User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
An investigational oral drug that targets RAS, the dominant oncogenic driver in pancreatic cancer, nearly doubled overall survival compared with common second-line chemotherapy regimens in patients with previously treated metastatic disease.
In the 500-patient phase 3 RASolute 302 trial, patients who received daraxonrasib, a RAS(ON) multi-selective inhibitor, lived a median of 13.2 months compared with 6.7 months for those who received chemotherapy. The drug also doubled progression-free survival, tripled the response rate, and delayed deterioration in both pain and quality of life.
“Results from RASolute 302 support daraxonrasib as a new standard of care for patients with previously treated metastatic pancreatic cancer,” said lead investigator Brian Wolpin, MD, MPH, director of the Hale Family Center for Pancreatic Cancer Research, Dana-Farber Cancer Institute in Boston, who presented the findings at the American Society of Clinical Oncology (ASCO) 2026. The study was also published simultaneously on May 31 in The New England Journal of Medicine.
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with most patients presenting with advanced disease at diagnosis. For those whose cancer progresses after first-line chemotherapy, no single standard second-line treatment has been established. Available regimens typically produce median progression-free survival of 3-4 months and a median overall survival of 6-7 months.
More than 90% of pancreatic cancers harbor activating RAS mutations — most commonly KRAS G12D and G12V — that drive tumor growth. The first approved KRAS inhibitors — sotorasib and adagrasib for certain lung and colorectal cancers — target KRAS G12C, which is rare in pancreatic cancer. So far, no RAS-targeted therapy has been approved for pancreatic cancer.
Daraxonrasib takes a broader approach. The oral agent can target a range of RAS variants, including mutant and wild-type RAS, by binding the active form of RAS and blocking downstream signaling.
In the phase 3 open-label RASolute 302 trial, researchers randomized 500 patients with previously treated metastatic pancreatic cancer 1:1 to receive daraxonrasib 300 mg orally once daily or investigator’s choice of chemotherapy. The most used regimens in the control arm were gemcitabine plus nab-paclitaxel (56.5%) and liposomal irinotecan plus fluorouracil and leucovorin (32.7%).
Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Documented RAS mutational status was required, and nearly 92% of patients had RAS G12 mutations.
The dual primary endpoints were overall survival and progression-free survival in the RAS G12 population. Key secondary endpoints included the same measures in the overall population as well as objective response rate and patient-reported quality of life. The median follow-up was 8.5 months.
In the RAS G12 population, daraxonrasib reduced the risk for death by 60% (hazard ratio [HR], 0.40; P < .001). The median overall survival was 13.2 months with daraxonrasib vs 6.6 months with chemotherapy. Results were nearly identical in the overall population: 13.2 vs 6.7 months (HR, 0.40; P < .001).
Progression-free survival in the RAS G12 population was 7.3 vs 3.5 months (HR, 0.45; P < .001). The objective response rate was 33.2% vs 11.8%.
Daraxonrasib significantly delayed deterioration in pain (median, 9.0 vs 3.7 months; HR, 0.51; P < .001) and global quality of life (5.6 vs 2.4 months; HR, 0.60; P < .001).
Treatment-related adverse events of grade ≥ 3 occurred in 43.6% of patients on daraxonrasib vs 57.5% of patients on chemotherapy. The most common high-grade events with daraxonrasib were rash (13.7%) and stomatitis (12.0%), whereas neutropenia (27.6%) and anemia (16.4%) were most common with chemotherapy.
One patient in the daraxonrasib arm died from treatment-related pneumonitis. Discontinuation due to adverse events occurred in 1.2% of patients on daraxonrasib vs 11.2% of patients on chemotherapy.
The median duration of treatment was 6.2 months with daraxonrasib vs 1.5-3.2 months across chemotherapy regimens, and 42% of patients on daraxonrasib remained on treatment at data cutoff, Wolpin said.
The open-label design is a limitation. About 15% of patients randomized to chemotherapy never received treatment, largely after learning their treatment assignment, although all were included in the intention-to-treat analysis.
Invited discussant Jennifer Knox, MD, a medical oncologist at Princess Margaret Cancer Center and professor of medicine at the University of Toronto in Toronto, Ontario, Canada, called daraxonrasib a game changer, describing the drug as “probably the most exciting strategy in five decades” for pancreatic cancer.
“It is an absolutely beautiful curve,” Knox said of the overall survival data, noting that the Kaplan-Meier curves separated early and continued to widen over time — a pattern rarely seen in pancreatic cancer trials where initial separation between treatment arms typically narrows as patients in both groups deteriorate.
Knox highlighted the pain and quality-of-life data as “the most important endpoints for our patients,” given the severity of the disease.
She pointed out, however, that the combination of rash and stomatitis will be “challenging” in practice, and called for improved supportive care and closer collaboration with dermatology as oncologists gain experience with this first-in-class drug.
Knox said RAS-targeted therapy “should dominate trials across the full spectrum of pancreatic cancer clinical presentations,” pointing to first-line combination trials already underway and noting that treating a larger, earlier population would have the greatest impact.
“This is the first glimpse at the real power of targeting RAS in pancreas cancer,” Knox said.
The study was funded by Revolution Medicines. Wolpin reported consulting or advisory roles with Revolution Medicines, Mirati Therapeutics, Ipsen, and others, and institutional research funding from Revolution Medicines, Agios, Amgen, AstraZeneca, Lilly, and Novartis. Knox reported receiving honoraria from Astellas Pharma, AstraZeneca, Incyte, and Ipsen, and consulting or advisory roles with AstraZeneca/MedImmune, Incyte, and Ipsen.
A version of this article was previously published on Medscape.com.
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
RAS Drug Nearly Doubles Survival in Metastatic Pancreatic Cancer
Rare Blood Diseases the Focus of AVAHO Virtual Session
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
Fact vs Fallacy: Challenging the Norms of Cancer Care Fallacies in Medicine
Fact vs Fallacy: Challenging the Norms of Cancer Care Fallacies in Medicine
This transcript has been edited for clarity.
Hello, everyone. This is Dr Bishal Gyawali, from Queens University, Kingston, Canada. Today, I’m back with you to talk about some of the fallacies that I have seen in medicine, oncology, and the drug regulatory space. I wanted to clarify some of these fallacies.
In my last video, I talked about the FDA denying the approval of a new cancer drug. Let me start with one of the fallacies that is pertinent to that, which is that some people make an argument that patients are dying from a certain condition, such as cancer, or even any other disease besides cancer. That is an absolutely true statement, but that does not necessarily mean there should be a lower bar for drug approvals or we should be approving any drug that has a hint of benefit.
In fact, if we have increased mortality rates and our patients are dying from a certain condition, that means we actually need to have good drugs. We need to have drugs that prevent mortality. We need to have drugs that improve outcomes. Just having any drug out there, if we lower our threshold and are letting any drug be used in these patients because the argument is that people are dying, then in fact, it can have negative consequences.
First, there will be opportunity costs. If you can get any lousy drug into the market and make billions of dollars out of it, then there is no strong motivation to produce drugs that actually remarkably improve outcomes.
Second, patients will also be misled. It’s the patient’s opportunity cost in that they will use whatever time they have remaining to pursue these treatments that were not going to improve their outcomes anyway. This is time they could have better spent either in pursuing better treatments, if those treatments are out there, or to prioritize their time accordingly. This rather gives them a false hope, which can be harmful in the long term.
The first fallacy is that just because people are dying does not necessarily mean we should have more new drugs with a lower bar for approval.
The second fallacy I want to talk about, which is also related to this, is that if a certain cancer is rare, the bar for new drug approval should be quite low.
Of course, rare cancers are a special category, and rare cancers should be treated differently from a regulatory perspective. Absolutely. If the cancer is rare, we cannot have trials with large sample sizes to generate evidence. That problem is there, but that does not necessarily translate to the decision that we should approve anything, even something with a small hint of benefit.
There are other methods to make sure that, even in rare cancers, we can generate good-quality evidence. In fact, from an equity perspective, why should patients with rare cancer not deserve drugs that have good-quality evidence?
We can’t tell someone that, “Your cancer is rare, so you should get drugs that only have a benefit in terms of response rate whereas other cancers that are not rare will have drugs based on survival.”
Going back to the point about the difficulty in doing big trials in patients with rare cancers, that is absolutely true and there should be regulatory flexibility in this. I think accelerated approval is a pathway that allows for this regulatory flexibility, which allows access to these drugs early on based on earlier signals of benefit. You can continue to generate evidence in the future and confirm the clinical benefit.
There are also other nuances to this. One is that we should also make sure that this regulatory flexibility with rare cancers should not be misused. What do I mean by that? First, all rare cancers are not the same. There are some cancers that are ultra rare, and then there are some cancers that technically might fit the definition of rare, but trials are possible. Case in point: adrenocortical cancer. It is considered to be a very rare cancer, but there have been randomized trials in adrenocortical cancer.
Our efforts should be to maximize our collaboration globally so that a cancer that is rare locally will still not be so rare globally when we collect all these patients.
In certain situations, like let’s say, based on the molecular subtypes, any common cancer can be sliced and diced into a rare subtype: MSI-high, BRAF-negative, HER2-positive, right-sided colon cancer. If you start to slice cancers into these smaller and smaller molecular subtypes, you can consider anything as a rare cancer. That should not be misused as an excuse to get away from doing proper trials and generating adequate evidence for our patients.
The third fallacy I want to talk about is that increasing cancer incidence in a certain subgroup of population does not automatically translate into, “We should start screening this subgroup of population.”
A certain cancer — let’s say cancer X or cancer Y — is increasing in a young population, so therefore, we should lower the age of the screening of young populations. This cancer is increasing in this ethnic population, so therefore we should start screening this ethnic population more frequently. This cancer is increasing in this type of minority, so therefore, we should start screening this minority more.
No, it does not work like that. Increasing incidence will make us concerned, of course, but that does not necessarily translate into, “We should start screening them.” In order for a screening test to be useful, it has to fulfill a number of criteria.
The goal is not to detect cancers. The goal is to detect cancers that are not indolent enough that they would have never caused problems, nor speed up the diagnosis of aggressive cancers that are going to be lethal pretty soon anyway. The goal is to detect those cancers in the middle, so that by detecting early, we can intervene and improve the outcomes and improve the mortality from that cancer.
This type of intervention requires a thoughtful consideration of the increasing incidence of the cancer, of course, but also the utility of the screening test in that subgroup of population; the life expectancy of this subgroup of patients with and without cancer; the interventions available to address that increasing burden of cancer; and whether by intervening we are going to reduce the mortality rates.
Just because we can detect cancers does not mean we should detect cancers. That’s the third fallacy I wanted to talk about.
The fourth fallacy is related to when someone is asking for more evidence for anything. There is a new drug for this cancer,so what is the evidence? Or there is this new intervention that will detect ctDNA or whatever before the cancer relapses, or before the cancer even shows up as a screening test.
Whenever there is any treatment that is being promoted and someone asks for evidence, people sometimes try to make personal attacks by saying, “Oh, so, you’re okay with patients dying. You don’t want to save lives.”
Absolutely we want to save lives. That’s why we’re in this field, and that’s why we’re asking for more evidence. You should not consider someone who is asking for evidence as evil or that this person does not want this new drug, or this person does not want this innovation. No, that person actually wants to make sure that that innovation actually helps people. That’s why that person is asking for more evidence.
If we stop asking for evidence, then our whole practice becomes based on emotions, faith, and trust rather than science. You could extrapolate it to the other extreme, like if you are not asking for evidence. If it is interpreted as someone who is asking for evidence is evil, or someone who does not want patients to get new drugs, then you could extrapolate these to people also making claims about alternative medicines or ivermectin nowadays, and claiming that this cures cancer.
Science is science. You need to be the same no matter the circumstances. If you are asking for data for ivermectin, you should also be asking data for your cancer drug that you think is going to work. We should always ask for evidence.
Asking for evidence is not a sign that whoever is asking for evidence does not want the patient to have access to the drug. It is showing that the person who is asking for evidence actually wants to make sure that the patients who get this drug are actually being helped by the drug rather than being harmed.
I’m talking about 5 fallacies today. The final, fifth fallacy is that clinical expertise does not equal expertise in making public health decisions or even expertise in critical appraisal. Someone can be a fantastic breast cancer doctor, the best oncologist for breast cancer. That does not automatically make that person the best person to evaluate clinical trials of breast cancer drugs.
Someone can be a fantastic colon cancer doctor. That does not make that person automatically the best person to evaluate whether or not colonoscopy or colon cancer screening is indicated in a certain patient population.
These population-level decisions — including should this drug be approved, should this drug be funded, and should this screening test be made a public health measure, all of these public health decisions that are done at a population level — require different expertise in critical appraisal, clinical epidemiology, and public health.
Just because someone is a fantastic clinician does not make that person a fantastic public health expert. I see on social media often that a famous doctor with expertise in their domain, let’s say a famous neurosurgeon, might say, “I think brain tumors are increasing in incidence in young persons, so we should be targeting an MRI screening for everyone over the age of 30.”
I’m just making this up, but we see examples of things not dissimilar to this. Just because someone is a neurosurgeon does not make them an expert on brain tumor epidemiology, surveillance, or screening. We should separate clinical expertise from public health expertise.
Thank you.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Hello, everyone. This is Dr Bishal Gyawali, from Queens University, Kingston, Canada. Today, I’m back with you to talk about some of the fallacies that I have seen in medicine, oncology, and the drug regulatory space. I wanted to clarify some of these fallacies.
In my last video, I talked about the FDA denying the approval of a new cancer drug. Let me start with one of the fallacies that is pertinent to that, which is that some people make an argument that patients are dying from a certain condition, such as cancer, or even any other disease besides cancer. That is an absolutely true statement, but that does not necessarily mean there should be a lower bar for drug approvals or we should be approving any drug that has a hint of benefit.
In fact, if we have increased mortality rates and our patients are dying from a certain condition, that means we actually need to have good drugs. We need to have drugs that prevent mortality. We need to have drugs that improve outcomes. Just having any drug out there, if we lower our threshold and are letting any drug be used in these patients because the argument is that people are dying, then in fact, it can have negative consequences.
First, there will be opportunity costs. If you can get any lousy drug into the market and make billions of dollars out of it, then there is no strong motivation to produce drugs that actually remarkably improve outcomes.
Second, patients will also be misled. It’s the patient’s opportunity cost in that they will use whatever time they have remaining to pursue these treatments that were not going to improve their outcomes anyway. This is time they could have better spent either in pursuing better treatments, if those treatments are out there, or to prioritize their time accordingly. This rather gives them a false hope, which can be harmful in the long term.
The first fallacy is that just because people are dying does not necessarily mean we should have more new drugs with a lower bar for approval.
The second fallacy I want to talk about, which is also related to this, is that if a certain cancer is rare, the bar for new drug approval should be quite low.
Of course, rare cancers are a special category, and rare cancers should be treated differently from a regulatory perspective. Absolutely. If the cancer is rare, we cannot have trials with large sample sizes to generate evidence. That problem is there, but that does not necessarily translate to the decision that we should approve anything, even something with a small hint of benefit.
There are other methods to make sure that, even in rare cancers, we can generate good-quality evidence. In fact, from an equity perspective, why should patients with rare cancer not deserve drugs that have good-quality evidence?
We can’t tell someone that, “Your cancer is rare, so you should get drugs that only have a benefit in terms of response rate whereas other cancers that are not rare will have drugs based on survival.”
Going back to the point about the difficulty in doing big trials in patients with rare cancers, that is absolutely true and there should be regulatory flexibility in this. I think accelerated approval is a pathway that allows for this regulatory flexibility, which allows access to these drugs early on based on earlier signals of benefit. You can continue to generate evidence in the future and confirm the clinical benefit.
There are also other nuances to this. One is that we should also make sure that this regulatory flexibility with rare cancers should not be misused. What do I mean by that? First, all rare cancers are not the same. There are some cancers that are ultra rare, and then there are some cancers that technically might fit the definition of rare, but trials are possible. Case in point: adrenocortical cancer. It is considered to be a very rare cancer, but there have been randomized trials in adrenocortical cancer.
Our efforts should be to maximize our collaboration globally so that a cancer that is rare locally will still not be so rare globally when we collect all these patients.
In certain situations, like let’s say, based on the molecular subtypes, any common cancer can be sliced and diced into a rare subtype: MSI-high, BRAF-negative, HER2-positive, right-sided colon cancer. If you start to slice cancers into these smaller and smaller molecular subtypes, you can consider anything as a rare cancer. That should not be misused as an excuse to get away from doing proper trials and generating adequate evidence for our patients.
The third fallacy I want to talk about is that increasing cancer incidence in a certain subgroup of population does not automatically translate into, “We should start screening this subgroup of population.”
A certain cancer — let’s say cancer X or cancer Y — is increasing in a young population, so therefore, we should lower the age of the screening of young populations. This cancer is increasing in this ethnic population, so therefore we should start screening this ethnic population more frequently. This cancer is increasing in this type of minority, so therefore, we should start screening this minority more.
No, it does not work like that. Increasing incidence will make us concerned, of course, but that does not necessarily translate into, “We should start screening them.” In order for a screening test to be useful, it has to fulfill a number of criteria.
The goal is not to detect cancers. The goal is to detect cancers that are not indolent enough that they would have never caused problems, nor speed up the diagnosis of aggressive cancers that are going to be lethal pretty soon anyway. The goal is to detect those cancers in the middle, so that by detecting early, we can intervene and improve the outcomes and improve the mortality from that cancer.
This type of intervention requires a thoughtful consideration of the increasing incidence of the cancer, of course, but also the utility of the screening test in that subgroup of population; the life expectancy of this subgroup of patients with and without cancer; the interventions available to address that increasing burden of cancer; and whether by intervening we are going to reduce the mortality rates.
Just because we can detect cancers does not mean we should detect cancers. That’s the third fallacy I wanted to talk about.
The fourth fallacy is related to when someone is asking for more evidence for anything. There is a new drug for this cancer,so what is the evidence? Or there is this new intervention that will detect ctDNA or whatever before the cancer relapses, or before the cancer even shows up as a screening test.
Whenever there is any treatment that is being promoted and someone asks for evidence, people sometimes try to make personal attacks by saying, “Oh, so, you’re okay with patients dying. You don’t want to save lives.”
Absolutely we want to save lives. That’s why we’re in this field, and that’s why we’re asking for more evidence. You should not consider someone who is asking for evidence as evil or that this person does not want this new drug, or this person does not want this innovation. No, that person actually wants to make sure that that innovation actually helps people. That’s why that person is asking for more evidence.
If we stop asking for evidence, then our whole practice becomes based on emotions, faith, and trust rather than science. You could extrapolate it to the other extreme, like if you are not asking for evidence. If it is interpreted as someone who is asking for evidence is evil, or someone who does not want patients to get new drugs, then you could extrapolate these to people also making claims about alternative medicines or ivermectin nowadays, and claiming that this cures cancer.
Science is science. You need to be the same no matter the circumstances. If you are asking for data for ivermectin, you should also be asking data for your cancer drug that you think is going to work. We should always ask for evidence.
Asking for evidence is not a sign that whoever is asking for evidence does not want the patient to have access to the drug. It is showing that the person who is asking for evidence actually wants to make sure that the patients who get this drug are actually being helped by the drug rather than being harmed.
I’m talking about 5 fallacies today. The final, fifth fallacy is that clinical expertise does not equal expertise in making public health decisions or even expertise in critical appraisal. Someone can be a fantastic breast cancer doctor, the best oncologist for breast cancer. That does not automatically make that person the best person to evaluate clinical trials of breast cancer drugs.
Someone can be a fantastic colon cancer doctor. That does not make that person automatically the best person to evaluate whether or not colonoscopy or colon cancer screening is indicated in a certain patient population.
These population-level decisions — including should this drug be approved, should this drug be funded, and should this screening test be made a public health measure, all of these public health decisions that are done at a population level — require different expertise in critical appraisal, clinical epidemiology, and public health.
Just because someone is a fantastic clinician does not make that person a fantastic public health expert. I see on social media often that a famous doctor with expertise in their domain, let’s say a famous neurosurgeon, might say, “I think brain tumors are increasing in incidence in young persons, so we should be targeting an MRI screening for everyone over the age of 30.”
I’m just making this up, but we see examples of things not dissimilar to this. Just because someone is a neurosurgeon does not make them an expert on brain tumor epidemiology, surveillance, or screening. We should separate clinical expertise from public health expertise.
Thank you.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Hello, everyone. This is Dr Bishal Gyawali, from Queens University, Kingston, Canada. Today, I’m back with you to talk about some of the fallacies that I have seen in medicine, oncology, and the drug regulatory space. I wanted to clarify some of these fallacies.
In my last video, I talked about the FDA denying the approval of a new cancer drug. Let me start with one of the fallacies that is pertinent to that, which is that some people make an argument that patients are dying from a certain condition, such as cancer, or even any other disease besides cancer. That is an absolutely true statement, but that does not necessarily mean there should be a lower bar for drug approvals or we should be approving any drug that has a hint of benefit.
In fact, if we have increased mortality rates and our patients are dying from a certain condition, that means we actually need to have good drugs. We need to have drugs that prevent mortality. We need to have drugs that improve outcomes. Just having any drug out there, if we lower our threshold and are letting any drug be used in these patients because the argument is that people are dying, then in fact, it can have negative consequences.
First, there will be opportunity costs. If you can get any lousy drug into the market and make billions of dollars out of it, then there is no strong motivation to produce drugs that actually remarkably improve outcomes.
Second, patients will also be misled. It’s the patient’s opportunity cost in that they will use whatever time they have remaining to pursue these treatments that were not going to improve their outcomes anyway. This is time they could have better spent either in pursuing better treatments, if those treatments are out there, or to prioritize their time accordingly. This rather gives them a false hope, which can be harmful in the long term.
The first fallacy is that just because people are dying does not necessarily mean we should have more new drugs with a lower bar for approval.
The second fallacy I want to talk about, which is also related to this, is that if a certain cancer is rare, the bar for new drug approval should be quite low.
Of course, rare cancers are a special category, and rare cancers should be treated differently from a regulatory perspective. Absolutely. If the cancer is rare, we cannot have trials with large sample sizes to generate evidence. That problem is there, but that does not necessarily translate to the decision that we should approve anything, even something with a small hint of benefit.
There are other methods to make sure that, even in rare cancers, we can generate good-quality evidence. In fact, from an equity perspective, why should patients with rare cancer not deserve drugs that have good-quality evidence?
We can’t tell someone that, “Your cancer is rare, so you should get drugs that only have a benefit in terms of response rate whereas other cancers that are not rare will have drugs based on survival.”
