User login
Genetic ‘barcode’ could help track malaria
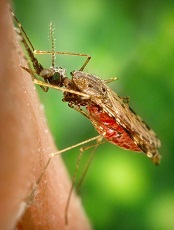
Credit: James Gathany
A genetic “barcode” for malaria parasites could be used to track and contain the spread of the disease, according to research published in Nature Communications.
Investigators analyzed the DNA of more than 700 Plasmodium falciparum parasites taken from patients in East and West Africa, South East Asia, Oceania, and South America.
And this revealed several short genetic sequences that were distinct in the DNA of parasites from certain geographic regions.
The team used this information to design a genetic barcode of 23 single-nucleotide polymorphisms that can be used to identify the source of new malaria infections.
“Being able to determine the geographic origin of malaria parasites has enormous potential in containing drug-resistance and eliminating malaria,” said study author Taane Clark, DPhil, of the London School of Hygiene & Tropical Medicine in the UK.
“Our work represents a breakthrough in the genetic barcoding of P falciparum, as it reveals very specific and accurate sequences for different geographic settings. We are currently extending the barcode to include other populations, such as India, Central America, southern Africa, and the Caribbean, and plan to include genetic markers for other types malaria, such as P vivax.”
Previous candidates for malaria genetic barcodes have relied on identifying DNA markers found in the parasite’s cell nucleus, which shows too much genetic variation between individual parasites to be used accurately.
But Dr Clark and his colleagues studied the DNA found in 2 parts of the parasite’s cells outside of the nucleus—the mitochondria and the apicolasts, which are only inherited through maternal lines, so their genes remain much more stable over generations.
By identifying short sequences in the DNA of the parasite’s mitochondria and apicoplasts that were specific for different geographic locations, the investigators were able to design a genetic barcode that is 92% predictive, stable, and geographically informative over time.
“By taking finger-prick bloodspots from malaria patients and using rapid gene sequencing technologies on small amounts of parasite material, local agencies could use this new barcode to quickly and accurately identify where a form of the parasite may have come from and help in programs of malaria elimination and resistance containment,” said study author Cally Roper, PhD, also of the London School of Hygiene & Tropical Medicine.
The investigators noted, however, that this barcode is limited because their study lacks representation of the Indian sub-continent, Central America, southern Africa, and the Caribbean, owing to the scarcity of sequence data from these regions.
Additionally, there’s a need to study more samples from East Africa, a region of high genetic diversity, high migration, and poor predictive ability. ![]()
Credit: James Gathany
A genetic “barcode” for malaria parasites could be used to track and contain the spread of the disease, according to research published in Nature Communications.
Investigators analyzed the DNA of more than 700 Plasmodium falciparum parasites taken from patients in East and West Africa, South East Asia, Oceania, and South America.
And this revealed several short genetic sequences that were distinct in the DNA of parasites from certain geographic regions.
The team used this information to design a genetic barcode of 23 single-nucleotide polymorphisms that can be used to identify the source of new malaria infections.
“Being able to determine the geographic origin of malaria parasites has enormous potential in containing drug-resistance and eliminating malaria,” said study author Taane Clark, DPhil, of the London School of Hygiene & Tropical Medicine in the UK.
“Our work represents a breakthrough in the genetic barcoding of P falciparum, as it reveals very specific and accurate sequences for different geographic settings. We are currently extending the barcode to include other populations, such as India, Central America, southern Africa, and the Caribbean, and plan to include genetic markers for other types malaria, such as P vivax.”
Previous candidates for malaria genetic barcodes have relied on identifying DNA markers found in the parasite’s cell nucleus, which shows too much genetic variation between individual parasites to be used accurately.
But Dr Clark and his colleagues studied the DNA found in 2 parts of the parasite’s cells outside of the nucleus—the mitochondria and the apicolasts, which are only inherited through maternal lines, so their genes remain much more stable over generations.
By identifying short sequences in the DNA of the parasite’s mitochondria and apicoplasts that were specific for different geographic locations, the investigators were able to design a genetic barcode that is 92% predictive, stable, and geographically informative over time.
