User login
Estrogen replacement therapy in endometrial cancer survivors
In the United States, uterine cancer is the fourth most common cancer among women, behind breast, lung/bronchus, and colorectal cancer. There are expected to be almost 66,000 new cases of uterine cancer in 2022.1 The majority of uterine cancers are endometrioid in histology and tend to be low grade, diagnosed at an early stage, and have a good prognosis. While our molecular understanding of endometrial cancers (EC) has changed significantly in recent years, low-grade endometrioid adenocarcinomas have historically been described as type 1 ECs. Type 1 ECs are typically caused by excess estrogen exposure (often unopposed or lacking progesterone protection) and are preceded by endometrial hyperplasia. Excess estrogen can come from exogenous sources (such as unopposed estrogen replacement therapy or tamoxifen, a commonly used treatment in estrogen receptor–positive breast cancer that acts as an estrogen agonist in the endometrium in postmenopausal patients) or endogenous ones (such as obesity).
Peripheral adipose tissue converts androgens into estrogens; paired with the decreased levels of sex hormone–binding globulin seen in obesity, there is more unbound or free serum estrogen (specifically estradiol) in obese women. Estrogen acts on the endometrium to cause proliferation and, if unopposed or imbalanced in relation to progesterone exposure, can ultimately lead to hyperplasia and malignancy.

If excess and unopposed estrogen exposure are major risk factors for the development of EC, is it safe to consider estrogen replacement therapy (ERT) in patients after EC treatment?
The short answer is the data are limited, but in a patient with a history of low-risk early-stage EC who undergoes appropriate counseling, it is likely safe to consider ERT.
Among EC survivors, there has been only one prospective randomized controlled trial that assessed the effect of recurrence rate and survival in women on ERT after EC treatment.2 Patients with stage I or occult stage II endometrial adenocarcinoma treated with at least a total hysterectomy and bilateral salpingo-oophorectomy were randomized to ERT versus placebo for 3 years of treatment, with therapy starting once recovered and within 20 weeks after surgery. Trial participation required an indication for ERT, such as vasomotor symptoms, vaginal atrophy, or increased risk of cardiovascular disease or osteoporosis.
The trial accrued 1,236 patients, falling short of its goal of 2,108 patients after enrollment decreased following the publication of the Women’s Health Initiative results in 2002. This publication prompted a review of the ERT study protocol that found that between decreased accrual and lower than expected recurrence rate, goal accrual would be impossible. Of those enrolled, participants were overwhelmingly white (84%-85%), 41-70 years old (80%-82%), and had stage IA or IB disease (88%). Median follow-up was almost 3 years.
Twenty-six (2.1%) patients experienced cancer recurrence, with similar rates in both groups. Three-year progression-free and overall survival were high overall among all study participants (94.8% and 96.5%). Unfortunately, because the study was closed early, definitive conclusions about the noninferiority of ERT versus placebo regarding oncologic outcomes in early-stage endometrial adenocarcinoma could not be made.
A subsequent meta-analysis looked at the effect of hormone therapy (HT) on recurrence rate in EC survivors.3 Five observational studies were included along with the previously discussed randomized controlled trial. Among 1,975 participants across six studies, there were cancer recurrences in 19 of 896 (2.1%) HT users and 64 of 1,079 (5.9%) controls. HT did not negatively affect cancer recurrence or overall survival. There was significant heterogeneity between studies as to dosing, duration, and type of HT given (some used estrogen-only replacement, others used estrogen and progesterone replacement, and some used both estrogen only and the combination of estrogen and progesterone replacement). Among the five nonrandomized studies included, a protective effect of combined HT on EC recurrence was noted. One study included patients with stage III disease, but only four patients received HT in this cohort.
Given the data we have, ERT does not appear to significantly affect oncologic outcomes in low-risk, early-stage EC survivors. We do not have data to support this same assertion in more advanced, high-risk disease. Before initiation of any ERT in an EC survivor, there should be a detailed discussion to weigh the risks and benefits of starting therapy. The goal of treatment should be to use the lowest dose of ERT possible to treat symptoms, with planned surveillance visits for symptom check-in and assessment of readiness to start tapering treatment.
Footnote: vaginal estrogen therapy
There are no randomized trials assessing the safety of vaginal estrogen preparations or their effect on oncologic outcomes in EC survivors. Observational data from the Women’s Health Initiative showed no increased risk of endometrial cancer in patients who used vaginal estrogen with an intact uterus.4 A recently published retrospective study among 244 gynecologic cancer survivors found low rates of disease recurrence and adverse outcomes among women who used vaginal estrogen for genitourinary symptoms.5 Among EC survivors, the incidence of recurrence was 2.4% for patients with stage I/II disease and 4.3% for stage III/IV disease, with a median follow-up of 80.2 months. While there appears to be some systemic absorption with vaginal estrogen use, this can be quite challenging to measure because of the current sensitivity of serum estradiol and estrone assays. Given the significantly lower serum levels with vaginal estrogen preparations compared with ERT, vaginal estrogen use appears to be safe in EC survivors.
Dr. Tucker is assistant professor of gynecologic oncology at the University of North Carolina at Chapel Hill.
References
1. Cancer Stat Facts: Uterine Cancer. National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Accessed 12 Aug. 2022. https://seer.cancer.gov/statfacts/html/corp.html.
2. Barakat RR et al. J Clin Oncol. 2006;24(4):587-92.
3. Shim SH et al. Eur J Cancer. 2014;50(9):1628-37.
4. Crandall CJ et al. Menopause. 2018 Jan;25(1):11-20.
5. Chambers LM et al. Int J Gynecol Cancer. 2020 Apr;30(4):515-24.
In the United States, uterine cancer is the fourth most common cancer among women, behind breast, lung/bronchus, and colorectal cancer. There are expected to be almost 66,000 new cases of uterine cancer in 2022.1 The majority of uterine cancers are endometrioid in histology and tend to be low grade, diagnosed at an early stage, and have a good prognosis. While our molecular understanding of endometrial cancers (EC) has changed significantly in recent years, low-grade endometrioid adenocarcinomas have historically been described as type 1 ECs. Type 1 ECs are typically caused by excess estrogen exposure (often unopposed or lacking progesterone protection) and are preceded by endometrial hyperplasia. Excess estrogen can come from exogenous sources (such as unopposed estrogen replacement therapy or tamoxifen, a commonly used treatment in estrogen receptor–positive breast cancer that acts as an estrogen agonist in the endometrium in postmenopausal patients) or endogenous ones (such as obesity).
Peripheral adipose tissue converts androgens into estrogens; paired with the decreased levels of sex hormone–binding globulin seen in obesity, there is more unbound or free serum estrogen (specifically estradiol) in obese women. Estrogen acts on the endometrium to cause proliferation and, if unopposed or imbalanced in relation to progesterone exposure, can ultimately lead to hyperplasia and malignancy.
If excess and unopposed estrogen exposure are major risk factors for the development of EC, is it safe to consider estrogen replacement therapy (ERT) in patients after EC treatment?
The short answer is the data are limited, but in a patient with a history of low-risk early-stage EC who undergoes appropriate counseling, it is likely safe to consider ERT.
Among EC survivors, there has been only one prospective randomized controlled trial that assessed the effect of recurrence rate and survival in women on ERT after EC treatment.2 Patients with stage I or occult stage II endometrial adenocarcinoma treated with at least a total hysterectomy and bilateral salpingo-oophorectomy were randomized to ERT versus placebo for 3 years of treatment, with therapy starting once recovered and within 20 weeks after surgery. Trial participation required an indication for ERT, such as vasomotor symptoms, vaginal atrophy, or increased risk of cardiovascular disease or osteoporosis.
The trial accrued 1,236 patients, falling short of its goal of 2,108 patients after enrollment decreased following the publication of the Women’s Health Initiative results in 2002. This publication prompted a review of the ERT study protocol that found that between decreased accrual and lower than expected recurrence rate, goal accrual would be impossible. Of those enrolled, participants were overwhelmingly white (84%-85%), 41-70 years old (80%-82%), and had stage IA or IB disease (88%). Median follow-up was almost 3 years.
Twenty-six (2.1%) patients experienced cancer recurrence, with similar rates in both groups. Three-year progression-free and overall survival were high overall among all study participants (94.8% and 96.5%). Unfortunately, because the study was closed early, definitive conclusions about the noninferiority of ERT versus placebo regarding oncologic outcomes in early-stage endometrial adenocarcinoma could not be made.
A subsequent meta-analysis looked at the effect of hormone therapy (HT) on recurrence rate in EC survivors.3 Five observational studies were included along with the previously discussed randomized controlled trial. Among 1,975 participants across six studies, there were cancer recurrences in 19 of 896 (2.1%) HT users and 64 of 1,079 (5.9%) controls. HT did not negatively affect cancer recurrence or overall survival. There was significant heterogeneity between studies as to dosing, duration, and type of HT given (some used estrogen-only replacement, others used estrogen and progesterone replacement, and some used both estrogen only and the combination of estrogen and progesterone replacement). Among the five nonrandomized studies included, a protective effect of combined HT on EC recurrence was noted. One study included patients with stage III disease, but only four patients received HT in this cohort.
Given the data we have, ERT does not appear to significantly affect oncologic outcomes in low-risk, early-stage EC survivors. We do not have data to support this same assertion in more advanced, high-risk disease. Before initiation of any ERT in an EC survivor, there should be a detailed discussion to weigh the risks and benefits of starting therapy. The goal of treatment should be to use the lowest dose of ERT possible to treat symptoms, with planned surveillance visits for symptom check-in and assessment of readiness to start tapering treatment.
Footnote: vaginal estrogen therapy
There are no randomized trials assessing the safety of vaginal estrogen preparations or their effect on oncologic outcomes in EC survivors. Observational data from the Women’s Health Initiative showed no increased risk of endometrial cancer in patients who used vaginal estrogen with an intact uterus.4 A recently published retrospective study among 244 gynecologic cancer survivors found low rates of disease recurrence and adverse outcomes among women who used vaginal estrogen for genitourinary symptoms.5 Among EC survivors, the incidence of recurrence was 2.4% for patients with stage I/II disease and 4.3% for stage III/IV disease, with a median follow-up of 80.2 months. While there appears to be some systemic absorption with vaginal estrogen use, this can be quite challenging to measure because of the current sensitivity of serum estradiol and estrone assays. Given the significantly lower serum levels with vaginal estrogen preparations compared with ERT, vaginal estrogen use appears to be safe in EC survivors.
Dr. Tucker is assistant professor of gynecologic oncology at the University of North Carolina at Chapel Hill.
References
1. Cancer Stat Facts: Uterine Cancer. National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Accessed 12 Aug. 2022. https://seer.cancer.gov/statfacts/html/corp.html.
2. Barakat RR et al. J Clin Oncol. 2006;24(4):587-92.
3. Shim SH et al. Eur J Cancer. 2014;50(9):1628-37.
4. Crandall CJ et al. Menopause. 2018 Jan;25(1):11-20.
5. Chambers LM et al. Int J Gynecol Cancer. 2020 Apr;30(4):515-24.
In the United States, uterine cancer is the fourth most common cancer among women, behind breast, lung/bronchus, and colorectal cancer. There are expected to be almost 66,000 new cases of uterine cancer in 2022.1 The majority of uterine cancers are endometrioid in histology and tend to be low grade, diagnosed at an early stage, and have a good prognosis. While our molecular understanding of endometrial cancers (EC) has changed significantly in recent years, low-grade endometrioid adenocarcinomas have historically been described as type 1 ECs. Type 1 ECs are typically caused by excess estrogen exposure (often unopposed or lacking progesterone protection) and are preceded by endometrial hyperplasia. Excess estrogen can come from exogenous sources (such as unopposed estrogen replacement therapy or tamoxifen, a commonly used treatment in estrogen receptor–positive breast cancer that acts as an estrogen agonist in the endometrium in postmenopausal patients) or endogenous ones (such as obesity).
Peripheral adipose tissue converts androgens into estrogens; paired with the decreased levels of sex hormone–binding globulin seen in obesity, there is more unbound or free serum estrogen (specifically estradiol) in obese women. Estrogen acts on the endometrium to cause proliferation and, if unopposed or imbalanced in relation to progesterone exposure, can ultimately lead to hyperplasia and malignancy.
If excess and unopposed estrogen exposure are major risk factors for the development of EC, is it safe to consider estrogen replacement therapy (ERT) in patients after EC treatment?
The short answer is the data are limited, but in a patient with a history of low-risk early-stage EC who undergoes appropriate counseling, it is likely safe to consider ERT.
Among EC survivors, there has been only one prospective randomized controlled trial that assessed the effect of recurrence rate and survival in women on ERT after EC treatment.2 Patients with stage I or occult stage II endometrial adenocarcinoma treated with at least a total hysterectomy and bilateral salpingo-oophorectomy were randomized to ERT versus placebo for 3 years of treatment, with therapy starting once recovered and within 20 weeks after surgery. Trial participation required an indication for ERT, such as vasomotor symptoms, vaginal atrophy, or increased risk of cardiovascular disease or osteoporosis.
The trial accrued 1,236 patients, falling short of its goal of 2,108 patients after enrollment decreased following the publication of the Women’s Health Initiative results in 2002. This publication prompted a review of the ERT study protocol that found that between decreased accrual and lower than expected recurrence rate, goal accrual would be impossible. Of those enrolled, participants were overwhelmingly white (84%-85%), 41-70 years old (80%-82%), and had stage IA or IB disease (88%). Median follow-up was almost 3 years.
Twenty-six (2.1%) patients experienced cancer recurrence, with similar rates in both groups. Three-year progression-free and overall survival were high overall among all study participants (94.8% and 96.5%). Unfortunately, because the study was closed early, definitive conclusions about the noninferiority of ERT versus placebo regarding oncologic outcomes in early-stage endometrial adenocarcinoma could not be made.
A subsequent meta-analysis looked at the effect of hormone therapy (HT) on recurrence rate in EC survivors.3 Five observational studies were included along with the previously discussed randomized controlled trial. Among 1,975 participants across six studies, there were cancer recurrences in 19 of 896 (2.1%) HT users and 64 of 1,079 (5.9%) controls. HT did not negatively affect cancer recurrence or overall survival. There was significant heterogeneity between studies as to dosing, duration, and type of HT given (some used estrogen-only replacement, others used estrogen and progesterone replacement, and some used both estrogen only and the combination of estrogen and progesterone replacement). Among the five nonrandomized studies included, a protective effect of combined HT on EC recurrence was noted. One study included patients with stage III disease, but only four patients received HT in this cohort.
Given the data we have, ERT does not appear to significantly affect oncologic outcomes in low-risk, early-stage EC survivors. We do not have data to support this same assertion in more advanced, high-risk disease. Before initiation of any ERT in an EC survivor, there should be a detailed discussion to weigh the risks and benefits of starting therapy. The goal of treatment should be to use the lowest dose of ERT possible to treat symptoms, with planned surveillance visits for symptom check-in and assessment of readiness to start tapering treatment.
Footnote: vaginal estrogen therapy
There are no randomized trials assessing the safety of vaginal estrogen preparations or their effect on oncologic outcomes in EC survivors. Observational data from the Women’s Health Initiative showed no increased risk of endometrial cancer in patients who used vaginal estrogen with an intact uterus.4 A recently published retrospective study among 244 gynecologic cancer survivors found low rates of disease recurrence and adverse outcomes among women who used vaginal estrogen for genitourinary symptoms.5 Among EC survivors, the incidence of recurrence was 2.4% for patients with stage I/II disease and 4.3% for stage III/IV disease, with a median follow-up of 80.2 months. While there appears to be some systemic absorption with vaginal estrogen use, this can be quite challenging to measure because of the current sensitivity of serum estradiol and estrone assays. Given the significantly lower serum levels with vaginal estrogen preparations compared with ERT, vaginal estrogen use appears to be safe in EC survivors.
Dr. Tucker is assistant professor of gynecologic oncology at the University of North Carolina at Chapel Hill.
References
1. Cancer Stat Facts: Uterine Cancer. National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Accessed 12 Aug. 2022. https://seer.cancer.gov/statfacts/html/corp.html.
2. Barakat RR et al. J Clin Oncol. 2006;24(4):587-92.
3. Shim SH et al. Eur J Cancer. 2014;50(9):1628-37.
4. Crandall CJ et al. Menopause. 2018 Jan;25(1):11-20.
5. Chambers LM et al. Int J Gynecol Cancer. 2020 Apr;30(4):515-24.
Obesity drug shortage triggers frustrations, workarounds
The glucagon-like peptide-1 (GLP-1) agonist semaglutide formulated for treating obesity (Wegovy) had a roaring takeoff a little more than a year ago, with surging patient demand after the U.S. Food and Drug Administration approved it in June 2021. But starting doses of the Wegovy form of semaglutide went missing in action starting late 2021 and continue to date, frustrating patients and their health care providers.
The arrival of Wegovy last year was hailed by obesity medicine specialists and others as a “game changer” for treating people with obesity because of semaglutide’s proven safety and efficacy at the subcutaneous dose of 2.4 mg delivered once a week to produce at least 15% weight loss in half the people who received it, as documented last year in results from one of the drug’s pivotal clinical trials.
But during the months following semaglutide’s approval for treating obesity (it also received an FDA marketing nod in late 2017 as Ozempic for treating type 2 diabetes), a worldwide shortage of Wegovy, including in the United States, emerged.
A manufacturing glitch shut down the primary location for production of U.S.-bound Wegovy injector pens for several months starting in late 2021, according to a December report from Novo Nordisk, the company that makes and markets the agent. (The Wegovy production issue appears to have had a very modest impact, especially in U.S. pharmacies, on the supply of semaglutide formulated as Ozempic, also marketed by Novo Nordisk, although Wegovy supply and demand have dramatically limited Ozempic availability in Australia.)
‘Unprecedented demand’ for Wegovy derailed when plant went offline
The supply side for Wegovy became so hopelessly broken that just months after U.S. sales began and immediately skyrocketed, Novo Nordisk made the remarkable decision to pull starting doses of Wegovy from the market to make it much harder to initiate patients (semaglutide and other GLP-1 agonists require gradual dose ramp-up to avoid gastrointestinal side effects), and the company publicly implored clinicians to not start new patients on the agent, which is where the status remains as of early August 2022.
Novo Nordisk’s financial report for the second quarter of 2022, released on Aug. 3, said the company “expects to make all Wegovy dose strengths available in the United States towards the end of 2022.”
A Dear Health Care Provider letter that Novo Nordisk posted on its U.S. Wegovy website last spring cited “unprecedented demand” that exceeded every prior product launch in the company’s history. It forced Novo Nordisk to pull the plug on all U.S. promotion of Wegovy and compelled the company to ask U.S. clinicians to halt new patient starts.
“I stopped offering Wegovy to new patients” since about the beginning of 2022, says Lauren D. Oshman, MD, a family and obesity medicine specialist at the University of Michigan, Ann Arbor. “It’s very frustrating to not have patients [with obesity] receive the optimal treatment available.” Although she adds that she tries to match obesity treatments to each patient’s clinical needs, and a GLP-1 agonist is not the first choice for every person with obesity.
“It was a disastrous rollout,” says Catherine W. Varney, DO, a family and obesity medicine specialist at the University of Virginia, Charlottesville. “It’s frustrating to know that the treatment is there but not being able to use it,” she said in an interview.
“I had about 800 patients on Wegovy” when the supply dropped earlier this year, and “I couldn’t handle the volume of messages that I got from patients,” recalls Angela Fitch, MD, associate director of the Massachusetts General Hospital Weight Center, Boston. “It was painful,” she said in an interview.
“Frustrating and chaotic,” is the description from Ivania M. Rizo, MD, director of obesity medicine at Boston Medical Center.
The liraglutide/Saxenda workaround
The upshot is that people with obesity and their health care providers have been busy devising workarounds to try to meet the intense demand for this drug-assisted approach to appetite control and weight loss. Their tactics run a wide gamut based on the crazy-quilt diversity of health insurance coverage across America.
Because the bottleneck for starting Wegovy resulted from unavailable starting doses (dosing starts at 0.25 mg delivered subcutaneously once a week, eventually ramping up to a maximum of 2.4 mg weekly), one option was to start patients on a different GLP-1 agonist, such as liraglutide (Saxenda, approved for obesity).
Starting a patient on liraglutide involves the same sort of up-titration and acclimation to a GLP-1 agonist that semaglutide requires, and transition between these agents seems feasible for at least some. It also means daily injections of liraglutide rather than the weekly schedule for semaglutide, although some patients prefer maintaining a daily dosing schedule. Another limitation of liraglutide is that evidence shows it is not nearly as effective for weight loss as semaglutide.
Results from the head-to-head STEP 8 trial, published in JAMA, showed an average weight loss from baseline of about 16% with semaglutide and about 6% with liraglutide (and about 2% with placebo).
A ‘reasonable’ evidence base, but more work
Changing from Saxenda to Wegovy, or from Wegovy to Saxenda, “would be reasonably evidence-based medicine,” said Dr. Oshman in an interview. She has managed a Wegovy-to-Saxenda switch for a “handful” of patients to deal with Wegovy shortages, but she has not yet moved anyone to Wegovy after a Saxenda initiation.
“No prospective study has looked at this transition,” but dose equivalence tables exist based on expert opinion, noted Dr. Oshman, as in this 2020 report.
Dr. Varney has several patients on the Saxenda-to-Wegovy track. She up-titrates patients on Saxenda to the maximum daily dose of 3.0 mg and then switches them to the 1.7 mg weekly dose of Wegovy, one of the “destination” Wegovy doses that has remained generally available during the shortage. But Dr. Varney’s experience is that only half of her patients made the changeover smoothly, with the others having “severe gastrointestinal distress,” including vomiting, she notes.
Dr. Fitch has also successfully used this Saxenda-to-Wegovy approach for some of her patients, but it hasn’t been easy.
“It’s more work and more prior authorizations. It’s harder and adds a layer of stress,” but, Dr. Fitch adds, “people are willing to work on it because the weight loss is worth it.”
The liraglutide to semaglutide shuffle is “doable,” says Dr. Rizo, “but I’m looking forward to not having to do it and being able to just start Wegovy.”
The tirzepatide coupon program works ‘off label’ for obesity
Another workaround depends on the FDA approval in May for tirzepatide (Mounjaro) for type 2 diabetes. Tirzepatide is a related GLP-1 agonist that also adds a second incretin-like agonist activity that mimics the glucose-dependent insulinotropic polypeptide.
Soon after approval, Lilly, the company that markets tirzepatide, started a U.S. coupon program geared exclusively to people with commercial insurance. Within certain refill and dollar limits, the program lets patients buy tirzepatide at pharmacies at an out-of-pocket cost of $25 for a 4-week supply (tirzepatide is also dosed by weekly subcutaneous injections). The program will extend into 2023.
Novo Nordisk offered U.S. patients with commercial insurance a similar discount when Wegovy first hit the U.S. market in 2021, but the program closed down once the supply shortage began.
Despite tirzepatide’s current approval only for type 2 diabetes, Dr. Varney has been successfully prescribing it to patients without diabetes off-label for weight loss.
“The coupons still work even when tirzepatide is used off-label,” she notes. And while the drug’s rollout is still only a couple of months old, so far, it’s gone “beautifully” with no hints of supply issues, she says.
But a major drawback to relying on an introductory coupon program that makes these agents affordable to patients is their ability to maintain treatment once the discounts inevitably end.
