User login
Gene editing strategy may control insect-borne diseases
Credit: James Gathany
Scientists have proposed that gene drives might be used to combat malaria and other insect-borne diseases, control invasive species, and promote sustainable agriculture.
Engineered gene drives are genetic systems that circumvent traditional rules of sexual reproduction and greatly increase the odds that the drive will be passed on to offspring.
This enables the spread of specified genetic alterations through targeted wild populations over many generations.
Gene drives represent a potentially powerful tool to confront regional or global challenges, including the control of invasive species and eradication of insect-borne diseases such as malaria and dengue.
The idea is not new, but a team of researchers has now outlined a technically feasible way to build gene drives that might spread almost any genomic change through populations of sexually reproducing species.
“We all rely on healthy ecosystems and share a responsibility to keep them intact for future generations,” said Kevin Esvelt, PhD, of the Wyss Institute at Harvard University in Boston.
“Given the broad potential of gene drives to address ecological problems, we hope to initiate a transparent, inclusive, and informed public discussion—well in advance of any testing—to collectively decide how we might use this technology for the betterment of humanity and the environment.”
Dr Esvelt and his colleagues have initiated this discussion by publishing papers on gene drives in Science and eLife.
The eLife paper describes the proposed technical methods of building gene drives in different species, defines their theoretical capabilities and limitations, and outlines possible applications.
The Science paper provides an initial assessment of potential environmental and security effects, an analysis of regulatory coverage, and recommendations to ensure responsible development and testing prior to use.
The new technical work in eLife builds upon research by Austin Burt, PhD, of Imperial College London in the UK, who, more than a decade ago, first proposed using a type of gene drive based on cutting DNA to alter populations.
The authors noted that the gene editing tool CRISPR—which is used to precisely insert, replace, and regulate genes—now makes it feasible to create gene drives that work in many different species.
“Our proposal represents a potentially powerful ecosystem management tool for global sustainability, but one that carries with it new concerns, as with any emerging technology,” said George Church, PhD, also of the Wyss Institute.
Dr Esvelt noted that the genomic changes made by gene drives should be reversible. The team has outlined in the eLife publication numerous precautionary measures intended to guide the safe and responsible development of gene drives, many of which were not possible with earlier technologies.
“If the public ever considers making use of a gene drive, we will need to develop appropriate safeguards,” he said. “Ensuring that we have a working reversal drive on hand to quickly undo the proposed genomic change would be one such precaution.”
Because the drives can spread traits only over generations, they will be most effective in species that reproduce quickly or can be released in large numbers, the researchers noted.
For insects, it could take only a couple of years to see a desired change in the population at large, while slower-reproducing organisms would require much longer. Altering human populations would require many centuries.
Gene drives could strike a powerful blow against malaria by altering mosquito populations so they can no longer spread the disease, according to the researchers.
Gene drives might also be used to rid local environments of invasive species or to pave the way toward more sustainable agriculture by reversing mutations that allow particular weed species, such as horseweed, to resist herbicides that are important for no-till farming.
However, the innovative nature of gene drives poses regulatory challenges.
“Simply put, gene drives do not fit comfortably within existing US regulations and international conventions,” said Kenneth Oye, PhD, of the Massachusetts Institute of Technology in Cambridge.
“For example, animal applications of gene drives would be regulated by the FDA as veterinary medicines. Potential implications of gene drives fall beyond the purview of the lists of bacteriological and viral agents that now define security regimes. We’ll need both regulatory reform and public engagement before we can consider beneficial uses. That is why we are excited about getting the conversation on gene drives going early.” ![]()
Credit: James Gathany
Scientists have proposed that gene drives might be used to combat malaria and other insect-borne diseases, control invasive species, and promote sustainable agriculture.
Engineered gene drives are genetic systems that circumvent traditional rules of sexual reproduction and greatly increase the odds that the drive will be passed on to offspring.
This enables the spread of specified genetic alterations through targeted wild populations over many generations.
Gene drives represent a potentially powerful tool to confront regional or global challenges, including the control of invasive species and eradication of insect-borne diseases such as malaria and dengue.
The idea is not new, but a team of researchers has now outlined a technically feasible way to build gene drives that might spread almost any genomic change through populations of sexually reproducing species.
“We all rely on healthy ecosystems and share a responsibility to keep them intact for future generations,” said Kevin Esvelt, PhD, of the Wyss Institute at Harvard University in Boston.
“Given the broad potential of gene drives to address ecological problems, we hope to initiate a transparent, inclusive, and informed public discussion—well in advance of any testing—to collectively decide how we might use this technology for the betterment of humanity and the environment.”
Dr Esvelt and his colleagues have initiated this discussion by publishing papers on gene drives in Science and eLife.
The eLife paper describes the proposed technical methods of building gene drives in different species, defines their theoretical capabilities and limitations, and outlines possible applications.
The Science paper provides an initial assessment of potential environmental and security effects, an analysis of regulatory coverage, and recommendations to ensure responsible development and testing prior to use.
The new technical work in eLife builds upon research by Austin Burt, PhD, of Imperial College London in the UK, who, more than a decade ago, first proposed using a type of gene drive based on cutting DNA to alter populations.
The authors noted that the gene editing tool CRISPR—which is used to precisely insert, replace, and regulate genes—now makes it feasible to create gene drives that work in many different species.
“Our proposal represents a potentially powerful ecosystem management tool for global sustainability, but one that carries with it new concerns, as with any emerging technology,” said George Church, PhD, also of the Wyss Institute.
Dr Esvelt noted that the genomic changes made by gene drives should be reversible. The team has outlined in the eLife publication numerous precautionary measures intended to guide the safe and responsible development of gene drives, many of which were not possible with earlier technologies.
“If the public ever considers making use of a gene drive, we will need to develop appropriate safeguards,” he said. “Ensuring that we have a working reversal drive on hand to quickly undo the proposed genomic change would be one such precaution.”
Because the drives can spread traits only over generations, they will be most effective in species that reproduce quickly or can be released in large numbers, the researchers noted.
For insects, it could take only a couple of years to see a desired change in the population at large, while slower-reproducing organisms would require much longer. Altering human populations would require many centuries.
Gene drives could strike a powerful blow against malaria by altering mosquito populations so they can no longer spread the disease, according to the researchers.
Gene drives might also be used to rid local environments of invasive species or to pave the way toward more sustainable agriculture by reversing mutations that allow particular weed species, such as horseweed, to resist herbicides that are important for no-till farming.
However, the innovative nature of gene drives poses regulatory challenges.
“Simply put, gene drives do not fit comfortably within existing US regulations and international conventions,” said Kenneth Oye, PhD, of the Massachusetts Institute of Technology in Cambridge.
“For example, animal applications of gene drives would be regulated by the FDA as veterinary medicines. Potential implications of gene drives fall beyond the purview of the lists of bacteriological and viral agents that now define security regimes. We’ll need both regulatory reform and public engagement before we can consider beneficial uses. That is why we are excited about getting the conversation on gene drives going early.” ![]()
Credit: James Gathany
Scientists have proposed that gene drives might be used to combat malaria and other insect-borne diseases, control invasive species, and promote sustainable agriculture.
Engineered gene drives are genetic systems that circumvent traditional rules of sexual reproduction and greatly increase the odds that the drive will be passed on to offspring.
This enables the spread of specified genetic alterations through targeted wild populations over many generations.
Gene drives represent a potentially powerful tool to confront regional or global challenges, including the control of invasive species and eradication of insect-borne diseases such as malaria and dengue.
The idea is not new, but a team of researchers has now outlined a technically feasible way to build gene drives that might spread almost any genomic change through populations of sexually reproducing species.
