User login
Palliative care helpful but underutilized for blood cancer patients
Specialty palliative care interventions improve outcomes in patients with hematologic malignancies but are underutilized, according to findings from a systematic literature review.
Outcomes that were improved, as demonstrated by 16 studies that met inclusion criteria for the review, included symptom management, inpatient mortality, health care utilization, health care costs, and caregiver-reported outcomes, Elizabeth Elliott, DO, a hematology and oncology fellow at the Cardinal Bernardin Cancer Center, Loyola University, Maywood, Ill., and colleagues reported.
The findings were published online in the Journal of Pain and Symptom Management.
Palliative care needs
Patients with hematologic malignancies, including leukemia, myeloma, and lymphoma, have a high need for supportive care, the authors noted, adding that, although its use has increased over time, palliative care (PC) is often provided late in the disease course – sometimes only in the final days of life.
“Compared with their solid tumor counterparts, patients with hematologic malignancies experience higher symptom burdens, have higher rates of cancer-directed care near death, and are more likely to die while hospitalized than at home or in hospice,” they wrote. “Despite this need, specialist palliative care is less commonly utilized in patients with hematologic malignancies than other cancer types.”
Given the high health care utilization among patients with hematologic malignancies, earlier and more widespread utilization of PC in this population may significantly reduce health care costs, they added.
Palliative care benefits
Of 5,345 studies published between 2005 and 2020 and screened for the current review, 16 met inclusion criteria, including 10 retrospective cohort studies; 4 prospective cohort studies; and 2 randomized, controlled studies.
Nine studies included only patients with hematologic malignancies and seven included both patients with solid tumors and patients with hematologic malignancies. Each study assessed as being of moderate quality.
Benefits of PC as demonstrated in the studies included:
Symptom management: One study, for example, showed that an integrated psychological and PC intervention improved traumatic stress levels, degree and number of physical symptoms, pain intensity, depressive symptoms, and quality of life, compared with no intervention. Another showed that the percentage of patients reporting moderate to severe pain improved from 57% to 18% with a PC intervention, and the number reporting depressive episodes improved from 13% to 5%.
Reduced in-patient death: Findings from eight studies showed that 21.9%-83% of those receiving PC died at home, compared with 6.0%-8.9% of controls. Two studies showed that PC provided at least 20 days prior to death decreased the likelihood of inpatient death and death in an ICU, compared with controls, and one showed that the rate of in-hospital deaths was 30% for those with home PC or hospice, compared with 80% of controls.
Health care utilization: The studies showed that hospitalization occurred in 45%-76.3% of hematologic malignancy patients who received PC, compared with 98% of controls. The odds ratio for hospitalization among acute leukemia patients receiving PC was 0.64, compared with 2.53 among those in a historical control group.
Caregiver-reported outcomes: One randomized, controlled study showed that PC was associated with smaller increases in depression scores, improved coping, and improved scores in multiple quality of life domains in caregivers versus controls.
Survival: One study showed that a larger percentage of hematologic malignancy patients who died 1-6 months after diagnosis had not received PC (28% vs. 23%), whereas more of those who died 6-12 months or 12 or more months after diagnosis had received PC (23.9 vs. 14.9% and 42.5% vs. 22.0%).
Health care costs: Two studies showed a decrease in inpatient costs after a palliative care consultation. Decreases in hospitalization costs were $2,321 and $1,506 for less medically complex patients and $3,515 and $5,617 for more medically complex patient.
Improving PC utilization
One potential strategy to promote earlier referrals to PC is improved education for hematologists, the authors said, citing a study showing that 98% of oncology fellows at one center reported improvement in their ability to assess and manage patient symptoms after completion of a 4-week mandatory PC rotation.
“Another strategy to improve referrals to PC of hematologic malignancies patients could be the creation of programs which facilitate collaboration between PC providers and hematologists, such as the palliative and supportive care special interest group within the American Society for Transplantation and Cellular Therapy,” they wrote.
A third strategy “could be to provide a concurrent care model, in which cancer directed therapy (such as transfusions) is provided at the same time as hospice care,” they added, explaining that such an approach was shown in a study of patients with advanced non–small cell lung cancer to be associated with less aggressive medical treatment and lower costs.
The authors also stressed that patient with solid tumors and those with hematologic malignancies have differing supportive care needs and health care utilization, but several studies included in the current review included both types of cancer.
“Further studies investigating PC use exclusively in patients with hematologic malignancies are needed. Our results demonstrate a strong argument for hematologists to refer their patients early and often for specialized PC,” they concluded.
Indeed, when PC is integrated within hematologic malignancies, impacts occur that are similar to those seen in a variety of other diseases and include improved symptom control, enhanced caregiver experience, and reduced burdens on the health care system, Toby C Campbell, MD, said in an interview.
“The benefits of providing palliative care concurrent with standard cancer care is felt by all the major stakeholders in this care: the patients, their caregivers, and the health care system around them,” said Dr. Campbell, a thoracic medical oncologist and professor in the division of hematology, medical oncology, and palliative care at the University of Wisconsin–Madison.
Overcoming challenges
However, this is “new territory” for most programs, added Dr. Campbell, who also is the University of Wisconsin health chief of palliative care and holds the Ellen and Peter O. Johnson Chair in Palliative Care .
“The palliative care clinicians have a lot of learning to do if they’re going to enter this space and provide expert care,” he said, adding that expert care is what is needed and what was studied in this review. “Providing palliative care to patients with hematologic malignancies has a unique pace and a number of subspecialized therapeutic options with which the palliative care clinician must become familiar.”
Examples include bone marrow transplantation with prolonged hospitalizations and transfusion support, he said.
“Palliative care programs, in order to provide high quality care, will need to familiarize themselves with these therapies and develop close partnership with hematologists to integrate seamlessly into the patient’s care,” he added. “At some centers, culture changes will be necessary concurrent with the clinical practice change of integrating palliative care and it is the responsibility of the palliative care clinicians to bring their very best to these new relationships and patient populations.”
The authors reported having no disclosures. Dr. Campbell also reported having no disclosures.
Specialty palliative care interventions improve outcomes in patients with hematologic malignancies but are underutilized, according to findings from a systematic literature review.
Outcomes that were improved, as demonstrated by 16 studies that met inclusion criteria for the review, included symptom management, inpatient mortality, health care utilization, health care costs, and caregiver-reported outcomes, Elizabeth Elliott, DO, a hematology and oncology fellow at the Cardinal Bernardin Cancer Center, Loyola University, Maywood, Ill., and colleagues reported.
The findings were published online in the Journal of Pain and Symptom Management.
Palliative care needs
Patients with hematologic malignancies, including leukemia, myeloma, and lymphoma, have a high need for supportive care, the authors noted, adding that, although its use has increased over time, palliative care (PC) is often provided late in the disease course – sometimes only in the final days of life.
“Compared with their solid tumor counterparts, patients with hematologic malignancies experience higher symptom burdens, have higher rates of cancer-directed care near death, and are more likely to die while hospitalized than at home or in hospice,” they wrote. “Despite this need, specialist palliative care is less commonly utilized in patients with hematologic malignancies than other cancer types.”
Given the high health care utilization among patients with hematologic malignancies, earlier and more widespread utilization of PC in this population may significantly reduce health care costs, they added.
Palliative care benefits
Of 5,345 studies published between 2005 and 2020 and screened for the current review, 16 met inclusion criteria, including 10 retrospective cohort studies; 4 prospective cohort studies; and 2 randomized, controlled studies.
Nine studies included only patients with hematologic malignancies and seven included both patients with solid tumors and patients with hematologic malignancies. Each study assessed as being of moderate quality.