“We try to only prescribe agents that patients can continue to access,” says Dr. Fitch, who has had some patients with commercial insurance start on Wegovy with coupon discounts only to later lose access.
Many commercial U.S. insurers do not cover obesity treatments, a decision often driven by the employers who sponsor the coverage, she notes.
Study results have documented that when people with obesity stop taking a GLP-1 agonist their lost weight rebounds, as in a study that tracked people who stopped taking semaglutide.
Dr. Fitch has had success prescribing tirzepatide to patients with obesity but without diabetes who have certain types of Medicare drug coverage policies, which often do not deny off-label drug coverage. That approach works until patients reach the “donut hole” in their drug coverage and are faced with a certain level of out-of-pocket costs that can balloon to several thousand dollars.
Even more workarounds
Other approaches patients have used to acquire Wegovy include purchasing it in other countries, such as Canada or Brazil, says Dr. Fitch. But prices outside the United States, while substantially lower, can still be a barrier for many patients, notes Dr. Oshman.
Semaglutide in Canada goes for about $300 for a 4-week supply, roughly a quarter the U.S. price, she says, but is “still too high for many of my patients.”
Intense patient demand sometimes bordering on desperation has prompted some to seek semaglutide from private compounding pharmacies, a step clinicians regard as downright dangerous.
“Semaglutide from compounding pharmacies is not known to be safe. We feel strongly that it’s not something that people should do,” says Dr. Fitch.
“Compounding pharmacies have no FDA regulation. People don’t know what they’re getting. It’s dangerous,” agrees Dr. Varney. Physicians who refer people for privately compounded semaglutide “are taking advantage of desperate people,” she adds.
Although it seems likely that Novo Nordisk will soon sort out the supply problems and Wegovy will once again become more widely available, some of the issues patients have had with access to the weight loss medication stem from more systemic issues in the United States health insurance landscape: an unwillingness by payers to cover the costs of weight loss medications, a shortcoming that also exists for Medicare and Medicaid.
“We need to make obesity treatment a standard benefit, and not something that can be carved out,” says Dr. Fitch. People with obesity “deserve access to effective treatments for their disease,” she declares.
Dr. Oshman, Dr. Varney, and Dr. Rizo have reported no relevant financial relationships. Dr. Fitch has reported being an advisor to Jenny Craig.
A version of this article first appeared on Medscape.com.
The glucagon-like peptide-1 (GLP-1) agonist semaglutide formulated for treating obesity (Wegovy) had a roaring takeoff a little more than a year ago, with surging patient demand after the U.S. Food and Drug Administration approved it in June 2021. But starting doses of the Wegovy form of semaglutide went missing in action starting late 2021 and continue to date, frustrating patients and their health care providers.
The arrival of Wegovy last year was hailed by obesity medicine specialists and others as a “game changer” for treating people with obesity because of semaglutide’s proven safety and efficacy at the subcutaneous dose of 2.4 mg delivered once a week to produce at least 15% weight loss in half the people who received it, as documented last year in results from one of the drug’s pivotal clinical trials.
But during the months following semaglutide’s approval for treating obesity (it also received an FDA marketing nod in late 2017 as Ozempic for treating type 2 diabetes), a worldwide shortage of Wegovy, including in the United States, emerged.
A manufacturing glitch shut down the primary location for production of U.S.-bound Wegovy injector pens for several months starting in late 2021, according to a December report from Novo Nordisk, the company that makes and markets the agent. (The Wegovy production issue appears to have had a very modest impact, especially in U.S. pharmacies, on the supply of semaglutide formulated as Ozempic, also marketed by Novo Nordisk, although Wegovy supply and demand have dramatically limited Ozempic availability in Australia.)
‘Unprecedented demand’ for Wegovy derailed when plant went offline
The supply side for Wegovy became so hopelessly broken that just months after U.S. sales began and immediately skyrocketed, Novo Nordisk made the remarkable decision to pull starting doses of Wegovy from the market to make it much harder to initiate patients (semaglutide and other GLP-1 agonists require gradual dose ramp-up to avoid gastrointestinal side effects), and the company publicly implored clinicians to not start new patients on the agent, which is where the status remains as of early August 2022.
Novo Nordisk’s financial report for the second quarter of 2022, released on Aug. 3, said the company “expects to make all Wegovy dose strengths available in the United States towards the end of 2022.”
A Dear Health Care Provider letter that Novo Nordisk posted on its U.S. Wegovy website last spring cited “unprecedented demand” that exceeded every prior product launch in the company’s history. It forced Novo Nordisk to pull the plug on all U.S. promotion of Wegovy and compelled the company to ask U.S. clinicians to halt new patient starts.
“I stopped offering Wegovy to new patients” since about the beginning of 2022, says Lauren D. Oshman, MD, a family and obesity medicine specialist at the University of Michigan, Ann Arbor. “It’s very frustrating to not have patients [with obesity] receive the optimal treatment available.” Although she adds that she tries to match obesity treatments to each patient’s clinical needs, and a GLP-1 agonist is not the first choice for every person with obesity.
“It was a disastrous rollout,” says Catherine W. Varney, DO, a family and obesity medicine specialist at the University of Virginia, Charlottesville. “It’s frustrating to know that the treatment is there but not being able to use it,” she said in an interview.
“I had about 800 patients on Wegovy” when the supply dropped earlier this year, and “I couldn’t handle the volume of messages that I got from patients,” recalls Angela Fitch, MD, associate director of the Massachusetts General Hospital Weight Center, Boston. “It was painful,” she said in an interview.
“Frustrating and chaotic,” is the description from Ivania M. Rizo, MD, director of obesity medicine at Boston Medical Center.
The liraglutide/Saxenda workaround
The upshot is that people with obesity and their health care providers have been busy devising workarounds to try to meet the intense demand for this drug-assisted approach to appetite control and weight loss. Their tactics run a wide gamut based on the crazy-quilt diversity of health insurance coverage across America.
Because the bottleneck for starting Wegovy resulted from unavailable starting doses (dosing starts at 0.25 mg delivered subcutaneously once a week, eventually ramping up to a maximum of 2.4 mg weekly), one option was to start patients on a different GLP-1 agonist, such as liraglutide (Saxenda, approved for obesity).
Starting a patient on liraglutide involves the same sort of up-titration and acclimation to a GLP-1 agonist that semaglutide requires, and transition between these agents seems feasible for at least some. It also means daily injections of liraglutide rather than the weekly schedule for semaglutide, although some patients prefer maintaining a daily dosing schedule. Another limitation of liraglutide is that evidence shows it is not nearly as effective for weight loss as semaglutide.
Results from the head-to-head STEP 8 trial, published in JAMA, showed an average weight loss from baseline of about 16% with semaglutide and about 6% with liraglutide (and about 2% with placebo).
A ‘reasonable’ evidence base, but more work
Changing from Saxenda to Wegovy, or from Wegovy to Saxenda, “would be reasonably evidence-based medicine,” said Dr. Oshman in an interview. She has managed a Wegovy-to-Saxenda switch for a “handful” of patients to deal with Wegovy shortages, but she has not yet moved anyone to Wegovy after a Saxenda initiation.
“No prospective study has looked at this transition,” but dose equivalence tables exist based on expert opinion, noted Dr. Oshman, as in this 2020 report.
Dr. Varney has several patients on the Saxenda-to-Wegovy track. She up-titrates patients on Saxenda to the maximum daily dose of 3.0 mg and then switches them to the 1.7 mg weekly dose of Wegovy, one of the “destination” Wegovy doses that has remained generally available during the shortage. But Dr. Varney’s experience is that only half of her patients made the changeover smoothly, with the others having “severe gastrointestinal distress,” including vomiting, she notes.
Dr. Fitch has also successfully used this Saxenda-to-Wegovy approach for some of her patients, but it hasn’t been easy.
“It’s more work and more prior authorizations. It’s harder and adds a layer of stress,” but, Dr. Fitch adds, “people are willing to work on it because the weight loss is worth it.”
The liraglutide to semaglutide shuffle is “doable,” says Dr. Rizo, “but I’m looking forward to not having to do it and being able to just start Wegovy.”
The tirzepatide coupon program works ‘off label’ for obesity
Another workaround depends on the FDA approval in May for tirzepatide (Mounjaro) for type 2 diabetes. Tirzepatide is a related GLP-1 agonist that also adds a second incretin-like agonist activity that mimics the glucose-dependent insulinotropic polypeptide.
Soon after approval, Lilly, the company that markets tirzepatide, started a U.S. coupon program geared exclusively to people with commercial insurance. Within certain refill and dollar limits, the program lets patients buy tirzepatide at pharmacies at an out-of-pocket cost of $25 for a 4-week supply (tirzepatide is also dosed by weekly subcutaneous injections). The program will extend into 2023.
Novo Nordisk offered U.S. patients with commercial insurance a similar discount when Wegovy first hit the U.S. market in 2021, but the program closed down once the supply shortage began.
Despite tirzepatide’s current approval only for type 2 diabetes, Dr. Varney has been successfully prescribing it to patients without diabetes off-label for weight loss.
“The coupons still work even when tirzepatide is used off-label,” she notes. And while the drug’s rollout is still only a couple of months old, so far, it’s gone “beautifully” with no hints of supply issues, she says.
But a major drawback to relying on an introductory coupon program that makes these agents affordable to patients is their ability to maintain treatment once the discounts inevitably end.
“We try to only prescribe agents that patients can continue to access,” says Dr. Fitch, who has had some patients with commercial insurance start on Wegovy with coupon discounts only to later lose access.
Many commercial U.S. insurers do not cover obesity treatments, a decision often driven by the employers who sponsor the coverage, she notes.
Study results have documented that when people with obesity stop taking a GLP-1 agonist their lost weight rebounds, as in a study that tracked people who stopped taking semaglutide.
Dr. Fitch has had success prescribing tirzepatide to patients with obesity but without diabetes who have certain types of Medicare drug coverage policies, which often do not deny off-label drug coverage. That approach works until patients reach the “donut hole” in their drug coverage and are faced with a certain level of out-of-pocket costs that can balloon to several thousand dollars.
Even more workarounds
Other approaches patients have used to acquire Wegovy include purchasing it in other countries, such as Canada or Brazil, says Dr. Fitch. But prices outside the United States, while substantially lower, can still be a barrier for many patients, notes Dr. Oshman.
Semaglutide in Canada goes for about $300 for a 4-week supply, roughly a quarter the U.S. price, she says, but is “still too high for many of my patients.”
Intense patient demand sometimes bordering on desperation has prompted some to seek semaglutide from private compounding pharmacies, a step clinicians regard as downright dangerous.
“Semaglutide from compounding pharmacies is not known to be safe. We feel strongly that it’s not something that people should do,” says Dr. Fitch.
“Compounding pharmacies have no FDA regulation. People don’t know what they’re getting. It’s dangerous,” agrees Dr. Varney. Physicians who refer people for privately compounded semaglutide “are taking advantage of desperate people,” she adds.
Although it seems likely that Novo Nordisk will soon sort out the supply problems and Wegovy will once again become more widely available, some of the issues patients have had with access to the weight loss medication stem from more systemic issues in the United States health insurance landscape: an unwillingness by payers to cover the costs of weight loss medications, a shortcoming that also exists for Medicare and Medicaid.
“We need to make obesity treatment a standard benefit, and not something that can be carved out,” says Dr. Fitch. People with obesity “deserve access to effective treatments for their disease,” she declares.
Dr. Oshman, Dr. Varney, and Dr. Rizo have reported no relevant financial relationships. Dr. Fitch has reported being an advisor to Jenny Craig.
A version of this article first appeared on Medscape.com.
The glucagon-like peptide-1 (GLP-1) agonist semaglutide formulated for treating obesity (Wegovy) had a roaring takeoff a little more than a year ago, with surging patient demand after the U.S. Food and Drug Administration approved it in June 2021. But starting doses of the Wegovy form of semaglutide went missing in action starting late 2021 and continue to date, frustrating patients and their health care providers.
The arrival of Wegovy last year was hailed by obesity medicine specialists and others as a “game changer” for treating people with obesity because of semaglutide’s proven safety and efficacy at the subcutaneous dose of 2.4 mg delivered once a week to produce at least 15% weight loss in half the people who received it, as documented last year in results from one of the drug’s pivotal clinical trials.
But during the months following semaglutide’s approval for treating obesity (it also received an FDA marketing nod in late 2017 as Ozempic for treating type 2 diabetes), a worldwide shortage of Wegovy, including in the United States, emerged.
A manufacturing glitch shut down the primary location for production of U.S.-bound Wegovy injector pens for several months starting in late 2021, according to a December report from Novo Nordisk, the company that makes and markets the agent. (The Wegovy production issue appears to have had a very modest impact, especially in U.S. pharmacies, on the supply of semaglutide formulated as Ozempic, also marketed by Novo Nordisk, although Wegovy supply and demand have dramatically limited Ozempic availability in Australia.)
‘Unprecedented demand’ for Wegovy derailed when plant went offline
The supply side for Wegovy became so hopelessly broken that just months after U.S. sales began and immediately skyrocketed, Novo Nordisk made the remarkable decision to pull starting doses of Wegovy from the market to make it much harder to initiate patients (semaglutide and other GLP-1 agonists require gradual dose ramp-up to avoid gastrointestinal side effects), and the company publicly implored clinicians to not start new patients on the agent, which is where the status remains as of early August 2022.
Novo Nordisk’s financial report for the second quarter of 2022, released on Aug. 3, said the company “expects to make all Wegovy dose strengths available in the United States towards the end of 2022.”
A Dear Health Care Provider letter that Novo Nordisk posted on its U.S. Wegovy website last spring cited “unprecedented demand” that exceeded every prior product launch in the company’s history. It forced Novo Nordisk to pull the plug on all U.S. promotion of Wegovy and compelled the company to ask U.S. clinicians to halt new patient starts.
“I stopped offering Wegovy to new patients” since about the beginning of 2022, says Lauren D. Oshman, MD, a family and obesity medicine specialist at the University of Michigan, Ann Arbor. “It’s very frustrating to not have patients [with obesity] receive the optimal treatment available.” Although she adds that she tries to match obesity treatments to each patient’s clinical needs, and a GLP-1 agonist is not the first choice for every person with obesity.
“It was a disastrous rollout,” says Catherine W. Varney, DO, a family and obesity medicine specialist at the University of Virginia, Charlottesville. “It’s frustrating to know that the treatment is there but not being able to use it,” she said in an interview.
“I had about 800 patients on Wegovy” when the supply dropped earlier this year, and “I couldn’t handle the volume of messages that I got from patients,” recalls Angela Fitch, MD, associate director of the Massachusetts General Hospital Weight Center, Boston. “It was painful,” she said in an interview.
“Frustrating and chaotic,” is the description from Ivania M. Rizo, MD, director of obesity medicine at Boston Medical Center.
The liraglutide/Saxenda workaround
The upshot is that people with obesity and their health care providers have been busy devising workarounds to try to meet the intense demand for this drug-assisted approach to appetite control and weight loss. Their tactics run a wide gamut based on the crazy-quilt diversity of health insurance coverage across America.
Because the bottleneck for starting Wegovy resulted from unavailable starting doses (dosing starts at 0.25 mg delivered subcutaneously once a week, eventually ramping up to a maximum of 2.4 mg weekly), one option was to start patients on a different GLP-1 agonist, such as liraglutide (Saxenda, approved for obesity).
Starting a patient on liraglutide involves the same sort of up-titration and acclimation to a GLP-1 agonist that semaglutide requires, and transition between these agents seems feasible for at least some. It also means daily injections of liraglutide rather than the weekly schedule for semaglutide, although some patients prefer maintaining a daily dosing schedule. Another limitation of liraglutide is that evidence shows it is not nearly as effective for weight loss as semaglutide.
Results from the head-to-head STEP 8 trial, published in JAMA, showed an average weight loss from baseline of about 16% with semaglutide and about 6% with liraglutide (and about 2% with placebo).
A ‘reasonable’ evidence base, but more work
Changing from Saxenda to Wegovy, or from Wegovy to Saxenda, “would be reasonably evidence-based medicine,” said Dr. Oshman in an interview. She has managed a Wegovy-to-Saxenda switch for a “handful” of patients to deal with Wegovy shortages, but she has not yet moved anyone to Wegovy after a Saxenda initiation.
“No prospective study has looked at this transition,” but dose equivalence tables exist based on expert opinion, noted Dr. Oshman, as in this 2020 report.
Dr. Varney has several patients on the Saxenda-to-Wegovy track. She up-titrates patients on Saxenda to the maximum daily dose of 3.0 mg and then switches them to the 1.7 mg weekly dose of Wegovy, one of the “destination” Wegovy doses that has remained generally available during the shortage. But Dr. Varney’s experience is that only half of her patients made the changeover smoothly, with the others having “severe gastrointestinal distress,” including vomiting, she notes.
Dr. Fitch has also successfully used this Saxenda-to-Wegovy approach for some of her patients, but it hasn’t been easy.
“It’s more work and more prior authorizations. It’s harder and adds a layer of stress,” but, Dr. Fitch adds, “people are willing to work on it because the weight loss is worth it.”
The liraglutide to semaglutide shuffle is “doable,” says Dr. Rizo, “but I’m looking forward to not having to do it and being able to just start Wegovy.”
The tirzepatide coupon program works ‘off label’ for obesity
Another workaround depends on the FDA approval in May for tirzepatide (Mounjaro) for type 2 diabetes. Tirzepatide is a related GLP-1 agonist that also adds a second incretin-like agonist activity that mimics the glucose-dependent insulinotropic polypeptide.
Soon after approval, Lilly, the company that markets tirzepatide, started a U.S. coupon program geared exclusively to people with commercial insurance. Within certain refill and dollar limits, the program lets patients buy tirzepatide at pharmacies at an out-of-pocket cost of $25 for a 4-week supply (tirzepatide is also dosed by weekly subcutaneous injections). The program will extend into 2023.
Novo Nordisk offered U.S. patients with commercial insurance a similar discount when Wegovy first hit the U.S. market in 2021, but the program closed down once the supply shortage began.
Despite tirzepatide’s current approval only for type 2 diabetes, Dr. Varney has been successfully prescribing it to patients without diabetes off-label for weight loss.
“The coupons still work even when tirzepatide is used off-label,” she notes. And while the drug’s rollout is still only a couple of months old, so far, it’s gone “beautifully” with no hints of supply issues, she says.
But a major drawback to relying on an introductory coupon program that makes these agents affordable to patients is their ability to maintain treatment once the discounts inevitably end.
“We try to only prescribe agents that patients can continue to access,” says Dr. Fitch, who has had some patients with commercial insurance start on Wegovy with coupon discounts only to later lose access.
Many commercial U.S. insurers do not cover obesity treatments, a decision often driven by the employers who sponsor the coverage, she notes.
Study results have documented that when people with obesity stop taking a GLP-1 agonist their lost weight rebounds, as in a study that tracked people who stopped taking semaglutide.
Dr. Fitch has had success prescribing tirzepatide to patients with obesity but without diabetes who have certain types of Medicare drug coverage policies, which often do not deny off-label drug coverage. That approach works until patients reach the “donut hole” in their drug coverage and are faced with a certain level of out-of-pocket costs that can balloon to several thousand dollars.
Even more workarounds
Other approaches patients have used to acquire Wegovy include purchasing it in other countries, such as Canada or Brazil, says Dr. Fitch. But prices outside the United States, while substantially lower, can still be a barrier for many patients, notes Dr. Oshman.
Semaglutide in Canada goes for about $300 for a 4-week supply, roughly a quarter the U.S. price, she says, but is “still too high for many of my patients.”
Intense patient demand sometimes bordering on desperation has prompted some to seek semaglutide from private compounding pharmacies, a step clinicians regard as downright dangerous.
“Semaglutide from compounding pharmacies is not known to be safe. We feel strongly that it’s not something that people should do,” says Dr. Fitch.
“Compounding pharmacies have no FDA regulation. People don’t know what they’re getting. It’s dangerous,” agrees Dr. Varney. Physicians who refer people for privately compounded semaglutide “are taking advantage of desperate people,” she adds.
Although it seems likely that Novo Nordisk will soon sort out the supply problems and Wegovy will once again become more widely available, some of the issues patients have had with access to the weight loss medication stem from more systemic issues in the United States health insurance landscape: an unwillingness by payers to cover the costs of weight loss medications, a shortcoming that also exists for Medicare and Medicaid.
“We need to make obesity treatment a standard benefit, and not something that can be carved out,” says Dr. Fitch. People with obesity “deserve access to effective treatments for their disease,” she declares.
Dr. Oshman, Dr. Varney, and Dr. Rizo have reported no relevant financial relationships. Dr. Fitch has reported being an advisor to Jenny Craig.
A version of this article first appeared on Medscape.com.
FDA approves Enhertu (trastuzumab deruxtecan) for HER2 lung cancer
Patients with lung cancer now have another treatment option: If their tumors are found to carry HER2 mutations, they can now be treated with trastuzumab deruxtecan (Enhertu), a drug that specifically targets that defect.
This product is already approved for used in HER2-positive breast cancer and gastric cancer. 
The FDA also approved companion diagnostic tests to detect HER2 mutations: Life Technologies Corporation’s Oncomine Dx Target Test for use in lung tissue and Guardant Health’s Guardant360 CDx for use on plasma samples. The agency notes that if no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Specifically, the new indication is used in patients with unresectable or metastatic non–small cell lung cancer (NSCLC) whose tumors have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have already received a prior systemic therapy.
About 3% of nonsquamous NSCLC tumors carry mutations in the HER2 gene, and they are associated with female sex, never-smokers, and a poor prognosis.
“HER2 mutant non–small cell lung cancer is an aggressive form of disease, which commonly affects young patients who have faced limited treatment options and a poor prognosis to date,” said Dave Fredrickson, executive vice-president of the oncology business unit at AstraZeneca.
The new approval “provides these patients with the opportunity to benefit from a targeted therapy and highlights the importance of testing for predictive markers, including HER2 in lung cancer, at the time of diagnosis to ensure patients receive the most appropriate treatment for their specific disease,” he commented in a company press release.
This is an accelerated approval, based on overall response rate data from the DESTINY-Lung02 phase 2 trial, the company noted. An interim efficacy analysis in a prespecified patient cohort showed that trastuzumab deruxtecan (at 5.4 mg/kg) demonstrated a confirmed overall response rate of 57.7% (n = 52; 95% confidence interval, 43.2%-71.3%) in patients with HER2-mutant unresectable or metastatic nonsquamous NSCLC who had received one prior systemic therapy as assessed by blinded independent central review. Complete responses were seen in 1.9% of patients (n = 1) and partial responses in 55.8% of patients (n = 29), with a median duration of response of 8.7 months (95% CI, 7.1-NE).
The FDA noted that for the 52 patients in the primary efficacy population of the DESTINY-Lung02 trial, the median age was 58 years (range, 30-78 years); 69% were female; and 79% were Asian, 12% were White, and 10% were of other races.
Clinical data welcomed by experts
Clinical data are already available from the DESTINY-Lung 01 trial, and the results were welcomed enthusiastically by experts when they were published in the New England Journal of Medicine earlier this year.
“These results establish the new standard of care for patients with NSCLC harboring HER2 mutations,” Antonio Passaro, MD, PhD, from the European Institute of Oncology IRCCS, Milan, and Solange Peters, MD, PhD, from Lausanne (Switzerland) University Hospital, wrote in an accompanying editorial.