“We all rely on healthy ecosystems and share a responsibility to keep them intact for future generations,” said Kevin Esvelt, PhD, of the Wyss Institute at Harvard University in Boston.
“Given the broad potential of gene drives to address ecological problems, we hope to initiate a transparent, inclusive, and informed public discussion—well in advance of any testing—to collectively decide how we might use this technology for the betterment of humanity and the environment.”
Dr Esvelt and his colleagues have initiated this discussion by publishing papers on gene drives in Science and eLife.
The eLife paper describes the proposed technical methods of building gene drives in different species, defines their theoretical capabilities and limitations, and outlines possible applications.
The Science paper provides an initial assessment of potential environmental and security effects, an analysis of regulatory coverage, and recommendations to ensure responsible development and testing prior to use.
The new technical work in eLife builds upon research by Austin Burt, PhD, of Imperial College London in the UK, who, more than a decade ago, first proposed using a type of gene drive based on cutting DNA to alter populations.
The authors noted that the gene editing tool CRISPR—which is used to precisely insert, replace, and regulate genes—now makes it feasible to create gene drives that work in many different species.
“Our proposal represents a potentially powerful ecosystem management tool for global sustainability, but one that carries with it new concerns, as with any emerging technology,” said George Church, PhD, also of the Wyss Institute.
Dr Esvelt noted that the genomic changes made by gene drives should be reversible. The team has outlined in the eLife publication numerous precautionary measures intended to guide the safe and responsible development of gene drives, many of which were not possible with earlier technologies.
“If the public ever considers making use of a gene drive, we will need to develop appropriate safeguards,” he said. “Ensuring that we have a working reversal drive on hand to quickly undo the proposed genomic change would be one such precaution.”
Because the drives can spread traits only over generations, they will be most effective in species that reproduce quickly or can be released in large numbers, the researchers noted.
For insects, it could take only a couple of years to see a desired change in the population at large, while slower-reproducing organisms would require much longer. Altering human populations would require many centuries.
Gene drives could strike a powerful blow against malaria by altering mosquito populations so they can no longer spread the disease, according to the researchers.
Gene drives might also be used to rid local environments of invasive species or to pave the way toward more sustainable agriculture by reversing mutations that allow particular weed species, such as horseweed, to resist herbicides that are important for no-till farming.
However, the innovative nature of gene drives poses regulatory challenges.
“Simply put, gene drives do not fit comfortably within existing US regulations and international conventions,” said Kenneth Oye, PhD, of the Massachusetts Institute of Technology in Cambridge.
“For example, animal applications of gene drives would be regulated by the FDA as veterinary medicines. Potential implications of gene drives fall beyond the purview of the lists of bacteriological and viral agents that now define security regimes. We’ll need both regulatory reform and public engagement before we can consider beneficial uses. That is why we are excited about getting the conversation on gene drives going early.” ![]()
Teams find new way to kill malaria parasite
red blood cell; Credit: St Jude
Children’s Research Hospital
Two groups of researchers have found they can kill the malaria parasite by targeting a protein complex.
The research showed that a protein complex known as the Plasmodium translocon of exported proteins (PTEX) is needed for the export of malaria-parasite proteins into the cytoplasm of infected red blood cells, and such export is essential for parasite survival.
When the researchers disrupted passage of the proteins in cell cultures, malaria parasites stopped growing and died.
“The malaria parasite secretes hundreds of diverse proteins to seize control of red blood cells,” said Josh R. Beck, PhD, of the Washington University School of Medicine in St Louis.
“We’ve been searching for a single step that all those various proteins have to take to be secreted, and this looks like just such a bottleneck.”
He and his colleagues detailed their findings in a letter to Nature.
The researchers focused on heat shock protein 101 (HSP101), a component of PTEX. Previous studies had suggested that HSP101 might be involved in protein secretion.
So Dr Beck and his colleagues disabled HSP101 in cell cultures, expecting to block the discharge of some malarial proteins. To their surprise, they stopped all of them.
“We think this is a very promising target for drug development,” said study author Daniel Goldberg, MD, PhD, also of Washington University.
“We’re a long way from getting a new drug, but, in the short term, we may look at screening a variety of compounds to see if they have the potential to block HSP101.”
The researchers think HSP101 may ready malarial proteins for secretion through a pore that opens into the red blood cell. Part of this preparation may involve unfolding the proteins into a linear form that allows them to more easily pass through the pore. HSP101 may also give the proteins a biochemical kick that pushes them through the pore.
A separate study published in the same issue of Nature also highlights the importance of PTEX to the malaria parasite’s survival.
Brendan Elsworth, of the Macfarlane Burnet Institute for Medical Research and Public Health in Melbourne, Australia, and his colleagues neutralized the malaria parasite by disabling either HSP101 or PTEX150, another component of PTEX.
“That suggests there are multiple components of the process that we may be able to target with drugs,” Dr Beck said. “In addition, many of the proteins involved in secretion are unlike any human proteins, which means we may be able to disable them without adversely affecting important human proteins.” ![]()
red blood cell; Credit: St Jude
Children’s Research Hospital
Two groups of researchers have found they can kill the malaria parasite by targeting a protein complex.
The research showed that a protein complex known as the Plasmodium translocon of exported proteins (PTEX) is needed for the export of malaria-parasite proteins into the cytoplasm of infected red blood cells, and such export is essential for parasite survival.
When the researchers disrupted passage of the proteins in cell cultures, malaria parasites stopped growing and died.
“The malaria parasite secretes hundreds of diverse proteins to seize control of red blood cells,” said Josh R. Beck, PhD, of the Washington University School of Medicine in St Louis.
“We’ve been searching for a single step that all those various proteins have to take to be secreted, and this looks like just such a bottleneck.”
He and his colleagues detailed their findings in a letter to Nature.
The researchers focused on heat shock protein 101 (HSP101), a component of PTEX. Previous studies had suggested that HSP101 might be involved in protein secretion.
So Dr Beck and his colleagues disabled HSP101 in cell cultures, expecting to block the discharge of some malarial proteins. To their surprise, they stopped all of them.
“We think this is a very promising target for drug development,” said study author Daniel Goldberg, MD, PhD, also of Washington University.
“We’re a long way from getting a new drug, but, in the short term, we may look at screening a variety of compounds to see if they have the potential to block HSP101.”
The researchers think HSP101 may ready malarial proteins for secretion through a pore that opens into the red blood cell. Part of this preparation may involve unfolding the proteins into a linear form that allows them to more easily pass through the pore. HSP101 may also give the proteins a biochemical kick that pushes them through the pore.
A separate study published in the same issue of Nature also highlights the importance of PTEX to the malaria parasite’s survival.
Brendan Elsworth, of the Macfarlane Burnet Institute for Medical Research and Public Health in Melbourne, Australia, and his colleagues neutralized the malaria parasite by disabling either HSP101 or PTEX150, another component of PTEX.
“That suggests there are multiple components of the process that we may be able to target with drugs,” Dr Beck said. “In addition, many of the proteins involved in secretion are unlike any human proteins, which means we may be able to disable them without adversely affecting important human proteins.” ![]()
red blood cell; Credit: St Jude
Children’s Research Hospital
Two groups of researchers have found they can kill the malaria parasite by targeting a protein complex.
The research showed that a protein complex known as the Plasmodium translocon of exported proteins (PTEX) is needed for the export of malaria-parasite proteins into the cytoplasm of infected red blood cells, and such export is essential for parasite survival.
When the researchers disrupted passage of the proteins in cell cultures, malaria parasites stopped growing and died.
