User login
FDA rejects poziotinib for certain types of lung cancer
The clinical data the company submitted were deemed insufficient for approval, and additional data including a randomized clinical trial would be needed, the agency said.
The move is not a surprise, as the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 9-4 against approval when it met to discuss the drug in September, as reported at the time by this news organization.
Poziotinib was developed for patients with previously treated locally advanced or metastatic NSCLC harboring HER2 exon 20 insertion mutations, which occur in about 2% of patients with NSCLC.
Poziotinib is a potent oral pan-HER tyrosine kinase inhibitor with activity in patients with these mutations. Clinical data from the ZENITH20 Trial reported last year showed an overall response rate of 43.8%, and the drug was described as showing “clinically meaningful efficacy for treatment-naive NSCLC HER2 exon 20 mutations with [daily] dosing.”
“We continue to believe that poziotinib could present a meaningful treatment option for patients with this rare form of lung cancer, for whom other therapies have failed,” commented Tom Riga, president and chief executive officer of Spectrum Pharmaceuticals.
However, following multiple interactions with the FDA, “we have made the strategic decision to immediately deprioritize the poziotinib program,” he said. The change is effective immediately, and the company is now in the process of reducing its R&D workforce by approximately 75%.
Drug development criticized
At the ODAC meeting, several panelists were openly critical of the approach Spectrum took in developing the drug. The FDA’s top cancer official, Richard Pazdur, MD, characterized Spectrum’s work as “poor drug development” and likened it to “building a house on quicksand.”
The FDA panel detailed several ways they felt that the poziotinib application fell short of the benchmarks needed for accelerated approval.
To win such a speedy clearance, a company needs to show that a drug provides a meaningful therapeutic benefit over existing treatments. The panel argued that, so far, poziotinib appears to be inferior to a product already available for HER2-mutant NSCLC, trastuzumab deruxtecan (Enhertu), which received accelerated approval in August.
The FDA staff contrasted a reported overall response rate for poziotinib, which was estimated at 28% (from data discussed at the meeting), with the overall response rate for trastuzumab deruxtecan, which is 58%.
Harpreet Singh, MD, a director in the FDA’s oncology division, asked the panel to consider what they would do as a physician treating a patient with this mutation, given the choices that are now available.
“That’s something we’re asking the committee to consider … to think about the context of what’s available to you in the clinic,” Dr. Singh said.
Dr. Singh said she expected that patients and physicians would prefer a drug such as trastuzumab deruxtecan, which has a more established record, regardless of the fact that treatment with poziotinib is more convenient because it is given as a tablet.
Dr. Singh and other staff also raised concerns about side effects of poziotinib, including diarrhea, as well as difficulty determining the right dose.
Katherine Scilla, MD, one of the nine ODAC panelists to vote “no,” echoed these views. Although Dr. Scilla, an oncologist at the University of Maryland, Baltimore, sympathized with the need for options for people with this rare form of lung cancer, she was not persuaded by the data on poziotinib that were presented to support accelerated approval.
“I’m not sure that this represents a meaningful therapeutic benefit over other agents,” she said at the time.
A version of this article first appeared on Medscape.com.
The clinical data the company submitted were deemed insufficient for approval, and additional data including a randomized clinical trial would be needed, the agency said.
The move is not a surprise, as the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 9-4 against approval when it met to discuss the drug in September, as reported at the time by this news organization.
Poziotinib was developed for patients with previously treated locally advanced or metastatic NSCLC harboring HER2 exon 20 insertion mutations, which occur in about 2% of patients with NSCLC.
Poziotinib is a potent oral pan-HER tyrosine kinase inhibitor with activity in patients with these mutations. Clinical data from the ZENITH20 Trial reported last year showed an overall response rate of 43.8%, and the drug was described as showing “clinically meaningful efficacy for treatment-naive NSCLC HER2 exon 20 mutations with [daily] dosing.”
“We continue to believe that poziotinib could present a meaningful treatment option for patients with this rare form of lung cancer, for whom other therapies have failed,” commented Tom Riga, president and chief executive officer of Spectrum Pharmaceuticals.
However, following multiple interactions with the FDA, “we have made the strategic decision to immediately deprioritize the poziotinib program,” he said. The change is effective immediately, and the company is now in the process of reducing its R&D workforce by approximately 75%.
Drug development criticized
At the ODAC meeting, several panelists were openly critical of the approach Spectrum took in developing the drug. The FDA’s top cancer official, Richard Pazdur, MD, characterized Spectrum’s work as “poor drug development” and likened it to “building a house on quicksand.”
The FDA panel detailed several ways they felt that the poziotinib application fell short of the benchmarks needed for accelerated approval.
To win such a speedy clearance, a company needs to show that a drug provides a meaningful therapeutic benefit over existing treatments. The panel argued that, so far, poziotinib appears to be inferior to a product already available for HER2-mutant NSCLC, trastuzumab deruxtecan (Enhertu), which received accelerated approval in August.
The FDA staff contrasted a reported overall response rate for poziotinib, which was estimated at 28% (from data discussed at the meeting), with the overall response rate for trastuzumab deruxtecan, which is 58%.
Harpreet Singh, MD, a director in the FDA’s oncology division, asked the panel to consider what they would do as a physician treating a patient with this mutation, given the choices that are now available.
“That’s something we’re asking the committee to consider … to think about the context of what’s available to you in the clinic,” Dr. Singh said.
Dr. Singh said she expected that patients and physicians would prefer a drug such as trastuzumab deruxtecan, which has a more established record, regardless of the fact that treatment with poziotinib is more convenient because it is given as a tablet.
Dr. Singh and other staff also raised concerns about side effects of poziotinib, including diarrhea, as well as difficulty determining the right dose.
Katherine Scilla, MD, one of the nine ODAC panelists to vote “no,” echoed these views. Although Dr. Scilla, an oncologist at the University of Maryland, Baltimore, sympathized with the need for options for people with this rare form of lung cancer, she was not persuaded by the data on poziotinib that were presented to support accelerated approval.
“I’m not sure that this represents a meaningful therapeutic benefit over other agents,” she said at the time.
A version of this article first appeared on Medscape.com.
The clinical data the company submitted were deemed insufficient for approval, and additional data including a randomized clinical trial would be needed, the agency said.
The move is not a surprise, as the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 9-4 against approval when it met to discuss the drug in September, as reported at the time by this news organization.
Poziotinib was developed for patients with previously treated locally advanced or metastatic NSCLC harboring HER2 exon 20 insertion mutations, which occur in about 2% of patients with NSCLC.
Poziotinib is a potent oral pan-HER tyrosine kinase inhibitor with activity in patients with these mutations. Clinical data from the ZENITH20 Trial reported last year showed an overall response rate of 43.8%, and the drug was described as showing “clinically meaningful efficacy for treatment-naive NSCLC HER2 exon 20 mutations with [daily] dosing.”
“We continue to believe that poziotinib could present a meaningful treatment option for patients with this rare form of lung cancer, for whom other therapies have failed,” commented Tom Riga, president and chief executive officer of Spectrum Pharmaceuticals.
However, following multiple interactions with the FDA, “we have made the strategic decision to immediately deprioritize the poziotinib program,” he said. The change is effective immediately, and the company is now in the process of reducing its R&D workforce by approximately 75%.
Drug development criticized
At the ODAC meeting, several panelists were openly critical of the approach Spectrum took in developing the drug. The FDA’s top cancer official, Richard Pazdur, MD, characterized Spectrum’s work as “poor drug development” and likened it to “building a house on quicksand.”
The FDA panel detailed several ways they felt that the poziotinib application fell short of the benchmarks needed for accelerated approval.
To win such a speedy clearance, a company needs to show that a drug provides a meaningful therapeutic benefit over existing treatments. The panel argued that, so far, poziotinib appears to be inferior to a product already available for HER2-mutant NSCLC, trastuzumab deruxtecan (Enhertu), which received accelerated approval in August.
The FDA staff contrasted a reported overall response rate for poziotinib, which was estimated at 28% (from data discussed at the meeting), with the overall response rate for trastuzumab deruxtecan, which is 58%.
Harpreet Singh, MD, a director in the FDA’s oncology division, asked the panel to consider what they would do as a physician treating a patient with this mutation, given the choices that are now available.
“That’s something we’re asking the committee to consider … to think about the context of what’s available to you in the clinic,” Dr. Singh said.
Dr. Singh said she expected that patients and physicians would prefer a drug such as trastuzumab deruxtecan, which has a more established record, regardless of the fact that treatment with poziotinib is more convenient because it is given as a tablet.
Dr. Singh and other staff also raised concerns about side effects of poziotinib, including diarrhea, as well as difficulty determining the right dose.
Katherine Scilla, MD, one of the nine ODAC panelists to vote “no,” echoed these views. Although Dr. Scilla, an oncologist at the University of Maryland, Baltimore, sympathized with the need for options for people with this rare form of lung cancer, she was not persuaded by the data on poziotinib that were presented to support accelerated approval.
“I’m not sure that this represents a meaningful therapeutic benefit over other agents,” she said at the time.
A version of this article first appeared on Medscape.com.
Atezolizumab (Tecentriq) bladder cancer indication withdrawn in United States
The drug is an anti–PD-L1 inhibitor immunotherapy, and continues to be approved for use in lung and liver cancer and melanoma.
The manufacturer, Genentech, announced that it was voluntarily withdrawing the U.S. indication for atezolizumab that covered its use in adults with locally advanced or metastatic urothelial carcinoma (bladder cancer) who are not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 or are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status.
The company said that it made the decision after consultation with the Food and Drug Administration.
“While we are disappointed with this withdrawal, we understand the need to uphold the principles of the FDA’s Accelerated Approval Program, which brings innovative medicines to patients sooner,” said Levi Garraway, MD, PhD, Genentech chief medical officer and head of Global Product Development.
Atezolizumab had been granted an accelerated approval for this indication back in 2016, based on response rate data from the IMvigor210 trial.
The company was obliged to conduct a follow-up trial to show clinical benefit, and launched IMvigor130, which it described as “the designated postmarketing requirement to convert the accelerated approval to regular approval.”
The bladder cancer indication for atezolizumab was discussed (alongside several other indications for different immunotherapy drugs) at a historic 3-day meeting of the FDA’s oncologic Drugs Advisory Committee in April 2021. At the time, ODAC voted 10-1 in favor of maintaining the indication for atezolizumab for the first-line treatment of cisplatin-ineligible patients with advanced/metastatic urothelial carcinoma, pending final overall survival results from the IMvigor130 trial.
Genentech has now said that this trial “did not meet the coprimary endpoint of overall survival for atezolizumab plus chemotherapy compared with chemotherapy alone” when used for the first-line treatment of patients with previously untreated advanced bladder cancer.
These data will be presented at an upcoming medical meeting, the company added.
“There is a considerable unmet need for effective and tolerable treatments for people living with advanced bladder cancer and so we regret that the IMvigor130 trial did not cross the statistical threshold for overall survival,” Dr. Garraway commented.
A version of this article first appeared on Medscape.com.
The drug is an anti–PD-L1 inhibitor immunotherapy, and continues to be approved for use in lung and liver cancer and melanoma.
The manufacturer, Genentech, announced that it was voluntarily withdrawing the U.S. indication for atezolizumab that covered its use in adults with locally advanced or metastatic urothelial carcinoma (bladder cancer) who are not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 or are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status.
The company said that it made the decision after consultation with the Food and Drug Administration.
“While we are disappointed with this withdrawal, we understand the need to uphold the principles of the FDA’s Accelerated Approval Program, which brings innovative medicines to patients sooner,” said Levi Garraway, MD, PhD, Genentech chief medical officer and head of Global Product Development.
Atezolizumab had been granted an accelerated approval for this indication back in 2016, based on response rate data from the IMvigor210 trial.
The company was obliged to conduct a follow-up trial to show clinical benefit, and launched IMvigor130, which it described as “the designated postmarketing requirement to convert the accelerated approval to regular approval.”
The bladder cancer indication for atezolizumab was discussed (alongside several other indications for different immunotherapy drugs) at a historic 3-day meeting of the FDA’s oncologic Drugs Advisory Committee in April 2021. At the time, ODAC voted 10-1 in favor of maintaining the indication for atezolizumab for the first-line treatment of cisplatin-ineligible patients with advanced/metastatic urothelial carcinoma, pending final overall survival results from the IMvigor130 trial.
Genentech has now said that this trial “did not meet the coprimary endpoint of overall survival for atezolizumab plus chemotherapy compared with chemotherapy alone” when used for the first-line treatment of patients with previously untreated advanced bladder cancer.
These data will be presented at an upcoming medical meeting, the company added.
“There is a considerable unmet need for effective and tolerable treatments for people living with advanced bladder cancer and so we regret that the IMvigor130 trial did not cross the statistical threshold for overall survival,” Dr. Garraway commented.
A version of this article first appeared on Medscape.com.
The drug is an anti–PD-L1 inhibitor immunotherapy, and continues to be approved for use in lung and liver cancer and melanoma.
The manufacturer, Genentech, announced that it was voluntarily withdrawing the U.S. indication for atezolizumab that covered its use in adults with locally advanced or metastatic urothelial carcinoma (bladder cancer) who are not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 or are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status.
The company said that it made the decision after consultation with the Food and Drug Administration.
“While we are disappointed with this withdrawal, we understand the need to uphold the principles of the FDA’s Accelerated Approval Program, which brings innovative medicines to patients sooner,” said Levi Garraway, MD, PhD, Genentech chief medical officer and head of Global Product Development.
Atezolizumab had been granted an accelerated approval for this indication back in 2016, based on response rate data from the IMvigor210 trial.
The company was obliged to conduct a follow-up trial to show clinical benefit, and launched IMvigor130, which it described as “the designated postmarketing requirement to convert the accelerated approval to regular approval.”
The bladder cancer indication for atezolizumab was discussed (alongside several other indications for different immunotherapy drugs) at a historic 3-day meeting of the FDA’s oncologic Drugs Advisory Committee in April 2021. At the time, ODAC voted 10-1 in favor of maintaining the indication for atezolizumab for the first-line treatment of cisplatin-ineligible patients with advanced/metastatic urothelial carcinoma, pending final overall survival results from the IMvigor130 trial.
Genentech has now said that this trial “did not meet the coprimary endpoint of overall survival for atezolizumab plus chemotherapy compared with chemotherapy alone” when used for the first-line treatment of patients with previously untreated advanced bladder cancer.
These data will be presented at an upcoming medical meeting, the company added.
“There is a considerable unmet need for effective and tolerable treatments for people living with advanced bladder cancer and so we regret that the IMvigor130 trial did not cross the statistical threshold for overall survival,” Dr. Garraway commented.
A version of this article first appeared on Medscape.com.
NSAIDs for knee osteoarthritis may worsen pain over time
CHICAGO – Taking NSAIDs for knee osteoarthritis may worsen inflammation and pain over time, suggest new data revealed at the annual meeting of the Radiological Society of North America.
Johanna Luitjens, MD, a postdoctoral scholar in the department of radiology and biomedical Imaging at the University of California, San Francisco, told this news organization that NSAIDs are frequently used to treat OA pain because inflammation is one of the main drivers of OA, but whether they actually help outcomes has been unclear. Her study suggests that they don’t help – and may actually worsen – outcomes.
In particular, this study looked at the impact of NSAIDs on synovitis – the inflammation of the membrane lining the knee joint – by using MRI-based structural biomarkers.
OA, the most common form of arthritis, affects more than 32 million adults in the United States and more than 500 million people worldwide.
No approved therapy to reduce OA progression
Little is known of the long-term effects of NSAIDs on OA progression. Currently, there’s no approved therapy to cure OA or to reduce its advance.
Dr. Luitjens noted, however, that the synovial membrane mediates development and progression of OA and may be a good therapeutic target.

Researchers studied participants from the Osteoarthritis Initiative (OAI) cohort with moderate to severe OA who used NSAIDs regularly for at least 1 year between baseline and 4-year follow-up. All participants had high-quality 3T MRI of the knee at baseline and after 4 years. Images were scored for biomarkers of inflammation, including cartilage thickness and composition.
Dr. Luitjens and associates studied 721 participants who matched the inclusion criteria (129 with and 592 participants without regular NSAID use). The available data did not further specify amounts of NSAIDs used.
At baseline, significantly higher signal intensity in the infrapatellar fat pad (IFP) was seen in patients who used NSAID, compared with controls (adjusted difference in score, 0.26; 95% confidence interval, –0.5 to –0.129; P = .039).
In addition, at the end of the study period, there was a significantly greater increase in signal intensity of IFP (adjusted difference in score, 0.46; 95% CI, 0.2-0.72; P < .001) and higher increase in effusion synovitis (adjusted difference in score, 0.27; 95% CI, 0.06-0.47; P = .01) in NSAID users, compared with controls.
IFP size and synovial proliferation score did not different significantly between groups at the start of the study and showed no significant change over time.
The results showed no long-term benefit of NSAID use. Joint inflammation and cartilage quality were worse at baseline in the participants taking NSAIDs, compared with the control group, and worsened at 4-year follow-up.
Design limits strength
Amanda E. Nelson, MD, associate professor of medicine, division of rheumatology, allergy, and immunology at the University of North Carolina at Chapel Hill, cautioned against assuming causality, pointing out that the OAI is an observational cohort study. (Dr. Nelson was not involved in the OAI or Dr. Luitjens’ analysis.)

“[The OAI is] large and well known, but it wasn’t designed to compare these groups, and this was a small subset,” she said in an interview. Without randomization, it’s hard to judge the results.
“It may be that people on NSAIDs for the duration of the study had more pain and had more disease to begin with, or had more symptoms or had failed other treatments,” she said, adding that the effect sizes were small.
Measures such as the IFP are ranked 0-3, so “the clinical difference of a 0.26 difference on a 0-3 scale is a bit uncertain,” she said.
Dr. Luitjens said that the researchers tried to adjust for potential confounders but agreed that randomized controlled trials are needed to better advise physicians and patients on the benefits or harms of using NSAIDs for OA.
Weighing the risks in older adults
Una Makris, MD, associate professor of internal medicine in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, noted that NSAIDs are “not always the safest option.”

“We are still in desperate need of disease-modifying drugs in OA with rigorous randomized trials to show efficacy for outcomes that are most meaningful to patients,” Dr. Makris, who was not involved in the study, told this news organization.
“OA is most common in older adults, those often with multiple comorbidities, so we must always weigh the risks – including known adverse effects which can be amplified in older adults – and benefits with the goal of improved function and less pain,” Dr. Makris said.
NSAID use also should be considered in the context of body mass index, cardiovascular risk, prior trauma or injury, other medication use, and behavioral factors, including physical activity, she said.
Dr. Luitjens, Dr. Nelson, and Dr. Makris reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – Taking NSAIDs for knee osteoarthritis may worsen inflammation and pain over time, suggest new data revealed at the annual meeting of the Radiological Society of North America.