This trial involved 91 patients, all treated with trastuzumab deruxtecan (at 6.4 mg/kg of body weight every 3 weeks). The median duration of treatment was 6.9 months, and the median follow-up was 13.1 months.
The results showed a 55% centrally confirmed objective response, and median duration of response was 9.3 months.
In addition, the investigators reported a median progression-free survival of 8.2 months and a median overall survival of almost 18 months, both of which they described as “encouraging” in this patient population.
However, the results also highlighted a problem with the drug in this patient population. Notably, 26% of patients experienced interstitial lung disease, which resulted in death in two patients. The drug was also withdrawn in 16 patients and interrupted in 8 patients because of this adverse event.
Editorialists Dr. Passaro and Dr. Peters described this finding as “a concern” and note that “the incidence of interstitial lung disease is significantly higher among patients with lung cancer than among those with breast or gastric cancers, which may indicate a role of smoking-related damage.”
They also highlighted the need for an “investigation of the clinical efficacy of a reduced dose of trastuzumab deruxtecan,” and so the dose was reduced for the DESTINY-Lung02 trial.
A version of this article first appeared on Medscape.com.
Patients with lung cancer now have another treatment option: If their tumors are found to carry HER2 mutations, they can now be treated with trastuzumab deruxtecan (Enhertu), a drug that specifically targets that defect.
This product is already approved for used in HER2-positive breast cancer and gastric cancer.
The FDA also approved companion diagnostic tests to detect HER2 mutations: Life Technologies Corporation’s Oncomine Dx Target Test for use in lung tissue and Guardant Health’s Guardant360 CDx for use on plasma samples. The agency notes that if no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Specifically, the new indication is used in patients with unresectable or metastatic non–small cell lung cancer (NSCLC) whose tumors have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have already received a prior systemic therapy.
About 3% of nonsquamous NSCLC tumors carry mutations in the HER2 gene, and they are associated with female sex, never-smokers, and a poor prognosis.
“HER2 mutant non–small cell lung cancer is an aggressive form of disease, which commonly affects young patients who have faced limited treatment options and a poor prognosis to date,” said Dave Fredrickson, executive vice-president of the oncology business unit at AstraZeneca.
The new approval “provides these patients with the opportunity to benefit from a targeted therapy and highlights the importance of testing for predictive markers, including HER2 in lung cancer, at the time of diagnosis to ensure patients receive the most appropriate treatment for their specific disease,” he commented in a company press release.
This is an accelerated approval, based on overall response rate data from the DESTINY-Lung02 phase 2 trial, the company noted. An interim efficacy analysis in a prespecified patient cohort showed that trastuzumab deruxtecan (at 5.4 mg/kg) demonstrated a confirmed overall response rate of 57.7% (n = 52; 95% confidence interval, 43.2%-71.3%) in patients with HER2-mutant unresectable or metastatic nonsquamous NSCLC who had received one prior systemic therapy as assessed by blinded independent central review. Complete responses were seen in 1.9% of patients (n = 1) and partial responses in 55.8% of patients (n = 29), with a median duration of response of 8.7 months (95% CI, 7.1-NE).
The FDA noted that for the 52 patients in the primary efficacy population of the DESTINY-Lung02 trial, the median age was 58 years (range, 30-78 years); 69% were female; and 79% were Asian, 12% were White, and 10% were of other races.
Clinical data welcomed by experts
Clinical data are already available from the DESTINY-Lung 01 trial, and the results were welcomed enthusiastically by experts when they were published in the New England Journal of Medicine earlier this year.
“These results establish the new standard of care for patients with NSCLC harboring HER2 mutations,” Antonio Passaro, MD, PhD, from the European Institute of Oncology IRCCS, Milan, and Solange Peters, MD, PhD, from Lausanne (Switzerland) University Hospital, wrote in an accompanying editorial.
This trial involved 91 patients, all treated with trastuzumab deruxtecan (at 6.4 mg/kg of body weight every 3 weeks). The median duration of treatment was 6.9 months, and the median follow-up was 13.1 months.
The results showed a 55% centrally confirmed objective response, and median duration of response was 9.3 months.
In addition, the investigators reported a median progression-free survival of 8.2 months and a median overall survival of almost 18 months, both of which they described as “encouraging” in this patient population.
However, the results also highlighted a problem with the drug in this patient population. Notably, 26% of patients experienced interstitial lung disease, which resulted in death in two patients. The drug was also withdrawn in 16 patients and interrupted in 8 patients because of this adverse event.
Editorialists Dr. Passaro and Dr. Peters described this finding as “a concern” and note that “the incidence of interstitial lung disease is significantly higher among patients with lung cancer than among those with breast or gastric cancers, which may indicate a role of smoking-related damage.”
They also highlighted the need for an “investigation of the clinical efficacy of a reduced dose of trastuzumab deruxtecan,” and so the dose was reduced for the DESTINY-Lung02 trial.
A version of this article first appeared on Medscape.com.
Patients with lung cancer now have another treatment option: If their tumors are found to carry HER2 mutations, they can now be treated with trastuzumab deruxtecan (Enhertu), a drug that specifically targets that defect.
This product is already approved for used in HER2-positive breast cancer and gastric cancer.
The FDA also approved companion diagnostic tests to detect HER2 mutations: Life Technologies Corporation’s Oncomine Dx Target Test for use in lung tissue and Guardant Health’s Guardant360 CDx for use on plasma samples. The agency notes that if no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Specifically, the new indication is used in patients with unresectable or metastatic non–small cell lung cancer (NSCLC) whose tumors have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have already received a prior systemic therapy.
About 3% of nonsquamous NSCLC tumors carry mutations in the HER2 gene, and they are associated with female sex, never-smokers, and a poor prognosis.
“HER2 mutant non–small cell lung cancer is an aggressive form of disease, which commonly affects young patients who have faced limited treatment options and a poor prognosis to date,” said Dave Fredrickson, executive vice-president of the oncology business unit at AstraZeneca.
The new approval “provides these patients with the opportunity to benefit from a targeted therapy and highlights the importance of testing for predictive markers, including HER2 in lung cancer, at the time of diagnosis to ensure patients receive the most appropriate treatment for their specific disease,” he commented in a company press release.
This is an accelerated approval, based on overall response rate data from the DESTINY-Lung02 phase 2 trial, the company noted. An interim efficacy analysis in a prespecified patient cohort showed that trastuzumab deruxtecan (at 5.4 mg/kg) demonstrated a confirmed overall response rate of 57.7% (n = 52; 95% confidence interval, 43.2%-71.3%) in patients with HER2-mutant unresectable or metastatic nonsquamous NSCLC who had received one prior systemic therapy as assessed by blinded independent central review. Complete responses were seen in 1.9% of patients (n = 1) and partial responses in 55.8% of patients (n = 29), with a median duration of response of 8.7 months (95% CI, 7.1-NE).
The FDA noted that for the 52 patients in the primary efficacy population of the DESTINY-Lung02 trial, the median age was 58 years (range, 30-78 years); 69% were female; and 79% were Asian, 12% were White, and 10% were of other races.
Clinical data welcomed by experts
Clinical data are already available from the DESTINY-Lung 01 trial, and the results were welcomed enthusiastically by experts when they were published in the New England Journal of Medicine earlier this year.
“These results establish the new standard of care for patients with NSCLC harboring HER2 mutations,” Antonio Passaro, MD, PhD, from the European Institute of Oncology IRCCS, Milan, and Solange Peters, MD, PhD, from Lausanne (Switzerland) University Hospital, wrote in an accompanying editorial.
This trial involved 91 patients, all treated with trastuzumab deruxtecan (at 6.4 mg/kg of body weight every 3 weeks). The median duration of treatment was 6.9 months, and the median follow-up was 13.1 months.
The results showed a 55% centrally confirmed objective response, and median duration of response was 9.3 months.
In addition, the investigators reported a median progression-free survival of 8.2 months and a median overall survival of almost 18 months, both of which they described as “encouraging” in this patient population.
However, the results also highlighted a problem with the drug in this patient population. Notably, 26% of patients experienced interstitial lung disease, which resulted in death in two patients. The drug was also withdrawn in 16 patients and interrupted in 8 patients because of this adverse event.
Editorialists Dr. Passaro and Dr. Peters described this finding as “a concern” and note that “the incidence of interstitial lung disease is significantly higher among patients with lung cancer than among those with breast or gastric cancers, which may indicate a role of smoking-related damage.”
They also highlighted the need for an “investigation of the clinical efficacy of a reduced dose of trastuzumab deruxtecan,” and so the dose was reduced for the DESTINY-Lung02 trial.
A version of this article first appeared on Medscape.com.
Value of a Pharmacy-Adjudicated Community Care Prior Authorization Drug Request Service
Veterans’ access to medical care was expanded outside of US Department of Veterans Affairs (VA) facilities with the inception of the 2014 Veterans Access, Choice, and Accountability Act (Choice Act).1 This legislation aimed to remove barriers some veterans were experiencing, specifically access to health care. In subsequent years, approximately 17% of veterans receiving care from the VA did so under the Choice Act.2 The Choice Act positively impacted medical care access for veterans but presented new challenges for VA pharmacies processing community care (CC) prescriptions, including limited access to outside health records, lack of interface between CC prescribers and the VA order entry system, and limited awareness of the VA national formulary.3,4 These factors made it difficult for VA pharmacies to assess prescriptions for clinical appropriateness, evaluate patient safety parameters, and manage expenditures.
In 2019, the Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act, which expanded CC support and better defined which veterans are able to receive care outside the VA, updated the Choice Act.4,5 However, VA pharmacies faced challenges in managing pharmacy drug costs and ensuring clinical appropriateness of prescription drug therapy. As a result, VA pharmacy departments have adjusted how they allocate workload, time, and funds.5
Pharmacists improve clinical outcomes and reduce health care costs by decreasing medication errors, unnecessary prescribing, and adverse drug events.6-12 Pharmacist-driven formulary management through evaluation of prior authorization drug requests (PADRs) has shown economic value.13,14 VA pharmacy review of community care PADRs is important because outside health care professionals (HCPs) might not be familiar with the VA formulary. This could lead to high volume of PADRs that do not meet criteria and could result in increased potential for medication misuse, adverse drug events, medication errors, and cost to the health system. It is imperative that CC orders are evaluated as critically as traditional orders.
The value of a centralized CC pharmacy team has not been assessed in the literature. The primary objective of this study was to assess the direct cost savings achieved through a centralized CC PADR process. Secondary objectives were to characterize the CC PADRs submitted to the site, including approval rate, reason for nonapproval, which medications were requested and by whom, and to compare CC prescriptions with other high-complexity (1a) VA facilities.
Community Care Pharmacy
VA health systems are stratified according to complexity, which reflects size, patient population, and services offered. This study was conducted at the Durham Veterans Affairs Health Care System (DVAHCS), North Carolina, a high-complexity, 251-bed, tertiary care referral, teaching, and research system. DVAHCS provides general and specialty medical, surgical, inpatient psychiatric, and ambulatory services, and serves as a major referral center.
DVAHCS created a centralized pharmacy team for processing CC prescriptions and managing customer service. This team’s goal is to increase CC prescription processing efficiency and transparency, ensure accountability of the health care team, and promote veteran-centric customer service. The pharmacy team includes a pharmacist program manager and a dedicated CC pharmacist with administrative support from a health benefits assistant and 4 pharmacy technicians. The CC pharmacy team assesses every new prescription to ensure the veteran is authorized to receive care in the community. Once eligibility is verified, a pharmacy technician or pharmacist evaluates the prescription to ensure it contains all required information, then contacts the prescriber for any missing data. If clinically appropriate, the pharmacist processes the prescription.
In 2020, the CC pharmacy team implemented a new process for reviewing and documenting CC prescriptions that require a PADR. The closed national VA formulary is set up so that all nonformulary medications and some formulary medications, including those that are restricted because of safety and/or cost, require a PADR.15 After a CC pharmacy technician confirms a veteran’s eligibility, the technician assesses whether the requested medication requires submitting a PADR to the VA internal electronic health record. The PADR is then adjudicated by a formulary management pharmacist, CC program manager, or CC pharmacist who reviews health records to determine whether the CC prescription meets VA medication use policy requirements.
If additional information is needed or an alternate medication is suggested, the pharmacist comments back on the PADR and a CC pharmacy technician contacts the prescriber. The PADR is canceled administratively then resubmitted once all information is obtained. While waiting for a response from the prescriber, the CC pharmacy technician contacts that veteran to give an update on the prescription status, as appropriate. Once there is sufficient information to adjudicate the PADR, the outcome is documented, and if approved, the order is processed.
Methods
The DVAHCS Institutional Review Board approved this retrospective review of CC PADRs submitted from June 1, 2020, through November 30, 2020. CC PADRs were excluded if they were duplicates or were reactivated administratively but had an initial submission date before the study period. Local data were collected for nonapproved CC PADRs including drug requested, dosage and directions, medication specialty, alternative drug recommended, drug acquisition cost, PADR submission date, PADR completion date, PADR nonapproval rationale, and documented time spent per PADR. Additional data was obtained for CC prescriptions at all 42 high-complexity VA facilities from the VA national CC prescription database for the study time interval and included total PADRs, PADR approval status, total CC prescription cost, and total CC fills.
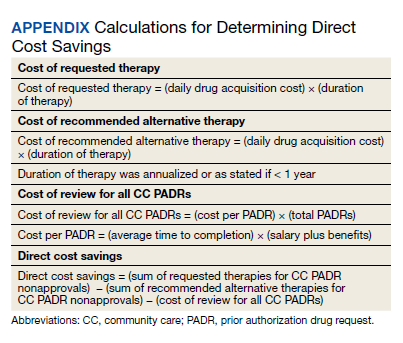
Direct cost savings were calculated by assessing the cost of requested therapy that was not approved minus the cost of recommended therapy and cost to review all PADRs, as described by Britt and colleagues.13 The cost of the requested and recommended therapy was calculated based on VA drug acquisition cost at time of data collection and multiplied by the expected duration of therapy up to 1 year. For each CC prescription, duration of therapy was based on the duration limit in the prescription or annualized if no duration limit was documented. Cost of PADR review was calculated based on the total time pharmacists and pharmacy technicians documented for each step of the review process for a representative sample of 100 nonapproved PADRs and then multiplied by the salary plus benefits of an entry-level pharmacist and pharmacy technician.16 The eAppendix describes specific equations used for determining direct cost savings. Descriptive statistics were used to evaluate study results.
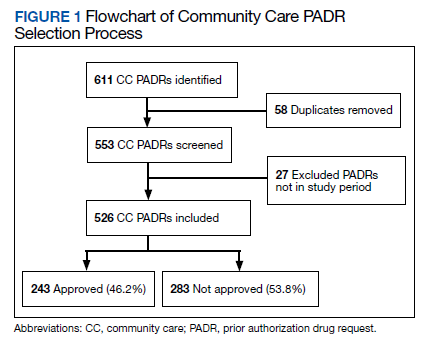
Results
During the 6-month study period, 611 CC PADRs were submitted to the pharmacy and 526 met inclusion criteria (Figure 1). Of those, 243 (46.2%) were approved and 283 (53.8%) were not approved. The cost of requested therapies for nonapproved CC PADRs totaled $584,565.48 and the cost of all recommended therapies was $57,473.59. The mean time per CC PADR was 24 minutes; 16 minutes for pharmacists and 8 minutes for pharmacy technicians. Given an hourly wage (plus benefits) of $67.25 for a pharmacist and $25.53 for a pharmacy technician, the total cost of review per CC PADR was $21.33. After subtracting the costs of all recommended therapies and review of all included CC PADRs, the process generated $515,872.31 in direct cost savings. After factoring in administrative lag time, such as HCP communication, an average of 8 calendar days was needed to complete a nonapproved PADR.
The most common rationale for PADR nonapproval was that the formulary alternative was not exhausted. Ondansetron orally disintegrating tablets was the most commonly nonapproved medication and azelastine was the most commonly approved medication. Dulaglutide was the most expensive nonapproved and tafamidis was the most expensive approved PADR (Table 1). Gastroenterology, endocrinology, and neurology were the top specialties for nonapproved PADRs while neurology, pulmonology, and endocrinology were the top specialties for approved PADRs (Table 2).
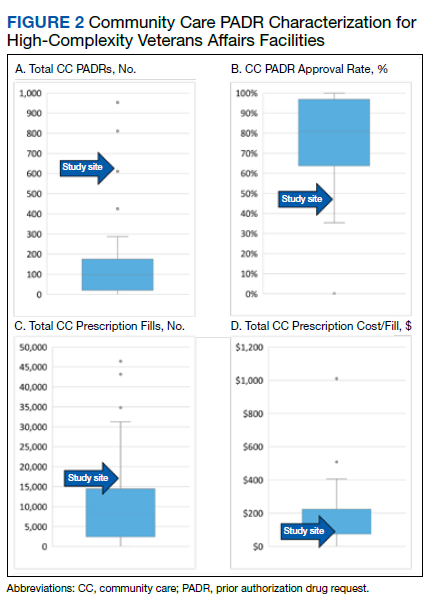
Several high-complexity VA facilities had no reported data; we used the median for the analysis to account for these outliers (Figure 2). The median (IQR) adjudicated CC PADRs for all facilities was 97 (20-175), median (IQR) CC PADR approval rate was 80.9% (63.7%-96.8%), median (IQR) total CC prescriptions was 8440 (2464-14,466), and median (IQR) cost per fill was $136.05 ($76.27-$221.28).
Discussion
This study demonstrated direct cost savings of $515,872.31 over 6 months with theadjudication of CC PADRs by a centralized CC pharmacy team. This could result in > $1,000,000 of cost savings per fiscal year.
The CC PADRs observed at DVAHCS had a 46.2% approval rate; almost one-half the approval rate of 84.1% of all PADRs submitted to the study site by VA HCPs captured by Britt and colleagues.13 Results from this study showed that coordination of care for nonapproved CC PADRs between the VA pharmacy and non-VA prescriber took an average of 8 calendar days. The noted CC PADR approval rate and administrative burden might be because of lack of familiarity of non-VA providers regarding the VA national formulary. The National VA Pharmacy Benefits Management determines the formulary using cost-effectiveness criteria that considers the medical literature and VA-specific contract pricing and prepares extensive guidance for restricted medications via relevant criteria for use.15 HCPs outside the VA might not know this information is available online. Because gastroenterology, endocrinology, and neurology specialty medications were among the most frequently nonapproved PADRs, VA formulary education could begin with CC HCPs in these practice areas.
This study showed that the CC PADR process was not solely driven by cost, but also included patient safety. Nonapproval rationale for some requests included submission without an indication, submission by a prescriber that did not have the authority to prescribe a type of medication, or contraindication based on patient-specific factors.
Compared with other VA high-complexity facilities, DVAHCS was among the top health care systems for total volume of CC prescriptions (n = 16,096) and among the lowest for cost/fill ($75.74). Similarly, DVAHCS was among the top sites for total adjudicated CC PADRs within the 6-month study period (n = 611) and the lowest approval rate (44.2%). This study shows that despite high volumes of overall CC prescriptions and CC PADRs, it is possible to maintain a low overall CC prescription cost/fill compared with other similarly complex sites across the country. Wide variance in reported results exists across high-complexity VA facilities because some sites had low to no CC fills and/or CC PADRs. This is likely a result of administrative differences when handling CC prescriptions and presents an opportunity to standardize this process nationally.
Limitations
CC PADRs were assessed during the COVID-19 pandemic, which might have resulted in lower-than-normal CC prescription and PADR volumes, therefore underestimating the potential for direct cost savings. Entry-level salary was used to demonstrate cost savings potential from the perspective of a newly hired CC team; however, the cost savings might have been less if the actual salaries of site personnel were higher. National contract pricing data were gathered at the time of data collection and might have been different than at the time of PADR submission. Chronic medication prescriptions were annualized, which could overestimate cost savings if the medication was discontinued or changed to an alternative therapy within that time period.
The study’s exclusion criteria could only be applied locally and did not include data received from the VA CC prescription database. This can be seen by the discrepancy in CC PADR approval rates from the local and national data (46.2% vs 44.2%, respectively) and CC PADR volume. High-complexity VA facility data were captured without assessing the CC prescription process at each site. As a result, definitive conclusions cannot be made regarding the impact of a centralized CC pharmacy team compared with other facilities.
Conclusions
Adjudication of CC PADRs by a centralized CC pharmacy team over a 6-month period provided > $500,000 in direct cost savings to a VA health care system. Considering the CC PADR approval rate seen in this study, the VA could allocate resources to educate CC providers about the VA formulary to increase the PADR approval rate and reduce administrative burden for VA pharmacies and prescribers. Future research should evaluate CC prescription handling practices at other VA facilities to compare the effectiveness among varying approaches and develop recommendations for a nationally standardized process.
Acknowledgments
Concept and design (AJJ, JNB, RBB, LAM, MD, MGH); acquisition of data (AJJ, MGH); analysis and interpretation of data (AJJ, JNB, RBB, LAM, MD, MGH); drafting of the manuscript (AJJ); critical revision of the manuscript for important intellectual content (AJJ, JNB, RBB, LAM, MD, MGH); statistical analysis (AJJ); administrative, technical, or logistic support (LAM, MGH); and supervision (MGH).
1. Gellad WF, Cunningham FE, Good CB, et al. Pharmacy use in the first year of the Veterans Choice Program: a mixed-methods evaluation. Med Care. 2017(7 suppl 1);55:S26. doi:10.1097/MLR.0000000000000661
2. Mattocks KM, Yehia B. Evaluating the veterans choice program: lessons for developing a high-performing integrated network. Med Care. 2017(7 suppl 1);55:S1-S3. doi:10.1097/MLR.0000000000000743.