“The malaria parasite secretes hundreds of diverse proteins to seize control of red blood cells,” said Josh R. Beck, PhD, of the Washington University School of Medicine in St Louis.
“We’ve been searching for a single step that all those various proteins have to take to be secreted, and this looks like just such a bottleneck.”
He and his colleagues detailed their findings in a letter to Nature.
The researchers focused on heat shock protein 101 (HSP101), a component of PTEX. Previous studies had suggested that HSP101 might be involved in protein secretion.
So Dr Beck and his colleagues disabled HSP101 in cell cultures, expecting to block the discharge of some malarial proteins. To their surprise, they stopped all of them.
“We think this is a very promising target for drug development,” said study author Daniel Goldberg, MD, PhD, also of Washington University.
“We’re a long way from getting a new drug, but, in the short term, we may look at screening a variety of compounds to see if they have the potential to block HSP101.”
The researchers think HSP101 may ready malarial proteins for secretion through a pore that opens into the red blood cell. Part of this preparation may involve unfolding the proteins into a linear form that allows them to more easily pass through the pore. HSP101 may also give the proteins a biochemical kick that pushes them through the pore.
A separate study published in the same issue of Nature also highlights the importance of PTEX to the malaria parasite’s survival.
Brendan Elsworth, of the Macfarlane Burnet Institute for Medical Research and Public Health in Melbourne, Australia, and his colleagues neutralized the malaria parasite by disabling either HSP101 or PTEX150, another component of PTEX.
“That suggests there are multiple components of the process that we may be able to target with drugs,” Dr Beck said. “In addition, many of the proteins involved in secretion are unlike any human proteins, which means we may be able to disable them without adversely affecting important human proteins.” ![]()
Nanoparticles could improve cancer diagnosis
Self-assembling nanoparticles may help physicians diagnose cancers earlier, according to a study published in Angewandte Chemie.
The nanoparticles boost the effectiveness of magnetic resonance imaging (MRI) by specifically seeking out CXCR4 receptors, which are found in cancerous cells.
The iron oxide nanoparticles are coated with peptide ligands that target tumor sites. When the particles find a tumor, they begin to interact with the cancerous cells.
Cancer-specific matrix metalloproteinase biomarkers prompt the nanoparticles to self-assemble into larger particles. And these larger particles are more visible on an MRI scan.
Researchers used cancer cells and mouse models to compare the effects of the self-assembling nanoparticles in MRI scanning against commonly used imaging agents. The nanoparticles produced a more powerful signal and created a clearer image of the tumor.
The team said the nanoparticles increase the sensitivity of MRI scans and could ultimately improve physicians’ ability to detect cancerous cells at much earlier stages of development.
“By improving the sensitivity of an MRI examination, our aim is to help doctors spot something that might be cancerous much more quickly,” said study author Nicholas Long, PhD, of Imperial College London in the UK. “This would enable patients to receive effective treatment sooner, which would hopefully improve survival rates from cancer.”
In addition to improving the sensitivity of MRI scans, the nanoparticles also appear to be safe. Before testing and injecting the particles into mice, the researchers had to ensure the particles would not become so big as to cause damage.
The team injected the particles into a saline solution inside a petri dish and monitored their growth over a 4-hour period. The nanoparticles grew from 100 nm to 800 nm, which was still small enough not to cause any harm.
Now, the researchers are working to enhance the effectiveness of the nanoparticles. And they hope to test their design in a human trial within the next 3 to 5 years.
“We would like to improve the design to make it even easier for doctors to spot a tumor and for surgeons to then operate on it,” Dr Long said. “We’re now trying to add an extra optical signal so that the nanoparticle would light up with a luminescent probe once it had found its target. So, combined with the better MRI signal, it will make it even easier to identify tumors.” ![]()
Self-assembling nanoparticles may help physicians diagnose cancers earlier, according to a study published in Angewandte Chemie.
The nanoparticles boost the effectiveness of magnetic resonance imaging (MRI) by specifically seeking out CXCR4 receptors, which are found in cancerous cells.
The iron oxide nanoparticles are coated with peptide ligands that target tumor sites. When the particles find a tumor, they begin to interact with the cancerous cells.
Cancer-specific matrix metalloproteinase biomarkers prompt the nanoparticles to self-assemble into larger particles. And these larger particles are more visible on an MRI scan.
Researchers used cancer cells and mouse models to compare the effects of the self-assembling nanoparticles in MRI scanning against commonly used imaging agents. The nanoparticles produced a more powerful signal and created a clearer image of the tumor.
The team said the nanoparticles increase the sensitivity of MRI scans and could ultimately improve physicians’ ability to detect cancerous cells at much earlier stages of development.
“By improving the sensitivity of an MRI examination, our aim is to help doctors spot something that might be cancerous much more quickly,” said study author Nicholas Long, PhD, of Imperial College London in the UK. “This would enable patients to receive effective treatment sooner, which would hopefully improve survival rates from cancer.”
In addition to improving the sensitivity of MRI scans, the nanoparticles also appear to be safe. Before testing and injecting the particles into mice, the researchers had to ensure the particles would not become so big as to cause damage.
The team injected the particles into a saline solution inside a petri dish and monitored their growth over a 4-hour period. The nanoparticles grew from 100 nm to 800 nm, which was still small enough not to cause any harm.
Now, the researchers are working to enhance the effectiveness of the nanoparticles. And they hope to test their design in a human trial within the next 3 to 5 years.
“We would like to improve the design to make it even easier for doctors to spot a tumor and for surgeons to then operate on it,” Dr Long said. “We’re now trying to add an extra optical signal so that the nanoparticle would light up with a luminescent probe once it had found its target. So, combined with the better MRI signal, it will make it even easier to identify tumors.” ![]()
Self-assembling nanoparticles may help physicians diagnose cancers earlier, according to a study published in Angewandte Chemie.
The nanoparticles boost the effectiveness of magnetic resonance imaging (MRI) by specifically seeking out CXCR4 receptors, which are found in cancerous cells.
The iron oxide nanoparticles are coated with peptide ligands that target tumor sites. When the particles find a tumor, they begin to interact with the cancerous cells.
Cancer-specific matrix metalloproteinase biomarkers prompt the nanoparticles to self-assemble into larger particles. And these larger particles are more visible on an MRI scan.
Researchers used cancer cells and mouse models to compare the effects of the self-assembling nanoparticles in MRI scanning against commonly used imaging agents. The nanoparticles produced a more powerful signal and created a clearer image of the tumor.
The team said the nanoparticles increase the sensitivity of MRI scans and could ultimately improve physicians’ ability to detect cancerous cells at much earlier stages of development.
“By improving the sensitivity of an MRI examination, our aim is to help doctors spot something that might be cancerous much more quickly,” said study author Nicholas Long, PhD, of Imperial College London in the UK. “This would enable patients to receive effective treatment sooner, which would hopefully improve survival rates from cancer.”
In addition to improving the sensitivity of MRI scans, the nanoparticles also appear to be safe. Before testing and injecting the particles into mice, the researchers had to ensure the particles would not become so big as to cause damage.
The team injected the particles into a saline solution inside a petri dish and monitored their growth over a 4-hour period. The nanoparticles grew from 100 nm to 800 nm, which was still small enough not to cause any harm.
Now, the researchers are working to enhance the effectiveness of the nanoparticles. And they hope to test their design in a human trial within the next 3 to 5 years.