Going back to the point about the difficulty in doing big trials in patients with rare cancers, that is absolutely true and there should be regulatory flexibility in this. I think accelerated approval is a pathway that allows for this regulatory flexibility, which allows access to these drugs early on based on earlier signals of benefit. You can continue to generate evidence in the future and confirm the clinical benefit.
There are also other nuances to this. One is that we should also make sure that this regulatory flexibility with rare cancers should not be misused. What do I mean by that? First, all rare cancers are not the same. There are some cancers that are ultra rare, and then there are some cancers that technically might fit the definition of rare, but trials are possible. Case in point: adrenocortical cancer. It is considered to be a very rare cancer, but there have been randomized trials in adrenocortical cancer.
Our efforts should be to maximize our collaboration globally so that a cancer that is rare locally will still not be so rare globally when we collect all these patients.
In certain situations, like let’s say, based on the molecular subtypes, any common cancer can be sliced and diced into a rare subtype: MSI-high, BRAF-negative, HER2-positive, right-sided colon cancer. If you start to slice cancers into these smaller and smaller molecular subtypes, you can consider anything as a rare cancer. That should not be misused as an excuse to get away from doing proper trials and generating adequate evidence for our patients.
The third fallacy I want to talk about is that increasing cancer incidence in a certain subgroup of population does not automatically translate into, “We should start screening this subgroup of population.”
A certain cancer — let’s say cancer X or cancer Y — is increasing in a young population, so therefore, we should lower the age of the screening of young populations. This cancer is increasing in this ethnic population, so therefore we should start screening this ethnic population more frequently. This cancer is increasing in this type of minority, so therefore, we should start screening this minority more.
No, it does not work like that. Increasing incidence will make us concerned, of course, but that does not necessarily translate into, “We should start screening them.” In order for a screening test to be useful, it has to fulfill a number of criteria.
The goal is not to detect cancers. The goal is to detect cancers that are not indolent enough that they would have never caused problems, nor speed up the diagnosis of aggressive cancers that are going to be lethal pretty soon anyway. The goal is to detect those cancers in the middle, so that by detecting early, we can intervene and improve the outcomes and improve the mortality from that cancer.
This type of intervention requires a thoughtful consideration of the increasing incidence of the cancer, of course, but also the utility of the screening test in that subgroup of population; the life expectancy of this subgroup of patients with and without cancer; the interventions available to address that increasing burden of cancer; and whether by intervening we are going to reduce the mortality rates.
Just because we can detect cancers does not mean we should detect cancers. That’s the third fallacy I wanted to talk about.
The fourth fallacy is related to when someone is asking for more evidence for anything. There is a new drug for this cancer,so what is the evidence? Or there is this new intervention that will detect ctDNA or whatever before the cancer relapses, or before the cancer even shows up as a screening test.
Whenever there is any treatment that is being promoted and someone asks for evidence, people sometimes try to make personal attacks by saying, “Oh, so, you’re okay with patients dying. You don’t want to save lives.”
Absolutely we want to save lives. That’s why we’re in this field, and that’s why we’re asking for more evidence. You should not consider someone who is asking for evidence as evil or that this person does not want this new drug, or this person does not want this innovation. No, that person actually wants to make sure that that innovation actually helps people. That’s why that person is asking for more evidence.
If we stop asking for evidence, then our whole practice becomes based on emotions, faith, and trust rather than science. You could extrapolate it to the other extreme, like if you are not asking for evidence. If it is interpreted as someone who is asking for evidence is evil, or someone who does not want patients to get new drugs, then you could extrapolate these to people also making claims about alternative medicines or ivermectin nowadays, and claiming that this cures cancer.
Science is science. You need to be the same no matter the circumstances. If you are asking for data for ivermectin, you should also be asking data for your cancer drug that you think is going to work. We should always ask for evidence.
Asking for evidence is not a sign that whoever is asking for evidence does not want the patient to have access to the drug. It is showing that the person who is asking for evidence actually wants to make sure that the patients who get this drug are actually being helped by the drug rather than being harmed.
I’m talking about 5 fallacies today. The final, fifth fallacy is that clinical expertise does not equal expertise in making public health decisions or even expertise in critical appraisal. Someone can be a fantastic breast cancer doctor, the best oncologist for breast cancer. That does not automatically make that person the best person to evaluate clinical trials of breast cancer drugs.
Someone can be a fantastic colon cancer doctor. That does not make that person automatically the best person to evaluate whether or not colonoscopy or colon cancer screening is indicated in a certain patient population.
These population-level decisions — including should this drug be approved, should this drug be funded, and should this screening test be made a public health measure, all of these public health decisions that are done at a population level — require different expertise in critical appraisal, clinical epidemiology, and public health.
Just because someone is a fantastic clinician does not make that person a fantastic public health expert. I see on social media often that a famous doctor with expertise in their domain, let’s say a famous neurosurgeon, might say, “I think brain tumors are increasing in incidence in young persons, so we should be targeting an MRI screening for everyone over the age of 30.”
I’m just making this up, but we see examples of things not dissimilar to this. Just because someone is a neurosurgeon does not make them an expert on brain tumor epidemiology, surveillance, or screening. We should separate clinical expertise from public health expertise.
Thank you.
A version of this article first appeared on Medscape.com.
Fact vs Fallacy: Challenging the Norms of Cancer Care Fallacies in Medicine
Fact vs Fallacy: Challenging the Norms of Cancer Care Fallacies in Medicine
The Fastest Way to Better Anticoagulants May Be a Land Snail
The Fastest Way to Better Anticoagulants May Be a Land Snail
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The Fastest Way to Better Anticoagulants May Be a Land Snail
The Fastest Way to Better Anticoagulants May Be a Land Snail
Can Fasting Around Chemo Improve Ovarian Cancer Outcomes?
Can Fasting Around Chemo Improve Ovarian Cancer Outcomes?
A few days of fasting around chemotherapy sessions may improve treatment response and outcomes for some patients with advanced ovarian cancer, a small phase 2 trial suggests.
The study, of 36 patients with stage III or IV high-grade ovarian cancer, found that those randomly assigned to fast for 36 hours before chemotherapy and 24 hours afterward had stronger pathologic responses to chemotherapy and longer progression-free survival than patients who ate normally during treatment.
The findings, reported at a press briefing ahead the American Society of Clinical Oncology (ASCO) 2026, hint at a straightforward measure to potentially improve patients’ outcomes.
The working theory is that short-term fasting boosts chemotherapy response by lowering insulin and IGF-1 levels, both of which are implicated in tumor growth and chemotherapy resistance, said study presenter Claudia Marchetti, MD, of Agostino Gemelli University in Rome, Italy.
Speaking at the briefing, ASCO President Eric Small, MD, of the University of California San Francisco, called the study “a great example of a very simple intervention that has benefit and can be undertaken and implemented anywhere in the world.”
“It’s not an expensive new drug,” he said, “and yet it has the potential to really have an impact on this cancer.”
Ovarian cancer affects more than 324,000 women worldwide each year and causes more than 206,000 deaths annually. Around 80% of patients are diagnosed at an advanced stage, and up to 60% receive neoadjuvant chemotherapy to reduce tumor size and facilitate surgery.
Despite advances in surgery and chemotherapy, patients with advanced disease still face poor outcomes. There is, Marchetti said, “an urgent need for safe, low-cost, and easily implementable strategies that can enhance treatment efficacy and improve patient prognosis.”
Given evidence on the role of insulin in tumor growth and chemotherapy response, her team hypothesized that short bouts of fasting around the time of treatment might have benefits.
To test that idea, the researchers recruited 36 patients with newly diagnosed stage III or IV high-grade serous ovarian carcinoma who were not candidates for primary cytoreduction. All were in good general health, with a mean age of 62 years.
All patients received 3 rounds of carboplatin and paclitaxel before surgery. Prior to starting chemotherapy, half were randomly assigned to fast for 36 hours before and 24 hours after chemotherapy, whereas the other half ate normally throughout treatment.
Patients in the fasting group consumed no more than 350 calories per day during the fasting window. They were allowed to have unrestricted water, herbal tea, limited vegetable juice, and small amounts of light vegetable broth. (A ketogenic diet group had initially been planned but was closed early because of poor patient compliance.)
The study met its primary endpoint of change in insulin levels during chemotherapy, Marchetti reported. Baseline insulin levels were comparable between the 2 groups, but after 3 rounds of chemotherapy, they’d dipped by an average of 1.12 µIU/mL in the fasting group and increased by 9.76 µIU/mL in the control group (P = .01).
Fasting also improved clinical outcomes. Specifically, Marchetti said, 59% of fasting patients achieved a chemotherapy response score of 3 — indicating complete or near-complete tumor response before surgery — compared with 17% of patients in the control group.
Median progression-free survival was significantly longer in the fasting group, at 38 vs 24 months.
Importantly, Marchetti said, the fasting protocol was feasible, well tolerated, and safe: All patients assigned to the fasting group completed treatment, and rates of chemotherapy-related toxicities were similar between the 2 groups.
Additional analyses shed more light on the possible mechanisms underlying the fasting group’s improved outcomes: The researchers found that those patients tended to have lower levels of circulating suppressor granulocyte and monocyte populations that have been linked to tumor immune escape, which suggests, Marchetti said, fasting may have set the stage for a “more favorable immune environment” during chemotherapy.
However, she cautioned that much more research is needed. Her team is planning a larger multicenter trial to validate the current findings, and longer-term follow-up is necessary to see whether fasting ultimately impacts patients’ survival, Marchetti said.
In a statement, Eleonora Teplinsky, MD, an ASCO expert in gynecologic cancers, said these early findings are “encouraging, support earlier data, and highlight a promising area of cancer research.”
But she, too, emphasized the need for larger clinical trials to build on the results.
The study had no commercial funding. Marchetti disclosed having relationships with Arquer Diagnostics, AstraZeneca, Clovis Oncology, and other companies. Small disclosed having relationships with Janssen, Johnson & Johnson, and others. Teplinsky had no disclosures.
A version of this article first appeared on Medscape.com.
A few days of fasting around chemotherapy sessions may improve treatment response and outcomes for some patients with advanced ovarian cancer, a small phase 2 trial suggests.
The study, of 36 patients with stage III or IV high-grade ovarian cancer, found that those randomly assigned to fast for 36 hours before chemotherapy and 24 hours afterward had stronger pathologic responses to chemotherapy and longer progression-free survival than patients who ate normally during treatment.
The findings, reported at a press briefing ahead the American Society of Clinical Oncology (ASCO) 2026, hint at a straightforward measure to potentially improve patients’ outcomes.
The working theory is that short-term fasting boosts chemotherapy response by lowering insulin and IGF-1 levels, both of which are implicated in tumor growth and chemotherapy resistance, said study presenter Claudia Marchetti, MD, of Agostino Gemelli University in Rome, Italy.
Speaking at the briefing, ASCO President Eric Small, MD, of the University of California San Francisco, called the study “a great example of a very simple intervention that has benefit and can be undertaken and implemented anywhere in the world.”
“It’s not an expensive new drug,” he said, “and yet it has the potential to really have an impact on this cancer.”
Ovarian cancer affects more than 324,000 women worldwide each year and causes more than 206,000 deaths annually. Around 80% of patients are diagnosed at an advanced stage, and up to 60% receive neoadjuvant chemotherapy to reduce tumor size and facilitate surgery.
Despite advances in surgery and chemotherapy, patients with advanced disease still face poor outcomes. There is, Marchetti said, “an urgent need for safe, low-cost, and easily implementable strategies that can enhance treatment efficacy and improve patient prognosis.”
Given evidence on the role of insulin in tumor growth and chemotherapy response, her team hypothesized that short bouts of fasting around the time of treatment might have benefits.
To test that idea, the researchers recruited 36 patients with newly diagnosed stage III or IV high-grade serous ovarian carcinoma who were not candidates for primary cytoreduction. All were in good general health, with a mean age of 62 years.
All patients received 3 rounds of carboplatin and paclitaxel before surgery. Prior to starting chemotherapy, half were randomly assigned to fast for 36 hours before and 24 hours after chemotherapy, whereas the other half ate normally throughout treatment.
Patients in the fasting group consumed no more than 350 calories per day during the fasting window. They were allowed to have unrestricted water, herbal tea, limited vegetable juice, and small amounts of light vegetable broth. (A ketogenic diet group had initially been planned but was closed early because of poor patient compliance.)
The study met its primary endpoint of change in insulin levels during chemotherapy, Marchetti reported. Baseline insulin levels were comparable between the 2 groups, but after 3 rounds of chemotherapy, they’d dipped by an average of 1.12 µIU/mL in the fasting group and increased by 9.76 µIU/mL in the control group (P = .01).
Fasting also improved clinical outcomes. Specifically, Marchetti said, 59% of fasting patients achieved a chemotherapy response score of 3 — indicating complete or near-complete tumor response before surgery — compared with 17% of patients in the control group.
Median progression-free survival was significantly longer in the fasting group, at 38 vs 24 months.
Importantly, Marchetti said, the fasting protocol was feasible, well tolerated, and safe: All patients assigned to the fasting group completed treatment, and rates of chemotherapy-related toxicities were similar between the 2 groups.
Additional analyses shed more light on the possible mechanisms underlying the fasting group’s improved outcomes: The researchers found that those patients tended to have lower levels of circulating suppressor granulocyte and monocyte populations that have been linked to tumor immune escape, which suggests, Marchetti said, fasting may have set the stage for a “more favorable immune environment” during chemotherapy.
However, she cautioned that much more research is needed. Her team is planning a larger multicenter trial to validate the current findings, and longer-term follow-up is necessary to see whether fasting ultimately impacts patients’ survival, Marchetti said.
In a statement, Eleonora Teplinsky, MD, an ASCO expert in gynecologic cancers, said these early findings are “encouraging, support earlier data, and highlight a promising area of cancer research.”
But she, too, emphasized the need for larger clinical trials to build on the results.
The study had no commercial funding. Marchetti disclosed having relationships with Arquer Diagnostics, AstraZeneca, Clovis Oncology, and other companies. Small disclosed having relationships with Janssen, Johnson & Johnson, and others. Teplinsky had no disclosures.
A version of this article first appeared on Medscape.com.
A few days of fasting around chemotherapy sessions may improve treatment response and outcomes for some patients with advanced ovarian cancer, a small phase 2 trial suggests.
The study, of 36 patients with stage III or IV high-grade ovarian cancer, found that those randomly assigned to fast for 36 hours before chemotherapy and 24 hours afterward had stronger pathologic responses to chemotherapy and longer progression-free survival than patients who ate normally during treatment.
The findings, reported at a press briefing ahead the American Society of Clinical Oncology (ASCO) 2026, hint at a straightforward measure to potentially improve patients’ outcomes.
The working theory is that short-term fasting boosts chemotherapy response by lowering insulin and IGF-1 levels, both of which are implicated in tumor growth and chemotherapy resistance, said study presenter Claudia Marchetti, MD, of Agostino Gemelli University in Rome, Italy.
Speaking at the briefing, ASCO President Eric Small, MD, of the University of California San Francisco, called the study “a great example of a very simple intervention that has benefit and can be undertaken and implemented anywhere in the world.”
“It’s not an expensive new drug,” he said, “and yet it has the potential to really have an impact on this cancer.”
Ovarian cancer affects more than 324,000 women worldwide each year and causes more than 206,000 deaths annually. Around 80% of patients are diagnosed at an advanced stage, and up to 60% receive neoadjuvant chemotherapy to reduce tumor size and facilitate surgery.
Despite advances in surgery and chemotherapy, patients with advanced disease still face poor outcomes. There is, Marchetti said, “an urgent need for safe, low-cost, and easily implementable strategies that can enhance treatment efficacy and improve patient prognosis.”
Given evidence on the role of insulin in tumor growth and chemotherapy response, her team hypothesized that short bouts of fasting around the time of treatment might have benefits.
To test that idea, the researchers recruited 36 patients with newly diagnosed stage III or IV high-grade serous ovarian carcinoma who were not candidates for primary cytoreduction. All were in good general health, with a mean age of 62 years.
All patients received 3 rounds of carboplatin and paclitaxel before surgery. Prior to starting chemotherapy, half were randomly assigned to fast for 36 hours before and 24 hours after chemotherapy, whereas the other half ate normally throughout treatment.
Patients in the fasting group consumed no more than 350 calories per day during the fasting window. They were allowed to have unrestricted water, herbal tea, limited vegetable juice, and small amounts of light vegetable broth. (A ketogenic diet group had initially been planned but was closed early because of poor patient compliance.)
The study met its primary endpoint of change in insulin levels during chemotherapy, Marchetti reported. Baseline insulin levels were comparable between the 2 groups, but after 3 rounds of chemotherapy, they’d dipped by an average of 1.12 µIU/mL in the fasting group and increased by 9.76 µIU/mL in the control group (P = .01).
Fasting also improved clinical outcomes. Specifically, Marchetti said, 59% of fasting patients achieved a chemotherapy response score of 3 — indicating complete or near-complete tumor response before surgery — compared with 17% of patients in the control group.
Median progression-free survival was significantly longer in the fasting group, at 38 vs 24 months.
Importantly, Marchetti said, the fasting protocol was feasible, well tolerated, and safe: All patients assigned to the fasting group completed treatment, and rates of chemotherapy-related toxicities were similar between the 2 groups.
Additional analyses shed more light on the possible mechanisms underlying the fasting group’s improved outcomes: The researchers found that those patients tended to have lower levels of circulating suppressor granulocyte and monocyte populations that have been linked to tumor immune escape, which suggests, Marchetti said, fasting may have set the stage for a “more favorable immune environment” during chemotherapy.
However, she cautioned that much more research is needed. Her team is planning a larger multicenter trial to validate the current findings, and longer-term follow-up is necessary to see whether fasting ultimately impacts patients’ survival, Marchetti said.
In a statement, Eleonora Teplinsky, MD, an ASCO expert in gynecologic cancers, said these early findings are “encouraging, support earlier data, and highlight a promising area of cancer research.”
But she, too, emphasized the need for larger clinical trials to build on the results.
The study had no commercial funding. Marchetti disclosed having relationships with Arquer Diagnostics, AstraZeneca, Clovis Oncology, and other companies. Small disclosed having relationships with Janssen, Johnson & Johnson, and others. Teplinsky had no disclosures.
A version of this article first appeared on Medscape.com.
Can Fasting Around Chemo Improve Ovarian Cancer Outcomes?
Can Fasting Around Chemo Improve Ovarian Cancer Outcomes?
Nurse Practitioner-Led Outreach Boosts Cancer Screening Rates Among Women Veterans in Rural Settings
Nurse Practitioner-Led Outreach Boosts Cancer Screening Rates Among Women Veterans in Rural Settings
TOPLINE:
Telephone outreach by a nurse practitioner (NP) providing counseling and care coordination reduced the gaps in breast and cervical cancer screenings among women veterans in rural areas, according to a retrospective study.
METHODOLOGY:
- Researchers conducted a retrospective chart review of 55 women veterans who received interventions related to breast or cervical cancer screening at a rural Veterans Health Administration health care system.
- A Boost team, including an NP, a medical director, a program coordinator, and a program evaluation team, was established to provide care coordination and counseling for these participants.
- The NP conducted outreach by telephone to these participants receiving care at five community-based outpatient clinics located in rural counties and helped coordinate access to screening appointments through the Office of Community Care.
- Outcomes included the number of veterans due for breast or cervical cancer screening at the time of outreach and the number of mammograms and Pap smears completed in the 12-month period following the intervention.
TAKEAWAY:
- Of the 55 veterans who received Boost interventions related to cancer screening, 35 (64%) were due for breast cancer screening and 27 (49%) were due for cervical cancer screening before the intervention.
- Following the Boost intervention, the number of veterans due for breast cancer and cervical cancer screenings decreased to 18 (32%) and 16 (29%), respectively.
- Among veterans due for breast cancer screening, 29 (83%) received counseling regarding screening and 17 (59%) of counseled participants completed mammography; however, among those due for cervical cancer screening, 22 (81%) received counseling and 11 (50%) completed screening.
- None of the veterans who were due for screening but did not receive counseling completed their screening, demonstrating the critical role of clinician-provided education and counseling.
IN PRACTICE:
“We hope to expand Boost outreach from one NP working part-time across two health systems to a national partnership of licensed independent providers conducting clinician-initiated outreach to a broader and geographically more diverse group of veterans,” the authors wrote.
SOURCE:
This study was led by Lina Vadlamani, MD, MBA, San Francisco Internal Medicine Residency Program, University of California, San Francisco. It was published online on April 24, 2026, in Military Medicine.
LIMITATIONS:
This study was a secondary analysis in which participants were not randomly assigned, limiting causal inferences. Veterans who answered the phone and engaged with the NP were likely easier to reach and potentially more proactive about their health than those who did not engage, and this selection bias may have limited the generalizability of the findings.
DISCLOSURES:
This study was funded by the Department of Veterans Affairs, Veterans Health Administration, and Office of Rural Health. The authors reported having no relevant conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Telephone outreach by a nurse practitioner (NP) providing counseling and care coordination reduced the gaps in breast and cervical cancer screenings among women veterans in rural areas, according to a retrospective study.
METHODOLOGY:
- Researchers conducted a retrospective chart review of 55 women veterans who received interventions related to breast or cervical cancer screening at a rural Veterans Health Administration health care system.
- A Boost team, including an NP, a medical director, a program coordinator, and a program evaluation team, was established to provide care coordination and counseling for these participants.
- The NP conducted outreach by telephone to these participants receiving care at five community-based outpatient clinics located in rural counties and helped coordinate access to screening appointments through the Office of Community Care.
- Outcomes included the number of veterans due for breast or cervical cancer screening at the time of outreach and the number of mammograms and Pap smears completed in the 12-month period following the intervention.
TAKEAWAY:
- Of the 55 veterans who received Boost interventions related to cancer screening, 35 (64%) were due for breast cancer screening and 27 (49%) were due for cervical cancer screening before the intervention.
- Following the Boost intervention, the number of veterans due for breast cancer and cervical cancer screenings decreased to 18 (32%) and 16 (29%), respectively.
- Among veterans due for breast cancer screening, 29 (83%) received counseling regarding screening and 17 (59%) of counseled participants completed mammography; however, among those due for cervical cancer screening, 22 (81%) received counseling and 11 (50%) completed screening.
- None of the veterans who were due for screening but did not receive counseling completed their screening, demonstrating the critical role of clinician-provided education and counseling.
IN PRACTICE:
“We hope to expand Boost outreach from one NP working part-time across two health systems to a national partnership of licensed independent providers conducting clinician-initiated outreach to a broader and geographically more diverse group of veterans,” the authors wrote.
SOURCE:
This study was led by Lina Vadlamani, MD, MBA, San Francisco Internal Medicine Residency Program, University of California, San Francisco. It was published online on April 24, 2026, in Military Medicine.
