User login
FDA issues warning about use of dermal fillers with needle-free devices
.

Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
.
Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
.
Specifically, the warning advises consumers and health care professionals “not to use needle-free devices such as hyaluron pens for injection of hyaluronic acid (HA) or other lip and facial fillers, collectively and commonly referred to as dermal fillers or fillers.”
According to the statement, the agency “is aware of serious injuries and in some cases, permanent harm to the skin, lips, or eyes with the use of needle-free devices for injection of fillers.”
Needle-free devices and lip and facial fillers for use with these devices are being sold directly to consumers online, and are promoted on social media “to increase lip volume, improve the appearance of wrinkles, change the shape of the nose, and other similar procedures,” according to the FDA warning.
The FDA points out that FDA-approved dermal fillers are for prescription use only, and should be administered only by licensed health care professionals using a syringe with a needle or cannula, and advises consumers not to buy or use lip or facial fillers sold directly to the public.
These products may be contaminated with infectious agents or chemicals. Moreover, “needle-free injection devices for aesthetic purposes do not provide enough control over where the injected product is placed,” the statement adds. In addition to infections, other risks include bleeding and bruising, formation of lumps, allergic reactions, blockage of a blood vessel (which can result in necrosis, blindness, or stroke), and transmission of diseases from sharing devices.
The FDA’s recommendations for health care providers include not using any aesthetic fillers with a needle-free device, and not using approved dermal fillers in such devices.
The American Society for Dermatologic Surgery Association (ASDSA) commended the FDA on the safety communication in a statement issued on October 11. In February, the ASDSA issued an alert about children using hyaluron pens to self-inject hyaluronic filler into the epidermal and upper dermal skin layers.
“I am pleased that the FDA has taken notice of this disturbing new trend, especially that of children using these devices on social media,” ASDSA president Mathew Avram, MD, JD, director of the Dermatology Laser and Cosmetic Center, at Massachusetts General Hospital, Boston, said in the statement. “The complexity of facial anatomy requires in-depth knowledge and expertise, and patients should always have medical procedures done by a physician who also has knowledge of adverse events,” he added, urging consumers to see a board-certified dermatologist before undergoing any cosmetic procedure.
In response to a query, an FDA spokesperson did not have an estimate of the number of reports of these adverse events.
People who have problems or are concerned about having had a filler injected with a needle-free device should contact a licensed health care provider. Consumers and health care professionals should report adverse events related to injection of fillers with a needle-free device to the FDA’s MedWatch program. In addition to MedWatch, adverse events can also be reported to the Cutaneous Procedures Adverse Events Reporting (CAPER) Registry, established earlier this year by the ASDSA with the department of dermatology at Northwestern University, Chicago.
*This story was updated on October 12.
In atopic dermatitis trial, abrocitinib offers faster itch relief than dupilumab
), in a multicenter randomized trial presented as a late breaker at the annual meeting of the European Academy of Dermatology and Venereology.

The earlier onset of action with the JAK inhibitor was achieved even though most patients in both arms were on topical corticosteroids, a design element that “is clinically relevant” for a practical comparison of these two agents, according to Kristian Reich, MD, PhD, Center for Translational Research in Inflammatory Skin Diseases, University Medical Center, Hamburg-Eppendorf, Germany.
The goal of this phase 3b trial, called JADE DARE, was to compare relative safety and efficacy of these strategies over the early course of treatment, he said.
Over 700 patients randomized
JADE DARE enrolled 727 patients over age 18 years who previously had an inadequate response to conventional topical therapies. All had moderate to severe AD defined by criteria such as body surface area greater than or equal to 10% and Eczema Area Severity Index (EASI) greater than or equal to 16. They were randomly assigned to 200 mg oral abrocitinib once daily or 300 mg subcutaneous dupilumab (after a loading dose of 600 mg) every 2 weeks. A double-dummy design preserved blinding.
The coprimary endpoints were at least a 4-point improvement in pruritus as measured with the Peak Pruritus Numerical Rating Scale (PP-NRS) score at week 2 and at least a 90% improvement in the EASI (EASI 90) at week 4.
The primary endpoint for pruritus at 2 weeks was reached by nearly twice as many patients randomly assigned to abrocitinib (46.2% vs. 25.5%; P < .001). The proportion of those meeting the EASI 90 endpoint at week 4 was also superior on abrocitinib (28.5% vs. 14.6%; P < .001)
Advantage for pruritus control dissipates
For the pruritus endpoint, the advantage of abrocitinib slowly diminished over time after the peak difference observed at 2 weeks. Although the advantage at week 4 (58.1% vs. 40.8%) and week 8 (65.8% vs. 52.7%) remained sizable, there were very small differences thereafter. However, Dr. Reich pointed out that the percentages continued to favor abrocitinib at least numerically through the 26 weeks of follow-up completed so far.
The pattern of response on EASI 90 was not the same. After demonstrating superiority at the 4-week timepoint, the advantage of abrocitinib persisted. When compared at week 16, which was a secondary endpoint of the JADE DARE trial, the advantage of abrocitinib remained significant (54.3% vs. 41.9%; P < .001). The advantage of abrocitinib narrowed but remained numerically superior at 26 weeks (54.6% vs. 47.6%).
Based on the data collected to date, “abrocitinib is clearly superior early on,” Dr. Reich said. Moreover, he reiterated that topical corticosteroids were allowed as background therapy in both arms.
“It is difficult to show an advantage for one active therapy over the other in patients on background corticosteroids,” Dr. Reich maintained.
Both drugs are well tolerated
The drugs were similarly well tolerated. Serious adverse events were uncommon in either arm. The rate of study dropouts due to an adverse event potentially related to treatment assignment was 3% in each group.
Nausea (19% vs. 2%), acne (13.5% vs. 2%), and headache (13% vs. 7.5%) were all more common in patients randomly assigned to abrocitinib. Conjunctivitis was more common in the group randomly assigned to dupilumab (10% vs. 2%).
The two deaths that occurred during this study were in the abrocitinib arm, but one was the result of COVID-19 infection and the other was a cardiovascular event in a patient with risk factors. Neither was considered to be treatment-related.
Abrocitinib’s relative selectivity for the JAK1 inhibitor is a potential differentiator from other currently available JAK inhibitors, although direct comparisons of these therapies for clinical activity in AD as well as most other diseases remains limited.
The relatively rapid relief of pruritus with the JAK inhibitor relative to the monoclonal antibody in the JADE DARE trial is likely to be perceived as clinically significant by patients with AD, according to Sonja Ständer, MD, professor of dermatology and neurodermatology at the University Hospital Münster, Germany.
“One of the highest needs of patients with atopic dermatitis is a rapid and profound relief of itch,” Dr. Ständer, who wrote a review article on AD earlier this year, said in an interview.
Although several current therapies are effective against pruritus, Dr. Ständer believes that the higher proportion of patients achieving itch control at 2 weeks on abrocitinib “will attract the attention of affected patients.”
However, she added that patients need to take both benefits and risks into account, indicating that clinical utility cannot be judged on a single outcome. In selecting one drug over the others, she advised “a balanced use of therapies.”
Abrocitinib was first approved in the United Kingdom in early September, followed by Japan last Thursday, for the treatment of moderate to severe AD in patients ages 12 and older. It is under review elsewhere, including in the United States and the European Union for AD.
In September, the FDA approved the first JAK inhibitor for treating AD – a topical JAK inhibitor, ruxolitinib.
Dr. Reich reports financial relationships with 20 pharmaceutical companies, including Pfizer, which provided funding for the JADE DARE trial. Dr. Ständer reports financial relationships with Beiersdorf AG, Galderma, Kliniska, Lilly, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
), in a multicenter randomized trial presented as a late breaker at the annual meeting of the European Academy of Dermatology and Venereology.