Benefits of PC as demonstrated in the studies included:
Symptom management: One study, for example, showed that an integrated psychological and PC intervention improved traumatic stress levels, degree and number of physical symptoms, pain intensity, depressive symptoms, and quality of life, compared with no intervention. Another showed that the percentage of patients reporting moderate to severe pain improved from 57% to 18% with a PC intervention, and the number reporting depressive episodes improved from 13% to 5%.
Reduced in-patient death: Findings from eight studies showed that 21.9%-83% of those receiving PC died at home, compared with 6.0%-8.9% of controls. Two studies showed that PC provided at least 20 days prior to death decreased the likelihood of inpatient death and death in an ICU, compared with controls, and one showed that the rate of in-hospital deaths was 30% for those with home PC or hospice, compared with 80% of controls.
Health care utilization: The studies showed that hospitalization occurred in 45%-76.3% of hematologic malignancy patients who received PC, compared with 98% of controls. The odds ratio for hospitalization among acute leukemia patients receiving PC was 0.64, compared with 2.53 among those in a historical control group.
Caregiver-reported outcomes: One randomized, controlled study showed that PC was associated with smaller increases in depression scores, improved coping, and improved scores in multiple quality of life domains in caregivers versus controls.
Survival: One study showed that a larger percentage of hematologic malignancy patients who died 1-6 months after diagnosis had not received PC (28% vs. 23%), whereas more of those who died 6-12 months or 12 or more months after diagnosis had received PC (23.9 vs. 14.9% and 42.5% vs. 22.0%).
Health care costs: Two studies showed a decrease in inpatient costs after a palliative care consultation. Decreases in hospitalization costs were $2,321 and $1,506 for less medically complex patients and $3,515 and $5,617 for more medically complex patient.
Improving PC utilization
One potential strategy to promote earlier referrals to PC is improved education for hematologists, the authors said, citing a study showing that 98% of oncology fellows at one center reported improvement in their ability to assess and manage patient symptoms after completion of a 4-week mandatory PC rotation.
“Another strategy to improve referrals to PC of hematologic malignancies patients could be the creation of programs which facilitate collaboration between PC providers and hematologists, such as the palliative and supportive care special interest group within the American Society for Transplantation and Cellular Therapy,” they wrote.
A third strategy “could be to provide a concurrent care model, in which cancer directed therapy (such as transfusions) is provided at the same time as hospice care,” they added, explaining that such an approach was shown in a study of patients with advanced non–small cell lung cancer to be associated with less aggressive medical treatment and lower costs.
The authors also stressed that patient with solid tumors and those with hematologic malignancies have differing supportive care needs and health care utilization, but several studies included in the current review included both types of cancer.
“Further studies investigating PC use exclusively in patients with hematologic malignancies are needed. Our results demonstrate a strong argument for hematologists to refer their patients early and often for specialized PC,” they concluded.
Indeed, when PC is integrated within hematologic malignancies, impacts occur that are similar to those seen in a variety of other diseases and include improved symptom control, enhanced caregiver experience, and reduced burdens on the health care system, Toby C Campbell, MD, said in an interview.
“The benefits of providing palliative care concurrent with standard cancer care is felt by all the major stakeholders in this care: the patients, their caregivers, and the health care system around them,” said Dr. Campbell, a thoracic medical oncologist and professor in the division of hematology, medical oncology, and palliative care at the University of Wisconsin–Madison.
Overcoming challenges
However, this is “new territory” for most programs, added Dr. Campbell, who also is the University of Wisconsin health chief of palliative care and holds the Ellen and Peter O. Johnson Chair in Palliative Care .
“The palliative care clinicians have a lot of learning to do if they’re going to enter this space and provide expert care,” he said, adding that expert care is what is needed and what was studied in this review. “Providing palliative care to patients with hematologic malignancies has a unique pace and a number of subspecialized therapeutic options with which the palliative care clinician must become familiar.”
Examples include bone marrow transplantation with prolonged hospitalizations and transfusion support, he said.
“Palliative care programs, in order to provide high quality care, will need to familiarize themselves with these therapies and develop close partnership with hematologists to integrate seamlessly into the patient’s care,” he added. “At some centers, culture changes will be necessary concurrent with the clinical practice change of integrating palliative care and it is the responsibility of the palliative care clinicians to bring their very best to these new relationships and patient populations.”
The authors reported having no disclosures. Dr. Campbell also reported having no disclosures.
Specialty palliative care interventions improve outcomes in patients with hematologic malignancies but are underutilized, according to findings from a systematic literature review.
Outcomes that were improved, as demonstrated by 16 studies that met inclusion criteria for the review, included symptom management, inpatient mortality, health care utilization, health care costs, and caregiver-reported outcomes, Elizabeth Elliott, DO, a hematology and oncology fellow at the Cardinal Bernardin Cancer Center, Loyola University, Maywood, Ill., and colleagues reported.
The findings were published online in the Journal of Pain and Symptom Management.
Palliative care needs
Patients with hematologic malignancies, including leukemia, myeloma, and lymphoma, have a high need for supportive care, the authors noted, adding that, although its use has increased over time, palliative care (PC) is often provided late in the disease course – sometimes only in the final days of life.
“Compared with their solid tumor counterparts, patients with hematologic malignancies experience higher symptom burdens, have higher rates of cancer-directed care near death, and are more likely to die while hospitalized than at home or in hospice,” they wrote. “Despite this need, specialist palliative care is less commonly utilized in patients with hematologic malignancies than other cancer types.”
Given the high health care utilization among patients with hematologic malignancies, earlier and more widespread utilization of PC in this population may significantly reduce health care costs, they added.
Palliative care benefits
Of 5,345 studies published between 2005 and 2020 and screened for the current review, 16 met inclusion criteria, including 10 retrospective cohort studies; 4 prospective cohort studies; and 2 randomized, controlled studies.
Nine studies included only patients with hematologic malignancies and seven included both patients with solid tumors and patients with hematologic malignancies. Each study assessed as being of moderate quality.
Benefits of PC as demonstrated in the studies included:
Symptom management: One study, for example, showed that an integrated psychological and PC intervention improved traumatic stress levels, degree and number of physical symptoms, pain intensity, depressive symptoms, and quality of life, compared with no intervention. Another showed that the percentage of patients reporting moderate to severe pain improved from 57% to 18% with a PC intervention, and the number reporting depressive episodes improved from 13% to 5%.
Reduced in-patient death: Findings from eight studies showed that 21.9%-83% of those receiving PC died at home, compared with 6.0%-8.9% of controls. Two studies showed that PC provided at least 20 days prior to death decreased the likelihood of inpatient death and death in an ICU, compared with controls, and one showed that the rate of in-hospital deaths was 30% for those with home PC or hospice, compared with 80% of controls.
Health care utilization: The studies showed that hospitalization occurred in 45%-76.3% of hematologic malignancy patients who received PC, compared with 98% of controls. The odds ratio for hospitalization among acute leukemia patients receiving PC was 0.64, compared with 2.53 among those in a historical control group.
Caregiver-reported outcomes: One randomized, controlled study showed that PC was associated with smaller increases in depression scores, improved coping, and improved scores in multiple quality of life domains in caregivers versus controls.
Survival: One study showed that a larger percentage of hematologic malignancy patients who died 1-6 months after diagnosis had not received PC (28% vs. 23%), whereas more of those who died 6-12 months or 12 or more months after diagnosis had received PC (23.9 vs. 14.9% and 42.5% vs. 22.0%).