3. Mattocks KM, Mengeling M, Sadler A, Baldor R, Bastian L. The Veterans Choice Act: a qualitative examination of rapid policy implementation in the Department of Veterans Affairs. Med Care. 2017;55(7 suppl 1):S71-S75. doi:10.1097/MLR.0000000000000667
4. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.08: VHA formulary management process. November 2, 2016. Accessed June 9, 2022. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3291
5. Massarweh NN, Itani KMF, Morris MS. The VA MISSION act and the future of veterans’ access to quality health care. JAMA. 2020;324:343-344. doi:10.1001/jama.2020.4505
6. Jourdan JP, Muzard A, Goyer I, et al. Impact of pharmacist interventions on clinical outcome and cost avoidance in a university teaching hospital. Int J Clin Pharm. 2018;40(6):1474-1481. doi:10.1007/s11096-018-0733-6
7. Lee AJ, Boro MS, Knapp KK, Meier JL, Korman NE. Clinical and economic outcomes of pharmacist recommendations in a Veterans Affairs medical center. Am J Health Syst Pharm. 2002;59(21):2070-2077. doi:10.1093/ajhp/59.21.2070
8. Dalton K, Byrne S. Role of the pharmacist in reducing healthcare costs: current insights. Integr Pharm Res Pract. 2017;6:37-46. doi:10.2147/IPRP.S108047
9. De Rijdt T, Willems L, Simoens S. Economic effects of clinical pharmacy interventions: a literature review. Am J Health Syst Pharm. 2008;65(12):1161-1172. doi:10.2146/ajhp070506
10. Perez A, Doloresco F, Hoffman J, et al. Economic evaluation of clinical pharmacy services: 2001-2005. Pharmacotherapy. 2009;29(1):128. doi:10.1592/phco.29.1.128
11. Nesbit TW, Shermock KM, Bobek MB, et al. Implementation and pharmacoeconomic analysis of a clinical staff pharmacist practice model. Am J Health Syst Pharm. 2001;58(9):784-790. doi:10.1093/ajhp/58.9.784
12. Yang S, Britt RB, Hashem MG, Brown JN. Outcomes of pharmacy-led hepatitis C direct-acting antiviral utilization management at a Veterans Affairs medical center. J Manag Care Pharm. 2017;23(3):364-369. doi:10.18553/jmcp.2017.23.3.364
13. Britt RB, Hashem MG, Bryan WE III, Kothapalli R, Brown JN. Economic outcomes associated with a pharmacist-adjudicated formulary consult service in a Veterans Affairs medical center. J Manag Care Pharm. 2016;22(9):1051-1061. doi:10.18553/jmcp.2016.22.9.1051
14. Jacob S, Britt RB, Bryan WE, Hashem MG, Hale JC, Brown JN. Economic outcomes associated with safety interventions by a pharmacist-adjudicated prior authorization consult service. J Manag Care Pharm. 2019;25(3):411-416. doi:10.18553/jmcp.2019.25.3.411
15. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063. doi:10.18553/jmcp.2016.22.9.1058
16. US Department of Veterans Affairs, Office of the Chief Human Capital Officer. Title 38 Pay Schedules. Updated January 26, 2022. Accessed June 9, 2022. https://www.va.gov/ohrm/pay
Veterans’ access to medical care was expanded outside of US Department of Veterans Affairs (VA) facilities with the inception of the 2014 Veterans Access, Choice, and Accountability Act (Choice Act).1 This legislation aimed to remove barriers some veterans were experiencing, specifically access to health care. In subsequent years, approximately 17% of veterans receiving care from the VA did so under the Choice Act.2 The Choice Act positively impacted medical care access for veterans but presented new challenges for VA pharmacies processing community care (CC) prescriptions, including limited access to outside health records, lack of interface between CC prescribers and the VA order entry system, and limited awareness of the VA national formulary.3,4 These factors made it difficult for VA pharmacies to assess prescriptions for clinical appropriateness, evaluate patient safety parameters, and manage expenditures.
In 2019, the Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act, which expanded CC support and better defined which veterans are able to receive care outside the VA, updated the Choice Act.4,5 However, VA pharmacies faced challenges in managing pharmacy drug costs and ensuring clinical appropriateness of prescription drug therapy. As a result, VA pharmacy departments have adjusted how they allocate workload, time, and funds.5
Pharmacists improve clinical outcomes and reduce health care costs by decreasing medication errors, unnecessary prescribing, and adverse drug events.6-12 Pharmacist-driven formulary management through evaluation of prior authorization drug requests (PADRs) has shown economic value.13,14 VA pharmacy review of community care PADRs is important because outside health care professionals (HCPs) might not be familiar with the VA formulary. This could lead to high volume of PADRs that do not meet criteria and could result in increased potential for medication misuse, adverse drug events, medication errors, and cost to the health system. It is imperative that CC orders are evaluated as critically as traditional orders.
The value of a centralized CC pharmacy team has not been assessed in the literature. The primary objective of this study was to assess the direct cost savings achieved through a centralized CC PADR process. Secondary objectives were to characterize the CC PADRs submitted to the site, including approval rate, reason for nonapproval, which medications were requested and by whom, and to compare CC prescriptions with other high-complexity (1a) VA facilities.
Community Care Pharmacy
VA health systems are stratified according to complexity, which reflects size, patient population, and services offered. This study was conducted at the Durham Veterans Affairs Health Care System (DVAHCS), North Carolina, a high-complexity, 251-bed, tertiary care referral, teaching, and research system. DVAHCS provides general and specialty medical, surgical, inpatient psychiatric, and ambulatory services, and serves as a major referral center.
DVAHCS created a centralized pharmacy team for processing CC prescriptions and managing customer service. This team’s goal is to increase CC prescription processing efficiency and transparency, ensure accountability of the health care team, and promote veteran-centric customer service. The pharmacy team includes a pharmacist program manager and a dedicated CC pharmacist with administrative support from a health benefits assistant and 4 pharmacy technicians. The CC pharmacy team assesses every new prescription to ensure the veteran is authorized to receive care in the community. Once eligibility is verified, a pharmacy technician or pharmacist evaluates the prescription to ensure it contains all required information, then contacts the prescriber for any missing data. If clinically appropriate, the pharmacist processes the prescription.
In 2020, the CC pharmacy team implemented a new process for reviewing and documenting CC prescriptions that require a PADR. The closed national VA formulary is set up so that all nonformulary medications and some formulary medications, including those that are restricted because of safety and/or cost, require a PADR.15 After a CC pharmacy technician confirms a veteran’s eligibility, the technician assesses whether the requested medication requires submitting a PADR to the VA internal electronic health record. The PADR is then adjudicated by a formulary management pharmacist, CC program manager, or CC pharmacist who reviews health records to determine whether the CC prescription meets VA medication use policy requirements.
If additional information is needed or an alternate medication is suggested, the pharmacist comments back on the PADR and a CC pharmacy technician contacts the prescriber. The PADR is canceled administratively then resubmitted once all information is obtained. While waiting for a response from the prescriber, the CC pharmacy technician contacts that veteran to give an update on the prescription status, as appropriate. Once there is sufficient information to adjudicate the PADR, the outcome is documented, and if approved, the order is processed.
Methods
The DVAHCS Institutional Review Board approved this retrospective review of CC PADRs submitted from June 1, 2020, through November 30, 2020. CC PADRs were excluded if they were duplicates or were reactivated administratively but had an initial submission date before the study period. Local data were collected for nonapproved CC PADRs including drug requested, dosage and directions, medication specialty, alternative drug recommended, drug acquisition cost, PADR submission date, PADR completion date, PADR nonapproval rationale, and documented time spent per PADR. Additional data was obtained for CC prescriptions at all 42 high-complexity VA facilities from the VA national CC prescription database for the study time interval and included total PADRs, PADR approval status, total CC prescription cost, and total CC fills.
Direct cost savings were calculated by assessing the cost of requested therapy that was not approved minus the cost of recommended therapy and cost to review all PADRs, as described by Britt and colleagues.13 The cost of the requested and recommended therapy was calculated based on VA drug acquisition cost at time of data collection and multiplied by the expected duration of therapy up to 1 year. For each CC prescription, duration of therapy was based on the duration limit in the prescription or annualized if no duration limit was documented. Cost of PADR review was calculated based on the total time pharmacists and pharmacy technicians documented for each step of the review process for a representative sample of 100 nonapproved PADRs and then multiplied by the salary plus benefits of an entry-level pharmacist and pharmacy technician.16 The eAppendix describes specific equations used for determining direct cost savings. Descriptive statistics were used to evaluate study results.
Results
During the 6-month study period, 611 CC PADRs were submitted to the pharmacy and 526 met inclusion criteria (Figure 1). Of those, 243 (46.2%) were approved and 283 (53.8%) were not approved. The cost of requested therapies for nonapproved CC PADRs totaled $584,565.48 and the cost of all recommended therapies was $57,473.59. The mean time per CC PADR was 24 minutes; 16 minutes for pharmacists and 8 minutes for pharmacy technicians. Given an hourly wage (plus benefits) of $67.25 for a pharmacist and $25.53 for a pharmacy technician, the total cost of review per CC PADR was $21.33. After subtracting the costs of all recommended therapies and review of all included CC PADRs, the process generated $515,872.31 in direct cost savings. After factoring in administrative lag time, such as HCP communication, an average of 8 calendar days was needed to complete a nonapproved PADR.
The most common rationale for PADR nonapproval was that the formulary alternative was not exhausted. Ondansetron orally disintegrating tablets was the most commonly nonapproved medication and azelastine was the most commonly approved medication. Dulaglutide was the most expensive nonapproved and tafamidis was the most expensive approved PADR (Table 1). Gastroenterology, endocrinology, and neurology were the top specialties for nonapproved PADRs while neurology, pulmonology, and endocrinology were the top specialties for approved PADRs (Table 2).
Several high-complexity VA facilities had no reported data; we used the median for the analysis to account for these outliers (Figure 2). The median (IQR) adjudicated CC PADRs for all facilities was 97 (20-175), median (IQR) CC PADR approval rate was 80.9% (63.7%-96.8%), median (IQR) total CC prescriptions was 8440 (2464-14,466), and median (IQR) cost per fill was $136.05 ($76.27-$221.28).
Discussion
This study demonstrated direct cost savings of $515,872.31 over 6 months with theadjudication of CC PADRs by a centralized CC pharmacy team. This could result in > $1,000,000 of cost savings per fiscal year.
The CC PADRs observed at DVAHCS had a 46.2% approval rate; almost one-half the approval rate of 84.1% of all PADRs submitted to the study site by VA HCPs captured by Britt and colleagues.13 Results from this study showed that coordination of care for nonapproved CC PADRs between the VA pharmacy and non-VA prescriber took an average of 8 calendar days. The noted CC PADR approval rate and administrative burden might be because of lack of familiarity of non-VA providers regarding the VA national formulary. The National VA Pharmacy Benefits Management determines the formulary using cost-effectiveness criteria that considers the medical literature and VA-specific contract pricing and prepares extensive guidance for restricted medications via relevant criteria for use.15 HCPs outside the VA might not know this information is available online. Because gastroenterology, endocrinology, and neurology specialty medications were among the most frequently nonapproved PADRs, VA formulary education could begin with CC HCPs in these practice areas.
This study showed that the CC PADR process was not solely driven by cost, but also included patient safety. Nonapproval rationale for some requests included submission without an indication, submission by a prescriber that did not have the authority to prescribe a type of medication, or contraindication based on patient-specific factors.
Compared with other VA high-complexity facilities, DVAHCS was among the top health care systems for total volume of CC prescriptions (n = 16,096) and among the lowest for cost/fill ($75.74). Similarly, DVAHCS was among the top sites for total adjudicated CC PADRs within the 6-month study period (n = 611) and the lowest approval rate (44.2%). This study shows that despite high volumes of overall CC prescriptions and CC PADRs, it is possible to maintain a low overall CC prescription cost/fill compared with other similarly complex sites across the country. Wide variance in reported results exists across high-complexity VA facilities because some sites had low to no CC fills and/or CC PADRs. This is likely a result of administrative differences when handling CC prescriptions and presents an opportunity to standardize this process nationally.
Limitations
CC PADRs were assessed during the COVID-19 pandemic, which might have resulted in lower-than-normal CC prescription and PADR volumes, therefore underestimating the potential for direct cost savings. Entry-level salary was used to demonstrate cost savings potential from the perspective of a newly hired CC team; however, the cost savings might have been less if the actual salaries of site personnel were higher. National contract pricing data were gathered at the time of data collection and might have been different than at the time of PADR submission. Chronic medication prescriptions were annualized, which could overestimate cost savings if the medication was discontinued or changed to an alternative therapy within that time period.
The study’s exclusion criteria could only be applied locally and did not include data received from the VA CC prescription database. This can be seen by the discrepancy in CC PADR approval rates from the local and national data (46.2% vs 44.2%, respectively) and CC PADR volume. High-complexity VA facility data were captured without assessing the CC prescription process at each site. As a result, definitive conclusions cannot be made regarding the impact of a centralized CC pharmacy team compared with other facilities.
Conclusions
Adjudication of CC PADRs by a centralized CC pharmacy team over a 6-month period provided > $500,000 in direct cost savings to a VA health care system. Considering the CC PADR approval rate seen in this study, the VA could allocate resources to educate CC providers about the VA formulary to increase the PADR approval rate and reduce administrative burden for VA pharmacies and prescribers. Future research should evaluate CC prescription handling practices at other VA facilities to compare the effectiveness among varying approaches and develop recommendations for a nationally standardized process.
Acknowledgments
Concept and design (AJJ, JNB, RBB, LAM, MD, MGH); acquisition of data (AJJ, MGH); analysis and interpretation of data (AJJ, JNB, RBB, LAM, MD, MGH); drafting of the manuscript (AJJ); critical revision of the manuscript for important intellectual content (AJJ, JNB, RBB, LAM, MD, MGH); statistical analysis (AJJ); administrative, technical, or logistic support (LAM, MGH); and supervision (MGH).
Veterans’ access to medical care was expanded outside of US Department of Veterans Affairs (VA) facilities with the inception of the 2014 Veterans Access, Choice, and Accountability Act (Choice Act).1 This legislation aimed to remove barriers some veterans were experiencing, specifically access to health care. In subsequent years, approximately 17% of veterans receiving care from the VA did so under the Choice Act.2 The Choice Act positively impacted medical care access for veterans but presented new challenges for VA pharmacies processing community care (CC) prescriptions, including limited access to outside health records, lack of interface between CC prescribers and the VA order entry system, and limited awareness of the VA national formulary.3,4 These factors made it difficult for VA pharmacies to assess prescriptions for clinical appropriateness, evaluate patient safety parameters, and manage expenditures.
In 2019, the Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act, which expanded CC support and better defined which veterans are able to receive care outside the VA, updated the Choice Act.4,5 However, VA pharmacies faced challenges in managing pharmacy drug costs and ensuring clinical appropriateness of prescription drug therapy. As a result, VA pharmacy departments have adjusted how they allocate workload, time, and funds.5
Pharmacists improve clinical outcomes and reduce health care costs by decreasing medication errors, unnecessary prescribing, and adverse drug events.6-12 Pharmacist-driven formulary management through evaluation of prior authorization drug requests (PADRs) has shown economic value.13,14 VA pharmacy review of community care PADRs is important because outside health care professionals (HCPs) might not be familiar with the VA formulary. This could lead to high volume of PADRs that do not meet criteria and could result in increased potential for medication misuse, adverse drug events, medication errors, and cost to the health system. It is imperative that CC orders are evaluated as critically as traditional orders.
The value of a centralized CC pharmacy team has not been assessed in the literature. The primary objective of this study was to assess the direct cost savings achieved through a centralized CC PADR process. Secondary objectives were to characterize the CC PADRs submitted to the site, including approval rate, reason for nonapproval, which medications were requested and by whom, and to compare CC prescriptions with other high-complexity (1a) VA facilities.
Community Care Pharmacy
VA health systems are stratified according to complexity, which reflects size, patient population, and services offered. This study was conducted at the Durham Veterans Affairs Health Care System (DVAHCS), North Carolina, a high-complexity, 251-bed, tertiary care referral, teaching, and research system. DVAHCS provides general and specialty medical, surgical, inpatient psychiatric, and ambulatory services, and serves as a major referral center.
DVAHCS created a centralized pharmacy team for processing CC prescriptions and managing customer service. This team’s goal is to increase CC prescription processing efficiency and transparency, ensure accountability of the health care team, and promote veteran-centric customer service. The pharmacy team includes a pharmacist program manager and a dedicated CC pharmacist with administrative support from a health benefits assistant and 4 pharmacy technicians. The CC pharmacy team assesses every new prescription to ensure the veteran is authorized to receive care in the community. Once eligibility is verified, a pharmacy technician or pharmacist evaluates the prescription to ensure it contains all required information, then contacts the prescriber for any missing data. If clinically appropriate, the pharmacist processes the prescription.
In 2020, the CC pharmacy team implemented a new process for reviewing and documenting CC prescriptions that require a PADR. The closed national VA formulary is set up so that all nonformulary medications and some formulary medications, including those that are restricted because of safety and/or cost, require a PADR.15 After a CC pharmacy technician confirms a veteran’s eligibility, the technician assesses whether the requested medication requires submitting a PADR to the VA internal electronic health record. The PADR is then adjudicated by a formulary management pharmacist, CC program manager, or CC pharmacist who reviews health records to determine whether the CC prescription meets VA medication use policy requirements.
If additional information is needed or an alternate medication is suggested, the pharmacist comments back on the PADR and a CC pharmacy technician contacts the prescriber. The PADR is canceled administratively then resubmitted once all information is obtained. While waiting for a response from the prescriber, the CC pharmacy technician contacts that veteran to give an update on the prescription status, as appropriate. Once there is sufficient information to adjudicate the PADR, the outcome is documented, and if approved, the order is processed.
Methods
The DVAHCS Institutional Review Board approved this retrospective review of CC PADRs submitted from June 1, 2020, through November 30, 2020. CC PADRs were excluded if they were duplicates or were reactivated administratively but had an initial submission date before the study period. Local data were collected for nonapproved CC PADRs including drug requested, dosage and directions, medication specialty, alternative drug recommended, drug acquisition cost, PADR submission date, PADR completion date, PADR nonapproval rationale, and documented time spent per PADR. Additional data was obtained for CC prescriptions at all 42 high-complexity VA facilities from the VA national CC prescription database for the study time interval and included total PADRs, PADR approval status, total CC prescription cost, and total CC fills.
Direct cost savings were calculated by assessing the cost of requested therapy that was not approved minus the cost of recommended therapy and cost to review all PADRs, as described by Britt and colleagues.13 The cost of the requested and recommended therapy was calculated based on VA drug acquisition cost at time of data collection and multiplied by the expected duration of therapy up to 1 year. For each CC prescription, duration of therapy was based on the duration limit in the prescription or annualized if no duration limit was documented. Cost of PADR review was calculated based on the total time pharmacists and pharmacy technicians documented for each step of the review process for a representative sample of 100 nonapproved PADRs and then multiplied by the salary plus benefits of an entry-level pharmacist and pharmacy technician.16 The eAppendix describes specific equations used for determining direct cost savings. Descriptive statistics were used to evaluate study results.
Results
During the 6-month study period, 611 CC PADRs were submitted to the pharmacy and 526 met inclusion criteria (Figure 1). Of those, 243 (46.2%) were approved and 283 (53.8%) were not approved. The cost of requested therapies for nonapproved CC PADRs totaled $584,565.48 and the cost of all recommended therapies was $57,473.59. The mean time per CC PADR was 24 minutes; 16 minutes for pharmacists and 8 minutes for pharmacy technicians. Given an hourly wage (plus benefits) of $67.25 for a pharmacist and $25.53 for a pharmacy technician, the total cost of review per CC PADR was $21.33. After subtracting the costs of all recommended therapies and review of all included CC PADRs, the process generated $515,872.31 in direct cost savings. After factoring in administrative lag time, such as HCP communication, an average of 8 calendar days was needed to complete a nonapproved PADR.
The most common rationale for PADR nonapproval was that the formulary alternative was not exhausted. Ondansetron orally disintegrating tablets was the most commonly nonapproved medication and azelastine was the most commonly approved medication. Dulaglutide was the most expensive nonapproved and tafamidis was the most expensive approved PADR (Table 1). Gastroenterology, endocrinology, and neurology were the top specialties for nonapproved PADRs while neurology, pulmonology, and endocrinology were the top specialties for approved PADRs (Table 2).
Several high-complexity VA facilities had no reported data; we used the median for the analysis to account for these outliers (Figure 2). The median (IQR) adjudicated CC PADRs for all facilities was 97 (20-175), median (IQR) CC PADR approval rate was 80.9% (63.7%-96.8%), median (IQR) total CC prescriptions was 8440 (2464-14,466), and median (IQR) cost per fill was $136.05 ($76.27-$221.28).
Discussion
This study demonstrated direct cost savings of $515,872.31 over 6 months with theadjudication of CC PADRs by a centralized CC pharmacy team. This could result in > $1,000,000 of cost savings per fiscal year.
The CC PADRs observed at DVAHCS had a 46.2% approval rate; almost one-half the approval rate of 84.1% of all PADRs submitted to the study site by VA HCPs captured by Britt and colleagues.13 Results from this study showed that coordination of care for nonapproved CC PADRs between the VA pharmacy and non-VA prescriber took an average of 8 calendar days. The noted CC PADR approval rate and administrative burden might be because of lack of familiarity of non-VA providers regarding the VA national formulary. The National VA Pharmacy Benefits Management determines the formulary using cost-effectiveness criteria that considers the medical literature and VA-specific contract pricing and prepares extensive guidance for restricted medications via relevant criteria for use.15 HCPs outside the VA might not know this information is available online. Because gastroenterology, endocrinology, and neurology specialty medications were among the most frequently nonapproved PADRs, VA formulary education could begin with CC HCPs in these practice areas.
This study showed that the CC PADR process was not solely driven by cost, but also included patient safety. Nonapproval rationale for some requests included submission without an indication, submission by a prescriber that did not have the authority to prescribe a type of medication, or contraindication based on patient-specific factors.
Compared with other VA high-complexity facilities, DVAHCS was among the top health care systems for total volume of CC prescriptions (n = 16,096) and among the lowest for cost/fill ($75.74). Similarly, DVAHCS was among the top sites for total adjudicated CC PADRs within the 6-month study period (n = 611) and the lowest approval rate (44.2%). This study shows that despite high volumes of overall CC prescriptions and CC PADRs, it is possible to maintain a low overall CC prescription cost/fill compared with other similarly complex sites across the country. Wide variance in reported results exists across high-complexity VA facilities because some sites had low to no CC fills and/or CC PADRs. This is likely a result of administrative differences when handling CC prescriptions and presents an opportunity to standardize this process nationally.
Limitations
CC PADRs were assessed during the COVID-19 pandemic, which might have resulted in lower-than-normal CC prescription and PADR volumes, therefore underestimating the potential for direct cost savings. Entry-level salary was used to demonstrate cost savings potential from the perspective of a newly hired CC team; however, the cost savings might have been less if the actual salaries of site personnel were higher. National contract pricing data were gathered at the time of data collection and might have been different than at the time of PADR submission. Chronic medication prescriptions were annualized, which could overestimate cost savings if the medication was discontinued or changed to an alternative therapy within that time period.
The study’s exclusion criteria could only be applied locally and did not include data received from the VA CC prescription database. This can be seen by the discrepancy in CC PADR approval rates from the local and national data (46.2% vs 44.2%, respectively) and CC PADR volume. High-complexity VA facility data were captured without assessing the CC prescription process at each site. As a result, definitive conclusions cannot be made regarding the impact of a centralized CC pharmacy team compared with other facilities.
Conclusions
Adjudication of CC PADRs by a centralized CC pharmacy team over a 6-month period provided > $500,000 in direct cost savings to a VA health care system. Considering the CC PADR approval rate seen in this study, the VA could allocate resources to educate CC providers about the VA formulary to increase the PADR approval rate and reduce administrative burden for VA pharmacies and prescribers. Future research should evaluate CC prescription handling practices at other VA facilities to compare the effectiveness among varying approaches and develop recommendations for a nationally standardized process.
Acknowledgments
Concept and design (AJJ, JNB, RBB, LAM, MD, MGH); acquisition of data (AJJ, MGH); analysis and interpretation of data (AJJ, JNB, RBB, LAM, MD, MGH); drafting of the manuscript (AJJ); critical revision of the manuscript for important intellectual content (AJJ, JNB, RBB, LAM, MD, MGH); statistical analysis (AJJ); administrative, technical, or logistic support (LAM, MGH); and supervision (MGH).
1. Gellad WF, Cunningham FE, Good CB, et al. Pharmacy use in the first year of the Veterans Choice Program: a mixed-methods evaluation. Med Care. 2017(7 suppl 1);55:S26. doi:10.1097/MLR.0000000000000661
2. Mattocks KM, Yehia B. Evaluating the veterans choice program: lessons for developing a high-performing integrated network. Med Care. 2017(7 suppl 1);55:S1-S3. doi:10.1097/MLR.0000000000000743.