The earlier onset of action with the JAK inhibitor was achieved even though most patients in both arms were on topical corticosteroids, a design element that “is clinically relevant” for a practical comparison of these two agents, according to Kristian Reich, MD, PhD, Center for Translational Research in Inflammatory Skin Diseases, University Medical Center, Hamburg-Eppendorf, Germany.
The goal of this phase 3b trial, called JADE DARE, was to compare relative safety and efficacy of these strategies over the early course of treatment, he said.
Over 700 patients randomized
JADE DARE enrolled 727 patients over age 18 years who previously had an inadequate response to conventional topical therapies. All had moderate to severe AD defined by criteria such as body surface area greater than or equal to 10% and Eczema Area Severity Index (EASI) greater than or equal to 16. They were randomly assigned to 200 mg oral abrocitinib once daily or 300 mg subcutaneous dupilumab (after a loading dose of 600 mg) every 2 weeks. A double-dummy design preserved blinding.
The coprimary endpoints were at least a 4-point improvement in pruritus as measured with the Peak Pruritus Numerical Rating Scale (PP-NRS) score at week 2 and at least a 90% improvement in the EASI (EASI 90) at week 4.
The primary endpoint for pruritus at 2 weeks was reached by nearly twice as many patients randomly assigned to abrocitinib (46.2% vs. 25.5%; P < .001). The proportion of those meeting the EASI 90 endpoint at week 4 was also superior on abrocitinib (28.5% vs. 14.6%; P < .001)
Advantage for pruritus control dissipates
For the pruritus endpoint, the advantage of abrocitinib slowly diminished over time after the peak difference observed at 2 weeks. Although the advantage at week 4 (58.1% vs. 40.8%) and week 8 (65.8% vs. 52.7%) remained sizable, there were very small differences thereafter. However, Dr. Reich pointed out that the percentages continued to favor abrocitinib at least numerically through the 26 weeks of follow-up completed so far.
The pattern of response on EASI 90 was not the same. After demonstrating superiority at the 4-week timepoint, the advantage of abrocitinib persisted. When compared at week 16, which was a secondary endpoint of the JADE DARE trial, the advantage of abrocitinib remained significant (54.3% vs. 41.9%; P < .001). The advantage of abrocitinib narrowed but remained numerically superior at 26 weeks (54.6% vs. 47.6%).
Based on the data collected to date, “abrocitinib is clearly superior early on,” Dr. Reich said. Moreover, he reiterated that topical corticosteroids were allowed as background therapy in both arms.
“It is difficult to show an advantage for one active therapy over the other in patients on background corticosteroids,” Dr. Reich maintained.
Both drugs are well tolerated
The drugs were similarly well tolerated. Serious adverse events were uncommon in either arm. The rate of study dropouts due to an adverse event potentially related to treatment assignment was 3% in each group.
Nausea (19% vs. 2%), acne (13.5% vs. 2%), and headache (13% vs. 7.5%) were all more common in patients randomly assigned to abrocitinib. Conjunctivitis was more common in the group randomly assigned to dupilumab (10% vs. 2%).
The two deaths that occurred during this study were in the abrocitinib arm, but one was the result of COVID-19 infection and the other was a cardiovascular event in a patient with risk factors. Neither was considered to be treatment-related.
Abrocitinib’s relative selectivity for the JAK1 inhibitor is a potential differentiator from other currently available JAK inhibitors, although direct comparisons of these therapies for clinical activity in AD as well as most other diseases remains limited.
The relatively rapid relief of pruritus with the JAK inhibitor relative to the monoclonal antibody in the JADE DARE trial is likely to be perceived as clinically significant by patients with AD, according to Sonja Ständer, MD, professor of dermatology and neurodermatology at the University Hospital Münster, Germany.
“One of the highest needs of patients with atopic dermatitis is a rapid and profound relief of itch,” Dr. Ständer, who wrote a review article on AD earlier this year, said in an interview.
Although several current therapies are effective against pruritus, Dr. Ständer believes that the higher proportion of patients achieving itch control at 2 weeks on abrocitinib “will attract the attention of affected patients.”
However, she added that patients need to take both benefits and risks into account, indicating that clinical utility cannot be judged on a single outcome. In selecting one drug over the others, she advised “a balanced use of therapies.”
Abrocitinib was first approved in the United Kingdom in early September, followed by Japan last Thursday, for the treatment of moderate to severe AD in patients ages 12 and older. It is under review elsewhere, including in the United States and the European Union for AD.
In September, the FDA approved the first JAK inhibitor for treating AD – a topical JAK inhibitor, ruxolitinib.
Dr. Reich reports financial relationships with 20 pharmaceutical companies, including Pfizer, which provided funding for the JADE DARE trial. Dr. Ständer reports financial relationships with Beiersdorf AG, Galderma, Kliniska, Lilly, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
), in a multicenter randomized trial presented as a late breaker at the annual meeting of the European Academy of Dermatology and Venereology.
The earlier onset of action with the JAK inhibitor was achieved even though most patients in both arms were on topical corticosteroids, a design element that “is clinically relevant” for a practical comparison of these two agents, according to Kristian Reich, MD, PhD, Center for Translational Research in Inflammatory Skin Diseases, University Medical Center, Hamburg-Eppendorf, Germany.
The goal of this phase 3b trial, called JADE DARE, was to compare relative safety and efficacy of these strategies over the early course of treatment, he said.
Over 700 patients randomized
JADE DARE enrolled 727 patients over age 18 years who previously had an inadequate response to conventional topical therapies. All had moderate to severe AD defined by criteria such as body surface area greater than or equal to 10% and Eczema Area Severity Index (EASI) greater than or equal to 16. They were randomly assigned to 200 mg oral abrocitinib once daily or 300 mg subcutaneous dupilumab (after a loading dose of 600 mg) every 2 weeks. A double-dummy design preserved blinding.
The coprimary endpoints were at least a 4-point improvement in pruritus as measured with the Peak Pruritus Numerical Rating Scale (PP-NRS) score at week 2 and at least a 90% improvement in the EASI (EASI 90) at week 4.
The primary endpoint for pruritus at 2 weeks was reached by nearly twice as many patients randomly assigned to abrocitinib (46.2% vs. 25.5%; P < .001). The proportion of those meeting the EASI 90 endpoint at week 4 was also superior on abrocitinib (28.5% vs. 14.6%; P < .001)
Advantage for pruritus control dissipates
For the pruritus endpoint, the advantage of abrocitinib slowly diminished over time after the peak difference observed at 2 weeks. Although the advantage at week 4 (58.1% vs. 40.8%) and week 8 (65.8% vs. 52.7%) remained sizable, there were very small differences thereafter. However, Dr. Reich pointed out that the percentages continued to favor abrocitinib at least numerically through the 26 weeks of follow-up completed so far.
The pattern of response on EASI 90 was not the same. After demonstrating superiority at the 4-week timepoint, the advantage of abrocitinib persisted. When compared at week 16, which was a secondary endpoint of the JADE DARE trial, the advantage of abrocitinib remained significant (54.3% vs. 41.9%; P < .001). The advantage of abrocitinib narrowed but remained numerically superior at 26 weeks (54.6% vs. 47.6%).
Based on the data collected to date, “abrocitinib is clearly superior early on,” Dr. Reich said. Moreover, he reiterated that topical corticosteroids were allowed as background therapy in both arms.
“It is difficult to show an advantage for one active therapy over the other in patients on background corticosteroids,” Dr. Reich maintained.
