User login
Pigmented Fungiform Papillae of the Tongue in an Indian Male
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
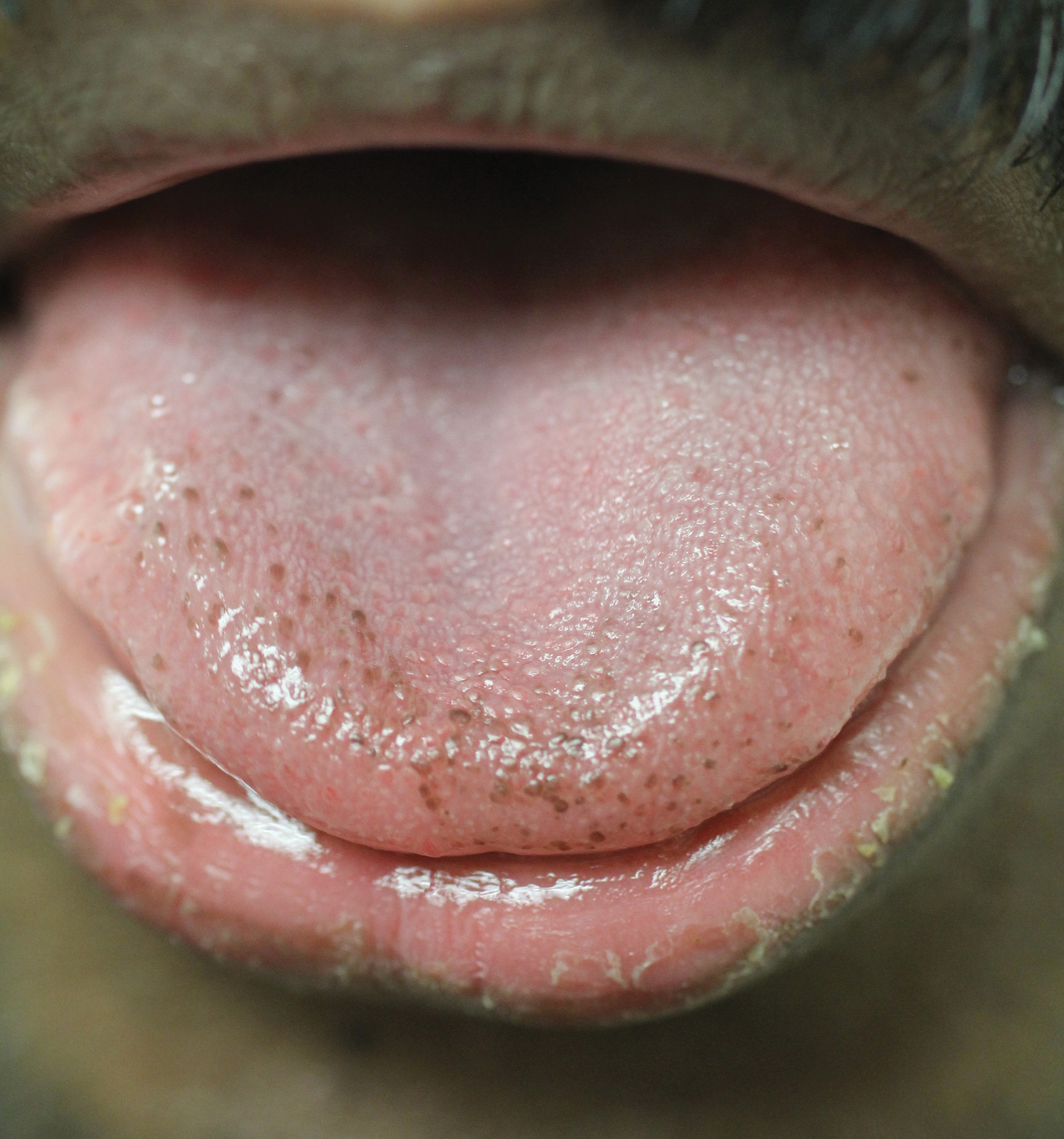
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
Practice Points
- Pigmented fungiform papillae of the tongue are common lingual hyperpigmented macules in patients with skin of color.
- It is important to be aware of this benign entity to provide reassurance to patients and avoid unnecessary testing.

Prurigo Pigmentosa Induced by Ketosis: Resolution Through Dietary Modification
To the Editor:
A 40-year-old white woman presented with a waxing and waning erythematous pruritic rash on the chest, back, and axillae of 3 years’ duration. The appearance of the rash coincided with an intentional weight loss of more than 100 lb, achieved through various diets, most recently a Paleolithic (paleo) diet that was high in protein; low in carbohydrates; and specifically restricted dairy, cereal grains, refined sugars, processed foods, white potatoes, salt, refined oils, and legumes.1 The patient had been monitoring blood glucose and ketone levels. Prior to presentation, she received various treatments including clotrimazole cream and topical steroids with no improvement.
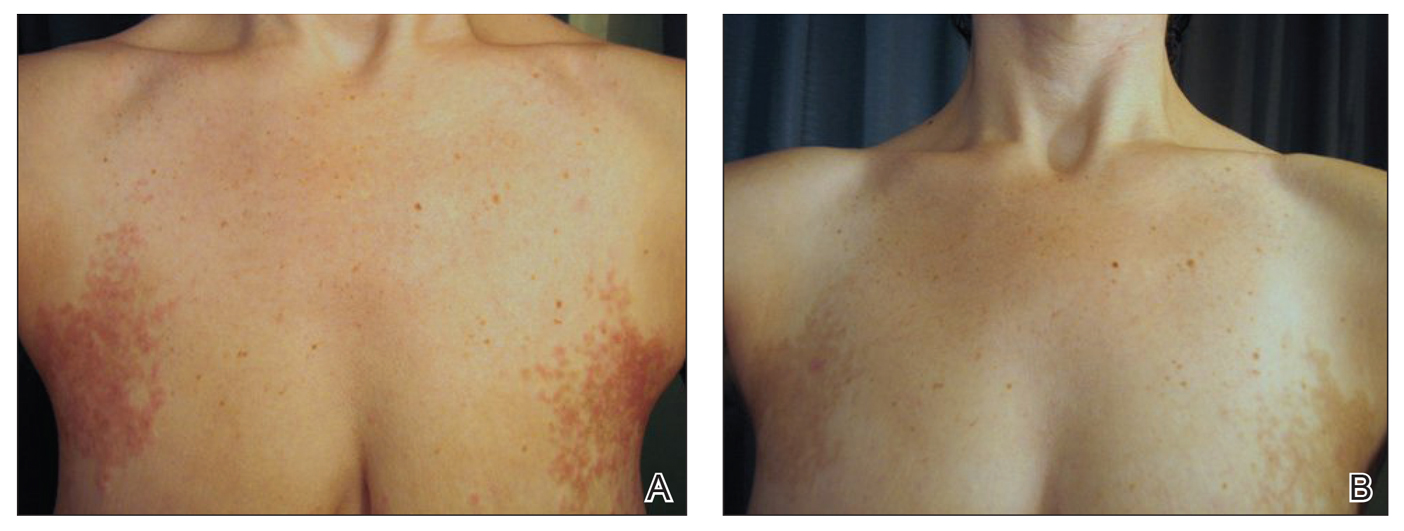
On physical examination, there were scaly, pink-red, reticulated papules and plaques coexisting with tan reticulated patches that were symmetrically distributed on the central back, lateral and central chest (Figure 1A), breasts, and inframammary areas. During the most severe flare-up, the blood ketones measured 1 mmol/L. There was no relevant medical history. She was of Spanish and Italian descent.
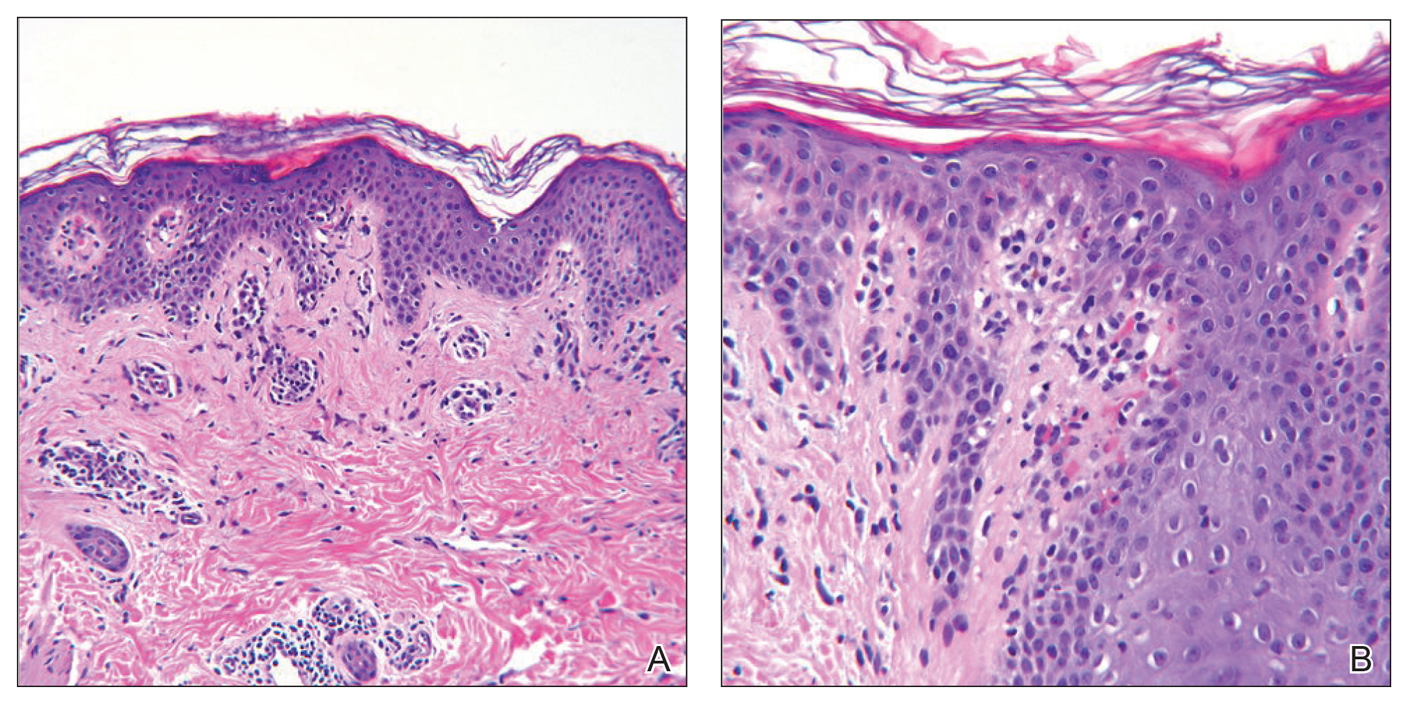
Histologic sections showed a sparse infiltrate of lymphocytes surrounding superficial dermal vessels and a mildly acanthotic epidermis with a focally parakeratotic stratum corneum (Figure 2A). Pigmentary incontinence and subtle interface changes were apparent, including rare necrotic keratinocytes (Figure 2B). No eosinophils or neutrophils were present.
After the initial presentation, carbohydrates were added back into her diet and both the ketosis and eruption remarkably resolved. When carbohydrate restriction was rechallenged, she again entered ketosis (0.5 mmol/L), followed by subsequent recurrence of the pruritic lesions. With re-introduction of carbohydrates, the eruption and ketosis once more resolved, leaving only postinflammatory reticulated hyperpigmentation (Figure 1B). Based on the clinical presentation, supportive histopathologic findings, and interesting response to ketones and diet modification, the patient was diagnosed with prurigo pigmentosa (PP).
Prurigo pigmentosa is a rare inflammatory dermatosis that was initially described in 1971 as “a peculiar pruriginous dermatosis with gross reticular pigmentation” by Nagashima et al.2 Prurigo pigmentosa is most frequently diagnosed in Japan, and since its discovery, it has been reported in more than 300 cases worldwide.2-4
Fewer than 50 non-Japanese cases have been reported, with the possibility of an additional ethnic predisposition among the Turkish and Sicilian populations, though only 6 cases have been reported in the United States.3-6 Prurigo pigmentosa tends to occur in the spring and summer months and is most common among young females, with a mean age of 24 years. The typical lesions of PP are symmetrically distributed on the trunk with a tendency to localize on the upper back, nape of the neck, and intermammary and inframammary regions. Eruptions have been reported to occur on additional areas; however, mucus membranes are always spared.6
Individual lesions differ in appearance depending on the stage of presentation and are categorized as early, fully developed, resolving, and late lesions.6 Pruritic macules and papules are present early in the disease state and resolve into crusted and/or scaly papules followed by pigmented macules. Early lesions tend to be intensely pruritic with signs of excoriation, while resolving lesions lack symptoms. Lesions last approximately 1 week but tend to reappear at the site where they were previously present, which allows for lesions of different ages to coexist, appearing in a reticular arrangement with hyperpigmented mottling lasting from a few weeks to months.6
Just as the clinical picture transpires rapidly within 1 week, so do the histopathologic findings.6 Early lesions are categorized by a superficial perivascular and interstitial infiltrate of neutrophils, spongiosis, ballooning, and necrotic keratinocytes. These early lesions are present for less than 48 hours, and these histopathologic findings are diagnostic of PP. Within 2 days, lymphocytes predominate in the dermal infiltrate, and a patchy lichenoid aspect is established in the fully developed lesion along with reticular and vacuolar alterations. Late lesions show a parakeratotic and hyperpigmented epidermis with melanophages present in the papillary and reticular dermis. At this last stage, the histopathologic features of PP are indistinguishable from any other disease that results in postinflammatory hyperpigmentation, making diagnosis difficult.6
A variety of therapeutic options are used in the treatment of PP, with the most effective agents being oral antibiotics including dapsone, minocycline, and doxycycline, all of which limit the local tissue inflammatory response and cytotoxic effects. Topical and systemic antihistamines as well as corticosteroids are ineffective and have not been shown to prevent the postinflammatory reticular pigmentation.6-10
Various underlying factors have been associated with PP, including friction, heat, sunlight, sweating, allergic contact sensitization, and ketosis due to nutritional deficiency or diabetes mellitus; however; the exact etiology remains ambiguous.2-7 The association with ketosis and nutrition is of particular interest in this case. Onset of PP has been reported to coincide with dieting, fasting, weight loss, anorexia nervosa, and diabetes mellitus.3,6-9 Roughly 50 patients with PP had ketosis subsequent to these metabolic disturbances.3,6-10 As of now, the only reported correlation between ketosis and PP is that upon diet modification, lesions resolved following ketone normalization, as was observed in our patient.3,6-8 Reports of PP in diabetic patients while in ketoacidosis describe resolution of lesions with insulin administration.6-9 The pathophysiology of ketosis and its association with PP is unclear; however, the similarities seen in the immune response of PP and that stimulated by ketosis may expose an associated mechanism.
Ketosis is a temporary condition characterized by elevated serum ketones that are used as an alternative energy source when blood glucose is low or insulin is deficient.11 The most common causes of ketosis are the physiologic responses to fasting, prolonged exercise, or a high-protein/low-carbohydrate diet, though pathologic causes include insulin-dependent diabetes mellitus, alcoholism, and salicylate overdose.11 In healthy individuals, blood ketone levels rarely approach 0.5 mmol/L. Prolonged fasting or restricting intake of carbohydrates to less than 40 g daily can induce mild ketosis that resolves with re-introduction of carbohydrates.11
Ketone bodies pass from the circulating blood into tissues or remain near the blood vessels, inducing cytotoxic effects and perivascular inflammation.10,11 Increased ketone bodies have been shown to upregulate intercellular adhesion molecule 1 (ICAM-1) and leukocyte function-associated antigen 1 (LFA-1), a phenomenon also seen in lesional keratinocytes of PP.12,13 Teraki et al13 observed that epidermal keratinocytes exhibited increased expression of ICAM-1 as well as intense expression of LFA-1 on dermal and epidermotropic leukocytes, which was thought to be due to cell-mediated cytotoxicity. Not only do increased ketone bodies upregulate ICAM-1 and LFA-1, but they also are involved in increasing many proinflammatory mediators that may be capable of inducing the response seen in PP keratinocytes.12,13
Intercellular adhesion molecule 1 is important in initiating cellular interactions in the immune response and is the ligand for LFA-1 found on most leukocytes.14 Increased ICAM-1/LFA-1 interaction is thought to be the major pathway by which leukocytes are able to attach to keratinocytes and endothelial cells, allowing for leukocyte tissue migration and specific immunologic reactions, including leukocyte-mediated cytotoxicity. Interestingly, glucocorticoids are ineffective in reducing the expression of ICAM-1 in cultured keratinocytes.14 This connection between ketosis and inflammation that results in leukocyte migration and ultimately keratinocyte cytotoxicity may well be fundamental to the pathophysiology of PP and may provide a possible explanation for the ineffectiveness of corticosteroid treatment.