“We would like to improve the design to make it even easier for doctors to spot a tumor and for surgeons to then operate on it,” Dr Long said. “We’re now trying to add an extra optical signal so that the nanoparticle would light up with a luminescent probe once it had found its target. So, combined with the better MRI signal, it will make it even easier to identify tumors.” ![]()
FDA approves product to treat attacks in HAE

The US Food and Drug Administration (FDA) has approved the first recombinant C1-esterase inhibitor product (Ruconest) for the treatment of acute attacks in adults and adolescents with hereditary angioedema (HAE).
HAE, which is caused by insufficient amounts of a plasma protein called C1-esterase inhibitor, affects approximately 6000 to 10,000 people in the US.
People with HAE can develop rapid swelling of the hands, feet, limbs, face, intestinal tract, or airway. These acute attacks can occur spontaneously or may be triggered by stress, surgery, or infection.
“Hereditary angioedema is a rare and potentially life-threatening disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research. “[The approval of Ruconest] provides an important treatment option for these patients.”
Ruconest is a human recombinant C1-esterase inhibitor purified from the milk of genetically modified rabbits. The product is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling.
Trial results have suggested Ruconest is superior to placebo in treating most HAE attacks. However, due to the limited number of patients with laryngeal attacks, Ruconest has not been established as an effective treatment for these attacks.
The FDA approval of Ruconest to treat HAE is based on results of a phase 3, randomized, controlled trial (RCT), which included an open-label extension (OLE) phase, and is supported by the results of 2 additional RCTs and 2 additional OLE studies.
The pivotal RCT and OLE studies included 44 subjects who experienced 170 HAE attacks. The primary efficacy endpoint was the time to the beginning of symptom relief, assessed using patient-reported responses to 2 questions about the change in overall severity of their HAE attack symptoms after the start of treatment.
The researchers assessed these responses at regular time points for each of the affected anatomical locations for up to 24 hours. To achieve the primary endpoint, a patient had to have a positive response to both questions, along with persistence of improvement at the next assessment time (ie, the same or a better response).
There was a statistically significant difference in the time to the beginning of symptom relief in the intent-to-treat population (n=75) between the Ruconest and placebo arms (P=0.031).
The median time to the beginning of symptom relief was 90 minutes for Ruconest-treated patients (n=44) and 152 minutes for placebo-treated patients (n=31).
The most common adverse events, reported in at least 2% of patients receiving Ruconest, were headache, nausea, and diarrhea.
Serious adverse events associated with the treatment include anaphylaxis and arterial and venous thromboembolic events in patients with risk factors, such as an indwelling venous catheter/access device, a prior history of thrombosis, underlying atherosclerosis, the use of oral contraceptives or certain androgens, morbid obesity, and immobility.
Ruconest is manufactured by Pharming Group NV, located in Leiden, the Netherlands, and will be distributed in the US by Santarus Inc., a wholly owned subsidiary of Salix Pharmaceuticals Inc., which is located in Raleigh, North Carolina.
Salix is planning to make Ruconest available to patients later this year. ![]()
The US Food and Drug Administration (FDA) has approved the first recombinant C1-esterase inhibitor product (Ruconest) for the treatment of acute attacks in adults and adolescents with hereditary angioedema (HAE).
HAE, which is caused by insufficient amounts of a plasma protein called C1-esterase inhibitor, affects approximately 6000 to 10,000 people in the US.
People with HAE can develop rapid swelling of the hands, feet, limbs, face, intestinal tract, or airway. These acute attacks can occur spontaneously or may be triggered by stress, surgery, or infection.
“Hereditary angioedema is a rare and potentially life-threatening disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research. “[The approval of Ruconest] provides an important treatment option for these patients.”
Ruconest is a human recombinant C1-esterase inhibitor purified from the milk of genetically modified rabbits. The product is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling.
Trial results have suggested Ruconest is superior to placebo in treating most HAE attacks. However, due to the limited number of patients with laryngeal attacks, Ruconest has not been established as an effective treatment for these attacks.
The FDA approval of Ruconest to treat HAE is based on results of a phase 3, randomized, controlled trial (RCT), which included an open-label extension (OLE) phase, and is supported by the results of 2 additional RCTs and 2 additional OLE studies.
The pivotal RCT and OLE studies included 44 subjects who experienced 170 HAE attacks. The primary efficacy endpoint was the time to the beginning of symptom relief, assessed using patient-reported responses to 2 questions about the change in overall severity of their HAE attack symptoms after the start of treatment.
The researchers assessed these responses at regular time points for each of the affected anatomical locations for up to 24 hours. To achieve the primary endpoint, a patient had to have a positive response to both questions, along with persistence of improvement at the next assessment time (ie, the same or a better response).
There was a statistically significant difference in the time to the beginning of symptom relief in the intent-to-treat population (n=75) between the Ruconest and placebo arms (P=0.031).
The median time to the beginning of symptom relief was 90 minutes for Ruconest-treated patients (n=44) and 152 minutes for placebo-treated patients (n=31).
The most common adverse events, reported in at least 2% of patients receiving Ruconest, were headache, nausea, and diarrhea.
Serious adverse events associated with the treatment include anaphylaxis and arterial and venous thromboembolic events in patients with risk factors, such as an indwelling venous catheter/access device, a prior history of thrombosis, underlying atherosclerosis, the use of oral contraceptives or certain androgens, morbid obesity, and immobility.
Ruconest is manufactured by Pharming Group NV, located in Leiden, the Netherlands, and will be distributed in the US by Santarus Inc., a wholly owned subsidiary of Salix Pharmaceuticals Inc., which is located in Raleigh, North Carolina.
Salix is planning to make Ruconest available to patients later this year. ![]()
The US Food and Drug Administration (FDA) has approved the first recombinant C1-esterase inhibitor product (Ruconest) for the treatment of acute attacks in adults and adolescents with hereditary angioedema (HAE).
HAE, which is caused by insufficient amounts of a plasma protein called C1-esterase inhibitor, affects approximately 6000 to 10,000 people in the US.
People with HAE can develop rapid swelling of the hands, feet, limbs, face, intestinal tract, or airway. These acute attacks can occur spontaneously or may be triggered by stress, surgery, or infection.
“Hereditary angioedema is a rare and potentially life-threatening disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research. “[The approval of Ruconest] provides an important treatment option for these patients.”
Ruconest is a human recombinant C1-esterase inhibitor purified from the milk of genetically modified rabbits. The product is intended to restore the level of functional C1-esterase inhibitor in a patient’s plasma, thereby treating the acute attack of swelling.
Trial results have suggested Ruconest is superior to placebo in treating most HAE attacks. However, due to the limited number of patients with laryngeal attacks, Ruconest has not been established as an effective treatment for these attacks.
The FDA approval of Ruconest to treat HAE is based on results of a phase 3, randomized, controlled trial (RCT), which included an open-label extension (OLE) phase, and is supported by the results of 2 additional RCTs and 2 additional OLE studies.
The pivotal RCT and OLE studies included 44 subjects who experienced 170 HAE attacks. The primary efficacy endpoint was the time to the beginning of symptom relief, assessed using patient-reported responses to 2 questions about the change in overall severity of their HAE attack symptoms after the start of treatment.
The researchers assessed these responses at regular time points for each of the affected anatomical locations for up to 24 hours. To achieve the primary endpoint, a patient had to have a positive response to both questions, along with persistence of improvement at the next assessment time (ie, the same or a better response).
There was a statistically significant difference in the time to the beginning of symptom relief in the intent-to-treat population (n=75) between the Ruconest and placebo arms (P=0.031).
The median time to the beginning of symptom relief was 90 minutes for Ruconest-treated patients (n=44) and 152 minutes for placebo-treated patients (n=31).
The most common adverse events, reported in at least 2% of patients receiving Ruconest, were headache, nausea, and diarrhea.