3. Mattocks KM, Mengeling M, Sadler A, Baldor R, Bastian L. The Veterans Choice Act: a qualitative examination of rapid policy implementation in the Department of Veterans Affairs. Med Care. 2017;55(7 suppl 1):S71-S75. doi:10.1097/MLR.0000000000000667
4. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.08: VHA formulary management process. November 2, 2016. Accessed June 9, 2022. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3291
5. Massarweh NN, Itani KMF, Morris MS. The VA MISSION act and the future of veterans’ access to quality health care. JAMA. 2020;324:343-344. doi:10.1001/jama.2020.4505
6. Jourdan JP, Muzard A, Goyer I, et al. Impact of pharmacist interventions on clinical outcome and cost avoidance in a university teaching hospital. Int J Clin Pharm. 2018;40(6):1474-1481. doi:10.1007/s11096-018-0733-6
7. Lee AJ, Boro MS, Knapp KK, Meier JL, Korman NE. Clinical and economic outcomes of pharmacist recommendations in a Veterans Affairs medical center. Am J Health Syst Pharm. 2002;59(21):2070-2077. doi:10.1093/ajhp/59.21.2070
8. Dalton K, Byrne S. Role of the pharmacist in reducing healthcare costs: current insights. Integr Pharm Res Pract. 2017;6:37-46. doi:10.2147/IPRP.S108047
9. De Rijdt T, Willems L, Simoens S. Economic effects of clinical pharmacy interventions: a literature review. Am J Health Syst Pharm. 2008;65(12):1161-1172. doi:10.2146/ajhp070506
10. Perez A, Doloresco F, Hoffman J, et al. Economic evaluation of clinical pharmacy services: 2001-2005. Pharmacotherapy. 2009;29(1):128. doi:10.1592/phco.29.1.128
11. Nesbit TW, Shermock KM, Bobek MB, et al. Implementation and pharmacoeconomic analysis of a clinical staff pharmacist practice model. Am J Health Syst Pharm. 2001;58(9):784-790. doi:10.1093/ajhp/58.9.784
12. Yang S, Britt RB, Hashem MG, Brown JN. Outcomes of pharmacy-led hepatitis C direct-acting antiviral utilization management at a Veterans Affairs medical center. J Manag Care Pharm. 2017;23(3):364-369. doi:10.18553/jmcp.2017.23.3.364
13. Britt RB, Hashem MG, Bryan WE III, Kothapalli R, Brown JN. Economic outcomes associated with a pharmacist-adjudicated formulary consult service in a Veterans Affairs medical center. J Manag Care Pharm. 2016;22(9):1051-1061. doi:10.18553/jmcp.2016.22.9.1051
14. Jacob S, Britt RB, Bryan WE, Hashem MG, Hale JC, Brown JN. Economic outcomes associated with safety interventions by a pharmacist-adjudicated prior authorization consult service. J Manag Care Pharm. 2019;25(3):411-416. doi:10.18553/jmcp.2019.25.3.411
15. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063. doi:10.18553/jmcp.2016.22.9.1058
16. US Department of Veterans Affairs, Office of the Chief Human Capital Officer. Title 38 Pay Schedules. Updated January 26, 2022. Accessed June 9, 2022. https://www.va.gov/ohrm/pay
1. Gellad WF, Cunningham FE, Good CB, et al. Pharmacy use in the first year of the Veterans Choice Program: a mixed-methods evaluation. Med Care. 2017(7 suppl 1);55:S26. doi:10.1097/MLR.0000000000000661
2. Mattocks KM, Yehia B. Evaluating the veterans choice program: lessons for developing a high-performing integrated network. Med Care. 2017(7 suppl 1);55:S1-S3. doi:10.1097/MLR.0000000000000743.
3. Mattocks KM, Mengeling M, Sadler A, Baldor R, Bastian L. The Veterans Choice Act: a qualitative examination of rapid policy implementation in the Department of Veterans Affairs. Med Care. 2017;55(7 suppl 1):S71-S75. doi:10.1097/MLR.0000000000000667
4. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.08: VHA formulary management process. November 2, 2016. Accessed June 9, 2022. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3291
5. Massarweh NN, Itani KMF, Morris MS. The VA MISSION act and the future of veterans’ access to quality health care. JAMA. 2020;324:343-344. doi:10.1001/jama.2020.4505
6. Jourdan JP, Muzard A, Goyer I, et al. Impact of pharmacist interventions on clinical outcome and cost avoidance in a university teaching hospital. Int J Clin Pharm. 2018;40(6):1474-1481. doi:10.1007/s11096-018-0733-6
7. Lee AJ, Boro MS, Knapp KK, Meier JL, Korman NE. Clinical and economic outcomes of pharmacist recommendations in a Veterans Affairs medical center. Am J Health Syst Pharm. 2002;59(21):2070-2077. doi:10.1093/ajhp/59.21.2070
8. Dalton K, Byrne S. Role of the pharmacist in reducing healthcare costs: current insights. Integr Pharm Res Pract. 2017;6:37-46. doi:10.2147/IPRP.S108047
9. De Rijdt T, Willems L, Simoens S. Economic effects of clinical pharmacy interventions: a literature review. Am J Health Syst Pharm. 2008;65(12):1161-1172. doi:10.2146/ajhp070506
10. Perez A, Doloresco F, Hoffman J, et al. Economic evaluation of clinical pharmacy services: 2001-2005. Pharmacotherapy. 2009;29(1):128. doi:10.1592/phco.29.1.128
11. Nesbit TW, Shermock KM, Bobek MB, et al. Implementation and pharmacoeconomic analysis of a clinical staff pharmacist practice model. Am J Health Syst Pharm. 2001;58(9):784-790. doi:10.1093/ajhp/58.9.784
12. Yang S, Britt RB, Hashem MG, Brown JN. Outcomes of pharmacy-led hepatitis C direct-acting antiviral utilization management at a Veterans Affairs medical center. J Manag Care Pharm. 2017;23(3):364-369. doi:10.18553/jmcp.2017.23.3.364
13. Britt RB, Hashem MG, Bryan WE III, Kothapalli R, Brown JN. Economic outcomes associated with a pharmacist-adjudicated formulary consult service in a Veterans Affairs medical center. J Manag Care Pharm. 2016;22(9):1051-1061. doi:10.18553/jmcp.2016.22.9.1051
14. Jacob S, Britt RB, Bryan WE, Hashem MG, Hale JC, Brown JN. Economic outcomes associated with safety interventions by a pharmacist-adjudicated prior authorization consult service. J Manag Care Pharm. 2019;25(3):411-416. doi:10.18553/jmcp.2019.25.3.411
15. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063. doi:10.18553/jmcp.2016.22.9.1058
16. US Department of Veterans Affairs, Office of the Chief Human Capital Officer. Title 38 Pay Schedules. Updated January 26, 2022. Accessed June 9, 2022. https://www.va.gov/ohrm/pay
Positive phase 3 results for novel schizophrenia drug
The investigational agent xanomeline-trospium (KarXT, Karuna Therapeutics), which combines a muscarinic receptor agonist with an anticholinergic agent, helps improve psychosis symptoms and is not associated with weight gain or sedation in adults with schizophrenia, new research shows.
Top-line results from the phase 3 EMERGENT-2 trial showed a significantly greater reduction from baseline on Positive and Negative Syndrome Scale (PANSS) total scores for those receiving the active drug than for those receiving placebo, meeting its primary endpoint.
and potentially usher in the first new class of medicine for these patients in more than 50 years,” Steve Paul, MD, chief executive officer, president, and chairman of Karuna Therapeutics, said in a press release.
Primary outcome met
About 20%-33% of patients with schizophrenia do not respond to conventional treatments, the company noted. Many have poor functional status and quality of life despite lifelong treatment with current antipsychotic agents.
Unlike current therapies, KarXT doesn’t rely on the dopaminergic or serotonergic pathways. It comprises the muscarinic agonist xanomeline and the muscarinic antagonist trospium and is designed to preferentially stimulate muscarinic receptors in the central nervous system.
Results from a phase 2 trial of almost 200 patients with schizophrenia were published last year in the New England Journal of Medicine. The findings showed that those who received xanomeline-trospium had a significantly greater reduction in psychosis symptoms than those who received placebo.
In the current phase 3 EMERGENT-2 trial, investigators included 252 adults aged 18-65 years who were diagnosed with schizophrenia and were experiencing symptoms of psychosis. Patients were randomly assigned to receive either a flexible dose of xanomeline-trospium or placebo twice daily.
The primary endpoint was change from baseline in the PANSS total score at week 5. Results showed a statistically significant and clinically meaningful 9.6-point reduction in the PANSS total score in participants taking the active drug, compared with those taking placebo (–21.2 vs. –11.6, respectively; P < .0001; Cohen’s d effect size, 0.61).
In addition, there was an early and sustained significant reduction of schizophrenia symptoms, as assessed by the PANSS total score, starting at week 2. This reduction was maintained through all trial timepoints.
Safety profile
The novel drug also met key secondary endpoints. In the active treatment group, there was a significant reduction on the PANSS subscales in both positive symptoms of schizophrenia, such as hallucinations or delusions, and negative symptoms, such as difficulty enjoying life or withdrawal from others.
Overall, the agent was generally well tolerated. The treatment-emergent adverse events (TEAEs) rate for xanomeline-trospium and placebo was 75% versus 58%, respectively.
The most common TEAEs for the active treatment were all mild-to-moderate in severity and included constipation, dyspepsia, nausea, vomiting, headache, increases in blood pressure, dizziness, gastroesophageal reflux disease, abdominal discomfort, and diarrhea.
As in prior trials, an increase in heart rate was also associated with the active treatment and decreased in magnitude by the end of the current study.
Discontinuation rates related to TEAEs were similar between xanomeline-trospium (7%) and placebo (6%), as were rates of serious TEAEs (2% in each group) – which included suicidal ideation, worsening of schizophrenia symptoms, and appendicitis.
Notably, the drug was not associated with common problematic adverse events of current therapies, such as weight gain, sedation, and movement disorders.
Karuna plans to submit a New Drug Application with the U.S. Food and Drug Administration for KarXT in mid-2023. In addition to schizophrenia, the drug is in development for the treatment of other psychiatric and neurological conditions, including Alzheimer’s disease.
A version of this article first appeared on Medscape.com.
The investigational agent xanomeline-trospium (KarXT, Karuna Therapeutics), which combines a muscarinic receptor agonist with an anticholinergic agent, helps improve psychosis symptoms and is not associated with weight gain or sedation in adults with schizophrenia, new research shows.
Top-line results from the phase 3 EMERGENT-2 trial showed a significantly greater reduction from baseline on Positive and Negative Syndrome Scale (PANSS) total scores for those receiving the active drug than for those receiving placebo, meeting its primary endpoint.
and potentially usher in the first new class of medicine for these patients in more than 50 years,” Steve Paul, MD, chief executive officer, president, and chairman of Karuna Therapeutics, said in a press release.
Primary outcome met
About 20%-33% of patients with schizophrenia do not respond to conventional treatments, the company noted. Many have poor functional status and quality of life despite lifelong treatment with current antipsychotic agents.
Unlike current therapies, KarXT doesn’t rely on the dopaminergic or serotonergic pathways. It comprises the muscarinic agonist xanomeline and the muscarinic antagonist trospium and is designed to preferentially stimulate muscarinic receptors in the central nervous system.
Results from a phase 2 trial of almost 200 patients with schizophrenia were published last year in the New England Journal of Medicine. The findings showed that those who received xanomeline-trospium had a significantly greater reduction in psychosis symptoms than those who received placebo.
In the current phase 3 EMERGENT-2 trial, investigators included 252 adults aged 18-65 years who were diagnosed with schizophrenia and were experiencing symptoms of psychosis. Patients were randomly assigned to receive either a flexible dose of xanomeline-trospium or placebo twice daily.
The primary endpoint was change from baseline in the PANSS total score at week 5. Results showed a statistically significant and clinically meaningful 9.6-point reduction in the PANSS total score in participants taking the active drug, compared with those taking placebo (–21.2 vs. –11.6, respectively; P < .0001; Cohen’s d effect size, 0.61).
In addition, there was an early and sustained significant reduction of schizophrenia symptoms, as assessed by the PANSS total score, starting at week 2. This reduction was maintained through all trial timepoints.
Safety profile
The novel drug also met key secondary endpoints. In the active treatment group, there was a significant reduction on the PANSS subscales in both positive symptoms of schizophrenia, such as hallucinations or delusions, and negative symptoms, such as difficulty enjoying life or withdrawal from others.
Overall, the agent was generally well tolerated. The treatment-emergent adverse events (TEAEs) rate for xanomeline-trospium and placebo was 75% versus 58%, respectively.
The most common TEAEs for the active treatment were all mild-to-moderate in severity and included constipation, dyspepsia, nausea, vomiting, headache, increases in blood pressure, dizziness, gastroesophageal reflux disease, abdominal discomfort, and diarrhea.
As in prior trials, an increase in heart rate was also associated with the active treatment and decreased in magnitude by the end of the current study.
Discontinuation rates related to TEAEs were similar between xanomeline-trospium (7%) and placebo (6%), as were rates of serious TEAEs (2% in each group) – which included suicidal ideation, worsening of schizophrenia symptoms, and appendicitis.
Notably, the drug was not associated with common problematic adverse events of current therapies, such as weight gain, sedation, and movement disorders.
Karuna plans to submit a New Drug Application with the U.S. Food and Drug Administration for KarXT in mid-2023. In addition to schizophrenia, the drug is in development for the treatment of other psychiatric and neurological conditions, including Alzheimer’s disease.
A version of this article first appeared on Medscape.com.
The investigational agent xanomeline-trospium (KarXT, Karuna Therapeutics), which combines a muscarinic receptor agonist with an anticholinergic agent, helps improve psychosis symptoms and is not associated with weight gain or sedation in adults with schizophrenia, new research shows.
Top-line results from the phase 3 EMERGENT-2 trial showed a significantly greater reduction from baseline on Positive and Negative Syndrome Scale (PANSS) total scores for those receiving the active drug than for those receiving placebo, meeting its primary endpoint.
and potentially usher in the first new class of medicine for these patients in more than 50 years,” Steve Paul, MD, chief executive officer, president, and chairman of Karuna Therapeutics, said in a press release.
Primary outcome met
About 20%-33% of patients with schizophrenia do not respond to conventional treatments, the company noted. Many have poor functional status and quality of life despite lifelong treatment with current antipsychotic agents.
Unlike current therapies, KarXT doesn’t rely on the dopaminergic or serotonergic pathways. It comprises the muscarinic agonist xanomeline and the muscarinic antagonist trospium and is designed to preferentially stimulate muscarinic receptors in the central nervous system.
Results from a phase 2 trial of almost 200 patients with schizophrenia were published last year in the New England Journal of Medicine. The findings showed that those who received xanomeline-trospium had a significantly greater reduction in psychosis symptoms than those who received placebo.
In the current phase 3 EMERGENT-2 trial, investigators included 252 adults aged 18-65 years who were diagnosed with schizophrenia and were experiencing symptoms of psychosis. Patients were randomly assigned to receive either a flexible dose of xanomeline-trospium or placebo twice daily.
The primary endpoint was change from baseline in the PANSS total score at week 5. Results showed a statistically significant and clinically meaningful 9.6-point reduction in the PANSS total score in participants taking the active drug, compared with those taking placebo (–21.2 vs. –11.6, respectively; P < .0001; Cohen’s d effect size, 0.61).
In addition, there was an early and sustained significant reduction of schizophrenia symptoms, as assessed by the PANSS total score, starting at week 2. This reduction was maintained through all trial timepoints.
Safety profile
The novel drug also met key secondary endpoints. In the active treatment group, there was a significant reduction on the PANSS subscales in both positive symptoms of schizophrenia, such as hallucinations or delusions, and negative symptoms, such as difficulty enjoying life or withdrawal from others.
Overall, the agent was generally well tolerated. The treatment-emergent adverse events (TEAEs) rate for xanomeline-trospium and placebo was 75% versus 58%, respectively.
The most common TEAEs for the active treatment were all mild-to-moderate in severity and included constipation, dyspepsia, nausea, vomiting, headache, increases in blood pressure, dizziness, gastroesophageal reflux disease, abdominal discomfort, and diarrhea.
As in prior trials, an increase in heart rate was also associated with the active treatment and decreased in magnitude by the end of the current study.
Discontinuation rates related to TEAEs were similar between xanomeline-trospium (7%) and placebo (6%), as were rates of serious TEAEs (2% in each group) – which included suicidal ideation, worsening of schizophrenia symptoms, and appendicitis.
Notably, the drug was not associated with common problematic adverse events of current therapies, such as weight gain, sedation, and movement disorders.
Karuna plans to submit a New Drug Application with the U.S. Food and Drug Administration for KarXT in mid-2023. In addition to schizophrenia, the drug is in development for the treatment of other psychiatric and neurological conditions, including Alzheimer’s disease.
A version of this article first appeared on Medscape.com.
FDA authorizes intradermal use of Jynneos vaccine for monkeypox
The Food and Drug Administration on Aug. 9 authorized intradermal administration of the Jynneos vaccine for the treatment of monkeypox. The process, approved specifically for high-risk patients, was passed under the administration’s Emergency Use Authorization. It follows the decision on Aug. 4 by the U.S. Department of Health and Human Services to declare monkeypox a public health emergency. Intradermal administration will allow providers to get five doses out of a one-dose vial.
This news organization will update this article as more information becomes available.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Aug. 9 authorized intradermal administration of the Jynneos vaccine for the treatment of monkeypox. The process, approved specifically for high-risk patients, was passed under the administration’s Emergency Use Authorization. It follows the decision on Aug. 4 by the U.S. Department of Health and Human Services to declare monkeypox a public health emergency. Intradermal administration will allow providers to get five doses out of a one-dose vial.
This news organization will update this article as more information becomes available.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on Aug. 9 authorized intradermal administration of the Jynneos vaccine for the treatment of monkeypox. The process, approved specifically for high-risk patients, was passed under the administration’s Emergency Use Authorization. It follows the decision on Aug. 4 by the U.S. Department of Health and Human Services to declare monkeypox a public health emergency. Intradermal administration will allow providers to get five doses out of a one-dose vial.
This news organization will update this article as more information becomes available.
A version of this article first appeared on Medscape.com.
NAMS affirms value of hormone therapy for menopausal women
Hormone therapy remains a topic for debate, but a constant in the 2 decades since the Women’s Health Initiative has been the demonstrated effectiveness for relief of vasomotor symptoms and reduction of fracture risk in menopausal women, according to the latest hormone therapy position statement of the North American Menopause Society.
“Healthcare professionals caring for menopausal women should understand the basic concepts of relative risk and absolute risk,” wrote Stephanie S. Faubion, MD, director of the Mayo Clinic Center for Women’s Health and medical director of NAMS, and members of the NAMS 2022 Hormone Therapy Position Statement Advisory Panel in Menopause.

The authors noted that the risks of hormone therapy vary considerably based on type, dose, duration, route of administration, timing of the start of therapy, and whether or not a progestogen is included.
The 2022 statement was commissioned to review new literature and identify the strength of recommendations and quality of evidence since the previous statement in 2017.
The current statement represents not so much a practice-changing update, “but rather that the literature has filled out in some areas,” Dr. Faubion said in an interview. “The recommendations overall haven’t changed,” she said. “The position statement reiterates that hormone therapy, which is significantly underutilized, remains a safe and effective treatment for menopause symptoms, which remain undertreated, with the benefits outweighing the risks for most healthy women who are within 10 years of menopause onset and under the age of 60 years,” she emphasized. “Individualizing therapy is key to maximizing benefits and minimizing risks,” she added.
Overall, the authors confirmed that hormone therapy remains the most effective treatment for vasomotor symptoms (VMS) and the genitourinary syndrome of menopause (GSM), and has been shown to prevent bone loss and fracture. The risks of hormone therapy differ depending on type, dose, duration of use, route of administration, timing of initiation, and whether a progestogen is used.
Risks and benefits should be stratified by age and time since the start of menopause, according to the statement.
For women younger than 60 years or within 10 years of the onset of menopause who have no contraindications, the potential benefits outweigh the risks in most cases for use of hormone therapy to manage vasomotor symptoms and to help prevent bone loss and reduce fracture risk.
For women who begin hormone therapy more than 10 or 20 years from the start of menopause, or who are aged 60 years and older, the risk-benefit ratio may be less favorable because of the increased absolute risk of coronary heart disease, stroke, venous thromboembolism, and dementia. However, strategies such as lower doses and transdermal administration may reduce this risk, according to the statement.
The authors continue to recommend that longer durations of hormone therapy be for documented indications, such as VMS relief, and that patients on longer duration of therapy be reassessed periodically as part of a shared decision-making process. Women with persistent VMS or quality of life issues, or those at risk for osteoporosis, may continue hormone therapy beyond age 60 or 65 years after appropriate evaluation and risk-benefit counseling.
Women with ongoing GSM without indications for systemic therapy whose GSM persists after over-the-counter therapies may try low-dose vaginal estrogen or other nonestrogen therapies regardless of age and for an extended duration if needed, according to the statement.
Challenges, research gaps, and goals
“Barriers to the use of hormone therapy include lack of access to high quality care,” Dr. Faubion said in an interview. The NAMS website, menopause.org, features an option to search for a NAMS-certified provider by ZIP code, she noted.
“Coverage of hormone therapy is highly variable and depends on the insurance company, but most women have access to one form or another with insurance coverage,” she said. “We need to continue to advocate for adequate coverage of menopause symptom treatments, including hormone therapy, so that women’s symptoms – which can significantly affect quality of life – are adequately managed.
“Additional research is needed on the thrombotic risk (venous thromboembolism, pulmonary embolism, and stroke) of oral versus transdermal therapies (including different formulations, doses, and durations of therapy),” Dr. Faubion told this news organization. “More clinical trial data are needed to confirm or refute the potential beneficial effects of hormone therapy on coronary heart disease and all-cause mortality when initiated in perimenopause or early postmenopause,” she said.
Other areas for research include “the breast effects of different estrogen preparations, including the role for selective estrogen receptor modulator (SERM) and tissue selective estrogen complex therapies, optimal progestogen or SERM regimens to prevent endometrial hyperplasia, the relationship between vasomotor symptoms and the risk for heart disease and cognitive changes, and the risks of premature ovarian insufficiency,” Dr. Faubion emphasized.
Looking ahead, “Studies are needed on the effects of longer use of low-dose vaginal estrogen therapy after breast or endometrial cancer, extended use of hormone therapy in women who are early initiators, improved tools to personalize or individualize benefits and risks of hormone therapy, and the role of aging and genetics,” said Dr. Faubion. Other areas for further research include “the long-term benefits and risks on women’s health of lifestyle modification or complementary or nonhormone therapies, if chosen in addition to or over hormone therapy for vasomotor symptoms, bone health, and cardiovascular disease risk reduction,” she added.
The complete statement was published in Menopause: The Journal of the North American Menopause Society.
The position statement received no outside funding. The authors had no financial conflicts to disclose.
Hormone therapy remains a topic for debate, but a constant in the 2 decades since the Women’s Health Initiative has been the demonstrated effectiveness for relief of vasomotor symptoms and reduction of fracture risk in menopausal women, according to the latest hormone therapy position statement of the North American Menopause Society.