LIMITATIONS:
This study was a secondary analysis in which participants were not randomly assigned, limiting causal inferences. Veterans who answered the phone and engaged with the NP were likely easier to reach and potentially more proactive about their health than those who did not engage, and this selection bias may have limited the generalizability of the findings.
DISCLOSURES:
This study was funded by the Department of Veterans Affairs, Veterans Health Administration, and Office of Rural Health. The authors reported having no relevant conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Telephone outreach by a nurse practitioner (NP) providing counseling and care coordination reduced the gaps in breast and cervical cancer screenings among women veterans in rural areas, according to a retrospective study.
METHODOLOGY:
- Researchers conducted a retrospective chart review of 55 women veterans who received interventions related to breast or cervical cancer screening at a rural Veterans Health Administration health care system.
- A Boost team, including an NP, a medical director, a program coordinator, and a program evaluation team, was established to provide care coordination and counseling for these participants.
- The NP conducted outreach by telephone to these participants receiving care at five community-based outpatient clinics located in rural counties and helped coordinate access to screening appointments through the Office of Community Care.
- Outcomes included the number of veterans due for breast or cervical cancer screening at the time of outreach and the number of mammograms and Pap smears completed in the 12-month period following the intervention.
TAKEAWAY:
- Of the 55 veterans who received Boost interventions related to cancer screening, 35 (64%) were due for breast cancer screening and 27 (49%) were due for cervical cancer screening before the intervention.
- Following the Boost intervention, the number of veterans due for breast cancer and cervical cancer screenings decreased to 18 (32%) and 16 (29%), respectively.
- Among veterans due for breast cancer screening, 29 (83%) received counseling regarding screening and 17 (59%) of counseled participants completed mammography; however, among those due for cervical cancer screening, 22 (81%) received counseling and 11 (50%) completed screening.
- None of the veterans who were due for screening but did not receive counseling completed their screening, demonstrating the critical role of clinician-provided education and counseling.
IN PRACTICE:
“We hope to expand Boost outreach from one NP working part-time across two health systems to a national partnership of licensed independent providers conducting clinician-initiated outreach to a broader and geographically more diverse group of veterans,” the authors wrote.
SOURCE:
This study was led by Lina Vadlamani, MD, MBA, San Francisco Internal Medicine Residency Program, University of California, San Francisco. It was published online on April 24, 2026, in Military Medicine.
LIMITATIONS:
This study was a secondary analysis in which participants were not randomly assigned, limiting causal inferences. Veterans who answered the phone and engaged with the NP were likely easier to reach and potentially more proactive about their health than those who did not engage, and this selection bias may have limited the generalizability of the findings.
DISCLOSURES:
This study was funded by the Department of Veterans Affairs, Veterans Health Administration, and Office of Rural Health. The authors reported having no relevant conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
Nurse Practitioner-Led Outreach Boosts Cancer Screening Rates Among Women Veterans in Rural Settings
Nurse Practitioner-Led Outreach Boosts Cancer Screening Rates Among Women Veterans in Rural Settings
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
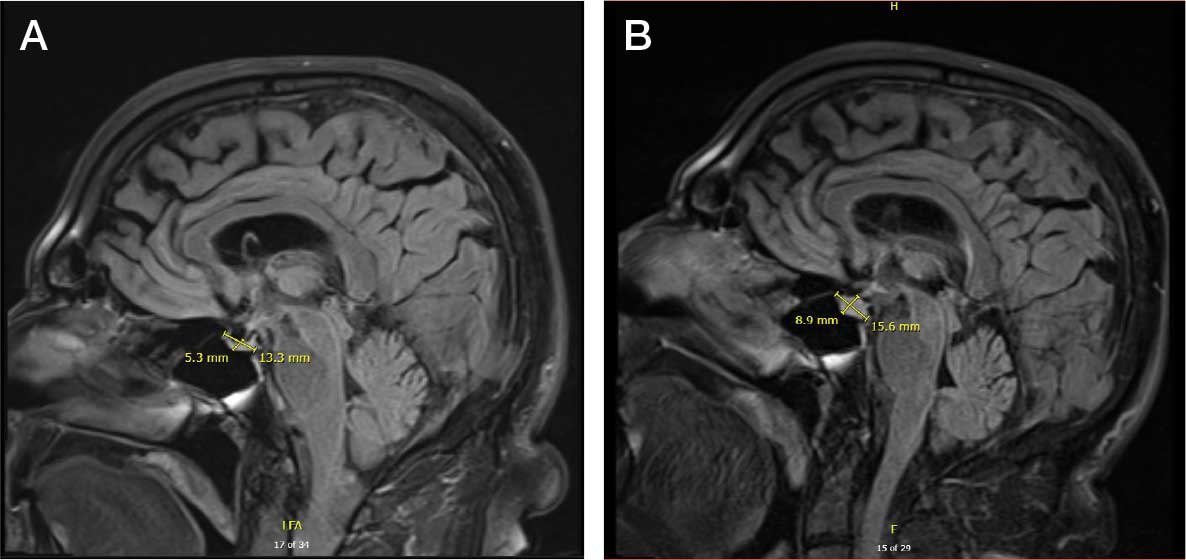
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.

No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
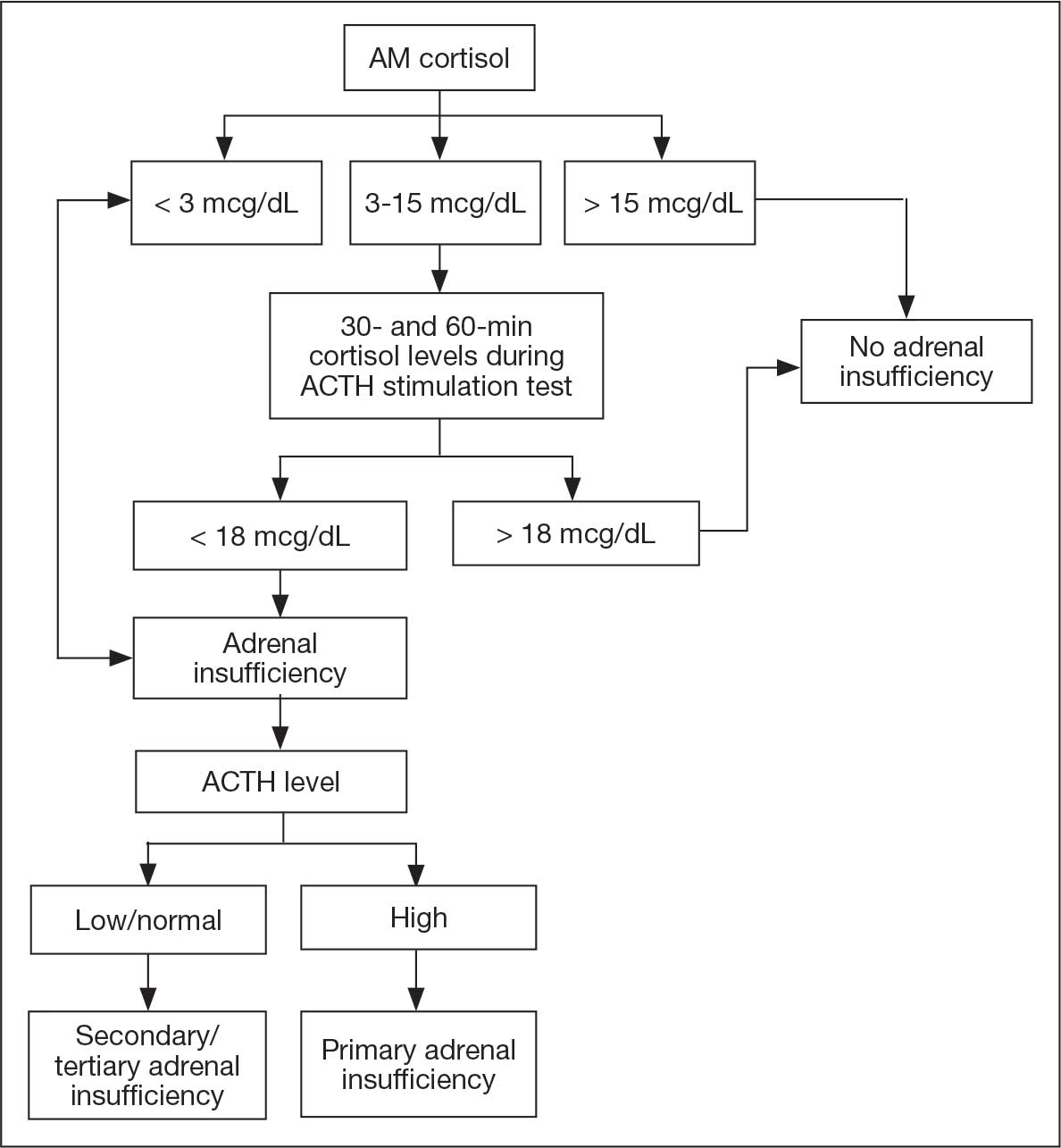
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
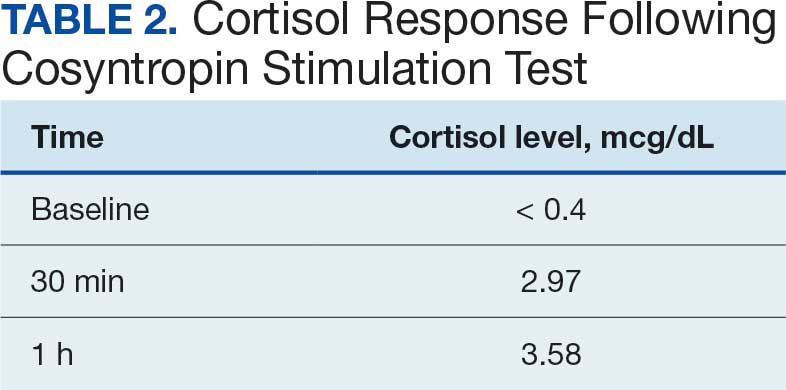
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
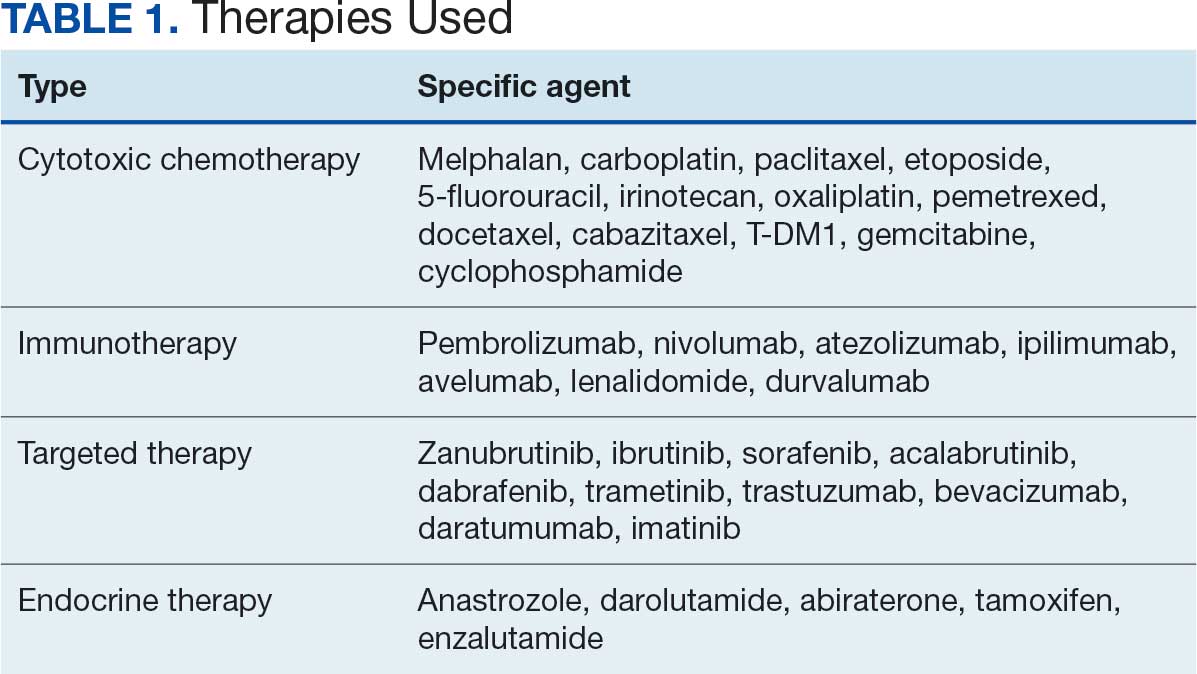
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
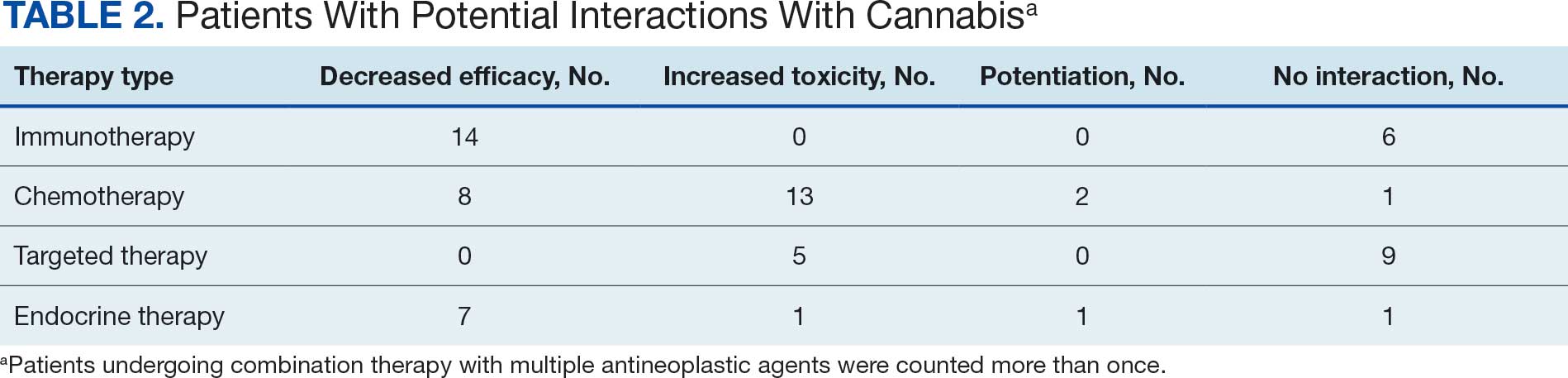
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review

Early Outcomes of Stereotactic Body Radiotherapy for Localized Prostate Cancer: A Retrospective Analysis
Early Outcomes of Stereotactic Body Radiotherapy for Localized Prostate Cancer: A Retrospective Analysis
Prostate cancer is the most common cancer in US males, with an estimated 313,780 new cases and 35,770 deaths in 2025.1 Several treatment options are available for localized prostate cancer that have similar outcomes, including active surveillance for low-risk cancers, surgery, or radiotherapy.2,3 Conventional fractionation radiotherapy (CFRT) with 40 to 45 fractions over 8 to 9 weeks has been used for decades. Over the past 2 decades, moderate hypofractionation schedules with 2.4 to 3.4 Gy per fraction over 20 to 28 fractions have become standard, as many noninferiority randomized clinical trials (RCTs) such as CHHiP (UK),4 PROFIT (Canada and Europe),5 NRG Oncology RTOG 0415 (US),6 HYPRO (Netherlands),7,8 and HYPO-RT-PC (Sweden and Denmark),9 have shown the noninferiority of moderately hypofractionated radiotherapy compared with CFRT. Notably, most of these noninferiority studies primarily included patients with low- or intermediate-risk prostate cancer, except for the HYPO-RT-PC trial,9 which also included patients with intermediate- and high-risk prostate cancer.
These noninferiority studies, along with technological advances in radiotherapy, such as intensity-modulated radiotherapy (IMRT), volumetric modulated arc therapy (VMAT), and image-guided radiotherapy (IGRT), paved the path to ultrahypofractionated stereotactic body radiotherapy (SBRT) that is delivered in 5 fractions of ≥ 6 Gy. This high dose per fraction may have a radiobiologic advantage over conventional fractionation. The relatively low a/ß ratio of prostate cancer, estimated to be between 1 and 2, suggests that tumor cells may be particularly sensitive to the high doses per fraction delivered in SBRT.10-13 Compared with CFRT, SBRT-induced tumor cell death may also be mediated through different pathways; this pathway appears to be generated in a dose-dependent manner, particularly with doses > 8 Gy per fraction.14,15 Additionally, the higher a/ß ratio for the surrounding organs at risk, such as the bladder and rectum, theoretically allows for an improved therapeutic ratio window that maximizes tumor control while minimizing damage to healthy tissues.
A substantial body of evidence from prospective studies and meta-analyses supports the use of SBRT for localized prostate cancer. HYPO-RT-PC, a significant phase 3 noninferiority study, enrolled 1200 patients with intermediate (89%) and high-risk (11%) prostate cancer randomized between 2 arms, including CFRT to 78 Gy in 39 fractions and SBRT to 42.7 Gy in 7 fractions, treated 3 days weekly. After a median follow-up of 60 months, the estimated 5-year biochemical relapse-free survival rate was 84% in both groups.9 This trial was notable because it was the first randomized study to demonstrate that SBRT was noninferior to CFRT in intermediate- and high-risk prostate cancer patients. Another pivotal phase 3 trial, the PACE-B study, enrolled 874 patients to compare SBRT (36.25 Gy to the prostate gland, with a secondary dose of 40 Gy to the gross tumor volume where applicable, in 5 fractions) with CFRT (78 Gy in 39 fractions) and moderately hypofractionated radiotherapy (HFRT) (62 Gy in 20 fractions) in patients with low- or intermediate-risk prostate cancer. With a 74-month median follow-up, the study reported 5-year biochemical free rates of 94.6% for CFRT and 95.8% for SBRT, confirming the noninferiority of SBRT to CFRT.15
SBRT offers short, effective, and convenient treatment to many patients with localized prostate cancer. While previous guidelines were more restrictive, the March 2026 National Comprehensive Cancer Network (NCCN) guidelines now list SBRT as a preferred treatment modality for high-risk prostate cancer.16
Given the growing body of evidence supporting the efficacy and safety of SBRT, we implemented an SBRT program in 2014 at a tertiary care center for veterans. This retrospective study was undertaken to evaluate the early efficacy and toxicity of SBRT in patients with localized prostate cancer treated at our institution, including patients across all risk stratifications.
METHODS
We identified 242 patients diagnosed with prostate cancer who underwent SBRT treatment between November 2014 and October 2024 at Overland Park Veterans Affairs Radiation Oncology Clinic. For the final analysis, 46 patients with < 2 years of follow-up and 22 patients who died from causes other than prostate cancer were excluded, resulting in a cohort of 174 patients with ≥ 24-month follow-up.
Treatment
Patients eligible for staging underwent imaging according to NCCN guidelines, including computed tomography (CT) of the abdomen and pelvis, bone scintigraphy, or, in recent years, prostate-specific membrane antigen positron emission tomography, primarily used for unfavorable intermediate-risk (UIR) and high-risk (HR) cancers. Patients with a negative staging work-up for nodal or skeletal disease were included. Prior to planning the CT simulation, patients were given bowel preparation instructions, including a low-fiber and low-gas-producing diet, simethicone, and enemas, the night before and morning of the simulation. Patients were instructed to arrive with a comfortably full bladder, having not voided for 2 to 3 hours prior to the procedure. At Kansas City Veterans Affairs Medical Center (KCVAMC), SBRT treatment was generally restricted to patients with a baseline American Urological Association symptom score of 15 to 20 out of 35 and a prostate gland size < 80 mL to minimize the risk of acute urinary toxicity. We did not use intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast agents for planning CT simulation.
Patients were placed in a supine position, and a vacuum bag was used for immobilization. Following the CT simulation, the images were transferred to the Eclipse treatment planning system. The clinical target volume (CTV) encompassed the prostate and the proximal 1.0 cm of the seminal vesicles for Gleason score (GS) 1 to 2, and the entire seminal vesicle was included for GS 3 to 5, which is consistent with KCVAMC practice and established safety protocols. The planning target volume (PTV) was created by uniformly expanding the CTV by 5 to 7 mm, except for the posterior margin, which was limited to 3 to 5 mm. When elective nodal radiotherapy was planned for HR prostate cancer, the pelvic field for CT simulation started at the L-2 upper border, with the lower border extending to the lesser trochanter. The pelvic nodes were delineated per Radiation Therapy Oncology Group (RTOG) guidelines.17 The CTV nodes (CTVn), including common iliac, external and internal iliac nodes, obturator, and presacral nodes, were created by uniformly expanding the CTVn by 2 to 3 mm. Slice-by-slice corrections were made to avoid bowel overlap in these patients.
The use of androgen deprivation therapy (ADT) for a duration of 6 to 24 months was prescribed for patients with UIR or HR prostate cancer per NCCN guidelines.16 The prescribed dose to the PTV was 36.25 to 40 Gy (40 Gy was mostly used as a boost to the dominant lesion) in 5 fractions, with each fraction ranging from 7.25 to 8 Gy. For elective nodal radiotherapy in patients at HR, the prescribed dose was 25 Gy in 5 fractions. All patients were planned for VMAT, which aims to deliver ≥ 95% of the prescription dose to 95% of the PTV. Once the physician approved the treatment plan and physics quality assessment was completed, treatments commenced on an every-other-day schedule. Patients received the same bowel preparation instructions for each treatment as for the planning CT simulation. Daily treatment accuracy was confirmed via daily 3-dimensional cone-beam CT (CBCT) for IGRT. No fiducials or hydrogel rectal spacers were used.
Follow-up Schedule and Toxicity Assessment
Follow-up assessments were conducted 4 to 6 weeks after radiation therapy and then repeated every 6 months for 2 to 5 years, and annually thereafter. At each follow-up visit, patients were evaluated for genitourinary (GU) and gastrointestinal (GI) toxicity, according to RTOG toxicity criteria. Prostate-specific antigen (PSA) levels were monitored; in patients receiving ADT, testosterone levels were also checked.
Statistical Analysis
Biochemical failure was defined using the Phoenix definition (nadir PSA + 2 ng/mL). Differences between dose cohorts were assessed using the log-rank test for survival outcomes and X2 testing for categorical variables. GU and GI toxicities were summarized as cumulative incidences of RTOG grade ≥ II events. Statistical significance was set at P < .05.