Serious adverse events associated with the treatment include anaphylaxis and arterial and venous thromboembolic events in patients with risk factors, such as an indwelling venous catheter/access device, a prior history of thrombosis, underlying atherosclerosis, the use of oral contraceptives or certain androgens, morbid obesity, and immobility.
Ruconest is manufactured by Pharming Group NV, located in Leiden, the Netherlands, and will be distributed in the US by Santarus Inc., a wholly owned subsidiary of Salix Pharmaceuticals Inc., which is located in Raleigh, North Carolina.
Salix is planning to make Ruconest available to patients later this year. ![]()
Assay can detect counterfeit malaria drugs

Credit: CDC
A new assay can be used to determine if a product actually contains the antimalarial drug artesunate, according to a paper published in the journal Talanta.
The testing system looks about as simple, and is almost as cheap, as a sheet of paper.
But it’s actually a colorimetric assay consumers could use to tell whether or not they are getting the medication they paid for—artesunate.
The assay also verifies that an adequate level of the drug is present.
“There are laboratory methods to analyze medications such as this, but they often are not available or widely used in the developing world, where malaria kills thousands of people every year,” said study author Vincent Remcho, PhD, of Oregon State University in Corvallis.
“What we need are inexpensive, accurate assays that can detect adulterated pharmaceuticals in the field, simple enough that anyone can use them. Our technology should provide that.”
The technology is an application of microfluidics in which a film is impressed onto paper that can then detect the presence and level of artesunate in a product.
A single pill can be crushed and dissolved in water. When a drop of the solution is placed on the paper, it turns yellow if the drug is present. The intensity of the color indicates the level of the drug, which can be compared to a simple color chart.
The system can also include another step. The researchers created an iPhone app that could be used to measure the color and tell with an even higher degree of accuracy both the presence and level of artesunate.
“This is conceptually similar to what we do with integrated circuit chips in computers, but we’re pushing fluids around instead of electrons, to reveal chemical information that’s useful to us,” Dr Remcho said. “Chemical communication is how Mother Nature does it, and the long-term applications of this approach really are mind-blowing.”
Aside from ensuring patients receive the appropriate treatment, the assay could help government officials combat the larger problem of drug counterfeiting. Researchers have found that, in some places in the developing world, more than 80% of outlets are selling counterfeit pharmaceuticals.
Dr Remcho and his colleagues also believe their technique could be expanded for a wide range of other medical conditions, pharmaceutical and diagnostic tests, pathogen detection, environmental analysis, and other uses. ![]()
Credit: CDC
A new assay can be used to determine if a product actually contains the antimalarial drug artesunate, according to a paper published in the journal Talanta.
The testing system looks about as simple, and is almost as cheap, as a sheet of paper.
But it’s actually a colorimetric assay consumers could use to tell whether or not they are getting the medication they paid for—artesunate.
The assay also verifies that an adequate level of the drug is present.
“There are laboratory methods to analyze medications such as this, but they often are not available or widely used in the developing world, where malaria kills thousands of people every year,” said study author Vincent Remcho, PhD, of Oregon State University in Corvallis.
“What we need are inexpensive, accurate assays that can detect adulterated pharmaceuticals in the field, simple enough that anyone can use them. Our technology should provide that.”
The technology is an application of microfluidics in which a film is impressed onto paper that can then detect the presence and level of artesunate in a product.
A single pill can be crushed and dissolved in water. When a drop of the solution is placed on the paper, it turns yellow if the drug is present. The intensity of the color indicates the level of the drug, which can be compared to a simple color chart.
The system can also include another step. The researchers created an iPhone app that could be used to measure the color and tell with an even higher degree of accuracy both the presence and level of artesunate.
“This is conceptually similar to what we do with integrated circuit chips in computers, but we’re pushing fluids around instead of electrons, to reveal chemical information that’s useful to us,” Dr Remcho said. “Chemical communication is how Mother Nature does it, and the long-term applications of this approach really are mind-blowing.”
Aside from ensuring patients receive the appropriate treatment, the assay could help government officials combat the larger problem of drug counterfeiting. Researchers have found that, in some places in the developing world, more than 80% of outlets are selling counterfeit pharmaceuticals.
Dr Remcho and his colleagues also believe their technique could be expanded for a wide range of other medical conditions, pharmaceutical and diagnostic tests, pathogen detection, environmental analysis, and other uses. ![]()
Credit: CDC
A new assay can be used to determine if a product actually contains the antimalarial drug artesunate, according to a paper published in the journal Talanta.
The testing system looks about as simple, and is almost as cheap, as a sheet of paper.
But it’s actually a colorimetric assay consumers could use to tell whether or not they are getting the medication they paid for—artesunate.
The assay also verifies that an adequate level of the drug is present.
“There are laboratory methods to analyze medications such as this, but they often are not available or widely used in the developing world, where malaria kills thousands of people every year,” said study author Vincent Remcho, PhD, of Oregon State University in Corvallis.
“What we need are inexpensive, accurate assays that can detect adulterated pharmaceuticals in the field, simple enough that anyone can use them. Our technology should provide that.”
The technology is an application of microfluidics in which a film is impressed onto paper that can then detect the presence and level of artesunate in a product.
A single pill can be crushed and dissolved in water. When a drop of the solution is placed on the paper, it turns yellow if the drug is present. The intensity of the color indicates the level of the drug, which can be compared to a simple color chart.
The system can also include another step. The researchers created an iPhone app that could be used to measure the color and tell with an even higher degree of accuracy both the presence and level of artesunate.
“This is conceptually similar to what we do with integrated circuit chips in computers, but we’re pushing fluids around instead of electrons, to reveal chemical information that’s useful to us,” Dr Remcho said. “Chemical communication is how Mother Nature does it, and the long-term applications of this approach really are mind-blowing.”
Aside from ensuring patients receive the appropriate treatment, the assay could help government officials combat the larger problem of drug counterfeiting. Researchers have found that, in some places in the developing world, more than 80% of outlets are selling counterfeit pharmaceuticals.
Dr Remcho and his colleagues also believe their technique could be expanded for a wide range of other medical conditions, pharmaceutical and diagnostic tests, pathogen detection, environmental analysis, and other uses. ![]()
Model reveals how to target cancer’s weaknesses

Credit: PNAS
A new model suggests we should be targeting cancers’ weaknesses instead of their strengths.
An article in BioEssays proposes that cancers form when recently evolved genes are damaged, and cancer cells have to revert to using older, inappropriate genetic pathways.
So we should create treatments that take advantage of capabilities humans have developed more recently—such as the adaptive immune system—instead of trying to target older capabilities—such as the innate immune system and cell proliferation.
“The rapid proliferation of cancer cells is an ancient, default capability that became regulated during the evolution of multicellularity about a billion years ago,” said study author Charley Lineweaver, PhD, of The Australian National University in Canberra.
“Our model suggests that cancer progression is the accumulation of damage to the more recently acquired genes. Without the regulation of these recent genes, cell physiology reverts to earlier programs, such as unregulated cell proliferation.”
To develop their model, Dr Lineweaver and his colleagues turned to knowledge uncovered by genome sequencing in a range of our distant relatives, including fish, coral, and sponges.
This knowledge has allowed scientists to establish the order in which genes evolved and is the basis of the new therapeutic implications of the model, Dr Lineweaver said.
He noted that the standard model of cancer development suggests that selection produces the acquired capabilities of cancer—such as sustained proliferative signaling and evading apoptosis—and they evolve during the lifetime of the patient.