Middleton and Norris15 observed that individual keratinocyte strains show considerable variability in ICAM-1 expression that was found to be attributable to genetic polymorphisms. The presence of a particular polymorphism affecting ICAM-1 expression on human keratinocytes may explain the apparent ethnogeographic predisposition of PP as well as the ease at which ICAM-1 is expressed in the presence of ketones.
We describe a case of a 40-year-old white woman who was diagnosed with PP that was prompted by a 100-lb weight loss and self-induced ketosis while following a paleo diet with carbohydrate restriction. Successful treatment was attained through diet modification alone. This interesting case was another instance in which the pathophysiology of PP was attributed to ketosis. Because not all patients that are in ketosis have PP, larger prospective cohort studies are needed to further elucidate the association of PP and ketosis.
- What is the paleo diet? The Paleo Diet website. http://thepaleodiet.com/the-paleo-diet-premise. Accessed March 9, 2019.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation [in Japanese]. Japanese J Dermatol. 1971;81:38-39.
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet [published online December 30, 2013]. Pediatr Dermatol. 2015;32:248-251.
- Baykal C, Buyukbabani N, Akinturk S, et al. Prurigo pigmentosa: not an uncommon disease in the Turkish population. Int J Dermatol. 2006;45:1164-1168.
- Whang T, Kirkorian Y, Krishtul A, et al. Prurigo pigmentosa: report of two cases in the United States and review of the literature. Dermatology Online J. 2011;17:2.
- Böer A, Ackerman AB. Prurigo Pigmentosa (Nagashima Disease): Textbook and Atlas of a Distinctive Inflammatory Disease of the Skin. New York, NY: Ardor Scribendi Ltd; 2004.
- Teraki Y, Teraki E, Kawashima M, at al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511.
- Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2011;26:1149-1153.
- Yokozeki M, Watanabe J, Hotsubo T, et al. Prurigo pigmentosa disappeared following improvement of diabetic ketosis by insulin. J Dermatol. 2003;30:257-258.
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
- VanItallie TB, Nufert TH. Ketones: metabolism’s ugly duckling. Annu Rev Nutr. 2003;61:327-341.
- Rains JL, Jain SK. Hyperketonemia increases monocyte adhesion to endothelial cells and is mediated by LFA-1 expression in monocytes and ICAM-1 expression in endothelial cells. Am J Physiol Endocrinol Metab. 2011;301:e298-e306.
- Teraki Y, Shiohara T, Nagashima M, et al. Prurigo pigmentosa: role of ICAM-1 in the localization of the eruption. Br J Dermatol. 1991;125:360-363.
- Kashihara-Sawami M, Norris DA. The state of differentiation of cultured human keratinocytes determines the level of intercellular adhesion molecule-1 (ICAM-1) expression induced by gamma interferon. J Invest Dermatol. 1992;98:741-747.
- Middleton MH, Norris DA. Cytokine-induced ICAM-1 expression in human keratinocytes is highly variable in keratinocyte strains from different donors. J Invest Dermatol. 1995;104:489-496.
To the Editor:
A 40-year-old white woman presented with a waxing and waning erythematous pruritic rash on the chest, back, and axillae of 3 years’ duration. The appearance of the rash coincided with an intentional weight loss of more than 100 lb, achieved through various diets, most recently a Paleolithic (paleo) diet that was high in protein; low in carbohydrates; and specifically restricted dairy, cereal grains, refined sugars, processed foods, white potatoes, salt, refined oils, and legumes.1 The patient had been monitoring blood glucose and ketone levels. Prior to presentation, she received various treatments including clotrimazole cream and topical steroids with no improvement.
On physical examination, there were scaly, pink-red, reticulated papules and plaques coexisting with tan reticulated patches that were symmetrically distributed on the central back, lateral and central chest (Figure 1A), breasts, and inframammary areas. During the most severe flare-up, the blood ketones measured 1 mmol/L. There was no relevant medical history. She was of Spanish and Italian descent.
Histologic sections showed a sparse infiltrate of lymphocytes surrounding superficial dermal vessels and a mildly acanthotic epidermis with a focally parakeratotic stratum corneum (Figure 2A). Pigmentary incontinence and subtle interface changes were apparent, including rare necrotic keratinocytes (Figure 2B). No eosinophils or neutrophils were present.
After the initial presentation, carbohydrates were added back into her diet and both the ketosis and eruption remarkably resolved. When carbohydrate restriction was rechallenged, she again entered ketosis (0.5 mmol/L), followed by subsequent recurrence of the pruritic lesions. With re-introduction of carbohydrates, the eruption and ketosis once more resolved, leaving only postinflammatory reticulated hyperpigmentation (Figure 1B). Based on the clinical presentation, supportive histopathologic findings, and interesting response to ketones and diet modification, the patient was diagnosed with prurigo pigmentosa (PP).
Prurigo pigmentosa is a rare inflammatory dermatosis that was initially described in 1971 as “a peculiar pruriginous dermatosis with gross reticular pigmentation” by Nagashima et al.2 Prurigo pigmentosa is most frequently diagnosed in Japan, and since its discovery, it has been reported in more than 300 cases worldwide.2-4
Fewer than 50 non-Japanese cases have been reported, with the possibility of an additional ethnic predisposition among the Turkish and Sicilian populations, though only 6 cases have been reported in the United States.3-6 Prurigo pigmentosa tends to occur in the spring and summer months and is most common among young females, with a mean age of 24 years. The typical lesions of PP are symmetrically distributed on the trunk with a tendency to localize on the upper back, nape of the neck, and intermammary and inframammary regions. Eruptions have been reported to occur on additional areas; however, mucus membranes are always spared.6
Individual lesions differ in appearance depending on the stage of presentation and are categorized as early, fully developed, resolving, and late lesions.6 Pruritic macules and papules are present early in the disease state and resolve into crusted and/or scaly papules followed by pigmented macules. Early lesions tend to be intensely pruritic with signs of excoriation, while resolving lesions lack symptoms. Lesions last approximately 1 week but tend to reappear at the site where they were previously present, which allows for lesions of different ages to coexist, appearing in a reticular arrangement with hyperpigmented mottling lasting from a few weeks to months.6
Just as the clinical picture transpires rapidly within 1 week, so do the histopathologic findings.6 Early lesions are categorized by a superficial perivascular and interstitial infiltrate of neutrophils, spongiosis, ballooning, and necrotic keratinocytes. These early lesions are present for less than 48 hours, and these histopathologic findings are diagnostic of PP. Within 2 days, lymphocytes predominate in the dermal infiltrate, and a patchy lichenoid aspect is established in the fully developed lesion along with reticular and vacuolar alterations. Late lesions show a parakeratotic and hyperpigmented epidermis with melanophages present in the papillary and reticular dermis. At this last stage, the histopathologic features of PP are indistinguishable from any other disease that results in postinflammatory hyperpigmentation, making diagnosis difficult.6
A variety of therapeutic options are used in the treatment of PP, with the most effective agents being oral antibiotics including dapsone, minocycline, and doxycycline, all of which limit the local tissue inflammatory response and cytotoxic effects. Topical and systemic antihistamines as well as corticosteroids are ineffective and have not been shown to prevent the postinflammatory reticular pigmentation.6-10
Various underlying factors have been associated with PP, including friction, heat, sunlight, sweating, allergic contact sensitization, and ketosis due to nutritional deficiency or diabetes mellitus; however; the exact etiology remains ambiguous.2-7 The association with ketosis and nutrition is of particular interest in this case. Onset of PP has been reported to coincide with dieting, fasting, weight loss, anorexia nervosa, and diabetes mellitus.3,6-9 Roughly 50 patients with PP had ketosis subsequent to these metabolic disturbances.3,6-10 As of now, the only reported correlation between ketosis and PP is that upon diet modification, lesions resolved following ketone normalization, as was observed in our patient.3,6-8 Reports of PP in diabetic patients while in ketoacidosis describe resolution of lesions with insulin administration.6-9 The pathophysiology of ketosis and its association with PP is unclear; however, the similarities seen in the immune response of PP and that stimulated by ketosis may expose an associated mechanism.
Ketosis is a temporary condition characterized by elevated serum ketones that are used as an alternative energy source when blood glucose is low or insulin is deficient.11 The most common causes of ketosis are the physiologic responses to fasting, prolonged exercise, or a high-protein/low-carbohydrate diet, though pathologic causes include insulin-dependent diabetes mellitus, alcoholism, and salicylate overdose.11 In healthy individuals, blood ketone levels rarely approach 0.5 mmol/L. Prolonged fasting or restricting intake of carbohydrates to less than 40 g daily can induce mild ketosis that resolves with re-introduction of carbohydrates.11
Ketone bodies pass from the circulating blood into tissues or remain near the blood vessels, inducing cytotoxic effects and perivascular inflammation.10,11 Increased ketone bodies have been shown to upregulate intercellular adhesion molecule 1 (ICAM-1) and leukocyte function-associated antigen 1 (LFA-1), a phenomenon also seen in lesional keratinocytes of PP.12,13 Teraki et al13 observed that epidermal keratinocytes exhibited increased expression of ICAM-1 as well as intense expression of LFA-1 on dermal and epidermotropic leukocytes, which was thought to be due to cell-mediated cytotoxicity. Not only do increased ketone bodies upregulate ICAM-1 and LFA-1, but they also are involved in increasing many proinflammatory mediators that may be capable of inducing the response seen in PP keratinocytes.12,13
Intercellular adhesion molecule 1 is important in initiating cellular interactions in the immune response and is the ligand for LFA-1 found on most leukocytes.14 Increased ICAM-1/LFA-1 interaction is thought to be the major pathway by which leukocytes are able to attach to keratinocytes and endothelial cells, allowing for leukocyte tissue migration and specific immunologic reactions, including leukocyte-mediated cytotoxicity. Interestingly, glucocorticoids are ineffective in reducing the expression of ICAM-1 in cultured keratinocytes.14 This connection between ketosis and inflammation that results in leukocyte migration and ultimately keratinocyte cytotoxicity may well be fundamental to the pathophysiology of PP and may provide a possible explanation for the ineffectiveness of corticosteroid treatment.
Middleton and Norris15 observed that individual keratinocyte strains show considerable variability in ICAM-1 expression that was found to be attributable to genetic polymorphisms. The presence of a particular polymorphism affecting ICAM-1 expression on human keratinocytes may explain the apparent ethnogeographic predisposition of PP as well as the ease at which ICAM-1 is expressed in the presence of ketones.
We describe a case of a 40-year-old white woman who was diagnosed with PP that was prompted by a 100-lb weight loss and self-induced ketosis while following a paleo diet with carbohydrate restriction. Successful treatment was attained through diet modification alone. This interesting case was another instance in which the pathophysiology of PP was attributed to ketosis. Because not all patients that are in ketosis have PP, larger prospective cohort studies are needed to further elucidate the association of PP and ketosis.
To the Editor:
A 40-year-old white woman presented with a waxing and waning erythematous pruritic rash on the chest, back, and axillae of 3 years’ duration. The appearance of the rash coincided with an intentional weight loss of more than 100 lb, achieved through various diets, most recently a Paleolithic (paleo) diet that was high in protein; low in carbohydrates; and specifically restricted dairy, cereal grains, refined sugars, processed foods, white potatoes, salt, refined oils, and legumes.1 The patient had been monitoring blood glucose and ketone levels. Prior to presentation, she received various treatments including clotrimazole cream and topical steroids with no improvement.
On physical examination, there were scaly, pink-red, reticulated papules and plaques coexisting with tan reticulated patches that were symmetrically distributed on the central back, lateral and central chest (Figure 1A), breasts, and inframammary areas. During the most severe flare-up, the blood ketones measured 1 mmol/L. There was no relevant medical history. She was of Spanish and Italian descent.
Histologic sections showed a sparse infiltrate of lymphocytes surrounding superficial dermal vessels and a mildly acanthotic epidermis with a focally parakeratotic stratum corneum (Figure 2A). Pigmentary incontinence and subtle interface changes were apparent, including rare necrotic keratinocytes (Figure 2B). No eosinophils or neutrophils were present.
After the initial presentation, carbohydrates were added back into her diet and both the ketosis and eruption remarkably resolved. When carbohydrate restriction was rechallenged, she again entered ketosis (0.5 mmol/L), followed by subsequent recurrence of the pruritic lesions. With re-introduction of carbohydrates, the eruption and ketosis once more resolved, leaving only postinflammatory reticulated hyperpigmentation (Figure 1B). Based on the clinical presentation, supportive histopathologic findings, and interesting response to ketones and diet modification, the patient was diagnosed with prurigo pigmentosa (PP).
Prurigo pigmentosa is a rare inflammatory dermatosis that was initially described in 1971 as “a peculiar pruriginous dermatosis with gross reticular pigmentation” by Nagashima et al.2 Prurigo pigmentosa is most frequently diagnosed in Japan, and since its discovery, it has been reported in more than 300 cases worldwide.2-4
Fewer than 50 non-Japanese cases have been reported, with the possibility of an additional ethnic predisposition among the Turkish and Sicilian populations, though only 6 cases have been reported in the United States.3-6 Prurigo pigmentosa tends to occur in the spring and summer months and is most common among young females, with a mean age of 24 years. The typical lesions of PP are symmetrically distributed on the trunk with a tendency to localize on the upper back, nape of the neck, and intermammary and inframammary regions. Eruptions have been reported to occur on additional areas; however, mucus membranes are always spared.6
Individual lesions differ in appearance depending on the stage of presentation and are categorized as early, fully developed, resolving, and late lesions.6 Pruritic macules and papules are present early in the disease state and resolve into crusted and/or scaly papules followed by pigmented macules. Early lesions tend to be intensely pruritic with signs of excoriation, while resolving lesions lack symptoms. Lesions last approximately 1 week but tend to reappear at the site where they were previously present, which allows for lesions of different ages to coexist, appearing in a reticular arrangement with hyperpigmented mottling lasting from a few weeks to months.6
Just as the clinical picture transpires rapidly within 1 week, so do the histopathologic findings.6 Early lesions are categorized by a superficial perivascular and interstitial infiltrate of neutrophils, spongiosis, ballooning, and necrotic keratinocytes. These early lesions are present for less than 48 hours, and these histopathologic findings are diagnostic of PP. Within 2 days, lymphocytes predominate in the dermal infiltrate, and a patchy lichenoid aspect is established in the fully developed lesion along with reticular and vacuolar alterations. Late lesions show a parakeratotic and hyperpigmented epidermis with melanophages present in the papillary and reticular dermis. At this last stage, the histopathologic features of PP are indistinguishable from any other disease that results in postinflammatory hyperpigmentation, making diagnosis difficult.6
A variety of therapeutic options are used in the treatment of PP, with the most effective agents being oral antibiotics including dapsone, minocycline, and doxycycline, all of which limit the local tissue inflammatory response and cytotoxic effects. Topical and systemic antihistamines as well as corticosteroids are ineffective and have not been shown to prevent the postinflammatory reticular pigmentation.6-10
Various underlying factors have been associated with PP, including friction, heat, sunlight, sweating, allergic contact sensitization, and ketosis due to nutritional deficiency or diabetes mellitus; however; the exact etiology remains ambiguous.2-7 The association with ketosis and nutrition is of particular interest in this case. Onset of PP has been reported to coincide with dieting, fasting, weight loss, anorexia nervosa, and diabetes mellitus.3,6-9 Roughly 50 patients with PP had ketosis subsequent to these metabolic disturbances.3,6-10 As of now, the only reported correlation between ketosis and PP is that upon diet modification, lesions resolved following ketone normalization, as was observed in our patient.3,6-8 Reports of PP in diabetic patients while in ketoacidosis describe resolution of lesions with insulin administration.6-9 The pathophysiology of ketosis and its association with PP is unclear; however, the similarities seen in the immune response of PP and that stimulated by ketosis may expose an associated mechanism.