Health care costs: Two studies showed a decrease in inpatient costs after a palliative care consultation. Decreases in hospitalization costs were $2,321 and $1,506 for less medically complex patients and $3,515 and $5,617 for more medically complex patient.
Improving PC utilization
One potential strategy to promote earlier referrals to PC is improved education for hematologists, the authors said, citing a study showing that 98% of oncology fellows at one center reported improvement in their ability to assess and manage patient symptoms after completion of a 4-week mandatory PC rotation.
“Another strategy to improve referrals to PC of hematologic malignancies patients could be the creation of programs which facilitate collaboration between PC providers and hematologists, such as the palliative and supportive care special interest group within the American Society for Transplantation and Cellular Therapy,” they wrote.
A third strategy “could be to provide a concurrent care model, in which cancer directed therapy (such as transfusions) is provided at the same time as hospice care,” they added, explaining that such an approach was shown in a study of patients with advanced non–small cell lung cancer to be associated with less aggressive medical treatment and lower costs.
The authors also stressed that patient with solid tumors and those with hematologic malignancies have differing supportive care needs and health care utilization, but several studies included in the current review included both types of cancer.
“Further studies investigating PC use exclusively in patients with hematologic malignancies are needed. Our results demonstrate a strong argument for hematologists to refer their patients early and often for specialized PC,” they concluded.
Indeed, when PC is integrated within hematologic malignancies, impacts occur that are similar to those seen in a variety of other diseases and include improved symptom control, enhanced caregiver experience, and reduced burdens on the health care system, Toby C Campbell, MD, said in an interview.
“The benefits of providing palliative care concurrent with standard cancer care is felt by all the major stakeholders in this care: the patients, their caregivers, and the health care system around them,” said Dr. Campbell, a thoracic medical oncologist and professor in the division of hematology, medical oncology, and palliative care at the University of Wisconsin–Madison.
Overcoming challenges
However, this is “new territory” for most programs, added Dr. Campbell, who also is the University of Wisconsin health chief of palliative care and holds the Ellen and Peter O. Johnson Chair in Palliative Care .
“The palliative care clinicians have a lot of learning to do if they’re going to enter this space and provide expert care,” he said, adding that expert care is what is needed and what was studied in this review. “Providing palliative care to patients with hematologic malignancies has a unique pace and a number of subspecialized therapeutic options with which the palliative care clinician must become familiar.”
Examples include bone marrow transplantation with prolonged hospitalizations and transfusion support, he said.