Both drugs are well tolerated
The drugs were similarly well tolerated. Serious adverse events were uncommon in either arm. The rate of study dropouts due to an adverse event potentially related to treatment assignment was 3% in each group.
Nausea (19% vs. 2%), acne (13.5% vs. 2%), and headache (13% vs. 7.5%) were all more common in patients randomly assigned to abrocitinib. Conjunctivitis was more common in the group randomly assigned to dupilumab (10% vs. 2%).
The two deaths that occurred during this study were in the abrocitinib arm, but one was the result of COVID-19 infection and the other was a cardiovascular event in a patient with risk factors. Neither was considered to be treatment-related.
Abrocitinib’s relative selectivity for the JAK1 inhibitor is a potential differentiator from other currently available JAK inhibitors, although direct comparisons of these therapies for clinical activity in AD as well as most other diseases remains limited.
The relatively rapid relief of pruritus with the JAK inhibitor relative to the monoclonal antibody in the JADE DARE trial is likely to be perceived as clinically significant by patients with AD, according to Sonja Ständer, MD, professor of dermatology and neurodermatology at the University Hospital Münster, Germany.
“One of the highest needs of patients with atopic dermatitis is a rapid and profound relief of itch,” Dr. Ständer, who wrote a review article on AD earlier this year, said in an interview.
Although several current therapies are effective against pruritus, Dr. Ständer believes that the higher proportion of patients achieving itch control at 2 weeks on abrocitinib “will attract the attention of affected patients.”
However, she added that patients need to take both benefits and risks into account, indicating that clinical utility cannot be judged on a single outcome. In selecting one drug over the others, she advised “a balanced use of therapies.”
Abrocitinib was first approved in the United Kingdom in early September, followed by Japan last Thursday, for the treatment of moderate to severe AD in patients ages 12 and older. It is under review elsewhere, including in the United States and the European Union for AD.
In September, the FDA approved the first JAK inhibitor for treating AD – a topical JAK inhibitor, ruxolitinib.
Dr. Reich reports financial relationships with 20 pharmaceutical companies, including Pfizer, which provided funding for the JADE DARE trial. Dr. Ständer reports financial relationships with Beiersdorf AG, Galderma, Kliniska, Lilly, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
Infant with edematous, erythematous toe
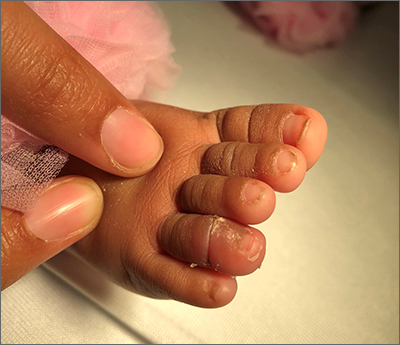
History, presentation, and clinical suspicion led to the diagnosis of hair tourniquet syndrome.
Hair tourniquet syndrome was first described in 1612 by French surgeon Jacques Guillemeau.1 It typically occurs in infants when a long hair gets tightly wrapped around tissue. It most commonly affects the digits, but the penis, labia, or clitoris may also be involved. If left untreated, this condition may lead to serious complications including ischemia and necrosis of the site, and more rarely, bone erosion.
Clinicians who work with children should be aware of this condition, as early diagnosis and treatment can prevent adverse outcomes. Diagnosis requires a high-level of clinical suspicion. Use of ultrasound guidance to confirm the presence of a foreign body may aid in prompt diagnosis.2
Treatment involves release of the constricting hair(s). Hair removal cream may be used if the skin barrier is not compromised. If the hair is visible, clinicians may also attempt to remove it with tweezers. If the hair is deeply embedded within the skin, as in this case, surgical dissection may be necessary.
For this patient, the physician used local anesthesia and surgical loupes to remove 3 strands of hair from beneath newly epithelialized tissue. The digit immediately turned warm and pink. Two minutes later, capillary-refill time was normal. The mother was counseled that women often lose more hair than usual during the postpartum period, and that as a result, it’s important to watch for strands of hair that may get wrapped around the baby’s fingers or toes. Follow-up, 1 month later, showed a healed lesion on a well-perfused and nontender toe.
Image courtesy of Omar Osmani, MD, Spine and Orthopedic Center of New Mexico, Roswell. Text courtesy of Sabah Osmani, BA, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Zimmerman LN, Wagner AJ. Clitoral hair tourniquet: a case report and review of the literature. Int J Pediatr Res. 2015;1:1-2. doi: 10.23937/2469-5769/1510007
2. Sebaratnam DF, Hernández‐Martín Á. Utility of ultrasonography in hair-thread tourniquet syndrome. Pediatr Dermatolo. 2018;35:e138–e139. doi: 10.1111/pde.13400
History, presentation, and clinical suspicion led to the diagnosis of hair tourniquet syndrome.
Hair tourniquet syndrome was first described in 1612 by French surgeon Jacques Guillemeau.1 It typically occurs in infants when a long hair gets tightly wrapped around tissue. It most commonly affects the digits, but the penis, labia, or clitoris may also be involved. If left untreated, this condition may lead to serious complications including ischemia and necrosis of the site, and more rarely, bone erosion.
Clinicians who work with children should be aware of this condition, as early diagnosis and treatment can prevent adverse outcomes. Diagnosis requires a high-level of clinical suspicion. Use of ultrasound guidance to confirm the presence of a foreign body may aid in prompt diagnosis.2
Treatment involves release of the constricting hair(s). Hair removal cream may be used if the skin barrier is not compromised. If the hair is visible, clinicians may also attempt to remove it with tweezers. If the hair is deeply embedded within the skin, as in this case, surgical dissection may be necessary.
For this patient, the physician used local anesthesia and surgical loupes to remove 3 strands of hair from beneath newly epithelialized tissue. The digit immediately turned warm and pink. Two minutes later, capillary-refill time was normal. The mother was counseled that women often lose more hair than usual during the postpartum period, and that as a result, it’s important to watch for strands of hair that may get wrapped around the baby’s fingers or toes. Follow-up, 1 month later, showed a healed lesion on a well-perfused and nontender toe.
Image courtesy of Omar Osmani, MD, Spine and Orthopedic Center of New Mexico, Roswell. Text courtesy of Sabah Osmani, BA, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
History, presentation, and clinical suspicion led to the diagnosis of hair tourniquet syndrome.
Hair tourniquet syndrome was first described in 1612 by French surgeon Jacques Guillemeau.1 It typically occurs in infants when a long hair gets tightly wrapped around tissue. It most commonly affects the digits, but the penis, labia, or clitoris may also be involved. If left untreated, this condition may lead to serious complications including ischemia and necrosis of the site, and more rarely, bone erosion.
Clinicians who work with children should be aware of this condition, as early diagnosis and treatment can prevent adverse outcomes. Diagnosis requires a high-level of clinical suspicion. Use of ultrasound guidance to confirm the presence of a foreign body may aid in prompt diagnosis.2
Treatment involves release of the constricting hair(s). Hair removal cream may be used if the skin barrier is not compromised. If the hair is visible, clinicians may also attempt to remove it with tweezers. If the hair is deeply embedded within the skin, as in this case, surgical dissection may be necessary.
For this patient, the physician used local anesthesia and surgical loupes to remove 3 strands of hair from beneath newly epithelialized tissue. The digit immediately turned warm and pink. Two minutes later, capillary-refill time was normal. The mother was counseled that women often lose more hair than usual during the postpartum period, and that as a result, it’s important to watch for strands of hair that may get wrapped around the baby’s fingers or toes. Follow-up, 1 month later, showed a healed lesion on a well-perfused and nontender toe.