“By taking finger-prick bloodspots from malaria patients and using rapid gene sequencing technologies on small amounts of parasite material, local agencies could use this new barcode to quickly and accurately identify where a form of the parasite may have come from and help in programs of malaria elimination and resistance containment,” said study author Cally Roper, PhD, also of the London School of Hygiene & Tropical Medicine.
The investigators noted, however, that this barcode is limited because their study lacks representation of the Indian sub-continent, Central America, southern Africa, and the Caribbean, owing to the scarcity of sequence data from these regions.
Additionally, there’s a need to study more samples from East Africa, a region of high genetic diversity, high migration, and poor predictive ability. ![]()
Credit: James Gathany
A genetic “barcode” for malaria parasites could be used to track and contain the spread of the disease, according to research published in Nature Communications.
Investigators analyzed the DNA of more than 700 Plasmodium falciparum parasites taken from patients in East and West Africa, South East Asia, Oceania, and South America.
And this revealed several short genetic sequences that were distinct in the DNA of parasites from certain geographic regions.
The team used this information to design a genetic barcode of 23 single-nucleotide polymorphisms that can be used to identify the source of new malaria infections.
“Being able to determine the geographic origin of malaria parasites has enormous potential in containing drug-resistance and eliminating malaria,” said study author Taane Clark, DPhil, of the London School of Hygiene & Tropical Medicine in the UK.
“Our work represents a breakthrough in the genetic barcoding of P falciparum, as it reveals very specific and accurate sequences for different geographic settings. We are currently extending the barcode to include other populations, such as India, Central America, southern Africa, and the Caribbean, and plan to include genetic markers for other types malaria, such as P vivax.”
Previous candidates for malaria genetic barcodes have relied on identifying DNA markers found in the parasite’s cell nucleus, which shows too much genetic variation between individual parasites to be used accurately.
But Dr Clark and his colleagues studied the DNA found in 2 parts of the parasite’s cells outside of the nucleus—the mitochondria and the apicolasts, which are only inherited through maternal lines, so their genes remain much more stable over generations.
By identifying short sequences in the DNA of the parasite’s mitochondria and apicoplasts that were specific for different geographic locations, the investigators were able to design a genetic barcode that is 92% predictive, stable, and geographically informative over time.
“By taking finger-prick bloodspots from malaria patients and using rapid gene sequencing technologies on small amounts of parasite material, local agencies could use this new barcode to quickly and accurately identify where a form of the parasite may have come from and help in programs of malaria elimination and resistance containment,” said study author Cally Roper, PhD, also of the London School of Hygiene & Tropical Medicine.
The investigators noted, however, that this barcode is limited because their study lacks representation of the Indian sub-continent, Central America, southern Africa, and the Caribbean, owing to the scarcity of sequence data from these regions.
Additionally, there’s a need to study more samples from East Africa, a region of high genetic diversity, high migration, and poor predictive ability. ![]()
Method allows for high-resolution cell imaging

filaments (red), microtubules
(green), and nuclei (blue)
National Institutes of Health
A new technique allows scientists to view the cell cytoskeleton with “unprecedented resolution,” according to a paper published in Nature Methods.
A group of researchers exploited the properties of a fluorescent molecule they developed to generate 2 probes for imaging the cytoskeleton.
The team said these probes can easily enter live cells, are non-toxic, have long-lasting signals, and offer better image resolution than current imaging techniques.
This research began when Kai Johnsson, PhD, of École Polytechnique Fédérale de Lausanne in Switzerland, and his colleagues developed a fluorescent molecule called silicon-rhodamine (SiR).
The molecule switches “on” only when it binds to the charged surface of a protein like those found on the cytoskeleton. When SiR switches on, it emits light at far-red wavelengths.
The challenge was getting SiR to bind specifically to the cytoskeleton’s proteins, actin and tubulin. To achieve this, the researchers fused SiR molecules with compounds that bind tubulin or actin.
The resulting hybrid molecules consist of a SiR molecule, which provides the fluorescent signal, and a molecule of a natural compound that can bind the target protein.
One such compound was docetaxel, an anticancer drug that binds tubulin, and the other was jasplakinolide, which specifically binds the cytoskeletal form of actin. Both compounds, which are used here in very low, non-toxic concentrations, can easily pass through the cell’s membrane and into the cell itself.