“Palliative care programs, in order to provide high quality care, will need to familiarize themselves with these therapies and develop close partnership with hematologists to integrate seamlessly into the patient’s care,” he added. “At some centers, culture changes will be necessary concurrent with the clinical practice change of integrating palliative care and it is the responsibility of the palliative care clinicians to bring their very best to these new relationships and patient populations.”
The authors reported having no disclosures. Dr. Campbell also reported having no disclosures.
FROM THE JOURNAL OF PAIN AND SYMPTOM MANAGEMENT
VEXAS: A novel rheumatologic, hematologic syndrome that’s making waves
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
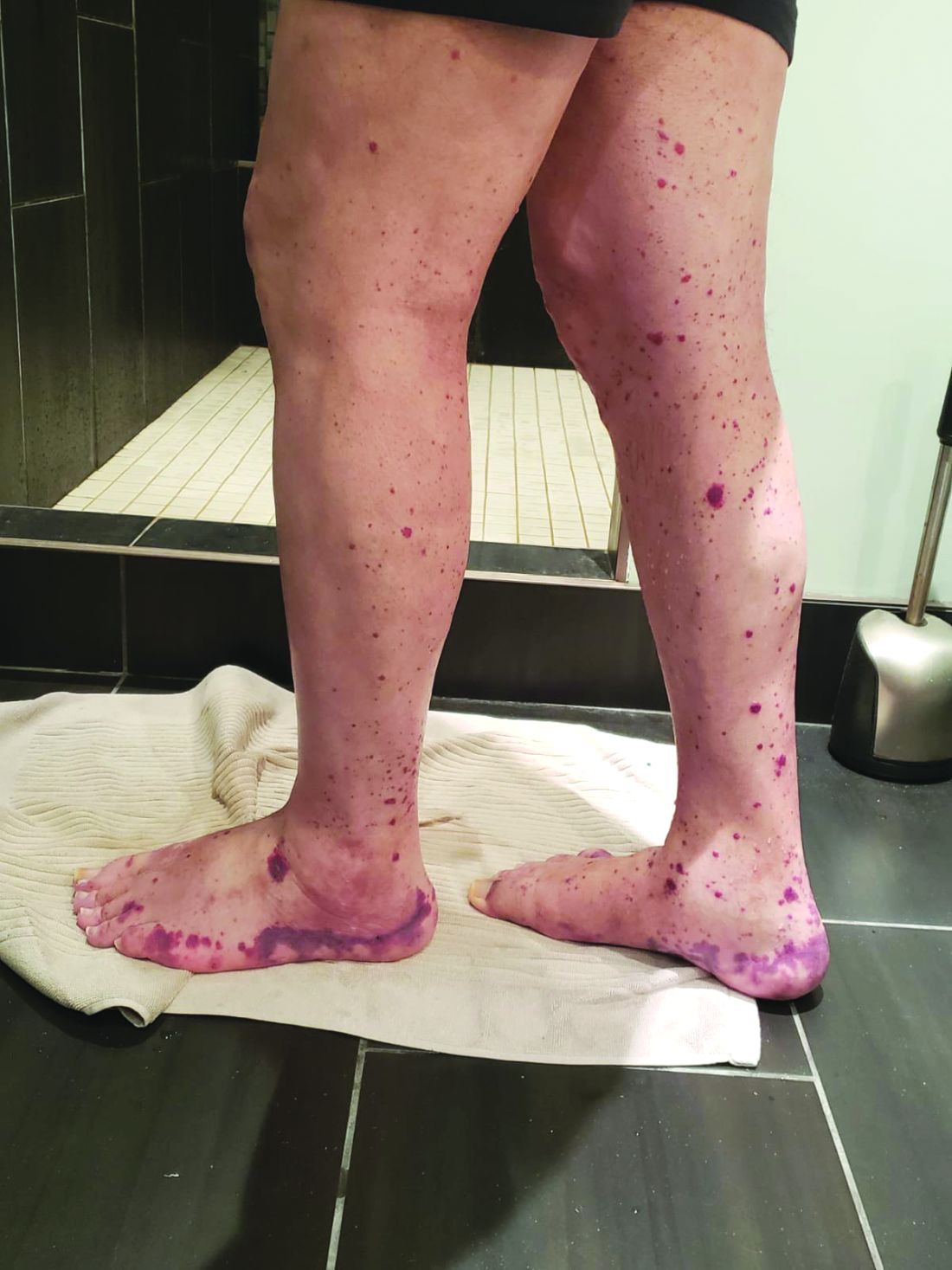
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
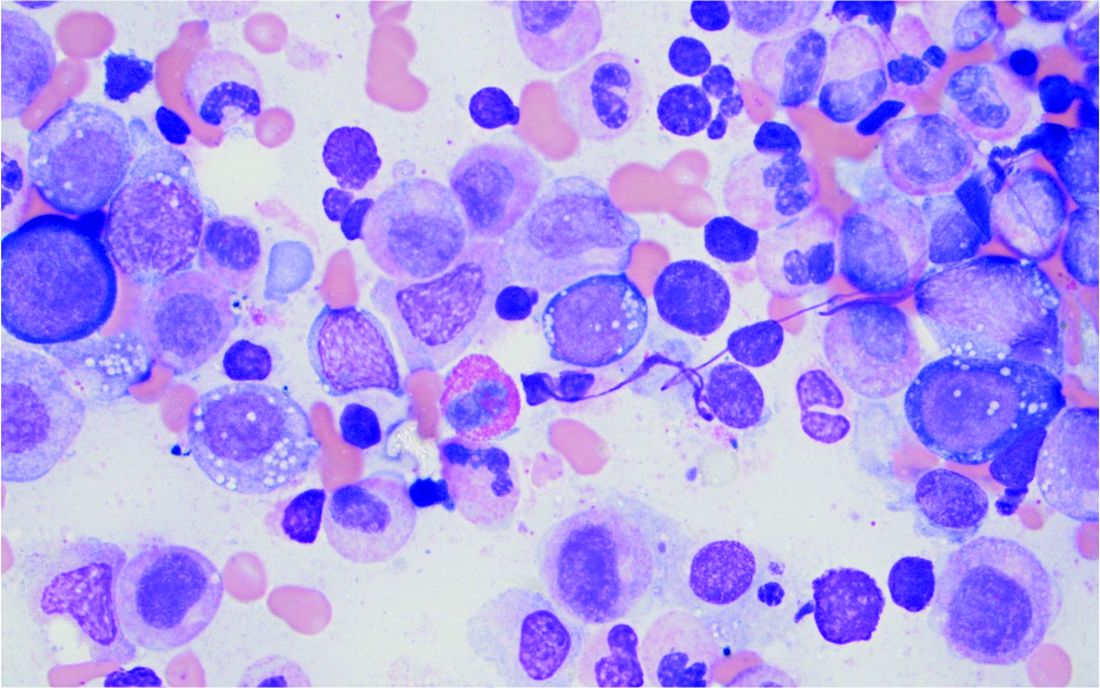
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
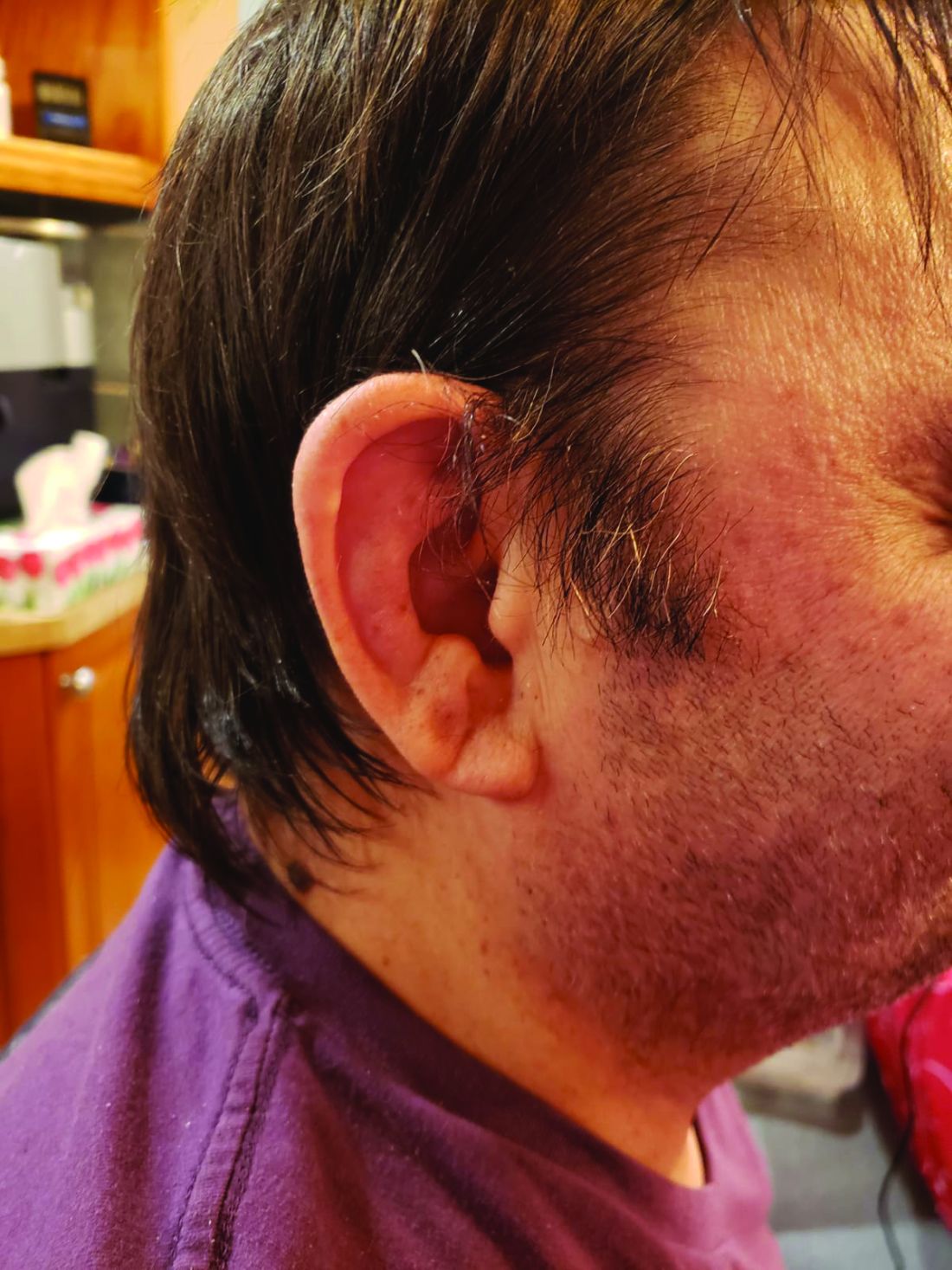
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Real-world outcomes of caplacizumab for iTTP comparable to clinical trial results
Real-world data for caplacizumab outcomes matched those seen in randomized controlled trials (RCTs) for the treatment of immune-mediated thrombotic thrombocytopenic purpura (iTTP), according to the results of a retrospective study.
Data collected from 2018 to 2020 were assessed for 85 patients (4 of them children) receiving caplacizumab at 22 United Kingdom hospitals, according to a report published online in Blood.
Researchers Tina Dutt, PhD, from the Liverpool (England) University Hospitals NHS Foundation Trust, and her colleagues compared patient characteristics and outcomes in these real-world clinical settings to those of caplacizumab trial endpoint results and to historical outcomes in the precaplacizumab era.
Acquired thrombotic thrombocytopenic purpura is an immune-mediated deficiency of the von Willebrand factor–cleaving protease (ADAMTS13), which allows unrestrained adhesion of von Willebrand factor multimers to platelets, leading to thrombocytopenia, hemolytic anemia, and tissue ischemia.
Standard management of iTTP has focused on the replacement of ADAMTS13 and the removal of autoantibodies using plasma exchange and immunosuppression, an approach which has reduced the mortality of acute TTP from greater than 90% to between 10% and 20%, according to the report.
Caplacizumab is a novel anti–von Willebrand factor immunoglobulin fragment that inhibits this interaction between von Willebrand factor multimers and platelets and is now added to the standard treatment regimen. The drug has been assessed in two pivotal multicenter RCTs that led to European Union and U.S. Food and Drug Administration approval.
Benefits and risk
Eighty-four of 85 patients received steroid and rituximab as well as plasma exchange along with caplacizumab treatment. All patients had ADAMTS13 activity at presentation less than 20 IU/dL, with 99% of patients (84/85) having ADAMTS13 activity less than 10 IU/dL, confirming a clinical diagnosis of acute TTP, according to the researchers.