But Dr Lineweaver’s model suggests the capabilities of cancer are acquired atavisms. They are activated during early embryogenesis and wound healing and reactivated inappropriately during carcinogenesis.
The most recent capabilities—mammalian and vertebrate capabilities—are the least entrenched in cancer. So they should be targeted with therapy.
The older capabilities—last eukaryotic common ancestor (LECA) capabilities, stem eukaryote capabilities, and the earliest evolved capabilities—are maintained in cancer and are therefore difficult to target.
For example, some human ATP binding cassette (ABC) transporters are ancient, and some are quite recent. Dr Lineweaver and his colleagues found that older ABC proteins were more likely to be active in cancer.
So the researchers believe we should create treatments that can be expelled by the newer ABC transporters. That way, normal cells will expel the treatment, but cancer cells will not.
Another potential treatment avenue, according to Dr Lineweaver, is targeting the adaptive immune system.
“The adaptive immune system that humans have has evolved relatively recently, and it seems cancer cells do not have the ability to talk to and be protected by it,” he noted.
“The new therapeutic strategies we are proposing target these weaknesses. These strategies are very different from current therapies, which attack cancer’s strength—its ability to proliferate rapidly.” ![]()
Credit: PNAS
A new model suggests we should be targeting cancers’ weaknesses instead of their strengths.
An article in BioEssays proposes that cancers form when recently evolved genes are damaged, and cancer cells have to revert to using older, inappropriate genetic pathways.
So we should create treatments that take advantage of capabilities humans have developed more recently—such as the adaptive immune system—instead of trying to target older capabilities—such as the innate immune system and cell proliferation.
“The rapid proliferation of cancer cells is an ancient, default capability that became regulated during the evolution of multicellularity about a billion years ago,” said study author Charley Lineweaver, PhD, of The Australian National University in Canberra.
“Our model suggests that cancer progression is the accumulation of damage to the more recently acquired genes. Without the regulation of these recent genes, cell physiology reverts to earlier programs, such as unregulated cell proliferation.”
To develop their model, Dr Lineweaver and his colleagues turned to knowledge uncovered by genome sequencing in a range of our distant relatives, including fish, coral, and sponges.
This knowledge has allowed scientists to establish the order in which genes evolved and is the basis of the new therapeutic implications of the model, Dr Lineweaver said.
He noted that the standard model of cancer development suggests that selection produces the acquired capabilities of cancer—such as sustained proliferative signaling and evading apoptosis—and they evolve during the lifetime of the patient.
But Dr Lineweaver’s model suggests the capabilities of cancer are acquired atavisms. They are activated during early embryogenesis and wound healing and reactivated inappropriately during carcinogenesis.
The most recent capabilities—mammalian and vertebrate capabilities—are the least entrenched in cancer. So they should be targeted with therapy.
The older capabilities—last eukaryotic common ancestor (LECA) capabilities, stem eukaryote capabilities, and the earliest evolved capabilities—are maintained in cancer and are therefore difficult to target.
For example, some human ATP binding cassette (ABC) transporters are ancient, and some are quite recent. Dr Lineweaver and his colleagues found that older ABC proteins were more likely to be active in cancer.
So the researchers believe we should create treatments that can be expelled by the newer ABC transporters. That way, normal cells will expel the treatment, but cancer cells will not.
Another potential treatment avenue, according to Dr Lineweaver, is targeting the adaptive immune system.
“The adaptive immune system that humans have has evolved relatively recently, and it seems cancer cells do not have the ability to talk to and be protected by it,” he noted.
“The new therapeutic strategies we are proposing target these weaknesses. These strategies are very different from current therapies, which attack cancer’s strength—its ability to proliferate rapidly.” ![]()
Credit: PNAS
A new model suggests we should be targeting cancers’ weaknesses instead of their strengths.
An article in BioEssays proposes that cancers form when recently evolved genes are damaged, and cancer cells have to revert to using older, inappropriate genetic pathways.
So we should create treatments that take advantage of capabilities humans have developed more recently—such as the adaptive immune system—instead of trying to target older capabilities—such as the innate immune system and cell proliferation.
“The rapid proliferation of cancer cells is an ancient, default capability that became regulated during the evolution of multicellularity about a billion years ago,” said study author Charley Lineweaver, PhD, of The Australian National University in Canberra.
“Our model suggests that cancer progression is the accumulation of damage to the more recently acquired genes. Without the regulation of these recent genes, cell physiology reverts to earlier programs, such as unregulated cell proliferation.”
To develop their model, Dr Lineweaver and his colleagues turned to knowledge uncovered by genome sequencing in a range of our distant relatives, including fish, coral, and sponges.
This knowledge has allowed scientists to establish the order in which genes evolved and is the basis of the new therapeutic implications of the model, Dr Lineweaver said.
He noted that the standard model of cancer development suggests that selection produces the acquired capabilities of cancer—such as sustained proliferative signaling and evading apoptosis—and they evolve during the lifetime of the patient.
But Dr Lineweaver’s model suggests the capabilities of cancer are acquired atavisms. They are activated during early embryogenesis and wound healing and reactivated inappropriately during carcinogenesis.
The most recent capabilities—mammalian and vertebrate capabilities—are the least entrenched in cancer. So they should be targeted with therapy.
The older capabilities—last eukaryotic common ancestor (LECA) capabilities, stem eukaryote capabilities, and the earliest evolved capabilities—are maintained in cancer and are therefore difficult to target.
For example, some human ATP binding cassette (ABC) transporters are ancient, and some are quite recent. Dr Lineweaver and his colleagues found that older ABC proteins were more likely to be active in cancer.
So the researchers believe we should create treatments that can be expelled by the newer ABC transporters. That way, normal cells will expel the treatment, but cancer cells will not.
Another potential treatment avenue, according to Dr Lineweaver, is targeting the adaptive immune system.
“The adaptive immune system that humans have has evolved relatively recently, and it seems cancer cells do not have the ability to talk to and be protected by it,” he noted.
“The new therapeutic strategies we are proposing target these weaknesses. These strategies are very different from current therapies, which attack cancer’s strength—its ability to proliferate rapidly.” ![]()
How federal budget cuts are affecting research

Credit: Rhoda Baer
A new report suggests recent budget cuts to federal health programs in the US have had some negative consequences for hematology researchers.
The Coalition for Health Funding, an alliance of more than 90 public health advocacy organizations, invited scientists, public health advocates, and others to share stories of how they have been hurt by the budget cuts.
The resulting report is titled “Faces of Austerity, How Budget Cuts Hurt America’s Health.”
It details the negative effects the cuts have had on scientific discovery and innovation, scientists and health practitioners, health and social services, and government programs designed to respond to health hazards and natural disasters.
Among the stories included in the report are 2 from members of the American Society of Hematology (ASH), who detail how a decade of flat funding for the National Institutes of Health (NIH) and a 5% budget cut in 2013 have shuttered labs and jeopardized tomorrow’s treatments.
“Most people I know have been affected,” said Debra Newman, PhD, an investigator at BloodCenter of Wisconsin in Milwaukee.
“Their research funding has decreased and, consequently, so has the size of their laboratories because they cannot afford to employ the same number of staff. Talented investigators have started to leave research and go on to other things because they can’t support a research operation without money to run it.”
The other ASH member story is that of Christopher Porter, MD, a pediatric hematologist/oncologist at Children’s Hospital Colorado in Aurora. Despite receiving an excellent score on an NIH grant application, Dr Porter was denied funding in 2013 amid budget cuts.
“My lab had been able to report exciting preliminary data, but we really needed supplemental funds to keep this project moving,” he said. “While our initial application to NIH scored high enough to have received funding in previous years, it was not within the current funding range.”