RESULTS
One hundred seventy-four patients were included in the retrospective review. Patients had a median follow-up of 45 months (range, 24-111) (Figure). The median age at treatment was 74 years (range, 51-88), and the median pretreatment PSA level was 11.9 ng/mL (range, 0.6-69.5). Twenty-six patients (14.9%) had a GS 1, 77 (44.3%) had GS 2, 41 (23.6%) had GS 3, 18 (10.3%) had GS 4, and 12 (6.9%) had GS 5. Fifty-one patients (29.3%) received elective pelvic nodal radiotherapy, and 93 patients (53.4%) received ADT (Table 1).
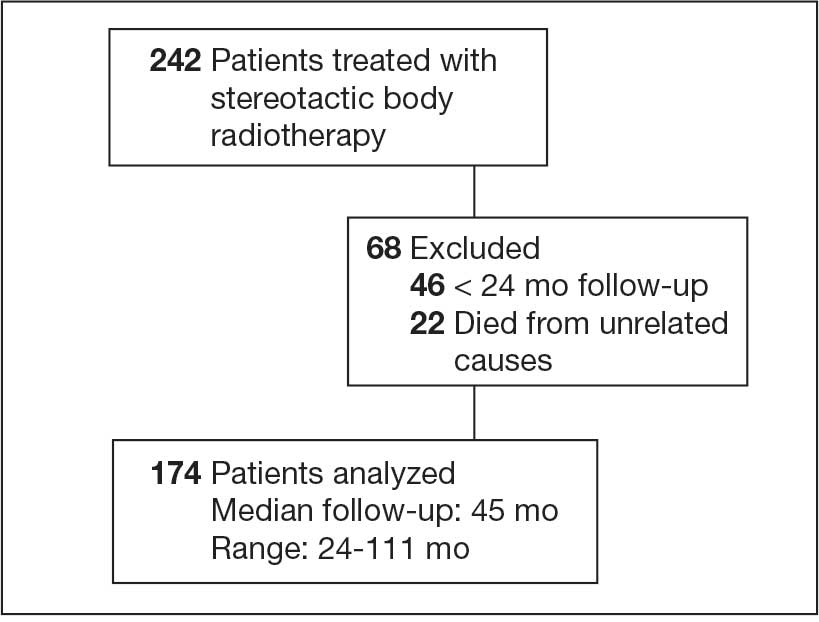
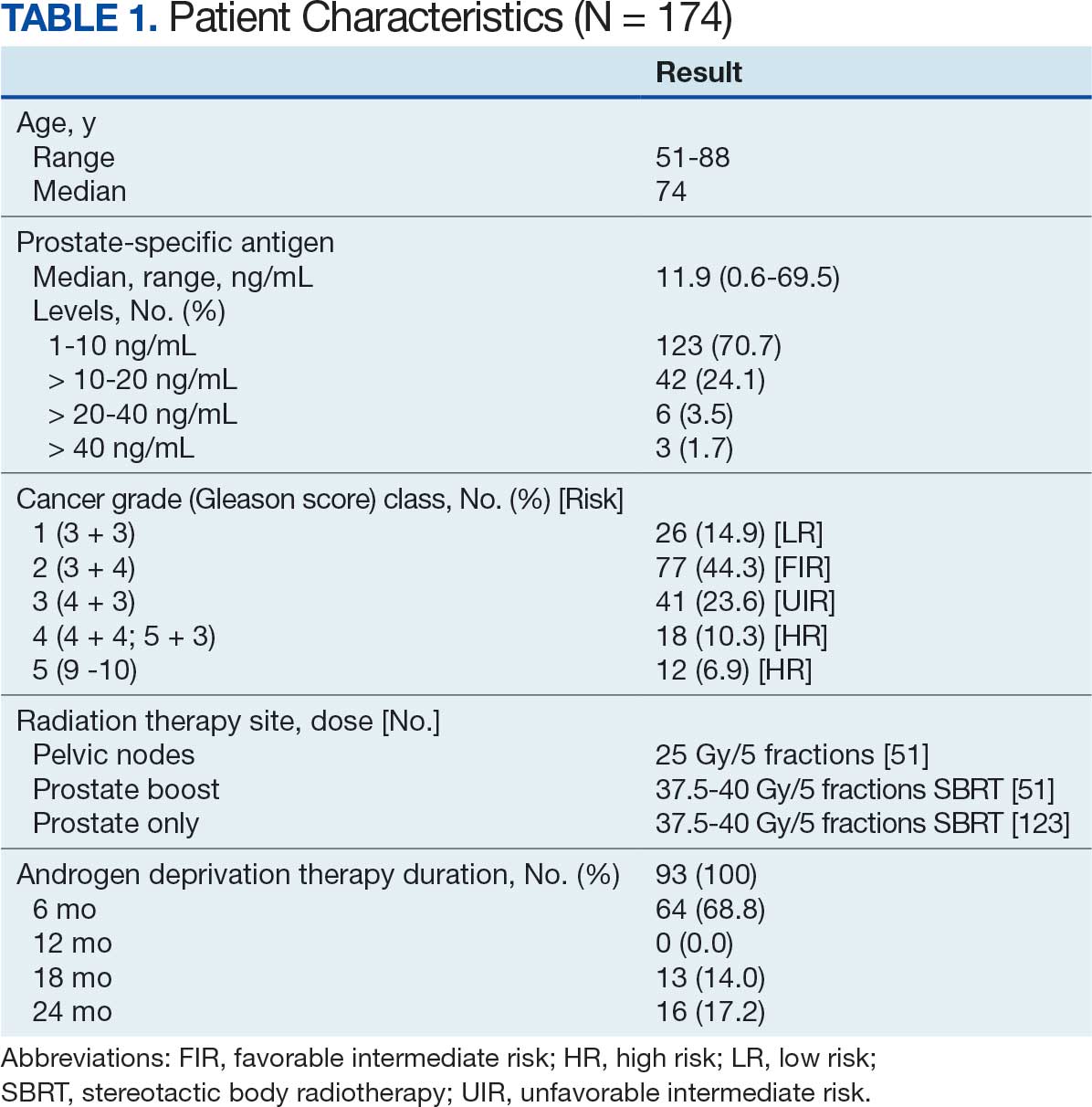
At 24 months follow-up, 6 patients (3.4%) had biochemical failures. One patient died from metastatic prostate cancer, and 5 patients are living with biochemical failure (Table 2). The actuarial 5-year overall survival (OS) rate was 99.4%, and the 5-year disease-free survival (DFS) rate was 96.6%. We performed a subanalysis comparing outcomes of the 36.25 Gy vs 40 Gy SBRT cohorts. There was no statistically significant difference in DFS, OS, or the cumulative incidence of grade II/III toxicity between patients treated with 40 Gy vs 36.25 Gy. Outcomes stratified by NCCN risk groups (low, intermediate, high/very high) are detailed in Table 3. As expected, DFS was slightly lower in the high-risk group, but overall disease control remained high across all stratifications.
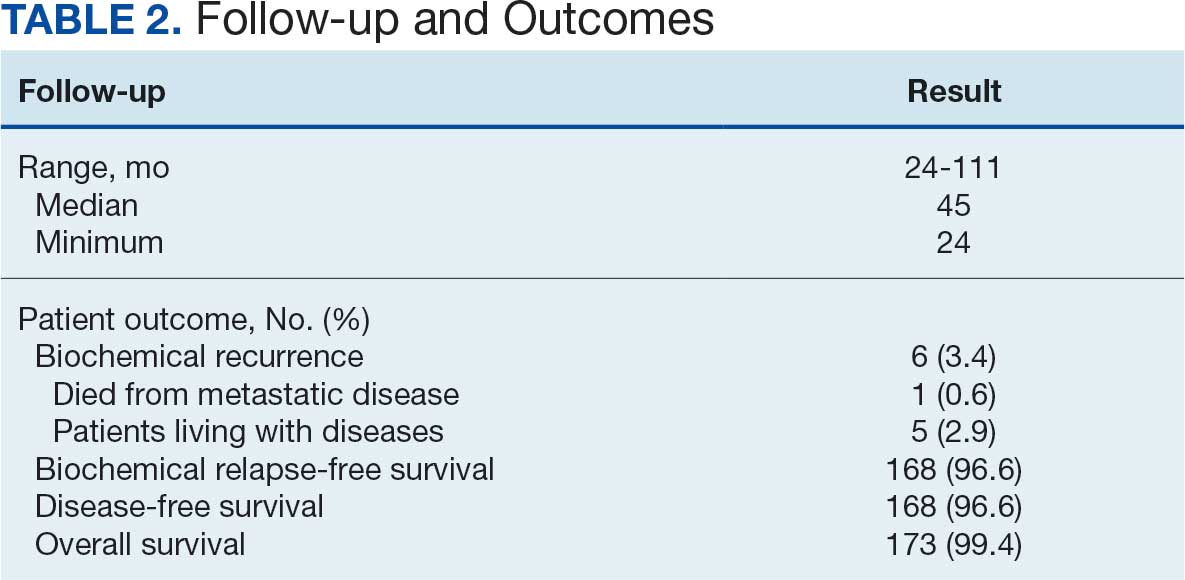
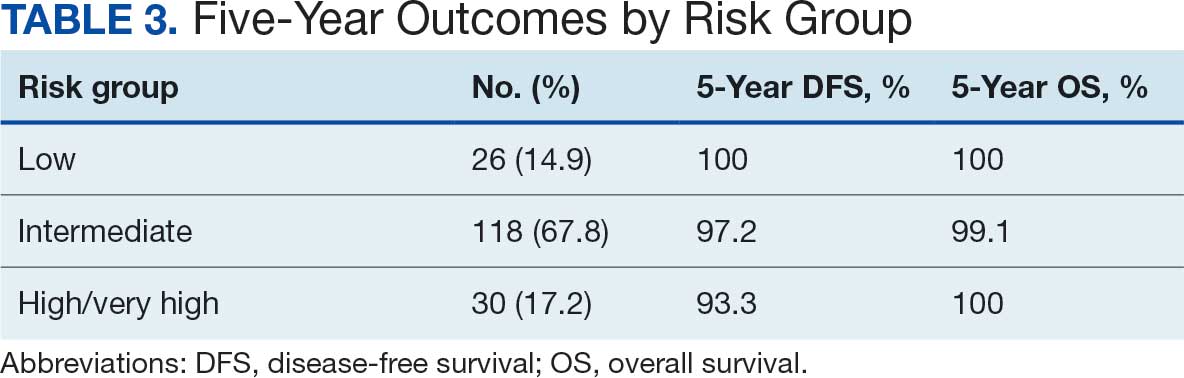
The cumulative incidence of RTOG grade II and higher GU toxicity was 28.2% (Table 4). This included 46 patients (26.4%) with grade II GU toxicity and 2 patients (1.2%) who developed grade III GU complications (1 requiring self-catheterization and another a suprapubic catheter for urinary retention). One patient (0.6%) treated with a 40 Gy dose regimen experienced a grade IV GU complication in the form of a rectovesical fistula necessitating surgical intervention.
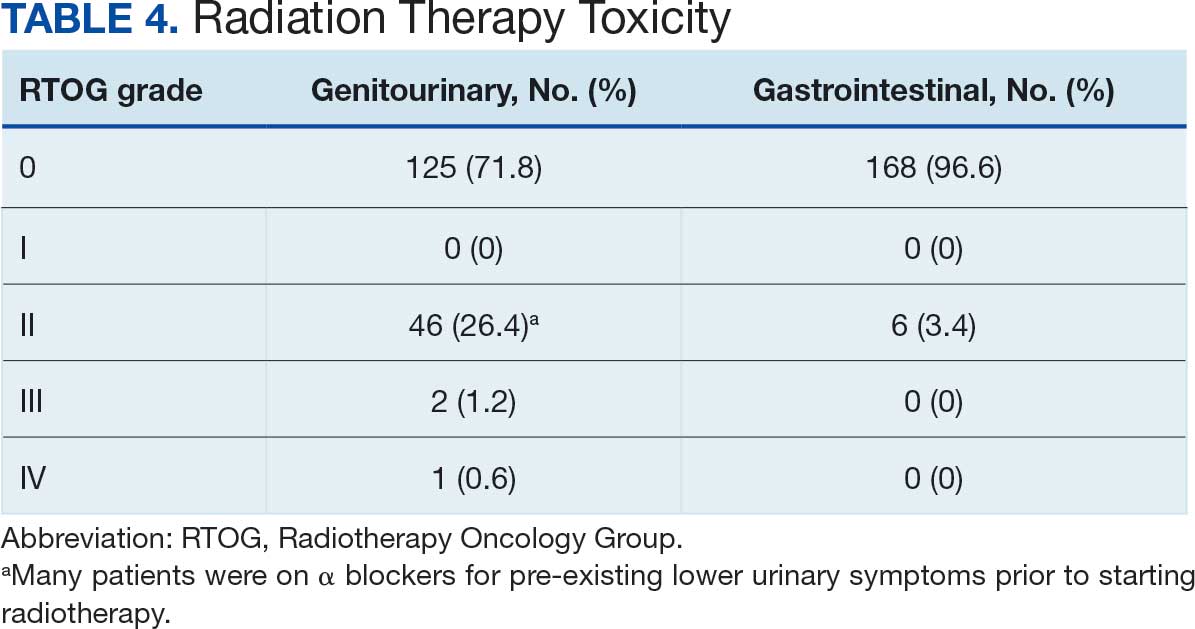
The cumulative incidence of RTOG grade II or higher GI toxicity was 3.4%, and no grade III or IV gastrointestinal toxicities were observed during the follow-up period. Importantly, intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast were not routinely used in this cohort of patients.
The high rates of actuarial 5-year DFS and OS observed suggest a favorable initial response to the SBRT regimen employed at KCVAMC. However, given the potential for late recurrence in patients with prostate cancer, longer follow-up is essential to determine the durability of these outcomes. The observed GU toxicity rate of 28.2% for grade II and higher events warrants careful consideration and compares with other published data on SBRT for prostate cancer.15 The occurrence of a grade IV rectovesical fistula, although rare, is a notable adverse event that warrants discussion in the context of the treatment approach. The low incidence of grade II or higher GI toxicity is an encouraging finding, particularly given that hydrogel rectal spacers are not routinely used to minimize rectal exposure.
DISCUSSION
The primary objective of this retrospective study was to evaluate the outcomes of SBRT for patients with localized prostate cancer treated at KCVAMC and to compare these results with those reported in the literature. Our findings demonstrate promising intermediate-term efficacy, with an estimated 5-year DFS of 96.6% and OS of 99.4% at a median follow-up of 45 months. Furthermore, the observed toxicity profile appears acceptable, with a cumulative grade II and higher GU toxicity rate of 28.2% and a grade II or higher GI toxicity rate of 3.4%. Notably, these outcomes were achieved without the routine use of intraprostatic fiducials or hydrogel rectal spacers.
Two pivotal randomized phase 3 trials have established the noninferiority of ultrahypofractionated radiotherapy (UHRT) with SBRT over conventional fractionation. The HYPO-RT-PC trial compared SBRT (42.7 Gy in 7 fractions) with conventional fractionation (78 Gy in 39 fractions) in intermediate- and high-risk patients with prostate cancer and reported a 5-year biochemical relapse-free survival of 84% in both arms.9 The PACE-B trial, which included patients at low- and intermediate-risk, compared SBRT (36.25 Gy in 5 fractions) with conventional or moderate HFRT and reported a 5-year biochemical control rate of 95.8% in the SBRT arm and 94.6% in the control arm.15
A comprehensive review and meta-analysis of 7 phase 3 studies involving 6795 patients compared different radiotherapy regimens, namely, UHRT, HFRT, and CFRT, and reported that after 5 years, the DFS rates were 85.1% for CFRT, 86% for HFRT, and 85% for UHRT, with no significant difference in toxicity among the 3 different treatment approaches.18 This suggests that shorter, more intense radiotherapy schedules (UHRT and HFRT) may be as effective and safe as traditional, longer courses of radiation.
There are multiple published nonrandomized prospective trials in which thousands of patients with extreme hypofractionation have been treated with different doses, fractions, and techniques. While heterogeneity and limited long-term follow-up in the existing evidence are acknowledged, these data suggest that prostate SBRT provides appropriate biochemical control with few high-grade toxicities, supporting its ongoing global use and justifying further prospective investigations. Comparative data are shown in Table 5. Several ongoing studies are evaluating noninferiority, superiority, and cost-effectiveness using different methodologies (Table 6).9,15,19-24
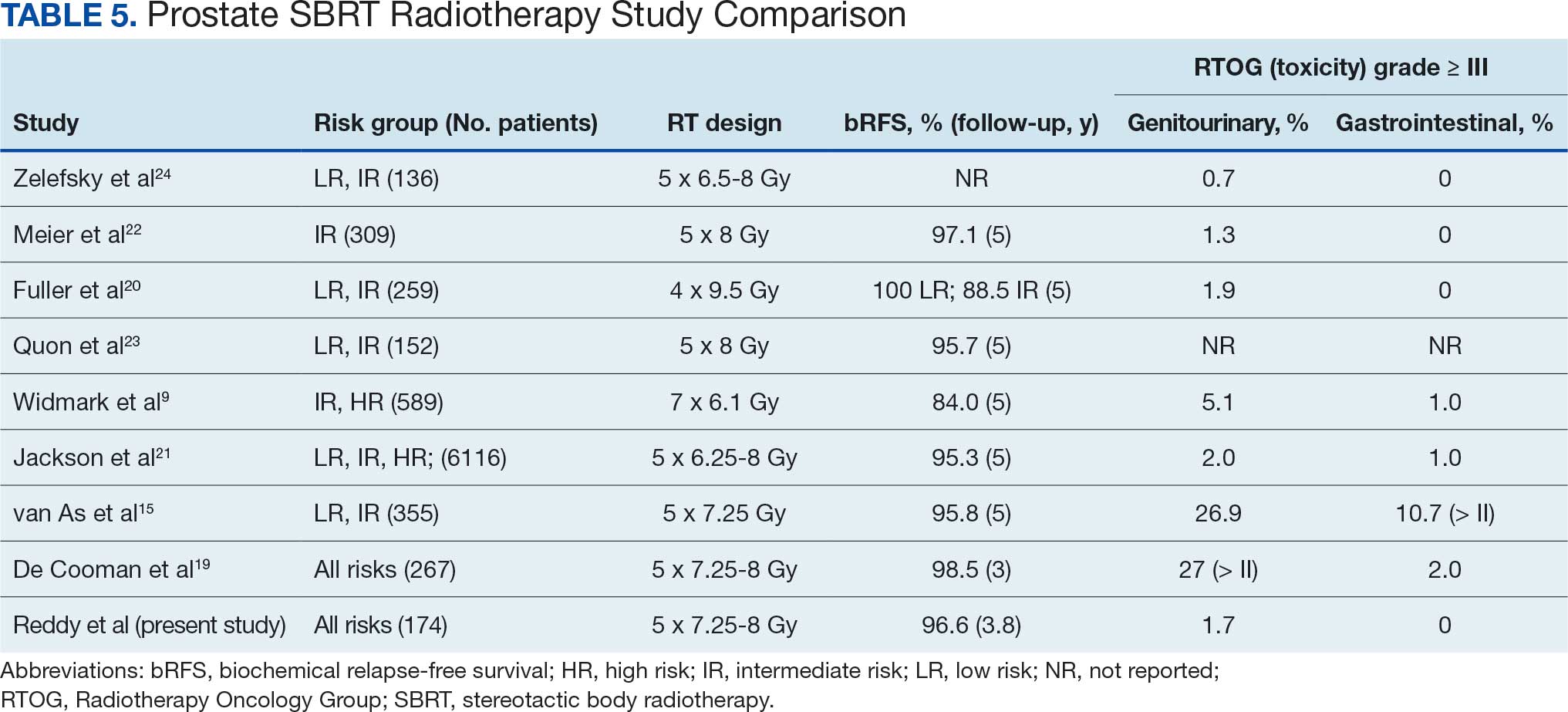
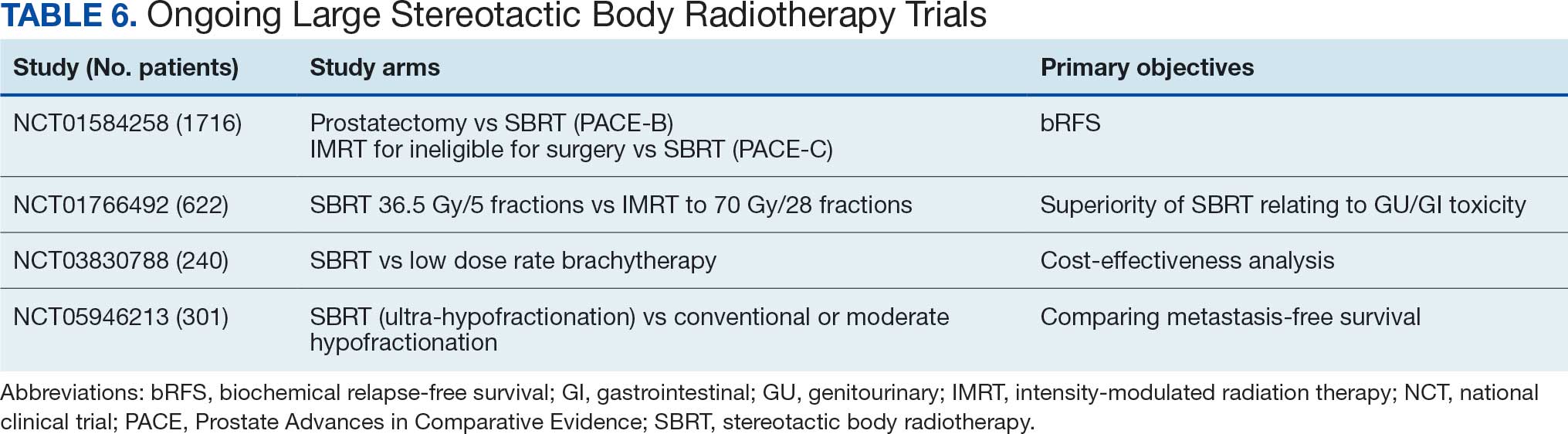
This study’s efficacy outcomes, particularly the high DFS rate, are consistent with the findings from these landmark trials, suggesting that the SBRT regimen used at KCVAMC is effective in achieving early disease control despite 17.2% of patients having high-risk disease. The GU toxicity observed in this study, with a 28.2% rate of grade II or higher events, is also comparable with the 26.9% reported in the 5-fraction SBRT arm of the PACE-B trial, which had a longer median follow-up of 74 months.15 It is important to note that a portion of these grade II events occurred in patients who were already on a blockers for pre-existing lower urinary tract symptoms before starting radiotherapy, which may inflate the observed cumulative acute toxicity score.