Image courtesy of Omar Osmani, MD, Spine and Orthopedic Center of New Mexico, Roswell. Text courtesy of Sabah Osmani, BA, University of New Mexico School of Medicine, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
1. Zimmerman LN, Wagner AJ. Clitoral hair tourniquet: a case report and review of the literature. Int J Pediatr Res. 2015;1:1-2. doi: 10.23937/2469-5769/1510007
2. Sebaratnam DF, Hernández‐Martín Á. Utility of ultrasonography in hair-thread tourniquet syndrome. Pediatr Dermatolo. 2018;35:e138–e139. doi: 10.1111/pde.13400
1. Zimmerman LN, Wagner AJ. Clitoral hair tourniquet: a case report and review of the literature. Int J Pediatr Res. 2015;1:1-2. doi: 10.23937/2469-5769/1510007
2. Sebaratnam DF, Hernández‐Martín Á. Utility of ultrasonography in hair-thread tourniquet syndrome. Pediatr Dermatolo. 2018;35:e138–e139. doi: 10.1111/pde.13400
JAK inhibitor provides impressive hair growth for patients with alopecia areata
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ruxolitinib cream meets primary endpoints in phase 3 vitiligo trial
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
COVID-19: Two more cases of mucosal skin ulcers reported in male teens
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Extension study finds dupilumab effective for up to 1 year in teens with AD
in a phase 3, open-label extension trial, researchers reported.

At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.

University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
in a phase 3, open-label extension trial, researchers reported.
At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.
University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
in a phase 3, open-label extension trial, researchers reported.
At 1 year, 86% of 50 remaining patients with weights under 60 kg (132 lb) had achieved 75% improvement on the Eczema Area and Severity Index (EASI-75, and 77% of 51 remaining patients with weights over 60 kg reached that level of clearance. Only 5 (1.7%) of 294 patients had serious treatment-emergent adverse events (TEAEs).
The findings back up a perception that patients can stay on dupilumab for some time instead of having to switch from one biologic to another after a few years, study coauthor Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, said in an interview. He added that the drug’s long-term safety profile is “very reassuring.”
The industry-funded findings of the study were released in a poster at the 2021 meeting of the World Congress of Pediatric Dermatology.
The FDA approved dupilumab (Dupixent), an interleukin-4 receptor alpha antagonist, for treating AD in adults in 2017; it is now approved for treating patients ages 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topicals.
The new study tracked patients who received at least 300 mg dupilumab subcutaneously every 4 weeks. The dose could be increased if needed to improve clinical response to once every 2 weeks (200 mg if baseline weight was <60 kg; 300 mg if ≥60 kg).
At 52 weeks, 37% of 52 patients with weights under 60 kg reached an Investigator Global Assessment (IGA) of 0/1, a level that had been fairly steady since week 16 (n = 146). Among 51 heavier patients, 49% reached an IGA of 0/1 at 52 weeks; this percentage grew steadily since baseline.
The mean percentage change in EASI was –87% in the lower-weight group (n = 50) at 52 weeks and –80.1% in the larger-weight group (n = 51). The majority of the reduction in EASI occurred in the first 4 weeks of treatment.
At 52 weeks, the mean Children’s Dermatology Life Quality Index level, which judges the effect of AD on life, was judged as “small” (low) in 71 patients. At baseline, the mean level among 189 patients was “moderate.” The levels dipped below “moderate” at week 4 and never rose above “small” after that.
“Treatment-emergent adverse events reported in ≥5% of patients were nasopharyngitis (21.1%), AD (19.4%), upper respiratory tract infection (12.4%), headache (9.4%), and oropharyngeal pain (5.7%),” the investigators wrote in the poster. They add that 6.7% of patients experienced injection-site reactions, and 8.7% of patients experienced treatment-emergent “narrow conjunctivitis,” which includes conjunctivitis, allergic conjunctivitis, bacterial conjunctivitis, viral conjunctivitis, and atopic keratoconjunctivitis.
Dr. Simpson noted that cases of conjunctivitis fell over time. It’s not clear why this adverse effect appears, he said.
He said that the findings reflect his own experience in clinic. Many of his adolescent patients took part in early dupilumab trials, he said, and dozens have been taking the drug for more than 5 years. “They just seem to get better and better,” he said.
University of Minnesota, Minneapolis, dermatologist Sheilagh Maguiness, MD, who wasn’t involved with the study, said in an interview that dupilumab remains “the safest, most effective and evidence-based therapy we had for children with moderate to severe atopic dermatitis.”
The new study’s findings are “very reassuring,” she said, and similar to those in a 2021 report that tracked long-term use of the drug in children aged 6-11.
Like Dr. Simpson, Dr. Maguiness said many pediatric patients at her clinic have stayed on the drug for more than 5 years. They still have “sustained improvement in skin disease and in their quality of life as well”
There are, however, still questions about dupilumab treatment. “For children who have responded well, when could we consider dose reduction or discontinuation? I have done this successfully just a handful of times, but I would love to see data about what percentage of pediatric patients experience rebound disease after coming off the drug and after what duration of treatment,” she said. “Another mystery that will be very interesting to unravel is the question as to whether or not early treatment with dupilumab may attenuate other atopic diseases.”
Dr. Maguiness added that “another issue specific to pediatric use of dupilumab is the recommendation surrounding vaccinations. This is an issue that should be studied in terms of antibody response and safety surrounding vaccinations, particularly as we are eagerly awaiting a pediatric FDA approval for the COVID-19 vaccine in children.”
She also urged colleagues to push back against insurers who resist paying for dupilumab. “Whether prescribing this medication on or off label, insurance companies are often requiring patients to try and fail other traditional immunosuppressive medications such as methotrexate, cyclosporine, or to pursue phototherapy,” she said. “Oftentimes, these are not practical or even safe options for children for a multitude of reasons. Don’t be shy about advocating for your patients by second- or even third-level appeals to try and gain approval for children who are in need of treatment.”
The study was funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The study authors reported various disclosures. Dr. Simpson reported investigator and consultant fee relationships from various pharmaceutical companies. Dr. Maguiness was an investigator for one of the initial pediatric dupilumab trials.
A version of this article first appeared on Medscape.com.
Nonsteroidal topical found effective for psoriasis in 52-week study
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.
Treatment with presented as a late-breaker at the virtual annual congress of the European Academy of Dermatology and Venereology.
The drug has several unique features with meaningful clinical differences from other topical psoriasis therapies, according to Linda Stein Gold, MD, director of dermatology clinical research, Henry Ford Health System, Detroit.
“The currently available nonsteroidal topical therapies are typically associated with significant irritation. We did not see that with tapinarof,” said Dr. Stein Gold. This is one of several reasons she believes this drug will be a valuable addition if it receives regulatory approval.
Tapinarof is a small-molecule aryl hydrocarbon receptor (AhR) modulating agent. AhR is widely expressed in immune cells, including macrophages, mast cells, and antigen-presenting cells. It is believed that modulation of AhR signaling by tapinarof reverses immune dysregulation that is involved in the formation of psoriatic lesions.
The newly presented PSOARING 3 data with tapinarof 1% build on the data from the 12-week PSOARING 1 and PSOARING 2 trials, which were released in August 2020 but have yet to be published.
The primary endpoint in both of the 12-week trials, each of which enrolled about 500 patients with plaque psoriasis, was a Physician Global Assessment (PGA) score of 0 (clear) or 1 (almost clear). Relative to a placebo response rate of about 6% in both trials, the proportion of patients who achieved scores of 0/1 with tapinarof 1% was 35.4% and 40.2% in the PSOARING 1 and PSOARING 2 trials, respectively (P < .0001 vs. placebo in both studies).