The median time to platelet count normalization (3 days), the median duration of plasma exchange (7 days), and the median hospital stay (12 days) were all comparable with the RCT data, according to the researchers. In addition, the median duration of plasma exchange and time from beginning plasma exchange to platelet count normalization were favorable, compared with historical outcomes (P < .05).
TTP recurred in 5 of the 85 patients, all of whom had persistent ADAMTS13 activity less than 5 IU/dL.
There were 31 adverse events reported in 26 patients, 17 of these (55%) were bleeding episodes, and 5 of 31 (16%) were thrombotic events (2 unrelated to caplacizumab). The overall mortality was 6% (five patients), with no deaths attributed to caplacizumab. In four of the five deaths, caplacizumab was introduced more than 48 hours after plasma exchange initiation (range 3-21 days).
“This real-world evidence from the largest series of TTP patients receiving caplacizumab, outside of the pivotal studies, provides confirmation of the therapeutic benefits of caplacizumab and its inherent bleeding risk,” the researchers concluded.
Dr. Dutt and several of her colleagues reported receiving honoraria from Sanofi for serving on advisory boards, as well as speaker fees from Sanofi and Alexion.
Real-world data for caplacizumab outcomes matched those seen in randomized controlled trials (RCTs) for the treatment of immune-mediated thrombotic thrombocytopenic purpura (iTTP), according to the results of a retrospective study.
Data collected from 2018 to 2020 were assessed for 85 patients (4 of them children) receiving caplacizumab at 22 United Kingdom hospitals, according to a report published online in Blood.
Researchers Tina Dutt, PhD, from the Liverpool (England) University Hospitals NHS Foundation Trust, and her colleagues compared patient characteristics and outcomes in these real-world clinical settings to those of caplacizumab trial endpoint results and to historical outcomes in the precaplacizumab era.
Acquired thrombotic thrombocytopenic purpura is an immune-mediated deficiency of the von Willebrand factor–cleaving protease (ADAMTS13), which allows unrestrained adhesion of von Willebrand factor multimers to platelets, leading to thrombocytopenia, hemolytic anemia, and tissue ischemia.
Standard management of iTTP has focused on the replacement of ADAMTS13 and the removal of autoantibodies using plasma exchange and immunosuppression, an approach which has reduced the mortality of acute TTP from greater than 90% to between 10% and 20%, according to the report.
Caplacizumab is a novel anti–von Willebrand factor immunoglobulin fragment that inhibits this interaction between von Willebrand factor multimers and platelets and is now added to the standard treatment regimen. The drug has been assessed in two pivotal multicenter RCTs that led to European Union and U.S. Food and Drug Administration approval.
Benefits and risk
Eighty-four of 85 patients received steroid and rituximab as well as plasma exchange along with caplacizumab treatment. All patients had ADAMTS13 activity at presentation less than 20 IU/dL, with 99% of patients (84/85) having ADAMTS13 activity less than 10 IU/dL, confirming a clinical diagnosis of acute TTP, according to the researchers.
The median time to platelet count normalization (3 days), the median duration of plasma exchange (7 days), and the median hospital stay (12 days) were all comparable with the RCT data, according to the researchers. In addition, the median duration of plasma exchange and time from beginning plasma exchange to platelet count normalization were favorable, compared with historical outcomes (P < .05).
TTP recurred in 5 of the 85 patients, all of whom had persistent ADAMTS13 activity less than 5 IU/dL.
There were 31 adverse events reported in 26 patients, 17 of these (55%) were bleeding episodes, and 5 of 31 (16%) were thrombotic events (2 unrelated to caplacizumab). The overall mortality was 6% (five patients), with no deaths attributed to caplacizumab. In four of the five deaths, caplacizumab was introduced more than 48 hours after plasma exchange initiation (range 3-21 days).
“This real-world evidence from the largest series of TTP patients receiving caplacizumab, outside of the pivotal studies, provides confirmation of the therapeutic benefits of caplacizumab and its inherent bleeding risk,” the researchers concluded.
Dr. Dutt and several of her colleagues reported receiving honoraria from Sanofi for serving on advisory boards, as well as speaker fees from Sanofi and Alexion.
Real-world data for caplacizumab outcomes matched those seen in randomized controlled trials (RCTs) for the treatment of immune-mediated thrombotic thrombocytopenic purpura (iTTP), according to the results of a retrospective study.
Data collected from 2018 to 2020 were assessed for 85 patients (4 of them children) receiving caplacizumab at 22 United Kingdom hospitals, according to a report published online in Blood.
Researchers Tina Dutt, PhD, from the Liverpool (England) University Hospitals NHS Foundation Trust, and her colleagues compared patient characteristics and outcomes in these real-world clinical settings to those of caplacizumab trial endpoint results and to historical outcomes in the precaplacizumab era.
Acquired thrombotic thrombocytopenic purpura is an immune-mediated deficiency of the von Willebrand factor–cleaving protease (ADAMTS13), which allows unrestrained adhesion of von Willebrand factor multimers to platelets, leading to thrombocytopenia, hemolytic anemia, and tissue ischemia.
Standard management of iTTP has focused on the replacement of ADAMTS13 and the removal of autoantibodies using plasma exchange and immunosuppression, an approach which has reduced the mortality of acute TTP from greater than 90% to between 10% and 20%, according to the report.
Caplacizumab is a novel anti–von Willebrand factor immunoglobulin fragment that inhibits this interaction between von Willebrand factor multimers and platelets and is now added to the standard treatment regimen. The drug has been assessed in two pivotal multicenter RCTs that led to European Union and U.S. Food and Drug Administration approval.
Benefits and risk
Eighty-four of 85 patients received steroid and rituximab as well as plasma exchange along with caplacizumab treatment. All patients had ADAMTS13 activity at presentation less than 20 IU/dL, with 99% of patients (84/85) having ADAMTS13 activity less than 10 IU/dL, confirming a clinical diagnosis of acute TTP, according to the researchers.
The median time to platelet count normalization (3 days), the median duration of plasma exchange (7 days), and the median hospital stay (12 days) were all comparable with the RCT data, according to the researchers. In addition, the median duration of plasma exchange and time from beginning plasma exchange to platelet count normalization were favorable, compared with historical outcomes (P < .05).
TTP recurred in 5 of the 85 patients, all of whom had persistent ADAMTS13 activity less than 5 IU/dL.
There were 31 adverse events reported in 26 patients, 17 of these (55%) were bleeding episodes, and 5 of 31 (16%) were thrombotic events (2 unrelated to caplacizumab). The overall mortality was 6% (five patients), with no deaths attributed to caplacizumab. In four of the five deaths, caplacizumab was introduced more than 48 hours after plasma exchange initiation (range 3-21 days).
“This real-world evidence from the largest series of TTP patients receiving caplacizumab, outside of the pivotal studies, provides confirmation of the therapeutic benefits of caplacizumab and its inherent bleeding risk,” the researchers concluded.
Dr. Dutt and several of her colleagues reported receiving honoraria from Sanofi for serving on advisory boards, as well as speaker fees from Sanofi and Alexion.
FROM BLOOD
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
High-dose chemo no better than standard dose for B-cell lymphoma
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
FROM THE LANCET HEMATOLOGY
Close joint health monitoring essential with new hemophilia therapies
Novel therapies have transformed the treatment of hemophilia in recent decades, but these new approaches also raise new challenges for clinicians who monitor joint health in persons with hemophilia, a specialist said.
“Patient-reported outcomes should be combined with other, more objective outcome measures for joint health monitoring, and joint ultrasound is a promising tool for objective joint health monitoring, although, due to its relatively recent introduction in clinical practice, we lack objective data and standardization,” said Roberta Gualtierotti, MD, PhD, from the Università Degli Studi of Milan.