A critical comparison is how SBRT toxicity aligns with moderate hypofractionation (eg, 60 Gy in 20 fractions or 70 Gy in 28 fractions as reported by others).4,6 Our observed grade III and higher GU toxicity rate (1.7%) and grade III and higher GI toxicity rate (0%) are highly favorable when compared with historical moderate hypofractionation data, which typically report grade III GU toxicity in the range of 2% to 3% and grade III GI toxicity around 1% to 2%. This suggests that despite the higher dose per fraction, SBRT does not necessarily lead to increased severe acute toxicity, potentially offering a superior therapeutic ratio for GI and GU sparing.
However, the occurrence of a grade IV rectovesical fistula in 1 patient (0.6%)—who received the 40 Gy dose—was a serious complication that warrants careful consideration. This rare, but severe, complication in the higher dose cohort underscores the potential for increased organ-at-risk toxicity, particularly in the absence of a hydrogel rectal spacer, which is designed to mitigate high-dose rectal exposure. While the overall rate of significant GU toxicity remains low, this event highlights the potential risks associated with SBRT. Hydrogel rectal spacers are designed to increase the distance between the prostate and the rectum, which can reduce the rectal radiation dose and potentially mitigate the risk of such fistulas. The low rate of grade II or worse GI toxicity (3.4%) in our study is noteworthy, especially considering that hydrogel spacers were not routinely used. This finding aligns with the 2.5% GI toxicity rate reported in the SBRT arm of the PACE-B trial, suggesting that careful treatment planning and delivery techniques, such as VMAT-IMRT and daily CBCT for IGRT, may contribute to minimizing GI toxicity even without the use of rectal spacers.15 The exclusive use of 3-dimensional CBCT for IGRT in our study, without the use of fiducial markers, suggests that accurate target localization can be achieved with this approach, contributing to the observed efficacy and reduced toxicity.
Strengths and Limitations
This study’s retrospective, single-center design may have introduced selection bias. The median follow-up of 45 months, while substantial, is still relatively short for assessing very late toxicities and long-term oncologic outcomes in prostate cancer, which is known for late recurrences. Additionally, the lack of a direct comparison group within KCVAMC limits the ability to definitively attribute the observed outcomes solely to SBRT treatment. However, the strengths of this study include the inclusion of a consecutive series of veteran patients with localized prostate cancer across all risk categories, providing a real-world perspective on SBRT outcomes in a diverse patient population. Furthermore, the detailed assessment of efficacy and toxicity via standardized RTOG criteria enhances the comparability of our findings with those of other published prospective studies, despite the retrospective nature of the data.
CONCLUSIONS
This single-institution retrospective analysis revealed that short-term SBRT (36.25 to 40 Gy in 5 fractions), with a minimum follow-up of 24 months and a median follow-up of 45 months, for localized prostate cancer, including patients at HR, is associated with promising early efficacy and acceptable toxicity, even in the absence of routine fiducial or hydrogel spacer use. The favorable actuarial 5-year DFS and OS rates, coupled with a manageable toxicity profile, suggest that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer. However, a longer follow-up is necessary to confirm these findings and fully characterize the long-term efficacy and toxicity of this SBRT regimen. Nevertheless, the results contribute to the growing body of evidence suggesting that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer.
- Siegel RL, Kratzer TB, Giaquinto AN, et al. Cancer statistics, 2025. CA Cancer J Clin. 2025;75:10-45. doi:10.3322/caac.21871
- Donovan JL, Hamdy FC, Lane JA, et al. Patient-reported outcomes after monitoring, surgery, or radiotherapy for prostate cancer. N Engl J Med. 2016;375:1425-1437. doi:10.1056/NEJMoa1606221
- Hamdy FC, Donovan JL, Lane JA, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med. 2016;375:1415-1424. doi:10.1056/NEJMoa1606220
- Dearnaley D, Syndikus I, Mossop H, et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol. 2016;17:1047-1060. doi:10.1016/S1470-2045(16)30102-4
- Catton CN, Lukka H, Gu CS, et al. Randomized trial of a hypofractionated radiation regimen for the treatment of localized prostate cancer. J Clin Oncol. 2017;35:1884-1890. doi:10.1200/JCO.2016.71.7397
- Lee WR, Dignam JJ, Amin MB, et al. Long-term analysis of NRG Oncology RTOG 0415: a randomized phase III noninferiority study comparing two fractionation schedules in patients with low-risk prostate cancer. J Clin Oncol. 2024;42:2377-2381. doi:10.1200/JCO.23.02445
- de Vries KC, Wortel RC, Oomen-de Hoop E, et al. Hypofractionated versus conventionally fractionated radiation therapy for patients with intermediate- or high-risk, localized, prostate cancer: 7-year outcomes from the randomized, multicenter, open-label, phase 3 HYPRO trial. Int J Radiat Oncol Biol Phys. 2020;106:108-115. doi:10.1016/j.ijrobp.2019.09.007
- Incrocci L, Wortel RC, Alemayehu WG, et al. Hypofractionated versus conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016;17:1061-1069. doi:10.1016/S1470-2045(16)30070-5
- Widmark A, Gunnlaugsson A, Beckman L, et al. Ultra-hypofractionated versus conventionally fractionated radiotherapy for prostate cancer: 5-year outcomes of the HYPO-RT-PC randomised, non-inferiority, phase 3 trial. Lancet. 2019;394:385-395. doi:10.1016/S0140-6736(19)31131-6
- Brenner DJ, Hall EJ. Fractionation and protraction for radiotherapy of prostate carcinoma. Int J Radiat Oncol Biol Phys. 1999;43:1095-101. doi:10.1016/s0360-3016(98)00438-6
- Dasu A. Is the alpha/beta value for prostate tumours low enough to be safely used in clinical trials? Clin Oncol (R Coll Radiol). 2007;19:289-301. doi:10.1016/j.clon.2007.02.007
- Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155-1159. doi:10.1126/science.1082504
- Gulliford S, Hall E, Dearnaley D. Hypofractionation trials and radiobiology of prostate cancer. Oncoscience. 2017;4:27-28. doi:10.18632/oncoscience.347
- Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89-91. doi:10.1016/j.ccr.2005.07.014
- van As N, Griffin C, Tree A, et al. Phase 3 Trial of stereotactic body radiotherapy in localized prostate cancer. N Engl J Med. Oct 17 2024;391:1413-1425. doi:10.1056/NEJMoa2403365
- National Comprehensive Cancer Network. NCCN Guidelines Version 5. 2026 Prostate Cancer. Accessed March 24, 2026. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- Lawton CA, Michalski J, El-Naqa I, et al. RTOG GU radiation oncology specialists reach consensus on pelvic lymph node volumes for high-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2009;74:383-387. doi:10.1016/j.ijrobp.2008.08.002
- Lehrer EJ, Kishan AU, Yu JB, et al. Ultrahypofractionated versus hypofractionated and conventionally fractionated radiation therapy for localized prostate cancer: a systematic review and meta-analysis of phase III randomized trials. Radiother Oncol. 2020;148:235-242. doi:10.1016/j.radonc.2020.04.037
- De Cooman B, Debacker T, Adams T, et al. Stereotactic body radiotherapy (SBRT) as a treatment for localized prostate cancer: a retrospective analysis. Radiat Oncol. 2025;20:25. doi:10.1186/s13014-025-02598-8
- Fuller DB, Falchook AD, Crabtree T, et al. Phase 2 multicenter trial of heterogeneous-dosing stereotactic body radiotherapy for low- and intermediate-risk prostate cancer: 5-year outcomes. Eur Urol Oncol. 2018;1:540-547. doi:10.1016/j.euo.2018.06.013
- Jackson WC, Silva J, Hartman HE, et al. Stereotactic body radiation therapy for localized prostate cancer: a systematic review and meta-analysis of over 6,000 patients treated on prospective studies. Int J Radiat Oncol Biol Phys. 2019;104:778-789. doi:10.1016/j.ijrobp.2019.03.051
- Meier RM, Bloch DA, Cotrutz C, et al. Multicenter trial of stereotactic body radiation therapy for low- and intermediate-risk prostate cancer: survival and toxicity endpoints. nt J Radiat Oncol Biol Phys. 2018;102:296-303. doi:10.1016/j.ijrobp.2018.05.040
- Quon HC, Ong A, Cheung P, et al. Once-weekly versus every-other-day stereotactic body radiotherapy in patients with prostate cancer (PATRIOT): a phase 2 randomized trial. Radiother Oncol. 2018;127:206-212. doi:10.1016/j.radonc.2018.02.029
- Zelefsky MJ, Kollmeier M, McBride S, et al. Five-year outcomes of a phase 1 dose-escalation study using stereotactic body radiosurgery for patients with low-risk and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2019;104:42-49. doi:10.1016/j.ijrobp.2018.12.045
Prostate cancer is the most common cancer in US males, with an estimated 313,780 new cases and 35,770 deaths in 2025.1 Several treatment options are available for localized prostate cancer that have similar outcomes, including active surveillance for low-risk cancers, surgery, or radiotherapy.2,3 Conventional fractionation radiotherapy (CFRT) with 40 to 45 fractions over 8 to 9 weeks has been used for decades. Over the past 2 decades, moderate hypofractionation schedules with 2.4 to 3.4 Gy per fraction over 20 to 28 fractions have become standard, as many noninferiority randomized clinical trials (RCTs) such as CHHiP (UK),4 PROFIT (Canada and Europe),5 NRG Oncology RTOG 0415 (US),6 HYPRO (Netherlands),7,8 and HYPO-RT-PC (Sweden and Denmark),9 have shown the noninferiority of moderately hypofractionated radiotherapy compared with CFRT. Notably, most of these noninferiority studies primarily included patients with low- or intermediate-risk prostate cancer, except for the HYPO-RT-PC trial,9 which also included patients with intermediate- and high-risk prostate cancer.
These noninferiority studies, along with technological advances in radiotherapy, such as intensity-modulated radiotherapy (IMRT), volumetric modulated arc therapy (VMAT), and image-guided radiotherapy (IGRT), paved the path to ultrahypofractionated stereotactic body radiotherapy (SBRT) that is delivered in 5 fractions of ≥ 6 Gy. This high dose per fraction may have a radiobiologic advantage over conventional fractionation. The relatively low a/ß ratio of prostate cancer, estimated to be between 1 and 2, suggests that tumor cells may be particularly sensitive to the high doses per fraction delivered in SBRT.10-13 Compared with CFRT, SBRT-induced tumor cell death may also be mediated through different pathways; this pathway appears to be generated in a dose-dependent manner, particularly with doses > 8 Gy per fraction.14,15 Additionally, the higher a/ß ratio for the surrounding organs at risk, such as the bladder and rectum, theoretically allows for an improved therapeutic ratio window that maximizes tumor control while minimizing damage to healthy tissues.
A substantial body of evidence from prospective studies and meta-analyses supports the use of SBRT for localized prostate cancer. HYPO-RT-PC, a significant phase 3 noninferiority study, enrolled 1200 patients with intermediate (89%) and high-risk (11%) prostate cancer randomized between 2 arms, including CFRT to 78 Gy in 39 fractions and SBRT to 42.7 Gy in 7 fractions, treated 3 days weekly. After a median follow-up of 60 months, the estimated 5-year biochemical relapse-free survival rate was 84% in both groups.9 This trial was notable because it was the first randomized study to demonstrate that SBRT was noninferior to CFRT in intermediate- and high-risk prostate cancer patients. Another pivotal phase 3 trial, the PACE-B study, enrolled 874 patients to compare SBRT (36.25 Gy to the prostate gland, with a secondary dose of 40 Gy to the gross tumor volume where applicable, in 5 fractions) with CFRT (78 Gy in 39 fractions) and moderately hypofractionated radiotherapy (HFRT) (62 Gy in 20 fractions) in patients with low- or intermediate-risk prostate cancer. With a 74-month median follow-up, the study reported 5-year biochemical free rates of 94.6% for CFRT and 95.8% for SBRT, confirming the noninferiority of SBRT to CFRT.15
SBRT offers short, effective, and convenient treatment to many patients with localized prostate cancer. While previous guidelines were more restrictive, the March 2026 National Comprehensive Cancer Network (NCCN) guidelines now list SBRT as a preferred treatment modality for high-risk prostate cancer.16
Given the growing body of evidence supporting the efficacy and safety of SBRT, we implemented an SBRT program in 2014 at a tertiary care center for veterans. This retrospective study was undertaken to evaluate the early efficacy and toxicity of SBRT in patients with localized prostate cancer treated at our institution, including patients across all risk stratifications.
METHODS
We identified 242 patients diagnosed with prostate cancer who underwent SBRT treatment between November 2014 and October 2024 at Overland Park Veterans Affairs Radiation Oncology Clinic. For the final analysis, 46 patients with < 2 years of follow-up and 22 patients who died from causes other than prostate cancer were excluded, resulting in a cohort of 174 patients with ≥ 24-month follow-up.
Treatment
Patients eligible for staging underwent imaging according to NCCN guidelines, including computed tomography (CT) of the abdomen and pelvis, bone scintigraphy, or, in recent years, prostate-specific membrane antigen positron emission tomography, primarily used for unfavorable intermediate-risk (UIR) and high-risk (HR) cancers. Patients with a negative staging work-up for nodal or skeletal disease were included. Prior to planning the CT simulation, patients were given bowel preparation instructions, including a low-fiber and low-gas-producing diet, simethicone, and enemas, the night before and morning of the simulation. Patients were instructed to arrive with a comfortably full bladder, having not voided for 2 to 3 hours prior to the procedure. At Kansas City Veterans Affairs Medical Center (KCVAMC), SBRT treatment was generally restricted to patients with a baseline American Urological Association symptom score of 15 to 20 out of 35 and a prostate gland size < 80 mL to minimize the risk of acute urinary toxicity. We did not use intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast agents for planning CT simulation.
Patients were placed in a supine position, and a vacuum bag was used for immobilization. Following the CT simulation, the images were transferred to the Eclipse treatment planning system. The clinical target volume (CTV) encompassed the prostate and the proximal 1.0 cm of the seminal vesicles for Gleason score (GS) 1 to 2, and the entire seminal vesicle was included for GS 3 to 5, which is consistent with KCVAMC practice and established safety protocols. The planning target volume (PTV) was created by uniformly expanding the CTV by 5 to 7 mm, except for the posterior margin, which was limited to 3 to 5 mm. When elective nodal radiotherapy was planned for HR prostate cancer, the pelvic field for CT simulation started at the L-2 upper border, with the lower border extending to the lesser trochanter. The pelvic nodes were delineated per Radiation Therapy Oncology Group (RTOG) guidelines.17 The CTV nodes (CTVn), including common iliac, external and internal iliac nodes, obturator, and presacral nodes, were created by uniformly expanding the CTVn by 2 to 3 mm. Slice-by-slice corrections were made to avoid bowel overlap in these patients.
The use of androgen deprivation therapy (ADT) for a duration of 6 to 24 months was prescribed for patients with UIR or HR prostate cancer per NCCN guidelines.16 The prescribed dose to the PTV was 36.25 to 40 Gy (40 Gy was mostly used as a boost to the dominant lesion) in 5 fractions, with each fraction ranging from 7.25 to 8 Gy. For elective nodal radiotherapy in patients at HR, the prescribed dose was 25 Gy in 5 fractions. All patients were planned for VMAT, which aims to deliver ≥ 95% of the prescription dose to 95% of the PTV. Once the physician approved the treatment plan and physics quality assessment was completed, treatments commenced on an every-other-day schedule. Patients received the same bowel preparation instructions for each treatment as for the planning CT simulation. Daily treatment accuracy was confirmed via daily 3-dimensional cone-beam CT (CBCT) for IGRT. No fiducials or hydrogel rectal spacers were used.
Follow-up Schedule and Toxicity Assessment
Follow-up assessments were conducted 4 to 6 weeks after radiation therapy and then repeated every 6 months for 2 to 5 years, and annually thereafter. At each follow-up visit, patients were evaluated for genitourinary (GU) and gastrointestinal (GI) toxicity, according to RTOG toxicity criteria. Prostate-specific antigen (PSA) levels were monitored; in patients receiving ADT, testosterone levels were also checked.
Statistical Analysis
Biochemical failure was defined using the Phoenix definition (nadir PSA + 2 ng/mL). Differences between dose cohorts were assessed using the log-rank test for survival outcomes and X2 testing for categorical variables. GU and GI toxicities were summarized as cumulative incidences of RTOG grade ≥ II events. Statistical significance was set at P < .05.
RESULTS
One hundred seventy-four patients were included in the retrospective review. Patients had a median follow-up of 45 months (range, 24-111) (Figure). The median age at treatment was 74 years (range, 51-88), and the median pretreatment PSA level was 11.9 ng/mL (range, 0.6-69.5). Twenty-six patients (14.9%) had a GS 1, 77 (44.3%) had GS 2, 41 (23.6%) had GS 3, 18 (10.3%) had GS 4, and 12 (6.9%) had GS 5. Fifty-one patients (29.3%) received elective pelvic nodal radiotherapy, and 93 patients (53.4%) received ADT (Table 1).
At 24 months follow-up, 6 patients (3.4%) had biochemical failures. One patient died from metastatic prostate cancer, and 5 patients are living with biochemical failure (Table 2). The actuarial 5-year overall survival (OS) rate was 99.4%, and the 5-year disease-free survival (DFS) rate was 96.6%. We performed a subanalysis comparing outcomes of the 36.25 Gy vs 40 Gy SBRT cohorts. There was no statistically significant difference in DFS, OS, or the cumulative incidence of grade II/III toxicity between patients treated with 40 Gy vs 36.25 Gy. Outcomes stratified by NCCN risk groups (low, intermediate, high/very high) are detailed in Table 3. As expected, DFS was slightly lower in the high-risk group, but overall disease control remained high across all stratifications.
The cumulative incidence of RTOG grade II and higher GU toxicity was 28.2% (Table 4). This included 46 patients (26.4%) with grade II GU toxicity and 2 patients (1.2%) who developed grade III GU complications (1 requiring self-catheterization and another a suprapubic catheter for urinary retention). One patient (0.6%) treated with a 40 Gy dose regimen experienced a grade IV GU complication in the form of a rectovesical fistula necessitating surgical intervention.
The cumulative incidence of RTOG grade II or higher GI toxicity was 3.4%, and no grade III or IV gastrointestinal toxicities were observed during the follow-up period. Importantly, intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast were not routinely used in this cohort of patients.
The high rates of actuarial 5-year DFS and OS observed suggest a favorable initial response to the SBRT regimen employed at KCVAMC. However, given the potential for late recurrence in patients with prostate cancer, longer follow-up is essential to determine the durability of these outcomes. The observed GU toxicity rate of 28.2% for grade II and higher events warrants careful consideration and compares with other published data on SBRT for prostate cancer.15 The occurrence of a grade IV rectovesical fistula, although rare, is a notable adverse event that warrants discussion in the context of the treatment approach. The low incidence of grade II or higher GI toxicity is an encouraging finding, particularly given that hydrogel rectal spacers are not routinely used to minimize rectal exposure.
DISCUSSION
The primary objective of this retrospective study was to evaluate the outcomes of SBRT for patients with localized prostate cancer treated at KCVAMC and to compare these results with those reported in the literature. Our findings demonstrate promising intermediate-term efficacy, with an estimated 5-year DFS of 96.6% and OS of 99.4% at a median follow-up of 45 months. Furthermore, the observed toxicity profile appears acceptable, with a cumulative grade II and higher GU toxicity rate of 28.2% and a grade II or higher GI toxicity rate of 3.4%. Notably, these outcomes were achieved without the routine use of intraprostatic fiducials or hydrogel rectal spacers.
Two pivotal randomized phase 3 trials have established the noninferiority of ultrahypofractionated radiotherapy (UHRT) with SBRT over conventional fractionation. The HYPO-RT-PC trial compared SBRT (42.7 Gy in 7 fractions) with conventional fractionation (78 Gy in 39 fractions) in intermediate- and high-risk patients with prostate cancer and reported a 5-year biochemical relapse-free survival of 84% in both arms.9 The PACE-B trial, which included patients at low- and intermediate-risk, compared SBRT (36.25 Gy in 5 fractions) with conventional or moderate HFRT and reported a 5-year biochemical control rate of 95.8% in the SBRT arm and 94.6% in the control arm.15
A comprehensive review and meta-analysis of 7 phase 3 studies involving 6795 patients compared different radiotherapy regimens, namely, UHRT, HFRT, and CFRT, and reported that after 5 years, the DFS rates were 85.1% for CFRT, 86% for HFRT, and 85% for UHRT, with no significant difference in toxicity among the 3 different treatment approaches.18 This suggests that shorter, more intense radiotherapy schedules (UHRT and HFRT) may be as effective and safe as traditional, longer courses of radiation.
There are multiple published nonrandomized prospective trials in which thousands of patients with extreme hypofractionation have been treated with different doses, fractions, and techniques. While heterogeneity and limited long-term follow-up in the existing evidence are acknowledged, these data suggest that prostate SBRT provides appropriate biochemical control with few high-grade toxicities, supporting its ongoing global use and justifying further prospective investigations. Comparative data are shown in Table 5. Several ongoing studies are evaluating noninferiority, superiority, and cost-effectiveness using different methodologies (Table 6).9,15,19-24
This study’s efficacy outcomes, particularly the high DFS rate, are consistent with the findings from these landmark trials, suggesting that the SBRT regimen used at KCVAMC is effective in achieving early disease control despite 17.2% of patients having high-risk disease. The GU toxicity observed in this study, with a 28.2% rate of grade II or higher events, is also comparable with the 26.9% reported in the 5-fraction SBRT arm of the PACE-B trial, which had a longer median follow-up of 74 months.15 It is important to note that a portion of these grade II events occurred in patients who were already on a blockers for pre-existing lower urinary tract symptoms before starting radiotherapy, which may inflate the observed cumulative acute toxicity score.
A critical comparison is how SBRT toxicity aligns with moderate hypofractionation (eg, 60 Gy in 20 fractions or 70 Gy in 28 fractions as reported by others).4,6 Our observed grade III and higher GU toxicity rate (1.7%) and grade III and higher GI toxicity rate (0%) are highly favorable when compared with historical moderate hypofractionation data, which typically report grade III GU toxicity in the range of 2% to 3% and grade III GI toxicity around 1% to 2%. This suggests that despite the higher dose per fraction, SBRT does not necessarily lead to increased severe acute toxicity, potentially offering a superior therapeutic ratio for GI and GU sparing.