The probes, called SiR-tubulin and SiR-actin, were used to visualize the dynamics of the cytoskeleton in human skin cells. Because the light signal of the probes is emitted in the far-red spectrum, it is easy to isolate from background noise, which generates images of unprecedented resolution when used with super-resolution microscopy.
An additional advantage of the probes, according to the researchers, is their practicality.
“You just add them directly into your cell culture, and they are taken up by the cells,” Dr Johnsson said.
The probes don’t require any washing or preparation of the cells before administration or any subsequent washing steps, which helps in maintaining the stability of their environment and their natural biological functions.
“Up to now, no probes were available that would allow you to get high-quality images of microtubules and microfilaments in living cells without some kind of genetic modification,” Dr Johnsson said.
“With this work, we provide the biological community with 2 high-performing, high-contrast fluorogenic probes that emit in the non-phototoxic part of the light spectrum, and can be even used in tissues like whole-blood samples.” ![]()
filaments (red), microtubules
(green), and nuclei (blue)
National Institutes of Health
A new technique allows scientists to view the cell cytoskeleton with “unprecedented resolution,” according to a paper published in Nature Methods.
A group of researchers exploited the properties of a fluorescent molecule they developed to generate 2 probes for imaging the cytoskeleton.
The team said these probes can easily enter live cells, are non-toxic, have long-lasting signals, and offer better image resolution than current imaging techniques.
This research began when Kai Johnsson, PhD, of École Polytechnique Fédérale de Lausanne in Switzerland, and his colleagues developed a fluorescent molecule called silicon-rhodamine (SiR).
The molecule switches “on” only when it binds to the charged surface of a protein like those found on the cytoskeleton. When SiR switches on, it emits light at far-red wavelengths.
The challenge was getting SiR to bind specifically to the cytoskeleton’s proteins, actin and tubulin. To achieve this, the researchers fused SiR molecules with compounds that bind tubulin or actin.
The resulting hybrid molecules consist of a SiR molecule, which provides the fluorescent signal, and a molecule of a natural compound that can bind the target protein.
One such compound was docetaxel, an anticancer drug that binds tubulin, and the other was jasplakinolide, which specifically binds the cytoskeletal form of actin. Both compounds, which are used here in very low, non-toxic concentrations, can easily pass through the cell’s membrane and into the cell itself.
The probes, called SiR-tubulin and SiR-actin, were used to visualize the dynamics of the cytoskeleton in human skin cells. Because the light signal of the probes is emitted in the far-red spectrum, it is easy to isolate from background noise, which generates images of unprecedented resolution when used with super-resolution microscopy.
An additional advantage of the probes, according to the researchers, is their practicality.
“You just add them directly into your cell culture, and they are taken up by the cells,” Dr Johnsson said.
The probes don’t require any washing or preparation of the cells before administration or any subsequent washing steps, which helps in maintaining the stability of their environment and their natural biological functions.
“Up to now, no probes were available that would allow you to get high-quality images of microtubules and microfilaments in living cells without some kind of genetic modification,” Dr Johnsson said.
“With this work, we provide the biological community with 2 high-performing, high-contrast fluorogenic probes that emit in the non-phototoxic part of the light spectrum, and can be even used in tissues like whole-blood samples.” ![]()
filaments (red), microtubules
(green), and nuclei (blue)
National Institutes of Health
A new technique allows scientists to view the cell cytoskeleton with “unprecedented resolution,” according to a paper published in Nature Methods.
A group of researchers exploited the properties of a fluorescent molecule they developed to generate 2 probes for imaging the cytoskeleton.
The team said these probes can easily enter live cells, are non-toxic, have long-lasting signals, and offer better image resolution than current imaging techniques.
This research began when Kai Johnsson, PhD, of École Polytechnique Fédérale de Lausanne in Switzerland, and his colleagues developed a fluorescent molecule called silicon-rhodamine (SiR).
The molecule switches “on” only when it binds to the charged surface of a protein like those found on the cytoskeleton. When SiR switches on, it emits light at far-red wavelengths.
The challenge was getting SiR to bind specifically to the cytoskeleton’s proteins, actin and tubulin. To achieve this, the researchers fused SiR molecules with compounds that bind tubulin or actin.