For the key secondary endpoint of at least 75% improvement in the Psoriasis Area and Severity Index (PASI 75), the relative advantage for tapinarof over placebo was similar. The results were highly statistically significant (P < .0001) in both of the 12-week trials.
More than 90% of the patients who participated in PSOARING 1 and PSOARING 2 and were eligible for the open-label PSOARING 3 extension trial, according to Dr. Stein Gold.
For the 79 patients with a score of 0 at the time of enrollment, tapinarof 1% was reapplied only if the PGA score reached at least 2 during the course of the study. For the 680 patients who entered with a PGA score of at least 1, once-daily applications of tapinarof 1% cream were maintained until a PGA score of 0 was achieved.
In the outcome analysis, response was defined as the proportion of patients with an initial PGA score of at least2 who achieved PGA 0. A remittive effect was defined as duration of a PGA score of 0 or 1 while off therapy after achieving a PGA score of 0. Durability of response was defined as the proportion of patients who achieved a PGA sore of 0 or 1 at least once during the study while on therapy. This last outcome provided a test of tachyphylaxis.
“Overall, 40.9% of patients achieved complete disease clearance at least once during the trial, and 58.2% who entered the study with a PGA score of 2 or higher achieved a PGA score of 0 or 1,” Dr. Stein Gold reported.
For the 79 patients who entered PSOARING 3 with a PGA score of 0 and were off treatment, the median duration of a remittive effect was 115 days. For the patients who entered the trial with a higher PGA score but who achieved a score of 0 during the study (312 patients), the mean remittive effect after discontinuing therapy was 130 days.
There was no evidence of tachyphylaxis. Rather, “there was no loss of effect despite intermittent therapy observed over the course of the trial,” Dr. Stein Gold reported.
The most common treatment-emergent adverse events in PSOARING 3, as in the previous PSOARING studies, were folliculitis, which was observed in 24.0% of patients; contact dermatitis, which occurred in 5.9% of patients; and headache, which was reported in 2%. Rates of study drug discontinuations for folliculitis and contact dermatitis were 1.2% and 1.4%, respectively. Headache did not lead to any study discontinuations.
Calling tapinarof a “first-in-class nonsteroidal,” Dr. Stein Gold suggested that this is likely to be a useful adjunctive therapy for psoriasis control. It avoids the adverse events associated with long-term topical steroid use, and its tolerability might be particularly attractive for use in sensitive areas.
“This is likely to be very useful in patients who are looking for a topical therapy for skin folds or the face, where there is a need for well-tolerated topical treatments,” Dr. Stein Gold said.
There are a lot of reasons to be positive about a new, well-tolerated topical agent for psoriasis, particularly as an alternative to topical steroids, agreed Adam Friedman, MD, director of translational research and professor and chair of the department of dermatology at George Washington University, Washington. He considers the data with tapinarof promising in general, but he also likes any new, effective topical psoriasis therapy.
“Patients and physicians are always hungry for new options, especially psoriasis patients, given many have ‘been there and done that’ with topical steroids,” Dr. Friedman said.
“Topical steroids are not irritating, but long-term use beyond recommended dosing can lead to skin thinning, lightening, tachyphylaxis, and, if really abused, HPA [hypothalamic-pituitary-adrenal] axis suppression and adrenal insufficiency,” he observed.
A topical therapy with a durable effect is particularly intriguing.
“The other issue with topical steroids is that psoriatic plaques return rather easily after stopping. The data I have seen with tapinarof show more sustainability after cessation, owing to its mechanism of action,” Dr. Friedman said. Rather than its potential for application to sensitive areas, such as the face, the durability “to me is more interesting.”
He suspects that, owing to “the incurable steroid phobia that haunts many of our patients,” an effective nonsteroidal topical option is also likely to lead to better compliance with topical treatment over time.
“A well-tolerated nonsteroidal topical drug will probably find an important place in the future management of chronic inflammatory diseases,” Marius-Anton Ionescu, MD, PhD, a dermatologist at the Hôpital Saint Louis, Paris, said in an interview. He referred to the positive effects of treatment with tapinarof in clinical trials in adults with atopic dermatitis, in addition to psoriasis.
Tapinarof 1% is also being investigated in a phase 3 study involving patients with moderate to severe atopic dermatitis. In that study, patients are as young as age 2 years. The drug is under review at the Food and Drug Administration for the plaque psoriasis indication in adults.
Dr. Stein Gold has financial relationships with Arcutis, Amgen, Bristol-Myers Squibb, Eli Lilly, Leo Pharma Ortho Dermatologic, UCB, and Dermavant Sciences, which is developing tapinarof and is provided funding for the PSOARING 3 trial. Dr. Friedman reported financial relationships with Amgen, Biogen, Encore, Galderma, GlaxoSmithKline, IntraDerm, Johnson & Johnson, Nerium, Novartis, Oculus, Onset, Pfizer, Sanova, and Valeant Pharmaceuticals. Dr. Ionescu has been a speaker or investigator (honoraria) for Celgene, Novartis, Lilly, and Uriage Cosmetics.
A version of this article first appeared on Medscape.com.
Oteseconazole promising for recurrent yeast infections
A phase 3, randomized, double-blind, controlled trial has shown that oteseconazole (Mycovia Pharmaceuticals), an oral antifungal agent, is safe and effective in treating acute and recurrent yeast infections (vulvovaginal candidiasis [VVC]) and in preventing recurrence of acute VVC episodes.
Findings of the ultraVIOLET trial, which compared oteseconazole with the standard fluconazole, were presented at IDWeek 2021, an annual scientific meeting on infectious diseases, by lead author Mark G. Martens, MD, a professor in the department of obstetrics and gynecology at Drexel University College of Medicine in Philadelphia.
About 75% of all women will have a yeast infection in their lifetime, Dr. Martens noted. About 138 million women worldwide have recurring episodes (at least three acute episodes in the last year) of the debilitating condition.
“Recurrent vulvovaginal candidiasis typically requires treatment of the acute episode followed by long-term suppressive therapy with either weekly or biweekly fluconazole,” Dr. Martens said. However, when therapy stops, more than 50% of patients with recurrent VVC experience an infection within the next 6 months, which takes a significant toll on daily life.
Additionally, fluconazole has been linked with safety issues concerning chronic dosing, he said, citing liver toxicity, drug-drug interactions and “increased risk of miscarriage and birth defects when used during pregnancy.”
Topical treatments have been associated with messy application and burning, he noted.
For this study, researchers enrolled 219 women with a history of recurrent VVC at 51 U.S. sites. Participants were randomized either to 600 mg oteseconazole on day 1, 450 mg oteseconazole on day 2 or placebo capsules; or three sequential 150 mg doses (every 72 hours) of fluconazole together with matching placebo capsules.
In the maintenance phase, 185 women with resolved acute VVC (clinical signs and symptoms were scored below 3) on day 14 received 150 mg oteseconazole or placebo weekly for 11 weeks.
Oteseconazole was superior to fluconazole/placebo in the proportion of subjects with at least one culture-verified acute VVC episode through week 50 in the intent-to-treat population (P < .001) which included subjects who failed to clear their infection in the induction phase.
The average percentage of participants with at least one culture-verified acute VVC episode through week 50 was lower in the oteseconazole group (5.1%), compared with the fluconazole/placebo group (42.2%).
Oteseconazole was noninferior to fluconazole in the proportion of subjects with resolved acute VVC infections at day 14 – 93.2% for the oteseconazole group vs. 95.8% for the fluconazole/placebo group.