“Healthcare professionals caring for menopausal women should understand the basic concepts of relative risk and absolute risk,” wrote Stephanie S. Faubion, MD, director of the Mayo Clinic Center for Women’s Health and medical director of NAMS, and members of the NAMS 2022 Hormone Therapy Position Statement Advisory Panel in Menopause.
The authors noted that the risks of hormone therapy vary considerably based on type, dose, duration, route of administration, timing of the start of therapy, and whether or not a progestogen is included.
The 2022 statement was commissioned to review new literature and identify the strength of recommendations and quality of evidence since the previous statement in 2017.
The current statement represents not so much a practice-changing update, “but rather that the literature has filled out in some areas,” Dr. Faubion said in an interview. “The recommendations overall haven’t changed,” she said. “The position statement reiterates that hormone therapy, which is significantly underutilized, remains a safe and effective treatment for menopause symptoms, which remain undertreated, with the benefits outweighing the risks for most healthy women who are within 10 years of menopause onset and under the age of 60 years,” she emphasized. “Individualizing therapy is key to maximizing benefits and minimizing risks,” she added.
Overall, the authors confirmed that hormone therapy remains the most effective treatment for vasomotor symptoms (VMS) and the genitourinary syndrome of menopause (GSM), and has been shown to prevent bone loss and fracture. The risks of hormone therapy differ depending on type, dose, duration of use, route of administration, timing of initiation, and whether a progestogen is used.
Risks and benefits should be stratified by age and time since the start of menopause, according to the statement.
For women younger than 60 years or within 10 years of the onset of menopause who have no contraindications, the potential benefits outweigh the risks in most cases for use of hormone therapy to manage vasomotor symptoms and to help prevent bone loss and reduce fracture risk.
For women who begin hormone therapy more than 10 or 20 years from the start of menopause, or who are aged 60 years and older, the risk-benefit ratio may be less favorable because of the increased absolute risk of coronary heart disease, stroke, venous thromboembolism, and dementia. However, strategies such as lower doses and transdermal administration may reduce this risk, according to the statement.
The authors continue to recommend that longer durations of hormone therapy be for documented indications, such as VMS relief, and that patients on longer duration of therapy be reassessed periodically as part of a shared decision-making process. Women with persistent VMS or quality of life issues, or those at risk for osteoporosis, may continue hormone therapy beyond age 60 or 65 years after appropriate evaluation and risk-benefit counseling.
Women with ongoing GSM without indications for systemic therapy whose GSM persists after over-the-counter therapies may try low-dose vaginal estrogen or other nonestrogen therapies regardless of age and for an extended duration if needed, according to the statement.
Challenges, research gaps, and goals
“Barriers to the use of hormone therapy include lack of access to high quality care,” Dr. Faubion said in an interview. The NAMS website, menopause.org, features an option to search for a NAMS-certified provider by ZIP code, she noted.
“Coverage of hormone therapy is highly variable and depends on the insurance company, but most women have access to one form or another with insurance coverage,” she said. “We need to continue to advocate for adequate coverage of menopause symptom treatments, including hormone therapy, so that women’s symptoms – which can significantly affect quality of life – are adequately managed.
“Additional research is needed on the thrombotic risk (venous thromboembolism, pulmonary embolism, and stroke) of oral versus transdermal therapies (including different formulations, doses, and durations of therapy),” Dr. Faubion told this news organization. “More clinical trial data are needed to confirm or refute the potential beneficial effects of hormone therapy on coronary heart disease and all-cause mortality when initiated in perimenopause or early postmenopause,” she said.
Other areas for research include “the breast effects of different estrogen preparations, including the role for selective estrogen receptor modulator (SERM) and tissue selective estrogen complex therapies, optimal progestogen or SERM regimens to prevent endometrial hyperplasia, the relationship between vasomotor symptoms and the risk for heart disease and cognitive changes, and the risks of premature ovarian insufficiency,” Dr. Faubion emphasized.
Looking ahead, “Studies are needed on the effects of longer use of low-dose vaginal estrogen therapy after breast or endometrial cancer, extended use of hormone therapy in women who are early initiators, improved tools to personalize or individualize benefits and risks of hormone therapy, and the role of aging and genetics,” said Dr. Faubion. Other areas for further research include “the long-term benefits and risks on women’s health of lifestyle modification or complementary or nonhormone therapies, if chosen in addition to or over hormone therapy for vasomotor symptoms, bone health, and cardiovascular disease risk reduction,” she added.
The complete statement was published in Menopause: The Journal of the North American Menopause Society.
The position statement received no outside funding. The authors had no financial conflicts to disclose.
Hormone therapy remains a topic for debate, but a constant in the 2 decades since the Women’s Health Initiative has been the demonstrated effectiveness for relief of vasomotor symptoms and reduction of fracture risk in menopausal women, according to the latest hormone therapy position statement of the North American Menopause Society.
“Healthcare professionals caring for menopausal women should understand the basic concepts of relative risk and absolute risk,” wrote Stephanie S. Faubion, MD, director of the Mayo Clinic Center for Women’s Health and medical director of NAMS, and members of the NAMS 2022 Hormone Therapy Position Statement Advisory Panel in Menopause.
The authors noted that the risks of hormone therapy vary considerably based on type, dose, duration, route of administration, timing of the start of therapy, and whether or not a progestogen is included.
The 2022 statement was commissioned to review new literature and identify the strength of recommendations and quality of evidence since the previous statement in 2017.
The current statement represents not so much a practice-changing update, “but rather that the literature has filled out in some areas,” Dr. Faubion said in an interview. “The recommendations overall haven’t changed,” she said. “The position statement reiterates that hormone therapy, which is significantly underutilized, remains a safe and effective treatment for menopause symptoms, which remain undertreated, with the benefits outweighing the risks for most healthy women who are within 10 years of menopause onset and under the age of 60 years,” she emphasized. “Individualizing therapy is key to maximizing benefits and minimizing risks,” she added.
Overall, the authors confirmed that hormone therapy remains the most effective treatment for vasomotor symptoms (VMS) and the genitourinary syndrome of menopause (GSM), and has been shown to prevent bone loss and fracture. The risks of hormone therapy differ depending on type, dose, duration of use, route of administration, timing of initiation, and whether a progestogen is used.
Risks and benefits should be stratified by age and time since the start of menopause, according to the statement.
For women younger than 60 years or within 10 years of the onset of menopause who have no contraindications, the potential benefits outweigh the risks in most cases for use of hormone therapy to manage vasomotor symptoms and to help prevent bone loss and reduce fracture risk.
For women who begin hormone therapy more than 10 or 20 years from the start of menopause, or who are aged 60 years and older, the risk-benefit ratio may be less favorable because of the increased absolute risk of coronary heart disease, stroke, venous thromboembolism, and dementia. However, strategies such as lower doses and transdermal administration may reduce this risk, according to the statement.
The authors continue to recommend that longer durations of hormone therapy be for documented indications, such as VMS relief, and that patients on longer duration of therapy be reassessed periodically as part of a shared decision-making process. Women with persistent VMS or quality of life issues, or those at risk for osteoporosis, may continue hormone therapy beyond age 60 or 65 years after appropriate evaluation and risk-benefit counseling.
Women with ongoing GSM without indications for systemic therapy whose GSM persists after over-the-counter therapies may try low-dose vaginal estrogen or other nonestrogen therapies regardless of age and for an extended duration if needed, according to the statement.
Challenges, research gaps, and goals
“Barriers to the use of hormone therapy include lack of access to high quality care,” Dr. Faubion said in an interview. The NAMS website, menopause.org, features an option to search for a NAMS-certified provider by ZIP code, she noted.
“Coverage of hormone therapy is highly variable and depends on the insurance company, but most women have access to one form or another with insurance coverage,” she said. “We need to continue to advocate for adequate coverage of menopause symptom treatments, including hormone therapy, so that women’s symptoms – which can significantly affect quality of life – are adequately managed.
“Additional research is needed on the thrombotic risk (venous thromboembolism, pulmonary embolism, and stroke) of oral versus transdermal therapies (including different formulations, doses, and durations of therapy),” Dr. Faubion told this news organization. “More clinical trial data are needed to confirm or refute the potential beneficial effects of hormone therapy on coronary heart disease and all-cause mortality when initiated in perimenopause or early postmenopause,” she said.
Other areas for research include “the breast effects of different estrogen preparations, including the role for selective estrogen receptor modulator (SERM) and tissue selective estrogen complex therapies, optimal progestogen or SERM regimens to prevent endometrial hyperplasia, the relationship between vasomotor symptoms and the risk for heart disease and cognitive changes, and the risks of premature ovarian insufficiency,” Dr. Faubion emphasized.
Looking ahead, “Studies are needed on the effects of longer use of low-dose vaginal estrogen therapy after breast or endometrial cancer, extended use of hormone therapy in women who are early initiators, improved tools to personalize or individualize benefits and risks of hormone therapy, and the role of aging and genetics,” said Dr. Faubion. Other areas for further research include “the long-term benefits and risks on women’s health of lifestyle modification or complementary or nonhormone therapies, if chosen in addition to or over hormone therapy for vasomotor symptoms, bone health, and cardiovascular disease risk reduction,” she added.
The complete statement was published in Menopause: The Journal of the North American Menopause Society.
The position statement received no outside funding. The authors had no financial conflicts to disclose.
FROM MENOPAUSE
Treatments explored to ease postviral symptoms of ME/CFS and long COVID
A variety of treatments, most already commercially available, are under investigation for treating the constellation of overlapping symptoms associated with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), “long COVID,” and dysautonomia.
At the virtual annual meeting of the International Association for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis, speakers presented data for a variety of approaches to ease symptoms common across postviral conditions, such as extreme fatigue, postexertional malaise (“crash”), cognitive dysfunction (“brain fog”), orthostatic intolerance including postural orthostatic tachycardia syndrome (POTS), and chronic pain. Most of the modalities are already commercially available for other indications, although some are costly and not covered by payers for these conditions.
“ ... In the past, patients were told ‘you have chronic fatigue syndrome but there’s nothing we can do for it.’ That certainly is not the case. There aren’t cures, but there are many management techniques to improve symptoms,” Charles W. Lapp, MD, medical director of the Hunter-Hopkins Center, Charlotte, N.C., said in an interview.
A current mainstay of treatment for ME/CFS – including that triggered by COVID-19 – is activity pacing, in which patients learn to stay within their “energy envelopes” in order to avoid postexertional malaise, a worsening of all symptoms with exertion. The use of “graded exercise” is no longer recommended, per U.K. and U.S. guidelines.
Data for the following approaches were presented at the IACFS/ME conference:
Pyridostigmine (mestinon, others)
Pyridostigmine, an acetylcholinesterase inhibitor, is approved for the treatment of muscle weakness resulting from myasthenia gravis and is available in generic form. It has previously been shown to produce significant improvement in both symptom burden and heart rate response in POTS.
At the IACFS/ME conference, David M. Systrom, MD, a pulmonary and critical care medicine specialist at Brigham and Women’s Hospital and director of the Massachusetts General Hospital Cardiopulmonary laboratory, both in Boston, summarized his group’s study in patients with ME/CFS using pyridostigmine as both a potential treatment for improving exercise capacity and a proof-of-concept that neurovascular dysregulation underlies exertional intolerance in the condition.
A total of 45 patients were randomized to 60 mg oral pyridostigmine or placebo after an invasive cardiopulmonary exercise test, and a second test performed 50 minutes later. Peak VO2 increased after pyridostigmine but decreased after placebo (+13.3 mL/min vs. –40.2 mL/min, P < .05). Cardiac output and right atrial pressure were also significantly improved with pyridostigmine and worse with placebo.
“We suggest that treatable neurovascular dysregulation underlies acute exercise intolerance in ME/CFS. ... Pyridostigmine may be a useful repurposed off-label treatment [for] a subset of patients with exercise intolerance,” Dr. Systrom said.
Asked to comment, Dr. Lapp said: “We’ve used Mestinon for years because it helps with POTS and also with neurally mediated hypotension. Systrom is taking it to a new level because he’s shown that it increases preload to the heart.” However, he noted that it’s unclear whether the drug will help patients who don’t have POTS specifically. On the other hand, patients rarely experience side effects from the drug.
Since the generic tablets come only in 60-mg doses, and the starting dose is 30 mg three times a day, he advised cutting the tablets in half during titration up to 60 mg three times a day.
Oxaloacetate (benaGene)
David Lyons Kaufman, MD, of the Center for Complex Diseases, Mountain View, Calif., summarized data from his group’s recently published open-label, nonrandomized, “proof-of-concept” study on use of the commercially available nutritional supplement anhydrous enol-oxaloacetate for treating mental and physical fatigue in 76 patients with longstanding ME/CFS and 43 with long-COVID fatigue.
Oxaloacetate is a major step in the Krebs cycle within the mitochondria that are depleted in patients with ME/CFS. It is also an energy metabolite that has multiple effects in cells and mitochondria, Dr. Kaufman explained.
Doses ranging from 500 mg twice daily up to 1,000 mg three times a day were given for 6 weeks. Up to 33% of the patients with ME/CFS and up to 46.8% of the long-COVID group achieved clinical efficacy as measured by physical and mental fatigue scores, compared with just 5.9% of historical ME/CFS controls. All doses showed highly significant improvements.
The only adverse effects were occasional dyspepsia, which was avoided by taking the supplement with food, and insomnia, resolved by having them dose at breakfast and lunch, Dr. Kaufman said.
Following those preliminary data, there is now an ongoing 90-day, randomized, placebo-controlled clinical trial of 80 patients with ME/CFS using 2,000 mg anhydrous enol-oxaloacetate per day. Endpoints include multiple objective measures.
“We have a health care crisis with long COVID, and we’ve had this smoldering crisis with ME/CFS for decades that’s never been addressed. ME/CFS and long COVID, if not identical, are certainly overlapping. ... We have to pursue these translational medicine pilot studies as rapidly as possible,” Dr. Kaufman remarked.
Dr. Lapp told this news organization that it makes sense to use constituents of the Krebs cycle to improve mitochondrial function, but the problem with oxaloacetate is its cost. Dr. Kaufman mentioned that based on the preliminary trial, the therapeutic “sweet spot” appeared to be 1,000 mg twice daily. The manufacturer’s website lists the price for a single bottle of 30 250-mg capsules at $49, or $42 if purchased via a monthly subscription.
“It’s a benign drug, and it’s over the counter. I would give it to any patient who’s got a big wallet,” Dr. Lapp quipped, adding: “If they’ve got the money, they can order it tonight.”
Inspiritol
Inspiritol is an investigational “nebulized, inhaled, multimechanism medication designed to treat the major symptoms of respiratory distress with antioxidant, anti-inflammatory, and broad-spectrum antiviral and antibacterial properties. Inspiritol is composed of both endogenously produced and naturally occurring, well-tolerated biochemicals,” according to the company website.
The hypothesis, Liisa K. Selin, MD, PhD, professor of pathology at the University of Massachusetts, Worcester, said at the meeting, is that “ME/CFS and long COVID-19 result from an aberrant response to an immunological trigger like infection, which results in a permanently dysregulated immune system as a result of overactivation of CD8 T cells and subsequent exhaustion.”
Inspiritol, containing five antioxidants, acts as an immune modulator to reverse the CD8 T cell exhaustion and improve symptoms. Administration by inhaler delivers it directly to the brain from the lung. It was originally designed for use in chronic obstructive pulmonary disease and asthma and has shown efficacy for acute COVID-19, Dr. Selin said.
In a preliminary study, four patients with ME/CFS and five with long COVID have been treated with Inspiritol for 2-15 months, and all have self-reported improved symptoms. Cough has been the only reported side effect.
The company is pursuing an Investigational New Drug Application for the product with the Food and Drug Administration and has several patents pending. Dr. Lapp called Inspiritol “very interesting,” and said that reversal of CD8 “exhaustion” also would appear to be a promising approach. However, he noted, “the problem is that we don’t know what’s in it.”
Stellate ganglion block
Injection of local anesthetic near the stellate ganglion to block activity of the entire cervical sympathetic chain has been used for nearly a century to treat a variety of sympathetically mediated conditions, including complex regional pain syndrome (CRPS), shingles, and phantom-limb pain. More recently, it has been used in a variety of other conditions, including PTSD, Raynaud’s disease, menopausal hot flashes, and hyperhidrosis.
Insurance companies typically cover it for CRPS, neuropathic upper-extremity pain, hyperhidrosis, and Raynaud’s, said Luke Liu, MD, an anesthesiologist who is founder and chief executive officer of Alaska-based pain management company Neuroversion.
Deborah Duricka, PhD, also with Neuroversion, presented results from a now-published case series of 11 patients with long COVID who underwent stellate ganglion block by a board-certified anesthesiologist, first on one side at the level of C6, then on the contralateral side the following day.
Clinically meaningful benefits were seen in at least five of the patients in fatigue, memory problems, problems concentrating, rapid heartbeat, orthostatic intolerance, sleep problems, postexertional malaise, anxiety, and depression.
The hypothetical mechanism, she said, is that “sympathetic block prevents sympathetically driven vasoconstriction in carotid and vertebral arteries.”
Dr. Liu presented another case series of five patients with ME/CFS who underwent the procedure with ultrasound guidance, again on one side and the other side the next day. All had upper-limb autonomic issues such as Raynaud’s and/or neuropathic pain that had been refractory to more conventional treatments.
All five patients reported improvements in symptoms of ME/CFS, including energy level, cognition, pain, and postexertional malaise. One patient reported “feeling well for the first time in decades.” However, that patient relapsed after a mild viral illness 3.5 months after treatment. Some of the patients have required further treatments.
Dr. Lapp commented that, although the procedure is generally safe when performed by an experienced clinician, “Any time you do an injection like that, there’s a high risk that you could nick an artery or a vein or hit an essential nerve in the neck. That’s why it has to be done under fluoroscopy or ultrasound.”
He said he’s had a few patients undergo the procedure, mostly for CRPS, and they seem to have benefited from it. “It might increase cerebral blood flow and preload to the heart, so it might decrease ME/CFS symptoms and help with POTS as well.”
Nonetheless, Dr. Lapp said he wouldn’t consider stellate ganglion block as first-line treatment for ME/CFS or long COVID. “I think it would be for the treatment-resistant patient, when you’ve gone through all the treatments that we know and addressed all the comorbidities and they’re still not getting better.”
But, he added, it is a standard procedure. “Any pain clinic can do a stellate block.”
Transcutaneous auricular vagus nerve stimulation
Nicola Clague-Baker, PhD, a physiotherapist at the University of Liverpool (England), presented findings from an international survey of people with ME/CFS regarding their experience with transcutaneous auricular vagus nerve stimulation (taVNS) to manage their autonomic symptoms. The technique involves stimulation of the autonomic nervous system via the vagus nerve using electrodes applied to part of the ear. The theory is that the technique stimulates the parasympathetic nervous system and improves autonomic balance.
Two small previous trials showing benefit of vagus nerve stimulation for people with ME/CFS used more invasive and less comfortable methods of applying the stimulation rather than to the ear, Dr. Clague-Baker and colleagues noted in a poster. It has also been used successfully in treating POTS, another conference speaker noted.
A total of 131 people with ME/CFS (called simply “ME” in the United Kingdom) responded to a survey advertised on social media and websites. The majority (60%) were from the United Kingdom while the rest were from Europe, Australia, and North America. Most were female, and slightly more than half had lived with ME for 10 or more years.
The majority (72%) were still using taVNS, while 28% had stopped using it. Only 9% had used the modality for longer than a year. Respondents identified more than 30 benefits in symptoms and activities, with improvements in postexertional malaise (39%) and brain fog (37%) being the most common. One reported significant reduction in constipation.
However, respondents also mentioned more than 20 short- and long-term negatives, including headaches (15%) and long-term irritation at the site (9%). One participant reported a “big improvement in neuropathic pain, but not so much for muscles and joints.”
Overall, 80% reported that they would continue using taVNS and 67% said they would recommend it to others with ME, and 56% said that the system was mildly to very beneficial.
Dr. Lapp noted that several types of transcutaneous electrical nerve stimulation units with ear clips are sold online, and he’s seen them work well for migraine treatment. However, he cautioned that some patients have had side effects from the treatment, such as headaches and dizziness. “It’s putting an electrical current through your brain. In my mind, it’s another last-ditch measure.”
Dr. Lapp reported no financial disclosures.
A version of this article first appeared on Medscape.com.
A variety of treatments, most already commercially available, are under investigation for treating the constellation of overlapping symptoms associated with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), “long COVID,” and dysautonomia.
At the virtual annual meeting of the International Association for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis, speakers presented data for a variety of approaches to ease symptoms common across postviral conditions, such as extreme fatigue, postexertional malaise (“crash”), cognitive dysfunction (“brain fog”), orthostatic intolerance including postural orthostatic tachycardia syndrome (POTS), and chronic pain. Most of the modalities are already commercially available for other indications, although some are costly and not covered by payers for these conditions.
“ ... In the past, patients were told ‘you have chronic fatigue syndrome but there’s nothing we can do for it.’ That certainly is not the case. There aren’t cures, but there are many management techniques to improve symptoms,” Charles W. Lapp, MD, medical director of the Hunter-Hopkins Center, Charlotte, N.C., said in an interview.
A current mainstay of treatment for ME/CFS – including that triggered by COVID-19 – is activity pacing, in which patients learn to stay within their “energy envelopes” in order to avoid postexertional malaise, a worsening of all symptoms with exertion. The use of “graded exercise” is no longer recommended, per U.K. and U.S. guidelines.
Data for the following approaches were presented at the IACFS/ME conference:
Pyridostigmine (mestinon, others)
Pyridostigmine, an acetylcholinesterase inhibitor, is approved for the treatment of muscle weakness resulting from myasthenia gravis and is available in generic form. It has previously been shown to produce significant improvement in both symptom burden and heart rate response in POTS.
At the IACFS/ME conference, David M. Systrom, MD, a pulmonary and critical care medicine specialist at Brigham and Women’s Hospital and director of the Massachusetts General Hospital Cardiopulmonary laboratory, both in Boston, summarized his group’s study in patients with ME/CFS using pyridostigmine as both a potential treatment for improving exercise capacity and a proof-of-concept that neurovascular dysregulation underlies exertional intolerance in the condition.
A total of 45 patients were randomized to 60 mg oral pyridostigmine or placebo after an invasive cardiopulmonary exercise test, and a second test performed 50 minutes later. Peak VO2 increased after pyridostigmine but decreased after placebo (+13.3 mL/min vs. –40.2 mL/min, P < .05). Cardiac output and right atrial pressure were also significantly improved with pyridostigmine and worse with placebo.
“We suggest that treatable neurovascular dysregulation underlies acute exercise intolerance in ME/CFS. ... Pyridostigmine may be a useful repurposed off-label treatment [for] a subset of patients with exercise intolerance,” Dr. Systrom said.
Asked to comment, Dr. Lapp said: “We’ve used Mestinon for years because it helps with POTS and also with neurally mediated hypotension. Systrom is taking it to a new level because he’s shown that it increases preload to the heart.” However, he noted that it’s unclear whether the drug will help patients who don’t have POTS specifically. On the other hand, patients rarely experience side effects from the drug.
Since the generic tablets come only in 60-mg doses, and the starting dose is 30 mg three times a day, he advised cutting the tablets in half during titration up to 60 mg three times a day.
Oxaloacetate (benaGene)
David Lyons Kaufman, MD, of the Center for Complex Diseases, Mountain View, Calif., summarized data from his group’s recently published open-label, nonrandomized, “proof-of-concept” study on use of the commercially available nutritional supplement anhydrous enol-oxaloacetate for treating mental and physical fatigue in 76 patients with longstanding ME/CFS and 43 with long-COVID fatigue.