However, the occurrence of a grade IV rectovesical fistula in 1 patient (0.6%)—who received the 40 Gy dose—was a serious complication that warrants careful consideration. This rare, but severe, complication in the higher dose cohort underscores the potential for increased organ-at-risk toxicity, particularly in the absence of a hydrogel rectal spacer, which is designed to mitigate high-dose rectal exposure. While the overall rate of significant GU toxicity remains low, this event highlights the potential risks associated with SBRT. Hydrogel rectal spacers are designed to increase the distance between the prostate and the rectum, which can reduce the rectal radiation dose and potentially mitigate the risk of such fistulas. The low rate of grade II or worse GI toxicity (3.4%) in our study is noteworthy, especially considering that hydrogel spacers were not routinely used. This finding aligns with the 2.5% GI toxicity rate reported in the SBRT arm of the PACE-B trial, suggesting that careful treatment planning and delivery techniques, such as VMAT-IMRT and daily CBCT for IGRT, may contribute to minimizing GI toxicity even without the use of rectal spacers.15 The exclusive use of 3-dimensional CBCT for IGRT in our study, without the use of fiducial markers, suggests that accurate target localization can be achieved with this approach, contributing to the observed efficacy and reduced toxicity.
Strengths and Limitations
This study’s retrospective, single-center design may have introduced selection bias. The median follow-up of 45 months, while substantial, is still relatively short for assessing very late toxicities and long-term oncologic outcomes in prostate cancer, which is known for late recurrences. Additionally, the lack of a direct comparison group within KCVAMC limits the ability to definitively attribute the observed outcomes solely to SBRT treatment. However, the strengths of this study include the inclusion of a consecutive series of veteran patients with localized prostate cancer across all risk categories, providing a real-world perspective on SBRT outcomes in a diverse patient population. Furthermore, the detailed assessment of efficacy and toxicity via standardized RTOG criteria enhances the comparability of our findings with those of other published prospective studies, despite the retrospective nature of the data.
CONCLUSIONS
This single-institution retrospective analysis revealed that short-term SBRT (36.25 to 40 Gy in 5 fractions), with a minimum follow-up of 24 months and a median follow-up of 45 months, for localized prostate cancer, including patients at HR, is associated with promising early efficacy and acceptable toxicity, even in the absence of routine fiducial or hydrogel spacer use. The favorable actuarial 5-year DFS and OS rates, coupled with a manageable toxicity profile, suggest that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer. However, a longer follow-up is necessary to confirm these findings and fully characterize the long-term efficacy and toxicity of this SBRT regimen. Nevertheless, the results contribute to the growing body of evidence suggesting that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer.
Prostate cancer is the most common cancer in US males, with an estimated 313,780 new cases and 35,770 deaths in 2025.1 Several treatment options are available for localized prostate cancer that have similar outcomes, including active surveillance for low-risk cancers, surgery, or radiotherapy.2,3 Conventional fractionation radiotherapy (CFRT) with 40 to 45 fractions over 8 to 9 weeks has been used for decades. Over the past 2 decades, moderate hypofractionation schedules with 2.4 to 3.4 Gy per fraction over 20 to 28 fractions have become standard, as many noninferiority randomized clinical trials (RCTs) such as CHHiP (UK),4 PROFIT (Canada and Europe),5 NRG Oncology RTOG 0415 (US),6 HYPRO (Netherlands),7,8 and HYPO-RT-PC (Sweden and Denmark),9 have shown the noninferiority of moderately hypofractionated radiotherapy compared with CFRT. Notably, most of these noninferiority studies primarily included patients with low- or intermediate-risk prostate cancer, except for the HYPO-RT-PC trial,9 which also included patients with intermediate- and high-risk prostate cancer.
These noninferiority studies, along with technological advances in radiotherapy, such as intensity-modulated radiotherapy (IMRT), volumetric modulated arc therapy (VMAT), and image-guided radiotherapy (IGRT), paved the path to ultrahypofractionated stereotactic body radiotherapy (SBRT) that is delivered in 5 fractions of ≥ 6 Gy. This high dose per fraction may have a radiobiologic advantage over conventional fractionation. The relatively low a/ß ratio of prostate cancer, estimated to be between 1 and 2, suggests that tumor cells may be particularly sensitive to the high doses per fraction delivered in SBRT.10-13 Compared with CFRT, SBRT-induced tumor cell death may also be mediated through different pathways; this pathway appears to be generated in a dose-dependent manner, particularly with doses > 8 Gy per fraction.14,15 Additionally, the higher a/ß ratio for the surrounding organs at risk, such as the bladder and rectum, theoretically allows for an improved therapeutic ratio window that maximizes tumor control while minimizing damage to healthy tissues.
A substantial body of evidence from prospective studies and meta-analyses supports the use of SBRT for localized prostate cancer. HYPO-RT-PC, a significant phase 3 noninferiority study, enrolled 1200 patients with intermediate (89%) and high-risk (11%) prostate cancer randomized between 2 arms, including CFRT to 78 Gy in 39 fractions and SBRT to 42.7 Gy in 7 fractions, treated 3 days weekly. After a median follow-up of 60 months, the estimated 5-year biochemical relapse-free survival rate was 84% in both groups.9 This trial was notable because it was the first randomized study to demonstrate that SBRT was noninferior to CFRT in intermediate- and high-risk prostate cancer patients. Another pivotal phase 3 trial, the PACE-B study, enrolled 874 patients to compare SBRT (36.25 Gy to the prostate gland, with a secondary dose of 40 Gy to the gross tumor volume where applicable, in 5 fractions) with CFRT (78 Gy in 39 fractions) and moderately hypofractionated radiotherapy (HFRT) (62 Gy in 20 fractions) in patients with low- or intermediate-risk prostate cancer. With a 74-month median follow-up, the study reported 5-year biochemical free rates of 94.6% for CFRT and 95.8% for SBRT, confirming the noninferiority of SBRT to CFRT.15
SBRT offers short, effective, and convenient treatment to many patients with localized prostate cancer. While previous guidelines were more restrictive, the March 2026 National Comprehensive Cancer Network (NCCN) guidelines now list SBRT as a preferred treatment modality for high-risk prostate cancer.16
Given the growing body of evidence supporting the efficacy and safety of SBRT, we implemented an SBRT program in 2014 at a tertiary care center for veterans. This retrospective study was undertaken to evaluate the early efficacy and toxicity of SBRT in patients with localized prostate cancer treated at our institution, including patients across all risk stratifications.
METHODS
We identified 242 patients diagnosed with prostate cancer who underwent SBRT treatment between November 2014 and October 2024 at Overland Park Veterans Affairs Radiation Oncology Clinic. For the final analysis, 46 patients with < 2 years of follow-up and 22 patients who died from causes other than prostate cancer were excluded, resulting in a cohort of 174 patients with ≥ 24-month follow-up.
Treatment
Patients eligible for staging underwent imaging according to NCCN guidelines, including computed tomography (CT) of the abdomen and pelvis, bone scintigraphy, or, in recent years, prostate-specific membrane antigen positron emission tomography, primarily used for unfavorable intermediate-risk (UIR) and high-risk (HR) cancers. Patients with a negative staging work-up for nodal or skeletal disease were included. Prior to planning the CT simulation, patients were given bowel preparation instructions, including a low-fiber and low-gas-producing diet, simethicone, and enemas, the night before and morning of the simulation. Patients were instructed to arrive with a comfortably full bladder, having not voided for 2 to 3 hours prior to the procedure. At Kansas City Veterans Affairs Medical Center (KCVAMC), SBRT treatment was generally restricted to patients with a baseline American Urological Association symptom score of 15 to 20 out of 35 and a prostate gland size < 80 mL to minimize the risk of acute urinary toxicity. We did not use intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast agents for planning CT simulation.
Patients were placed in a supine position, and a vacuum bag was used for immobilization. Following the CT simulation, the images were transferred to the Eclipse treatment planning system. The clinical target volume (CTV) encompassed the prostate and the proximal 1.0 cm of the seminal vesicles for Gleason score (GS) 1 to 2, and the entire seminal vesicle was included for GS 3 to 5, which is consistent with KCVAMC practice and established safety protocols. The planning target volume (PTV) was created by uniformly expanding the CTV by 5 to 7 mm, except for the posterior margin, which was limited to 3 to 5 mm. When elective nodal radiotherapy was planned for HR prostate cancer, the pelvic field for CT simulation started at the L-2 upper border, with the lower border extending to the lesser trochanter. The pelvic nodes were delineated per Radiation Therapy Oncology Group (RTOG) guidelines.17 The CTV nodes (CTVn), including common iliac, external and internal iliac nodes, obturator, and presacral nodes, were created by uniformly expanding the CTVn by 2 to 3 mm. Slice-by-slice corrections were made to avoid bowel overlap in these patients.
The use of androgen deprivation therapy (ADT) for a duration of 6 to 24 months was prescribed for patients with UIR or HR prostate cancer per NCCN guidelines.16 The prescribed dose to the PTV was 36.25 to 40 Gy (40 Gy was mostly used as a boost to the dominant lesion) in 5 fractions, with each fraction ranging from 7.25 to 8 Gy. For elective nodal radiotherapy in patients at HR, the prescribed dose was 25 Gy in 5 fractions. All patients were planned for VMAT, which aims to deliver ≥ 95% of the prescription dose to 95% of the PTV. Once the physician approved the treatment plan and physics quality assessment was completed, treatments commenced on an every-other-day schedule. Patients received the same bowel preparation instructions for each treatment as for the planning CT simulation. Daily treatment accuracy was confirmed via daily 3-dimensional cone-beam CT (CBCT) for IGRT. No fiducials or hydrogel rectal spacers were used.
Follow-up Schedule and Toxicity Assessment
Follow-up assessments were conducted 4 to 6 weeks after radiation therapy and then repeated every 6 months for 2 to 5 years, and annually thereafter. At each follow-up visit, patients were evaluated for genitourinary (GU) and gastrointestinal (GI) toxicity, according to RTOG toxicity criteria. Prostate-specific antigen (PSA) levels were monitored; in patients receiving ADT, testosterone levels were also checked.
Statistical Analysis
Biochemical failure was defined using the Phoenix definition (nadir PSA + 2 ng/mL). Differences between dose cohorts were assessed using the log-rank test for survival outcomes and X2 testing for categorical variables. GU and GI toxicities were summarized as cumulative incidences of RTOG grade ≥ II events. Statistical significance was set at P < .05.
RESULTS
One hundred seventy-four patients were included in the retrospective review. Patients had a median follow-up of 45 months (range, 24-111) (Figure). The median age at treatment was 74 years (range, 51-88), and the median pretreatment PSA level was 11.9 ng/mL (range, 0.6-69.5). Twenty-six patients (14.9%) had a GS 1, 77 (44.3%) had GS 2, 41 (23.6%) had GS 3, 18 (10.3%) had GS 4, and 12 (6.9%) had GS 5. Fifty-one patients (29.3%) received elective pelvic nodal radiotherapy, and 93 patients (53.4%) received ADT (Table 1).
At 24 months follow-up, 6 patients (3.4%) had biochemical failures. One patient died from metastatic prostate cancer, and 5 patients are living with biochemical failure (Table 2). The actuarial 5-year overall survival (OS) rate was 99.4%, and the 5-year disease-free survival (DFS) rate was 96.6%. We performed a subanalysis comparing outcomes of the 36.25 Gy vs 40 Gy SBRT cohorts. There was no statistically significant difference in DFS, OS, or the cumulative incidence of grade II/III toxicity between patients treated with 40 Gy vs 36.25 Gy. Outcomes stratified by NCCN risk groups (low, intermediate, high/very high) are detailed in Table 3. As expected, DFS was slightly lower in the high-risk group, but overall disease control remained high across all stratifications.
The cumulative incidence of RTOG grade II and higher GU toxicity was 28.2% (Table 4). This included 46 patients (26.4%) with grade II GU toxicity and 2 patients (1.2%) who developed grade III GU complications (1 requiring self-catheterization and another a suprapubic catheter for urinary retention). One patient (0.6%) treated with a 40 Gy dose regimen experienced a grade IV GU complication in the form of a rectovesical fistula necessitating surgical intervention.
The cumulative incidence of RTOG grade II or higher GI toxicity was 3.4%, and no grade III or IV gastrointestinal toxicities were observed during the follow-up period. Importantly, intraprostatic fiducials, hydrogel rectal spacers, or intravenous contrast were not routinely used in this cohort of patients.
The high rates of actuarial 5-year DFS and OS observed suggest a favorable initial response to the SBRT regimen employed at KCVAMC. However, given the potential for late recurrence in patients with prostate cancer, longer follow-up is essential to determine the durability of these outcomes. The observed GU toxicity rate of 28.2% for grade II and higher events warrants careful consideration and compares with other published data on SBRT for prostate cancer.15 The occurrence of a grade IV rectovesical fistula, although rare, is a notable adverse event that warrants discussion in the context of the treatment approach. The low incidence of grade II or higher GI toxicity is an encouraging finding, particularly given that hydrogel rectal spacers are not routinely used to minimize rectal exposure.
DISCUSSION
The primary objective of this retrospective study was to evaluate the outcomes of SBRT for patients with localized prostate cancer treated at KCVAMC and to compare these results with those reported in the literature. Our findings demonstrate promising intermediate-term efficacy, with an estimated 5-year DFS of 96.6% and OS of 99.4% at a median follow-up of 45 months. Furthermore, the observed toxicity profile appears acceptable, with a cumulative grade II and higher GU toxicity rate of 28.2% and a grade II or higher GI toxicity rate of 3.4%. Notably, these outcomes were achieved without the routine use of intraprostatic fiducials or hydrogel rectal spacers.
Two pivotal randomized phase 3 trials have established the noninferiority of ultrahypofractionated radiotherapy (UHRT) with SBRT over conventional fractionation. The HYPO-RT-PC trial compared SBRT (42.7 Gy in 7 fractions) with conventional fractionation (78 Gy in 39 fractions) in intermediate- and high-risk patients with prostate cancer and reported a 5-year biochemical relapse-free survival of 84% in both arms.9 The PACE-B trial, which included patients at low- and intermediate-risk, compared SBRT (36.25 Gy in 5 fractions) with conventional or moderate HFRT and reported a 5-year biochemical control rate of 95.8% in the SBRT arm and 94.6% in the control arm.15
A comprehensive review and meta-analysis of 7 phase 3 studies involving 6795 patients compared different radiotherapy regimens, namely, UHRT, HFRT, and CFRT, and reported that after 5 years, the DFS rates were 85.1% for CFRT, 86% for HFRT, and 85% for UHRT, with no significant difference in toxicity among the 3 different treatment approaches.18 This suggests that shorter, more intense radiotherapy schedules (UHRT and HFRT) may be as effective and safe as traditional, longer courses of radiation.
There are multiple published nonrandomized prospective trials in which thousands of patients with extreme hypofractionation have been treated with different doses, fractions, and techniques. While heterogeneity and limited long-term follow-up in the existing evidence are acknowledged, these data suggest that prostate SBRT provides appropriate biochemical control with few high-grade toxicities, supporting its ongoing global use and justifying further prospective investigations. Comparative data are shown in Table 5. Several ongoing studies are evaluating noninferiority, superiority, and cost-effectiveness using different methodologies (Table 6).9,15,19-24
This study’s efficacy outcomes, particularly the high DFS rate, are consistent with the findings from these landmark trials, suggesting that the SBRT regimen used at KCVAMC is effective in achieving early disease control despite 17.2% of patients having high-risk disease. The GU toxicity observed in this study, with a 28.2% rate of grade II or higher events, is also comparable with the 26.9% reported in the 5-fraction SBRT arm of the PACE-B trial, which had a longer median follow-up of 74 months.15 It is important to note that a portion of these grade II events occurred in patients who were already on a blockers for pre-existing lower urinary tract symptoms before starting radiotherapy, which may inflate the observed cumulative acute toxicity score.
A critical comparison is how SBRT toxicity aligns with moderate hypofractionation (eg, 60 Gy in 20 fractions or 70 Gy in 28 fractions as reported by others).4,6 Our observed grade III and higher GU toxicity rate (1.7%) and grade III and higher GI toxicity rate (0%) are highly favorable when compared with historical moderate hypofractionation data, which typically report grade III GU toxicity in the range of 2% to 3% and grade III GI toxicity around 1% to 2%. This suggests that despite the higher dose per fraction, SBRT does not necessarily lead to increased severe acute toxicity, potentially offering a superior therapeutic ratio for GI and GU sparing.
However, the occurrence of a grade IV rectovesical fistula in 1 patient (0.6%)—who received the 40 Gy dose—was a serious complication that warrants careful consideration. This rare, but severe, complication in the higher dose cohort underscores the potential for increased organ-at-risk toxicity, particularly in the absence of a hydrogel rectal spacer, which is designed to mitigate high-dose rectal exposure. While the overall rate of significant GU toxicity remains low, this event highlights the potential risks associated with SBRT. Hydrogel rectal spacers are designed to increase the distance between the prostate and the rectum, which can reduce the rectal radiation dose and potentially mitigate the risk of such fistulas. The low rate of grade II or worse GI toxicity (3.4%) in our study is noteworthy, especially considering that hydrogel spacers were not routinely used. This finding aligns with the 2.5% GI toxicity rate reported in the SBRT arm of the PACE-B trial, suggesting that careful treatment planning and delivery techniques, such as VMAT-IMRT and daily CBCT for IGRT, may contribute to minimizing GI toxicity even without the use of rectal spacers.15 The exclusive use of 3-dimensional CBCT for IGRT in our study, without the use of fiducial markers, suggests that accurate target localization can be achieved with this approach, contributing to the observed efficacy and reduced toxicity.
Strengths and Limitations
This study’s retrospective, single-center design may have introduced selection bias. The median follow-up of 45 months, while substantial, is still relatively short for assessing very late toxicities and long-term oncologic outcomes in prostate cancer, which is known for late recurrences. Additionally, the lack of a direct comparison group within KCVAMC limits the ability to definitively attribute the observed outcomes solely to SBRT treatment. However, the strengths of this study include the inclusion of a consecutive series of veteran patients with localized prostate cancer across all risk categories, providing a real-world perspective on SBRT outcomes in a diverse patient population. Furthermore, the detailed assessment of efficacy and toxicity via standardized RTOG criteria enhances the comparability of our findings with those of other published prospective studies, despite the retrospective nature of the data.
CONCLUSIONS
This single-institution retrospective analysis revealed that short-term SBRT (36.25 to 40 Gy in 5 fractions), with a minimum follow-up of 24 months and a median follow-up of 45 months, for localized prostate cancer, including patients at HR, is associated with promising early efficacy and acceptable toxicity, even in the absence of routine fiducial or hydrogel spacer use. The favorable actuarial 5-year DFS and OS rates, coupled with a manageable toxicity profile, suggest that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer. However, a longer follow-up is necessary to confirm these findings and fully characterize the long-term efficacy and toxicity of this SBRT regimen. Nevertheless, the results contribute to the growing body of evidence suggesting that SBRT is a safe and convenient treatment option for many patients with localized prostate cancer.