The percentages of women who had at least one treatment-emergent adverse event (TEAE) were similar – 54% in the oteseconazole group and 64% in the fluconazole/placebo group. Most TEAEs were mild or moderate and there were no drug-related SAEs or adverse effects on liver function.
“There was no difference in the two groups in he baseline characteristics of age, race, and history of diabetes,” he said.
Oluwatosin Goje, MD, an ob.gyn. with the Cleveland Clinic told this news organization that the drug may offer another option for women who don’t respond to azoles.
“The CDC guidelines say, and I agree, that most episodes of recurrent VVC that are caused by Candida albicans will respond to topical azoles, to oral azoles, to the known drugs that are available. You just may have to use them for a prolonged period of time,” Dr. Goje said. But some patients won’t respond to azoles, the currently available drugs, and topical treatments – so new options are welcome for them, she noted.
She pointed out that the U.S. Food and Drug Administration in June approved ibrexafungerp (Brexafemme), the first oral nonazole treatment for vaginal yeast infections. It was the first approved medicine in a novel antifungal class in more than 2 decades.
Dr. Goje, who runs a large clinic with substantial numbers of women with recurrent yeast infections, said the psychosocial problems women with recurrent yeast infections face – and the time off work and money spent trying to get temporary relief from over-the-counter medications – is underestimated.
“Women have long suffered vaginitis. It can be a lot of social and economic burden. So anything in the toolbox to help women is welcome,” Dr. Goje said.
The study was sponsored by Mycovia Pharmaceuticals. Dr. Martens reports no relevant financial relationships. Several coauthors are either employees of Mycovia or receive support from the company. Dr. Goje has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A phase 3, randomized, double-blind, controlled trial has shown that oteseconazole (Mycovia Pharmaceuticals), an oral antifungal agent, is safe and effective in treating acute and recurrent yeast infections (vulvovaginal candidiasis [VVC]) and in preventing recurrence of acute VVC episodes.
Findings of the ultraVIOLET trial, which compared oteseconazole with the standard fluconazole, were presented at IDWeek 2021, an annual scientific meeting on infectious diseases, by lead author Mark G. Martens, MD, a professor in the department of obstetrics and gynecology at Drexel University College of Medicine in Philadelphia.
About 75% of all women will have a yeast infection in their lifetime, Dr. Martens noted. About 138 million women worldwide have recurring episodes (at least three acute episodes in the last year) of the debilitating condition.
“Recurrent vulvovaginal candidiasis typically requires treatment of the acute episode followed by long-term suppressive therapy with either weekly or biweekly fluconazole,” Dr. Martens said. However, when therapy stops, more than 50% of patients with recurrent VVC experience an infection within the next 6 months, which takes a significant toll on daily life.
Additionally, fluconazole has been linked with safety issues concerning chronic dosing, he said, citing liver toxicity, drug-drug interactions and “increased risk of miscarriage and birth defects when used during pregnancy.”
Topical treatments have been associated with messy application and burning, he noted.
For this study, researchers enrolled 219 women with a history of recurrent VVC at 51 U.S. sites. Participants were randomized either to 600 mg oteseconazole on day 1, 450 mg oteseconazole on day 2 or placebo capsules; or three sequential 150 mg doses (every 72 hours) of fluconazole together with matching placebo capsules.
In the maintenance phase, 185 women with resolved acute VVC (clinical signs and symptoms were scored below 3) on day 14 received 150 mg oteseconazole or placebo weekly for 11 weeks.
Oteseconazole was superior to fluconazole/placebo in the proportion of subjects with at least one culture-verified acute VVC episode through week 50 in the intent-to-treat population (P < .001) which included subjects who failed to clear their infection in the induction phase.
The average percentage of participants with at least one culture-verified acute VVC episode through week 50 was lower in the oteseconazole group (5.1%), compared with the fluconazole/placebo group (42.2%).
Oteseconazole was noninferior to fluconazole in the proportion of subjects with resolved acute VVC infections at day 14 – 93.2% for the oteseconazole group vs. 95.8% for the fluconazole/placebo group.
The percentages of women who had at least one treatment-emergent adverse event (TEAE) were similar – 54% in the oteseconazole group and 64% in the fluconazole/placebo group. Most TEAEs were mild or moderate and there were no drug-related SAEs or adverse effects on liver function.
“There was no difference in the two groups in he baseline characteristics of age, race, and history of diabetes,” he said.
Oluwatosin Goje, MD, an ob.gyn. with the Cleveland Clinic told this news organization that the drug may offer another option for women who don’t respond to azoles.
“The CDC guidelines say, and I agree, that most episodes of recurrent VVC that are caused by Candida albicans will respond to topical azoles, to oral azoles, to the known drugs that are available. You just may have to use them for a prolonged period of time,” Dr. Goje said. But some patients won’t respond to azoles, the currently available drugs, and topical treatments – so new options are welcome for them, she noted.
She pointed out that the U.S. Food and Drug Administration in June approved ibrexafungerp (Brexafemme), the first oral nonazole treatment for vaginal yeast infections. It was the first approved medicine in a novel antifungal class in more than 2 decades.
Dr. Goje, who runs a large clinic with substantial numbers of women with recurrent yeast infections, said the psychosocial problems women with recurrent yeast infections face – and the time off work and money spent trying to get temporary relief from over-the-counter medications – is underestimated.
“Women have long suffered vaginitis. It can be a lot of social and economic burden. So anything in the toolbox to help women is welcome,” Dr. Goje said.
The study was sponsored by Mycovia Pharmaceuticals. Dr. Martens reports no relevant financial relationships. Several coauthors are either employees of Mycovia or receive support from the company. Dr. Goje has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A phase 3, randomized, double-blind, controlled trial has shown that oteseconazole (Mycovia Pharmaceuticals), an oral antifungal agent, is safe and effective in treating acute and recurrent yeast infections (vulvovaginal candidiasis [VVC]) and in preventing recurrence of acute VVC episodes.
Findings of the ultraVIOLET trial, which compared oteseconazole with the standard fluconazole, were presented at IDWeek 2021, an annual scientific meeting on infectious diseases, by lead author Mark G. Martens, MD, a professor in the department of obstetrics and gynecology at Drexel University College of Medicine in Philadelphia.
About 75% of all women will have a yeast infection in their lifetime, Dr. Martens noted. About 138 million women worldwide have recurring episodes (at least three acute episodes in the last year) of the debilitating condition.
“Recurrent vulvovaginal candidiasis typically requires treatment of the acute episode followed by long-term suppressive therapy with either weekly or biweekly fluconazole,” Dr. Martens said. However, when therapy stops, more than 50% of patients with recurrent VVC experience an infection within the next 6 months, which takes a significant toll on daily life.
Additionally, fluconazole has been linked with safety issues concerning chronic dosing, he said, citing liver toxicity, drug-drug interactions and “increased risk of miscarriage and birth defects when used during pregnancy.”
Topical treatments have been associated with messy application and burning, he noted.
For this study, researchers enrolled 219 women with a history of recurrent VVC at 51 U.S. sites. Participants were randomized either to 600 mg oteseconazole on day 1, 450 mg oteseconazole on day 2 or placebo capsules; or three sequential 150 mg doses (every 72 hours) of fluconazole together with matching placebo capsules.
In the maintenance phase, 185 women with resolved acute VVC (clinical signs and symptoms were scored below 3) on day 14 received 150 mg oteseconazole or placebo weekly for 11 weeks.
Oteseconazole was superior to fluconazole/placebo in the proportion of subjects with at least one culture-verified acute VVC episode through week 50 in the intent-to-treat population (P < .001) which included subjects who failed to clear their infection in the induction phase.