Oxaloacetate is a major step in the Krebs cycle within the mitochondria that are depleted in patients with ME/CFS. It is also an energy metabolite that has multiple effects in cells and mitochondria, Dr. Kaufman explained.
Doses ranging from 500 mg twice daily up to 1,000 mg three times a day were given for 6 weeks. Up to 33% of the patients with ME/CFS and up to 46.8% of the long-COVID group achieved clinical efficacy as measured by physical and mental fatigue scores, compared with just 5.9% of historical ME/CFS controls. All doses showed highly significant improvements.
The only adverse effects were occasional dyspepsia, which was avoided by taking the supplement with food, and insomnia, resolved by having them dose at breakfast and lunch, Dr. Kaufman said.
Following those preliminary data, there is now an ongoing 90-day, randomized, placebo-controlled clinical trial of 80 patients with ME/CFS using 2,000 mg anhydrous enol-oxaloacetate per day. Endpoints include multiple objective measures.
“We have a health care crisis with long COVID, and we’ve had this smoldering crisis with ME/CFS for decades that’s never been addressed. ME/CFS and long COVID, if not identical, are certainly overlapping. ... We have to pursue these translational medicine pilot studies as rapidly as possible,” Dr. Kaufman remarked.
Dr. Lapp told this news organization that it makes sense to use constituents of the Krebs cycle to improve mitochondrial function, but the problem with oxaloacetate is its cost. Dr. Kaufman mentioned that based on the preliminary trial, the therapeutic “sweet spot” appeared to be 1,000 mg twice daily. The manufacturer’s website lists the price for a single bottle of 30 250-mg capsules at $49, or $42 if purchased via a monthly subscription.
“It’s a benign drug, and it’s over the counter. I would give it to any patient who’s got a big wallet,” Dr. Lapp quipped, adding: “If they’ve got the money, they can order it tonight.”
Inspiritol
Inspiritol is an investigational “nebulized, inhaled, multimechanism medication designed to treat the major symptoms of respiratory distress with antioxidant, anti-inflammatory, and broad-spectrum antiviral and antibacterial properties. Inspiritol is composed of both endogenously produced and naturally occurring, well-tolerated biochemicals,” according to the company website.
The hypothesis, Liisa K. Selin, MD, PhD, professor of pathology at the University of Massachusetts, Worcester, said at the meeting, is that “ME/CFS and long COVID-19 result from an aberrant response to an immunological trigger like infection, which results in a permanently dysregulated immune system as a result of overactivation of CD8 T cells and subsequent exhaustion.”
Inspiritol, containing five antioxidants, acts as an immune modulator to reverse the CD8 T cell exhaustion and improve symptoms. Administration by inhaler delivers it directly to the brain from the lung. It was originally designed for use in chronic obstructive pulmonary disease and asthma and has shown efficacy for acute COVID-19, Dr. Selin said.
In a preliminary study, four patients with ME/CFS and five with long COVID have been treated with Inspiritol for 2-15 months, and all have self-reported improved symptoms. Cough has been the only reported side effect.
The company is pursuing an Investigational New Drug Application for the product with the Food and Drug Administration and has several patents pending. Dr. Lapp called Inspiritol “very interesting,” and said that reversal of CD8 “exhaustion” also would appear to be a promising approach. However, he noted, “the problem is that we don’t know what’s in it.”
Stellate ganglion block
Injection of local anesthetic near the stellate ganglion to block activity of the entire cervical sympathetic chain has been used for nearly a century to treat a variety of sympathetically mediated conditions, including complex regional pain syndrome (CRPS), shingles, and phantom-limb pain. More recently, it has been used in a variety of other conditions, including PTSD, Raynaud’s disease, menopausal hot flashes, and hyperhidrosis.
Insurance companies typically cover it for CRPS, neuropathic upper-extremity pain, hyperhidrosis, and Raynaud’s, said Luke Liu, MD, an anesthesiologist who is founder and chief executive officer of Alaska-based pain management company Neuroversion.
Deborah Duricka, PhD, also with Neuroversion, presented results from a now-published case series of 11 patients with long COVID who underwent stellate ganglion block by a board-certified anesthesiologist, first on one side at the level of C6, then on the contralateral side the following day.
Clinically meaningful benefits were seen in at least five of the patients in fatigue, memory problems, problems concentrating, rapid heartbeat, orthostatic intolerance, sleep problems, postexertional malaise, anxiety, and depression.
The hypothetical mechanism, she said, is that “sympathetic block prevents sympathetically driven vasoconstriction in carotid and vertebral arteries.”
Dr. Liu presented another case series of five patients with ME/CFS who underwent the procedure with ultrasound guidance, again on one side and the other side the next day. All had upper-limb autonomic issues such as Raynaud’s and/or neuropathic pain that had been refractory to more conventional treatments.
All five patients reported improvements in symptoms of ME/CFS, including energy level, cognition, pain, and postexertional malaise. One patient reported “feeling well for the first time in decades.” However, that patient relapsed after a mild viral illness 3.5 months after treatment. Some of the patients have required further treatments.
Dr. Lapp commented that, although the procedure is generally safe when performed by an experienced clinician, “Any time you do an injection like that, there’s a high risk that you could nick an artery or a vein or hit an essential nerve in the neck. That’s why it has to be done under fluoroscopy or ultrasound.”
He said he’s had a few patients undergo the procedure, mostly for CRPS, and they seem to have benefited from it. “It might increase cerebral blood flow and preload to the heart, so it might decrease ME/CFS symptoms and help with POTS as well.”
Nonetheless, Dr. Lapp said he wouldn’t consider stellate ganglion block as first-line treatment for ME/CFS or long COVID. “I think it would be for the treatment-resistant patient, when you’ve gone through all the treatments that we know and addressed all the comorbidities and they’re still not getting better.”
But, he added, it is a standard procedure. “Any pain clinic can do a stellate block.”
Transcutaneous auricular vagus nerve stimulation
Nicola Clague-Baker, PhD, a physiotherapist at the University of Liverpool (England), presented findings from an international survey of people with ME/CFS regarding their experience with transcutaneous auricular vagus nerve stimulation (taVNS) to manage their autonomic symptoms. The technique involves stimulation of the autonomic nervous system via the vagus nerve using electrodes applied to part of the ear. The theory is that the technique stimulates the parasympathetic nervous system and improves autonomic balance.
Two small previous trials showing benefit of vagus nerve stimulation for people with ME/CFS used more invasive and less comfortable methods of applying the stimulation rather than to the ear, Dr. Clague-Baker and colleagues noted in a poster. It has also been used successfully in treating POTS, another conference speaker noted.
A total of 131 people with ME/CFS (called simply “ME” in the United Kingdom) responded to a survey advertised on social media and websites. The majority (60%) were from the United Kingdom while the rest were from Europe, Australia, and North America. Most were female, and slightly more than half had lived with ME for 10 or more years.
The majority (72%) were still using taVNS, while 28% had stopped using it. Only 9% had used the modality for longer than a year. Respondents identified more than 30 benefits in symptoms and activities, with improvements in postexertional malaise (39%) and brain fog (37%) being the most common. One reported significant reduction in constipation.
However, respondents also mentioned more than 20 short- and long-term negatives, including headaches (15%) and long-term irritation at the site (9%). One participant reported a “big improvement in neuropathic pain, but not so much for muscles and joints.”
Overall, 80% reported that they would continue using taVNS and 67% said they would recommend it to others with ME, and 56% said that the system was mildly to very beneficial.
Dr. Lapp noted that several types of transcutaneous electrical nerve stimulation units with ear clips are sold online, and he’s seen them work well for migraine treatment. However, he cautioned that some patients have had side effects from the treatment, such as headaches and dizziness. “It’s putting an electrical current through your brain. In my mind, it’s another last-ditch measure.”
Dr. Lapp reported no financial disclosures.
A version of this article first appeared on Medscape.com.
A variety of treatments, most already commercially available, are under investigation for treating the constellation of overlapping symptoms associated with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), “long COVID,” and dysautonomia.
At the virtual annual meeting of the International Association for Chronic Fatigue Syndrome/Myalgic Encephalomyelitis, speakers presented data for a variety of approaches to ease symptoms common across postviral conditions, such as extreme fatigue, postexertional malaise (“crash”), cognitive dysfunction (“brain fog”), orthostatic intolerance including postural orthostatic tachycardia syndrome (POTS), and chronic pain. Most of the modalities are already commercially available for other indications, although some are costly and not covered by payers for these conditions.
“ ... In the past, patients were told ‘you have chronic fatigue syndrome but there’s nothing we can do for it.’ That certainly is not the case. There aren’t cures, but there are many management techniques to improve symptoms,” Charles W. Lapp, MD, medical director of the Hunter-Hopkins Center, Charlotte, N.C., said in an interview.
A current mainstay of treatment for ME/CFS – including that triggered by COVID-19 – is activity pacing, in which patients learn to stay within their “energy envelopes” in order to avoid postexertional malaise, a worsening of all symptoms with exertion. The use of “graded exercise” is no longer recommended, per U.K. and U.S. guidelines.
Data for the following approaches were presented at the IACFS/ME conference:
Pyridostigmine (mestinon, others)
Pyridostigmine, an acetylcholinesterase inhibitor, is approved for the treatment of muscle weakness resulting from myasthenia gravis and is available in generic form. It has previously been shown to produce significant improvement in both symptom burden and heart rate response in POTS.
At the IACFS/ME conference, David M. Systrom, MD, a pulmonary and critical care medicine specialist at Brigham and Women’s Hospital and director of the Massachusetts General Hospital Cardiopulmonary laboratory, both in Boston, summarized his group’s study in patients with ME/CFS using pyridostigmine as both a potential treatment for improving exercise capacity and a proof-of-concept that neurovascular dysregulation underlies exertional intolerance in the condition.
A total of 45 patients were randomized to 60 mg oral pyridostigmine or placebo after an invasive cardiopulmonary exercise test, and a second test performed 50 minutes later. Peak VO2 increased after pyridostigmine but decreased after placebo (+13.3 mL/min vs. –40.2 mL/min, P < .05). Cardiac output and right atrial pressure were also significantly improved with pyridostigmine and worse with placebo.
“We suggest that treatable neurovascular dysregulation underlies acute exercise intolerance in ME/CFS. ... Pyridostigmine may be a useful repurposed off-label treatment [for] a subset of patients with exercise intolerance,” Dr. Systrom said.
Asked to comment, Dr. Lapp said: “We’ve used Mestinon for years because it helps with POTS and also with neurally mediated hypotension. Systrom is taking it to a new level because he’s shown that it increases preload to the heart.” However, he noted that it’s unclear whether the drug will help patients who don’t have POTS specifically. On the other hand, patients rarely experience side effects from the drug.
Since the generic tablets come only in 60-mg doses, and the starting dose is 30 mg three times a day, he advised cutting the tablets in half during titration up to 60 mg three times a day.
Oxaloacetate (benaGene)
David Lyons Kaufman, MD, of the Center for Complex Diseases, Mountain View, Calif., summarized data from his group’s recently published open-label, nonrandomized, “proof-of-concept” study on use of the commercially available nutritional supplement anhydrous enol-oxaloacetate for treating mental and physical fatigue in 76 patients with longstanding ME/CFS and 43 with long-COVID fatigue.
Oxaloacetate is a major step in the Krebs cycle within the mitochondria that are depleted in patients with ME/CFS. It is also an energy metabolite that has multiple effects in cells and mitochondria, Dr. Kaufman explained.
Doses ranging from 500 mg twice daily up to 1,000 mg three times a day were given for 6 weeks. Up to 33% of the patients with ME/CFS and up to 46.8% of the long-COVID group achieved clinical efficacy as measured by physical and mental fatigue scores, compared with just 5.9% of historical ME/CFS controls. All doses showed highly significant improvements.
The only adverse effects were occasional dyspepsia, which was avoided by taking the supplement with food, and insomnia, resolved by having them dose at breakfast and lunch, Dr. Kaufman said.
Following those preliminary data, there is now an ongoing 90-day, randomized, placebo-controlled clinical trial of 80 patients with ME/CFS using 2,000 mg anhydrous enol-oxaloacetate per day. Endpoints include multiple objective measures.
“We have a health care crisis with long COVID, and we’ve had this smoldering crisis with ME/CFS for decades that’s never been addressed. ME/CFS and long COVID, if not identical, are certainly overlapping. ... We have to pursue these translational medicine pilot studies as rapidly as possible,” Dr. Kaufman remarked.
Dr. Lapp told this news organization that it makes sense to use constituents of the Krebs cycle to improve mitochondrial function, but the problem with oxaloacetate is its cost. Dr. Kaufman mentioned that based on the preliminary trial, the therapeutic “sweet spot” appeared to be 1,000 mg twice daily. The manufacturer’s website lists the price for a single bottle of 30 250-mg capsules at $49, or $42 if purchased via a monthly subscription.
“It’s a benign drug, and it’s over the counter. I would give it to any patient who’s got a big wallet,” Dr. Lapp quipped, adding: “If they’ve got the money, they can order it tonight.”
Inspiritol
Inspiritol is an investigational “nebulized, inhaled, multimechanism medication designed to treat the major symptoms of respiratory distress with antioxidant, anti-inflammatory, and broad-spectrum antiviral and antibacterial properties. Inspiritol is composed of both endogenously produced and naturally occurring, well-tolerated biochemicals,” according to the company website.
The hypothesis, Liisa K. Selin, MD, PhD, professor of pathology at the University of Massachusetts, Worcester, said at the meeting, is that “ME/CFS and long COVID-19 result from an aberrant response to an immunological trigger like infection, which results in a permanently dysregulated immune system as a result of overactivation of CD8 T cells and subsequent exhaustion.”
Inspiritol, containing five antioxidants, acts as an immune modulator to reverse the CD8 T cell exhaustion and improve symptoms. Administration by inhaler delivers it directly to the brain from the lung. It was originally designed for use in chronic obstructive pulmonary disease and asthma and has shown efficacy for acute COVID-19, Dr. Selin said.
In a preliminary study, four patients with ME/CFS and five with long COVID have been treated with Inspiritol for 2-15 months, and all have self-reported improved symptoms. Cough has been the only reported side effect.
The company is pursuing an Investigational New Drug Application for the product with the Food and Drug Administration and has several patents pending. Dr. Lapp called Inspiritol “very interesting,” and said that reversal of CD8 “exhaustion” also would appear to be a promising approach. However, he noted, “the problem is that we don’t know what’s in it.”
Stellate ganglion block
Injection of local anesthetic near the stellate ganglion to block activity of the entire cervical sympathetic chain has been used for nearly a century to treat a variety of sympathetically mediated conditions, including complex regional pain syndrome (CRPS), shingles, and phantom-limb pain. More recently, it has been used in a variety of other conditions, including PTSD, Raynaud’s disease, menopausal hot flashes, and hyperhidrosis.
Insurance companies typically cover it for CRPS, neuropathic upper-extremity pain, hyperhidrosis, and Raynaud’s, said Luke Liu, MD, an anesthesiologist who is founder and chief executive officer of Alaska-based pain management company Neuroversion.
Deborah Duricka, PhD, also with Neuroversion, presented results from a now-published case series of 11 patients with long COVID who underwent stellate ganglion block by a board-certified anesthesiologist, first on one side at the level of C6, then on the contralateral side the following day.
Clinically meaningful benefits were seen in at least five of the patients in fatigue, memory problems, problems concentrating, rapid heartbeat, orthostatic intolerance, sleep problems, postexertional malaise, anxiety, and depression.
The hypothetical mechanism, she said, is that “sympathetic block prevents sympathetically driven vasoconstriction in carotid and vertebral arteries.”
Dr. Liu presented another case series of five patients with ME/CFS who underwent the procedure with ultrasound guidance, again on one side and the other side the next day. All had upper-limb autonomic issues such as Raynaud’s and/or neuropathic pain that had been refractory to more conventional treatments.
All five patients reported improvements in symptoms of ME/CFS, including energy level, cognition, pain, and postexertional malaise. One patient reported “feeling well for the first time in decades.” However, that patient relapsed after a mild viral illness 3.5 months after treatment. Some of the patients have required further treatments.
Dr. Lapp commented that, although the procedure is generally safe when performed by an experienced clinician, “Any time you do an injection like that, there’s a high risk that you could nick an artery or a vein or hit an essential nerve in the neck. That’s why it has to be done under fluoroscopy or ultrasound.”
He said he’s had a few patients undergo the procedure, mostly for CRPS, and they seem to have benefited from it. “It might increase cerebral blood flow and preload to the heart, so it might decrease ME/CFS symptoms and help with POTS as well.”
Nonetheless, Dr. Lapp said he wouldn’t consider stellate ganglion block as first-line treatment for ME/CFS or long COVID. “I think it would be for the treatment-resistant patient, when you’ve gone through all the treatments that we know and addressed all the comorbidities and they’re still not getting better.”
But, he added, it is a standard procedure. “Any pain clinic can do a stellate block.”
Transcutaneous auricular vagus nerve stimulation
Nicola Clague-Baker, PhD, a physiotherapist at the University of Liverpool (England), presented findings from an international survey of people with ME/CFS regarding their experience with transcutaneous auricular vagus nerve stimulation (taVNS) to manage their autonomic symptoms. The technique involves stimulation of the autonomic nervous system via the vagus nerve using electrodes applied to part of the ear. The theory is that the technique stimulates the parasympathetic nervous system and improves autonomic balance.
Two small previous trials showing benefit of vagus nerve stimulation for people with ME/CFS used more invasive and less comfortable methods of applying the stimulation rather than to the ear, Dr. Clague-Baker and colleagues noted in a poster. It has also been used successfully in treating POTS, another conference speaker noted.
A total of 131 people with ME/CFS (called simply “ME” in the United Kingdom) responded to a survey advertised on social media and websites. The majority (60%) were from the United Kingdom while the rest were from Europe, Australia, and North America. Most were female, and slightly more than half had lived with ME for 10 or more years.
The majority (72%) were still using taVNS, while 28% had stopped using it. Only 9% had used the modality for longer than a year. Respondents identified more than 30 benefits in symptoms and activities, with improvements in postexertional malaise (39%) and brain fog (37%) being the most common. One reported significant reduction in constipation.
However, respondents also mentioned more than 20 short- and long-term negatives, including headaches (15%) and long-term irritation at the site (9%). One participant reported a “big improvement in neuropathic pain, but not so much for muscles and joints.”
Overall, 80% reported that they would continue using taVNS and 67% said they would recommend it to others with ME, and 56% said that the system was mildly to very beneficial.
Dr. Lapp noted that several types of transcutaneous electrical nerve stimulation units with ear clips are sold online, and he’s seen them work well for migraine treatment. However, he cautioned that some patients have had side effects from the treatment, such as headaches and dizziness. “It’s putting an electrical current through your brain. In my mind, it’s another last-ditch measure.”
Dr. Lapp reported no financial disclosures.
A version of this article first appeared on Medscape.com.
FROM IACFSME 2022
Topical ruxolitinib quickly relieves atopic dermatitis itch in Black patients
“Ruxolitinib cream monotherapy over 8 weeks was associated with rapid and considerable itch relief in Black or African American patients with AD and was well tolerated,” the study authors wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology.
AD can behave differently in different racial groups and can be especially bothersome in Black patients. AD has a prevalence of about 20% in Black children and 5%-10% in Black adults. Black children are roughly twice as likely to be diagnosed with AD, and to have severe AD, than White children, according to the authors.
Lead author Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, and colleagues used pooled data from two identically designed phase 3 studies to describe the effects of the cream formulation of the Janus kinase (JAK) 1 and JAK 2 inhibitor ruxolitinib on itch in Black patients.
Topical ruxolitinib (Opzelura), 1.5%, was approved last September for treating AD in non-immunocompromised patients with mild to moderate AD, ages 12 years and older. In July 2022, it was approved for the treatment of nonsegmental vitiligo in the same age group.
FDA approval for AD was based on the results of the TRuE-AD1 and TRuE-AD2 double-blind randomized trials, which enrolled about 1,200 patients over age 12 with AD. These patients included 292 Black teenagers and adults between aged 12-71 years who had AD for 2 years or longer, with an Investigator’s Global Assessment (IGA) score of 2 or 3, with 3%-20% affected body surface area, excluding the scalp.
Of the 292 patients, those in the two treatment groups (n = 231) applied ruxolitinib cream twice a day for 8 weeks (0.75% in 118 patients and 1.5% in 113 patients) and 61 applied the vehicle. They used electronic diaries to record the worst level of itch they had experienced each day, from 0 (no itch) to 10 (worst imaginable itch). The main results were as follows:
- Mean itch numerical rating scale (NRS) scores at baseline were 5.3 and 5.4 for ruxolitinib cream 0.75% and 1.5%, respectively, and 5.7 for vehicle. Within about 12 hours of first application, mean itch NRS scores dropped –0.6 and –0.7 from baseline among those treated with ruxolitinib cream 0.75% and 1.5%, respectively, compared with –0.2 for those on the vehicle. At day 4, the decreases were –1.4 and –1.6 for ruxolitinib cream 0.75% and 1.5%, respectively, versus –0.6 for the vehicle (P = .026 and P = .005, respectively, vs. vehicle).
- At day 2, among the 187 patients with a baseline itch NRS score 4 or higher, more patients achieved 4-point or greater itch NRS improvement: 6.1% and 16.4% for ruxolitinib cream 0.75% and 1.5%, respectively versus 0% for vehicle. At day 7, the differences were 15.9% and 26.6% versus 3%, respectively. And by week 8, they increased to 30.1% and 43.2% versus 17.5% (P = .212 and P = .009), respectively.
- At week 2, 19% of patients in the 0.75% formulation group and 19.4% of patients in the 1.5% formulation group, compared with 5.3% in the vehicle group, reported no days of itch on question 1 of the Patient-Oriented Eczema Measure (POEM) questionnaire that evaluated various aspects of the disease over the previous week. By week 8, the differences grew to 34% and 30.8% versus 12.2%, respectively.
- Adverse events, reported by 14.4% and 22.1% of patients on 0.75% and 1.5% ruxolitinib, respectively, and by 32.8% of patients who received the vehicle, were headaches, upper respiratory tract infection, and application site pain.
Ruxolitinib may be an alternative to systemic immunosuppressives
Asked to comment on the results, Amy J. McMichael, MD, professor of dermatology at Wake Forest University School of Medicine, Winston-Salem, N.C., called itch “one of the major life disruptors in atopic dermatitis.”
Providers often assume that patients of different races respond similarly to treatment, but that is not always true, she noted in an email.
“This study proves ruxolitinib’s effectiveness in Black patients, who often have more severe atopic dermatitis signs and symptoms,” said Dr. McMichael, who was not involved in the study. “The fact that atopic dermatitis in patients of color has been singled out to examine efficacy is a great way to show that the findings are not just in those who have thinner plaques and potentially less longstanding thickening of the skin from scratching (lichenification),” she added.
Dr. McMichael welcomed the lack of systemic side effects and quick relief of itch with this treatment, noting that the effect on itch “is rare with other treatments and extremely rare with other topical medications.”
The effect of topical ruxolitinib on pruritus “was interesting and surprising because very few available topical medications can control itch,” she explained. “The strongest topical steroids can help with pruritus, but they have the risk for skin thinning (atrophy),” while topical ruxolitinib is not associated with skin atrophy.