- Siegel RL, Kratzer TB, Giaquinto AN, et al. Cancer statistics, 2025. CA Cancer J Clin. 2025;75:10-45. doi:10.3322/caac.21871
- Donovan JL, Hamdy FC, Lane JA, et al. Patient-reported outcomes after monitoring, surgery, or radiotherapy for prostate cancer. N Engl J Med. 2016;375:1425-1437. doi:10.1056/NEJMoa1606221
- Hamdy FC, Donovan JL, Lane JA, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med. 2016;375:1415-1424. doi:10.1056/NEJMoa1606220
- Dearnaley D, Syndikus I, Mossop H, et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol. 2016;17:1047-1060. doi:10.1016/S1470-2045(16)30102-4
- Catton CN, Lukka H, Gu CS, et al. Randomized trial of a hypofractionated radiation regimen for the treatment of localized prostate cancer. J Clin Oncol. 2017;35:1884-1890. doi:10.1200/JCO.2016.71.7397
- Lee WR, Dignam JJ, Amin MB, et al. Long-term analysis of NRG Oncology RTOG 0415: a randomized phase III noninferiority study comparing two fractionation schedules in patients with low-risk prostate cancer. J Clin Oncol. 2024;42:2377-2381. doi:10.1200/JCO.23.02445
- de Vries KC, Wortel RC, Oomen-de Hoop E, et al. Hypofractionated versus conventionally fractionated radiation therapy for patients with intermediate- or high-risk, localized, prostate cancer: 7-year outcomes from the randomized, multicenter, open-label, phase 3 HYPRO trial. Int J Radiat Oncol Biol Phys. 2020;106:108-115. doi:10.1016/j.ijrobp.2019.09.007
- Incrocci L, Wortel RC, Alemayehu WG, et al. Hypofractionated versus conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016;17:1061-1069. doi:10.1016/S1470-2045(16)30070-5
- Widmark A, Gunnlaugsson A, Beckman L, et al. Ultra-hypofractionated versus conventionally fractionated radiotherapy for prostate cancer: 5-year outcomes of the HYPO-RT-PC randomised, non-inferiority, phase 3 trial. Lancet. 2019;394:385-395. doi:10.1016/S0140-6736(19)31131-6
- Brenner DJ, Hall EJ. Fractionation and protraction for radiotherapy of prostate carcinoma. Int J Radiat Oncol Biol Phys. 1999;43:1095-101. doi:10.1016/s0360-3016(98)00438-6
- Dasu A. Is the alpha/beta value for prostate tumours low enough to be safely used in clinical trials? Clin Oncol (R Coll Radiol). 2007;19:289-301. doi:10.1016/j.clon.2007.02.007
- Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155-1159. doi:10.1126/science.1082504
- Gulliford S, Hall E, Dearnaley D. Hypofractionation trials and radiobiology of prostate cancer. Oncoscience. 2017;4:27-28. doi:10.18632/oncoscience.347
- Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89-91. doi:10.1016/j.ccr.2005.07.014
- van As N, Griffin C, Tree A, et al. Phase 3 Trial of stereotactic body radiotherapy in localized prostate cancer. N Engl J Med. Oct 17 2024;391:1413-1425. doi:10.1056/NEJMoa2403365
- National Comprehensive Cancer Network. NCCN Guidelines Version 5. 2026 Prostate Cancer. Accessed March 24, 2026. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- Lawton CA, Michalski J, El-Naqa I, et al. RTOG GU radiation oncology specialists reach consensus on pelvic lymph node volumes for high-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2009;74:383-387. doi:10.1016/j.ijrobp.2008.08.002
- Lehrer EJ, Kishan AU, Yu JB, et al. Ultrahypofractionated versus hypofractionated and conventionally fractionated radiation therapy for localized prostate cancer: a systematic review and meta-analysis of phase III randomized trials. Radiother Oncol. 2020;148:235-242. doi:10.1016/j.radonc.2020.04.037
- De Cooman B, Debacker T, Adams T, et al. Stereotactic body radiotherapy (SBRT) as a treatment for localized prostate cancer: a retrospective analysis. Radiat Oncol. 2025;20:25. doi:10.1186/s13014-025-02598-8
- Fuller DB, Falchook AD, Crabtree T, et al. Phase 2 multicenter trial of heterogeneous-dosing stereotactic body radiotherapy for low- and intermediate-risk prostate cancer: 5-year outcomes. Eur Urol Oncol. 2018;1:540-547. doi:10.1016/j.euo.2018.06.013
- Jackson WC, Silva J, Hartman HE, et al. Stereotactic body radiation therapy for localized prostate cancer: a systematic review and meta-analysis of over 6,000 patients treated on prospective studies. Int J Radiat Oncol Biol Phys. 2019;104:778-789. doi:10.1016/j.ijrobp.2019.03.051
- Meier RM, Bloch DA, Cotrutz C, et al. Multicenter trial of stereotactic body radiation therapy for low- and intermediate-risk prostate cancer: survival and toxicity endpoints. nt J Radiat Oncol Biol Phys. 2018;102:296-303. doi:10.1016/j.ijrobp.2018.05.040
- Quon HC, Ong A, Cheung P, et al. Once-weekly versus every-other-day stereotactic body radiotherapy in patients with prostate cancer (PATRIOT): a phase 2 randomized trial. Radiother Oncol. 2018;127:206-212. doi:10.1016/j.radonc.2018.02.029
- Zelefsky MJ, Kollmeier M, McBride S, et al. Five-year outcomes of a phase 1 dose-escalation study using stereotactic body radiosurgery for patients with low-risk and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2019;104:42-49. doi:10.1016/j.ijrobp.2018.12.045
- Siegel RL, Kratzer TB, Giaquinto AN, et al. Cancer statistics, 2025. CA Cancer J Clin. 2025;75:10-45. doi:10.3322/caac.21871
- Donovan JL, Hamdy FC, Lane JA, et al. Patient-reported outcomes after monitoring, surgery, or radiotherapy for prostate cancer. N Engl J Med. 2016;375:1425-1437. doi:10.1056/NEJMoa1606221
- Hamdy FC, Donovan JL, Lane JA, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med. 2016;375:1415-1424. doi:10.1056/NEJMoa1606220
- Dearnaley D, Syndikus I, Mossop H, et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol. 2016;17:1047-1060. doi:10.1016/S1470-2045(16)30102-4
- Catton CN, Lukka H, Gu CS, et al. Randomized trial of a hypofractionated radiation regimen for the treatment of localized prostate cancer. J Clin Oncol. 2017;35:1884-1890. doi:10.1200/JCO.2016.71.7397
- Lee WR, Dignam JJ, Amin MB, et al. Long-term analysis of NRG Oncology RTOG 0415: a randomized phase III noninferiority study comparing two fractionation schedules in patients with low-risk prostate cancer. J Clin Oncol. 2024;42:2377-2381. doi:10.1200/JCO.23.02445
- de Vries KC, Wortel RC, Oomen-de Hoop E, et al. Hypofractionated versus conventionally fractionated radiation therapy for patients with intermediate- or high-risk, localized, prostate cancer: 7-year outcomes from the randomized, multicenter, open-label, phase 3 HYPRO trial. Int J Radiat Oncol Biol Phys. 2020;106:108-115. doi:10.1016/j.ijrobp.2019.09.007
- Incrocci L, Wortel RC, Alemayehu WG, et al. Hypofractionated versus conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016;17:1061-1069. doi:10.1016/S1470-2045(16)30070-5
- Widmark A, Gunnlaugsson A, Beckman L, et al. Ultra-hypofractionated versus conventionally fractionated radiotherapy for prostate cancer: 5-year outcomes of the HYPO-RT-PC randomised, non-inferiority, phase 3 trial. Lancet. 2019;394:385-395. doi:10.1016/S0140-6736(19)31131-6
- Brenner DJ, Hall EJ. Fractionation and protraction for radiotherapy of prostate carcinoma. Int J Radiat Oncol Biol Phys. 1999;43:1095-101. doi:10.1016/s0360-3016(98)00438-6
- Dasu A. Is the alpha/beta value for prostate tumours low enough to be safely used in clinical trials? Clin Oncol (R Coll Radiol). 2007;19:289-301. doi:10.1016/j.clon.2007.02.007
- Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155-1159. doi:10.1126/science.1082504
- Gulliford S, Hall E, Dearnaley D. Hypofractionation trials and radiobiology of prostate cancer. Oncoscience. 2017;4:27-28. doi:10.18632/oncoscience.347
- Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89-91. doi:10.1016/j.ccr.2005.07.014
- van As N, Griffin C, Tree A, et al. Phase 3 Trial of stereotactic body radiotherapy in localized prostate cancer. N Engl J Med. Oct 17 2024;391:1413-1425. doi:10.1056/NEJMoa2403365
- National Comprehensive Cancer Network. NCCN Guidelines Version 5. 2026 Prostate Cancer. Accessed March 24, 2026. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- Lawton CA, Michalski J, El-Naqa I, et al. RTOG GU radiation oncology specialists reach consensus on pelvic lymph node volumes for high-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2009;74:383-387. doi:10.1016/j.ijrobp.2008.08.002
- Lehrer EJ, Kishan AU, Yu JB, et al. Ultrahypofractionated versus hypofractionated and conventionally fractionated radiation therapy for localized prostate cancer: a systematic review and meta-analysis of phase III randomized trials. Radiother Oncol. 2020;148:235-242. doi:10.1016/j.radonc.2020.04.037
- De Cooman B, Debacker T, Adams T, et al. Stereotactic body radiotherapy (SBRT) as a treatment for localized prostate cancer: a retrospective analysis. Radiat Oncol. 2025;20:25. doi:10.1186/s13014-025-02598-8
- Fuller DB, Falchook AD, Crabtree T, et al. Phase 2 multicenter trial of heterogeneous-dosing stereotactic body radiotherapy for low- and intermediate-risk prostate cancer: 5-year outcomes. Eur Urol Oncol. 2018;1:540-547. doi:10.1016/j.euo.2018.06.013
- Jackson WC, Silva J, Hartman HE, et al. Stereotactic body radiation therapy for localized prostate cancer: a systematic review and meta-analysis of over 6,000 patients treated on prospective studies. Int J Radiat Oncol Biol Phys. 2019;104:778-789. doi:10.1016/j.ijrobp.2019.03.051
- Meier RM, Bloch DA, Cotrutz C, et al. Multicenter trial of stereotactic body radiation therapy for low- and intermediate-risk prostate cancer: survival and toxicity endpoints. nt J Radiat Oncol Biol Phys. 2018;102:296-303. doi:10.1016/j.ijrobp.2018.05.040
- Quon HC, Ong A, Cheung P, et al. Once-weekly versus every-other-day stereotactic body radiotherapy in patients with prostate cancer (PATRIOT): a phase 2 randomized trial. Radiother Oncol. 2018;127:206-212. doi:10.1016/j.radonc.2018.02.029
- Zelefsky MJ, Kollmeier M, McBride S, et al. Five-year outcomes of a phase 1 dose-escalation study using stereotactic body radiosurgery for patients with low-risk and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2019;104:42-49. doi:10.1016/j.ijrobp.2018.12.045
Early Outcomes of Stereotactic Body Radiotherapy for Localized Prostate Cancer: A Retrospective Analysis
Early Outcomes of Stereotactic Body Radiotherapy for Localized Prostate Cancer: A Retrospective Analysis
Capturing Pathology Workload Associated With Precision Oncology
Capturing Pathology Workload Associated With Precision Oncology
Precision oncology (PO) is cancer treatment individualized to the special characteristics of a patient’s tumor. It has become standard care for most patients with advanced cancer. Advances in molecular cell biology and immunology have identified numerous targets and many therapies have been developed as a result. Molecular testing and targeted therapy are typically covered by insurance, even when inflation-adjusted price growth is considered.1 Barriers remain, however, and pathologists are uniquely qualified to address some of the challenges.2
Most US laboratories do not perform molecular diagnostic tests for PO, particularly comprehensive evaluation of multiple targets by next-generation sequencing, or other techniques. Instead, these tests are sent to reference laboratories. The workload associated with referral testing is an obstacle to increased use of such tests. Despite guideline recommendations, a minority of indicated tests are performed.3 This is true even when testing costs are covered by clinical trials or grants, such as those in the Veterans Health Administration (VHA).
The main characteristic of successful PO programs is a multidisciplinary commitment, including pathology involvement in molecular tumor boards and assistance with test selection, tissue collection, and result interpretation.2 This, however, adds to the workload for the pathology department, an underappreciated phenomenon in the context of pathology workforce shortages.4
Workforce shortages impact all occupations in the laboratory setting. Though the shortage of medical technologists in clinical pathology laboratories has repeatedly been identified as critical at the VHA as well as in the private sector, the same cannot be said for staff shortages in anatomic pathology laboratories. Thus, the hospital laboratory divisions are concerned with biopsy or resection tissue specimens as opposed to the bodily fluid specimens that predominate in clinical laboratories.5 The lack of accurate data on histopathology technicians and technologists has precluded the degree of recognition seen for medical technologists. In labor statistics, these occupations are often obscured by inclusion with other jobs in broad categories such as medical and clinical laboratory technologists and technicians.6 Vacancy—the principal metric used to assess medical laboratory workforce shortage—fails to account for positions that are eventually eliminated after remaining vacant for prolonged periods, positions not replaced as a result of ambitious efficiency measures, or positions that were never created due to insufficient funding, reasons for administrative disapproval, or coverage by laboratory professionals working extra shifts or second jobs.7
Increased demand for pathologists is suggested by a 42% increase in workload per pathologist over the last decade, while a shortage is suggested by decreases in absolute and population-adjusted numbers of pathologists.8,9 An influx of pathologists is not an expected remedy due to the global decline in medical graduates pursuing careers in the field.8
Approximations for required labor and potential revenue generation are necessary to justify creation of pathology positions. This work mostly has gone uncaptured due to the limitations of Current Procedural Terminology (CPT) codes. Few laboratories have consistently used the 88363, 88325, and G0452 CPT codes. The pathology clinical consultation CPT codes (80503-80506) released in 2022 enhance acquisition of this work. The new codes replace 80500 and 80502 and allow for more precise identification of any work requiring medical judgment that a pathologist does at the request of another qualified health care professional (HCP) or as required by federal or state regulation.
The codes can be used to bill for associated time spent reviewing health records, communicating with other HCPs, placing orders, and documentation. An HCP can bill according to level of medical decision-making (MDM) or time spent by the consulting pathologist. Code 80503 can be billed for 5 to 20 minutes of a pathologist's time, 80504 for 21 to 40 minutes, 80505 for 41 to 60 minutes, and 80506 for each additional 30 minutes after the first hour. Levels of MDM (low, moderate, and high) are defined as for other evaluation and management services. A consultation report must be generated and contain documentation of the consultation request, pathologist interpretation, and justification for the level of service associated with the chosen code. Relative value units (RVUs) and reimbursement associated with each as well as other consultation-related codes are available in Table 1.
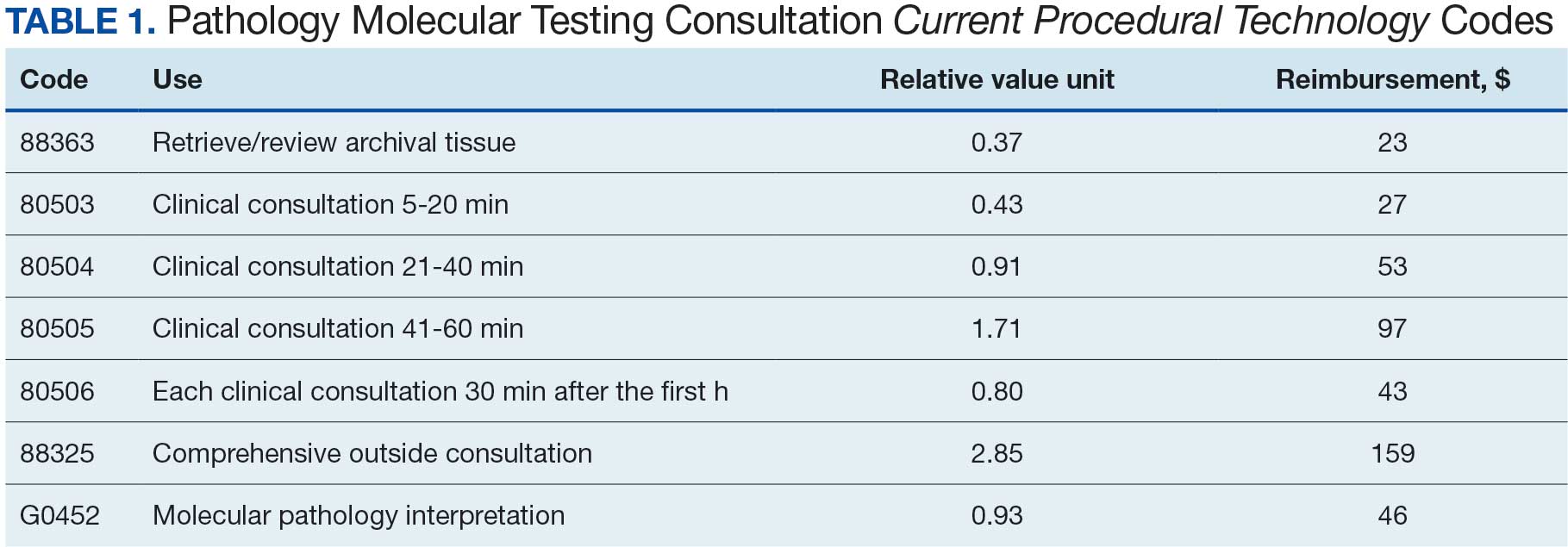
This article outlines how the pathology time investment associated with anatomic pathology molecular testing at the Kansas City Veterans Affairs Medical Center (KCVAMC) can be captured using the consultation process and new CPT codes. Staff included 4 pathologists, 3 histotechnologists, 1 histology supervisor, 1 grossing room technician, and 1 cytotechnologist, 1 cytology technician.
METHODS
The AP molecular testing consultation process at the KCVAMC was mapped, with the average time measured for each step (Figure). AP records for 2021 were reviewed to determine the number of AP molecular send out tests. Cumulative time investment was calculated in hours and a theoretical number of RVUs was calculated using the new pathology clinical consultation CPT codes (80503-80506). This theoretical number of RVUs was compared with the total AP RVUs generated in 2021 to determine a potential increase in RVUs with use of the new CPT codes to capture pathology work associated with AP molecular testing consultations.
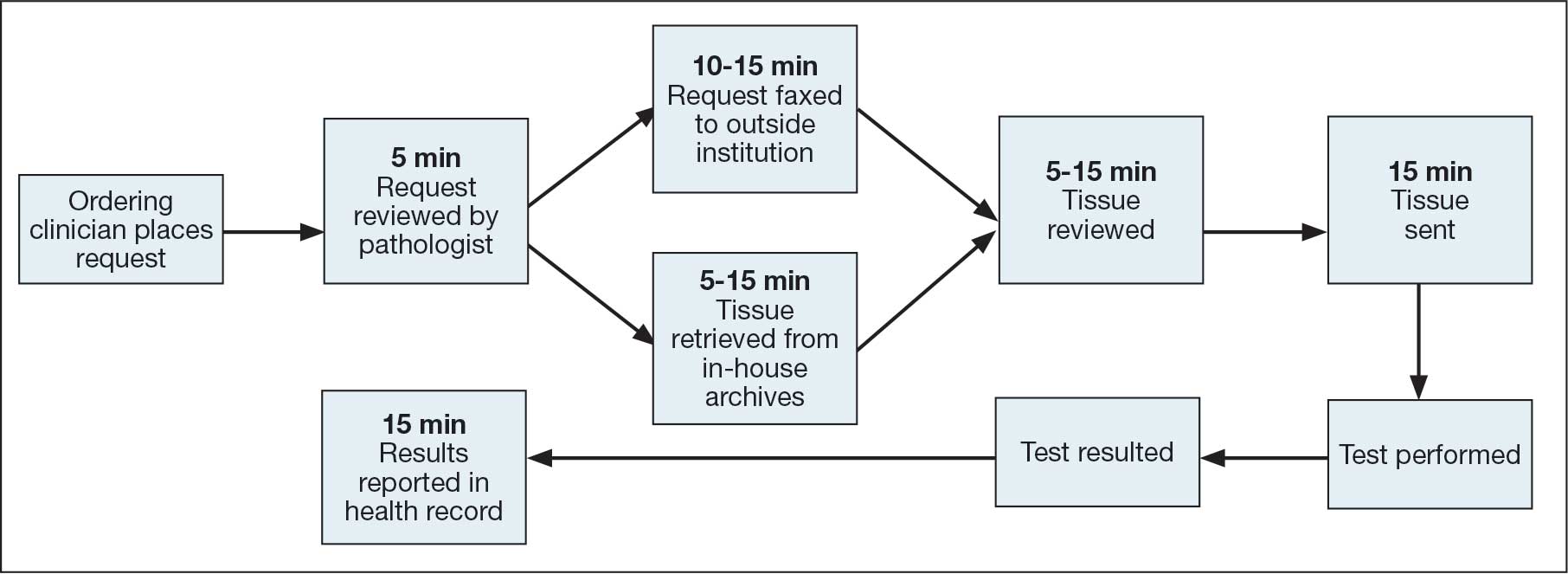
RESULTS
From 2021 to 2023, there were 21,021 AP cases at the KCVAMC. Basal cell carcinomas and squamous cell carcinomas of the skin were excluded because they comprise most cancer cases but almost never necessitate AP molecular test consultations. A total of 2118 cancer cases were included, representing 10.1% of all cases. Ancillary AP molecular send-out tests were performed on 1338 (6.4%) cases. Since ancillary tissue tests are requested by consultation at the KCVAMC, this resulted in 1338 consultations (Table 2).
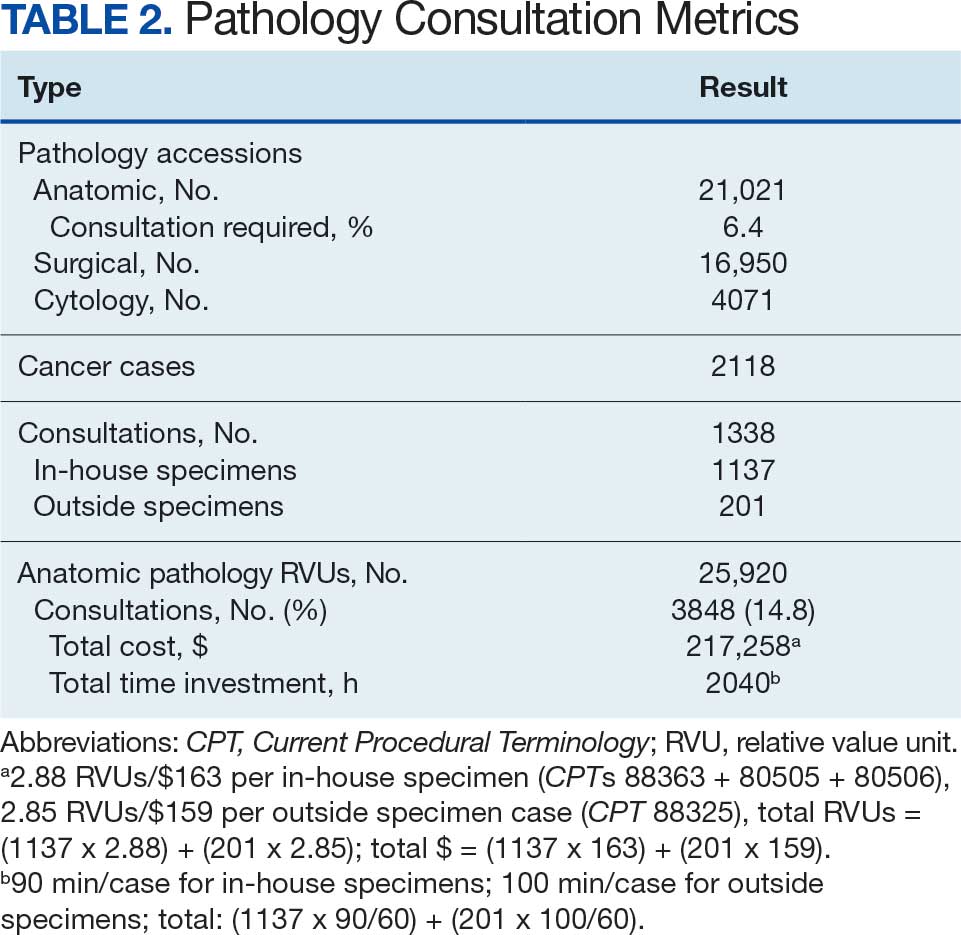
The time to complete a consultation was measured by calculating the mean time required to complete each step (Table 3). With in-house specimen consultations requiring 90 minutes each and outside specimen consultations requiring 100 minutes each, a total of 2040 hours of pathology staff time was necessary to complete associated consultations. Billing for this time with the new pathology clinical consultation CPT codes would generate 3847 RVUs, which would have equated to 14.8% (3847/25,920) of the anatomic pathology RVUs.

DISCUSSION
When considering the lengths of time for tasks associated with each consultation, it is important to remember that the volume (2-3 daily), was insufficient to meaningfully benefit from batching. Thus, waiting to perform a particular task until it was needed for multiple cases reduced the inefficiency associated with starting and switching between tasks. Both the Computerized Patient Record System and VistA had to be reopened, reauthenticated, and reloaded for each step that required use of the health record, printer, or fax machine. Faxes also required waiting for transmission and printed confirmation of successful transmission. As a result, the time values denoted for each step are likely underestimated, as the effect of interruptions is significant and not reflected in the estimates recorded.10
This analysis has demonstrated that PO entails a significant amount of work for pathology departments. To determine and maintain appropriate staffing models, this work must be captured and reimbursed. Unlike other pathology work, which is performed in-house and reimbursed for the associated test, a significant proportion of PO testing is sent out. Even if more reliable assays are developed, the physical processes of sending out samples and reporting test results cannot be outsourced. Independent and commensurate reimbursement methods are necessary to allow for this work and PO.
CMS included new pathology clinical consultation codes that may be used to bill for some of this work according to the 2022 physician fee schedule due to advocacy work by the College of American Pathologists and the American Medical Association CPT editorial panel.11
CONCLUSIONS
This analysis found that adoption of PO may present a significant amount of additional work for pathology departments. To determine and maintain appropriate staffing models, work completed by pathologists in this manner must be recorded and reimbursed. Pathologists need to be trained and encouraged to use these CPT codes and bill for the work described in this article. The increased revenue will allow for additional positions to alleviate the burdens imposed by understaffing so that pathology can function as a facilitator of PO rather than as a barrier to it.