Johanna Luitjens, MD, a postdoctoral scholar in the department of radiology and biomedical Imaging at the University of California, San Francisco, told this news organization that NSAIDs are frequently used to treat OA pain because inflammation is one of the main drivers of OA, but whether they actually help outcomes has been unclear. Her study suggests that they don’t help – and may actually worsen – outcomes.
In particular, this study looked at the impact of NSAIDs on synovitis – the inflammation of the membrane lining the knee joint – by using MRI-based structural biomarkers.
OA, the most common form of arthritis, affects more than 32 million adults in the United States and more than 500 million people worldwide.
No approved therapy to reduce OA progression
Little is known of the long-term effects of NSAIDs on OA progression. Currently, there’s no approved therapy to cure OA or to reduce its advance.
Dr. Luitjens noted, however, that the synovial membrane mediates development and progression of OA and may be a good therapeutic target.
Researchers studied participants from the Osteoarthritis Initiative (OAI) cohort with moderate to severe OA who used NSAIDs regularly for at least 1 year between baseline and 4-year follow-up. All participants had high-quality 3T MRI of the knee at baseline and after 4 years. Images were scored for biomarkers of inflammation, including cartilage thickness and composition.
Dr. Luitjens and associates studied 721 participants who matched the inclusion criteria (129 with and 592 participants without regular NSAID use). The available data did not further specify amounts of NSAIDs used.
At baseline, significantly higher signal intensity in the infrapatellar fat pad (IFP) was seen in patients who used NSAID, compared with controls (adjusted difference in score, 0.26; 95% confidence interval, –0.5 to –0.129; P = .039).
In addition, at the end of the study period, there was a significantly greater increase in signal intensity of IFP (adjusted difference in score, 0.46; 95% CI, 0.2-0.72; P < .001) and higher increase in effusion synovitis (adjusted difference in score, 0.27; 95% CI, 0.06-0.47; P = .01) in NSAID users, compared with controls.
IFP size and synovial proliferation score did not different significantly between groups at the start of the study and showed no significant change over time.
The results showed no long-term benefit of NSAID use. Joint inflammation and cartilage quality were worse at baseline in the participants taking NSAIDs, compared with the control group, and worsened at 4-year follow-up.
Design limits strength
Amanda E. Nelson, MD, associate professor of medicine, division of rheumatology, allergy, and immunology at the University of North Carolina at Chapel Hill, cautioned against assuming causality, pointing out that the OAI is an observational cohort study. (Dr. Nelson was not involved in the OAI or Dr. Luitjens’ analysis.)
“[The OAI is] large and well known, but it wasn’t designed to compare these groups, and this was a small subset,” she said in an interview. Without randomization, it’s hard to judge the results.
“It may be that people on NSAIDs for the duration of the study had more pain and had more disease to begin with, or had more symptoms or had failed other treatments,” she said, adding that the effect sizes were small.
Measures such as the IFP are ranked 0-3, so “the clinical difference of a 0.26 difference on a 0-3 scale is a bit uncertain,” she said.
Dr. Luitjens said that the researchers tried to adjust for potential confounders but agreed that randomized controlled trials are needed to better advise physicians and patients on the benefits or harms of using NSAIDs for OA.
Weighing the risks in older adults
Una Makris, MD, associate professor of internal medicine in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, noted that NSAIDs are “not always the safest option.”
“We are still in desperate need of disease-modifying drugs in OA with rigorous randomized trials to show efficacy for outcomes that are most meaningful to patients,” Dr. Makris, who was not involved in the study, told this news organization.
“OA is most common in older adults, those often with multiple comorbidities, so we must always weigh the risks – including known adverse effects which can be amplified in older adults – and benefits with the goal of improved function and less pain,” Dr. Makris said.
NSAID use also should be considered in the context of body mass index, cardiovascular risk, prior trauma or injury, other medication use, and behavioral factors, including physical activity, she said.
Dr. Luitjens, Dr. Nelson, and Dr. Makris reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – Taking NSAIDs for knee osteoarthritis may worsen inflammation and pain over time, suggest new data revealed at the annual meeting of the Radiological Society of North America.
Johanna Luitjens, MD, a postdoctoral scholar in the department of radiology and biomedical Imaging at the University of California, San Francisco, told this news organization that NSAIDs are frequently used to treat OA pain because inflammation is one of the main drivers of OA, but whether they actually help outcomes has been unclear. Her study suggests that they don’t help – and may actually worsen – outcomes.
In particular, this study looked at the impact of NSAIDs on synovitis – the inflammation of the membrane lining the knee joint – by using MRI-based structural biomarkers.
OA, the most common form of arthritis, affects more than 32 million adults in the United States and more than 500 million people worldwide.
No approved therapy to reduce OA progression
Little is known of the long-term effects of NSAIDs on OA progression. Currently, there’s no approved therapy to cure OA or to reduce its advance.
Dr. Luitjens noted, however, that the synovial membrane mediates development and progression of OA and may be a good therapeutic target.
Researchers studied participants from the Osteoarthritis Initiative (OAI) cohort with moderate to severe OA who used NSAIDs regularly for at least 1 year between baseline and 4-year follow-up. All participants had high-quality 3T MRI of the knee at baseline and after 4 years. Images were scored for biomarkers of inflammation, including cartilage thickness and composition.
Dr. Luitjens and associates studied 721 participants who matched the inclusion criteria (129 with and 592 participants without regular NSAID use). The available data did not further specify amounts of NSAIDs used.
At baseline, significantly higher signal intensity in the infrapatellar fat pad (IFP) was seen in patients who used NSAID, compared with controls (adjusted difference in score, 0.26; 95% confidence interval, –0.5 to –0.129; P = .039).
In addition, at the end of the study period, there was a significantly greater increase in signal intensity of IFP (adjusted difference in score, 0.46; 95% CI, 0.2-0.72; P < .001) and higher increase in effusion synovitis (adjusted difference in score, 0.27; 95% CI, 0.06-0.47; P = .01) in NSAID users, compared with controls.
IFP size and synovial proliferation score did not different significantly between groups at the start of the study and showed no significant change over time.
The results showed no long-term benefit of NSAID use. Joint inflammation and cartilage quality were worse at baseline in the participants taking NSAIDs, compared with the control group, and worsened at 4-year follow-up.
Design limits strength
Amanda E. Nelson, MD, associate professor of medicine, division of rheumatology, allergy, and immunology at the University of North Carolina at Chapel Hill, cautioned against assuming causality, pointing out that the OAI is an observational cohort study. (Dr. Nelson was not involved in the OAI or Dr. Luitjens’ analysis.)
“[The OAI is] large and well known, but it wasn’t designed to compare these groups, and this was a small subset,” she said in an interview. Without randomization, it’s hard to judge the results.
“It may be that people on NSAIDs for the duration of the study had more pain and had more disease to begin with, or had more symptoms or had failed other treatments,” she said, adding that the effect sizes were small.
Measures such as the IFP are ranked 0-3, so “the clinical difference of a 0.26 difference on a 0-3 scale is a bit uncertain,” she said.
Dr. Luitjens said that the researchers tried to adjust for potential confounders but agreed that randomized controlled trials are needed to better advise physicians and patients on the benefits or harms of using NSAIDs for OA.
Weighing the risks in older adults
Una Makris, MD, associate professor of internal medicine in the division of rheumatic diseases at the University of Texas Southwestern Medical Center, Dallas, noted that NSAIDs are “not always the safest option.”
“We are still in desperate need of disease-modifying drugs in OA with rigorous randomized trials to show efficacy for outcomes that are most meaningful to patients,” Dr. Makris, who was not involved in the study, told this news organization.
“OA is most common in older adults, those often with multiple comorbidities, so we must always weigh the risks – including known adverse effects which can be amplified in older adults – and benefits with the goal of improved function and less pain,” Dr. Makris said.
NSAID use also should be considered in the context of body mass index, cardiovascular risk, prior trauma or injury, other medication use, and behavioral factors, including physical activity, she said.
Dr. Luitjens, Dr. Nelson, and Dr. Makris reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT RSNA 2022
Vitamin D fails to stave off statin-related muscle symptoms
Vitamin D supplements do not prevent muscle symptoms in new statin users or affect the likelihood of discontinuing a statin due to muscle pain and discomfort, a substudy of the VITAL trial indicates.
Among more than 2,000 randomized participants, statin-associated muscle symptoms (SAMS) were reported by 31% assigned to vitamin D and 31% assigned to placebo.
The two groups were equally likely to stop taking a statin due to muscle symptoms, at 13%.
No significant difference was observed in SAMS (odds ratio [OR], 0.97; 95% confidence interval [CI], 0.80-1.18) or statin discontinuations (OR, 1.04; 95% CI, 0.80-1.35) after adjustment for baseline variables and other characteristics, namely age, sex, and African-American race, previously found to be associated with SAMS in VITAL.
“We actually thought when we started out that maybe we were going to show something, that maybe it was going to be that the people who got the vitamin D were least likely to have a problem with a statin than all those who didn’t get vitamin D, but that is not what we showed,” senior author Neil J. Stone, MD, Northwestern University, Chicago, told this news organization.
He noted that patients in the clinic with low levels of vitamin D often have muscle pain and discomfort and that previous unblinded studies suggested vitamin D might benefit patients with SAMS and reduce statin intolerance.
As previously reported, the double-blind VITAL trial showed no difference in the primary prevention of cardiovascular disease or cancer at 5 years among 25,871 middle-aged adults randomized to vitamin D3 at 2000 IU/d or placebo, regardless of their baseline vitamin D level.
Unlike previous studies showing a benefit with vitamin D on SAMS, importantly, VITAL participants were unaware of whether they were taking vitamin D or placebo and were not expecting any help with their muscle symptoms, first author Mark A. Hlatky, MD, Stanford (Calif.) University, pointed out in an interview.
As to how many statin users turn to the popular supplement for SAMS, he said that number couldn’t be pinned down, despite a lengthy search. “But I think it’s very common, because up to half of people stop taking their statins within a year and many of these do so because of statin-associated muscle symptoms, and we found it in about 30% of people who have them. I have them myself and was motivated to study it because I thought this was an interesting question.”
The results were published online in JAMA Cardiology.
SAMS by baseline 25-OHD
The substudy included 2,083 patients who initiated statin therapy after randomization and were surveyed in early 2016 about their statin use and muscle symptoms.
Two-thirds, or 1,397 patients, had 25-hydroxy vitamin D (25-OHD) measured at baseline, with 47% having levels < 30 ng/mL and 13% levels < 20 ng/mL.
Serum 25-OHD levels were virtually identical in the two treatment groups (mean, 30.4 ng/mL; median, 30.0 ng/mL). The frequency of SAMS did not differ between those assigned to vitamin D or placebo (28% vs. 31%).
The odds ratios for the association with vitamin D on SAMS were:
- 0.86 in all respondents with 25-OHD measured (95% CI, 0.69-1.09).
- 0.87 in those with levels ≥ 30 ng/mL (95% CI, 0.64-1.19).
- 0.85 with levels of 20-30 ng/mL (95% CI, 0.56-1.28).
- 0.93 with levels < 20 ng/mL (95% CI, 0.50-1.74).
The test for treatment effect modification by baseline serum 25-OHD level was not significant (P for interaction = .83).
In addition, the rate of muscle symptoms was similar between participants randomized to vitamin D and placebo when researchers used a cutpoint to define low 25-OHD of < 30 ng/mL (27% vs. 30%) or < 20 ng/mL (33% vs. 35%).
“We didn’t find any evidence at all that the people who came into the study with low levels of vitamin D did better with the supplement in this case,” Dr. Hlatky said. “So that wasn’t the reason we didn’t see anything.”
Critics may suggest the trial didn’t use a high enough dose of vitamin D, but both Dr. Hlatky and Dr. Stone say that’s unlikely to be a factor in the results because 2,000 IU/d is a substantial dose and well above the recommended adult daily dose of 600-800 IU.
They caution that the substudy wasn’t prespecified, was smaller than the parent trial, and did not have a protocol in place to detail SAMS. They also can’t rule out the possibility that vitamin D may have an effect in patients who have confirmed intolerance to multiple statins, especially after adjustment for the statin type and dose.
“If you’re taking vitamin D to keep from having statin-associated muscle symptoms, this very carefully done substudy with the various caveats doesn’t support that and that’s not something I would give my patients,” Dr. Stone said.
“The most important thing from a negative study is that it allows you to focus your attention on things that may be much more productive rather than assuming that just giving everybody vitamin D will take care of the statin issue,” he added. “Maybe the answer is going to be somewhere else, and there’ll be a lot of people I’m sure who will offer their advice as what the answer is but, I would argue, we want to see more studies to pin it down. So people can get some science behind what they do to try to reduce statin-associated muscle symptoms.”
Paul D. Thompson, MD, chief of cardiology emeritus at Hartford (Conn.) Hospital, and a SAMS expert who was not involved with the research, said, “This is a useful publication, and it’s smart in that it took advantage of a study that was already done.”
He acknowledged being skeptical of a beneficial effect of vitamin D supplementation on SAMS, because some previous data have been retracted, but said that potential treatments are best tested in patients with confirmed statin myalgia, as was the case in his team’s negative trial of CoQ10 supplementation.
That said, the present “study was able to at least give some of the best evidence so far that vitamin D doesn’t do anything to improve symptoms,” Dr. Thompson said. “So maybe it will cut down on so many vitamin D levels [being measured] and use of vitamin D when you don’t really need it.”
The study was sponsored by the Hyperlipidemia Research Fund at Northwestern University. The VITAL trial was supported by grants from the National Institutes of Health, and Quest Diagnostics performed the laboratory measurements at no additional costs. Dr. Hlatky reports no relevant financial relationships. Dr. Stone reports a grant from the Hyperlipidemia Research Fund at Northwestern and honorarium for educational activity for Knowledge to Practice. Dr. Thompson is on the executive committee for a study examining bempedoic acid in patients with statin-associated muscle symptoms.
A version of this article first appeared on Medscape.com.
Vitamin D supplements do not prevent muscle symptoms in new statin users or affect the likelihood of discontinuing a statin due to muscle pain and discomfort, a substudy of the VITAL trial indicates.
Among more than 2,000 randomized participants, statin-associated muscle symptoms (SAMS) were reported by 31% assigned to vitamin D and 31% assigned to placebo.
The two groups were equally likely to stop taking a statin due to muscle symptoms, at 13%.
No significant difference was observed in SAMS (odds ratio [OR], 0.97; 95% confidence interval [CI], 0.80-1.18) or statin discontinuations (OR, 1.04; 95% CI, 0.80-1.35) after adjustment for baseline variables and other characteristics, namely age, sex, and African-American race, previously found to be associated with SAMS in VITAL.
“We actually thought when we started out that maybe we were going to show something, that maybe it was going to be that the people who got the vitamin D were least likely to have a problem with a statin than all those who didn’t get vitamin D, but that is not what we showed,” senior author Neil J. Stone, MD, Northwestern University, Chicago, told this news organization.
He noted that patients in the clinic with low levels of vitamin D often have muscle pain and discomfort and that previous unblinded studies suggested vitamin D might benefit patients with SAMS and reduce statin intolerance.
As previously reported, the double-blind VITAL trial showed no difference in the primary prevention of cardiovascular disease or cancer at 5 years among 25,871 middle-aged adults randomized to vitamin D3 at 2000 IU/d or placebo, regardless of their baseline vitamin D level.
Unlike previous studies showing a benefit with vitamin D on SAMS, importantly, VITAL participants were unaware of whether they were taking vitamin D or placebo and were not expecting any help with their muscle symptoms, first author Mark A. Hlatky, MD, Stanford (Calif.) University, pointed out in an interview.
As to how many statin users turn to the popular supplement for SAMS, he said that number couldn’t be pinned down, despite a lengthy search. “But I think it’s very common, because up to half of people stop taking their statins within a year and many of these do so because of statin-associated muscle symptoms, and we found it in about 30% of people who have them. I have them myself and was motivated to study it because I thought this was an interesting question.”
The results were published online in JAMA Cardiology.
SAMS by baseline 25-OHD
The substudy included 2,083 patients who initiated statin therapy after randomization and were surveyed in early 2016 about their statin use and muscle symptoms.
Two-thirds, or 1,397 patients, had 25-hydroxy vitamin D (25-OHD) measured at baseline, with 47% having levels < 30 ng/mL and 13% levels < 20 ng/mL.
Serum 25-OHD levels were virtually identical in the two treatment groups (mean, 30.4 ng/mL; median, 30.0 ng/mL). The frequency of SAMS did not differ between those assigned to vitamin D or placebo (28% vs. 31%).
The odds ratios for the association with vitamin D on SAMS were:
- 0.86 in all respondents with 25-OHD measured (95% CI, 0.69-1.09).
- 0.87 in those with levels ≥ 30 ng/mL (95% CI, 0.64-1.19).
- 0.85 with levels of 20-30 ng/mL (95% CI, 0.56-1.28).
- 0.93 with levels < 20 ng/mL (95% CI, 0.50-1.74).
The test for treatment effect modification by baseline serum 25-OHD level was not significant (P for interaction = .83).
In addition, the rate of muscle symptoms was similar between participants randomized to vitamin D and placebo when researchers used a cutpoint to define low 25-OHD of < 30 ng/mL (27% vs. 30%) or < 20 ng/mL (33% vs. 35%).
“We didn’t find any evidence at all that the people who came into the study with low levels of vitamin D did better with the supplement in this case,” Dr. Hlatky said. “So that wasn’t the reason we didn’t see anything.”
Critics may suggest the trial didn’t use a high enough dose of vitamin D, but both Dr. Hlatky and Dr. Stone say that’s unlikely to be a factor in the results because 2,000 IU/d is a substantial dose and well above the recommended adult daily dose of 600-800 IU.
They caution that the substudy wasn’t prespecified, was smaller than the parent trial, and did not have a protocol in place to detail SAMS. They also can’t rule out the possibility that vitamin D may have an effect in patients who have confirmed intolerance to multiple statins, especially after adjustment for the statin type and dose.
“If you’re taking vitamin D to keep from having statin-associated muscle symptoms, this very carefully done substudy with the various caveats doesn’t support that and that’s not something I would give my patients,” Dr. Stone said.
“The most important thing from a negative study is that it allows you to focus your attention on things that may be much more productive rather than assuming that just giving everybody vitamin D will take care of the statin issue,” he added. “Maybe the answer is going to be somewhere else, and there’ll be a lot of people I’m sure who will offer their advice as what the answer is but, I would argue, we want to see more studies to pin it down. So people can get some science behind what they do to try to reduce statin-associated muscle symptoms.”
Paul D. Thompson, MD, chief of cardiology emeritus at Hartford (Conn.) Hospital, and a SAMS expert who was not involved with the research, said, “This is a useful publication, and it’s smart in that it took advantage of a study that was already done.”
He acknowledged being skeptical of a beneficial effect of vitamin D supplementation on SAMS, because some previous data have been retracted, but said that potential treatments are best tested in patients with confirmed statin myalgia, as was the case in his team’s negative trial of CoQ10 supplementation.