She reviewed the challenges and approaches to monitoring joint health in persons with hemophilia during the annual congress of the European Association for Haemophilia and Allied Disorders.
Over the last decades the target of hemophilia treatment has shifted from prolonging survival to improving joint health and quality of life, and care has improved with the introduction of novel therapies such as extended half-life replacement products, nonreplacement therapies, and gene therapy, she noted.
However, “due to different pharmacodynamics and pharmacokinetics profiles, the currently available therapies cannot be compared to each other on several levels,” Dr. Gualtierotti said.
Laboratory monitoring of replacement therapies with standard coagulation assays may be unreliable, and depending on the mechanism of action and type of administration of nonreplacement agents, patients may experience breakthrough bleeding, especially after traumatic injury, she said.
Until the specific noncoagulatory effects of factor VIII on bone and joint health is better understood, close monitoring of patients will be required, she added.
Outcome measures
Subjective measures of joint health include patient-reported bleeding rates and health-related quality of life. These are practical for home management, but patients may not be able to distinguish symptoms of acute joint bleeding from chronic arthritis pain, with the potential for either under- or overtreatment, and subjective reporting is likely to miss subclinical bleeding that can occur even when patients are on prophylaxis.
Health-related quality of life tools, whether generic or specific for hemophilia, are not sensitive to small improvements, and they are not always used in routine clinical practice.
Objective measures include physical examination with scoring according to the World Federation of Hemophilia (Gilbert Scale) score or Hemophilia Joint Health Score, but these measures have limited ability to identify early or subclinical joint abnormalities.
“Therefore, joint physical examination on its own is not a sufficient measure of treatment efficacy, and it should be used in combination with other tools more objective, such as imaging,” Dr. Gualtierotti said.
Get the picture?
Imaging with x-rays, MRI, and, recently in some centers, point-of-care ultrasound can provide clinicians with important real-time information about the joint stability and health.
Point-of-care ultrasound in particular offers promise as a practical tool, with no ionizing radiation and high sensitivity for synovial hyperplasia subclinical joint effusion. It’s relatively inexpensive, can be used to image multiple joints, and allows for ease of follow-up, she said. The technique requires specialized training, however, and there is a lack of prospective data about its utility in hemophilia.
Various ultrasound scoring systems have been proposed, and home-based ultrasound is currently being explored in several clinical trials, Dr. Gualtierotti noted.
Other avenues for remote joint health monitoring under consideration are serum or synovial biomarkers for joint bleeding and arthropathy that could be employed at bedside or at home, smartphone apps for breakthrough bleeding and patient-reported outcomes, and sensors for detecting abnormalities in gait that may signal joint dysfunction, she said.
Best practice
In the question-and-answer session following the talk, Fernando Zikan, MD, from the Federal University of Rio de Janeiro noted that, “in underdeveloped countries, we still find it very difficult to guide good practices for joint health control by the patient and family members. Which strategy do you think is fundamental for the patient to feel safe to notice changes in his body?”
“It would be useful to educate patients to come to the center whenever the patient has trauma and whenever an increase in his physical activity occurs. If this is far, a bedside ultrasound evaluation by the general practitioner could help avoid joint damage. Finally, a correct rehabilitation is fundamental,” Dr. Gualtierotti replied.
Asked by several others whether she used ultrasound in her daily practice, Dr. Gualtierotti said that “we use joint ultrasound in our clinical practice in the regular annual check-up examination and whenever the patient suspects and reports hemarthrosis.”
Dr. Gualtierotti reported participation in advisory boards for Biomarin, Pfizer, Bayer, and Takeda, and in educational seminars sponsored by Pfizer, Sobi, and Roche. She has received support for congress travel and/or attendance by Bayer and Pfizer. Dr. Zikan reported no relevant disclosures.
Novel therapies have transformed the treatment of hemophilia in recent decades, but these new approaches also raise new challenges for clinicians who monitor joint health in persons with hemophilia, a specialist said.
“Patient-reported outcomes should be combined with other, more objective outcome measures for joint health monitoring, and joint ultrasound is a promising tool for objective joint health monitoring, although, due to its relatively recent introduction in clinical practice, we lack objective data and standardization,” said Roberta Gualtierotti, MD, PhD, from the Università Degli Studi of Milan.
She reviewed the challenges and approaches to monitoring joint health in persons with hemophilia during the annual congress of the European Association for Haemophilia and Allied Disorders.
Over the last decades the target of hemophilia treatment has shifted from prolonging survival to improving joint health and quality of life, and care has improved with the introduction of novel therapies such as extended half-life replacement products, nonreplacement therapies, and gene therapy, she noted.
However, “due to different pharmacodynamics and pharmacokinetics profiles, the currently available therapies cannot be compared to each other on several levels,” Dr. Gualtierotti said.
Laboratory monitoring of replacement therapies with standard coagulation assays may be unreliable, and depending on the mechanism of action and type of administration of nonreplacement agents, patients may experience breakthrough bleeding, especially after traumatic injury, she said.
Until the specific noncoagulatory effects of factor VIII on bone and joint health is better understood, close monitoring of patients will be required, she added.
Outcome measures
Subjective measures of joint health include patient-reported bleeding rates and health-related quality of life. These are practical for home management, but patients may not be able to distinguish symptoms of acute joint bleeding from chronic arthritis pain, with the potential for either under- or overtreatment, and subjective reporting is likely to miss subclinical bleeding that can occur even when patients are on prophylaxis.
Health-related quality of life tools, whether generic or specific for hemophilia, are not sensitive to small improvements, and they are not always used in routine clinical practice.
Objective measures include physical examination with scoring according to the World Federation of Hemophilia (Gilbert Scale) score or Hemophilia Joint Health Score, but these measures have limited ability to identify early or subclinical joint abnormalities.
“Therefore, joint physical examination on its own is not a sufficient measure of treatment efficacy, and it should be used in combination with other tools more objective, such as imaging,” Dr. Gualtierotti said.
Get the picture?
Imaging with x-rays, MRI, and, recently in some centers, point-of-care ultrasound can provide clinicians with important real-time information about the joint stability and health.
Point-of-care ultrasound in particular offers promise as a practical tool, with no ionizing radiation and high sensitivity for synovial hyperplasia subclinical joint effusion. It’s relatively inexpensive, can be used to image multiple joints, and allows for ease of follow-up, she said. The technique requires specialized training, however, and there is a lack of prospective data about its utility in hemophilia.
Various ultrasound scoring systems have been proposed, and home-based ultrasound is currently being explored in several clinical trials, Dr. Gualtierotti noted.
Other avenues for remote joint health monitoring under consideration are serum or synovial biomarkers for joint bleeding and arthropathy that could be employed at bedside or at home, smartphone apps for breakthrough bleeding and patient-reported outcomes, and sensors for detecting abnormalities in gait that may signal joint dysfunction, she said.
Best practice
In the question-and-answer session following the talk, Fernando Zikan, MD, from the Federal University of Rio de Janeiro noted that, “in underdeveloped countries, we still find it very difficult to guide good practices for joint health control by the patient and family members. Which strategy do you think is fundamental for the patient to feel safe to notice changes in his body?”
“It would be useful to educate patients to come to the center whenever the patient has trauma and whenever an increase in his physical activity occurs. If this is far, a bedside ultrasound evaluation by the general practitioner could help avoid joint damage. Finally, a correct rehabilitation is fundamental,” Dr. Gualtierotti replied.
