchers failed to observe the dose-dependent response to the drug that is known to occur in humans.
Finally, the researchers did not test the drug on a range of animal models, focusing instead on juvenile female mice with a compromised immune system. Malignancies tested in a wider range of animal types, such as mice that have spontaneously developed tumors, showed less extreme effect sizes.
“Preclinical research is plagued by poor design and reporting practices, exposing patients to harmful and inactive agents, wasting time in the lab, and driving up the price of drugs,” Dr Kimmelman said.
“Our findings provide compelling reasons for developing and implementing guidelines for the design and reporting of preclinical studies in cancer, similar to those already in use for stroke, epilepsy, and cardiology.” ![]()
Poor study design and the tendency to publish positive—but not negative—results threaten the validity of preclinical research, according to an article published in eLife.
“Only a fraction of drugs that show promise in animals end up proving safe and effective in humans,” said study author Jonathan Kimmelman, PhD, of McGill University in Montreal, Quebec, Canada.
“An important reason is because studies in animals are often not well designed and because positive results have a higher chance of being published. They end up skewing what we think we know about the potential of a drug.”
Dr Kimmelman and his colleagues came to this conclusion after evaluating all published animal studies of sunitinib, a drug used to treat advanced kidney cancer, a rare type of stomach cancer, and rare tumors of the neuroendocrine system.
The investigators found evidence to suggest that studies reporting little or no anticancer effect were simply not published, leading anticancer effects of the drug to be overestimated by as much as 45%.
The team noted, however, that these findings do not raise any concerns about the clinical use of sunitinib.
Dr Kimmelman and his colleagues also found that few studies used practices like blinding or randomization. And it was often unclear how many animals had been tested because the sample size was not reported.
The drug was tested against different cancers, and all types tested showed statistically significant anticancer activity, a result that “strains credibility”, according to Dr Kimmelman.
He added that researchers failed to observe the dose-dependent response to the drug that is known to occur in humans.
Finally, the researchers did not test the drug on a range of animal models, focusing instead on juvenile female mice with a compromised immune system. Malignancies tested in a wider range of animal types, such as mice that have spontaneously developed tumors, showed less extreme effect sizes.
“Preclinical research is plagued by poor design and reporting practices, exposing patients to harmful and inactive agents, wasting time in the lab, and driving up the price of drugs,” Dr Kimmelman said.
“Our findings provide compelling reasons for developing and implementing guidelines for the design and reporting of preclinical studies in cancer, similar to those already in use for stroke, epilepsy, and cardiology.” ![]()
NCCN creates tool to aid treatment decisions

patient and her father
Photo by Rhoda Baer
The National Comprehensive Cancer Network (NCCN) has developed a new tool to accompany its clinical practice guidelines.
The tool—known as NCCN Evidence Blocks™—is designed to provide additional information about guideline recommendations and help inform treatment decisions.
NCCN has already added Evidence Blocks to its guidelines for chronic myelogenous leukemia and multiple myeloma.
The organization hopes to have Evidence Blocks for all of its guidelines by early 2017.
The Evidence Blocks provide a visual representation of 5 key value measures pertaining to guideline recommendations:
- Efficacy of treatment regimens
- Safety of regimens
- Quality and quantity of evidence supporting regimens
- Consistency of evidence supporting regimens
- Affordability of regimens. (This represents an estimate of overall total cost of a therapy, including but not limited to acquisition, administration, inpatient vs outpatient care, supportive care, infusions, toxicity monitoring, antiemetics and growth factors, and hospitalization.)
The Evidence Blocks are graphics of actual blocks that consist of 25 small squares. So each block has 5 rows and 5 columns.
Each of the 5 value measures—efficacy, safety, etc.—has a dedicated column within an Evidence Block, and each row of the Evidence Block represents a rating on a scale of 1 to 5. A score of 1 is unfavorable and a score of 5 is most favorable.
The rating of each value measure is shown by filling in the squares of the dedicated column—such as efficacy—up to the row that represents its assigned score—such as 4.
For example:
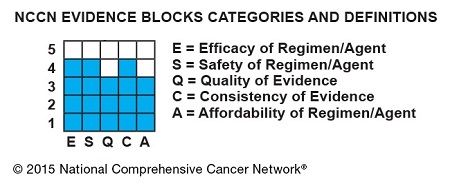
NCCN hopes this visual rating system will help patients and their physicians identify the optimal treatment based on clinical and economic considerations that are of the most value to the patient.
“Some patients will want an emerging therapy even with limited data,” said Robert W. Carlson, MD, chief executive officer of NCCN.
“Others will be most concerned about the expected side effects of the treatment indicated in the safety column. Still others may be very sensitive to cost. By considering the attributes of the range of possible therapies, the healthcare provider and the patient can discuss the benefits and drawbacks of each option and come to a decision most acceptable to the individual.”
By the end of 2015, NCCN expects to publish NCCN Evidence Blocks for systemic therapies (not surgery or radiation therapy) in the NCCN guidelines for breast, colon, non-small cell lung, and rectal cancers.
NCCN Evidence Blocks for systemic therapies are expected to be contained within the complete library of NCCN guidelines by the end of 2016.
In the near term, NCCN will continue to publish 2 sets of guidelines: those including NCCN Evidence Blocks and those without. The Evidence Blocks are not currently published in the NCCN Guidelines for Patients® and are intended for use in the US only.