Ketosis is a temporary condition characterized by elevated serum ketones that are used as an alternative energy source when blood glucose is low or insulin is deficient.11 The most common causes of ketosis are the physiologic responses to fasting, prolonged exercise, or a high-protein/low-carbohydrate diet, though pathologic causes include insulin-dependent diabetes mellitus, alcoholism, and salicylate overdose.11 In healthy individuals, blood ketone levels rarely approach 0.5 mmol/L. Prolonged fasting or restricting intake of carbohydrates to less than 40 g daily can induce mild ketosis that resolves with re-introduction of carbohydrates.11
Ketone bodies pass from the circulating blood into tissues or remain near the blood vessels, inducing cytotoxic effects and perivascular inflammation.10,11 Increased ketone bodies have been shown to upregulate intercellular adhesion molecule 1 (ICAM-1) and leukocyte function-associated antigen 1 (LFA-1), a phenomenon also seen in lesional keratinocytes of PP.12,13 Teraki et al13 observed that epidermal keratinocytes exhibited increased expression of ICAM-1 as well as intense expression of LFA-1 on dermal and epidermotropic leukocytes, which was thought to be due to cell-mediated cytotoxicity. Not only do increased ketone bodies upregulate ICAM-1 and LFA-1, but they also are involved in increasing many proinflammatory mediators that may be capable of inducing the response seen in PP keratinocytes.12,13
Intercellular adhesion molecule 1 is important in initiating cellular interactions in the immune response and is the ligand for LFA-1 found on most leukocytes.14 Increased ICAM-1/LFA-1 interaction is thought to be the major pathway by which leukocytes are able to attach to keratinocytes and endothelial cells, allowing for leukocyte tissue migration and specific immunologic reactions, including leukocyte-mediated cytotoxicity. Interestingly, glucocorticoids are ineffective in reducing the expression of ICAM-1 in cultured keratinocytes.14 This connection between ketosis and inflammation that results in leukocyte migration and ultimately keratinocyte cytotoxicity may well be fundamental to the pathophysiology of PP and may provide a possible explanation for the ineffectiveness of corticosteroid treatment.
Middleton and Norris15 observed that individual keratinocyte strains show considerable variability in ICAM-1 expression that was found to be attributable to genetic polymorphisms. The presence of a particular polymorphism affecting ICAM-1 expression on human keratinocytes may explain the apparent ethnogeographic predisposition of PP as well as the ease at which ICAM-1 is expressed in the presence of ketones.
We describe a case of a 40-year-old white woman who was diagnosed with PP that was prompted by a 100-lb weight loss and self-induced ketosis while following a paleo diet with carbohydrate restriction. Successful treatment was attained through diet modification alone. This interesting case was another instance in which the pathophysiology of PP was attributed to ketosis. Because not all patients that are in ketosis have PP, larger prospective cohort studies are needed to further elucidate the association of PP and ketosis.
- What is the paleo diet? The Paleo Diet website. http://thepaleodiet.com/the-paleo-diet-premise. Accessed March 9, 2019.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation [in Japanese]. Japanese J Dermatol. 1971;81:38-39.
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet [published online December 30, 2013]. Pediatr Dermatol. 2015;32:248-251.
- Baykal C, Buyukbabani N, Akinturk S, et al. Prurigo pigmentosa: not an uncommon disease in the Turkish population. Int J Dermatol. 2006;45:1164-1168.
- Whang T, Kirkorian Y, Krishtul A, et al. Prurigo pigmentosa: report of two cases in the United States and review of the literature. Dermatology Online J. 2011;17:2.
- Böer A, Ackerman AB. Prurigo Pigmentosa (Nagashima Disease): Textbook and Atlas of a Distinctive Inflammatory Disease of the Skin. New York, NY: Ardor Scribendi Ltd; 2004.
- Teraki Y, Teraki E, Kawashima M, at al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511.
- Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2011;26:1149-1153.
- Yokozeki M, Watanabe J, Hotsubo T, et al. Prurigo pigmentosa disappeared following improvement of diabetic ketosis by insulin. J Dermatol. 2003;30:257-258.
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
- VanItallie TB, Nufert TH. Ketones: metabolism’s ugly duckling. Annu Rev Nutr. 2003;61:327-341.
- Rains JL, Jain SK. Hyperketonemia increases monocyte adhesion to endothelial cells and is mediated by LFA-1 expression in monocytes and ICAM-1 expression in endothelial cells. Am J Physiol Endocrinol Metab. 2011;301:e298-e306.
- Teraki Y, Shiohara T, Nagashima M, et al. Prurigo pigmentosa: role of ICAM-1 in the localization of the eruption. Br J Dermatol. 1991;125:360-363.
- Kashihara-Sawami M, Norris DA. The state of differentiation of cultured human keratinocytes determines the level of intercellular adhesion molecule-1 (ICAM-1) expression induced by gamma interferon. J Invest Dermatol. 1992;98:741-747.
- Middleton MH, Norris DA. Cytokine-induced ICAM-1 expression in human keratinocytes is highly variable in keratinocyte strains from different donors. J Invest Dermatol. 1995;104:489-496.
- What is the paleo diet? The Paleo Diet website. http://thepaleodiet.com/the-paleo-diet-premise. Accessed March 9, 2019.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation [in Japanese]. Japanese J Dermatol. 1971;81:38-39.
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet [published online December 30, 2013]. Pediatr Dermatol. 2015;32:248-251.
- Baykal C, Buyukbabani N, Akinturk S, et al. Prurigo pigmentosa: not an uncommon disease in the Turkish population. Int J Dermatol. 2006;45:1164-1168.
- Whang T, Kirkorian Y, Krishtul A, et al. Prurigo pigmentosa: report of two cases in the United States and review of the literature. Dermatology Online J. 2011;17:2.
- Böer A, Ackerman AB. Prurigo Pigmentosa (Nagashima Disease): Textbook and Atlas of a Distinctive Inflammatory Disease of the Skin. New York, NY: Ardor Scribendi Ltd; 2004.
- Teraki Y, Teraki E, Kawashima M, at al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511.
- Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2011;26:1149-1153.
- Yokozeki M, Watanabe J, Hotsubo T, et al. Prurigo pigmentosa disappeared following improvement of diabetic ketosis by insulin. J Dermatol. 2003;30:257-258.
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
- VanItallie TB, Nufert TH. Ketones: metabolism’s ugly duckling. Annu Rev Nutr. 2003;61:327-341.
- Rains JL, Jain SK. Hyperketonemia increases monocyte adhesion to endothelial cells and is mediated by LFA-1 expression in monocytes and ICAM-1 expression in endothelial cells. Am J Physiol Endocrinol Metab. 2011;301:e298-e306.
- Teraki Y, Shiohara T, Nagashima M, et al. Prurigo pigmentosa: role of ICAM-1 in the localization of the eruption. Br J Dermatol. 1991;125:360-363.
- Kashihara-Sawami M, Norris DA. The state of differentiation of cultured human keratinocytes determines the level of intercellular adhesion molecule-1 (ICAM-1) expression induced by gamma interferon. J Invest Dermatol. 1992;98:741-747.
- Middleton MH, Norris DA. Cytokine-induced ICAM-1 expression in human keratinocytes is highly variable in keratinocyte strains from different donors. J Invest Dermatol. 1995;104:489-496.
Practice Points
- Ketosis can be associated with a specific rash known as prurigo pigmentosa (PP).
- Resolution of PP is related to re-introduction of carbohydrates into the diet.
- Consider asking about dietary modifications in patients presenting with a new rash.
Radiographic Changes of Osteomyelitis in a Patient With Periungual Lichen Planus
To the Editor:
A 60-year-old woman presented for evaluation of a 1-year history of left hallux nail plate dystrophy and proximal nail fold inflammation. Her medical history included Cushing disease with associated uncontrolled diabetes mellitus (DM) and a remote history of cutaneous lichen planus (LP) that resolved 15 years prior to presentation. She noted improvement during intravenous courses of antibiotics for other infections.
Examination of the left hallux revealed onycholysis, loss of the nail plate, and a yellow fibrinous base alongside erosion, erythema, and edema of the proximal toenail fold (Figure 1). The left second toe pad was markedly tender to palpation with scant exudate expressed from underneath the nail bed. Two biopsies of the hallux were performed. The proximal nail fold specimen revealed mild epidermal hyperplasia, and the nail bed demonstrated a nonspecific ulcer that was negative for acid-fast bacilli and fungi.
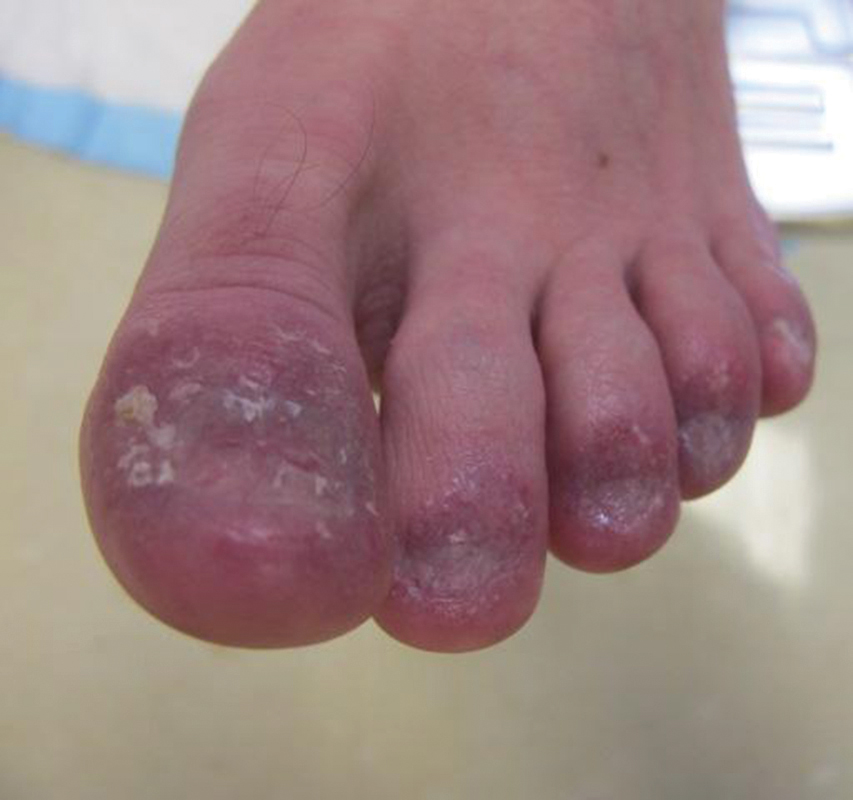
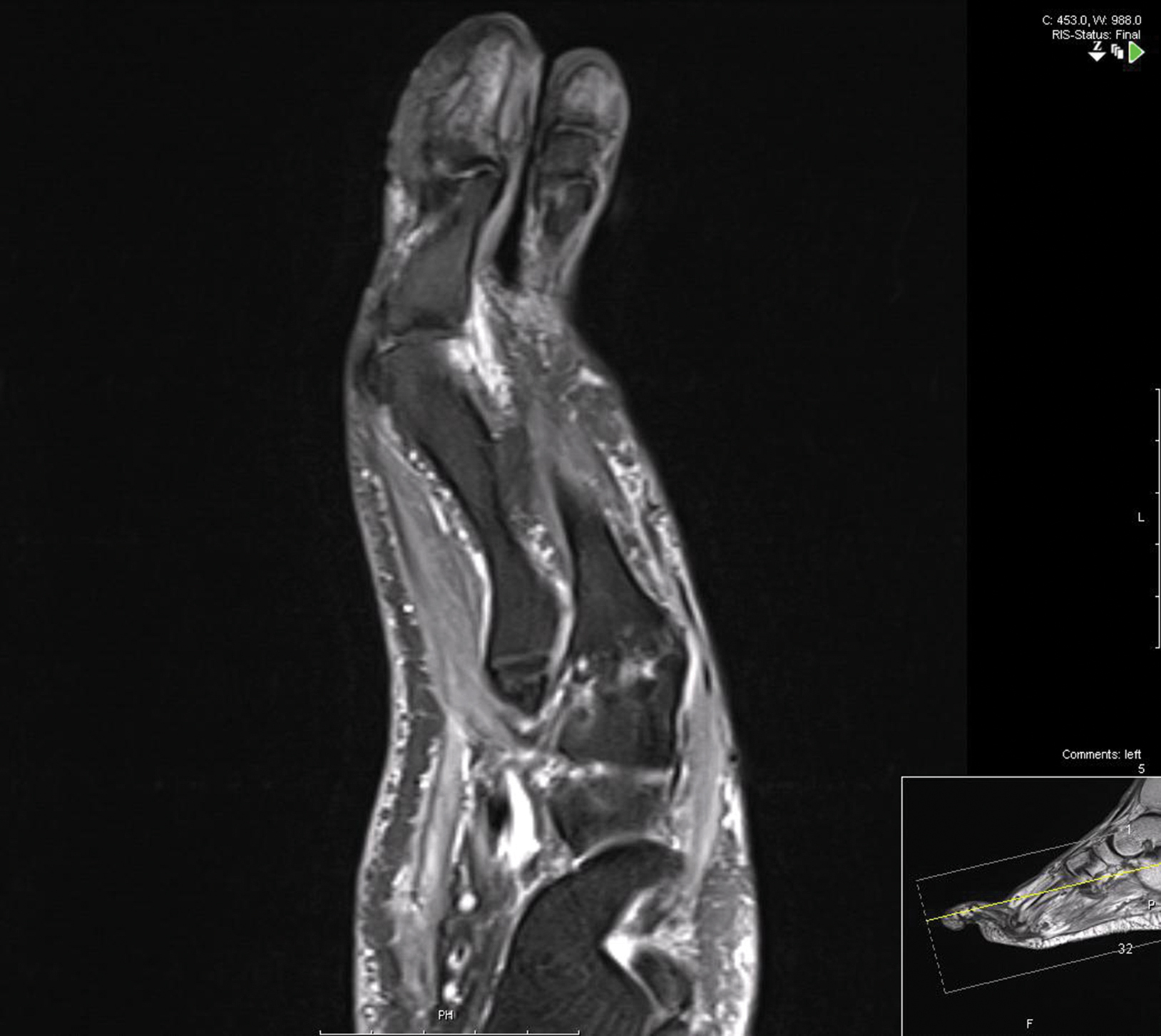
Treatment over 2 months with cephalexin yielded improvement in both erythema and edema. Initial and repeat nail plate cultures grew ampicillin- and penicillin-sensitive Enterococcus faecalis. Magnetic resonance imaging was performed to evaluate for osteomyelitis because of lack of resolution. Results demonstrated osteomyelitis of the distal tuft of the left hallux and the distal phalanx of the second toe (Figure 2). Vascular surgery evaluation revealed no evidence of large vessel arterial insufficiency. She was started on amoxicillin for superficial Enterococcus and ciprofloxacin for underlying enteric bacilli. The persistence of infection was attributed to microvascular disease secondary to the patient's associated DM. Months later, due to suspected worsening of osteomyelitis, she underwent treatment with oral fluconazole to cover potential fungal co-infection and intravenous vancomycin and piperacillin-tazobactam for broad-spectrum antibacterial coverage. She was eventually transitioned to antimicrobial agents including amoxicillin-clavulanate potassium and topical mupirocin with improvement in periungual erythema and edema.