Drs Newman and Porter are among the first recipients of ASH Bridge Grants, awards first offered in 2012 for investigators who applied for competitive grants from NIH but were denied funding due to cuts. The awards are intended to “bridge” investigators to their next NIH grant.
While such supplementary grant funding programs are helpful, they cannot replace critical NIH funding that has been cut for hematology research, according to ASH.
“When biomedical research is under-funded, everybody loses,” said ASH President Linda J. Burns, MD, of the University of Minnesota.
“Scientists are forced to slow or suspend research because they no longer have the resources to continue searching for new treatments, and even cures, for some of the world’s deadliest diseases. We continue to urge Congress to support a balanced approach to deficit reduction that does not include further cuts to critical biomedical research and public health and safety programs.”
“Faces of Austerity” is available online at www.cutshurt.org. A related report, “Faces of Austerity: How Budget Cuts Have Made Us Sicker, Poorer, and Less Safe,” was published last November. ![]()
Credit: Rhoda Baer
A new report suggests recent budget cuts to federal health programs in the US have had some negative consequences for hematology researchers.
The Coalition for Health Funding, an alliance of more than 90 public health advocacy organizations, invited scientists, public health advocates, and others to share stories of how they have been hurt by the budget cuts.
The resulting report is titled “Faces of Austerity, How Budget Cuts Hurt America’s Health.”
It details the negative effects the cuts have had on scientific discovery and innovation, scientists and health practitioners, health and social services, and government programs designed to respond to health hazards and natural disasters.
Among the stories included in the report are 2 from members of the American Society of Hematology (ASH), who detail how a decade of flat funding for the National Institutes of Health (NIH) and a 5% budget cut in 2013 have shuttered labs and jeopardized tomorrow’s treatments.
“Most people I know have been affected,” said Debra Newman, PhD, an investigator at BloodCenter of Wisconsin in Milwaukee.
“Their research funding has decreased and, consequently, so has the size of their laboratories because they cannot afford to employ the same number of staff. Talented investigators have started to leave research and go on to other things because they can’t support a research operation without money to run it.”
The other ASH member story is that of Christopher Porter, MD, a pediatric hematologist/oncologist at Children’s Hospital Colorado in Aurora. Despite receiving an excellent score on an NIH grant application, Dr Porter was denied funding in 2013 amid budget cuts.
“My lab had been able to report exciting preliminary data, but we really needed supplemental funds to keep this project moving,” he said. “While our initial application to NIH scored high enough to have received funding in previous years, it was not within the current funding range.”
Drs Newman and Porter are among the first recipients of ASH Bridge Grants, awards first offered in 2012 for investigators who applied for competitive grants from NIH but were denied funding due to cuts. The awards are intended to “bridge” investigators to their next NIH grant.
While such supplementary grant funding programs are helpful, they cannot replace critical NIH funding that has been cut for hematology research, according to ASH.
“When biomedical research is under-funded, everybody loses,” said ASH President Linda J. Burns, MD, of the University of Minnesota.
“Scientists are forced to slow or suspend research because they no longer have the resources to continue searching for new treatments, and even cures, for some of the world’s deadliest diseases. We continue to urge Congress to support a balanced approach to deficit reduction that does not include further cuts to critical biomedical research and public health and safety programs.”
“Faces of Austerity” is available online at www.cutshurt.org. A related report, “Faces of Austerity: How Budget Cuts Have Made Us Sicker, Poorer, and Less Safe,” was published last November. ![]()
Credit: Rhoda Baer
A new report suggests recent budget cuts to federal health programs in the US have had some negative consequences for hematology researchers.
The Coalition for Health Funding, an alliance of more than 90 public health advocacy organizations, invited scientists, public health advocates, and others to share stories of how they have been hurt by the budget cuts.
The resulting report is titled “Faces of Austerity, How Budget Cuts Hurt America’s Health.”
It details the negative effects the cuts have had on scientific discovery and innovation, scientists and health practitioners, health and social services, and government programs designed to respond to health hazards and natural disasters.
Among the stories included in the report are 2 from members of the American Society of Hematology (ASH), who detail how a decade of flat funding for the National Institutes of Health (NIH) and a 5% budget cut in 2013 have shuttered labs and jeopardized tomorrow’s treatments.
“Most people I know have been affected,” said Debra Newman, PhD, an investigator at BloodCenter of Wisconsin in Milwaukee.
“Their research funding has decreased and, consequently, so has the size of their laboratories because they cannot afford to employ the same number of staff. Talented investigators have started to leave research and go on to other things because they can’t support a research operation without money to run it.”
The other ASH member story is that of Christopher Porter, MD, a pediatric hematologist/oncologist at Children’s Hospital Colorado in Aurora. Despite receiving an excellent score on an NIH grant application, Dr Porter was denied funding in 2013 amid budget cuts.
“My lab had been able to report exciting preliminary data, but we really needed supplemental funds to keep this project moving,” he said. “While our initial application to NIH scored high enough to have received funding in previous years, it was not within the current funding range.”
Drs Newman and Porter are among the first recipients of ASH Bridge Grants, awards first offered in 2012 for investigators who applied for competitive grants from NIH but were denied funding due to cuts. The awards are intended to “bridge” investigators to their next NIH grant.
While such supplementary grant funding programs are helpful, they cannot replace critical NIH funding that has been cut for hematology research, according to ASH.
“When biomedical research is under-funded, everybody loses,” said ASH President Linda J. Burns, MD, of the University of Minnesota.
“Scientists are forced to slow or suspend research because they no longer have the resources to continue searching for new treatments, and even cures, for some of the world’s deadliest diseases. We continue to urge Congress to support a balanced approach to deficit reduction that does not include further cuts to critical biomedical research and public health and safety programs.”
“Faces of Austerity” is available online at www.cutshurt.org. A related report, “Faces of Austerity: How Budget Cuts Have Made Us Sicker, Poorer, and Less Safe,” was published last November.
Method forces cells to devour dying neighbors
engulfed dying cells (purple)
Credit: Toru Komatsu
A two-pronged approach can prompt phagocytosis in inert cells, according to a paper published in Science Signaling.
Researchers manipulated HeLa cells, which typically cannot perform phagocytosis, by activating one protein inside the cells and expressing another protein on the cells’ surface. This forced the cells to engulf apoptotic Jurkat T cells.
So the researchers believe this technique could be used as a targeted therapy, with engineered cells consuming unwanted cells.
“Our goal is to build artificial cells programmed to eat up dangerous junk in the body, which could be anything from bacteria to the amyloid-beta plaques that cause Alzheimer’s to the body’s own rogue cancer cells,” said study author Takanari Inoue, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“By figuring out how to get normally inert cells to recognize and engulf dying cells, we’ve taken an important step in that direction.”
Dr Inoue and his colleagues set out to “strip down” phagocytosis, determining the minimum tools one cell needs to eat another. Their first task was to induce the HeLa cells to attach to nearby dying cells—apoptotic Jurkat T cells—by getting the right receptors to the HeLa cells’ surface.
The researchers knew that part of a receptor protein called MFG-E8 would recognize and stick to a distress signal on the surface of dying cells, and coaxing the HeLa cells to make the protein fragment was straightforward.
To get the fragment, termed C2, onto the outside of the cells, the team found a way to stick it to another protein that was bound for the cell’s surface, thus taking advantage of the cell’s own transportation system.
As a result, up to 6 apoptotic Jurkat T cells stuck to each HeLa cell. The bad news was that the HeLa cells weren’t actually eating the T cells.