The resulting hybrid molecules consist of a SiR molecule, which provides the fluorescent signal, and a molecule of a natural compound that can bind the target protein.
One such compound was docetaxel, an anticancer drug that binds tubulin, and the other was jasplakinolide, which specifically binds the cytoskeletal form of actin. Both compounds, which are used here in very low, non-toxic concentrations, can easily pass through the cell’s membrane and into the cell itself.
The probes, called SiR-tubulin and SiR-actin, were used to visualize the dynamics of the cytoskeleton in human skin cells. Because the light signal of the probes is emitted in the far-red spectrum, it is easy to isolate from background noise, which generates images of unprecedented resolution when used with super-resolution microscopy.
An additional advantage of the probes, according to the researchers, is their practicality.
“You just add them directly into your cell culture, and they are taken up by the cells,” Dr Johnsson said.
The probes don’t require any washing or preparation of the cells before administration or any subsequent washing steps, which helps in maintaining the stability of their environment and their natural biological functions.
“Up to now, no probes were available that would allow you to get high-quality images of microtubules and microfilaments in living cells without some kind of genetic modification,” Dr Johnsson said.
“With this work, we provide the biological community with 2 high-performing, high-contrast fluorogenic probes that emit in the non-phototoxic part of the light spectrum, and can be even used in tissues like whole-blood samples.” ![]()
Research reveals previously unidentified proteins
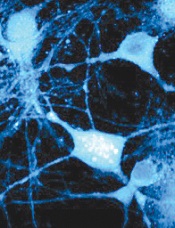
Credit: Lu Wei
By cataloging more than 18,000 proteins, scientists have produced an “almost-complete” inventory of the human proteome.
They discovered protein fragments encoded by DNA outside of currently known genes and found that many known genes have become non-functional.
They also described an important function for messenger RNA (mRNA) and showed that protein profiles could predict drug sensitivity.
The team reported their findings in Nature.
They also made the information freely available in ProteomicsDB. This database includes information on the types, distribution, and abundance of proteins in various cells, tissues, and body fluids.
Through their protein cataloguing, the researchers showed that each mRNA determines the number of protein copies to be produced by a cell. And this “copying key” is specific to each protein.
“It appears that every mRNA molecule knows the unit amount for its protein so it knows whether to produce 10, 100, or 1000 copies,” said study author Bernhard Küster, PhD, of Technische Universitaet Muenchen in Germany.
“Since we now know this ratio for a large number of proteins, we can infer protein abundance from mRNA abundance in just about every tissue, and vice versa.”
The researchers were also surprised to discover hundreds of protein fragments that are encoded by DNA outside of currently known genes. The team believes these “new” proteins may possess novel biological properties and functions that could be exploited for therapeutic purposes.
On the other hand, Dr Küster and his colleagues have been unable to locate roughly 2000 proteins that should exist, according to the gene map. The team also found evidence suggesting that many known genes have become non-functional.
“We might be watching evolution in action here,” Dr Küster said. “The human organism deactivates superfluous genes and tests new gene prototypes at the same time.”
That being the case, the researchers noted that it might never be possible to determine exactly how many proteins there are in the human body.
Lastly, Dr Küster and his colleagues confirmed the findings of previous studies, which showed that specific protein patterns can predict the efficacy of a given drug.
The team evaluated 24 cancer drugs and found their effectiveness against 35 cancer cell lines was strongly correlated with their protein profiles.
“This edges us a little bit closer to the individualized treatment of patients,” Dr Küster said. “If we knew the protein profile of a tumor in detail, we might be able to administer drugs in a more targeted way.” ![]()
Credit: Lu Wei
By cataloging more than 18,000 proteins, scientists have produced an “almost-complete” inventory of the human proteome.
They discovered protein fragments encoded by DNA outside of currently known genes and found that many known genes have become non-functional.
They also described an important function for messenger RNA (mRNA) and showed that protein profiles could predict drug sensitivity.
The team reported their findings in Nature.
They also made the information freely available in ProteomicsDB. This database includes information on the types, distribution, and abundance of proteins in various cells, tissues, and body fluids.
Through their protein cataloguing, the researchers showed that each mRNA determines the number of protein copies to be produced by a cell. And this “copying key” is specific to each protein.