On subsequent dermatologic evaluation after 1 month, she presented with pterygium and loss of all nail plates on the left foot. The nail bed now had a violaceous color and was studded with milia. The clinical findings were suggestive of LP, consistent with her history of LP. In light of these new findings, both topical corticosteroids and retinoids were utilized for treatment without remarkable benefit. The patient declined further management with systemic medications.
We report a case of nail LP associated with underlying radiographic osteomyelitis. Erosive nail LP has been associated with underlying osteomyelitis of the phalanx.1 Our patient developed these manifestations in the setting of Cushing disease, a unique finding given that many report improvement of LP with systemic corticosteroids.2,3 Tacrolimus, a calcineurin inhibitor, has been used in oral or topical formulations for lower extremity ulcers caused by LP as well as nail LP.1,4 Long-term prognosis of nail LP is poor, with high relapse rates and permanent damage to the nail unit.2 It is important to be aware that LP of the nail unit may cause radiographic changes of osteomyelitis that are not infectious in nature.
- Miller S. The effect of tacrolimus on lower extremity ulcers: a case study and review of the literature. Ostomy Wound Manage. 2008;54:36-42.
- Goettmann S, Zaraa I, Moulonguet I. Nail lichen planus: epidemiological, clinical, pathological, therapeutic and prognosis study of 67 cases. Eur Acad Dermatol Venereol. 2012;26:1304-1309.
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496.
- Ujiie H, Shibaki A, Akiyama M, et al. Successful treatment of nail lichen planus with topical tacrolimus. Acta Derm Venereol. 2010;90:218-219.
To the Editor:
A 60-year-old woman presented for evaluation of a 1-year history of left hallux nail plate dystrophy and proximal nail fold inflammation. Her medical history included Cushing disease with associated uncontrolled diabetes mellitus (DM) and a remote history of cutaneous lichen planus (LP) that resolved 15 years prior to presentation. She noted improvement during intravenous courses of antibiotics for other infections.
Examination of the left hallux revealed onycholysis, loss of the nail plate, and a yellow fibrinous base alongside erosion, erythema, and edema of the proximal toenail fold (Figure 1). The left second toe pad was markedly tender to palpation with scant exudate expressed from underneath the nail bed. Two biopsies of the hallux were performed. The proximal nail fold specimen revealed mild epidermal hyperplasia, and the nail bed demonstrated a nonspecific ulcer that was negative for acid-fast bacilli and fungi.
Treatment over 2 months with cephalexin yielded improvement in both erythema and edema. Initial and repeat nail plate cultures grew ampicillin- and penicillin-sensitive Enterococcus faecalis. Magnetic resonance imaging was performed to evaluate for osteomyelitis because of lack of resolution. Results demonstrated osteomyelitis of the distal tuft of the left hallux and the distal phalanx of the second toe (Figure 2). Vascular surgery evaluation revealed no evidence of large vessel arterial insufficiency. She was started on amoxicillin for superficial Enterococcus and ciprofloxacin for underlying enteric bacilli. The persistence of infection was attributed to microvascular disease secondary to the patient's associated DM. Months later, due to suspected worsening of osteomyelitis, she underwent treatment with oral fluconazole to cover potential fungal co-infection and intravenous vancomycin and piperacillin-tazobactam for broad-spectrum antibacterial coverage. She was eventually transitioned to antimicrobial agents including amoxicillin-clavulanate potassium and topical mupirocin with improvement in periungual erythema and edema.
On subsequent dermatologic evaluation after 1 month, she presented with pterygium and loss of all nail plates on the left foot. The nail bed now had a violaceous color and was studded with milia. The clinical findings were suggestive of LP, consistent with her history of LP. In light of these new findings, both topical corticosteroids and retinoids were utilized for treatment without remarkable benefit. The patient declined further management with systemic medications.
We report a case of nail LP associated with underlying radiographic osteomyelitis. Erosive nail LP has been associated with underlying osteomyelitis of the phalanx.1 Our patient developed these manifestations in the setting of Cushing disease, a unique finding given that many report improvement of LP with systemic corticosteroids.2,3 Tacrolimus, a calcineurin inhibitor, has been used in oral or topical formulations for lower extremity ulcers caused by LP as well as nail LP.1,4 Long-term prognosis of nail LP is poor, with high relapse rates and permanent damage to the nail unit.2 It is important to be aware that LP of the nail unit may cause radiographic changes of osteomyelitis that are not infectious in nature.
To the Editor:
A 60-year-old woman presented for evaluation of a 1-year history of left hallux nail plate dystrophy and proximal nail fold inflammation. Her medical history included Cushing disease with associated uncontrolled diabetes mellitus (DM) and a remote history of cutaneous lichen planus (LP) that resolved 15 years prior to presentation. She noted improvement during intravenous courses of antibiotics for other infections.
Examination of the left hallux revealed onycholysis, loss of the nail plate, and a yellow fibrinous base alongside erosion, erythema, and edema of the proximal toenail fold (Figure 1). The left second toe pad was markedly tender to palpation with scant exudate expressed from underneath the nail bed. Two biopsies of the hallux were performed. The proximal nail fold specimen revealed mild epidermal hyperplasia, and the nail bed demonstrated a nonspecific ulcer that was negative for acid-fast bacilli and fungi.
Treatment over 2 months with cephalexin yielded improvement in both erythema and edema. Initial and repeat nail plate cultures grew ampicillin- and penicillin-sensitive Enterococcus faecalis. Magnetic resonance imaging was performed to evaluate for osteomyelitis because of lack of resolution. Results demonstrated osteomyelitis of the distal tuft of the left hallux and the distal phalanx of the second toe (Figure 2). Vascular surgery evaluation revealed no evidence of large vessel arterial insufficiency. She was started on amoxicillin for superficial Enterococcus and ciprofloxacin for underlying enteric bacilli. The persistence of infection was attributed to microvascular disease secondary to the patient's associated DM. Months later, due to suspected worsening of osteomyelitis, she underwent treatment with oral fluconazole to cover potential fungal co-infection and intravenous vancomycin and piperacillin-tazobactam for broad-spectrum antibacterial coverage. She was eventually transitioned to antimicrobial agents including amoxicillin-clavulanate potassium and topical mupirocin with improvement in periungual erythema and edema.
On subsequent dermatologic evaluation after 1 month, she presented with pterygium and loss of all nail plates on the left foot. The nail bed now had a violaceous color and was studded with milia. The clinical findings were suggestive of LP, consistent with her history of LP. In light of these new findings, both topical corticosteroids and retinoids were utilized for treatment without remarkable benefit. The patient declined further management with systemic medications.
We report a case of nail LP associated with underlying radiographic osteomyelitis. Erosive nail LP has been associated with underlying osteomyelitis of the phalanx.1 Our patient developed these manifestations in the setting of Cushing disease, a unique finding given that many report improvement of LP with systemic corticosteroids.2,3 Tacrolimus, a calcineurin inhibitor, has been used in oral or topical formulations for lower extremity ulcers caused by LP as well as nail LP.1,4 Long-term prognosis of nail LP is poor, with high relapse rates and permanent damage to the nail unit.2 It is important to be aware that LP of the nail unit may cause radiographic changes of osteomyelitis that are not infectious in nature.
- Miller S. The effect of tacrolimus on lower extremity ulcers: a case study and review of the literature. Ostomy Wound Manage. 2008;54:36-42.
- Goettmann S, Zaraa I, Moulonguet I. Nail lichen planus: epidemiological, clinical, pathological, therapeutic and prognosis study of 67 cases. Eur Acad Dermatol Venereol. 2012;26:1304-1309.
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496.
- Ujiie H, Shibaki A, Akiyama M, et al. Successful treatment of nail lichen planus with topical tacrolimus. Acta Derm Venereol. 2010;90:218-219.
- Miller S. The effect of tacrolimus on lower extremity ulcers: a case study and review of the literature. Ostomy Wound Manage. 2008;54:36-42.
- Goettmann S, Zaraa I, Moulonguet I. Nail lichen planus: epidemiological, clinical, pathological, therapeutic and prognosis study of 67 cases. Eur Acad Dermatol Venereol. 2012;26:1304-1309.
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496.
- Ujiie H, Shibaki A, Akiyama M, et al. Successful treatment of nail lichen planus with topical tacrolimus. Acta Derm Venereol. 2010;90:218-219.
Practice Points
- Lichen planus (LP) is an inflammatory mucocutaneous disorder with variable presentations.
- With extensive nail involvement, nail LP may impart radiographic findings suggestive of osteomyelitis.
Concurrent Keratoacanthomas and Nonsarcoidal Granulomatous Reactions in New and Preexisting Tattoos
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
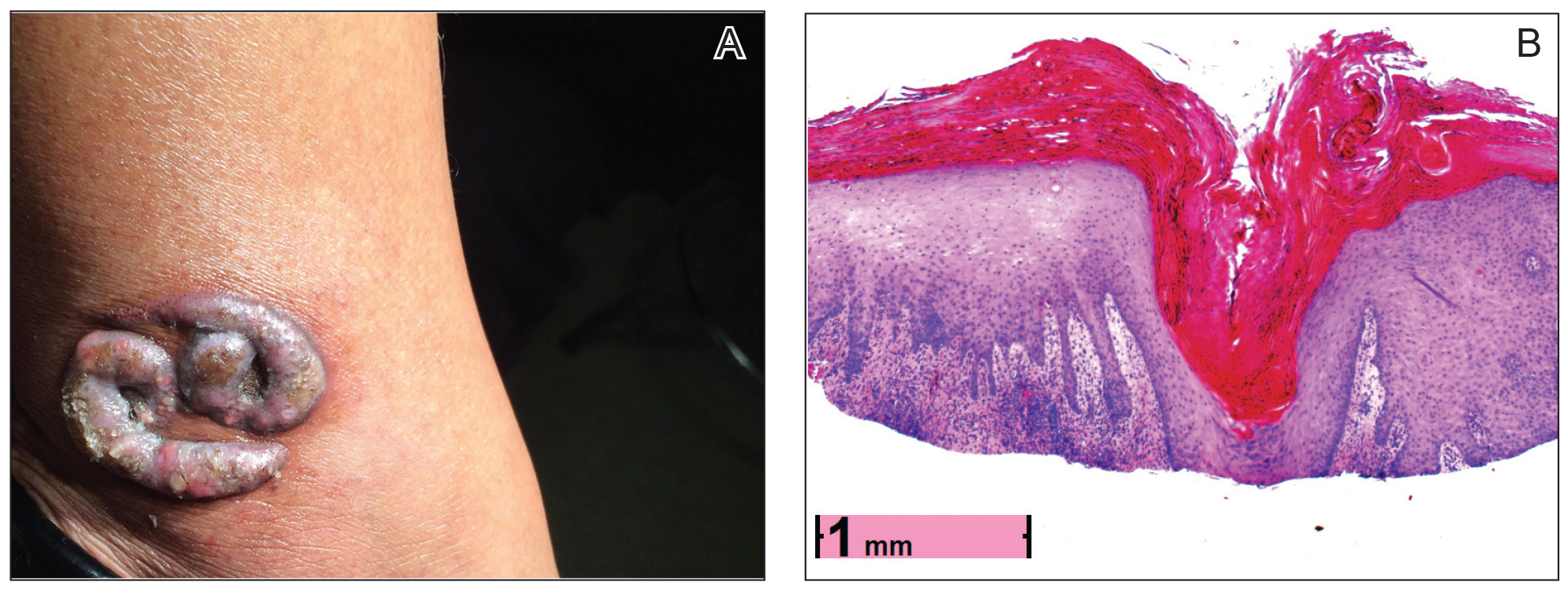
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.

contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
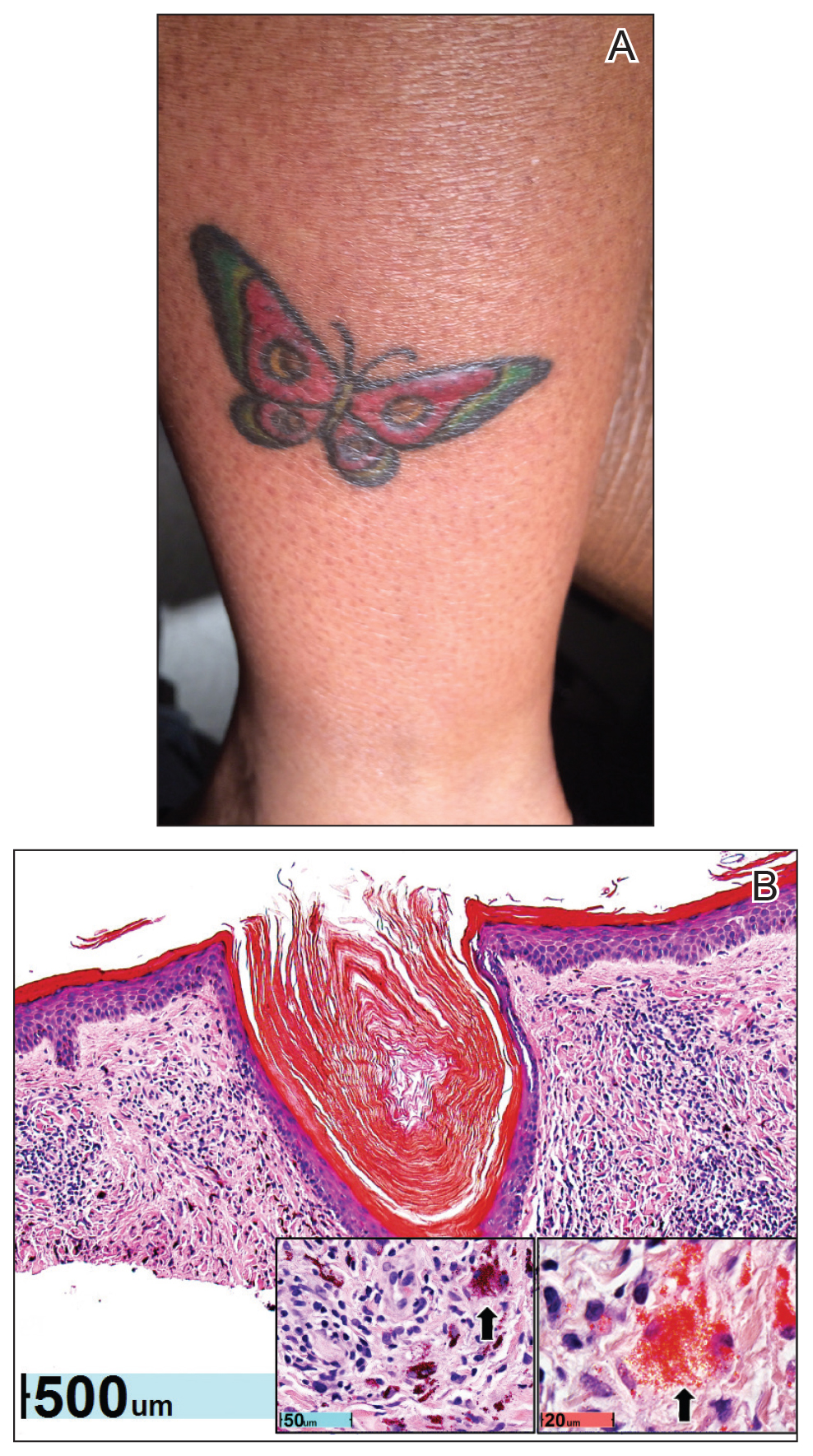
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
To the Editor:
Cutaneous reactions to tattoos are common and histologically diverse. As outlined by Jacob,1 these reactions can be categorized into 4 main groups: inoculative/infective, hypersensitive, neoplastic, and coincidental. A thorough history and physical examination can aid in distinguishing the type of cutaneous reaction, but diagnosis often requires histopathologic clarification. We report the case of a patient who presented with painful indurated nodules within red ink areas of new and preexisting tattoos.