“After topical steroids fail as first-line treatment, it is likely that more patients will be given this topical medication rather than be moved to immunosuppressive systemic medications,” she noted.
All study authors report relevant relationships with Incyte Corporation, which manufactures ruxolitinib and funded the study, and several authors report employment and shareholding interests in the company. Dr. McMichael reports no relevant relationship with the study.
A version of this article first appeared on Medscape.com.
“Ruxolitinib cream monotherapy over 8 weeks was associated with rapid and considerable itch relief in Black or African American patients with AD and was well tolerated,” the study authors wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology.
AD can behave differently in different racial groups and can be especially bothersome in Black patients. AD has a prevalence of about 20% in Black children and 5%-10% in Black adults. Black children are roughly twice as likely to be diagnosed with AD, and to have severe AD, than White children, according to the authors.
Lead author Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, and colleagues used pooled data from two identically designed phase 3 studies to describe the effects of the cream formulation of the Janus kinase (JAK) 1 and JAK 2 inhibitor ruxolitinib on itch in Black patients.
Topical ruxolitinib (Opzelura), 1.5%, was approved last September for treating AD in non-immunocompromised patients with mild to moderate AD, ages 12 years and older. In July 2022, it was approved for the treatment of nonsegmental vitiligo in the same age group.
FDA approval for AD was based on the results of the TRuE-AD1 and TRuE-AD2 double-blind randomized trials, which enrolled about 1,200 patients over age 12 with AD. These patients included 292 Black teenagers and adults between aged 12-71 years who had AD for 2 years or longer, with an Investigator’s Global Assessment (IGA) score of 2 or 3, with 3%-20% affected body surface area, excluding the scalp.
Of the 292 patients, those in the two treatment groups (n = 231) applied ruxolitinib cream twice a day for 8 weeks (0.75% in 118 patients and 1.5% in 113 patients) and 61 applied the vehicle. They used electronic diaries to record the worst level of itch they had experienced each day, from 0 (no itch) to 10 (worst imaginable itch). The main results were as follows:
- Mean itch numerical rating scale (NRS) scores at baseline were 5.3 and 5.4 for ruxolitinib cream 0.75% and 1.5%, respectively, and 5.7 for vehicle. Within about 12 hours of first application, mean itch NRS scores dropped –0.6 and –0.7 from baseline among those treated with ruxolitinib cream 0.75% and 1.5%, respectively, compared with –0.2 for those on the vehicle. At day 4, the decreases were –1.4 and –1.6 for ruxolitinib cream 0.75% and 1.5%, respectively, versus –0.6 for the vehicle (P = .026 and P = .005, respectively, vs. vehicle).
- At day 2, among the 187 patients with a baseline itch NRS score 4 or higher, more patients achieved 4-point or greater itch NRS improvement: 6.1% and 16.4% for ruxolitinib cream 0.75% and 1.5%, respectively versus 0% for vehicle. At day 7, the differences were 15.9% and 26.6% versus 3%, respectively. And by week 8, they increased to 30.1% and 43.2% versus 17.5% (P = .212 and P = .009), respectively.
- At week 2, 19% of patients in the 0.75% formulation group and 19.4% of patients in the 1.5% formulation group, compared with 5.3% in the vehicle group, reported no days of itch on question 1 of the Patient-Oriented Eczema Measure (POEM) questionnaire that evaluated various aspects of the disease over the previous week. By week 8, the differences grew to 34% and 30.8% versus 12.2%, respectively.
- Adverse events, reported by 14.4% and 22.1% of patients on 0.75% and 1.5% ruxolitinib, respectively, and by 32.8% of patients who received the vehicle, were headaches, upper respiratory tract infection, and application site pain.
Ruxolitinib may be an alternative to systemic immunosuppressives
Asked to comment on the results, Amy J. McMichael, MD, professor of dermatology at Wake Forest University School of Medicine, Winston-Salem, N.C., called itch “one of the major life disruptors in atopic dermatitis.”
Providers often assume that patients of different races respond similarly to treatment, but that is not always true, she noted in an email.
“This study proves ruxolitinib’s effectiveness in Black patients, who often have more severe atopic dermatitis signs and symptoms,” said Dr. McMichael, who was not involved in the study. “The fact that atopic dermatitis in patients of color has been singled out to examine efficacy is a great way to show that the findings are not just in those who have thinner plaques and potentially less longstanding thickening of the skin from scratching (lichenification),” she added.
Dr. McMichael welcomed the lack of systemic side effects and quick relief of itch with this treatment, noting that the effect on itch “is rare with other treatments and extremely rare with other topical medications.”
The effect of topical ruxolitinib on pruritus “was interesting and surprising because very few available topical medications can control itch,” she explained. “The strongest topical steroids can help with pruritus, but they have the risk for skin thinning (atrophy),” while topical ruxolitinib is not associated with skin atrophy.
“After topical steroids fail as first-line treatment, it is likely that more patients will be given this topical medication rather than be moved to immunosuppressive systemic medications,” she noted.
All study authors report relevant relationships with Incyte Corporation, which manufactures ruxolitinib and funded the study, and several authors report employment and shareholding interests in the company. Dr. McMichael reports no relevant relationship with the study.
A version of this article first appeared on Medscape.com.
“Ruxolitinib cream monotherapy over 8 weeks was associated with rapid and considerable itch relief in Black or African American patients with AD and was well tolerated,” the study authors wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology.
AD can behave differently in different racial groups and can be especially bothersome in Black patients. AD has a prevalence of about 20% in Black children and 5%-10% in Black adults. Black children are roughly twice as likely to be diagnosed with AD, and to have severe AD, than White children, according to the authors.
Lead author Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, and colleagues used pooled data from two identically designed phase 3 studies to describe the effects of the cream formulation of the Janus kinase (JAK) 1 and JAK 2 inhibitor ruxolitinib on itch in Black patients.
Topical ruxolitinib (Opzelura), 1.5%, was approved last September for treating AD in non-immunocompromised patients with mild to moderate AD, ages 12 years and older. In July 2022, it was approved for the treatment of nonsegmental vitiligo in the same age group.
FDA approval for AD was based on the results of the TRuE-AD1 and TRuE-AD2 double-blind randomized trials, which enrolled about 1,200 patients over age 12 with AD. These patients included 292 Black teenagers and adults between aged 12-71 years who had AD for 2 years or longer, with an Investigator’s Global Assessment (IGA) score of 2 or 3, with 3%-20% affected body surface area, excluding the scalp.
Of the 292 patients, those in the two treatment groups (n = 231) applied ruxolitinib cream twice a day for 8 weeks (0.75% in 118 patients and 1.5% in 113 patients) and 61 applied the vehicle. They used electronic diaries to record the worst level of itch they had experienced each day, from 0 (no itch) to 10 (worst imaginable itch). The main results were as follows:
- Mean itch numerical rating scale (NRS) scores at baseline were 5.3 and 5.4 for ruxolitinib cream 0.75% and 1.5%, respectively, and 5.7 for vehicle. Within about 12 hours of first application, mean itch NRS scores dropped –0.6 and –0.7 from baseline among those treated with ruxolitinib cream 0.75% and 1.5%, respectively, compared with –0.2 for those on the vehicle. At day 4, the decreases were –1.4 and –1.6 for ruxolitinib cream 0.75% and 1.5%, respectively, versus –0.6 for the vehicle (P = .026 and P = .005, respectively, vs. vehicle).
- At day 2, among the 187 patients with a baseline itch NRS score 4 or higher, more patients achieved 4-point or greater itch NRS improvement: 6.1% and 16.4% for ruxolitinib cream 0.75% and 1.5%, respectively versus 0% for vehicle. At day 7, the differences were 15.9% and 26.6% versus 3%, respectively. And by week 8, they increased to 30.1% and 43.2% versus 17.5% (P = .212 and P = .009), respectively.
- At week 2, 19% of patients in the 0.75% formulation group and 19.4% of patients in the 1.5% formulation group, compared with 5.3% in the vehicle group, reported no days of itch on question 1 of the Patient-Oriented Eczema Measure (POEM) questionnaire that evaluated various aspects of the disease over the previous week. By week 8, the differences grew to 34% and 30.8% versus 12.2%, respectively.
- Adverse events, reported by 14.4% and 22.1% of patients on 0.75% and 1.5% ruxolitinib, respectively, and by 32.8% of patients who received the vehicle, were headaches, upper respiratory tract infection, and application site pain.
Ruxolitinib may be an alternative to systemic immunosuppressives
Asked to comment on the results, Amy J. McMichael, MD, professor of dermatology at Wake Forest University School of Medicine, Winston-Salem, N.C., called itch “one of the major life disruptors in atopic dermatitis.”
Providers often assume that patients of different races respond similarly to treatment, but that is not always true, she noted in an email.
“This study proves ruxolitinib’s effectiveness in Black patients, who often have more severe atopic dermatitis signs and symptoms,” said Dr. McMichael, who was not involved in the study. “The fact that atopic dermatitis in patients of color has been singled out to examine efficacy is a great way to show that the findings are not just in those who have thinner plaques and potentially less longstanding thickening of the skin from scratching (lichenification),” she added.
Dr. McMichael welcomed the lack of systemic side effects and quick relief of itch with this treatment, noting that the effect on itch “is rare with other treatments and extremely rare with other topical medications.”
The effect of topical ruxolitinib on pruritus “was interesting and surprising because very few available topical medications can control itch,” she explained. “The strongest topical steroids can help with pruritus, but they have the risk for skin thinning (atrophy),” while topical ruxolitinib is not associated with skin atrophy.
“After topical steroids fail as first-line treatment, it is likely that more patients will be given this topical medication rather than be moved to immunosuppressive systemic medications,” she noted.
All study authors report relevant relationships with Incyte Corporation, which manufactures ruxolitinib and funded the study, and several authors report employment and shareholding interests in the company. Dr. McMichael reports no relevant relationship with the study.
A version of this article first appeared on Medscape.com.
FROM SID 2022
Antibiotic-resistant bacteria emerging in community settings
A new study from the Centers for Disease Control and Prevention found that
Traditionally, CRE has been thought of as a nosocomial infection, acquired in a hospital or other health care facility (nursing home, long-term acute care hospital, dialysis center, etc.). This is the first population-level study to show otherwise, with fully 10% of the CRE isolates found to be community acquired.
CREs are a group of multidrug-resistant bacteria considered an urgent health threat by the CDC because they can rapidly spread between patients, especially those who are most seriously ill and vulnerable, and because they are so difficult to treat. These patients often require treatment with toxic antibiotics, such as colistin, and carry a high mortality rate – up to 50% in some studies.
Overall, 30% of CREs carry a carbapenemase – an enzyme that can make them resistant to carbapenem antibiotics. The genes for this are readily transferable between bacteria and help account for their spread in hospitals.
But in this study, published in the American Journal of Infection Control, of the 12 isolates that underwent whole-genome sequencing, 42% of the CA-CRE isolates carried the carbapenemase gene. Lead author Sandra Bulens, MPH, a health scientist in the CDC’s division of health care quality promotion, said in an interview, “The findings highlight the potential for CP-CRE to move from health care settings into the community. The fact that 5 of the 12 isolates harbored a carbapenemase gene introduces new challenges for controlling spread of CP-CRE.”
CDC researchers analyzed data from eight U.S. metropolitan areas between 2012 and 2015 as part of the CDC’s Emerging Infections Program (EIP) health care–associated infections – community interface activity, which conducts surveillance for CRE and other drug-resistant gram-negative bacteria. Cases of CA-CRE were compared with HCA-CRE, with 1499 cases in 1,194 case-patients being analyzed. Though Klebsiella pneumoniae was the most common isolate, there were some differences between metropolitan areas.
The incidence of CRE cases per 100,000 population was 2.96 (95% confidence interval, 2.81-3.11) overall and 0.29 (95% CI, 0.25-0.25) for CA-CRE. Most CA-CRE cases were in White persons (73%) and women (84%). Urine cultures were the source of 98% of all CA-CRE cases, compared with 86% of HCA-CRE cases (P < .001). Though small numbers, the numbers of patients with CA-CRE without apparent underlying medical condition (n = 51; 37%) was greater when compared with patients with HCA-CRE (n = 36; 3%; P < .001).
Asked for independent comment, Lance Price, PhD, of George Washington University and the founding director of GW’s Antibiotic Resistance Action Center, Washington, said, “what’s striking about these data is that: ‘Who is the front line, at least in the United States for CRE?’ It’s women, older women. ... At some point, we have to frame drug resistance as a women’s health issue.”
Dr. Price noted that the 10% of patients with CA-CRE acquired it in the community. “I would argue that probably none of them had any idea, because there’s this silent community epidemic,” he said. “It’s asymptomatic carriage and transmission in the community. Somebody can be this walking reservoir of these really dangerous bacteria and have no idea.”
This is an increasingly serious problem for women, Dr. Price said, because, “with a community-acquired bladder infection, you’re going to call your doctor or go to an urgent care, and they’re not going to test you. They’re going to guess what you have, and they’re going to prescribe an antibiotic, and that antibiotic is going to fail. So then your bladder infection continues, and then you wait a few more days, and you start to get flank pain and kidney infection. ... If you start getting a fever, they might admit you. They are going to start treating you immediately, and they might miss it because you’ve got this organism that’s resistant to all the best antibiotics. ... The gateway to the blood is the UTI.”
Because of such empiric treatment and increasing resistance, the risk for treatment failure is quite high, especially for older women. Ms. Bulens, however, said that, “[although] 10% of CRE were in persons without health care risk factors, the proportion of all UTIs in this population that are CRE is going to be very, very small.”
This study involved cultures from 2012 to 2015. Before the pandemic, from 2012 to 2017, U.S. deaths from antibiotic resistance fell by 18% overall and by 30% in hospitals.
But in the first year of the COVID-19 pandemic, there was a 15% increase in infections and deaths from antibiotic-resistant (AMR), hospital-acquired bacteria. In 2020, 29,400 patients died from AMR infections. There was a 78% increase in carbapenem-resistant Acinetobacter baumannii health care–associated infections, a 35% increase in carbapenem-resistant Enterobacterales, and 32% increases in both multidrug-resistant Pseudomonas aeruginosa and extended-spectrum beta-lactamase–producing Enterobacterales. Aside from gram-negative bacteria, methicillin-resistant Staphylococcus aureus rose 13%, and Candida auris rose 60%. But owing to limited surveillance, recent sound figures are lacking.
Ms. Bulens and Dr. Price reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A new study from the Centers for Disease Control and Prevention found that
Traditionally, CRE has been thought of as a nosocomial infection, acquired in a hospital or other health care facility (nursing home, long-term acute care hospital, dialysis center, etc.). This is the first population-level study to show otherwise, with fully 10% of the CRE isolates found to be community acquired.
CREs are a group of multidrug-resistant bacteria considered an urgent health threat by the CDC because they can rapidly spread between patients, especially those who are most seriously ill and vulnerable, and because they are so difficult to treat. These patients often require treatment with toxic antibiotics, such as colistin, and carry a high mortality rate – up to 50% in some studies.
Overall, 30% of CREs carry a carbapenemase – an enzyme that can make them resistant to carbapenem antibiotics. The genes for this are readily transferable between bacteria and help account for their spread in hospitals.
But in this study, published in the American Journal of Infection Control, of the 12 isolates that underwent whole-genome sequencing, 42% of the CA-CRE isolates carried the carbapenemase gene. Lead author Sandra Bulens, MPH, a health scientist in the CDC’s division of health care quality promotion, said in an interview, “The findings highlight the potential for CP-CRE to move from health care settings into the community. The fact that 5 of the 12 isolates harbored a carbapenemase gene introduces new challenges for controlling spread of CP-CRE.”
CDC researchers analyzed data from eight U.S. metropolitan areas between 2012 and 2015 as part of the CDC’s Emerging Infections Program (EIP) health care–associated infections – community interface activity, which conducts surveillance for CRE and other drug-resistant gram-negative bacteria. Cases of CA-CRE were compared with HCA-CRE, with 1499 cases in 1,194 case-patients being analyzed. Though Klebsiella pneumoniae was the most common isolate, there were some differences between metropolitan areas.
The incidence of CRE cases per 100,000 population was 2.96 (95% confidence interval, 2.81-3.11) overall and 0.29 (95% CI, 0.25-0.25) for CA-CRE. Most CA-CRE cases were in White persons (73%) and women (84%). Urine cultures were the source of 98% of all CA-CRE cases, compared with 86% of HCA-CRE cases (P < .001). Though small numbers, the numbers of patients with CA-CRE without apparent underlying medical condition (n = 51; 37%) was greater when compared with patients with HCA-CRE (n = 36; 3%; P < .001).
Asked for independent comment, Lance Price, PhD, of George Washington University and the founding director of GW’s Antibiotic Resistance Action Center, Washington, said, “what’s striking about these data is that: ‘Who is the front line, at least in the United States for CRE?’ It’s women, older women. ... At some point, we have to frame drug resistance as a women’s health issue.”
Dr. Price noted that the 10% of patients with CA-CRE acquired it in the community. “I would argue that probably none of them had any idea, because there’s this silent community epidemic,” he said. “It’s asymptomatic carriage and transmission in the community. Somebody can be this walking reservoir of these really dangerous bacteria and have no idea.”
This is an increasingly serious problem for women, Dr. Price said, because, “with a community-acquired bladder infection, you’re going to call your doctor or go to an urgent care, and they’re not going to test you. They’re going to guess what you have, and they’re going to prescribe an antibiotic, and that antibiotic is going to fail. So then your bladder infection continues, and then you wait a few more days, and you start to get flank pain and kidney infection. ... If you start getting a fever, they might admit you. They are going to start treating you immediately, and they might miss it because you’ve got this organism that’s resistant to all the best antibiotics. ... The gateway to the blood is the UTI.”
Because of such empiric treatment and increasing resistance, the risk for treatment failure is quite high, especially for older women. Ms. Bulens, however, said that, “[although] 10% of CRE were in persons without health care risk factors, the proportion of all UTIs in this population that are CRE is going to be very, very small.”
This study involved cultures from 2012 to 2015. Before the pandemic, from 2012 to 2017, U.S. deaths from antibiotic resistance fell by 18% overall and by 30% in hospitals.
But in the first year of the COVID-19 pandemic, there was a 15% increase in infections and deaths from antibiotic-resistant (AMR), hospital-acquired bacteria. In 2020, 29,400 patients died from AMR infections. There was a 78% increase in carbapenem-resistant Acinetobacter baumannii health care–associated infections, a 35% increase in carbapenem-resistant Enterobacterales, and 32% increases in both multidrug-resistant Pseudomonas aeruginosa and extended-spectrum beta-lactamase–producing Enterobacterales. Aside from gram-negative bacteria, methicillin-resistant Staphylococcus aureus rose 13%, and Candida auris rose 60%. But owing to limited surveillance, recent sound figures are lacking.
Ms. Bulens and Dr. Price reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A new study from the Centers for Disease Control and Prevention found that
Traditionally, CRE has been thought of as a nosocomial infection, acquired in a hospital or other health care facility (nursing home, long-term acute care hospital, dialysis center, etc.). This is the first population-level study to show otherwise, with fully 10% of the CRE isolates found to be community acquired.
CREs are a group of multidrug-resistant bacteria considered an urgent health threat by the CDC because they can rapidly spread between patients, especially those who are most seriously ill and vulnerable, and because they are so difficult to treat. These patients often require treatment with toxic antibiotics, such as colistin, and carry a high mortality rate – up to 50% in some studies.
Overall, 30% of CREs carry a carbapenemase – an enzyme that can make them resistant to carbapenem antibiotics. The genes for this are readily transferable between bacteria and help account for their spread in hospitals.
But in this study, published in the American Journal of Infection Control, of the 12 isolates that underwent whole-genome sequencing, 42% of the CA-CRE isolates carried the carbapenemase gene. Lead author Sandra Bulens, MPH, a health scientist in the CDC’s division of health care quality promotion, said in an interview, “The findings highlight the potential for CP-CRE to move from health care settings into the community. The fact that 5 of the 12 isolates harbored a carbapenemase gene introduces new challenges for controlling spread of CP-CRE.”
CDC researchers analyzed data from eight U.S. metropolitan areas between 2012 and 2015 as part of the CDC’s Emerging Infections Program (EIP) health care–associated infections – community interface activity, which conducts surveillance for CRE and other drug-resistant gram-negative bacteria. Cases of CA-CRE were compared with HCA-CRE, with 1499 cases in 1,194 case-patients being analyzed. Though Klebsiella pneumoniae was the most common isolate, there were some differences between metropolitan areas.
The incidence of CRE cases per 100,000 population was 2.96 (95% confidence interval, 2.81-3.11) overall and 0.29 (95% CI, 0.25-0.25) for CA-CRE. Most CA-CRE cases were in White persons (73%) and women (84%). Urine cultures were the source of 98% of all CA-CRE cases, compared with 86% of HCA-CRE cases (P < .001). Though small numbers, the numbers of patients with CA-CRE without apparent underlying medical condition (n = 51; 37%) was greater when compared with patients with HCA-CRE (n = 36; 3%; P < .001).
Asked for independent comment, Lance Price, PhD, of George Washington University and the founding director of GW’s Antibiotic Resistance Action Center, Washington, said, “what’s striking about these data is that: ‘Who is the front line, at least in the United States for CRE?’ It’s women, older women. ... At some point, we have to frame drug resistance as a women’s health issue.”
Dr. Price noted that the 10% of patients with CA-CRE acquired it in the community. “I would argue that probably none of them had any idea, because there’s this silent community epidemic,” he said. “It’s asymptomatic carriage and transmission in the community. Somebody can be this walking reservoir of these really dangerous bacteria and have no idea.”
This is an increasingly serious problem for women, Dr. Price said, because, “with a community-acquired bladder infection, you’re going to call your doctor or go to an urgent care, and they’re not going to test you. They’re going to guess what you have, and they’re going to prescribe an antibiotic, and that antibiotic is going to fail. So then your bladder infection continues, and then you wait a few more days, and you start to get flank pain and kidney infection. ... If you start getting a fever, they might admit you. They are going to start treating you immediately, and they might miss it because you’ve got this organism that’s resistant to all the best antibiotics. ... The gateway to the blood is the UTI.”
Because of such empiric treatment and increasing resistance, the risk for treatment failure is quite high, especially for older women. Ms. Bulens, however, said that, “[although] 10% of CRE were in persons without health care risk factors, the proportion of all UTIs in this population that are CRE is going to be very, very small.”
This study involved cultures from 2012 to 2015. Before the pandemic, from 2012 to 2017, U.S. deaths from antibiotic resistance fell by 18% overall and by 30% in hospitals.
But in the first year of the COVID-19 pandemic, there was a 15% increase in infections and deaths from antibiotic-resistant (AMR), hospital-acquired bacteria. In 2020, 29,400 patients died from AMR infections. There was a 78% increase in carbapenem-resistant Acinetobacter baumannii health care–associated infections, a 35% increase in carbapenem-resistant Enterobacterales, and 32% increases in both multidrug-resistant Pseudomonas aeruginosa and extended-spectrum beta-lactamase–producing Enterobacterales. Aside from gram-negative bacteria, methicillin-resistant Staphylococcus aureus rose 13%, and Candida auris rose 60%. But owing to limited surveillance, recent sound figures are lacking.
Ms. Bulens and Dr. Price reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE AMERICAN JOURNAL OF INFECTION CONTROL