- Wilson LE, Greiner MA, Altomare I, et al. Rapid rise in the cost of targeted cancer therapies for Medicare patients with solid tumors from 2006 to 2015. J Geriatr Oncol. 2021;12:375-380. doi:10.1016/j.jgo.2020.11.007
- Ersek JL, Black LJ, Thompson MA, et al. Implementing precision medicine programs and clinical trials in the community-based oncology practice: barriers and best practices. Am Soc Clin Oncol Educ Book. 2018;38:188-196. doi:10.1200/EDBK_200633
- Inal C, Yilmaz E, Cheng H, et al. Effect of reflex testing by pathologists on molecular testing rates in lung cancer patients: experience from a community-based academic center. J Clin Oncol. 2014;32:8098. doi:10.1200/jco.2014.32.15_suppl.8098
- Robboy SJ, Gupta S, Crawford JM, et al. The pathologist workforce in the United States: II. an interactive modeling tool for analyzing future qualitative and quantitative staffing demands for services. Arch Pathol Lab Med. 2015;139:1413-1430. doi:10.5858/arpa.2014-0559-OA
- OIG determination of Veterans Health Administration’s occupational staffing shortages fiscal year 2021. Department of Veterans Affairs OIG. September 28, 2021. Accessed January 30, 2026. https://www.oversight.gov/report/VA/OIG-determination-veterans-health-administrations-occupational-staffing-shortages-fiscal
- Zanto S, Cremeans L, Deutsch-Keahey D, et al. Addressing the clinical laboratory workforce shortage. The American Society for Clinical Laboratory Science. July 2, 2020. Accessed January 30, 2026. https://ascls.org/addressing-the-clinical-laboratory-workforce-shortage/
- Bennett A, Garcia E, Schulze M, et al. Building a laboratory workforce to meet the future: ASCP Task Force on the Laboratory Professionals Workforce. Am J Clin Pathol. 2014;141:154-167. doi:10.1309/AJCPIV2OG8TEGHHZ
- Fielder T, Watts F, Howden C, et al. Why choose a pathology career? Arch Pathol Lab Med. 2022;146:903-910. doi:10.5858/arpa.2021-0118-OA
- Metter DM, Colgan TJ, Leung ST, et al. Trends in the US and Canadian pathologist workforces from 2007 to 2017. JAMA Netw Open. 2019;2:e194337. doi:10.1001/jamanetworkopen.2019.4337
- Schulte B. Work interruptions can cost you 6 hours a day. An efficiency expert explains how to avoid them. The Washington Post. June 1, 2015. Accessed January 30, 2026. https://www.washingtonpost.com/news/inspired-life/wp/2015/06/01/interruptions-at-work-can-cost-you-up-to-6-hours-a-day-heres-how-to-avoid-them/
- Fiegl C. Medicare adopts new clinical consult billing codes. College of American Pathologists Today. December 2021. Accessed January 30, 2026. https://www.captodayonline.com/medicare-adopts-new-clinical-consult-billing-code
Precision oncology (PO) is cancer treatment individualized to the special characteristics of a patient’s tumor. It has become standard care for most patients with advanced cancer. Advances in molecular cell biology and immunology have identified numerous targets and many therapies have been developed as a result. Molecular testing and targeted therapy are typically covered by insurance, even when inflation-adjusted price growth is considered.1 Barriers remain, however, and pathologists are uniquely qualified to address some of the challenges.2
Most US laboratories do not perform molecular diagnostic tests for PO, particularly comprehensive evaluation of multiple targets by next-generation sequencing, or other techniques. Instead, these tests are sent to reference laboratories. The workload associated with referral testing is an obstacle to increased use of such tests. Despite guideline recommendations, a minority of indicated tests are performed.3 This is true even when testing costs are covered by clinical trials or grants, such as those in the Veterans Health Administration (VHA).
The main characteristic of successful PO programs is a multidisciplinary commitment, including pathology involvement in molecular tumor boards and assistance with test selection, tissue collection, and result interpretation.2 This, however, adds to the workload for the pathology department, an underappreciated phenomenon in the context of pathology workforce shortages.4
Workforce shortages impact all occupations in the laboratory setting. Though the shortage of medical technologists in clinical pathology laboratories has repeatedly been identified as critical at the VHA as well as in the private sector, the same cannot be said for staff shortages in anatomic pathology laboratories. Thus, the hospital laboratory divisions are concerned with biopsy or resection tissue specimens as opposed to the bodily fluid specimens that predominate in clinical laboratories.5 The lack of accurate data on histopathology technicians and technologists has precluded the degree of recognition seen for medical technologists. In labor statistics, these occupations are often obscured by inclusion with other jobs in broad categories such as medical and clinical laboratory technologists and technicians.6 Vacancy—the principal metric used to assess medical laboratory workforce shortage—fails to account for positions that are eventually eliminated after remaining vacant for prolonged periods, positions not replaced as a result of ambitious efficiency measures, or positions that were never created due to insufficient funding, reasons for administrative disapproval, or coverage by laboratory professionals working extra shifts or second jobs.7
Increased demand for pathologists is suggested by a 42% increase in workload per pathologist over the last decade, while a shortage is suggested by decreases in absolute and population-adjusted numbers of pathologists.8,9 An influx of pathologists is not an expected remedy due to the global decline in medical graduates pursuing careers in the field.8
Approximations for required labor and potential revenue generation are necessary to justify creation of pathology positions. This work mostly has gone uncaptured due to the limitations of Current Procedural Terminology (CPT) codes. Few laboratories have consistently used the 88363, 88325, and G0452 CPT codes. The pathology clinical consultation CPT codes (80503-80506) released in 2022 enhance acquisition of this work. The new codes replace 80500 and 80502 and allow for more precise identification of any work requiring medical judgment that a pathologist does at the request of another qualified health care professional (HCP) or as required by federal or state regulation.
The codes can be used to bill for associated time spent reviewing health records, communicating with other HCPs, placing orders, and documentation. An HCP can bill according to level of medical decision-making (MDM) or time spent by the consulting pathologist. Code 80503 can be billed for 5 to 20 minutes of a pathologist's time, 80504 for 21 to 40 minutes, 80505 for 41 to 60 minutes, and 80506 for each additional 30 minutes after the first hour. Levels of MDM (low, moderate, and high) are defined as for other evaluation and management services. A consultation report must be generated and contain documentation of the consultation request, pathologist interpretation, and justification for the level of service associated with the chosen code. Relative value units (RVUs) and reimbursement associated with each as well as other consultation-related codes are available in Table 1.
This article outlines how the pathology time investment associated with anatomic pathology molecular testing at the Kansas City Veterans Affairs Medical Center (KCVAMC) can be captured using the consultation process and new CPT codes. Staff included 4 pathologists, 3 histotechnologists, 1 histology supervisor, 1 grossing room technician, and 1 cytotechnologist, 1 cytology technician.
METHODS
The AP molecular testing consultation process at the KCVAMC was mapped, with the average time measured for each step (Figure). AP records for 2021 were reviewed to determine the number of AP molecular send out tests. Cumulative time investment was calculated in hours and a theoretical number of RVUs was calculated using the new pathology clinical consultation CPT codes (80503-80506). This theoretical number of RVUs was compared with the total AP RVUs generated in 2021 to determine a potential increase in RVUs with use of the new CPT codes to capture pathology work associated with AP molecular testing consultations.
RESULTS
From 2021 to 2023, there were 21,021 AP cases at the KCVAMC. Basal cell carcinomas and squamous cell carcinomas of the skin were excluded because they comprise most cancer cases but almost never necessitate AP molecular test consultations. A total of 2118 cancer cases were included, representing 10.1% of all cases. Ancillary AP molecular send-out tests were performed on 1338 (6.4%) cases. Since ancillary tissue tests are requested by consultation at the KCVAMC, this resulted in 1338 consultations (Table 2).
The time to complete a consultation was measured by calculating the mean time required to complete each step (Table 3). With in-house specimen consultations requiring 90 minutes each and outside specimen consultations requiring 100 minutes each, a total of 2040 hours of pathology staff time was necessary to complete associated consultations. Billing for this time with the new pathology clinical consultation CPT codes would generate 3847 RVUs, which would have equated to 14.8% (3847/25,920) of the anatomic pathology RVUs.
DISCUSSION
When considering the lengths of time for tasks associated with each consultation, it is important to remember that the volume (2-3 daily), was insufficient to meaningfully benefit from batching. Thus, waiting to perform a particular task until it was needed for multiple cases reduced the inefficiency associated with starting and switching between tasks. Both the Computerized Patient Record System and VistA had to be reopened, reauthenticated, and reloaded for each step that required use of the health record, printer, or fax machine. Faxes also required waiting for transmission and printed confirmation of successful transmission. As a result, the time values denoted for each step are likely underestimated, as the effect of interruptions is significant and not reflected in the estimates recorded.10
This analysis has demonstrated that PO entails a significant amount of work for pathology departments. To determine and maintain appropriate staffing models, this work must be captured and reimbursed. Unlike other pathology work, which is performed in-house and reimbursed for the associated test, a significant proportion of PO testing is sent out. Even if more reliable assays are developed, the physical processes of sending out samples and reporting test results cannot be outsourced. Independent and commensurate reimbursement methods are necessary to allow for this work and PO.
CMS included new pathology clinical consultation codes that may be used to bill for some of this work according to the 2022 physician fee schedule due to advocacy work by the College of American Pathologists and the American Medical Association CPT editorial panel.11
CONCLUSIONS
This analysis found that adoption of PO may present a significant amount of additional work for pathology departments. To determine and maintain appropriate staffing models, work completed by pathologists in this manner must be recorded and reimbursed. Pathologists need to be trained and encouraged to use these CPT codes and bill for the work described in this article. The increased revenue will allow for additional positions to alleviate the burdens imposed by understaffing so that pathology can function as a facilitator of PO rather than as a barrier to it.
Precision oncology (PO) is cancer treatment individualized to the special characteristics of a patient’s tumor. It has become standard care for most patients with advanced cancer. Advances in molecular cell biology and immunology have identified numerous targets and many therapies have been developed as a result. Molecular testing and targeted therapy are typically covered by insurance, even when inflation-adjusted price growth is considered.1 Barriers remain, however, and pathologists are uniquely qualified to address some of the challenges.2
Most US laboratories do not perform molecular diagnostic tests for PO, particularly comprehensive evaluation of multiple targets by next-generation sequencing, or other techniques. Instead, these tests are sent to reference laboratories. The workload associated with referral testing is an obstacle to increased use of such tests. Despite guideline recommendations, a minority of indicated tests are performed.3 This is true even when testing costs are covered by clinical trials or grants, such as those in the Veterans Health Administration (VHA).
The main characteristic of successful PO programs is a multidisciplinary commitment, including pathology involvement in molecular tumor boards and assistance with test selection, tissue collection, and result interpretation.2 This, however, adds to the workload for the pathology department, an underappreciated phenomenon in the context of pathology workforce shortages.4
Workforce shortages impact all occupations in the laboratory setting. Though the shortage of medical technologists in clinical pathology laboratories has repeatedly been identified as critical at the VHA as well as in the private sector, the same cannot be said for staff shortages in anatomic pathology laboratories. Thus, the hospital laboratory divisions are concerned with biopsy or resection tissue specimens as opposed to the bodily fluid specimens that predominate in clinical laboratories.5 The lack of accurate data on histopathology technicians and technologists has precluded the degree of recognition seen for medical technologists. In labor statistics, these occupations are often obscured by inclusion with other jobs in broad categories such as medical and clinical laboratory technologists and technicians.6 Vacancy—the principal metric used to assess medical laboratory workforce shortage—fails to account for positions that are eventually eliminated after remaining vacant for prolonged periods, positions not replaced as a result of ambitious efficiency measures, or positions that were never created due to insufficient funding, reasons for administrative disapproval, or coverage by laboratory professionals working extra shifts or second jobs.7
Increased demand for pathologists is suggested by a 42% increase in workload per pathologist over the last decade, while a shortage is suggested by decreases in absolute and population-adjusted numbers of pathologists.8,9 An influx of pathologists is not an expected remedy due to the global decline in medical graduates pursuing careers in the field.8
Approximations for required labor and potential revenue generation are necessary to justify creation of pathology positions. This work mostly has gone uncaptured due to the limitations of Current Procedural Terminology (CPT) codes. Few laboratories have consistently used the 88363, 88325, and G0452 CPT codes. The pathology clinical consultation CPT codes (80503-80506) released in 2022 enhance acquisition of this work. The new codes replace 80500 and 80502 and allow for more precise identification of any work requiring medical judgment that a pathologist does at the request of another qualified health care professional (HCP) or as required by federal or state regulation.
The codes can be used to bill for associated time spent reviewing health records, communicating with other HCPs, placing orders, and documentation. An HCP can bill according to level of medical decision-making (MDM) or time spent by the consulting pathologist. Code 80503 can be billed for 5 to 20 minutes of a pathologist's time, 80504 for 21 to 40 minutes, 80505 for 41 to 60 minutes, and 80506 for each additional 30 minutes after the first hour. Levels of MDM (low, moderate, and high) are defined as for other evaluation and management services. A consultation report must be generated and contain documentation of the consultation request, pathologist interpretation, and justification for the level of service associated with the chosen code. Relative value units (RVUs) and reimbursement associated with each as well as other consultation-related codes are available in Table 1.
This article outlines how the pathology time investment associated with anatomic pathology molecular testing at the Kansas City Veterans Affairs Medical Center (KCVAMC) can be captured using the consultation process and new CPT codes. Staff included 4 pathologists, 3 histotechnologists, 1 histology supervisor, 1 grossing room technician, and 1 cytotechnologist, 1 cytology technician.
METHODS
The AP molecular testing consultation process at the KCVAMC was mapped, with the average time measured for each step (Figure). AP records for 2021 were reviewed to determine the number of AP molecular send out tests. Cumulative time investment was calculated in hours and a theoretical number of RVUs was calculated using the new pathology clinical consultation CPT codes (80503-80506). This theoretical number of RVUs was compared with the total AP RVUs generated in 2021 to determine a potential increase in RVUs with use of the new CPT codes to capture pathology work associated with AP molecular testing consultations.
RESULTS
From 2021 to 2023, there were 21,021 AP cases at the KCVAMC. Basal cell carcinomas and squamous cell carcinomas of the skin were excluded because they comprise most cancer cases but almost never necessitate AP molecular test consultations. A total of 2118 cancer cases were included, representing 10.1% of all cases. Ancillary AP molecular send-out tests were performed on 1338 (6.4%) cases. Since ancillary tissue tests are requested by consultation at the KCVAMC, this resulted in 1338 consultations (Table 2).
The time to complete a consultation was measured by calculating the mean time required to complete each step (Table 3). With in-house specimen consultations requiring 90 minutes each and outside specimen consultations requiring 100 minutes each, a total of 2040 hours of pathology staff time was necessary to complete associated consultations. Billing for this time with the new pathology clinical consultation CPT codes would generate 3847 RVUs, which would have equated to 14.8% (3847/25,920) of the anatomic pathology RVUs.
DISCUSSION
When considering the lengths of time for tasks associated with each consultation, it is important to remember that the volume (2-3 daily), was insufficient to meaningfully benefit from batching. Thus, waiting to perform a particular task until it was needed for multiple cases reduced the inefficiency associated with starting and switching between tasks. Both the Computerized Patient Record System and VistA had to be reopened, reauthenticated, and reloaded for each step that required use of the health record, printer, or fax machine. Faxes also required waiting for transmission and printed confirmation of successful transmission. As a result, the time values denoted for each step are likely underestimated, as the effect of interruptions is significant and not reflected in the estimates recorded.10
This analysis has demonstrated that PO entails a significant amount of work for pathology departments. To determine and maintain appropriate staffing models, this work must be captured and reimbursed. Unlike other pathology work, which is performed in-house and reimbursed for the associated test, a significant proportion of PO testing is sent out. Even if more reliable assays are developed, the physical processes of sending out samples and reporting test results cannot be outsourced. Independent and commensurate reimbursement methods are necessary to allow for this work and PO.
CMS included new pathology clinical consultation codes that may be used to bill for some of this work according to the 2022 physician fee schedule due to advocacy work by the College of American Pathologists and the American Medical Association CPT editorial panel.11
CONCLUSIONS
This analysis found that adoption of PO may present a significant amount of additional work for pathology departments. To determine and maintain appropriate staffing models, work completed by pathologists in this manner must be recorded and reimbursed. Pathologists need to be trained and encouraged to use these CPT codes and bill for the work described in this article. The increased revenue will allow for additional positions to alleviate the burdens imposed by understaffing so that pathology can function as a facilitator of PO rather than as a barrier to it.
- Wilson LE, Greiner MA, Altomare I, et al. Rapid rise in the cost of targeted cancer therapies for Medicare patients with solid tumors from 2006 to 2015. J Geriatr Oncol. 2021;12:375-380. doi:10.1016/j.jgo.2020.11.007
- Ersek JL, Black LJ, Thompson MA, et al. Implementing precision medicine programs and clinical trials in the community-based oncology practice: barriers and best practices. Am Soc Clin Oncol Educ Book. 2018;38:188-196. doi:10.1200/EDBK_200633
- Inal C, Yilmaz E, Cheng H, et al. Effect of reflex testing by pathologists on molecular testing rates in lung cancer patients: experience from a community-based academic center. J Clin Oncol. 2014;32:8098. doi:10.1200/jco.2014.32.15_suppl.8098
- Robboy SJ, Gupta S, Crawford JM, et al. The pathologist workforce in the United States: II. an interactive modeling tool for analyzing future qualitative and quantitative staffing demands for services. Arch Pathol Lab Med. 2015;139:1413-1430. doi:10.5858/arpa.2014-0559-OA
- OIG determination of Veterans Health Administration’s occupational staffing shortages fiscal year 2021. Department of Veterans Affairs OIG. September 28, 2021. Accessed January 30, 2026. https://www.oversight.gov/report/VA/OIG-determination-veterans-health-administrations-occupational-staffing-shortages-fiscal
- Zanto S, Cremeans L, Deutsch-Keahey D, et al. Addressing the clinical laboratory workforce shortage. The American Society for Clinical Laboratory Science. July 2, 2020. Accessed January 30, 2026. https://ascls.org/addressing-the-clinical-laboratory-workforce-shortage/
- Bennett A, Garcia E, Schulze M, et al. Building a laboratory workforce to meet the future: ASCP Task Force on the Laboratory Professionals Workforce. Am J Clin Pathol. 2014;141:154-167. doi:10.1309/AJCPIV2OG8TEGHHZ
- Fielder T, Watts F, Howden C, et al. Why choose a pathology career? Arch Pathol Lab Med. 2022;146:903-910. doi:10.5858/arpa.2021-0118-OA
- Metter DM, Colgan TJ, Leung ST, et al. Trends in the US and Canadian pathologist workforces from 2007 to 2017. JAMA Netw Open. 2019;2:e194337. doi:10.1001/jamanetworkopen.2019.4337
- Schulte B. Work interruptions can cost you 6 hours a day. An efficiency expert explains how to avoid them. The Washington Post. June 1, 2015. Accessed January 30, 2026. https://www.washingtonpost.com/news/inspired-life/wp/2015/06/01/interruptions-at-work-can-cost-you-up-to-6-hours-a-day-heres-how-to-avoid-them/
- Fiegl C. Medicare adopts new clinical consult billing codes. College of American Pathologists Today. December 2021. Accessed January 30, 2026. https://www.captodayonline.com/medicare-adopts-new-clinical-consult-billing-code
- Wilson LE, Greiner MA, Altomare I, et al. Rapid rise in the cost of targeted cancer therapies for Medicare patients with solid tumors from 2006 to 2015. J Geriatr Oncol. 2021;12:375-380. doi:10.1016/j.jgo.2020.11.007
- Ersek JL, Black LJ, Thompson MA, et al. Implementing precision medicine programs and clinical trials in the community-based oncology practice: barriers and best practices. Am Soc Clin Oncol Educ Book. 2018;38:188-196. doi:10.1200/EDBK_200633
- Inal C, Yilmaz E, Cheng H, et al. Effect of reflex testing by pathologists on molecular testing rates in lung cancer patients: experience from a community-based academic center. J Clin Oncol. 2014;32:8098. doi:10.1200/jco.2014.32.15_suppl.8098
- Robboy SJ, Gupta S, Crawford JM, et al. The pathologist workforce in the United States: II. an interactive modeling tool for analyzing future qualitative and quantitative staffing demands for services. Arch Pathol Lab Med. 2015;139:1413-1430. doi:10.5858/arpa.2014-0559-OA
- OIG determination of Veterans Health Administration’s occupational staffing shortages fiscal year 2021. Department of Veterans Affairs OIG. September 28, 2021. Accessed January 30, 2026. https://www.oversight.gov/report/VA/OIG-determination-veterans-health-administrations-occupational-staffing-shortages-fiscal
- Zanto S, Cremeans L, Deutsch-Keahey D, et al. Addressing the clinical laboratory workforce shortage. The American Society for Clinical Laboratory Science. July 2, 2020. Accessed January 30, 2026. https://ascls.org/addressing-the-clinical-laboratory-workforce-shortage/
- Bennett A, Garcia E, Schulze M, et al. Building a laboratory workforce to meet the future: ASCP Task Force on the Laboratory Professionals Workforce. Am J Clin Pathol. 2014;141:154-167. doi:10.1309/AJCPIV2OG8TEGHHZ
- Fielder T, Watts F, Howden C, et al. Why choose a pathology career? Arch Pathol Lab Med. 2022;146:903-910. doi:10.5858/arpa.2021-0118-OA
- Metter DM, Colgan TJ, Leung ST, et al. Trends in the US and Canadian pathologist workforces from 2007 to 2017. JAMA Netw Open. 2019;2:e194337. doi:10.1001/jamanetworkopen.2019.4337
- Schulte B. Work interruptions can cost you 6 hours a day. An efficiency expert explains how to avoid them. The Washington Post. June 1, 2015. Accessed January 30, 2026. https://www.washingtonpost.com/news/inspired-life/wp/2015/06/01/interruptions-at-work-can-cost-you-up-to-6-hours-a-day-heres-how-to-avoid-them/
- Fiegl C. Medicare adopts new clinical consult billing codes. College of American Pathologists Today. December 2021. Accessed January 30, 2026. https://www.captodayonline.com/medicare-adopts-new-clinical-consult-billing-code
Capturing Pathology Workload Associated With Precision Oncology
Capturing Pathology Workload Associated With Precision Oncology