A 48-year-old woman with no prior medical conditions presented with tender pruritic nodules at the site of a new tattoo and within recently retouched tattoos of 5 months’ duration. The tattoos were done at an “organic” tattoo parlor 8 months prior to presentation. Simultaneously, the patient also developed induration and pain in 2 older tattoos that had been done 10 years prior and had not been retouched.
Physical examination revealed 2 smooth and serpiginous nodules nested perfectly within the new red tattoo on the left medial ankle (Figure 1A). Examination of the retouched tattoos on the dorsum of the right foot revealed 4 discrete nodules within the red, heart-shaped areas of the tattoos (Figure 2A). Additionally, the red-inked portions of an older tattoo on the left lateral calf that were outlined in red ink also were raised and indurated (Figure 3A), and a tattoo on the right volar wrist, also in red ink, was indurated and tender to palpation. The remainder of the physical examination was normal.
contiguous dilated follicular infundibula with atypical keratinocytes that had hyperchromatic nuclei, consistent with a keratoacanthoma, as well as a lymphocytic infiltrate in the dermis above a dense infiltrate of lymphocytes and histiocytes (H&E, original magnification ×2.5 [original magnification ×6.2]).
The lesions continued to enlarge and become increasingly painful despite trials of fluticasone propionate cream 0.05%, clobetasol propionate gel 0.05%, a 7-day course of oral levofloxacin, and a 10-day course of oral amoxicillin-clavulanate. Ultimately, a shave biopsy from the new tattoo on the left medial ankle revealed an early keratoacanthoma (KA)(Figure 1B). Subsequent shave biopsies of the retouched tattoos on the dorsal foot and the preexisting tattoo on the calf revealed KAs and a granulomatous reaction, respectively (Figures 2B and 3B). The left ankle KA was treated with 2 injections of 5-fluorouracil without improvement. The patient ultimately underwent Mohs micrographic surgery of the left ankle KA and underwent total excision with skin graft.
The development of KAs within tattoos is a known but poorly understood phenomenon.2 Keratoacanthomas are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution. They typically present as solitary isolated nodules arising in sun-exposed areas of patients of either sex, with a predilection for individuals of Fitzpatrick skin types I and II and in areas of prior trauma or sun damage.3
Histologically, the proliferative phase is defined by keratin-filled invagination of the epidermis into the dermis, with areas of hyperkeratosis, acanthosis, and mitotic activity within the strands and nodules. A high degree of nuclear atypia underlines the diagnostic difficulty in distinguishing KAs from squamous cell carcinomas (SCCs). A fully developed KA has less prominent cellular atypia and a characteristic buttressing lip of epithelium extending over the edges of an irregular, keratin-filled crater. In the final involution stage of KAs, granulation tissue and fibrosis predominate and apoptotic cells may be noted.4
The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, treatment with BRAF inhibitors, and trauma. Keratoacanthoma incidence also has been associated with chronic scarring diseases such as discoid lupus erythematous5 and lichen planus.6 Although solitary lesions are more typical, multiple generalized KAs can arise at once, as observed in generalized eruptive KA of Grzybowski, a rare condition, as well as in the multiple self-healing epitheliomas seen in Ferguson-Smith disease.
Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Some contest that KAs are benign and self-limited reactive proliferations, whereas others propose they are malignant variants of SCC.3,4,7,8 This debate is compounded by the difficulty in distinguishing KAs from SCC when specimen sampling is inadequate and given documentation that SCCs can develop within KAs over time.7 There also is some concern regarding the remote possibility of aggressive infiltration and even metastasis. One systematic review by Savage and Maize8 attempted to clarify the biologic behavior and malignant potential of KAs. Their review of 445 cases of KA with reported follow-up led to the conclusion that KAs exhibit a benign natural course with no reliable reports of death or metastasis. This finding was in stark contrast to 429 cases of SCC, of which 61 cases (14.2%) resulted in metastasis despite treatment.8
Our patient’s presentation was unique compared to others already reported in the literature because of the simultaneous development of nonsarcoidal granulomatous dermatitis within the older and nonretouched tattoos. Nonsarcoidal granulomatous dermatitis, which encompasses inflammatory skin diseases with histiocytes, is a reactive cutaneous proliferation that also has been reported to occur within tattoos.9,10 Granulomatous tattoo reactions can be further subdivided as foreign body type or sarcoidal type. Foreign body reactions are distinguished by the presence of pigment-containing multinucleated giant cells (as seen in our patient), whereas the sarcoidal type contains compact nodules of epithelioid histiocytes with few lymphocytes.4
The concurrent development of 2 clinically and histologically distinct entities suggests that a similar overlapping pathogenesis underlies each. One hypothesis is that the introduction of exogenous dyes may have instigated an inflammatory foreign body reaction, with the red ink acting as the unifying offender. The formation of granulomas in the preexisting tattoos is likely explained by an exaggerated immune response in the form of a type IV delayed hypersensitivity reaction triggered by reintroduction of the antigen—the red ink—in a presensitized host. Secondly, the parallel development of KAs within the new and retouched tattoos could be a result of the traumatic direct inoculation of the foreign material to which the body was presensitized and subsequent attempt by the skin to degrade and remove it.11
This case provides an example of the development of multiple KAs via a reactive process. Many other similar cases have been described in the literature, including case reports of KAs arising in areas of trauma such as thermal burns, vaccination sites, scars, skin grafts, arthropod bites, and tattoos.2-4,8 Together, the trauma and immune response may lead to localized inflammation and/or cellular hyperplasia, ultimately predisposing the individual to the development of dermoepidermal proliferation. Moreover, the exaggerated keratinocyte proliferation in KAs in response to trauma is reminiscent of the Köbner phenomenon. Other lesions that demonstrate köbnerization also have been reported to occur within new tattoos, including psoriasis, lichen planus, molluscum contagiosum, and verruca vulgaris.1,3
Although KAs are not always a consequence of trauma among humans, trauma-induced KA has been proven as a reliable phenomenon among animal models; an older study showed consistent KA development after animal skin was traumatized from the application of chemical carcinogens.12 Keratoacanthomas within areas of trauma seem to develop rapidly—within a week to a year after trauma—while the development of trauma-related nonmelanoma skin cancers appears to take longer, approximately 1 to 50 years later.13
More research is needed to clarify the pathophysiology of KAs and its precise relationship to trauma and immunology, but our case adds additional weight to the idea that some KAs are primarily reactive phenomena, sharing features of other reactive cutaneous proliferations such as foreign body granulomas.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
- Jacob CI. Tattoo-associated dermatoses: a case report and review of the literature. Dermatol Surg. 2002;28:962-965.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Goldsmith LA, Katz SL, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia: Lippincott, 2005.
- Minicucci EM, Weber SA, Stolf HO, et al. Keratoacanthoma of the lower lip complicating discoid lupus erythematosus in a 14-year-old boy. Pediatr Dermatol. 2007;24:329-330.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Weedon DD, Malo J, Brooks D, et al. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32:423-426.
- Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol. 2014;36:422-429.
- Schwartz RA, Mathias CG, Miller CH, et al. Granulomatous reaction to purple tattoo pigment. Contact Derm. 1987;16:198-202.
- Bagley MP, Schwartz RA, Lambert WC. Hyperplastic reaction developing within a tattoo. granulomatous tattoo reaction, probably to mercuric sulfide (cinnabar). Arch Dermatol. 1987;123:1557, 1560-1561.
- Kluger N, Plantier F, Moguelet P, et al. Tattoos: natural history and histopathology of cutaneous reactions. Ann Dermatol Venereol. 2011;138:146-154.
- Ghadially FN, Barton BW, Kerridge DF. The etiology of keratoacanthoma. Cancer. 1963;16:603-611.
- Kluger N, Koljonen V. Tattoos, inks, and cancer. Lancet Oncol. 2012;13:e161-168.
Practice Points
- Keratoacanthomas (KAs) are common keratinizing, squamous cell lesions of follicular origin distinguished by their eruptive onset, rapid growth, and spontaneous involution.
- The etiology of KAs remains controversial, but several factors have been correlated with their development, including UV light exposure, chemical carcinogenesis, genetic predisposition, viruses (namely human papillomavirus infection), immunosuppression, scarring disorders, and trauma (including tattoos).
- Because of the unusual histology of KAs and their tendency to spontaneously regress, it is not totally understood where they fall on the benign vs malignant spectrum. Our case adds additional weight to the idea that some KAs are primarily reactive phenomena sharing features of other reactive cutaneous proliferations such as foreign body granulomas.

Chronic Lymphocytic Leukemia and Infiltrates Seen During Excision of Nonmelanoma Skin Cancer
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
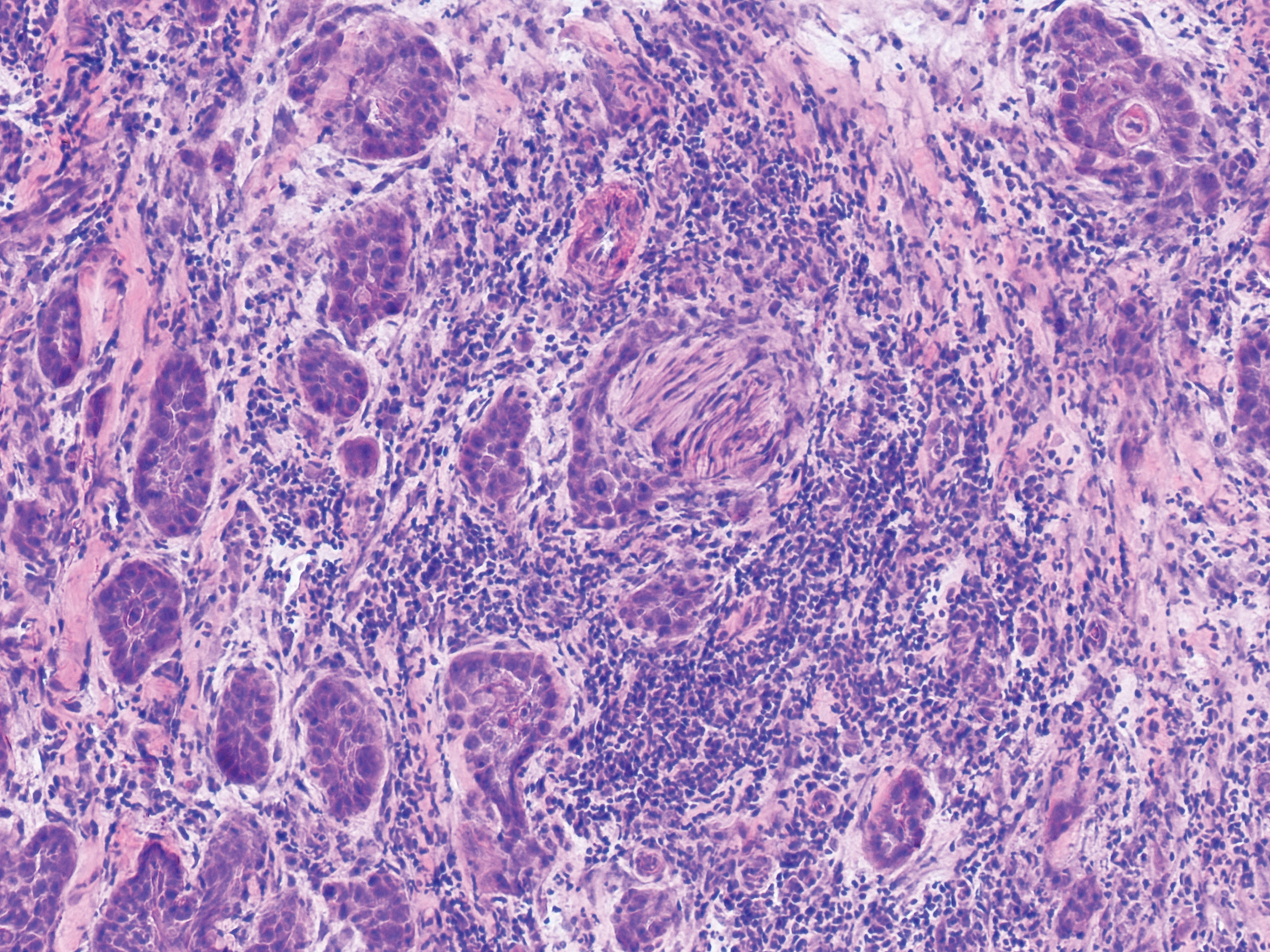
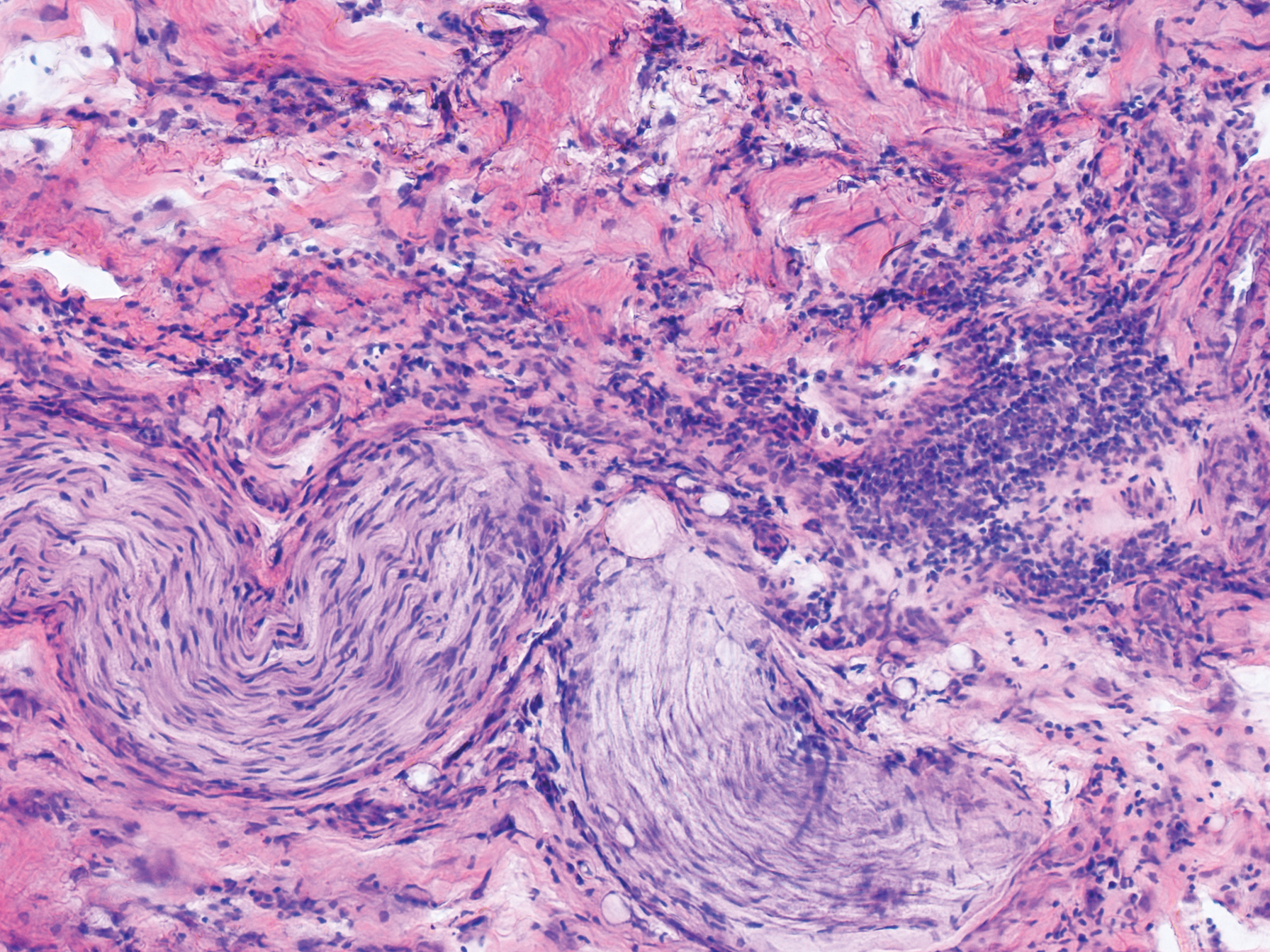
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7
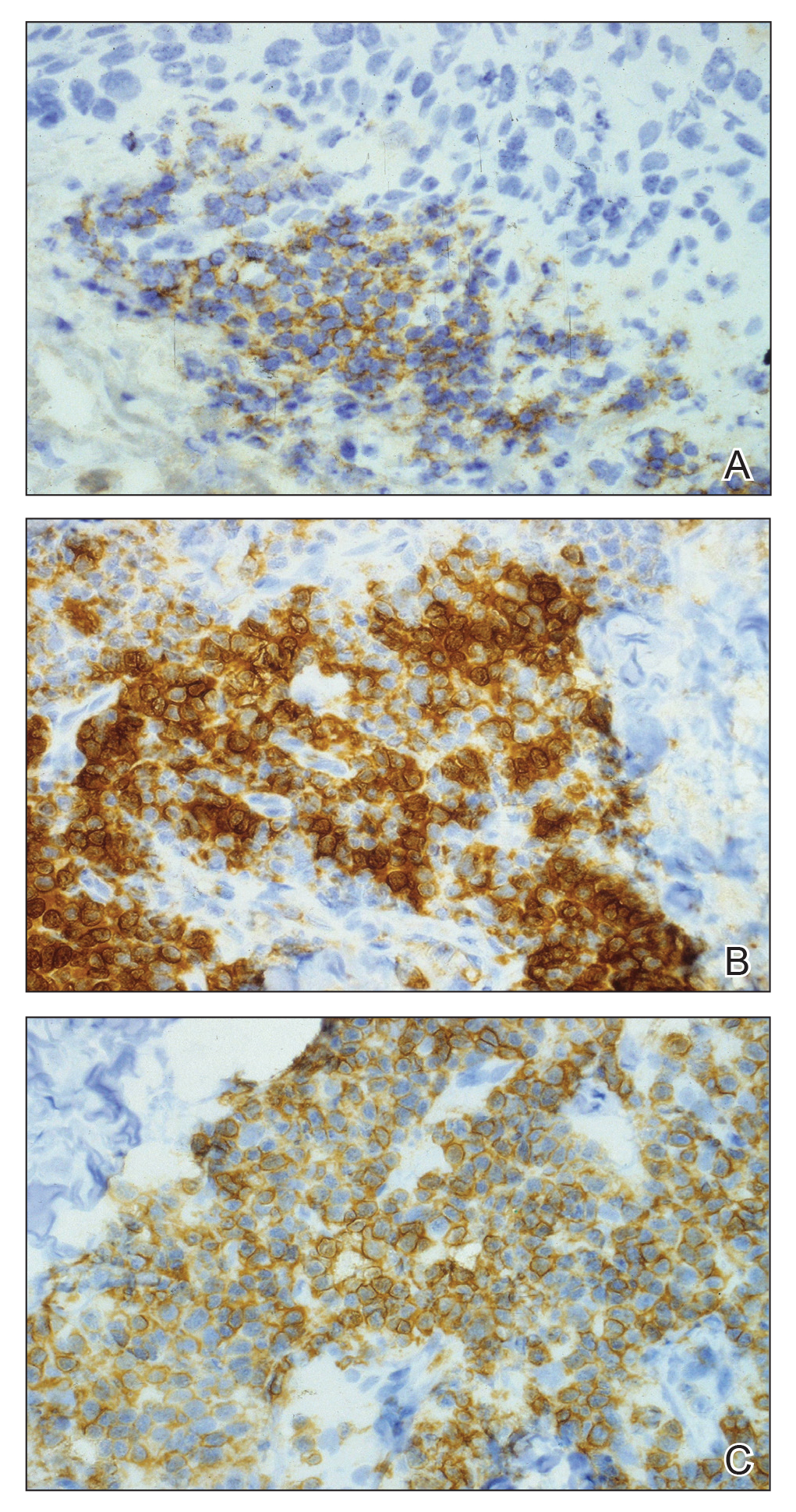

The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7
The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7
The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
Practice Points
- Chronic lymphocytic leukemia (CLL) may be seen during histologic examination of specimens during Mohs micrographic surgery as a monomorphic infiltrate of small mature lymphocytes with dense nuclei. Patients may be unaware of their diagnosis, which can be the presenting feature.