“It appears that every mRNA molecule knows the unit amount for its protein so it knows whether to produce 10, 100, or 1000 copies,” said study author Bernhard Küster, PhD, of Technische Universitaet Muenchen in Germany.
“Since we now know this ratio for a large number of proteins, we can infer protein abundance from mRNA abundance in just about every tissue, and vice versa.”
The researchers were also surprised to discover hundreds of protein fragments that are encoded by DNA outside of currently known genes. The team believes these “new” proteins may possess novel biological properties and functions that could be exploited for therapeutic purposes.
On the other hand, Dr Küster and his colleagues have been unable to locate roughly 2000 proteins that should exist, according to the gene map. The team also found evidence suggesting that many known genes have become non-functional.
“We might be watching evolution in action here,” Dr Küster said. “The human organism deactivates superfluous genes and tests new gene prototypes at the same time.”
That being the case, the researchers noted that it might never be possible to determine exactly how many proteins there are in the human body.
Lastly, Dr Küster and his colleagues confirmed the findings of previous studies, which showed that specific protein patterns can predict the efficacy of a given drug.
The team evaluated 24 cancer drugs and found their effectiveness against 35 cancer cell lines was strongly correlated with their protein profiles.
“This edges us a little bit closer to the individualized treatment of patients,” Dr Küster said. “If we knew the protein profile of a tumor in detail, we might be able to administer drugs in a more targeted way.” ![]()
Credit: Lu Wei
By cataloging more than 18,000 proteins, scientists have produced an “almost-complete” inventory of the human proteome.
They discovered protein fragments encoded by DNA outside of currently known genes and found that many known genes have become non-functional.
They also described an important function for messenger RNA (mRNA) and showed that protein profiles could predict drug sensitivity.
The team reported their findings in Nature.
They also made the information freely available in ProteomicsDB. This database includes information on the types, distribution, and abundance of proteins in various cells, tissues, and body fluids.
Through their protein cataloguing, the researchers showed that each mRNA determines the number of protein copies to be produced by a cell. And this “copying key” is specific to each protein.
“It appears that every mRNA molecule knows the unit amount for its protein so it knows whether to produce 10, 100, or 1000 copies,” said study author Bernhard Küster, PhD, of Technische Universitaet Muenchen in Germany.
“Since we now know this ratio for a large number of proteins, we can infer protein abundance from mRNA abundance in just about every tissue, and vice versa.”
The researchers were also surprised to discover hundreds of protein fragments that are encoded by DNA outside of currently known genes. The team believes these “new” proteins may possess novel biological properties and functions that could be exploited for therapeutic purposes.
On the other hand, Dr Küster and his colleagues have been unable to locate roughly 2000 proteins that should exist, according to the gene map. The team also found evidence suggesting that many known genes have become non-functional.
“We might be watching evolution in action here,” Dr Küster said. “The human organism deactivates superfluous genes and tests new gene prototypes at the same time.”
That being the case, the researchers noted that it might never be possible to determine exactly how many proteins there are in the human body.
Lastly, Dr Küster and his colleagues confirmed the findings of previous studies, which showed that specific protein patterns can predict the efficacy of a given drug.
The team evaluated 24 cancer drugs and found their effectiveness against 35 cancer cell lines was strongly correlated with their protein profiles.
“This edges us a little bit closer to the individualized treatment of patients,” Dr Küster said. “If we knew the protein profile of a tumor in detail, we might be able to administer drugs in a more targeted way.” ![]()
‘Nanodaisies’ deliver drug cocktail to leukemia cells

Credit: PNAS
Biomedical engineers have reported that daisy-shaped, nanoscale structures can deliver a cocktail of drugs directly to cancer cells.
The “nanodaisies” effectively delivered a 2-drug combination in a range of cell lines, including the leukemia cell line HL-60.
The drug-delivery vehicles also proved effective in a mouse model of lung cancer.
Zhen Gu, PhD, of North Carolina State University and the University of North Carolina at Chapel Hill, and his colleagues detailed these results in Biomaterials.
“We found that this technique was much better than conventional drug-delivery techniques at inhibiting the growth of lung cancer tumors in mice,” Dr Gu said.