Asked by several others whether she used ultrasound in her daily practice, Dr. Gualtierotti said that “we use joint ultrasound in our clinical practice in the regular annual check-up examination and whenever the patient suspects and reports hemarthrosis.”
Dr. Gualtierotti reported participation in advisory boards for Biomarin, Pfizer, Bayer, and Takeda, and in educational seminars sponsored by Pfizer, Sobi, and Roche. She has received support for congress travel and/or attendance by Bayer and Pfizer. Dr. Zikan reported no relevant disclosures.
Novel therapies have transformed the treatment of hemophilia in recent decades, but these new approaches also raise new challenges for clinicians who monitor joint health in persons with hemophilia, a specialist said.
“Patient-reported outcomes should be combined with other, more objective outcome measures for joint health monitoring, and joint ultrasound is a promising tool for objective joint health monitoring, although, due to its relatively recent introduction in clinical practice, we lack objective data and standardization,” said Roberta Gualtierotti, MD, PhD, from the Università Degli Studi of Milan.
She reviewed the challenges and approaches to monitoring joint health in persons with hemophilia during the annual congress of the European Association for Haemophilia and Allied Disorders.
Over the last decades the target of hemophilia treatment has shifted from prolonging survival to improving joint health and quality of life, and care has improved with the introduction of novel therapies such as extended half-life replacement products, nonreplacement therapies, and gene therapy, she noted.
However, “due to different pharmacodynamics and pharmacokinetics profiles, the currently available therapies cannot be compared to each other on several levels,” Dr. Gualtierotti said.
Laboratory monitoring of replacement therapies with standard coagulation assays may be unreliable, and depending on the mechanism of action and type of administration of nonreplacement agents, patients may experience breakthrough bleeding, especially after traumatic injury, she said.
Until the specific noncoagulatory effects of factor VIII on bone and joint health is better understood, close monitoring of patients will be required, she added.
Outcome measures
Subjective measures of joint health include patient-reported bleeding rates and health-related quality of life. These are practical for home management, but patients may not be able to distinguish symptoms of acute joint bleeding from chronic arthritis pain, with the potential for either under- or overtreatment, and subjective reporting is likely to miss subclinical bleeding that can occur even when patients are on prophylaxis.
Health-related quality of life tools, whether generic or specific for hemophilia, are not sensitive to small improvements, and they are not always used in routine clinical practice.
Objective measures include physical examination with scoring according to the World Federation of Hemophilia (Gilbert Scale) score or Hemophilia Joint Health Score, but these measures have limited ability to identify early or subclinical joint abnormalities.
“Therefore, joint physical examination on its own is not a sufficient measure of treatment efficacy, and it should be used in combination with other tools more objective, such as imaging,” Dr. Gualtierotti said.
Get the picture?
Imaging with x-rays, MRI, and, recently in some centers, point-of-care ultrasound can provide clinicians with important real-time information about the joint stability and health.
Point-of-care ultrasound in particular offers promise as a practical tool, with no ionizing radiation and high sensitivity for synovial hyperplasia subclinical joint effusion. It’s relatively inexpensive, can be used to image multiple joints, and allows for ease of follow-up, she said. The technique requires specialized training, however, and there is a lack of prospective data about its utility in hemophilia.
Various ultrasound scoring systems have been proposed, and home-based ultrasound is currently being explored in several clinical trials, Dr. Gualtierotti noted.
Other avenues for remote joint health monitoring under consideration are serum or synovial biomarkers for joint bleeding and arthropathy that could be employed at bedside or at home, smartphone apps for breakthrough bleeding and patient-reported outcomes, and sensors for detecting abnormalities in gait that may signal joint dysfunction, she said.
Best practice
In the question-and-answer session following the talk, Fernando Zikan, MD, from the Federal University of Rio de Janeiro noted that, “in underdeveloped countries, we still find it very difficult to guide good practices for joint health control by the patient and family members. Which strategy do you think is fundamental for the patient to feel safe to notice changes in his body?”
“It would be useful to educate patients to come to the center whenever the patient has trauma and whenever an increase in his physical activity occurs. If this is far, a bedside ultrasound evaluation by the general practitioner could help avoid joint damage. Finally, a correct rehabilitation is fundamental,” Dr. Gualtierotti replied.
Asked by several others whether she used ultrasound in her daily practice, Dr. Gualtierotti said that “we use joint ultrasound in our clinical practice in the regular annual check-up examination and whenever the patient suspects and reports hemarthrosis.”
Dr. Gualtierotti reported participation in advisory boards for Biomarin, Pfizer, Bayer, and Takeda, and in educational seminars sponsored by Pfizer, Sobi, and Roche. She has received support for congress travel and/or attendance by Bayer and Pfizer. Dr. Zikan reported no relevant disclosures.
FROM EAHAD 2021
Type 3 von Willebrand a rare but serious bleeding disorder
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
Type 3 von Willebrand disease (VWD) is rare, but this form of the disease is associated with severe bleeding, particularly in muscles and joints, a bleeding disorders expert said.
“There’s a virtually complete deficiency in von Willebrand factor [in type 3 disease], so usually it’s defined as below 5 or in some studies below 3 IU/dL, but also due to the very low levels of von Willebrand factor, there’s also a very low level of factor VIII,” said Jeroen Eikenboom, MD, PhD, from Leiden (the Netherlands) University Medical Center.
“The inheritance pattern is autosomal recessive, and the prevalence is about 1 in a million,” he said during the annual congress of the European Association for Haemophilia and Allied Disorders.
Erik Adolf von Willenbrand, MD, PhD, first described this form of VWD in a family from the Åland Islands, an autonomous region of Finland. The disease, later discovered to be caused in this family by a cytosine deletion in exon 18 of the von Willebrand factor (VWF) gene, was associated with fatal bleeding events in several family members.
“As VWF is the carrier protein of factor VIII, the very low VWF level leads to a strongly reduced factor VIII level, comparable to the levels seen in mild to moderate hemophilia A. As a consequence, VWD type 3 has the combined characteristics of a primary as well as a secondary hemostasis defect,” Dr. Eikenboom explained in an abstract accompanying his talk.
Compared with VWD type 1 or 2, type 3 VWD is associated with bleeding episodes more commonly seen in patients with hemophilia A, notably mucocutaneous bleeding, bleeding after trauma or during surgery, and bleeding into joints and/or muscles.
Treatment
The goals of treatment for patients with type 3 VWD are to correct the dual hemostasis defects of impaired platelet adhesion because of low VWF levels, and the intrinsic coagulation defect because of levels of factor VIII.
Desmopressin is not effective in type 3 VWD, Dr. Eikenboom said, so treatment requires the use of either plasma-derived VWF, with or without factor VIII, or recombinant VWF.
In the United States, the only standalone VWF concentrate approved by the Food and Drug Administration is a recombinant product (Vonvendi), Three other human plasma–derived concentrates containing both VWF and factor VIII are also licensed (Alpanate, Humate-P, Wilate).
Clinicians prescribing the combined factor concentrates need to be aware of differences in pharmacokinetics between the products.
For example, following infusion of Wilate, which has equal amounts of von Willebrand factor and factor VIII, there is an increase in circulation of both von Willebrand factor and factor VIII and a similar decline in each factor over time.
In contrast, following an infusion of Humate-P, which contains lower levels of factor VIII, “interestingly, you see a secondary rise of factor VIII in Humate-P–infused patients, whereas the secondary rise is not visible in the Wilate patients,” he said.
Approximately 22% of patients with type 3 VWD also receive prophylaxis with VWF concentrate, which has been shown to decrease the median annualized bleeding rate from 25% to 6.1%.