- An infiltrate of CLL may mimic aggressive behavior of nonmelanoma skin cancers including perineural invasion. A leukemic infiltrate may appear more dense and monomorphic. If needed, immunohistochemical staining of leukemic cells will show CD5 and CD23 positivity.
- Anecdotally, patients with CLL may not remember this pertinent medical history. Whether due to its asymptomatic nature or lack of treatment in early stages, direct questioning about CLL may be warranted if this characteristic infiltrate is encountered.
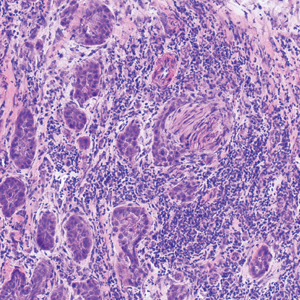
Cutaneous Collagenous Vasculopathy
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
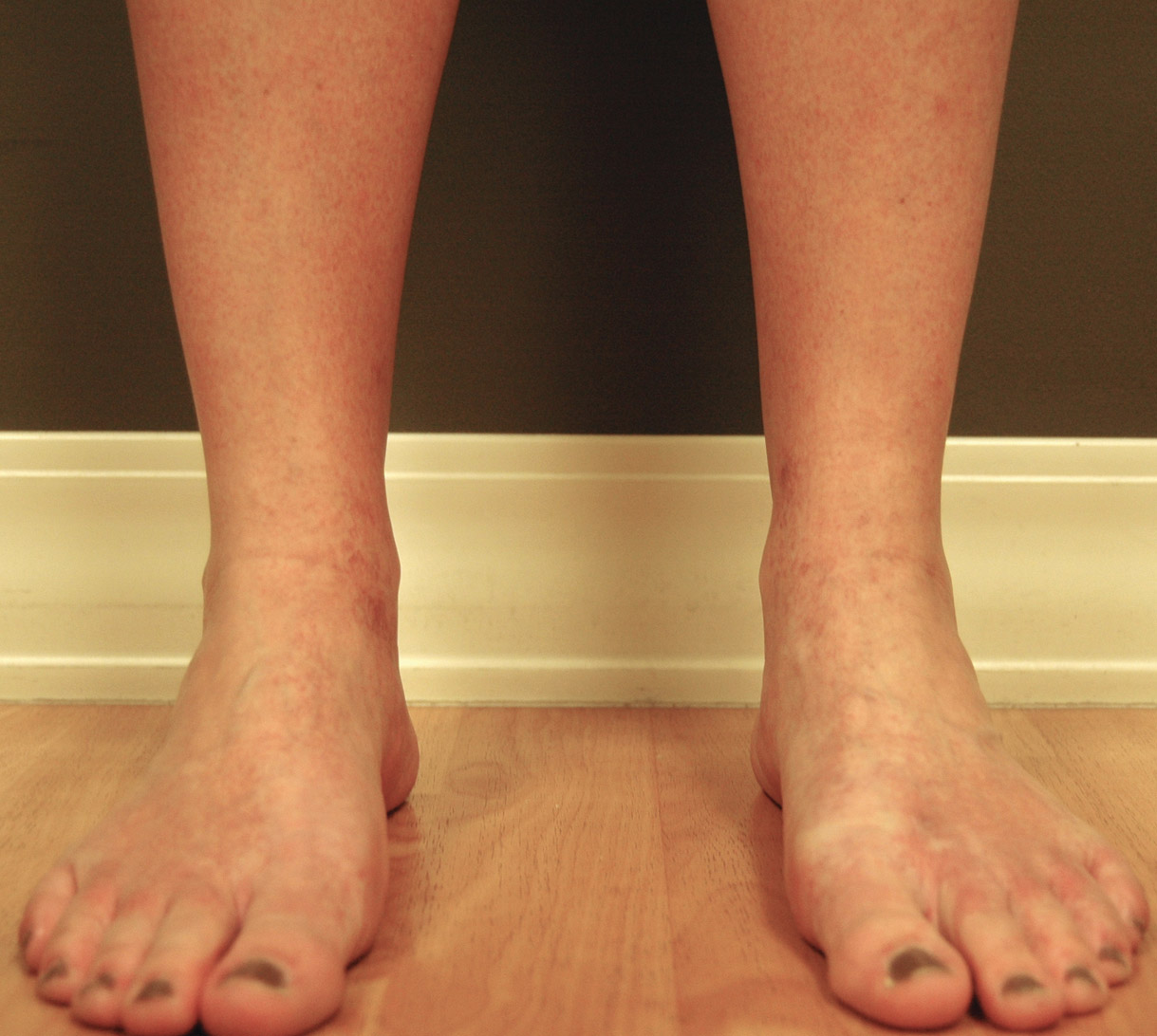
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
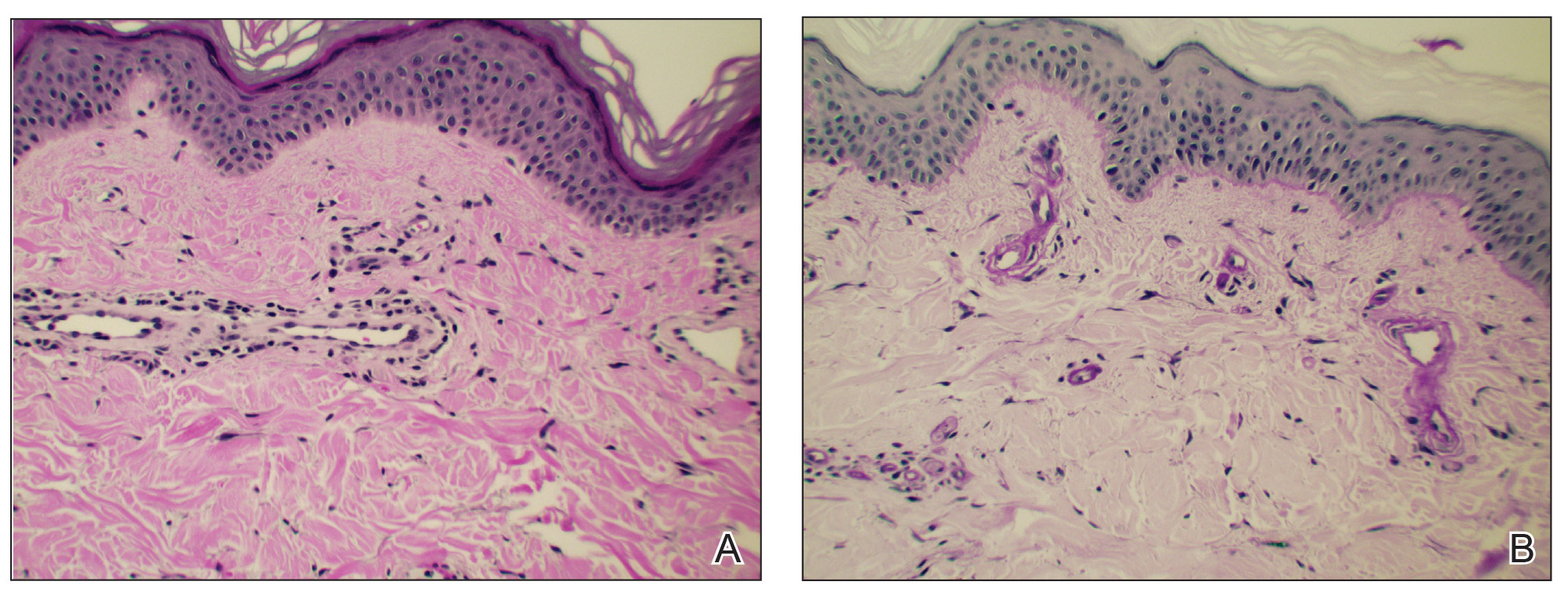
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
Practice Points
- In cutaneous collagenous vasculopathy (CCV), skin biopsy may demonstrate eosinophilic hyaline thickening of superficial dermal blood vessels with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes.
- Lack of mucous membrane and nail involvement differentiates CCV from hereditary hemorrhagic telangiectasia.
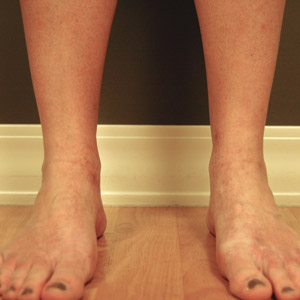
Elephantiasis Nostras Verrucosa Secondary to Scleroderma
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).

A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
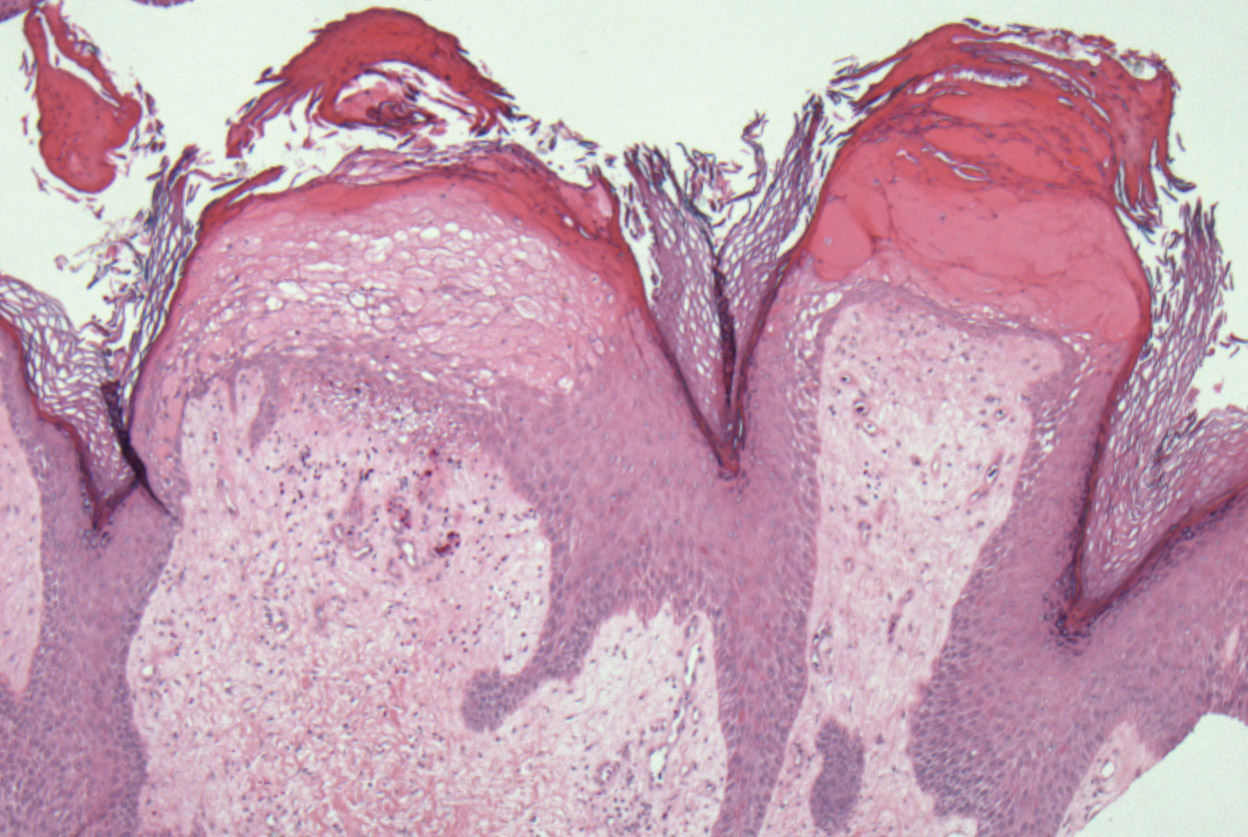
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
Practice Points
- Scleroderma rarely may lead to elephantiasis nostras verrucosa (ENV) of the upper extremities.