The average percentage of participants with at least one culture-verified acute VVC episode through week 50 was lower in the oteseconazole group (5.1%), compared with the fluconazole/placebo group (42.2%).
Oteseconazole was noninferior to fluconazole in the proportion of subjects with resolved acute VVC infections at day 14 – 93.2% for the oteseconazole group vs. 95.8% for the fluconazole/placebo group.
The percentages of women who had at least one treatment-emergent adverse event (TEAE) were similar – 54% in the oteseconazole group and 64% in the fluconazole/placebo group. Most TEAEs were mild or moderate and there were no drug-related SAEs or adverse effects on liver function.
“There was no difference in the two groups in he baseline characteristics of age, race, and history of diabetes,” he said.
Oluwatosin Goje, MD, an ob.gyn. with the Cleveland Clinic told this news organization that the drug may offer another option for women who don’t respond to azoles.
“The CDC guidelines say, and I agree, that most episodes of recurrent VVC that are caused by Candida albicans will respond to topical azoles, to oral azoles, to the known drugs that are available. You just may have to use them for a prolonged period of time,” Dr. Goje said. But some patients won’t respond to azoles, the currently available drugs, and topical treatments – so new options are welcome for them, she noted.
She pointed out that the U.S. Food and Drug Administration in June approved ibrexafungerp (Brexafemme), the first oral nonazole treatment for vaginal yeast infections. It was the first approved medicine in a novel antifungal class in more than 2 decades.
Dr. Goje, who runs a large clinic with substantial numbers of women with recurrent yeast infections, said the psychosocial problems women with recurrent yeast infections face – and the time off work and money spent trying to get temporary relief from over-the-counter medications – is underestimated.
“Women have long suffered vaginitis. It can be a lot of social and economic burden. So anything in the toolbox to help women is welcome,” Dr. Goje said.
The study was sponsored by Mycovia Pharmaceuticals. Dr. Martens reports no relevant financial relationships. Several coauthors are either employees of Mycovia or receive support from the company. Dr. Goje has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ketosis, including ketogenic diets, implicated in prurigo pigmentosa
, according to a dermatologist, who reviewed skin conditions common to patients of Asian descent at the Skin of Color Update 2021.

“Ketogenic diets are gaining popularity globally for weight loss. After 2-4 weeks [on a strict ketogenic diet], some patients start to notice very pruritic papules on their trunk, the so-called keto rash,” reported Hye Jin Chung, MD, director of the Asian Skin Clinic, Beth Israel Deaconess Medical Center, Boston. “Keto rash is actually prurigo pigmentosa.”
The exact pathogenesis of prurigo pigmentosa, a highly pruritic macular and papular rash with gross reticular pigmentation, is unclear, but Dr. Chung reported that the strong link with ketosis might explain why more cases are now being encountered outside of east Asia. Ketosis or conditions associated with a high risk for ketosis, such as anorexia nervosa, diabetes mellitus, or recent bariatric surgery, have been linked to prurigo pigmentosa in all skin types and ethnicities.
“I tell my residents that this is a disease you will never forget after your first case,” she said.
The differential diagnosis includes contact dermatitis and other inflammatory disorders, but Dr. Chung said that the reticular pattern of the lesions is a relatively unique feature. Confluent and reticulated papillomatosis (CARP) shares a pattern of reticulated lesions, but Dr. Chung said it lacks the inflammatory erythematous papules and the severe pruritus common to prurigo pigmentosa.
Histologically, the pattern evolves. It begins as a perivascular infiltration dominated by neutrophils and eosinophils with hyperkeratosis, acanthosis, and spongiosis. Over time, Dr. Chung said that the histologic picture shows an increasing degree of dyskeratosis as keratinocytes die.
Prurigo pigmentosa was first described 50 years ago by Masaji Nagashima, MD, who published a report on eight patients in Japan with a pruriginous truncal dermatosis featuring symmetrical pigmentation. Most subsequent reports were also from Japan or other east Asian countries, but it has since spread.
This global spread was captured in a recently published review of 115 published studies and case reports from 24 countries. In this review, the proportion of studies from Europe (36.5%) approached that of those from east Asia (38.2%), even if 76% of the patients for whom race was reported were of Asian ethnicity.
Of the 369 patients evaluated in these studies and case reports, 72.1% were female. The mean age was 25.6 years. In the studies originating outside of Asia, prurigo pigmentosa was reported in a spectrum of skin types and ethnicities, including Whites, Blacks, and Hispanics. The lowest reported incidence has been in the latter two groups, but the authors of the review speculated that this condition is likely being underdiagnosed in non-Asian individuals.
Dr. Chung agreed, and she cautioned that the consequences typically result in a significant delay for achieving disease control. In recounting a recent case of prurigo pigmentosa at her center, she said that the 59-year-old Asian patient had been initiated on topical steroids and oral antihistamines by her primary care physician before she was referred. This is a common and reasonable strategy for a highly pruritic rash potentially caused by contact dermatitis, but it is ineffective for this disorder.
“Prurigo pigmentosa requires anti-inflammatory agents,” she explained. She said that doxycycline and minocycline are the treatments of choice, but noted that there are also reports of efficacy with dapsone, macrolide antibiotics, and isotretinoin.
In her most recent case, she initiated the patient on 100 mg of doxycycline twice daily. There was significant improvement within 2 weeks, and the rash resolved within a month with no relapse in follow-up that now exceeds 12 months, Dr. Chung said.
According to Dr. Chung, Asian-Americans are the most rapidly growing ethnic group in the United States, making it increasingly important to be familiar with conditions common or unique to Asian skin, but prurigo pigmentosa is no longer confined to those of Asian descent. She encouraged clinicians to recognize this disorder to reduce the common delays to effective treatment.
The senior author of the recently published review of studies, Jensen Yeung, MD, of the department of dermatology, University of Toronto, agreed. He, too, believes that dermatologists need to increase their awareness of the signs and symptoms of prurigo pigmentosa – and not just in Asian patients or patients of Asian descent.
“This diagnosis is often missed,” he contended in an interview. “This condition has become more common in the past 5 years in my clinical experience.” He added that the increasing incidence might not just be related to better diagnostic accuracy, although the most significant of other possible explanations “is not yet well understood.”
Dr. Chung reports that she has no relevant financial relationships to disclose. Dr. Yeung reports financial relationships with more than 25 pharmaceutical companies, some of which produce treatments employed in the control of prurigo pigmentosa.
, according to a dermatologist, who reviewed skin conditions common to patients of Asian descent at the Skin of Color Update 2021.
“Ketogenic diets are gaining popularity globally for weight loss. After 2-4 weeks [on a strict ketogenic diet], some patients start to notice very pruritic papules on their trunk, the so-called keto rash,” reported Hye Jin Chung, MD, director of the Asian Skin Clinic, Beth Israel Deaconess Medical Center, Boston. “Keto rash is actually prurigo pigmentosa.”
The exact pathogenesis of prurigo pigmentosa, a highly pruritic macular and papular rash with gross reticular pigmentation, is unclear, but Dr. Chung reported that the strong link with ketosis might explain why more cases are now being encountered outside of east Asia. Ketosis or conditions associated with a high risk for ketosis, such as anorexia nervosa, diabetes mellitus, or recent bariatric surgery, have been linked to prurigo pigmentosa in all skin types and ethnicities.
“I tell my residents that this is a disease you will never forget after your first case,” she said.