“And based on in vitro tests in 9 different cell lines, the technique is also promising for use against leukemia, breast, prostate, liver, ovarian, and brain cancers.”
To make the “nanodaisies,” the researchers begin with a solution that contains a polymer called polyethylene glycol (PEG). The PEG forms long strands that have much shorter strands branching off to either side.
The researchers directly link the anticancer drug camptothecin (CPT) onto the shorter strands and introduce the anticancer drug doxorubicin (Dox) into the solution.
PEG is hydrophilic, but CPT and Dox are hydrophobic. As a result, the CPT and Dox cluster together in the solution, wrapping the PEG around themselves. This results in a daisy-shaped drug cocktail, only 50 nanometers in diameter, that can (in theory) be injected into a cancer patient.
Once injected, the nanodaisies float through the bloodstream until they are absorbed by cancer cells. In fact, one of the reasons the researchers chose to use PEG is because it has chemical properties that prolong the life of the drugs in the bloodstream. Once in a cancer cell, the drugs are released.
“Both drugs attack the cell’s nucleus but via different mechanisms,” said study author Wanyi Tai, PhD, who was previously a researcher in Dr Gu’s lab but is now at the University of Washington in Seattle.
“Combined, the drugs are more effective than either drug is by itself,” Dr Gu added. “We are very optimistic about this technique and are hoping to begin preclinical testing in the near future.” ![]()
Credit: PNAS
Biomedical engineers have reported that daisy-shaped, nanoscale structures can deliver a cocktail of drugs directly to cancer cells.
The “nanodaisies” effectively delivered a 2-drug combination in a range of cell lines, including the leukemia cell line HL-60.
The drug-delivery vehicles also proved effective in a mouse model of lung cancer.
Zhen Gu, PhD, of North Carolina State University and the University of North Carolina at Chapel Hill, and his colleagues detailed these results in Biomaterials.
“We found that this technique was much better than conventional drug-delivery techniques at inhibiting the growth of lung cancer tumors in mice,” Dr Gu said.
“And based on in vitro tests in 9 different cell lines, the technique is also promising for use against leukemia, breast, prostate, liver, ovarian, and brain cancers.”
To make the “nanodaisies,” the researchers begin with a solution that contains a polymer called polyethylene glycol (PEG). The PEG forms long strands that have much shorter strands branching off to either side.
The researchers directly link the anticancer drug camptothecin (CPT) onto the shorter strands and introduce the anticancer drug doxorubicin (Dox) into the solution.
PEG is hydrophilic, but CPT and Dox are hydrophobic. As a result, the CPT and Dox cluster together in the solution, wrapping the PEG around themselves. This results in a daisy-shaped drug cocktail, only 50 nanometers in diameter, that can (in theory) be injected into a cancer patient.
Once injected, the nanodaisies float through the bloodstream until they are absorbed by cancer cells. In fact, one of the reasons the researchers chose to use PEG is because it has chemical properties that prolong the life of the drugs in the bloodstream. Once in a cancer cell, the drugs are released.
“Both drugs attack the cell’s nucleus but via different mechanisms,” said study author Wanyi Tai, PhD, who was previously a researcher in Dr Gu’s lab but is now at the University of Washington in Seattle.
“Combined, the drugs are more effective than either drug is by itself,” Dr Gu added. “We are very optimistic about this technique and are hoping to begin preclinical testing in the near future.” ![]()
Credit: PNAS
Biomedical engineers have reported that daisy-shaped, nanoscale structures can deliver a cocktail of drugs directly to cancer cells.
The “nanodaisies” effectively delivered a 2-drug combination in a range of cell lines, including the leukemia cell line HL-60.
The drug-delivery vehicles also proved effective in a mouse model of lung cancer.
Zhen Gu, PhD, of North Carolina State University and the University of North Carolina at Chapel Hill, and his colleagues detailed these results in Biomaterials.
“We found that this technique was much better than conventional drug-delivery techniques at inhibiting the growth of lung cancer tumors in mice,” Dr Gu said.
“And based on in vitro tests in 9 different cell lines, the technique is also promising for use against leukemia, breast, prostate, liver, ovarian, and brain cancers.”
To make the “nanodaisies,” the researchers begin with a solution that contains a polymer called polyethylene glycol (PEG). The PEG forms long strands that have much shorter strands branching off to either side.