- Avoiding lymphostasis through compression and control of concomitant skin and soft tissue infections is important in the treatment and prevention of ENV.
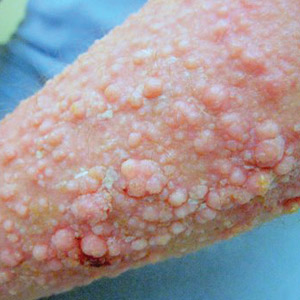
Neurofibromatosis Type 1 in the Setting of Systemic Lupus Erythematosus
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
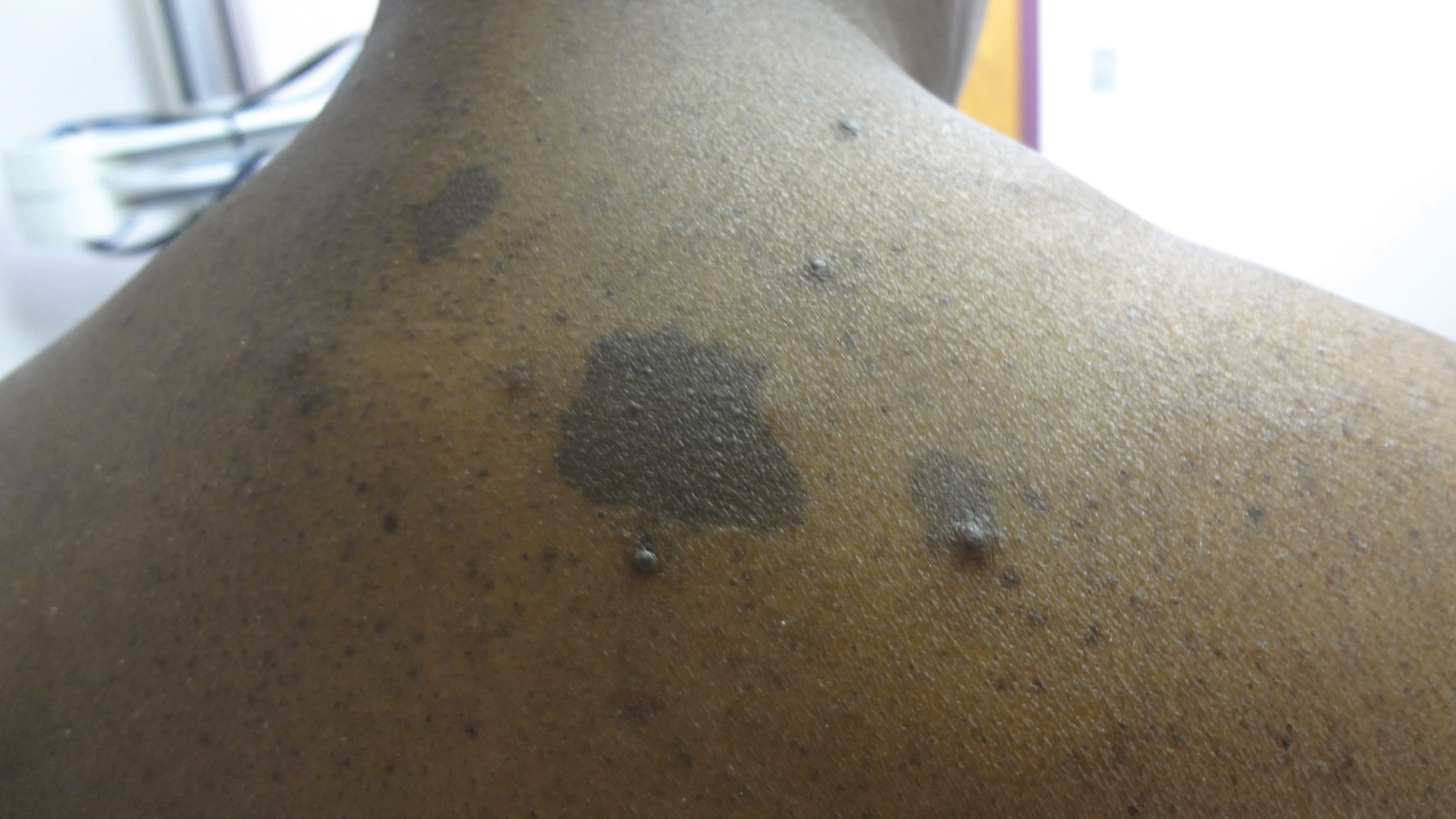
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.

We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
Practice Points
- Patients with neurofibromatosis type 1 (NF-1) benefit from early diagnosis and long-term follow-up.
- Patients with systemic lupus erythematosus (SLE) may develop different malignancies given the immune dysregulation. We recommend that patients with SLE undergo detailed skin examinations to check for subtle clues for NF-1.
- Similarly, patients with NF-1 can develop SLE later in life.
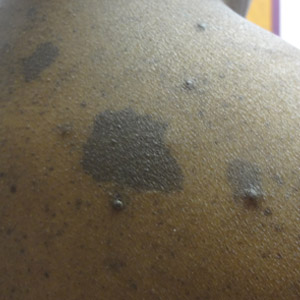
Locally Destructive Metastatic Basal Cell Carcinoma
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
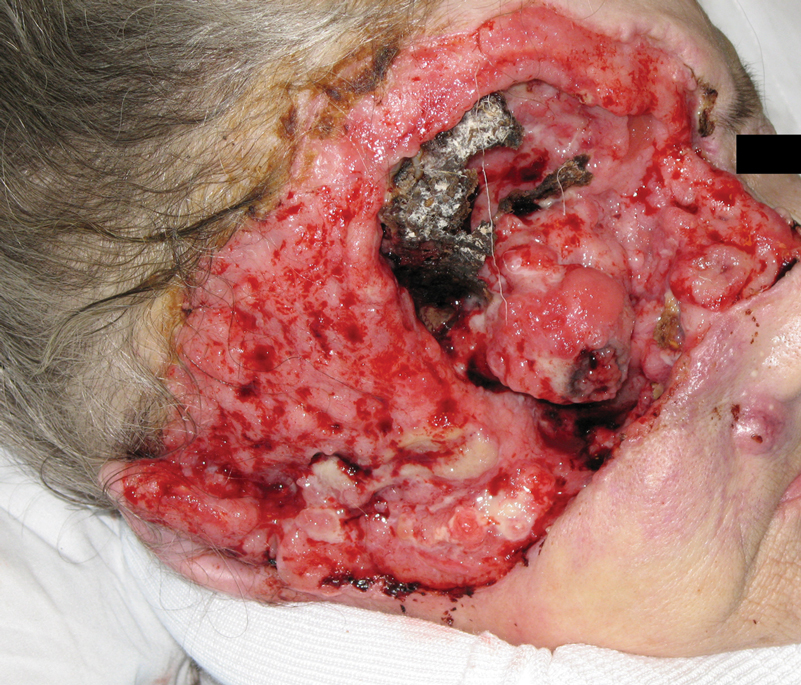
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
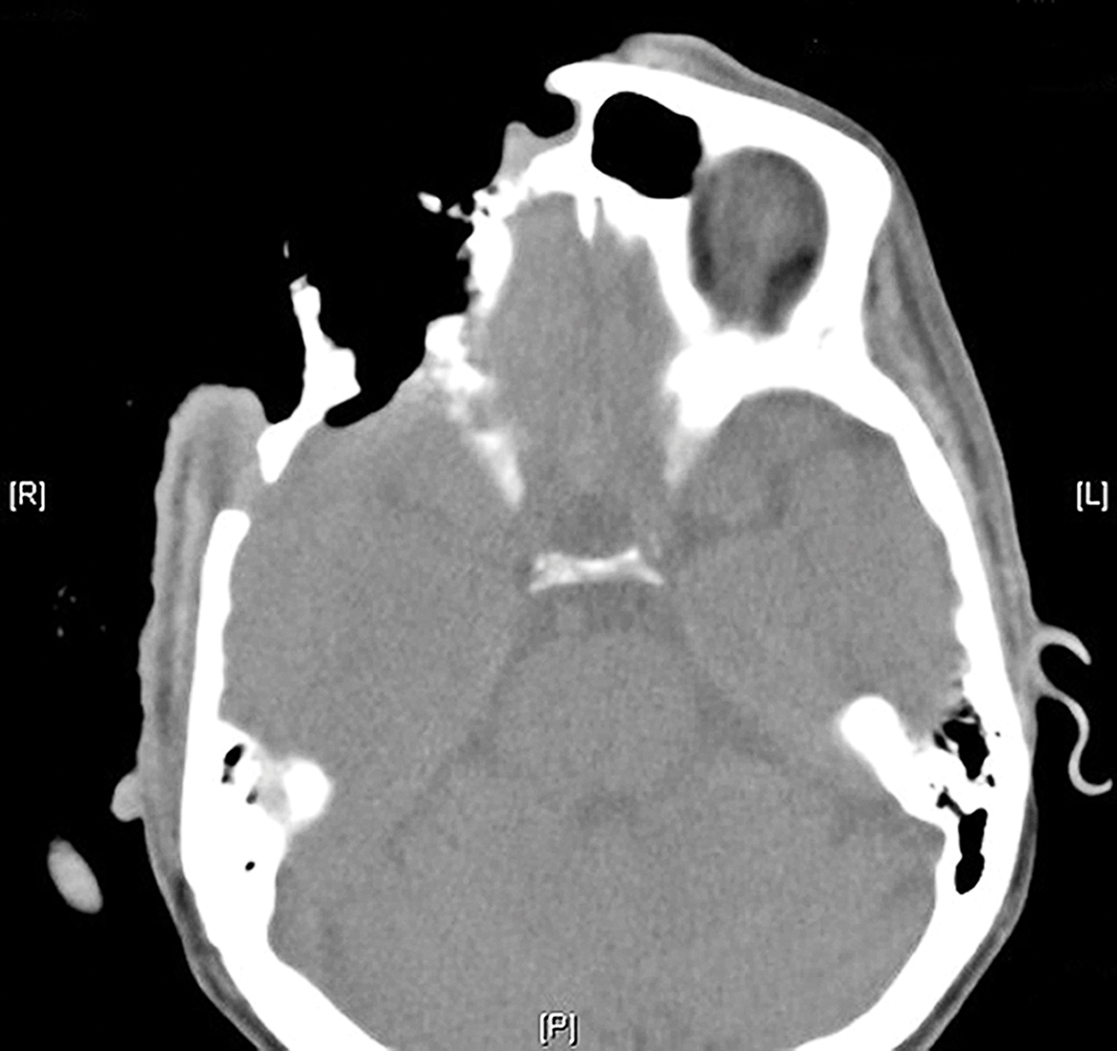
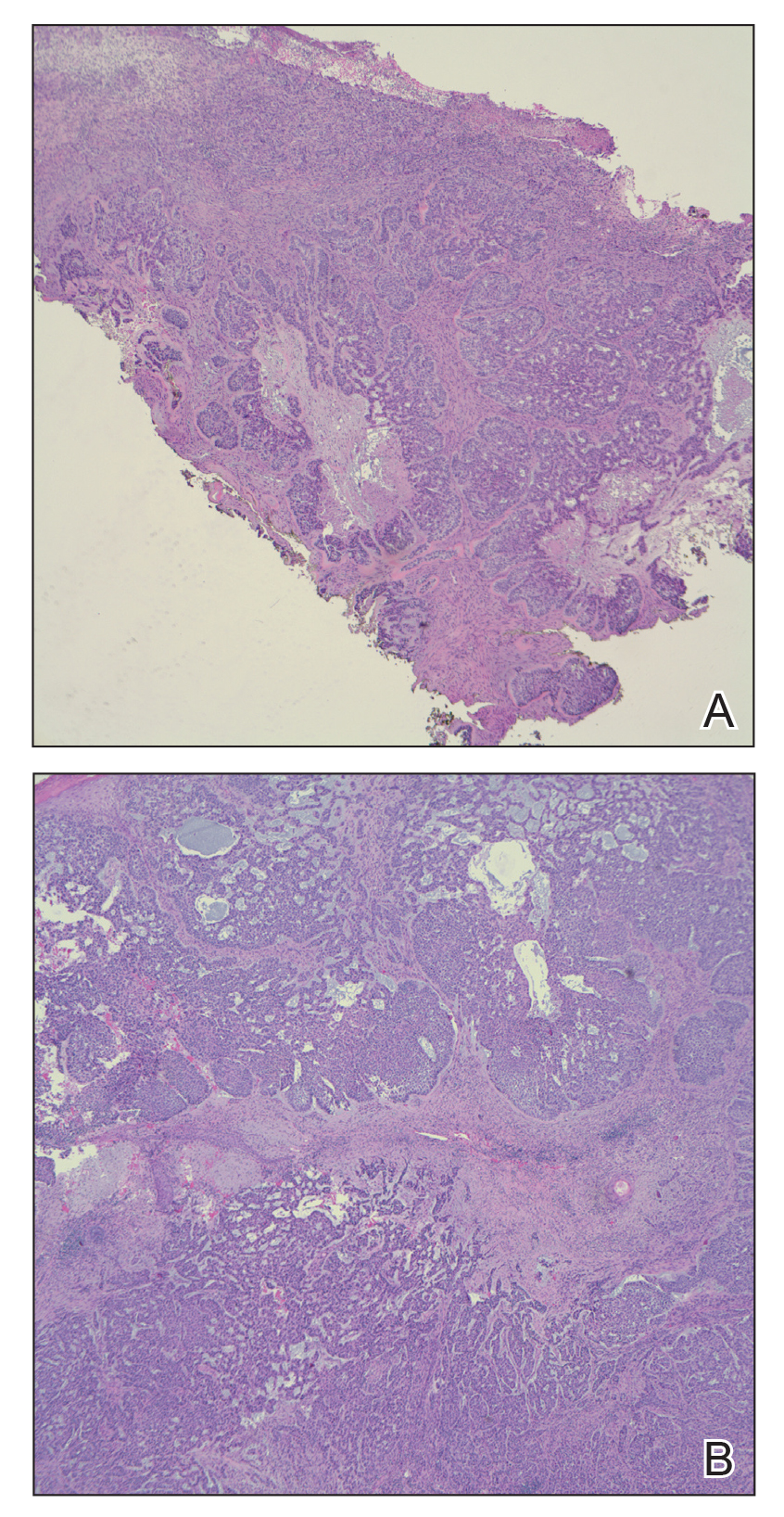
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
Practice Points
- Risk factors associated with metastatic spread of basal cell carcinoma (BCC) include larger tumor size, greater depth of invasion, long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.
- The median interval between onset of BCC and metastasis has been shown to be approximately 9 years.
- Vismodegib can be an effective oral therapy for patients with metastatic BCC, locally advanced BCC, recurrence following surgery, or those who are not candidates for surgery or radiation.