For more information about NCCN Evidence Blocks, visit NCCN.org/EvidenceBlocks. ![]()
patient and her father
Photo by Rhoda Baer
The National Comprehensive Cancer Network (NCCN) has developed a new tool to accompany its clinical practice guidelines.
The tool—known as NCCN Evidence Blocks™—is designed to provide additional information about guideline recommendations and help inform treatment decisions.
NCCN has already added Evidence Blocks to its guidelines for chronic myelogenous leukemia and multiple myeloma.
The organization hopes to have Evidence Blocks for all of its guidelines by early 2017.
The Evidence Blocks provide a visual representation of 5 key value measures pertaining to guideline recommendations:
- Efficacy of treatment regimens
- Safety of regimens
- Quality and quantity of evidence supporting regimens
- Consistency of evidence supporting regimens
- Affordability of regimens. (This represents an estimate of overall total cost of a therapy, including but not limited to acquisition, administration, inpatient vs outpatient care, supportive care, infusions, toxicity monitoring, antiemetics and growth factors, and hospitalization.)
The Evidence Blocks are graphics of actual blocks that consist of 25 small squares. So each block has 5 rows and 5 columns.
Each of the 5 value measures—efficacy, safety, etc.—has a dedicated column within an Evidence Block, and each row of the Evidence Block represents a rating on a scale of 1 to 5. A score of 1 is unfavorable and a score of 5 is most favorable.
The rating of each value measure is shown by filling in the squares of the dedicated column—such as efficacy—up to the row that represents its assigned score—such as 4.
For example:
NCCN hopes this visual rating system will help patients and their physicians identify the optimal treatment based on clinical and economic considerations that are of the most value to the patient.
“Some patients will want an emerging therapy even with limited data,” said Robert W. Carlson, MD, chief executive officer of NCCN.
“Others will be most concerned about the expected side effects of the treatment indicated in the safety column. Still others may be very sensitive to cost. By considering the attributes of the range of possible therapies, the healthcare provider and the patient can discuss the benefits and drawbacks of each option and come to a decision most acceptable to the individual.”
By the end of 2015, NCCN expects to publish NCCN Evidence Blocks for systemic therapies (not surgery or radiation therapy) in the NCCN guidelines for breast, colon, non-small cell lung, and rectal cancers.
NCCN Evidence Blocks for systemic therapies are expected to be contained within the complete library of NCCN guidelines by the end of 2016.
In the near term, NCCN will continue to publish 2 sets of guidelines: those including NCCN Evidence Blocks and those without. The Evidence Blocks are not currently published in the NCCN Guidelines for Patients® and are intended for use in the US only.
For more information about NCCN Evidence Blocks, visit NCCN.org/EvidenceBlocks. ![]()
patient and her father
Photo by Rhoda Baer
The National Comprehensive Cancer Network (NCCN) has developed a new tool to accompany its clinical practice guidelines.
The tool—known as NCCN Evidence Blocks™—is designed to provide additional information about guideline recommendations and help inform treatment decisions.
NCCN has already added Evidence Blocks to its guidelines for chronic myelogenous leukemia and multiple myeloma.
The organization hopes to have Evidence Blocks for all of its guidelines by early 2017.
The Evidence Blocks provide a visual representation of 5 key value measures pertaining to guideline recommendations:
- Efficacy of treatment regimens
- Safety of regimens
- Quality and quantity of evidence supporting regimens
- Consistency of evidence supporting regimens
- Affordability of regimens. (This represents an estimate of overall total cost of a therapy, including but not limited to acquisition, administration, inpatient vs outpatient care, supportive care, infusions, toxicity monitoring, antiemetics and growth factors, and hospitalization.)
The Evidence Blocks are graphics of actual blocks that consist of 25 small squares. So each block has 5 rows and 5 columns.
Each of the 5 value measures—efficacy, safety, etc.—has a dedicated column within an Evidence Block, and each row of the Evidence Block represents a rating on a scale of 1 to 5. A score of 1 is unfavorable and a score of 5 is most favorable.
The rating of each value measure is shown by filling in the squares of the dedicated column—such as efficacy—up to the row that represents its assigned score—such as 4.
For example:
NCCN hopes this visual rating system will help patients and their physicians identify the optimal treatment based on clinical and economic considerations that are of the most value to the patient.
“Some patients will want an emerging therapy even with limited data,” said Robert W. Carlson, MD, chief executive officer of NCCN.
“Others will be most concerned about the expected side effects of the treatment indicated in the safety column. Still others may be very sensitive to cost. By considering the attributes of the range of possible therapies, the healthcare provider and the patient can discuss the benefits and drawbacks of each option and come to a decision most acceptable to the individual.”
By the end of 2015, NCCN expects to publish NCCN Evidence Blocks for systemic therapies (not surgery or radiation therapy) in the NCCN guidelines for breast, colon, non-small cell lung, and rectal cancers.
NCCN Evidence Blocks for systemic therapies are expected to be contained within the complete library of NCCN guidelines by the end of 2016.
In the near term, NCCN will continue to publish 2 sets of guidelines: those including NCCN Evidence Blocks and those without. The Evidence Blocks are not currently published in the NCCN Guidelines for Patients® and are intended for use in the US only.
For more information about NCCN Evidence Blocks, visit NCCN.org/EvidenceBlocks. ![]()
FDA approves reversal agent for dabigatran
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab. ![]()
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab. ![]()
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab.
Health Canada approves drug for acquired hemophilia A
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
COMP recommends orphan designations for KTE-C19

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
Watchdog condemns FDA oversight of dabigatran
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.