The researchers directly link the anticancer drug camptothecin (CPT) onto the shorter strands and introduce the anticancer drug doxorubicin (Dox) into the solution.
PEG is hydrophilic, but CPT and Dox are hydrophobic. As a result, the CPT and Dox cluster together in the solution, wrapping the PEG around themselves. This results in a daisy-shaped drug cocktail, only 50 nanometers in diameter, that can (in theory) be injected into a cancer patient.
Once injected, the nanodaisies float through the bloodstream until they are absorbed by cancer cells. In fact, one of the reasons the researchers chose to use PEG is because it has chemical properties that prolong the life of the drugs in the bloodstream. Once in a cancer cell, the drugs are released.
“Both drugs attack the cell’s nucleus but via different mechanisms,” said study author Wanyi Tai, PhD, who was previously a researcher in Dr Gu’s lab but is now at the University of Washington in Seattle.
“Combined, the drugs are more effective than either drug is by itself,” Dr Gu added. “We are very optimistic about this technique and are hoping to begin preclinical testing in the near future.” ![]()
Study elucidates enzyme’s role in cell survival
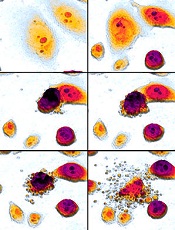
Credit: Egelberg
Investigators say they have solved the mystery of an enzyme’s role in cell survival, thereby offering clues as to how the immune system fights infection and pointing to possible strategies for treating cancers.
The enzyme, receptor-interacting protein kinase 1 (RIPK1), is known to play a pivotal role in survival after birth.
But the new research, published in Cell, reveals that RIPK1 inhibits the pathways that control apoptosis and necroptosis.
By removing different components of each pathway in different combinations, the investigators demonstrated that, after birth, RIPK1 helps cells maintain a balanced response to signals that promote either pathway.
“We are learning that, in disease, this balancing act can be perturbed to produce damage and cell death,” said study author Douglas Green, PhD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
The results resolve long-standing questions about RIPK1’s role in cell survival and provide clues about how the immune system might use these pathways to contain infections.
The findings have also prompted the researchers to launch an investigation into whether RIPK1 could be harnessed to kill cancer cells or provide insight into tumor development.
“This study fundamentally changes the way we think about RIPK1, a molecule that we care about because it is required for life,” Dr Green said. “The results helped us identify new pathways involved in regulating programmed cell death and suggest that we might be able to develop cancer therapies that target these pathways or engage them in other ways to advance treatment of a range of diseases.”
The report builds on previous research from Dr Green’s lab regarding regulation of the pathways that control apoptosis and necroptosis. The investigators knew that apoptosis is driven by the enzyme caspase-8, which forms a complex with FADD and other proteins.
And necroptosis involves a pathway orchestrated by the enzyme receptor-interacting protein kinase 3 (RIPK3). Before birth, RIPK1 works through RIPK3 to trigger cell death by necroptosis, but, until now, the enzyme’s primary role after birth was uncertain.
So the investigators bred mice lacking different combinations of genes for RIPK1, RIPK3, caspase-8, FADD and other components of both the apoptotic and necroptotic pathways.
Mice lacking RIPK1 died. Mice missing 2 genes—RIPK1 plus RIPK3 or RIPK1 plus caspase-8 or FADD—also died soon after birth.
However, mice survived and developed normally when the investigators removed 3 genes—RIPK1, RIPK3, and either caspase-8 or FADD.
“The fact that the mice survived was totally unexpected and made us rethink how these pathways work,” Dr Green said.
The results also demonstrated that other pathways must exist in cells to maintain a balanced response to signals pushing for cell death via apoptosis or necroptosis.
Evidence in this study, for example, suggested one possible new pathway that triggered necroptosis using interferon and other elements of the immune response to infections. ![]()
Credit: Egelberg
Investigators say they have solved the mystery of an enzyme’s role in cell survival, thereby offering clues as to how the immune system fights infection and pointing to possible strategies for treating cancers.
The enzyme, receptor-interacting protein kinase 1 (RIPK1), is known to play a pivotal role in survival after birth.