That said, the present “study was able to at least give some of the best evidence so far that vitamin D doesn’t do anything to improve symptoms,” Dr. Thompson said. “So maybe it will cut down on so many vitamin D levels [being measured] and use of vitamin D when you don’t really need it.”
The study was sponsored by the Hyperlipidemia Research Fund at Northwestern University. The VITAL trial was supported by grants from the National Institutes of Health, and Quest Diagnostics performed the laboratory measurements at no additional costs. Dr. Hlatky reports no relevant financial relationships. Dr. Stone reports a grant from the Hyperlipidemia Research Fund at Northwestern and honorarium for educational activity for Knowledge to Practice. Dr. Thompson is on the executive committee for a study examining bempedoic acid in patients with statin-associated muscle symptoms.
A version of this article first appeared on Medscape.com.
Vitamin D supplements do not prevent muscle symptoms in new statin users or affect the likelihood of discontinuing a statin due to muscle pain and discomfort, a substudy of the VITAL trial indicates.
Among more than 2,000 randomized participants, statin-associated muscle symptoms (SAMS) were reported by 31% assigned to vitamin D and 31% assigned to placebo.
The two groups were equally likely to stop taking a statin due to muscle symptoms, at 13%.
No significant difference was observed in SAMS (odds ratio [OR], 0.97; 95% confidence interval [CI], 0.80-1.18) or statin discontinuations (OR, 1.04; 95% CI, 0.80-1.35) after adjustment for baseline variables and other characteristics, namely age, sex, and African-American race, previously found to be associated with SAMS in VITAL.
“We actually thought when we started out that maybe we were going to show something, that maybe it was going to be that the people who got the vitamin D were least likely to have a problem with a statin than all those who didn’t get vitamin D, but that is not what we showed,” senior author Neil J. Stone, MD, Northwestern University, Chicago, told this news organization.
He noted that patients in the clinic with low levels of vitamin D often have muscle pain and discomfort and that previous unblinded studies suggested vitamin D might benefit patients with SAMS and reduce statin intolerance.
As previously reported, the double-blind VITAL trial showed no difference in the primary prevention of cardiovascular disease or cancer at 5 years among 25,871 middle-aged adults randomized to vitamin D3 at 2000 IU/d or placebo, regardless of their baseline vitamin D level.
Unlike previous studies showing a benefit with vitamin D on SAMS, importantly, VITAL participants were unaware of whether they were taking vitamin D or placebo and were not expecting any help with their muscle symptoms, first author Mark A. Hlatky, MD, Stanford (Calif.) University, pointed out in an interview.
As to how many statin users turn to the popular supplement for SAMS, he said that number couldn’t be pinned down, despite a lengthy search. “But I think it’s very common, because up to half of people stop taking their statins within a year and many of these do so because of statin-associated muscle symptoms, and we found it in about 30% of people who have them. I have them myself and was motivated to study it because I thought this was an interesting question.”
The results were published online in JAMA Cardiology.
SAMS by baseline 25-OHD
The substudy included 2,083 patients who initiated statin therapy after randomization and were surveyed in early 2016 about their statin use and muscle symptoms.
Two-thirds, or 1,397 patients, had 25-hydroxy vitamin D (25-OHD) measured at baseline, with 47% having levels < 30 ng/mL and 13% levels < 20 ng/mL.
Serum 25-OHD levels were virtually identical in the two treatment groups (mean, 30.4 ng/mL; median, 30.0 ng/mL). The frequency of SAMS did not differ between those assigned to vitamin D or placebo (28% vs. 31%).
The odds ratios for the association with vitamin D on SAMS were:
- 0.86 in all respondents with 25-OHD measured (95% CI, 0.69-1.09).
- 0.87 in those with levels ≥ 30 ng/mL (95% CI, 0.64-1.19).
- 0.85 with levels of 20-30 ng/mL (95% CI, 0.56-1.28).
- 0.93 with levels < 20 ng/mL (95% CI, 0.50-1.74).
The test for treatment effect modification by baseline serum 25-OHD level was not significant (P for interaction = .83).
In addition, the rate of muscle symptoms was similar between participants randomized to vitamin D and placebo when researchers used a cutpoint to define low 25-OHD of < 30 ng/mL (27% vs. 30%) or < 20 ng/mL (33% vs. 35%).
“We didn’t find any evidence at all that the people who came into the study with low levels of vitamin D did better with the supplement in this case,” Dr. Hlatky said. “So that wasn’t the reason we didn’t see anything.”
Critics may suggest the trial didn’t use a high enough dose of vitamin D, but both Dr. Hlatky and Dr. Stone say that’s unlikely to be a factor in the results because 2,000 IU/d is a substantial dose and well above the recommended adult daily dose of 600-800 IU.
They caution that the substudy wasn’t prespecified, was smaller than the parent trial, and did not have a protocol in place to detail SAMS. They also can’t rule out the possibility that vitamin D may have an effect in patients who have confirmed intolerance to multiple statins, especially after adjustment for the statin type and dose.
“If you’re taking vitamin D to keep from having statin-associated muscle symptoms, this very carefully done substudy with the various caveats doesn’t support that and that’s not something I would give my patients,” Dr. Stone said.
“The most important thing from a negative study is that it allows you to focus your attention on things that may be much more productive rather than assuming that just giving everybody vitamin D will take care of the statin issue,” he added. “Maybe the answer is going to be somewhere else, and there’ll be a lot of people I’m sure who will offer their advice as what the answer is but, I would argue, we want to see more studies to pin it down. So people can get some science behind what they do to try to reduce statin-associated muscle symptoms.”
Paul D. Thompson, MD, chief of cardiology emeritus at Hartford (Conn.) Hospital, and a SAMS expert who was not involved with the research, said, “This is a useful publication, and it’s smart in that it took advantage of a study that was already done.”
He acknowledged being skeptical of a beneficial effect of vitamin D supplementation on SAMS, because some previous data have been retracted, but said that potential treatments are best tested in patients with confirmed statin myalgia, as was the case in his team’s negative trial of CoQ10 supplementation.
That said, the present “study was able to at least give some of the best evidence so far that vitamin D doesn’t do anything to improve symptoms,” Dr. Thompson said. “So maybe it will cut down on so many vitamin D levels [being measured] and use of vitamin D when you don’t really need it.”
The study was sponsored by the Hyperlipidemia Research Fund at Northwestern University. The VITAL trial was supported by grants from the National Institutes of Health, and Quest Diagnostics performed the laboratory measurements at no additional costs. Dr. Hlatky reports no relevant financial relationships. Dr. Stone reports a grant from the Hyperlipidemia Research Fund at Northwestern and honorarium for educational activity for Knowledge to Practice. Dr. Thompson is on the executive committee for a study examining bempedoic acid in patients with statin-associated muscle symptoms.
A version of this article first appeared on Medscape.com.
Stage 3 melanoma attacked with immunotherapy and a virus-like particle
The result led researchers to call for a future study comparing the regimen against a suitable control group.
“We were very excited to see the ability of intratumoral vidutolimod to augment T-cell infiltrate. (Pathologic) response was associated with a dense infiltrate of CD8 T cells. We were also able to demonstrate for what I think may be the first time, that intratumoral CpG resulted in clear evidence of CD303+ plasmacytoid dendritic cells [pDCs],” said Diwakar Davar, MD, assistant professor of medicine at the University of Pittsburgh, during a presentation of the results at the annual meeting of the Society for Immunotherapy of Cancer. He noted that pDCs represent a very rare cell population, less than 0.4% of circulating peripheral blood mononuclear cells, and tend to be found in lymph nodes.
The current standard of care for stage 3 melanoma is up-front surgery followed by adjuvant therapy – anti–PD-1 therapy for patients with wild-type or BRAF-mutant cancers, and targeted therapy with BRAF/MEK inhibitors in patients with BRAF mutations. However, preclinical studies suggest that neoadjuvant immunotherapy could lead to a stronger antitumor T-cell response than adjuvant immunotherapy.
Vidutolimod targets the toll-like receptor 9 (TLR-9) endosomal receptor found in B cells and pDC cells. The formulation is a virus-like particle (VLP) that contains unmethylated cytosine guanine–rich oligonucleotides (CpG ODN). Bacterial and viral genomes tend to be enriched in CpG ODN, and this acts as a TLR-9 agonist. TLR-9 activation in turn triggers an interferon response, and this may help overcome PD-1 blockade resistance in metastatic melanoma.
The researchers conducted a nonrandomized, open-label trial that included 30 patients with stage 3 melanoma (14 women; median age, 61 years). Patients received neoadjuvant nivolumab and vidutolimod for 8 weeks, then were evaluated for surgery. Patients continued both drugs in the adjuvant setting for 48 weeks. 47% experienced complete pathologic response, 10% a major pathologic response, and 10% a partial pathologic response.
Analysis of resected samples revealed clear evidence of an immune response, Dr. Davar said during a press conference held in advance of the meeting. “Pathologic response was associated with compelling evidence of immune activation both peripherally and within the tumor, with clear evidence of pDC infiltrate and pDC activation – something that has not previously been seen in human specimens.”
The study regimen appeared safe, with no dose-limiting toxicities or grade 4 or 5 adverse events. He noted that the regimen is now being tested in the phase 2 ECOG-ACRIN trial.
The results are “very exciting,” said Pamela Ohashi, PhD, who commented on the study during the press conference. The virus-like nature of vidutolimod may be an important element of the therapy. “I think scientifically we would have predicted that the VLP carrying the CPG would be very good at activating the CD8 cells, which in fact is what you’re seeing. So I think it’s very exciting and has lots of potential for future combinations,” said Dr. Ohashi, who is director of the tumor immunotherapy program at the Princess Margaret Cancer Centre, Toronto.
The study was funded by Checkmate Pharmaceuticals. Dr. Davar has financial relationships with Checkmate Pharmaceuticals and Regeneron, which has acquired Checkmate Pharmaceuticals.
The result led researchers to call for a future study comparing the regimen against a suitable control group.
“We were very excited to see the ability of intratumoral vidutolimod to augment T-cell infiltrate. (Pathologic) response was associated with a dense infiltrate of CD8 T cells. We were also able to demonstrate for what I think may be the first time, that intratumoral CpG resulted in clear evidence of CD303+ plasmacytoid dendritic cells [pDCs],” said Diwakar Davar, MD, assistant professor of medicine at the University of Pittsburgh, during a presentation of the results at the annual meeting of the Society for Immunotherapy of Cancer. He noted that pDCs represent a very rare cell population, less than 0.4% of circulating peripheral blood mononuclear cells, and tend to be found in lymph nodes.
The current standard of care for stage 3 melanoma is up-front surgery followed by adjuvant therapy – anti–PD-1 therapy for patients with wild-type or BRAF-mutant cancers, and targeted therapy with BRAF/MEK inhibitors in patients with BRAF mutations. However, preclinical studies suggest that neoadjuvant immunotherapy could lead to a stronger antitumor T-cell response than adjuvant immunotherapy.
Vidutolimod targets the toll-like receptor 9 (TLR-9) endosomal receptor found in B cells and pDC cells. The formulation is a virus-like particle (VLP) that contains unmethylated cytosine guanine–rich oligonucleotides (CpG ODN). Bacterial and viral genomes tend to be enriched in CpG ODN, and this acts as a TLR-9 agonist. TLR-9 activation in turn triggers an interferon response, and this may help overcome PD-1 blockade resistance in metastatic melanoma.
The researchers conducted a nonrandomized, open-label trial that included 30 patients with stage 3 melanoma (14 women; median age, 61 years). Patients received neoadjuvant nivolumab and vidutolimod for 8 weeks, then were evaluated for surgery. Patients continued both drugs in the adjuvant setting for 48 weeks. 47% experienced complete pathologic response, 10% a major pathologic response, and 10% a partial pathologic response.
Analysis of resected samples revealed clear evidence of an immune response, Dr. Davar said during a press conference held in advance of the meeting. “Pathologic response was associated with compelling evidence of immune activation both peripherally and within the tumor, with clear evidence of pDC infiltrate and pDC activation – something that has not previously been seen in human specimens.”
The study regimen appeared safe, with no dose-limiting toxicities or grade 4 or 5 adverse events. He noted that the regimen is now being tested in the phase 2 ECOG-ACRIN trial.
The results are “very exciting,” said Pamela Ohashi, PhD, who commented on the study during the press conference. The virus-like nature of vidutolimod may be an important element of the therapy. “I think scientifically we would have predicted that the VLP carrying the CPG would be very good at activating the CD8 cells, which in fact is what you’re seeing. So I think it’s very exciting and has lots of potential for future combinations,” said Dr. Ohashi, who is director of the tumor immunotherapy program at the Princess Margaret Cancer Centre, Toronto.
The study was funded by Checkmate Pharmaceuticals. Dr. Davar has financial relationships with Checkmate Pharmaceuticals and Regeneron, which has acquired Checkmate Pharmaceuticals.
The result led researchers to call for a future study comparing the regimen against a suitable control group.
“We were very excited to see the ability of intratumoral vidutolimod to augment T-cell infiltrate. (Pathologic) response was associated with a dense infiltrate of CD8 T cells. We were also able to demonstrate for what I think may be the first time, that intratumoral CpG resulted in clear evidence of CD303+ plasmacytoid dendritic cells [pDCs],” said Diwakar Davar, MD, assistant professor of medicine at the University of Pittsburgh, during a presentation of the results at the annual meeting of the Society for Immunotherapy of Cancer. He noted that pDCs represent a very rare cell population, less than 0.4% of circulating peripheral blood mononuclear cells, and tend to be found in lymph nodes.
The current standard of care for stage 3 melanoma is up-front surgery followed by adjuvant therapy – anti–PD-1 therapy for patients with wild-type or BRAF-mutant cancers, and targeted therapy with BRAF/MEK inhibitors in patients with BRAF mutations. However, preclinical studies suggest that neoadjuvant immunotherapy could lead to a stronger antitumor T-cell response than adjuvant immunotherapy.
Vidutolimod targets the toll-like receptor 9 (TLR-9) endosomal receptor found in B cells and pDC cells. The formulation is a virus-like particle (VLP) that contains unmethylated cytosine guanine–rich oligonucleotides (CpG ODN). Bacterial and viral genomes tend to be enriched in CpG ODN, and this acts as a TLR-9 agonist. TLR-9 activation in turn triggers an interferon response, and this may help overcome PD-1 blockade resistance in metastatic melanoma.
The researchers conducted a nonrandomized, open-label trial that included 30 patients with stage 3 melanoma (14 women; median age, 61 years). Patients received neoadjuvant nivolumab and vidutolimod for 8 weeks, then were evaluated for surgery. Patients continued both drugs in the adjuvant setting for 48 weeks. 47% experienced complete pathologic response, 10% a major pathologic response, and 10% a partial pathologic response.
Analysis of resected samples revealed clear evidence of an immune response, Dr. Davar said during a press conference held in advance of the meeting. “Pathologic response was associated with compelling evidence of immune activation both peripherally and within the tumor, with clear evidence of pDC infiltrate and pDC activation – something that has not previously been seen in human specimens.”
The study regimen appeared safe, with no dose-limiting toxicities or grade 4 or 5 adverse events. He noted that the regimen is now being tested in the phase 2 ECOG-ACRIN trial.
The results are “very exciting,” said Pamela Ohashi, PhD, who commented on the study during the press conference. The virus-like nature of vidutolimod may be an important element of the therapy. “I think scientifically we would have predicted that the VLP carrying the CPG would be very good at activating the CD8 cells, which in fact is what you’re seeing. So I think it’s very exciting and has lots of potential for future combinations,” said Dr. Ohashi, who is director of the tumor immunotherapy program at the Princess Margaret Cancer Centre, Toronto.
The study was funded by Checkmate Pharmaceuticals. Dr. Davar has financial relationships with Checkmate Pharmaceuticals and Regeneron, which has acquired Checkmate Pharmaceuticals.
FROM SITC 2022
Blenrep for multiple myeloma withdrawn from U.S. market
A drug used in the treatment of relapsed/refractory multiple myeloma (RRMM) is in the process of being pulled off the U.S. market by its manufacturer.
The drug is belantamab mafodotin-blmf (Blenrep), an antibody drug conjugate that targets B-cell maturation antigen (BCMA).
The manufacturer, GSK, announced that it has started the process of withdrawing this drug from the market at the request of the U.S. Food and Drug Administration (FDA).
This request follows disappointing results from a large confirmatory trial, known as DREAMM-3, in which the drug failed to meet the primary endpoint of showing an improvement in progression-free survival (PFS).
The company was obliged to carry out this confirmatory trial after the FDA granted an accelerated approval for the drug in August 2020.
The accelerated approval was based on response data, and it was dependent on later trials’ confirming a clinical benefit. In this case, those trials did not confirm a clinical benefit.
“We respect the Agency’s approach to the accelerated approval regulations and associated process,” commented the GSK Chief Medical Officer Sabine Luik.
The company will continue to “work with the U.S. FDA on a path forward for this important treatment option for patients with multiple myeloma.”
Further clinical trials in the DREAMM program are still underway. Results from the DREAMM-7 and DREAMM-8 trials are expected in early 2023.
The company had high hopes for the drug when it was launched. At that time, belanatamab mafodotin-blmf was the only drug on the market that targeted BCMA, and so it was the first drug in its class.
However, it is no longer unique. In the 2 years that it has been available, several other products that target BCMA have been launched for use in the treatment of multiple myeloma. These include the two chimeric antigen receptor T-cell products, idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti), as well as the bispecific antibody teclistamab (Tecvayli).
For relapsed/refractory disease
Belantamab mafodotin-blmf was approved for use in patients with RRMM who had already undergone treatment with one of the three major classes of drugs, namely, an immunomodulatory agent, a proteasome inhibitor, and a CD-38 monoclonal antibody.
Patients who are currently taking the drug and would like to continue doing so will have the option to enroll in a compassionate use program to retain their access to treatment, the company said.
“GSK continues to believe, based on the totality of data available from the DREAMM (DRiving Excellence in Approaches to Multiple Myeloma) development program, that the benefit-risk profile of belantamab mafodotin remains favorable in this hard-to-treat RRMM patient population. Patients responding to belantamab mafodotin experienced durable clinical benefit, and safety remains consistent with the known safety profile,” the company said.
Details of DREAMM-3 results
DREAMM-3 was a phase 3 trial that compared single-agent belantamab mafodotin to pomalidomide (Pomalyst) in combination with low-dose dexamethasone (PomDex) for patients with RRMM.
The results for the primary endpoint of PFS did not reach statistical significance: median PFS was 11.2 vs. 7 months with PomDex (hazard ratio, 1.03; 95% confidence interval, 0.72-1.47).
At the time of the primary analysis, the overall survival (OS) data had only achieved 37.5% overall maturity. The median OS was 21.2 vs. 21.1 months with PomDex (HR, 1.14; 95% CI, 0.77-1.68).
A version of this article first appeared on Medscape.com.
A drug used in the treatment of relapsed/refractory multiple myeloma (RRMM) is in the process of being pulled off the U.S. market by its manufacturer.
The drug is belantamab mafodotin-blmf (Blenrep), an antibody drug conjugate that targets B-cell maturation antigen (BCMA).