The differential diagnosis includes contact dermatitis and other inflammatory disorders, but Dr. Chung said that the reticular pattern of the lesions is a relatively unique feature. Confluent and reticulated papillomatosis (CARP) shares a pattern of reticulated lesions, but Dr. Chung said it lacks the inflammatory erythematous papules and the severe pruritus common to prurigo pigmentosa.
Histologically, the pattern evolves. It begins as a perivascular infiltration dominated by neutrophils and eosinophils with hyperkeratosis, acanthosis, and spongiosis. Over time, Dr. Chung said that the histologic picture shows an increasing degree of dyskeratosis as keratinocytes die.
Prurigo pigmentosa was first described 50 years ago by Masaji Nagashima, MD, who published a report on eight patients in Japan with a pruriginous truncal dermatosis featuring symmetrical pigmentation. Most subsequent reports were also from Japan or other east Asian countries, but it has since spread.
This global spread was captured in a recently published review of 115 published studies and case reports from 24 countries. In this review, the proportion of studies from Europe (36.5%) approached that of those from east Asia (38.2%), even if 76% of the patients for whom race was reported were of Asian ethnicity.
Of the 369 patients evaluated in these studies and case reports, 72.1% were female. The mean age was 25.6 years. In the studies originating outside of Asia, prurigo pigmentosa was reported in a spectrum of skin types and ethnicities, including Whites, Blacks, and Hispanics. The lowest reported incidence has been in the latter two groups, but the authors of the review speculated that this condition is likely being underdiagnosed in non-Asian individuals.
Dr. Chung agreed, and she cautioned that the consequences typically result in a significant delay for achieving disease control. In recounting a recent case of prurigo pigmentosa at her center, she said that the 59-year-old Asian patient had been initiated on topical steroids and oral antihistamines by her primary care physician before she was referred. This is a common and reasonable strategy for a highly pruritic rash potentially caused by contact dermatitis, but it is ineffective for this disorder.
“Prurigo pigmentosa requires anti-inflammatory agents,” she explained. She said that doxycycline and minocycline are the treatments of choice, but noted that there are also reports of efficacy with dapsone, macrolide antibiotics, and isotretinoin.
In her most recent case, she initiated the patient on 100 mg of doxycycline twice daily. There was significant improvement within 2 weeks, and the rash resolved within a month with no relapse in follow-up that now exceeds 12 months, Dr. Chung said.
According to Dr. Chung, Asian-Americans are the most rapidly growing ethnic group in the United States, making it increasingly important to be familiar with conditions common or unique to Asian skin, but prurigo pigmentosa is no longer confined to those of Asian descent. She encouraged clinicians to recognize this disorder to reduce the common delays to effective treatment.
The senior author of the recently published review of studies, Jensen Yeung, MD, of the department of dermatology, University of Toronto, agreed. He, too, believes that dermatologists need to increase their awareness of the signs and symptoms of prurigo pigmentosa – and not just in Asian patients or patients of Asian descent.
“This diagnosis is often missed,” he contended in an interview. “This condition has become more common in the past 5 years in my clinical experience.” He added that the increasing incidence might not just be related to better diagnostic accuracy, although the most significant of other possible explanations “is not yet well understood.”
Dr. Chung reports that she has no relevant financial relationships to disclose. Dr. Yeung reports financial relationships with more than 25 pharmaceutical companies, some of which produce treatments employed in the control of prurigo pigmentosa.
, according to a dermatologist, who reviewed skin conditions common to patients of Asian descent at the Skin of Color Update 2021.
“Ketogenic diets are gaining popularity globally for weight loss. After 2-4 weeks [on a strict ketogenic diet], some patients start to notice very pruritic papules on their trunk, the so-called keto rash,” reported Hye Jin Chung, MD, director of the Asian Skin Clinic, Beth Israel Deaconess Medical Center, Boston. “Keto rash is actually prurigo pigmentosa.”
The exact pathogenesis of prurigo pigmentosa, a highly pruritic macular and papular rash with gross reticular pigmentation, is unclear, but Dr. Chung reported that the strong link with ketosis might explain why more cases are now being encountered outside of east Asia. Ketosis or conditions associated with a high risk for ketosis, such as anorexia nervosa, diabetes mellitus, or recent bariatric surgery, have been linked to prurigo pigmentosa in all skin types and ethnicities.
“I tell my residents that this is a disease you will never forget after your first case,” she said.
The differential diagnosis includes contact dermatitis and other inflammatory disorders, but Dr. Chung said that the reticular pattern of the lesions is a relatively unique feature. Confluent and reticulated papillomatosis (CARP) shares a pattern of reticulated lesions, but Dr. Chung said it lacks the inflammatory erythematous papules and the severe pruritus common to prurigo pigmentosa.
Histologically, the pattern evolves. It begins as a perivascular infiltration dominated by neutrophils and eosinophils with hyperkeratosis, acanthosis, and spongiosis. Over time, Dr. Chung said that the histologic picture shows an increasing degree of dyskeratosis as keratinocytes die.
Prurigo pigmentosa was first described 50 years ago by Masaji Nagashima, MD, who published a report on eight patients in Japan with a pruriginous truncal dermatosis featuring symmetrical pigmentation. Most subsequent reports were also from Japan or other east Asian countries, but it has since spread.
This global spread was captured in a recently published review of 115 published studies and case reports from 24 countries. In this review, the proportion of studies from Europe (36.5%) approached that of those from east Asia (38.2%), even if 76% of the patients for whom race was reported were of Asian ethnicity.
Of the 369 patients evaluated in these studies and case reports, 72.1% were female. The mean age was 25.6 years. In the studies originating outside of Asia, prurigo pigmentosa was reported in a spectrum of skin types and ethnicities, including Whites, Blacks, and Hispanics. The lowest reported incidence has been in the latter two groups, but the authors of the review speculated that this condition is likely being underdiagnosed in non-Asian individuals.
Dr. Chung agreed, and she cautioned that the consequences typically result in a significant delay for achieving disease control. In recounting a recent case of prurigo pigmentosa at her center, she said that the 59-year-old Asian patient had been initiated on topical steroids and oral antihistamines by her primary care physician before she was referred. This is a common and reasonable strategy for a highly pruritic rash potentially caused by contact dermatitis, but it is ineffective for this disorder.
“Prurigo pigmentosa requires anti-inflammatory agents,” she explained. She said that doxycycline and minocycline are the treatments of choice, but noted that there are also reports of efficacy with dapsone, macrolide antibiotics, and isotretinoin.
In her most recent case, she initiated the patient on 100 mg of doxycycline twice daily. There was significant improvement within 2 weeks, and the rash resolved within a month with no relapse in follow-up that now exceeds 12 months, Dr. Chung said.
According to Dr. Chung, Asian-Americans are the most rapidly growing ethnic group in the United States, making it increasingly important to be familiar with conditions common or unique to Asian skin, but prurigo pigmentosa is no longer confined to those of Asian descent. She encouraged clinicians to recognize this disorder to reduce the common delays to effective treatment.
The senior author of the recently published review of studies, Jensen Yeung, MD, of the department of dermatology, University of Toronto, agreed. He, too, believes that dermatologists need to increase their awareness of the signs and symptoms of prurigo pigmentosa – and not just in Asian patients or patients of Asian descent.
“This diagnosis is often missed,” he contended in an interview. “This condition has become more common in the past 5 years in my clinical experience.” He added that the increasing incidence might not just be related to better diagnostic accuracy, although the most significant of other possible explanations “is not yet well understood.”
Dr. Chung reports that she has no relevant financial relationships to disclose. Dr. Yeung reports financial relationships with more than 25 pharmaceutical companies, some of which produce treatments employed in the control of prurigo pigmentosa.
FROM SOC 2021