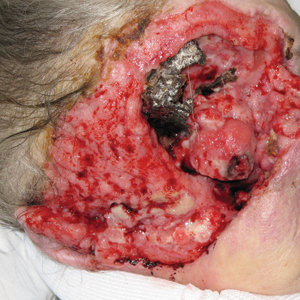
Multicentric Reticulohistiocytosis With Arthralgia and Red-Orange Papulonodules
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
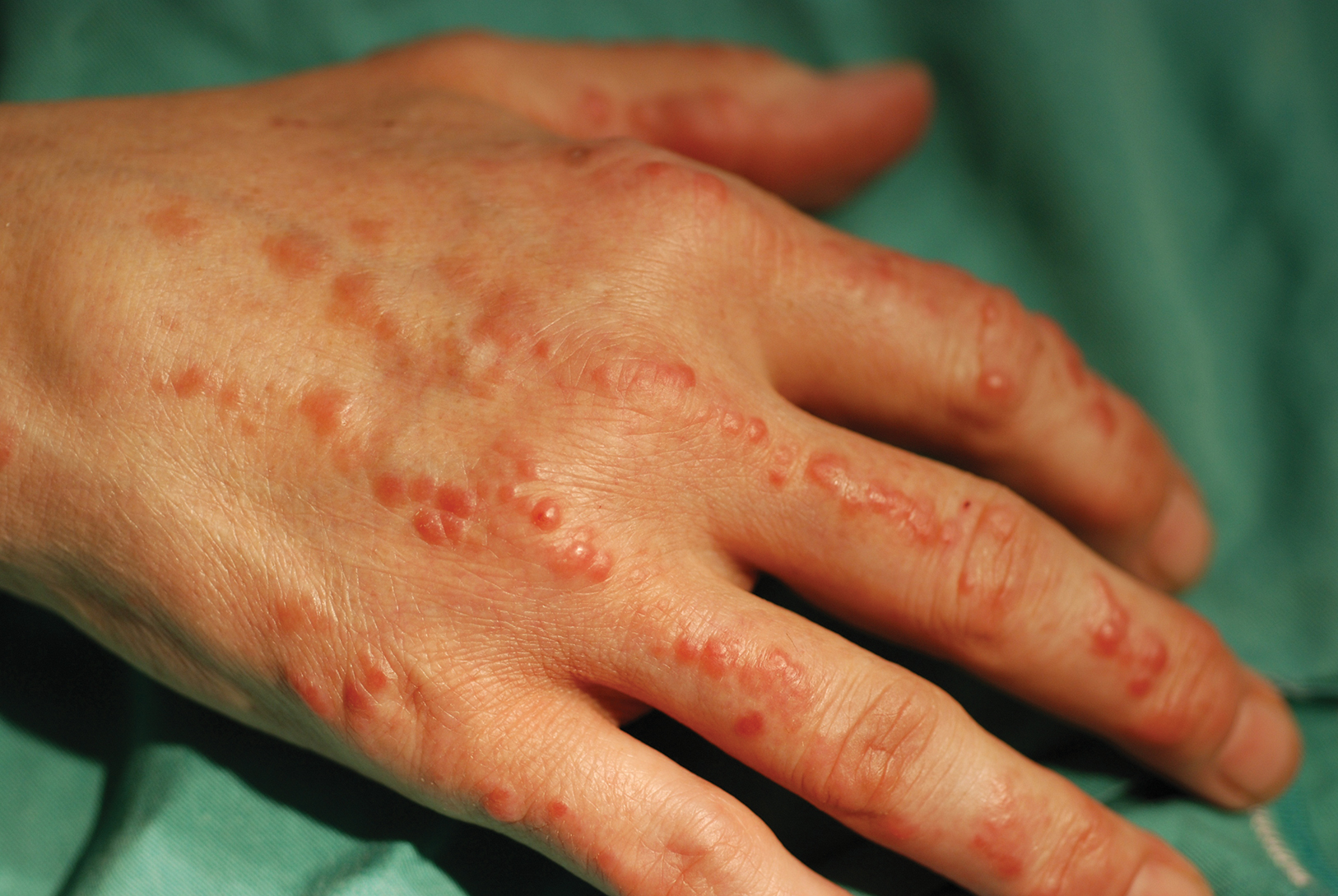
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
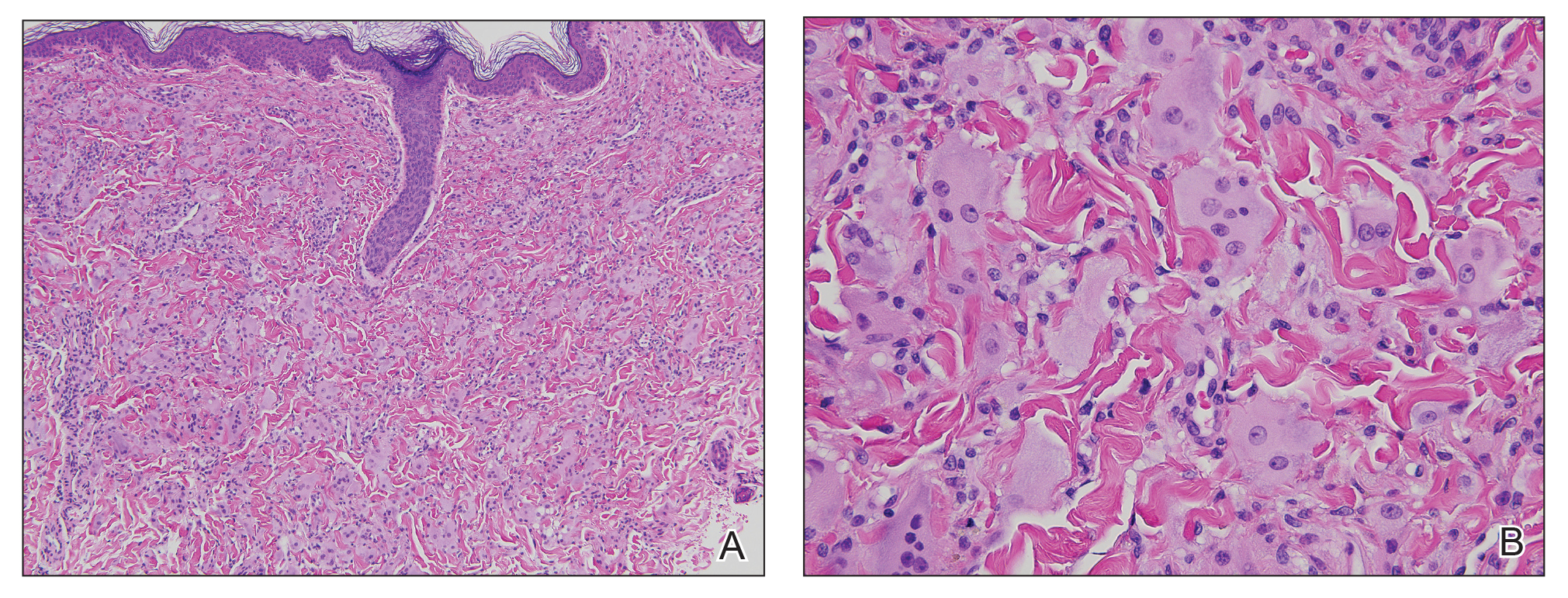
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
To the Editor:
A 50-year-old woman presented with an asymptomatic eruption on the dorsal aspect of the hands, abdomen, and face of 6 months’ duration. The eruption was associated with generalized arthralgia and fatigue. Within several weeks of onset of the cutaneous eruption, the patient developed swelling in the hands as well as worsening arthralgia. She was treated for presumed Lyme borreliosis but reported no improvement in the symptoms. She was then referred to dermatology for further management.
Physical examination revealed red-orange, edematous, monomorphic papulonodules scattered on the nasolabial folds, upper lip, and along the dorsal aspect of the hands and fingers (Figure 1). A brown rippled plaque was present on the left lower abdomen. The oral mucosa and nails were unremarkable. Laboratory studies showed elevated total cholesterol (244 mg/dL [reference range, <200 mg/dL]), low-density lipoproteins (130 mg/dL [reference range, 10–30 mg/dL]), aspartate aminotransferase (140 U/L [reference range, 10–30 U/L]), alanine aminotransferase (110 U/L [reference range, 10–40 U/L]), and total bilirubin (1.5 mg/dL [reference range, 0.3–1.2 mg/dL]). White blood cell count and C-reactive protein levels were within reference range. An antinuclear antibody titer of 1:80 with a homogenous pattern was found, and aldolase levels were elevated. Laboratory investigations for rheumatoid factor, Lyme disease, tuberculosis, hepatitis, and human immunodeficiency virus were negative. A chest radiograph was normal.
A punch biopsy from the right dorsal hand revealed a dermal proliferation of mononucleated and multinucleated epithelioid histiocytes with ample amounts of eosinophilic ground-glass cytoplasm (Figure 2). Immunohistochemistry revealed epithelioid histiocytes reactive for CD68, CD163, and factor XIIIA, and negative for S-100 and CD1a.
The patient was diagnosed with multicentric reticulohistiocytosis (MRH) and was initially treated with prednisone. Treatment was later augmented with etanercept and methotrexate with improvement in both the skin and joint symptoms.
Multicentric reticulohistiocytosis is a rare, non–Langerhans cell histiocytosis with both cutaneous and systemic features. Although case reports date back to the late 1800s, the term multicentric reticulohistiocytosis was first used in 1954.1 Multicentric reticulohistiocytosis is extremely uncommon and precludes thorough investigation of its etiology and management. The condition typically presents in the fifth to sixth decades of life and occurs more frequently in women with a female to male ratio estimated at 3 to 1.2,3 Pediatric cases have been reported but are exceedingly rare.4
Multicentric reticulohistiocytosis typically presents with a severe erosive arthropathy known as arthritis mutilans. Patients display a symmetric polyarthritis that commonly involves the elbows, wrists, and proximal and distal aspects of the interphalangeal joints. Onset and progression can be rapid, and the erosive nature leads to deformities in up to 45% of patients.2,5,6 Cutaneous findings arise an average of 3 years after the development of arthritis, though one-fifth of patients will initially present with cutaneous findings followed by the development of arthritis at any time.3,6 Clinical features include flesh-colored to reddish brown or yellow papulonodules that range in size from several millimeters to 2 cm. The lesions most commonly occur on the face (eg, ears, nose, paranasal cheeks), scalp, dorsal and lateral aspects of the hands and fingers, and overlying articular regions of the extremities. Characteristic periungual lesions classically are referred to as coral beads.4,6 Patients commonly report pruritus that may precede the development of the papules and nodules. Other cutaneous manifestations include xanthelasma, nail changes, and a photodistributed erythematous maculopapular eruption that may mimic dermatomyositis.6
Cutaneous findings of MRH can mimic rheumatoid nodules, gout, Gottron papules of dermatomyositis, lipoid proteinosis, sarcoidosis, lepromatous leprosy, granuloma annulare, xanthoma, xanthogranuloma, and fibroxanthoma.6,7 Histopathologic features may distinguish MRH from such entities. Findings include fairly well-circumscribed aggregates of large multinucleated giant cells with characteristic eosinophilic ground-glass cytoplasm. Histiocytes stain positively for CD68, HAM56, CD11b, and CD14, and variably for factor XIIIa. CD68, which is expressed by monocytes/macrophages, has been universally reported to be the most reliable marker of MRH. Negative staining for S-100 and CD1a supports a non-Langerhans origin for the involved histiocytes. If arthritic symptoms predominate, MRH must be distinguished from rheumatoid and psoriatic arthritis.6,7
Mucosal involvement occurs in approximately 50% of patients and includes the presence of nodules in the oral, nasal, and pharyngeal mucosae, as well as eye structures.2,3 Histiocytic infiltration has been documented in the heart, lungs, thyroid, liver, stomach, kidneys, muscle, bone marrow, and urogenital tract. Histiocytes also can invade the cartilage of the ears and nose causing disfigurement and characteristic leonine facies. Pathologic fractures may occur with bone involvement.5
Systemic features associated with MRH include hyperlipidemia, diabetes mellitus, thyroid disease, hypergammaglobulinemia, and various autoimmune diseases. Patients less frequently report fever and weight loss.2,5,6,8 Additionally, a positive tuberculin test occurs in 12% to 50% of patients.6 Various autoimmune diseases occur in 6% to 17% of cases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, dermatomyositis, Sjögren syndrome, and primary biliary cirrhosis.2,5,6,8 The most clinically salient feature of MRH is its association with malignant conditions, which occur in up to 31% of patients. A variety of cancers have been reported in association with MRH, including breast, cervical, ovarian, stomach, penile, lymphoma, mesothelioma, and melanoma.7
The etiology of MRH is unclear. Although onset may precede the development of a malignant condition and regress with treatment, it cannot be considered a true paraneoplastic disorder, as it has no association with a specific cancer and does not typically parallel the disease course.6,9 Reports of increased levels of inflammatory mediators released from macrophages and endothelial cells, specifically IL-12, IL-1β, IL-6, and tumor necrosis factor α (TNF-α), have been thought to drive the destruction of bone and cartilage.6 In particular, TNF-α acts to indirectly induce destruction by stimulating proteolytic activity in macrophages, similar to the pathogenesis of joint damage in rheumatoid arthritis.8 Osteoclastic activity may play a role in the pathogenesis of MRH, as multinucleated giant cells in MRH can mature into osteoclasts by receptor activated nuclear factor–κB ligand signaling. In addition, patients treated with bisphosphonates have had decreased lacunar resorption.2,8
Initial management of MRH should include screening for hyperlipidemia, hypergammaglobulinemia, hyperglycemia, thyroid dysfunction, and autoimmune diseases, as well as age-appropriate cancer screening. Imaging studies should evaluate for the presence of erosive arthritis. There are no well-defined treatment algorithms for MRH due to the rarity of the disease, and recommendations largely rely on case reports. Although spontaneous remission typically occurs within 5 to 10 years, the risk for joint destruction argues for early pharmacologic intervention. Current management includes the use of nonsteroidal anti-inflammatory drugs and various immunosuppressants including oral glucocorticoids, cyclophosphamide, chlorambucil, methotrexate, or azathioprine.2 A combination of methotrexate with cyclophosphamide or glucocorticoids also has shown efficacy.10 Anti–TNF-α agents, such as etanercept, adalimumab, and infliximab, have been used with some success.2 Tumor necrosis factor α inhibitors used in combination with oral glucocorticoids and methotrexate may have an increased benefit.2,9,11 Evidence suggesting that TNF-α plays a role in the destruction of bone and cartilage led to the successful use of infliximab in combination with oral glucocorticoids and methotrexate, which prevented possible development of antibodies to infliximab and increased its efficacy.12 Bisphosphonate use in combination with glucocorticoids and methotrexate may prevent joint destruction without the serious adverse events associated with anti–TNF-α agents.2,9,13,14
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
- Goltz RW, Laymon CW. Multicentric reticulohistiocytosis of the skin and synovia; reticulohistiocytoma or ganglioneuroma. AMA Arch Derma Syphilol. 1954;69:717-731.
- Islam AD, Naguwa SM, Cheema GS, et al. Multicentric reticulohistiocytosis: a rare yet challenging disease. Clin Rev Allergy Immunol. 2013;45:281-289.
- West KL, Sporn T, Puri PK. Multicentric reticulohistiocytosis: a unique case with pulmonary fibrosis. Arch Dermatol. 2012;148:228-232.
- Outland JD, Keiran SJ, Schikler KN, et al. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol. 2002;19:527-531.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Trotta F, Castellino G, Lo Monaco A. Multicentric reticulohistiocytosis. Best Pract Res Clin Rheumatol. 2004;18:759-772.
- Kalajian AH, Callen JP. Multicentric reticulohistiocytosis successfully treated with infliximab: an illustrative case and evaluation of cytokine expression supporting anti-tumor necrosis factor therapy. Arch Derm. 2008;144:1360-1366.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. case report, review of the literature, and proposal for treatment. Arthritis Rheum. 1996;39:171-174.
- Lovelace K, Loyd A, Adelson D, et al. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol. 2005;141:1167-1168.
- Lee MW, Lee EY, Jeong YI, et al. Successful treatment of multicentric reticulohistiocytosis with a combination of infliximab, prednisolone and methotrexate. Acta Derm Venereol. 2004;84:478-479.
- Adamopoulos IE, Wordsworth PB, Edwards JR, et al. Osteoclast differentiation and bone resorption in multicentric reticulohistiocytosis. Hum Pathol. 2006;37:1176-1185.
- Satoh M, Oyama N, Yamada H, et al. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol. 2008;35:168-171.
Practice Points
- Multicentric reticulohistiocytosis (MRH) is an important entity to recognize given its association with underlying malignancy and irreversible destructive arthritis.
- Diagnosis of MRH warrants extensive review of systems, age-appropriate cancer screening, and relevant systemic workup.
- Early pharmacologic intervention should be initiated with nonsteroidal anti-inflammatory agents or immunosuppressant agents.