The manufacturer, GSK, announced that it has started the process of withdrawing this drug from the market at the request of the U.S. Food and Drug Administration (FDA).
This request follows disappointing results from a large confirmatory trial, known as DREAMM-3, in which the drug failed to meet the primary endpoint of showing an improvement in progression-free survival (PFS).
The company was obliged to carry out this confirmatory trial after the FDA granted an accelerated approval for the drug in August 2020.
The accelerated approval was based on response data, and it was dependent on later trials’ confirming a clinical benefit. In this case, those trials did not confirm a clinical benefit.
“We respect the Agency’s approach to the accelerated approval regulations and associated process,” commented the GSK Chief Medical Officer Sabine Luik.
The company will continue to “work with the U.S. FDA on a path forward for this important treatment option for patients with multiple myeloma.”
Further clinical trials in the DREAMM program are still underway. Results from the DREAMM-7 and DREAMM-8 trials are expected in early 2023.
The company had high hopes for the drug when it was launched. At that time, belanatamab mafodotin-blmf was the only drug on the market that targeted BCMA, and so it was the first drug in its class.
However, it is no longer unique. In the 2 years that it has been available, several other products that target BCMA have been launched for use in the treatment of multiple myeloma. These include the two chimeric antigen receptor T-cell products, idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti), as well as the bispecific antibody teclistamab (Tecvayli).
For relapsed/refractory disease
Belantamab mafodotin-blmf was approved for use in patients with RRMM who had already undergone treatment with one of the three major classes of drugs, namely, an immunomodulatory agent, a proteasome inhibitor, and a CD-38 monoclonal antibody.
Patients who are currently taking the drug and would like to continue doing so will have the option to enroll in a compassionate use program to retain their access to treatment, the company said.
“GSK continues to believe, based on the totality of data available from the DREAMM (DRiving Excellence in Approaches to Multiple Myeloma) development program, that the benefit-risk profile of belantamab mafodotin remains favorable in this hard-to-treat RRMM patient population. Patients responding to belantamab mafodotin experienced durable clinical benefit, and safety remains consistent with the known safety profile,” the company said.
Details of DREAMM-3 results
DREAMM-3 was a phase 3 trial that compared single-agent belantamab mafodotin to pomalidomide (Pomalyst) in combination with low-dose dexamethasone (PomDex) for patients with RRMM.
The results for the primary endpoint of PFS did not reach statistical significance: median PFS was 11.2 vs. 7 months with PomDex (hazard ratio, 1.03; 95% confidence interval, 0.72-1.47).
At the time of the primary analysis, the overall survival (OS) data had only achieved 37.5% overall maturity. The median OS was 21.2 vs. 21.1 months with PomDex (HR, 1.14; 95% CI, 0.77-1.68).
A version of this article first appeared on Medscape.com.
A drug used in the treatment of relapsed/refractory multiple myeloma (RRMM) is in the process of being pulled off the U.S. market by its manufacturer.
The drug is belantamab mafodotin-blmf (Blenrep), an antibody drug conjugate that targets B-cell maturation antigen (BCMA).
The manufacturer, GSK, announced that it has started the process of withdrawing this drug from the market at the request of the U.S. Food and Drug Administration (FDA).
This request follows disappointing results from a large confirmatory trial, known as DREAMM-3, in which the drug failed to meet the primary endpoint of showing an improvement in progression-free survival (PFS).
The company was obliged to carry out this confirmatory trial after the FDA granted an accelerated approval for the drug in August 2020.
The accelerated approval was based on response data, and it was dependent on later trials’ confirming a clinical benefit. In this case, those trials did not confirm a clinical benefit.
“We respect the Agency’s approach to the accelerated approval regulations and associated process,” commented the GSK Chief Medical Officer Sabine Luik.
The company will continue to “work with the U.S. FDA on a path forward for this important treatment option for patients with multiple myeloma.”
Further clinical trials in the DREAMM program are still underway. Results from the DREAMM-7 and DREAMM-8 trials are expected in early 2023.
The company had high hopes for the drug when it was launched. At that time, belanatamab mafodotin-blmf was the only drug on the market that targeted BCMA, and so it was the first drug in its class.
However, it is no longer unique. In the 2 years that it has been available, several other products that target BCMA have been launched for use in the treatment of multiple myeloma. These include the two chimeric antigen receptor T-cell products, idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti), as well as the bispecific antibody teclistamab (Tecvayli).
For relapsed/refractory disease
Belantamab mafodotin-blmf was approved for use in patients with RRMM who had already undergone treatment with one of the three major classes of drugs, namely, an immunomodulatory agent, a proteasome inhibitor, and a CD-38 monoclonal antibody.
Patients who are currently taking the drug and would like to continue doing so will have the option to enroll in a compassionate use program to retain their access to treatment, the company said.
“GSK continues to believe, based on the totality of data available from the DREAMM (DRiving Excellence in Approaches to Multiple Myeloma) development program, that the benefit-risk profile of belantamab mafodotin remains favorable in this hard-to-treat RRMM patient population. Patients responding to belantamab mafodotin experienced durable clinical benefit, and safety remains consistent with the known safety profile,” the company said.
Details of DREAMM-3 results
DREAMM-3 was a phase 3 trial that compared single-agent belantamab mafodotin to pomalidomide (Pomalyst) in combination with low-dose dexamethasone (PomDex) for patients with RRMM.
The results for the primary endpoint of PFS did not reach statistical significance: median PFS was 11.2 vs. 7 months with PomDex (hazard ratio, 1.03; 95% confidence interval, 0.72-1.47).
At the time of the primary analysis, the overall survival (OS) data had only achieved 37.5% overall maturity. The median OS was 21.2 vs. 21.1 months with PomDex (HR, 1.14; 95% CI, 0.77-1.68).
A version of this article first appeared on Medscape.com.
IRONMAN galvanizes case for IV iron repletion in heart failure
CHICAGO – Another major study appears to back the use of intravenous iron repletion in patients with heart failure (HF) and iron deficiency, strengthening largely consistent evidence, researchers say, that the treatment may improve symptoms and prevent some HF-related hospital admissions.
To be sure, the IRONMAN trial, which compared intravenous iron versus usual care in such patients – most with reduced ejection fraction and not hospitalized – failed to show a benefit for its primary endpoint. The 18% reduction in risk for HF hospitalization or cardiovascular (CV) death seen in the trial, however encouraging, can only be called a trend (P = .07).
But the intervention showed signs of benefit for some secondary endpoints, including quality of life scores, and hinted at such an effect on HF hospitalization. Risk for the latter endpoint dropped 20% (P = .085) over a median follow-up of 2.7 years.
The findings “build upon the other data we have that correcting iron deficiency can help improve well-being, and particularly reduce the risk of hospitalization, in a broad range of [HF] patients,” said Paul Kalra, MD, of the University of Glasgow and Portsmouth (England) Hospitals University NHS Trust.
The tested regimen “was well tolerated with no safety concerns” and offers “reassurance about the long-term safety” of the intravenous iron it used, ferric derisomaltose (MonoFerric), in patients with HF, Dr. Kalra said at a media briefing on the trial.
The remarks preceded his formal presentation of IRONMAN at the American Heart Association scientific sessions. Dr. Kalra is also lead author on the trial’s publication in The Lancet.
IRONMAN strengthens the base of evidence supporting intravenous iron in HF with iron deficiency, especially chronic HF in outpatients, Dr. Kalra and others said. It also supports efficacy for a form of intravenous iron not previously tested in a major HF trial.
Still, “the totality of data are now supporting intravenous iron per se,” regardless of the iron agent used, said Dr. Kalra. But ferric derisomaltose may have dosing advantages, he observed, “and we’ve now got these long-term safety data.”
The strongest prior support for intravenous iron in HF came from hospitalized patients who received it as ferric carboxymaltose (Ferinject) and were followed only 12 months. That was in the AFFIRM-AHF trial, published 2 years ago, which also missed its primary endpoint – the same one used in IRONMAN. Some outcomes in the two trials were similar.
The risk for HF hospitalization or CV death for intravenous iron therapy, compared with usual care, in AFFIRM-AHF fell 21% (P = .059), missing significance but apparently driven by a 26% drop in risk for HF readmissions (P = .013). But neither that trial nor IRONMAN suggested a benefit for CV mortality on its own.
The COVID effect
In IRONMAN, Dr. Kalra said, usual care could include oral iron supplementation, which 17% of patients in the control group received. That could potentially have kept the intravenous iron group from making a better showing for the primary endpoint, he proposed.
And some iron doses and other treatments were missed by a substantial number of patients in both groups who entered the trial after the United Kingdom’s national lockdown in response to the COVID-19 pandemic, he observed. “Patients were not able to come into hospitals for research visits, or in fact when they were able, may not have wanted to.”
So, the group conducted a “prespecified” sensitivity analysis that excluded the 9% of patients enrolled by the end of March 2020, about the time of the first lockdown, and followed the remainder for another 6 months.
In that analysis, risk for HF hospitalization or CV death declined 24% in the intravenous iron group, a marginal but significant result (P = .047) that was dominated by an improvement in HF hospitalizations.
Effects on guidelines
The intravenous iron recommendations in the European HF guidelines refer only to ferric carboxymaltose without mentioning other forms, such as ferric derisomaltose, “but this is now a class effect given the similarities between AFFIRM-AHF and IRONMAN,” said Gregory D. Lewis, MD, Mass General Brigham, Boston, invited discussant for Dr. Kalra’s presentation at the AHA session.
“In the United States, we relegate IV iron to improvement in functional capacity as a comorbidity of heart failure. Perhaps this role will expand,” added Dr. Lewis, who is medical director of his center’s heart transplant program.
He also wondered aloud whether the purported clinical benefits of intravenous iron in HF patients with iron deficiency, not as yet supported by a significant primary-endpoint showing in one of the major trials, currently justify expansion of its use in practice.
“With the benefits of IV iron on exercise capacity and quality of life, and the safety of administering high doses of IV iron,” potentially reducing HF polypharmacy, he noted, “should we be considering IV iron more commonly for utilization in our patients even if we find that heart failure hospitalizations and mortality are only modestly improved?”
IRONMAN “asked whether there’s benefit to IV iron in the longer term,” Kiran Musunuru, MD, PhD, MPH, University of Pennsylvania,Philadelphia, observed at the media briefing. As the trial was reported, “that does in fact, seem to be the case,” said Dr. Musunuru, who was not involved in IRONMAN.
Therefore, he said, “this study reinforces the message that we should be routinely monitoring our heart failure patients for iron deficiency and supplementing them as needed.”
A commentary linked to the IRONMAN publication agreed. The trial “increases the evidence base for the treatment of iron deficiency with intravenous iron supplementation,” wrote the editorialists, led by Theresa A. McDonagh, MD, King’s College Hospital and School of Cardiovascular Sciences, London.
Patients with acute or chronic HF, iron deficiency, and reduced or mildly reduced ejection fractions “should be offered treatment with intravenous iron to reduce their risk of hospital admission for heart failure,” they concluded.
Mostly reduced-EF outpatients
The open-label, blinded-endpoint IRONMAN trial, conducted at 70 centers in the United Kingdom, entered adults with HF, ejection fractions 45% or lower within the previous 2 years, and iron deficiency defined as transferrin saturation less than 20% or serum ferritin levels below 100 mcg/L, the report states. They were either hospitalized for HF, had such a hospitalization within the past 6 months, or were outpatients with elevated natriuretic peptide levels; the third category accounted for two thirds of the trial population.
Of the 1,137 randomized patients, 569 were assigned to receive intravenous ferric derisomaltose at weight- and hemoglobin-adjusted dosages; 568 went to the usual-care group.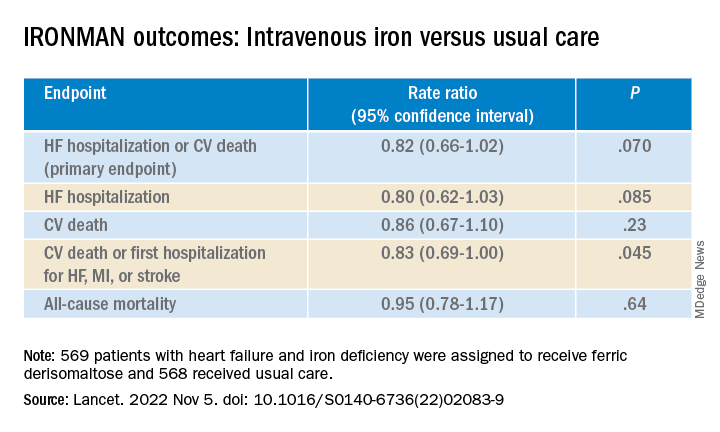
Those receiving intravenous iron visited the trial clinic 4 weeks later and then every 4 months. At those visits, they received a round of ferric derisomaltose if their ferritin levels were below 100 mcg/L, or 400 mcg/L or lower if transferrin saturation was below 25%, the published report states.
Mean scores on the Minnesota Living with Heart Failure Questionnaire improved by a marginally significant 3.33 points (P = .050) at 4 months in the intravenous iron group. The gain receded to a nonsignificant 2.57 points by 20 months (P = .23).
In COVID-related sensitivity analysis, the intravenous iron group showed a significant benefit for the primary endpoint and a trend for improved HF hospitalizations.
- HF hospitalization or CV death: RR, 0.76 (95% confidence interval, 0.58-1.00; P = .047)
- HF hospitalization: RR 0.76 (95% CI, 0.56-1.03; P = .077)
Fewer patients in the intravenous iron group experienced serious cardiac adverse events, 36% compared with 43% in for those on usual care, P = .016.
The recently updated European Society of Cardiology guidelines for HF made it a class 1 recommendation to assess iron status in every patient, Kalra observed. “It doesn›t specify how frequently, but I think we should be thinking about every 4-6 months.”
Dr. Kalra disclosed receiving research grants from Pharmacosmos; and consulting or lecturing for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Novartis, Pfizer, Pharmacosmos, Servier, and Vifor Pharma. Dr. Musunuru disclosed significant ownership interest in Verve Therapeutics and Variant Bio. Dr. Lewis disclosed relationships with NXT, American Regent, and RIVUS; and receiving research grants from Cytokinetics and Amgen.
A version of this article first appeared on Medscape.com.
CHICAGO – Another major study appears to back the use of intravenous iron repletion in patients with heart failure (HF) and iron deficiency, strengthening largely consistent evidence, researchers say, that the treatment may improve symptoms and prevent some HF-related hospital admissions.
To be sure, the IRONMAN trial, which compared intravenous iron versus usual care in such patients – most with reduced ejection fraction and not hospitalized – failed to show a benefit for its primary endpoint. The 18% reduction in risk for HF hospitalization or cardiovascular (CV) death seen in the trial, however encouraging, can only be called a trend (P = .07).
But the intervention showed signs of benefit for some secondary endpoints, including quality of life scores, and hinted at such an effect on HF hospitalization. Risk for the latter endpoint dropped 20% (P = .085) over a median follow-up of 2.7 years.
The findings “build upon the other data we have that correcting iron deficiency can help improve well-being, and particularly reduce the risk of hospitalization, in a broad range of [HF] patients,” said Paul Kalra, MD, of the University of Glasgow and Portsmouth (England) Hospitals University NHS Trust.
The tested regimen “was well tolerated with no safety concerns” and offers “reassurance about the long-term safety” of the intravenous iron it used, ferric derisomaltose (MonoFerric), in patients with HF, Dr. Kalra said at a media briefing on the trial.
The remarks preceded his formal presentation of IRONMAN at the American Heart Association scientific sessions. Dr. Kalra is also lead author on the trial’s publication in The Lancet.
IRONMAN strengthens the base of evidence supporting intravenous iron in HF with iron deficiency, especially chronic HF in outpatients, Dr. Kalra and others said. It also supports efficacy for a form of intravenous iron not previously tested in a major HF trial.
Still, “the totality of data are now supporting intravenous iron per se,” regardless of the iron agent used, said Dr. Kalra. But ferric derisomaltose may have dosing advantages, he observed, “and we’ve now got these long-term safety data.”
The strongest prior support for intravenous iron in HF came from hospitalized patients who received it as ferric carboxymaltose (Ferinject) and were followed only 12 months. That was in the AFFIRM-AHF trial, published 2 years ago, which also missed its primary endpoint – the same one used in IRONMAN. Some outcomes in the two trials were similar.
The risk for HF hospitalization or CV death for intravenous iron therapy, compared with usual care, in AFFIRM-AHF fell 21% (P = .059), missing significance but apparently driven by a 26% drop in risk for HF readmissions (P = .013). But neither that trial nor IRONMAN suggested a benefit for CV mortality on its own.
The COVID effect
In IRONMAN, Dr. Kalra said, usual care could include oral iron supplementation, which 17% of patients in the control group received. That could potentially have kept the intravenous iron group from making a better showing for the primary endpoint, he proposed.
And some iron doses and other treatments were missed by a substantial number of patients in both groups who entered the trial after the United Kingdom’s national lockdown in response to the COVID-19 pandemic, he observed. “Patients were not able to come into hospitals for research visits, or in fact when they were able, may not have wanted to.”
So, the group conducted a “prespecified” sensitivity analysis that excluded the 9% of patients enrolled by the end of March 2020, about the time of the first lockdown, and followed the remainder for another 6 months.
In that analysis, risk for HF hospitalization or CV death declined 24% in the intravenous iron group, a marginal but significant result (P = .047) that was dominated by an improvement in HF hospitalizations.
Effects on guidelines
The intravenous iron recommendations in the European HF guidelines refer only to ferric carboxymaltose without mentioning other forms, such as ferric derisomaltose, “but this is now a class effect given the similarities between AFFIRM-AHF and IRONMAN,” said Gregory D. Lewis, MD, Mass General Brigham, Boston, invited discussant for Dr. Kalra’s presentation at the AHA session.
“In the United States, we relegate IV iron to improvement in functional capacity as a comorbidity of heart failure. Perhaps this role will expand,” added Dr. Lewis, who is medical director of his center’s heart transplant program.
He also wondered aloud whether the purported clinical benefits of intravenous iron in HF patients with iron deficiency, not as yet supported by a significant primary-endpoint showing in one of the major trials, currently justify expansion of its use in practice.
“With the benefits of IV iron on exercise capacity and quality of life, and the safety of administering high doses of IV iron,” potentially reducing HF polypharmacy, he noted, “should we be considering IV iron more commonly for utilization in our patients even if we find that heart failure hospitalizations and mortality are only modestly improved?”
IRONMAN “asked whether there’s benefit to IV iron in the longer term,” Kiran Musunuru, MD, PhD, MPH, University of Pennsylvania,Philadelphia, observed at the media briefing. As the trial was reported, “that does in fact, seem to be the case,” said Dr. Musunuru, who was not involved in IRONMAN.
Therefore, he said, “this study reinforces the message that we should be routinely monitoring our heart failure patients for iron deficiency and supplementing them as needed.”