Fortunately, the researchers already had an idea about what to try next. Previous research had shown that activating the Rac gene could cause a cell to engulf beads stuck to its surface.
Sure enough, the team found that HeLa cells with both surface C2 and activated Rac swallowed the apoptotic cells readily.
“We’ve shown it’s possible to endow ordinary cells with the power to do something unique: take on the role of a specialized macrophage,” Dr Inoue said.
He cautioned, however, that the researchers don’t believe the engulfed cells are being broken down. Getting the HeLa cells to finish the process of phagocytosis will be one of the group’s next steps.
engulfed dying cells (purple)
Credit: Toru Komatsu
A two-pronged approach can prompt phagocytosis in inert cells, according to a paper published in Science Signaling.
Researchers manipulated HeLa cells, which typically cannot perform phagocytosis, by activating one protein inside the cells and expressing another protein on the cells’ surface. This forced the cells to engulf apoptotic Jurkat T cells.
So the researchers believe this technique could be used as a targeted therapy, with engineered cells consuming unwanted cells.
“Our goal is to build artificial cells programmed to eat up dangerous junk in the body, which could be anything from bacteria to the amyloid-beta plaques that cause Alzheimer’s to the body’s own rogue cancer cells,” said study author Takanari Inoue, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“By figuring out how to get normally inert cells to recognize and engulf dying cells, we’ve taken an important step in that direction.”
Dr Inoue and his colleagues set out to “strip down” phagocytosis, determining the minimum tools one cell needs to eat another. Their first task was to induce the HeLa cells to attach to nearby dying cells—apoptotic Jurkat T cells—by getting the right receptors to the HeLa cells’ surface.
The researchers knew that part of a receptor protein called MFG-E8 would recognize and stick to a distress signal on the surface of dying cells, and coaxing the HeLa cells to make the protein fragment was straightforward.
To get the fragment, termed C2, onto the outside of the cells, the team found a way to stick it to another protein that was bound for the cell’s surface, thus taking advantage of the cell’s own transportation system.
As a result, up to 6 apoptotic Jurkat T cells stuck to each HeLa cell. The bad news was that the HeLa cells weren’t actually eating the T cells.
Fortunately, the researchers already had an idea about what to try next. Previous research had shown that activating the Rac gene could cause a cell to engulf beads stuck to its surface.
Sure enough, the team found that HeLa cells with both surface C2 and activated Rac swallowed the apoptotic cells readily.
“We’ve shown it’s possible to endow ordinary cells with the power to do something unique: take on the role of a specialized macrophage,” Dr Inoue said.
He cautioned, however, that the researchers don’t believe the engulfed cells are being broken down. Getting the HeLa cells to finish the process of phagocytosis will be one of the group’s next steps.
engulfed dying cells (purple)
Credit: Toru Komatsu
A two-pronged approach can prompt phagocytosis in inert cells, according to a paper published in Science Signaling.
Researchers manipulated HeLa cells, which typically cannot perform phagocytosis, by activating one protein inside the cells and expressing another protein on the cells’ surface. This forced the cells to engulf apoptotic Jurkat T cells.
So the researchers believe this technique could be used as a targeted therapy, with engineered cells consuming unwanted cells.
“Our goal is to build artificial cells programmed to eat up dangerous junk in the body, which could be anything from bacteria to the amyloid-beta plaques that cause Alzheimer’s to the body’s own rogue cancer cells,” said study author Takanari Inoue, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“By figuring out how to get normally inert cells to recognize and engulf dying cells, we’ve taken an important step in that direction.”
Dr Inoue and his colleagues set out to “strip down” phagocytosis, determining the minimum tools one cell needs to eat another. Their first task was to induce the HeLa cells to attach to nearby dying cells—apoptotic Jurkat T cells—by getting the right receptors to the HeLa cells’ surface.
The researchers knew that part of a receptor protein called MFG-E8 would recognize and stick to a distress signal on the surface of dying cells, and coaxing the HeLa cells to make the protein fragment was straightforward.
To get the fragment, termed C2, onto the outside of the cells, the team found a way to stick it to another protein that was bound for the cell’s surface, thus taking advantage of the cell’s own transportation system.
As a result, up to 6 apoptotic Jurkat T cells stuck to each HeLa cell. The bad news was that the HeLa cells weren’t actually eating the T cells.
Fortunately, the researchers already had an idea about what to try next. Previous research had shown that activating the Rac gene could cause a cell to engulf beads stuck to its surface.
Sure enough, the team found that HeLa cells with both surface C2 and activated Rac swallowed the apoptotic cells readily.
“We’ve shown it’s possible to endow ordinary cells with the power to do something unique: take on the role of a specialized macrophage,” Dr Inoue said.
He cautioned, however, that the researchers don’t believe the engulfed cells are being broken down. Getting the HeLa cells to finish the process of phagocytosis will be one of the group’s next steps.
Pair details ‘promise and perils’ of antioxidants

Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets.
Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets.
Two researchers have offered an explanation as to why antioxidants are not effective in fighting cancers and suggested a way to change that.
The duo proposed that antioxidants from supplements or dietary sources are proving ineffective because they are not acting where reactive oxygen species (ROS) are produced.
So therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, may be more effective.
David Tuveson, MD, PhD, of the Cold Spring Harbor Laboratory in New York, and Navdeep S. Chandel, PhD, of the Feinberg School of Medicine at Northwestern University in Chicago, detailed these theories in a report published in The New England Journal of Medicine.
The pair’s insights are based on recent advances in understanding the cell system that establishes a natural balance between oxidizing and antioxidizing compounds.
Oxidants like hydrogen peroxide are manufactured within cells and are essential in small quantities. But oxidants are toxic in large amounts, and cells naturally generate their own antioxidants to neutralize oxidants.
It has seemed logical, therefore, to boost a person’s intake of antioxidants to counter the effects of hydrogen peroxide and other similarly toxic ROS. All the more because cancer cells are known to generate higher levels of ROS to help feed their abnormal growth.
However, Drs Tuveson and Chandel proposed that taking antioxidant pills or eating foods rich in antioxidants may be failing to show a beneficial effect against cancer because antioxidants do not act where tumor-promoting ROS are produced—at mitochondria.
Rather, supplements and dietary antioxidants tend to accumulate at scattered distant sites in the cell, “leaving tumor-promoting ROS relatively unperturbed.”
Therefore, the authors suggested therapies that directly inhibit the production of mitochondrial- and NADPH oxidase-derived ROS, or that scavenge ROS at these sites, will be more effective than dietary antioxidants.
An alternative approach
Drs Tuveson and Chandel also proposed an alternative approach: disabling antioxidants in cancer cells. They noted that quantities of both ROS and natural antioxidants are higher in cancer cells. The higher levels of antioxidants are a natural defense by cancer cells to keep their higher levels of oxidants in check so that growth can continue.
In fact, therapies that raise the levels of oxidants in cells can be beneficial, whereas those that act as antioxidants may further stimulate the cancer cells.
So the authors suggested that genetic or pharmacologic inhibition of antioxidant proteins—a concept tested successfully in rodent models of lung and pancreatic cancers—may be a useful therapeutic approach in humans.
The key challenge is to identify antioxidant proteins and pathways in cells that are used only by cancer cells and not by healthy cells. Impeding antioxidant production in healthy cells will upset the delicate redox balance upon which normal cellular function depends.
So it seems research is needed to profile antioxidant pathways in tumor and adjacent normal cells, to identify possible therapeutic targets.
Gene editing doesn’t increase mutations in iPSCs
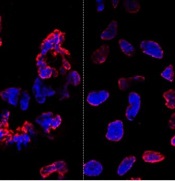
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()