Dr. Eikenboom cautioned that 5%-10% of patients with type 3 VWD may develop allo-antibodies against VWF concentrates, which can complicate treatment and carries risk of anaphylactic shock.
“It’s also been mentioned in literature that there may be an association with partial or complete von Willebrand factor gene deletions or nonsense mutations and the development of allo-antibodies,” he said.
Prophylaxis burdensome but helpful
Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee, who specializes in the treatment of patients with von Willebrand disease, follows a number of both girls and boys with type 3 VWD.
“Those are the people who will have bleeding into their joints, and for the girls, worse periods than some of those with other types of von Willebrand disease, and it is true that if you want to stop their bleeding, you cannot use desmopressin like we use in most other von Willebrand patients. They will need factor, although for the heavy menstrual bleeding you can use hormones or tranexamic acid – there are some other options for that,” she said in an interview.
She also noted that type 3 von Willebrand disease can be highly variable. For patients with especially frequent joint bleeding, her center recommends prophylaxis.
“Prophylaxis can be very burdensome for patients. You’re talking about IV therapy several times a week, but it’s very helpful for the joint bleeds. Episodic prophylaxis can be very helpful for heavy menstrual bleeding, and we actually have type 2, type 3, and some type 1 patients with bad enough nose bleeds that they end up on prophylaxis,” she said.
Patients with gastrointestinal bleeding are the most challenging to care for, she noted.
“You can put them on factor prophylaxis, but even that isn’t always enough to help some adults with bad GI bleeding, and we’re investigating other options for that,” she said.
Dr. Eikenboom disclosed research support from CSL Behring and honoraria (directed to his institution) for educational activities sponsored by Roche and Celgene. Dr. Flood reported having no conflicts of interest to disclose.
FROM EAHAD 2021
Bleeding disorder diagnoses delayed by years in girls and women
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
Diagnosis of bleeding disorders in girls and women can lag behind diagnosis in boys and men by more than a decade, meaning needless delays in treatment and poor quality of life for many with hemophilia or related conditions.
“There is increasing awareness about issues faced by women and girls with inherited bleeding disorders, but disparities still exist both in both access to diagnosis and treatment,” said Roseline D’Oiron, MD, from Hôpital Bicêtre in Paris.
“Diagnosis, when it is made, is often made late, particularly in women. Indeed, a recent study from the European Hemophilia Consortium including more than 700 women with bleeding disorders showed that the median age at diagnosis was 16 years old,” she said during the annual congress of the European Association for Haemophilia and Allied Disorders.
She said that delayed diagnosis of bleeding disorders in women and girls may be caused by a lack of knowledge by patients, families, and general practitioners about family history of bleeding disorders, abnormal bleeding events, and heavy menstrual bleeding. In addition, despite the frequency and severity of heavy bleeding events, patients, their families, and caregivers may underestimate the effect on the patient’s quality of life.
Disparities documented
Dr. D’Oiron pointed to several studies showing clear sex-based disparities in time to diagnosis. For example, a study published in Haemophilia showed that in 22 girls with hemophilia A or hemophilia B, the diagnosis of severe hemophilia was delayed by a median of 6.5 months compared with the diagnosis in boys, and a diagnosis of moderate hemophilia in girls was delayed by a median of 39 months.
In a second, single-center study comparing 44 women and girls with mild hemophilia (factor VIII or factor XI levels from 5 to 50 IU/dL) with 77 men and boys with mild hemophilia, the mean age at diagnosis was 31.63 years versus 19.18 years, respectively – a delay of 12.45 years.
A third study comparing 442 girls/women and 442 boys/men with mild hemophilia in France showed a difference of 6.07 years in diagnosis: the median age for girls/women at diagnosis was 16.91 years versus10.84 years for boys/men.
Why it matters
Dr. D’Oiron described the case of a patient named Clare, who first experienced, at age 8, 12 hours of bleeding following a dental procedure. At age 12.5, she began having heavy menstrual bleeding, causing her to miss school for a few days each month, to be feel tired, and have poor-quality sleep.
Despite repeated bleeding episodes, severe anemia, and iron deficiency, her hemophilia was not suspected until after her 16th birthday, and a definitive diagnosis of hemophilia in both Clare and her mother was finally made when Clare was past 17, when a nonsense variant factor in F8, the gene encoding for factor VIII, was detected.
“For Clare, it took more the 8 years after the first bleeding symptoms, and nearly 4 years after presenting with heavy menstrual bleeding to recognize that she had a bleeding problem,” she said.
In total, Clare had about 450 days of heavy menstrual bleeding, causing her to miss an estimated 140 days of school because of the delayed diagnosis and treatment.
“In my view, this is the main argument why it is urgent for these patients to achieve diagnosis early: this is to reduce the duration [of] a very poor quality of life,” Dr. D’Oiron said.
Barriers to diagnosis
Patients and families have reported difficulty distinguishing normal bleeding from abnormal symptoms, and girls may be reluctant to discuss their symptoms with their family or peers. In addition, primary care practitioners may not recognize the severity of the symptoms and therefore may not refer patients to hematologists for further workup.
These findings emphasize the need for improved tools to help patients differentiate between normal and abnormal bleeding, using symptom recognition–based language tools that can lead to early testing and application of accurate diagnostic tools, she said.
Standardization of definitions can help to improve screening and diagnosis, Dr. D’Oiron said, pointing to a recent study in Blood Advances proposing definitions for future research in von Willebrand disease.
For example, the authors of that study proposed a definition of heavy menstrual bleeding to include any of the following:
- Bleeding lasting 8 or more days
- Bleeding that consistently soaks through one or more sanitary protections every 2 hours on multiple days
- Requires use of more than one sanitary protection item at a time
- Requires changing sanitary protection during the night
- Is associated with repeat passing of blood clots
- Has a Pictorial Blood Assessment Chart score greater than 100.
Problem and solutions
Answering the question posed in the title of her talk, Dr. D’Oiron said: “Yes, we do have a problem with the diagnosis of bleeding disorders in women and girls, but we also have solutions.”
The solutions include family and patient outreach efforts; communication to improve awareness; inclusion of general practitioners in the circle of care; and early screening, diagnosis, and treatment.
A bleeding disorders specialist who was not involved in the study said that Dr. D’Oiron’s report closely reflects what she sees in the clinic.
“I do pediatrics, and usually what happens is that I see a teenager with heavy menstrual bleeding and we take her history, and we find out that Mom and multiple female family members have had horrible menstrual bleeding, possibly many of whom have had hysterectomies for it, and then diagnosing the parents and other family members after diagnosing the girl that we’re seeing” said Veronica H. Flood, MD, from the Medical College of Wisconsin, Milwaukee.
“It is unfortunately a very real thing,” she added.
Reasons for the delay likely include lack of awareness of bleeding disorders.
“If you present to a hematologist, we think about bleeding disorders, but if you present to a primary care physician, they don’t always have that on their radar,” she said.
Additionally, a girl from a family with a history of heavy menstrual bleeding may just assume that what she is experiencing is “normal,” despite the serious affect it has on her quality of life, Dr. Flood said.
Dr. D’Oiron’s research is supported by her institution, the French Hemophilia Association, FranceCoag and Mhemon, the European Hemophilia Consortium, and the World Federation of Hemophilia. She reported advisory board or invited speaker activities for multiple companies. Dr. Flood reported having no conflicts of interest to disclose.
FROM EAHAD 2021
‘Phenomenal’ results with CAR T cells in R/R multiple myeloma
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
CAR-T in children branching out to solid tumors
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
FROM TCT 2021