A commentary linked to the IRONMAN publication agreed. The trial “increases the evidence base for the treatment of iron deficiency with intravenous iron supplementation,” wrote the editorialists, led by Theresa A. McDonagh, MD, King’s College Hospital and School of Cardiovascular Sciences, London.
Patients with acute or chronic HF, iron deficiency, and reduced or mildly reduced ejection fractions “should be offered treatment with intravenous iron to reduce their risk of hospital admission for heart failure,” they concluded.
Mostly reduced-EF outpatients
The open-label, blinded-endpoint IRONMAN trial, conducted at 70 centers in the United Kingdom, entered adults with HF, ejection fractions 45% or lower within the previous 2 years, and iron deficiency defined as transferrin saturation less than 20% or serum ferritin levels below 100 mcg/L, the report states. They were either hospitalized for HF, had such a hospitalization within the past 6 months, or were outpatients with elevated natriuretic peptide levels; the third category accounted for two thirds of the trial population.
Of the 1,137 randomized patients, 569 were assigned to receive intravenous ferric derisomaltose at weight- and hemoglobin-adjusted dosages; 568 went to the usual-care group.
Those receiving intravenous iron visited the trial clinic 4 weeks later and then every 4 months. At those visits, they received a round of ferric derisomaltose if their ferritin levels were below 100 mcg/L, or 400 mcg/L or lower if transferrin saturation was below 25%, the published report states.
Mean scores on the Minnesota Living with Heart Failure Questionnaire improved by a marginally significant 3.33 points (P = .050) at 4 months in the intravenous iron group. The gain receded to a nonsignificant 2.57 points by 20 months (P = .23).
In COVID-related sensitivity analysis, the intravenous iron group showed a significant benefit for the primary endpoint and a trend for improved HF hospitalizations.
- HF hospitalization or CV death: RR, 0.76 (95% confidence interval, 0.58-1.00; P = .047)
- HF hospitalization: RR 0.76 (95% CI, 0.56-1.03; P = .077)
Fewer patients in the intravenous iron group experienced serious cardiac adverse events, 36% compared with 43% in for those on usual care, P = .016.
The recently updated European Society of Cardiology guidelines for HF made it a class 1 recommendation to assess iron status in every patient, Kalra observed. “It doesn›t specify how frequently, but I think we should be thinking about every 4-6 months.”
Dr. Kalra disclosed receiving research grants from Pharmacosmos; and consulting or lecturing for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Novartis, Pfizer, Pharmacosmos, Servier, and Vifor Pharma. Dr. Musunuru disclosed significant ownership interest in Verve Therapeutics and Variant Bio. Dr. Lewis disclosed relationships with NXT, American Regent, and RIVUS; and receiving research grants from Cytokinetics and Amgen.
A version of this article first appeared on Medscape.com.
CHICAGO – Another major study appears to back the use of intravenous iron repletion in patients with heart failure (HF) and iron deficiency, strengthening largely consistent evidence, researchers say, that the treatment may improve symptoms and prevent some HF-related hospital admissions.
To be sure, the IRONMAN trial, which compared intravenous iron versus usual care in such patients – most with reduced ejection fraction and not hospitalized – failed to show a benefit for its primary endpoint. The 18% reduction in risk for HF hospitalization or cardiovascular (CV) death seen in the trial, however encouraging, can only be called a trend (P = .07).
But the intervention showed signs of benefit for some secondary endpoints, including quality of life scores, and hinted at such an effect on HF hospitalization. Risk for the latter endpoint dropped 20% (P = .085) over a median follow-up of 2.7 years.
The findings “build upon the other data we have that correcting iron deficiency can help improve well-being, and particularly reduce the risk of hospitalization, in a broad range of [HF] patients,” said Paul Kalra, MD, of the University of Glasgow and Portsmouth (England) Hospitals University NHS Trust.
The tested regimen “was well tolerated with no safety concerns” and offers “reassurance about the long-term safety” of the intravenous iron it used, ferric derisomaltose (MonoFerric), in patients with HF, Dr. Kalra said at a media briefing on the trial.
The remarks preceded his formal presentation of IRONMAN at the American Heart Association scientific sessions. Dr. Kalra is also lead author on the trial’s publication in The Lancet.
IRONMAN strengthens the base of evidence supporting intravenous iron in HF with iron deficiency, especially chronic HF in outpatients, Dr. Kalra and others said. It also supports efficacy for a form of intravenous iron not previously tested in a major HF trial.
Still, “the totality of data are now supporting intravenous iron per se,” regardless of the iron agent used, said Dr. Kalra. But ferric derisomaltose may have dosing advantages, he observed, “and we’ve now got these long-term safety data.”
The strongest prior support for intravenous iron in HF came from hospitalized patients who received it as ferric carboxymaltose (Ferinject) and were followed only 12 months. That was in the AFFIRM-AHF trial, published 2 years ago, which also missed its primary endpoint – the same one used in IRONMAN. Some outcomes in the two trials were similar.
The risk for HF hospitalization or CV death for intravenous iron therapy, compared with usual care, in AFFIRM-AHF fell 21% (P = .059), missing significance but apparently driven by a 26% drop in risk for HF readmissions (P = .013). But neither that trial nor IRONMAN suggested a benefit for CV mortality on its own.
The COVID effect
In IRONMAN, Dr. Kalra said, usual care could include oral iron supplementation, which 17% of patients in the control group received. That could potentially have kept the intravenous iron group from making a better showing for the primary endpoint, he proposed.
And some iron doses and other treatments were missed by a substantial number of patients in both groups who entered the trial after the United Kingdom’s national lockdown in response to the COVID-19 pandemic, he observed. “Patients were not able to come into hospitals for research visits, or in fact when they were able, may not have wanted to.”
So, the group conducted a “prespecified” sensitivity analysis that excluded the 9% of patients enrolled by the end of March 2020, about the time of the first lockdown, and followed the remainder for another 6 months.
In that analysis, risk for HF hospitalization or CV death declined 24% in the intravenous iron group, a marginal but significant result (P = .047) that was dominated by an improvement in HF hospitalizations.
Effects on guidelines
The intravenous iron recommendations in the European HF guidelines refer only to ferric carboxymaltose without mentioning other forms, such as ferric derisomaltose, “but this is now a class effect given the similarities between AFFIRM-AHF and IRONMAN,” said Gregory D. Lewis, MD, Mass General Brigham, Boston, invited discussant for Dr. Kalra’s presentation at the AHA session.
“In the United States, we relegate IV iron to improvement in functional capacity as a comorbidity of heart failure. Perhaps this role will expand,” added Dr. Lewis, who is medical director of his center’s heart transplant program.
He also wondered aloud whether the purported clinical benefits of intravenous iron in HF patients with iron deficiency, not as yet supported by a significant primary-endpoint showing in one of the major trials, currently justify expansion of its use in practice.
“With the benefits of IV iron on exercise capacity and quality of life, and the safety of administering high doses of IV iron,” potentially reducing HF polypharmacy, he noted, “should we be considering IV iron more commonly for utilization in our patients even if we find that heart failure hospitalizations and mortality are only modestly improved?”
IRONMAN “asked whether there’s benefit to IV iron in the longer term,” Kiran Musunuru, MD, PhD, MPH, University of Pennsylvania,Philadelphia, observed at the media briefing. As the trial was reported, “that does in fact, seem to be the case,” said Dr. Musunuru, who was not involved in IRONMAN.
Therefore, he said, “this study reinforces the message that we should be routinely monitoring our heart failure patients for iron deficiency and supplementing them as needed.”
A commentary linked to the IRONMAN publication agreed. The trial “increases the evidence base for the treatment of iron deficiency with intravenous iron supplementation,” wrote the editorialists, led by Theresa A. McDonagh, MD, King’s College Hospital and School of Cardiovascular Sciences, London.
Patients with acute or chronic HF, iron deficiency, and reduced or mildly reduced ejection fractions “should be offered treatment with intravenous iron to reduce their risk of hospital admission for heart failure,” they concluded.
Mostly reduced-EF outpatients
The open-label, blinded-endpoint IRONMAN trial, conducted at 70 centers in the United Kingdom, entered adults with HF, ejection fractions 45% or lower within the previous 2 years, and iron deficiency defined as transferrin saturation less than 20% or serum ferritin levels below 100 mcg/L, the report states. They were either hospitalized for HF, had such a hospitalization within the past 6 months, or were outpatients with elevated natriuretic peptide levels; the third category accounted for two thirds of the trial population.
Of the 1,137 randomized patients, 569 were assigned to receive intravenous ferric derisomaltose at weight- and hemoglobin-adjusted dosages; 568 went to the usual-care group.
Those receiving intravenous iron visited the trial clinic 4 weeks later and then every 4 months. At those visits, they received a round of ferric derisomaltose if their ferritin levels were below 100 mcg/L, or 400 mcg/L or lower if transferrin saturation was below 25%, the published report states.
Mean scores on the Minnesota Living with Heart Failure Questionnaire improved by a marginally significant 3.33 points (P = .050) at 4 months in the intravenous iron group. The gain receded to a nonsignificant 2.57 points by 20 months (P = .23).
In COVID-related sensitivity analysis, the intravenous iron group showed a significant benefit for the primary endpoint and a trend for improved HF hospitalizations.
- HF hospitalization or CV death: RR, 0.76 (95% confidence interval, 0.58-1.00; P = .047)
- HF hospitalization: RR 0.76 (95% CI, 0.56-1.03; P = .077)
Fewer patients in the intravenous iron group experienced serious cardiac adverse events, 36% compared with 43% in for those on usual care, P = .016.
The recently updated European Society of Cardiology guidelines for HF made it a class 1 recommendation to assess iron status in every patient, Kalra observed. “It doesn›t specify how frequently, but I think we should be thinking about every 4-6 months.”
Dr. Kalra disclosed receiving research grants from Pharmacosmos; and consulting or lecturing for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Novartis, Pfizer, Pharmacosmos, Servier, and Vifor Pharma. Dr. Musunuru disclosed significant ownership interest in Verve Therapeutics and Variant Bio. Dr. Lewis disclosed relationships with NXT, American Regent, and RIVUS; and receiving research grants from Cytokinetics and Amgen.
A version of this article first appeared on Medscape.com.
AT AHA 2022
Optimize HF meds rapidly and fully after hospital discharge: STRONG-HF
CHICAGO – Clinicians who prescribe heart failure meds are holding the best hand they’ve ever had, but with so much underuse and suboptimal dosing in actual practice, it seems many may not appreciate the value of their cards. But a major randomized trial that has captured the field’s attention may embolden them to go all in.
Results showed that a strategy of early, rapid up-titration of multiple guideline-directed meds in patients hospitalized with heart failure, compared with a usual-care approach, cut their 6-month risk for death or HF readmission by a steep 34% (P = .002).
The drugs had been started and partly up-titrated in the hospital with the goal of full up-titration within 2 weeks after discharge.
Patients well tolerated the high-intensity approach, researchers said. Their quality-of-life scores improved (P < .0001) compared with the usual-care group, and adverse events were considered few and manageable in the international trial with more than 1,000 patients.
Safety on the high-intensity strategy depended on close patient monitoring at frequently planned clinic visits along with guidance for the up-titrations from clinical signs and natriuretic peptide levels, observed Alexandre Mebazaa, MD, PhD, University of Paris and Public Hospitals of Paris.
Dr. Mebazaa is principal investigator on the trial, called STRONG-HF, which he presented at the American Heart Association scientific sessions, held in Chicago and virtually. He is also lead author on the study’s same-day publication in the Lancet.
The high-intensity strategy’s superiority emerged early in the trial, which was halted early on the data safety monitoring board’s recommendation, with about 90% of follow-ups completed. The board “felt it was unethical to keep patients in usual care,” Dr. Mebazaa said at a press conference.
A dramatic change
The next step, he said, will be to educate the heart failure community on the high-intensity care technique so it can swiftly enter clinical practice. Currently in acute heart failure, “very few patients are monitored after discharge and treated with full doses of heart failure therapies.”
Adoption of the strategy “would be a dramatic change from what’s currently being done,” said Martin B. Leon, MD, NewYork-Presbyterian/Columbia University Irving Medical Center, New York, who moderated the press conference.
Only an estimated 5% of patients with HF in the United States receive full guideline-directed medical therapy, Dr. Leon said, “so the generalizability of this strategy, with careful follow-up that has safety involved in it, is absolutely crucial.”
But the potential impact of this high-intensity approach on resource use is unknown, raising questions about how widely and consistently it could be implemented, said Dr. Leon, who is not connected with STRONG-HF.
The trial called for in-hospital initiation of the three distinct drug classes that, at the time, were the core of guideline-directed HF therapy, with up-titration to 50% of recommended dosage by hospital discharge, and then to 100% within 2 weeks later.
The meds included a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system inhibitor (RASI). The latter could be an ACE inhibitor, angiotensin-receptor blocker (ARB), or angiotensin receptor-neprilysin inhibitor (ARNI).
How about a fourth drug?
Conspicuously absent from the list, for contemporary practice, was an SGLT2 inhibitor, a class that entered the HF guidelines well after STRONG-HF was designed. They would undoubtedly join the other three agents were the high-intensity strategy to enter practice, potentially changing its complexity and safety profile.
But Dr. Mebazaa and other experts don’t see that as a big challenge and would expect a smooth transition to a high-intensity approach that also includes the SGLT2 inhibitors.
STRONG-HF was necessary in part because many clinicians have been “reluctant” to take full advantage of three agents that had been the basis of guideline-directed therapy, he told this news organization.
That reluctance stemmed from concerns that beta-blockers might worsen the heart failure, ACE inhibitors could hurt the kidneys, or MRAs might cause hyperkalemia, Dr. Mebazaa said. The STRONG-HF high-intensity regimen, therefore, demanded multiple clinic visits for close follow-up.
But the SGLT2 inhibitors “are known to be rather safe drugs, at least much safer than the three others,” he said. So, it seems unlikely that their addition to a beta-blocker, RASI, and MRA in patients with HF would worsen the risk of adverse events.
John G.F. Cleland, MD, PhD, agrees. With addition of the fourth agent, “You may need to be a little bit more careful with renal function, just in that first couple of weeks,” he told this news organization. “But I think it would be easy to add an SGLT2 inhibitor into this regimen. And in general, there’s no titration with an SGLT2 inhibitor, so they’ll all be on full dose predischarge.”
Given the drugs’ diuretic-like action, moreover, some patients might be able to pull back on their loop diuretics, speculated Dr. Cleland, from the University of Glasgow’s School of Health and Wellbeing.
The prospect of a high-intensity strategy’s wide implementation in practice presents both “challenges and opportunities,” Amanda R. Vest, MBBS, MPH, Tufts University, Boston, told this news organization.
“There may be additional challenges in terms of ensuring we avoid hypotension or acute kidney injury in the up-titration phase,” said Dr. Vest, who is medical director of her center’s cardiac transplantation program but not connected with STRONG-HF.
“But it also gives us opportunities,” she added, “because there are some patients, especially in that vulnerable postdischarge phase, who are actually much more able to tolerate introduction of an SGLT2 inhibitor than, for example, an ACE inhibitor, ARB, or ARNI – or maybe a beta-blocker if they’ve been in a low cardiac-output state.” Effective dosing would depend on “the personalization and skill of the clinician in optimizing the medications in their correct sequence,” Dr. Vest said.
“It’s challenging to think that we would ever get to 100% up-titration,” she added, “and even in this excellent study, they didn’t get to 100%.” But as clinicians gain experience with the high-intensity strategy, especially as the SGLT2 inhibitors are included, “I think we can reasonably expect more progress to be made in these up-titration skills.”
No restrictions on LVEF
The researchers entered 1,078 patients hospitalized with acute HF in 14 countries across Africa, Europe, the Middle East, and South America, and randomly assigned them to the high-intensity management strategy or usual care.
About 60% of the patients were male and 77% were White. There were no entry restrictions based on left ventricular ejection fraction (LVEF), which exceeded 40% in almost a third of cases.
In the high-intensity care group’s 542 patients, the three agents were up-titrated to 50% of the maximum guideline-recommended dosage prior to hospital discharge, and to 100% within 2 weeks after discharge. Symptoms and laboratory biomarkers, including natriuretic peptides, were monitored closely at four planned clinical visits over the following 6 weeks.
The 536 patients assigned to usual care were discharged and managed according to local standards, with their meds handled by their own primary care doctors or cardiologists, the published report notes. They were reevaluated by STRONG-HF clinicians 90 days after discharge.
The number of clinic visits in the first 90 postdischarge days averaged 4.8 in the high-intensity care group and 1.0 for those receiving usual care. Full up-titration was far more likely in the high-intensity care group: 55% vs. 2% for RASI agents, 49% vs. 4% for beta-blockers, and 84% vs. 46% for MRAs.
They also fared significantly better on all measured parameters associated with decongestion, including weight, prevalence of peripheral edema, jugular venous pressure, NYHA functional class, and natriuretic peptide levels, the researchers said.
The primary endpoint of 180-day death from any cause or HF readmission was met by 15.2% of the high-intensity care group and 23.3% of usual-care patients, for an adjusted risk ratio (RR) of 0.66 (95% CI, 0.50-0.86; P = .0021).
Subgroup analyses saw no significant interactions by age, sex, race, geography, or baseline blood pressure, renal function, or LVEF. Patients with higher vs. lower baseline natriuretic peptide levels trend toward better responses to high-intensity care (P = .08)
The COVID effect
The group performed a sensitivity analysis that excluded deaths attributed to COVID-19 in STRONG-HF, which launched prior to the pandemic. The high-intensity strategy’s benefit for the primary endpoint grew, with an adjusted RR of 0.61 (95% CI, 0.46-0.82; P = .0005). There was no corresponding effect on death from any cause (P = .15).
Treatment-related adverse effects in the overall trial were seen in 41.1% of the high-intensity care group and in 29.5% of those assigned to usual care.
The higher rate in the high-intensity care arm “may be related to their higher number of [clinic] visits compared to usual care,” Dr. Mebazaa said. “However, serious adverse events and fatal adverse events were similar in both arms.”
Cardiac failure was the most common adverse event, developing in about 15% in both groups. It was followed by hypotension, hyperkalemia, and renal impairment, according to the published report.
Dr. Cleland cautioned that the risk of adverse events would potentially be higher should the high-intensity strategy become common clinical practice. The median age in STRONG-HF was 63, which is “10-15 years younger, on average, than the population with recently admitted heart failure that we see. There’s no doubt that older people have more multimorbidity.”
STRONG-HF was funded by Roche Diagnostics. Dr. Mebazaa discloses receiving grants from Roche Diagnostics, Abbott Laboratories, 4TEEN4, and Windtree Therapeutics; honoraria for lectures from Roche Diagnostics, Bayer, and Merck, Sharp & Dohme; and consulting for Corteria Pharmaceuticals, S-form Pharma, FIRE-1, Implicity, 4TEEN4, and Adrenomed; and to being a co-inventor on a patent involving combination therapy for patients having acute or persistent dyspnea.
Dr. Vest reports modest relationships with Boehringer Ingelheim, Corvia, and CareDx; and receiving research grants from the American Heart Association and the National Institutes of Health. Dr. Cleland discloses receiving honoraria from Idorsia; and research grants from Vifor Pharma, Medtronic, Bayer, and Bristol-Myers Squibb. Dr. Leon had no disclosures.
A version of this article first appeared on Medscape.com.
CHICAGO – Clinicians who prescribe heart failure meds are holding the best hand they’ve ever had, but with so much underuse and suboptimal dosing in actual practice, it seems many may not appreciate the value of their cards. But a major randomized trial that has captured the field’s attention may embolden them to go all in.
Results showed that a strategy of early, rapid up-titration of multiple guideline-directed meds in patients hospitalized with heart failure, compared with a usual-care approach, cut their 6-month risk for death or HF readmission by a steep 34% (P = .002).
The drugs had been started and partly up-titrated in the hospital with the goal of full up-titration within 2 weeks after discharge.
Patients well tolerated the high-intensity approach, researchers said. Their quality-of-life scores improved (P < .0001) compared with the usual-care group, and adverse events were considered few and manageable in the international trial with more than 1,000 patients.
Safety on the high-intensity strategy depended on close patient monitoring at frequently planned clinic visits along with guidance for the up-titrations from clinical signs and natriuretic peptide levels, observed Alexandre Mebazaa, MD, PhD, University of Paris and Public Hospitals of Paris.
Dr. Mebazaa is principal investigator on the trial, called STRONG-HF, which he presented at the American Heart Association scientific sessions, held in Chicago and virtually. He is also lead author on the study’s same-day publication in the Lancet.
The high-intensity strategy’s superiority emerged early in the trial, which was halted early on the data safety monitoring board’s recommendation, with about 90% of follow-ups completed. The board “felt it was unethical to keep patients in usual care,” Dr. Mebazaa said at a press conference.
A dramatic change
The next step, he said, will be to educate the heart failure community on the high-intensity care technique so it can swiftly enter clinical practice. Currently in acute heart failure, “very few patients are monitored after discharge and treated with full doses of heart failure therapies.”
Adoption of the strategy “would be a dramatic change from what’s currently being done,” said Martin B. Leon, MD, NewYork-Presbyterian/Columbia University Irving Medical Center, New York, who moderated the press conference.
Only an estimated 5% of patients with HF in the United States receive full guideline-directed medical therapy, Dr. Leon said, “so the generalizability of this strategy, with careful follow-up that has safety involved in it, is absolutely crucial.”
But the potential impact of this high-intensity approach on resource use is unknown, raising questions about how widely and consistently it could be implemented, said Dr. Leon, who is not connected with STRONG-HF.
The trial called for in-hospital initiation of the three distinct drug classes that, at the time, were the core of guideline-directed HF therapy, with up-titration to 50% of recommended dosage by hospital discharge, and then to 100% within 2 weeks later.
The meds included a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system inhibitor (RASI). The latter could be an ACE inhibitor, angiotensin-receptor blocker (ARB), or angiotensin receptor-neprilysin inhibitor (ARNI).
How about a fourth drug?
Conspicuously absent from the list, for contemporary practice, was an SGLT2 inhibitor, a class that entered the HF guidelines well after STRONG-HF was designed. They would undoubtedly join the other three agents were the high-intensity strategy to enter practice, potentially changing its complexity and safety profile.
But Dr. Mebazaa and other experts don’t see that as a big challenge and would expect a smooth transition to a high-intensity approach that also includes the SGLT2 inhibitors.
STRONG-HF was necessary in part because many clinicians have been “reluctant” to take full advantage of three agents that had been the basis of guideline-directed therapy, he told this news organization.
That reluctance stemmed from concerns that beta-blockers might worsen the heart failure, ACE inhibitors could hurt the kidneys, or MRAs might cause hyperkalemia, Dr. Mebazaa said. The STRONG-HF high-intensity regimen, therefore, demanded multiple clinic visits for close follow-up.
But the SGLT2 inhibitors “are known to be rather safe drugs, at least much safer than the three others,” he said. So, it seems unlikely that their addition to a beta-blocker, RASI, and MRA in patients with HF would worsen the risk of adverse events.
John G.F. Cleland, MD, PhD, agrees. With addition of the fourth agent, “You may need to be a little bit more careful with renal function, just in that first couple of weeks,” he told this news organization. “But I think it would be easy to add an SGLT2 inhibitor into this regimen. And in general, there’s no titration with an SGLT2 inhibitor, so they’ll all be on full dose predischarge.”
Given the drugs’ diuretic-like action, moreover, some patients might be able to pull back on their loop diuretics, speculated Dr. Cleland, from the University of Glasgow’s School of Health and Wellbeing.
The prospect of a high-intensity strategy’s wide implementation in practice presents both “challenges and opportunities,” Amanda R. Vest, MBBS, MPH, Tufts University, Boston, told this news organization.
“There may be additional challenges in terms of ensuring we avoid hypotension or acute kidney injury in the up-titration phase,” said Dr. Vest, who is medical director of her center’s cardiac transplantation program but not connected with STRONG-HF.
“But it also gives us opportunities,” she added, “because there are some patients, especially in that vulnerable postdischarge phase, who are actually much more able to tolerate introduction of an SGLT2 inhibitor than, for example, an ACE inhibitor, ARB, or ARNI – or maybe a beta-blocker if they’ve been in a low cardiac-output state.” Effective dosing would depend on “the personalization and skill of the clinician in optimizing the medications in their correct sequence,” Dr. Vest said.
“It’s challenging to think that we would ever get to 100% up-titration,” she added, “and even in this excellent study, they didn’t get to 100%.” But as clinicians gain experience with the high-intensity strategy, especially as the SGLT2 inhibitors are included, “I think we can reasonably expect more progress to be made in these up-titration skills.”
No restrictions on LVEF
The researchers entered 1,078 patients hospitalized with acute HF in 14 countries across Africa, Europe, the Middle East, and South America, and randomly assigned them to the high-intensity management strategy or usual care.
About 60% of the patients were male and 77% were White. There were no entry restrictions based on left ventricular ejection fraction (LVEF), which exceeded 40% in almost a third of cases.
In the high-intensity care group’s 542 patients, the three agents were up-titrated to 50% of the maximum guideline-recommended dosage prior to hospital discharge, and to 100% within 2 weeks after discharge. Symptoms and laboratory biomarkers, including natriuretic peptides, were monitored closely at four planned clinical visits over the following 6 weeks.
The 536 patients assigned to usual care were discharged and managed according to local standards, with their meds handled by their own primary care doctors or cardiologists, the published report notes. They were reevaluated by STRONG-HF clinicians 90 days after discharge.
The number of clinic visits in the first 90 postdischarge days averaged 4.8 in the high-intensity care group and 1.0 for those receiving usual care. Full up-titration was far more likely in the high-intensity care group: 55% vs. 2% for RASI agents, 49% vs. 4% for beta-blockers, and 84% vs. 46% for MRAs.
They also fared significantly better on all measured parameters associated with decongestion, including weight, prevalence of peripheral edema, jugular venous pressure, NYHA functional class, and natriuretic peptide levels, the researchers said.
The primary endpoint of 180-day death from any cause or HF readmission was met by 15.2% of the high-intensity care group and 23.3% of usual-care patients, for an adjusted risk ratio (RR) of 0.66 (95% CI, 0.50-0.86; P = .0021).
Subgroup analyses saw no significant interactions by age, sex, race, geography, or baseline blood pressure, renal function, or LVEF. Patients with higher vs. lower baseline natriuretic peptide levels trend toward better responses to high-intensity care (P = .08)
The COVID effect
The group performed a sensitivity analysis that excluded deaths attributed to COVID-19 in STRONG-HF, which launched prior to the pandemic. The high-intensity strategy’s benefit for the primary endpoint grew, with an adjusted RR of 0.61 (95% CI, 0.46-0.82; P = .0005). There was no corresponding effect on death from any cause (P = .15).
Treatment-related adverse effects in the overall trial were seen in 41.1% of the high-intensity care group and in 29.5% of those assigned to usual care.
The higher rate in the high-intensity care arm “may be related to their higher number of [clinic] visits compared to usual care,” Dr. Mebazaa said. “However, serious adverse events and fatal adverse events were similar in both arms.”
Cardiac failure was the most common adverse event, developing in about 15% in both groups. It was followed by hypotension, hyperkalemia, and renal impairment, according to the published report.
Dr. Cleland cautioned that the risk of adverse events would potentially be higher should the high-intensity strategy become common clinical practice. The median age in STRONG-HF was 63, which is “10-15 years younger, on average, than the population with recently admitted heart failure that we see. There’s no doubt that older people have more multimorbidity.”
STRONG-HF was funded by Roche Diagnostics. Dr. Mebazaa discloses receiving grants from Roche Diagnostics, Abbott Laboratories, 4TEEN4, and Windtree Therapeutics; honoraria for lectures from Roche Diagnostics, Bayer, and Merck, Sharp & Dohme; and consulting for Corteria Pharmaceuticals, S-form Pharma, FIRE-1, Implicity, 4TEEN4, and Adrenomed; and to being a co-inventor on a patent involving combination therapy for patients having acute or persistent dyspnea.
Dr. Vest reports modest relationships with Boehringer Ingelheim, Corvia, and CareDx; and receiving research grants from the American Heart Association and the National Institutes of Health. Dr. Cleland discloses receiving honoraria from Idorsia; and research grants from Vifor Pharma, Medtronic, Bayer, and Bristol-Myers Squibb. Dr. Leon had no disclosures.
A version of this article first appeared on Medscape.com.
CHICAGO – Clinicians who prescribe heart failure meds are holding the best hand they’ve ever had, but with so much underuse and suboptimal dosing in actual practice, it seems many may not appreciate the value of their cards. But a major randomized trial that has captured the field’s attention may embolden them to go all in.
Results showed that a strategy of early, rapid up-titration of multiple guideline-directed meds in patients hospitalized with heart failure, compared with a usual-care approach, cut their 6-month risk for death or HF readmission by a steep 34% (P = .002).
The drugs had been started and partly up-titrated in the hospital with the goal of full up-titration within 2 weeks after discharge.
Patients well tolerated the high-intensity approach, researchers said. Their quality-of-life scores improved (P < .0001) compared with the usual-care group, and adverse events were considered few and manageable in the international trial with more than 1,000 patients.
Safety on the high-intensity strategy depended on close patient monitoring at frequently planned clinic visits along with guidance for the up-titrations from clinical signs and natriuretic peptide levels, observed Alexandre Mebazaa, MD, PhD, University of Paris and Public Hospitals of Paris.
Dr. Mebazaa is principal investigator on the trial, called STRONG-HF, which he presented at the American Heart Association scientific sessions, held in Chicago and virtually. He is also lead author on the study’s same-day publication in the Lancet.
The high-intensity strategy’s superiority emerged early in the trial, which was halted early on the data safety monitoring board’s recommendation, with about 90% of follow-ups completed. The board “felt it was unethical to keep patients in usual care,” Dr. Mebazaa said at a press conference.
A dramatic change
The next step, he said, will be to educate the heart failure community on the high-intensity care technique so it can swiftly enter clinical practice. Currently in acute heart failure, “very few patients are monitored after discharge and treated with full doses of heart failure therapies.”
Adoption of the strategy “would be a dramatic change from what’s currently being done,” said Martin B. Leon, MD, NewYork-Presbyterian/Columbia University Irving Medical Center, New York, who moderated the press conference.
Only an estimated 5% of patients with HF in the United States receive full guideline-directed medical therapy, Dr. Leon said, “so the generalizability of this strategy, with careful follow-up that has safety involved in it, is absolutely crucial.”
But the potential impact of this high-intensity approach on resource use is unknown, raising questions about how widely and consistently it could be implemented, said Dr. Leon, who is not connected with STRONG-HF.
The trial called for in-hospital initiation of the three distinct drug classes that, at the time, were the core of guideline-directed HF therapy, with up-titration to 50% of recommended dosage by hospital discharge, and then to 100% within 2 weeks later.
The meds included a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system inhibitor (RASI). The latter could be an ACE inhibitor, angiotensin-receptor blocker (ARB), or angiotensin receptor-neprilysin inhibitor (ARNI).
How about a fourth drug?
Conspicuously absent from the list, for contemporary practice, was an SGLT2 inhibitor, a class that entered the HF guidelines well after STRONG-HF was designed. They would undoubtedly join the other three agents were the high-intensity strategy to enter practice, potentially changing its complexity and safety profile.
But Dr. Mebazaa and other experts don’t see that as a big challenge and would expect a smooth transition to a high-intensity approach that also includes the SGLT2 inhibitors.
STRONG-HF was necessary in part because many clinicians have been “reluctant” to take full advantage of three agents that had been the basis of guideline-directed therapy, he told this news organization.
That reluctance stemmed from concerns that beta-blockers might worsen the heart failure, ACE inhibitors could hurt the kidneys, or MRAs might cause hyperkalemia, Dr. Mebazaa said. The STRONG-HF high-intensity regimen, therefore, demanded multiple clinic visits for close follow-up.
But the SGLT2 inhibitors “are known to be rather safe drugs, at least much safer than the three others,” he said. So, it seems unlikely that their addition to a beta-blocker, RASI, and MRA in patients with HF would worsen the risk of adverse events.
John G.F. Cleland, MD, PhD, agrees. With addition of the fourth agent, “You may need to be a little bit more careful with renal function, just in that first couple of weeks,” he told this news organization. “But I think it would be easy to add an SGLT2 inhibitor into this regimen. And in general, there’s no titration with an SGLT2 inhibitor, so they’ll all be on full dose predischarge.”
Given the drugs’ diuretic-like action, moreover, some patients might be able to pull back on their loop diuretics, speculated Dr. Cleland, from the University of Glasgow’s School of Health and Wellbeing.
The prospect of a high-intensity strategy’s wide implementation in practice presents both “challenges and opportunities,” Amanda R. Vest, MBBS, MPH, Tufts University, Boston, told this news organization.
“There may be additional challenges in terms of ensuring we avoid hypotension or acute kidney injury in the up-titration phase,” said Dr. Vest, who is medical director of her center’s cardiac transplantation program but not connected with STRONG-HF.
“But it also gives us opportunities,” she added, “because there are some patients, especially in that vulnerable postdischarge phase, who are actually much more able to tolerate introduction of an SGLT2 inhibitor than, for example, an ACE inhibitor, ARB, or ARNI – or maybe a beta-blocker if they’ve been in a low cardiac-output state.” Effective dosing would depend on “the personalization and skill of the clinician in optimizing the medications in their correct sequence,” Dr. Vest said.
“It’s challenging to think that we would ever get to 100% up-titration,” she added, “and even in this excellent study, they didn’t get to 100%.” But as clinicians gain experience with the high-intensity strategy, especially as the SGLT2 inhibitors are included, “I think we can reasonably expect more progress to be made in these up-titration skills.”
No restrictions on LVEF
The researchers entered 1,078 patients hospitalized with acute HF in 14 countries across Africa, Europe, the Middle East, and South America, and randomly assigned them to the high-intensity management strategy or usual care.
About 60% of the patients were male and 77% were White. There were no entry restrictions based on left ventricular ejection fraction (LVEF), which exceeded 40% in almost a third of cases.
In the high-intensity care group’s 542 patients, the three agents were up-titrated to 50% of the maximum guideline-recommended dosage prior to hospital discharge, and to 100% within 2 weeks after discharge. Symptoms and laboratory biomarkers, including natriuretic peptides, were monitored closely at four planned clinical visits over the following 6 weeks.
The 536 patients assigned to usual care were discharged and managed according to local standards, with their meds handled by their own primary care doctors or cardiologists, the published report notes. They were reevaluated by STRONG-HF clinicians 90 days after discharge.
The number of clinic visits in the first 90 postdischarge days averaged 4.8 in the high-intensity care group and 1.0 for those receiving usual care. Full up-titration was far more likely in the high-intensity care group: 55% vs. 2% for RASI agents, 49% vs. 4% for beta-blockers, and 84% vs. 46% for MRAs.
They also fared significantly better on all measured parameters associated with decongestion, including weight, prevalence of peripheral edema, jugular venous pressure, NYHA functional class, and natriuretic peptide levels, the researchers said.
The primary endpoint of 180-day death from any cause or HF readmission was met by 15.2% of the high-intensity care group and 23.3% of usual-care patients, for an adjusted risk ratio (RR) of 0.66 (95% CI, 0.50-0.86; P = .0021).
Subgroup analyses saw no significant interactions by age, sex, race, geography, or baseline blood pressure, renal function, or LVEF. Patients with higher vs. lower baseline natriuretic peptide levels trend toward better responses to high-intensity care (P = .08)
The COVID effect
The group performed a sensitivity analysis that excluded deaths attributed to COVID-19 in STRONG-HF, which launched prior to the pandemic. The high-intensity strategy’s benefit for the primary endpoint grew, with an adjusted RR of 0.61 (95% CI, 0.46-0.82; P = .0005). There was no corresponding effect on death from any cause (P = .15).
Treatment-related adverse effects in the overall trial were seen in 41.1% of the high-intensity care group and in 29.5% of those assigned to usual care.
The higher rate in the high-intensity care arm “may be related to their higher number of [clinic] visits compared to usual care,” Dr. Mebazaa said. “However, serious adverse events and fatal adverse events were similar in both arms.”
Cardiac failure was the most common adverse event, developing in about 15% in both groups. It was followed by hypotension, hyperkalemia, and renal impairment, according to the published report.
Dr. Cleland cautioned that the risk of adverse events would potentially be higher should the high-intensity strategy become common clinical practice. The median age in STRONG-HF was 63, which is “10-15 years younger, on average, than the population with recently admitted heart failure that we see. There’s no doubt that older people have more multimorbidity.”
STRONG-HF was funded by Roche Diagnostics. Dr. Mebazaa discloses receiving grants from Roche Diagnostics, Abbott Laboratories, 4TEEN4, and Windtree Therapeutics; honoraria for lectures from Roche Diagnostics, Bayer, and Merck, Sharp & Dohme; and consulting for Corteria Pharmaceuticals, S-form Pharma, FIRE-1, Implicity, 4TEEN4, and Adrenomed; and to being a co-inventor on a patent involving combination therapy for patients having acute or persistent dyspnea.
Dr. Vest reports modest relationships with Boehringer Ingelheim, Corvia, and CareDx; and receiving research grants from the American Heart Association and the National Institutes of Health. Dr. Cleland discloses receiving honoraria from Idorsia; and research grants from Vifor Pharma, Medtronic, Bayer, and Bristol-Myers Squibb. Dr. Leon had no disclosures.
A version of this article first appeared on Medscape.com.
AT AHA 2022
Flu vaccination associated with reduced stroke risk
The risk of stroke was about 23% lower in the 6 months following a flu shot, regardless of the patient’s age, sex, or underlying health conditions.
“There is an established link between upper respiratory infection and both heart attack and stroke. This has been very salient in the past few years throughout the COVID-19 pandemic,” study author Jessalyn Holodinsky, PhD, a stroke epidemiologist and postdoctoral fellow in clinical neurosciences at the University of Calgary (Alta.) told this news organization.
“It is also known that the flu shot can reduce risk of heart attack and hospitalization for those with heart disease,” she said. “Given both of these [observations], we thought it prudent to study whether there is a link between vaccination for influenza and stroke.”
The study was published in the Lancet Public Health.
Large effect size
The investigators analyzed administrative data from 2009 through 2018 from the Alberta Health Care Insurance Plan, which covers all residents of Alberta. The province provides free seasonal influenza vaccines to residents under the insurance plan.
The research team looked for stroke events such as acute ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, and transient ischemic attack. They then analyzed the risk of stroke events among those with or without a flu shot in the previous 6 months. They accounted for multiple factors, including age, sex, income, location, and factors related to stroke risk, such as anticoagulant use, atrial fibrillation, chronic obstructive pulmonary disease, diabetes, and hypertension.
Among the 4.1 million adults included in the researchers’ analysis, about 1.8 million (43%) received at least one vaccination during the study period. Nearly 97,000 people received a flu vaccine in each year they were in the study, including 29,288 who received a shot in all 10 flu seasons included in the study.
About 38,000 stroke events were recorded, including about 34,000 (90%) first stroke events. Among the 10% of strokes that were recurrent events, the maximum number of stroke events in one person was nine.
Overall, patients who received at least one influenza vaccine were more likely to be older, be women, and have higher rates of comorbidities. The vaccinated group had a slightly higher proportion of people who lived in urban areas, but the income levels were similar between the vaccinated and unvaccinated groups.
The crude incidence of stroke was higher among people who had ever received an influenza vaccination, at 1.25%, compared with 0.52% among those who hadn’t been vaccinated. However, after adjusting for age, sex, underlying conditions, and socioeconomic status, recent flu vaccination (that is, in the previous 6 months) was associated with a 23% reduced risk of stroke.
The significant reduction in risk applied to all stroke types, particularly acute ischemic stroke and intracerebral hemorrhage. In addition, influenza vaccination was associated with a reduced risk across all ages and risk profiles, except patients without hypertension.
“What we were most surprised by was the sheer magnitude of the effect and that it existed across different adult age groups, for both sexes, and for those with and without risk factors for stroke,” said Dr. Holodinsky.
Vaccination was associated with a larger reduction in stroke risk in men than in women, perhaps because unvaccinated men had a significantly higher baseline risk for stroke than unvaccinated women, the study authors write.
Promoting cardiovascular health
In addition, vaccination was associated with a greater relative reduction in stroke risk in younger age groups, lower income groups, and those with diabetes, chronic obstructive pulmonary disease, and anticoagulant use.
Among 2.4 million people observed for the entire study period, vaccination protection increased with the number of vaccines received. People who were vaccinated serially each year had a significantly lower risk of stroke than those who received one shot.
Dr. Holodinsky and colleagues are conducting additional research into influenza vaccination, including stroke risk in children. They’re also investigating whether the reduced risk applies to other vaccinations for respiratory illnesses, such as COVID-19 and pneumonia.
“We hope that this added effect of vaccination encourages more adults to receive the flu shot,” she said. “One day, vaccinations might be considered a key pillar of cardiovascular health, along with diet, exercise, control of hypertension and high cholesterol, and smoking cessation.”
Future research should also investigate the reasons why adults – particularly people at high risk with underlying conditions – don’t receive recommended influenza vaccines, the study authors wrote.
‘Call to action’
Bahar Behrouzi, an MD-PhD candidate focused on clinical epidemiology at the Institute of Health Policy, Management, and Evaluation, University of Toronto, said: “There are a variety of observational studies around the world that show that flu vaccine uptake is low among the general population and high-risk persons. In studying these questions, our hope is that we can continue to build confidence in viral respiratory vaccines like the influenza vaccine by continuing to generate rigorous evidence with the latest data.”
Ms. Behrouzi, who wasn’t involved with this study, has researched influenza vaccination and cardiovascular risk. She and her colleagues have found that flu vaccines were associated with a 34% lower risk of major adverse cardiovascular events, including a 45% reduced risk among patients with recent acute coronary syndrome.
“The broader public health message is for people to advocate for themselves and get the seasonal flu vaccine, especially if they are part of an at-risk group,” she said. “In our studies, we have positioned this message as a call to action not only for the public, but also for health care professionals – particularly specialists such as cardiologists or neurologists – to encourage or remind them to engage in conversation about the broad benefits of vaccination beyond just preventing or reducing the severity of flu infection.”
The study was conducted without outside funding. Dr. Holodinsky and Ms. Behrouzi have reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
The risk of stroke was about 23% lower in the 6 months following a flu shot, regardless of the patient’s age, sex, or underlying health conditions.
“There is an established link between upper respiratory infection and both heart attack and stroke. This has been very salient in the past few years throughout the COVID-19 pandemic,” study author Jessalyn Holodinsky, PhD, a stroke epidemiologist and postdoctoral fellow in clinical neurosciences at the University of Calgary (Alta.) told this news organization.
“It is also known that the flu shot can reduce risk of heart attack and hospitalization for those with heart disease,” she said. “Given both of these [observations], we thought it prudent to study whether there is a link between vaccination for influenza and stroke.”
The study was published in the Lancet Public Health.
Large effect size
The investigators analyzed administrative data from 2009 through 2018 from the Alberta Health Care Insurance Plan, which covers all residents of Alberta. The province provides free seasonal influenza vaccines to residents under the insurance plan.
The research team looked for stroke events such as acute ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, and transient ischemic attack. They then analyzed the risk of stroke events among those with or without a flu shot in the previous 6 months. They accounted for multiple factors, including age, sex, income, location, and factors related to stroke risk, such as anticoagulant use, atrial fibrillation, chronic obstructive pulmonary disease, diabetes, and hypertension.
Among the 4.1 million adults included in the researchers’ analysis, about 1.8 million (43%) received at least one vaccination during the study period. Nearly 97,000 people received a flu vaccine in each year they were in the study, including 29,288 who received a shot in all 10 flu seasons included in the study.
About 38,000 stroke events were recorded, including about 34,000 (90%) first stroke events. Among the 10% of strokes that were recurrent events, the maximum number of stroke events in one person was nine.
Overall, patients who received at least one influenza vaccine were more likely to be older, be women, and have higher rates of comorbidities. The vaccinated group had a slightly higher proportion of people who lived in urban areas, but the income levels were similar between the vaccinated and unvaccinated groups.
The crude incidence of stroke was higher among people who had ever received an influenza vaccination, at 1.25%, compared with 0.52% among those who hadn’t been vaccinated. However, after adjusting for age, sex, underlying conditions, and socioeconomic status, recent flu vaccination (that is, in the previous 6 months) was associated with a 23% reduced risk of stroke.
The significant reduction in risk applied to all stroke types, particularly acute ischemic stroke and intracerebral hemorrhage. In addition, influenza vaccination was associated with a reduced risk across all ages and risk profiles, except patients without hypertension.
“What we were most surprised by was the sheer magnitude of the effect and that it existed across different adult age groups, for both sexes, and for those with and without risk factors for stroke,” said Dr. Holodinsky.
Vaccination was associated with a larger reduction in stroke risk in men than in women, perhaps because unvaccinated men had a significantly higher baseline risk for stroke than unvaccinated women, the study authors write.
Promoting cardiovascular health
In addition, vaccination was associated with a greater relative reduction in stroke risk in younger age groups, lower income groups, and those with diabetes, chronic obstructive pulmonary disease, and anticoagulant use.
Among 2.4 million people observed for the entire study period, vaccination protection increased with the number of vaccines received. People who were vaccinated serially each year had a significantly lower risk of stroke than those who received one shot.
Dr. Holodinsky and colleagues are conducting additional research into influenza vaccination, including stroke risk in children. They’re also investigating whether the reduced risk applies to other vaccinations for respiratory illnesses, such as COVID-19 and pneumonia.
“We hope that this added effect of vaccination encourages more adults to receive the flu shot,” she said. “One day, vaccinations might be considered a key pillar of cardiovascular health, along with diet, exercise, control of hypertension and high cholesterol, and smoking cessation.”
Future research should also investigate the reasons why adults – particularly people at high risk with underlying conditions – don’t receive recommended influenza vaccines, the study authors wrote.
‘Call to action’
Bahar Behrouzi, an MD-PhD candidate focused on clinical epidemiology at the Institute of Health Policy, Management, and Evaluation, University of Toronto, said: “There are a variety of observational studies around the world that show that flu vaccine uptake is low among the general population and high-risk persons. In studying these questions, our hope is that we can continue to build confidence in viral respiratory vaccines like the influenza vaccine by continuing to generate rigorous evidence with the latest data.”
Ms. Behrouzi, who wasn’t involved with this study, has researched influenza vaccination and cardiovascular risk. She and her colleagues have found that flu vaccines were associated with a 34% lower risk of major adverse cardiovascular events, including a 45% reduced risk among patients with recent acute coronary syndrome.
“The broader public health message is for people to advocate for themselves and get the seasonal flu vaccine, especially if they are part of an at-risk group,” she said. “In our studies, we have positioned this message as a call to action not only for the public, but also for health care professionals – particularly specialists such as cardiologists or neurologists – to encourage or remind them to engage in conversation about the broad benefits of vaccination beyond just preventing or reducing the severity of flu infection.”
The study was conducted without outside funding. Dr. Holodinsky and Ms. Behrouzi have reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
The risk of stroke was about 23% lower in the 6 months following a flu shot, regardless of the patient’s age, sex, or underlying health conditions.
“There is an established link between upper respiratory infection and both heart attack and stroke. This has been very salient in the past few years throughout the COVID-19 pandemic,” study author Jessalyn Holodinsky, PhD, a stroke epidemiologist and postdoctoral fellow in clinical neurosciences at the University of Calgary (Alta.) told this news organization.
“It is also known that the flu shot can reduce risk of heart attack and hospitalization for those with heart disease,” she said. “Given both of these [observations], we thought it prudent to study whether there is a link between vaccination for influenza and stroke.”
The study was published in the Lancet Public Health.
Large effect size
The investigators analyzed administrative data from 2009 through 2018 from the Alberta Health Care Insurance Plan, which covers all residents of Alberta. The province provides free seasonal influenza vaccines to residents under the insurance plan.
The research team looked for stroke events such as acute ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, and transient ischemic attack. They then analyzed the risk of stroke events among those with or without a flu shot in the previous 6 months. They accounted for multiple factors, including age, sex, income, location, and factors related to stroke risk, such as anticoagulant use, atrial fibrillation, chronic obstructive pulmonary disease, diabetes, and hypertension.
Among the 4.1 million adults included in the researchers’ analysis, about 1.8 million (43%) received at least one vaccination during the study period. Nearly 97,000 people received a flu vaccine in each year they were in the study, including 29,288 who received a shot in all 10 flu seasons included in the study.
About 38,000 stroke events were recorded, including about 34,000 (90%) first stroke events. Among the 10% of strokes that were recurrent events, the maximum number of stroke events in one person was nine.
Overall, patients who received at least one influenza vaccine were more likely to be older, be women, and have higher rates of comorbidities. The vaccinated group had a slightly higher proportion of people who lived in urban areas, but the income levels were similar between the vaccinated and unvaccinated groups.
The crude incidence of stroke was higher among people who had ever received an influenza vaccination, at 1.25%, compared with 0.52% among those who hadn’t been vaccinated. However, after adjusting for age, sex, underlying conditions, and socioeconomic status, recent flu vaccination (that is, in the previous 6 months) was associated with a 23% reduced risk of stroke.
The significant reduction in risk applied to all stroke types, particularly acute ischemic stroke and intracerebral hemorrhage. In addition, influenza vaccination was associated with a reduced risk across all ages and risk profiles, except patients without hypertension.
“What we were most surprised by was the sheer magnitude of the effect and that it existed across different adult age groups, for both sexes, and for those with and without risk factors for stroke,” said Dr. Holodinsky.
Vaccination was associated with a larger reduction in stroke risk in men than in women, perhaps because unvaccinated men had a significantly higher baseline risk for stroke than unvaccinated women, the study authors write.
Promoting cardiovascular health
In addition, vaccination was associated with a greater relative reduction in stroke risk in younger age groups, lower income groups, and those with diabetes, chronic obstructive pulmonary disease, and anticoagulant use.
Among 2.4 million people observed for the entire study period, vaccination protection increased with the number of vaccines received. People who were vaccinated serially each year had a significantly lower risk of stroke than those who received one shot.
Dr. Holodinsky and colleagues are conducting additional research into influenza vaccination, including stroke risk in children. They’re also investigating whether the reduced risk applies to other vaccinations for respiratory illnesses, such as COVID-19 and pneumonia.
“We hope that this added effect of vaccination encourages more adults to receive the flu shot,” she said. “One day, vaccinations might be considered a key pillar of cardiovascular health, along with diet, exercise, control of hypertension and high cholesterol, and smoking cessation.”
Future research should also investigate the reasons why adults – particularly people at high risk with underlying conditions – don’t receive recommended influenza vaccines, the study authors wrote.
‘Call to action’
Bahar Behrouzi, an MD-PhD candidate focused on clinical epidemiology at the Institute of Health Policy, Management, and Evaluation, University of Toronto, said: “There are a variety of observational studies around the world that show that flu vaccine uptake is low among the general population and high-risk persons. In studying these questions, our hope is that we can continue to build confidence in viral respiratory vaccines like the influenza vaccine by continuing to generate rigorous evidence with the latest data.”
Ms. Behrouzi, who wasn’t involved with this study, has researched influenza vaccination and cardiovascular risk. She and her colleagues have found that flu vaccines were associated with a 34% lower risk of major adverse cardiovascular events, including a 45% reduced risk among patients with recent acute coronary syndrome.
“The broader public health message is for people to advocate for themselves and get the seasonal flu vaccine, especially if they are part of an at-risk group,” she said. “In our studies, we have positioned this message as a call to action not only for the public, but also for health care professionals – particularly specialists such as cardiologists or neurologists – to encourage or remind them to engage in conversation about the broad benefits of vaccination beyond just preventing or reducing the severity of flu infection.”
The study was conducted without outside funding. Dr. Holodinsky and Ms. Behrouzi have reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM LANCET PUBLIC HEALTH
Tirzepatide cuts BP during obesity treatment
CHICAGO – compared with placebo, while causing modest increases in heart rate, in a prespecified substudy of the SURMOUNT-1 trial.
“The large effects on ambulatory 24-hour blood pressure raise the possibility that there may be important long-term benefits of [tirzepatide] on the complications of obesity,” said James A. de Lemos, MD, during a presentation at the American Heart Association scientific sessions.

“The findings are concordant with the [previously reported] office-based measurements, and the blood pressure reductions provide further evidence for the potential benefits of tirzepatide on cardiovascular health and outcomes,” said Dr. de Lemos, a cardiologist and professor at the University of Texas Southwestern Medical Center, Dallas.
The substudy included 600 of the 2,539 people enrolled in SURMOUNT-1, the first of two pivotal trials for tirzepatide (Mounjaro) in people without diabetes but with obesity or overweight (body mass index of 27-29 kg/m2) plus at least one weight-related complication. The primary endpoints of SURMOUNT-1 were the percent change in weight from baseline to 72 weeks on treatment with either of three different weekly injected doses of tirzepatide, compared with control subjects who received placebo, and the percentage of enrolled subjects achieving at least 5% loss in baseline weight, compared with the controls.
Tirzepatide treatment led to significant increases in both results, compared with controls, with the highest dose tested, 15 mg/week, resulting in an average 20.9% drop in weight from baseline after 72 weeks of treatment, and 91% of enrolled subjects on that dose achieving the 5% weight-loss threshold during the same time frame, in results published in 2022 in the New England Journal of Medicine.
24-hour ambulatory pressures from 494 people
The substudy enrolled 600 of the SURMOUNT-1 participants and involved 24-hour ambulatory BP and heart rate measurements at entry and after 36 weeks on treatment. Full results were available for 494 of these people. The substudy included only study participants who entered with a BP of less than 140/90 mm Hg. Enrollment in SURMOUNT-1 overall excluded people with a BP of 160/100 mm Hg or higher. The average BP among all enrolled participants was about 123/80 mm Hg, while heart rates averaged about 73 beats per minute.
Systolic BP measured with the ambulatory monitor fell from baseline by an average of 5.6, 8.8, and 6.2 mm Hg in the people who received tirzepatide in weekly doses of 5, 10, or 15 mg, respectively, and rose by an average 1.8 mm Hg among the controls, Dr. de Lemos reported. Diastolic BP dropped among the tirzepatide recipients by an average of 1.5, 2.4, and 0.0 mm Hg in the three ascending tirzepatide treatment arms, and rose by an average 0.5 mm Hg among the controls. All of the differences between the intervention groups and the controls were significant except for the change in diastolic BP among participants who received 15 mg of tirzepatide weekly.
The results showed that 36 weeks on tirzepatide treatment was associated with “arguably clinically meaningful” reductions in systolic and diastolic BPs, Dr. de Lemos said. “There is a lot of optimism that this will translate into clinical benefits.” He also noted that, “within the limits of cross-study comparisons, the blood pressure changes look favorable, compared with the single-incretin mechanism GLP-1 [glucagonlike peptide–1] receptor agonists.”
Heart rate fell by an average 1.8 bpm in the controls, and rose by an average 0.3, 0.5, and 3.6 bpm among the three groups receiving ascending weekly tirzepatide doses, effects that were “consistent with what’s been seen with the GLP-1 receptor agonists,” noted Dr. de Lemos.
Tirzepatide is known as a “twincretin” because it shares this GLP-1 receptor agonism and also has a second incretin agonist activity, to the receptor for the glucose-dependent insulinotropic polypeptide.
Lowering of blood pressure plateaus
Changes in BP over time during the 72 weeks on treatment, data first presented in the original report, showed that average systolic pressure in the people who received tirzepatide fell sharply during the first 24 weeks on treatment, and then leveled out with little further change over time. Furthermore, all three tirzepatide doses produced roughly similar systolic BP reductions. Changes in diastolic pressure over time showed a mostly similar pattern of reduction, although a modest ongoing decrease in average diastolic pressure continued beyond 24 weeks.

This pattern of a plateau in BP reduction has been seen before in studies using other treatments to produce weight loss, including bariatric surgery, said Naveed Sattar, MBChB, PhD, professor of metabolic medicine at the University of Glasgow, who was not involved in SURMOUNT-1. He attributed the plateau in BP reduction among tirzepatide-treated people to them hitting a wall in their BP nadir based on homeostatic limits. Dr. Sattar noted that most enrolled participants had normal BPs at entry based on the reported study averages.
“It’s hard to go lower, but the blood pressure reduction may be larger in people who start at higher pressure levels,” Dr. Sattar said in an interview.

Another inferred cap on BP reductions in the trial hypothesizes that the individual clinicians who managed the enrolled patients may have cut back on other BP-lowering agents as the pressures of the tirzepatide recipients fell to relatively low levels, suggested Darren McGuire, MD, a cardiologist and professor at UT Southwestern Medical Center, who also was not involved in the SURMOUNT-1 study.
Incretin agonists as antihypertensive drugs
The substantial BP-lowering seen with tirzepatide, as well as with other incretin agonist agents, suggests a new way to think about BP control in people with overweight or obesity, Dr. Sattar said.
“Until now, we haven’t had tools where people lose so much weight. Now that we have these tools [incretin agonists as well as bariatric surgery], we see substantial blood pressure reductions. It makes you think we should use weight-loss agents to lower blood pressure rather than a beta-blocker or angiotensin-converting enzyme inhibitor; then we’d also produce all the other benefits from weight loss,” Dr. Sattar suggested.
Dr. de Lemos said he sees signals that the BP reductions caused by tirzepatide and the GLP-1 receptor agonists may go beyond just weight-loss effects.
“There appears to be a larger blood pressure reduction than anticipated based on the change in weight,” he said during his presentation. “GLP-1 is active in most vascular tissues, so these [receptor agonist] agents likely have vascular or cardiac effects, or even effects on other tissues that may affect blood pressure.”
Heart rate increases were usually modest
The experiences with GLP-1 receptor agonists also suggest that the heart rate increases seen with tirzepatide treatment in SURMOUNT-1 will not have long-term effects. “The [Food and Drug Administration] mandated this heart rate substudy to make sure that the increase in heart rate was not larger than what would be anticipated” with a GLP-1 receptor agonist, Dr. de Lemos explained.
SURMOUNT-1 had a treatment-stopping rule to prevent a person’s heart rate from rising beyond 10 bpm from baseline. “Trivial numbers” of patients experienced a heart rate increase of this magnitude, he said. If used in routine practice, Dr. de Lemos said that he would closely investigate a patient with a heart rate increase greater than 10 mm Hg. The average increase seen with the highest dose, about 4 bpm above baseline, would generally not be concerning.
Tirzepatide received U.S. marketing approval from the FDA in May 2022 for treating people with type 2 diabetes. In October 2022, the FDA gave tirzepatide “Fast Track” designation for the pending application for approval of an indication to treat people with overweight or obesity who match the entry criteria for SURMOUNT-1 and for the second pivotal trial for this indication, SURMOUNT-2. According to a statement from Eli Lilly, the company that is developing and markets tirzepatide (Mounjaro), the FDA’s decision on the obesity indication will remain pending until the SURMOUNT-2 results are available, which the company expects will occur in 2023.
SURMOUNT-1 and SURMOUNT-2 were sponsored by Lilly, the company that markets tirzepatide. Dr. de Lemos has been a consultant to Lilly as well as to Amgen, AstraZeneca, Janssen, Novo Nordisk, Ortho, Quidel Cardiovascular, and Regeneron. Dr. Sattar has financial ties to Lilly, Afimmune, Amgen, AstraZeneca, Boehringer Ingelheim, Hammi, Merck Sharpe & Dohme, Novartis, Novo Nordisk, Pfizer, Roche, and Sanofi-Aventis. Dr. McGuire has ties to Lilly as well as to Altimmune, Applied Therapeutics, Bayer, Boehringer Ingelheim, CSL Behring, Lexicon, Merck, Metavant, Novo Nordisk, and Sanofi.
CHICAGO – compared with placebo, while causing modest increases in heart rate, in a prespecified substudy of the SURMOUNT-1 trial.
“The large effects on ambulatory 24-hour blood pressure raise the possibility that there may be important long-term benefits of [tirzepatide] on the complications of obesity,” said James A. de Lemos, MD, during a presentation at the American Heart Association scientific sessions.
“The findings are concordant with the [previously reported] office-based measurements, and the blood pressure reductions provide further evidence for the potential benefits of tirzepatide on cardiovascular health and outcomes,” said Dr. de Lemos, a cardiologist and professor at the University of Texas Southwestern Medical Center, Dallas.
The substudy included 600 of the 2,539 people enrolled in SURMOUNT-1, the first of two pivotal trials for tirzepatide (Mounjaro) in people without diabetes but with obesity or overweight (body mass index of 27-29 kg/m2) plus at least one weight-related complication. The primary endpoints of SURMOUNT-1 were the percent change in weight from baseline to 72 weeks on treatment with either of three different weekly injected doses of tirzepatide, compared with control subjects who received placebo, and the percentage of enrolled subjects achieving at least 5% loss in baseline weight, compared with the controls.
Tirzepatide treatment led to significant increases in both results, compared with controls, with the highest dose tested, 15 mg/week, resulting in an average 20.9% drop in weight from baseline after 72 weeks of treatment, and 91% of enrolled subjects on that dose achieving the 5% weight-loss threshold during the same time frame, in results published in 2022 in the New England Journal of Medicine.
24-hour ambulatory pressures from 494 people
The substudy enrolled 600 of the SURMOUNT-1 participants and involved 24-hour ambulatory BP and heart rate measurements at entry and after 36 weeks on treatment. Full results were available for 494 of these people. The substudy included only study participants who entered with a BP of less than 140/90 mm Hg. Enrollment in SURMOUNT-1 overall excluded people with a BP of 160/100 mm Hg or higher. The average BP among all enrolled participants was about 123/80 mm Hg, while heart rates averaged about 73 beats per minute.
Systolic BP measured with the ambulatory monitor fell from baseline by an average of 5.6, 8.8, and 6.2 mm Hg in the people who received tirzepatide in weekly doses of 5, 10, or 15 mg, respectively, and rose by an average 1.8 mm Hg among the controls, Dr. de Lemos reported. Diastolic BP dropped among the tirzepatide recipients by an average of 1.5, 2.4, and 0.0 mm Hg in the three ascending tirzepatide treatment arms, and rose by an average 0.5 mm Hg among the controls. All of the differences between the intervention groups and the controls were significant except for the change in diastolic BP among participants who received 15 mg of tirzepatide weekly.
The results showed that 36 weeks on tirzepatide treatment was associated with “arguably clinically meaningful” reductions in systolic and diastolic BPs, Dr. de Lemos said. “There is a lot of optimism that this will translate into clinical benefits.” He also noted that, “within the limits of cross-study comparisons, the blood pressure changes look favorable, compared with the single-incretin mechanism GLP-1 [glucagonlike peptide–1] receptor agonists.”
Heart rate fell by an average 1.8 bpm in the controls, and rose by an average 0.3, 0.5, and 3.6 bpm among the three groups receiving ascending weekly tirzepatide doses, effects that were “consistent with what’s been seen with the GLP-1 receptor agonists,” noted Dr. de Lemos.
Tirzepatide is known as a “twincretin” because it shares this GLP-1 receptor agonism and also has a second incretin agonist activity, to the receptor for the glucose-dependent insulinotropic polypeptide.
Lowering of blood pressure plateaus
Changes in BP over time during the 72 weeks on treatment, data first presented in the original report, showed that average systolic pressure in the people who received tirzepatide fell sharply during the first 24 weeks on treatment, and then leveled out with little further change over time. Furthermore, all three tirzepatide doses produced roughly similar systolic BP reductions. Changes in diastolic pressure over time showed a mostly similar pattern of reduction, although a modest ongoing decrease in average diastolic pressure continued beyond 24 weeks.
This pattern of a plateau in BP reduction has been seen before in studies using other treatments to produce weight loss, including bariatric surgery, said Naveed Sattar, MBChB, PhD, professor of metabolic medicine at the University of Glasgow, who was not involved in SURMOUNT-1. He attributed the plateau in BP reduction among tirzepatide-treated people to them hitting a wall in their BP nadir based on homeostatic limits. Dr. Sattar noted that most enrolled participants had normal BPs at entry based on the reported study averages.
“It’s hard to go lower, but the blood pressure reduction may be larger in people who start at higher pressure levels,” Dr. Sattar said in an interview.
Another inferred cap on BP reductions in the trial hypothesizes that the individual clinicians who managed the enrolled patients may have cut back on other BP-lowering agents as the pressures of the tirzepatide recipients fell to relatively low levels, suggested Darren McGuire, MD, a cardiologist and professor at UT Southwestern Medical Center, who also was not involved in the SURMOUNT-1 study.
Incretin agonists as antihypertensive drugs
The substantial BP-lowering seen with tirzepatide, as well as with other incretin agonist agents, suggests a new way to think about BP control in people with overweight or obesity, Dr. Sattar said.
“Until now, we haven’t had tools where people lose so much weight. Now that we have these tools [incretin agonists as well as bariatric surgery], we see substantial blood pressure reductions. It makes you think we should use weight-loss agents to lower blood pressure rather than a beta-blocker or angiotensin-converting enzyme inhibitor; then we’d also produce all the other benefits from weight loss,” Dr. Sattar suggested.
Dr. de Lemos said he sees signals that the BP reductions caused by tirzepatide and the GLP-1 receptor agonists may go beyond just weight-loss effects.
“There appears to be a larger blood pressure reduction than anticipated based on the change in weight,” he said during his presentation. “GLP-1 is active in most vascular tissues, so these [receptor agonist] agents likely have vascular or cardiac effects, or even effects on other tissues that may affect blood pressure.”
Heart rate increases were usually modest
The experiences with GLP-1 receptor agonists also suggest that the heart rate increases seen with tirzepatide treatment in SURMOUNT-1 will not have long-term effects. “The [Food and Drug Administration] mandated this heart rate substudy to make sure that the increase in heart rate was not larger than what would be anticipated” with a GLP-1 receptor agonist, Dr. de Lemos explained.
SURMOUNT-1 had a treatment-stopping rule to prevent a person’s heart rate from rising beyond 10 bpm from baseline. “Trivial numbers” of patients experienced a heart rate increase of this magnitude, he said. If used in routine practice, Dr. de Lemos said that he would closely investigate a patient with a heart rate increase greater than 10 mm Hg. The average increase seen with the highest dose, about 4 bpm above baseline, would generally not be concerning.
Tirzepatide received U.S. marketing approval from the FDA in May 2022 for treating people with type 2 diabetes. In October 2022, the FDA gave tirzepatide “Fast Track” designation for the pending application for approval of an indication to treat people with overweight or obesity who match the entry criteria for SURMOUNT-1 and for the second pivotal trial for this indication, SURMOUNT-2. According to a statement from Eli Lilly, the company that is developing and markets tirzepatide (Mounjaro), the FDA’s decision on the obesity indication will remain pending until the SURMOUNT-2 results are available, which the company expects will occur in 2023.
SURMOUNT-1 and SURMOUNT-2 were sponsored by Lilly, the company that markets tirzepatide. Dr. de Lemos has been a consultant to Lilly as well as to Amgen, AstraZeneca, Janssen, Novo Nordisk, Ortho, Quidel Cardiovascular, and Regeneron. Dr. Sattar has financial ties to Lilly, Afimmune, Amgen, AstraZeneca, Boehringer Ingelheim, Hammi, Merck Sharpe & Dohme, Novartis, Novo Nordisk, Pfizer, Roche, and Sanofi-Aventis. Dr. McGuire has ties to Lilly as well as to Altimmune, Applied Therapeutics, Bayer, Boehringer Ingelheim, CSL Behring, Lexicon, Merck, Metavant, Novo Nordisk, and Sanofi.
CHICAGO – compared with placebo, while causing modest increases in heart rate, in a prespecified substudy of the SURMOUNT-1 trial.
“The large effects on ambulatory 24-hour blood pressure raise the possibility that there may be important long-term benefits of [tirzepatide] on the complications of obesity,” said James A. de Lemos, MD, during a presentation at the American Heart Association scientific sessions.
“The findings are concordant with the [previously reported] office-based measurements, and the blood pressure reductions provide further evidence for the potential benefits of tirzepatide on cardiovascular health and outcomes,” said Dr. de Lemos, a cardiologist and professor at the University of Texas Southwestern Medical Center, Dallas.
The substudy included 600 of the 2,539 people enrolled in SURMOUNT-1, the first of two pivotal trials for tirzepatide (Mounjaro) in people without diabetes but with obesity or overweight (body mass index of 27-29 kg/m2) plus at least one weight-related complication. The primary endpoints of SURMOUNT-1 were the percent change in weight from baseline to 72 weeks on treatment with either of three different weekly injected doses of tirzepatide, compared with control subjects who received placebo, and the percentage of enrolled subjects achieving at least 5% loss in baseline weight, compared with the controls.
Tirzepatide treatment led to significant increases in both results, compared with controls, with the highest dose tested, 15 mg/week, resulting in an average 20.9% drop in weight from baseline after 72 weeks of treatment, and 91% of enrolled subjects on that dose achieving the 5% weight-loss threshold during the same time frame, in results published in 2022 in the New England Journal of Medicine.
24-hour ambulatory pressures from 494 people
The substudy enrolled 600 of the SURMOUNT-1 participants and involved 24-hour ambulatory BP and heart rate measurements at entry and after 36 weeks on treatment. Full results were available for 494 of these people. The substudy included only study participants who entered with a BP of less than 140/90 mm Hg. Enrollment in SURMOUNT-1 overall excluded people with a BP of 160/100 mm Hg or higher. The average BP among all enrolled participants was about 123/80 mm Hg, while heart rates averaged about 73 beats per minute.
Systolic BP measured with the ambulatory monitor fell from baseline by an average of 5.6, 8.8, and 6.2 mm Hg in the people who received tirzepatide in weekly doses of 5, 10, or 15 mg, respectively, and rose by an average 1.8 mm Hg among the controls, Dr. de Lemos reported. Diastolic BP dropped among the tirzepatide recipients by an average of 1.5, 2.4, and 0.0 mm Hg in the three ascending tirzepatide treatment arms, and rose by an average 0.5 mm Hg among the controls. All of the differences between the intervention groups and the controls were significant except for the change in diastolic BP among participants who received 15 mg of tirzepatide weekly.
The results showed that 36 weeks on tirzepatide treatment was associated with “arguably clinically meaningful” reductions in systolic and diastolic BPs, Dr. de Lemos said. “There is a lot of optimism that this will translate into clinical benefits.” He also noted that, “within the limits of cross-study comparisons, the blood pressure changes look favorable, compared with the single-incretin mechanism GLP-1 [glucagonlike peptide–1] receptor agonists.”
Heart rate fell by an average 1.8 bpm in the controls, and rose by an average 0.3, 0.5, and 3.6 bpm among the three groups receiving ascending weekly tirzepatide doses, effects that were “consistent with what’s been seen with the GLP-1 receptor agonists,” noted Dr. de Lemos.
Tirzepatide is known as a “twincretin” because it shares this GLP-1 receptor agonism and also has a second incretin agonist activity, to the receptor for the glucose-dependent insulinotropic polypeptide.
Lowering of blood pressure plateaus
Changes in BP over time during the 72 weeks on treatment, data first presented in the original report, showed that average systolic pressure in the people who received tirzepatide fell sharply during the first 24 weeks on treatment, and then leveled out with little further change over time. Furthermore, all three tirzepatide doses produced roughly similar systolic BP reductions. Changes in diastolic pressure over time showed a mostly similar pattern of reduction, although a modest ongoing decrease in average diastolic pressure continued beyond 24 weeks.
This pattern of a plateau in BP reduction has been seen before in studies using other treatments to produce weight loss, including bariatric surgery, said Naveed Sattar, MBChB, PhD, professor of metabolic medicine at the University of Glasgow, who was not involved in SURMOUNT-1. He attributed the plateau in BP reduction among tirzepatide-treated people to them hitting a wall in their BP nadir based on homeostatic limits. Dr. Sattar noted that most enrolled participants had normal BPs at entry based on the reported study averages.
“It’s hard to go lower, but the blood pressure reduction may be larger in people who start at higher pressure levels,” Dr. Sattar said in an interview.
Another inferred cap on BP reductions in the trial hypothesizes that the individual clinicians who managed the enrolled patients may have cut back on other BP-lowering agents as the pressures of the tirzepatide recipients fell to relatively low levels, suggested Darren McGuire, MD, a cardiologist and professor at UT Southwestern Medical Center, who also was not involved in the SURMOUNT-1 study.
Incretin agonists as antihypertensive drugs
The substantial BP-lowering seen with tirzepatide, as well as with other incretin agonist agents, suggests a new way to think about BP control in people with overweight or obesity, Dr. Sattar said.
“Until now, we haven’t had tools where people lose so much weight. Now that we have these tools [incretin agonists as well as bariatric surgery], we see substantial blood pressure reductions. It makes you think we should use weight-loss agents to lower blood pressure rather than a beta-blocker or angiotensin-converting enzyme inhibitor; then we’d also produce all the other benefits from weight loss,” Dr. Sattar suggested.
Dr. de Lemos said he sees signals that the BP reductions caused by tirzepatide and the GLP-1 receptor agonists may go beyond just weight-loss effects.
“There appears to be a larger blood pressure reduction than anticipated based on the change in weight,” he said during his presentation. “GLP-1 is active in most vascular tissues, so these [receptor agonist] agents likely have vascular or cardiac effects, or even effects on other tissues that may affect blood pressure.”
Heart rate increases were usually modest
The experiences with GLP-1 receptor agonists also suggest that the heart rate increases seen with tirzepatide treatment in SURMOUNT-1 will not have long-term effects. “The [Food and Drug Administration] mandated this heart rate substudy to make sure that the increase in heart rate was not larger than what would be anticipated” with a GLP-1 receptor agonist, Dr. de Lemos explained.
SURMOUNT-1 had a treatment-stopping rule to prevent a person’s heart rate from rising beyond 10 bpm from baseline. “Trivial numbers” of patients experienced a heart rate increase of this magnitude, he said. If used in routine practice, Dr. de Lemos said that he would closely investigate a patient with a heart rate increase greater than 10 mm Hg. The average increase seen with the highest dose, about 4 bpm above baseline, would generally not be concerning.
Tirzepatide received U.S. marketing approval from the FDA in May 2022 for treating people with type 2 diabetes. In October 2022, the FDA gave tirzepatide “Fast Track” designation for the pending application for approval of an indication to treat people with overweight or obesity who match the entry criteria for SURMOUNT-1 and for the second pivotal trial for this indication, SURMOUNT-2. According to a statement from Eli Lilly, the company that is developing and markets tirzepatide (Mounjaro), the FDA’s decision on the obesity indication will remain pending until the SURMOUNT-2 results are available, which the company expects will occur in 2023.
SURMOUNT-1 and SURMOUNT-2 were sponsored by Lilly, the company that markets tirzepatide. Dr. de Lemos has been a consultant to Lilly as well as to Amgen, AstraZeneca, Janssen, Novo Nordisk, Ortho, Quidel Cardiovascular, and Regeneron. Dr. Sattar has financial ties to Lilly, Afimmune, Amgen, AstraZeneca, Boehringer Ingelheim, Hammi, Merck Sharpe & Dohme, Novartis, Novo Nordisk, Pfizer, Roche, and Sanofi-Aventis. Dr. McGuire has ties to Lilly as well as to Altimmune, Applied Therapeutics, Bayer, Boehringer Ingelheim, CSL Behring, Lexicon, Merck, Metavant, Novo Nordisk, and Sanofi.
AT AHA 2022