But the new research, published in Cell, reveals that RIPK1 inhibits the pathways that control apoptosis and necroptosis.
By removing different components of each pathway in different combinations, the investigators demonstrated that, after birth, RIPK1 helps cells maintain a balanced response to signals that promote either pathway.
“We are learning that, in disease, this balancing act can be perturbed to produce damage and cell death,” said study author Douglas Green, PhD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
The results resolve long-standing questions about RIPK1’s role in cell survival and provide clues about how the immune system might use these pathways to contain infections.
The findings have also prompted the researchers to launch an investigation into whether RIPK1 could be harnessed to kill cancer cells or provide insight into tumor development.
“This study fundamentally changes the way we think about RIPK1, a molecule that we care about because it is required for life,” Dr Green said. “The results helped us identify new pathways involved in regulating programmed cell death and suggest that we might be able to develop cancer therapies that target these pathways or engage them in other ways to advance treatment of a range of diseases.”
The report builds on previous research from Dr Green’s lab regarding regulation of the pathways that control apoptosis and necroptosis. The investigators knew that apoptosis is driven by the enzyme caspase-8, which forms a complex with FADD and other proteins.
And necroptosis involves a pathway orchestrated by the enzyme receptor-interacting protein kinase 3 (RIPK3). Before birth, RIPK1 works through RIPK3 to trigger cell death by necroptosis, but, until now, the enzyme’s primary role after birth was uncertain.
So the investigators bred mice lacking different combinations of genes for RIPK1, RIPK3, caspase-8, FADD and other components of both the apoptotic and necroptotic pathways.
Mice lacking RIPK1 died. Mice missing 2 genes—RIPK1 plus RIPK3 or RIPK1 plus caspase-8 or FADD—also died soon after birth.
However, mice survived and developed normally when the investigators removed 3 genes—RIPK1, RIPK3, and either caspase-8 or FADD.
“The fact that the mice survived was totally unexpected and made us rethink how these pathways work,” Dr Green said.
The results also demonstrated that other pathways must exist in cells to maintain a balanced response to signals pushing for cell death via apoptosis or necroptosis.
Evidence in this study, for example, suggested one possible new pathway that triggered necroptosis using interferon and other elements of the immune response to infections. ![]()
Credit: Egelberg
Investigators say they have solved the mystery of an enzyme’s role in cell survival, thereby offering clues as to how the immune system fights infection and pointing to possible strategies for treating cancers.
The enzyme, receptor-interacting protein kinase 1 (RIPK1), is known to play a pivotal role in survival after birth.
But the new research, published in Cell, reveals that RIPK1 inhibits the pathways that control apoptosis and necroptosis.
By removing different components of each pathway in different combinations, the investigators demonstrated that, after birth, RIPK1 helps cells maintain a balanced response to signals that promote either pathway.
“We are learning that, in disease, this balancing act can be perturbed to produce damage and cell death,” said study author Douglas Green, PhD, of the St Jude Children’s Research Hospital in Memphis, Tennessee.
The results resolve long-standing questions about RIPK1’s role in cell survival and provide clues about how the immune system might use these pathways to contain infections.
The findings have also prompted the researchers to launch an investigation into whether RIPK1 could be harnessed to kill cancer cells or provide insight into tumor development.
“This study fundamentally changes the way we think about RIPK1, a molecule that we care about because it is required for life,” Dr Green said. “The results helped us identify new pathways involved in regulating programmed cell death and suggest that we might be able to develop cancer therapies that target these pathways or engage them in other ways to advance treatment of a range of diseases.”
The report builds on previous research from Dr Green’s lab regarding regulation of the pathways that control apoptosis and necroptosis. The investigators knew that apoptosis is driven by the enzyme caspase-8, which forms a complex with FADD and other proteins.
And necroptosis involves a pathway orchestrated by the enzyme receptor-interacting protein kinase 3 (RIPK3). Before birth, RIPK1 works through RIPK3 to trigger cell death by necroptosis, but, until now, the enzyme’s primary role after birth was uncertain.
So the investigators bred mice lacking different combinations of genes for RIPK1, RIPK3, caspase-8, FADD and other components of both the apoptotic and necroptotic pathways.
Mice lacking RIPK1 died. Mice missing 2 genes—RIPK1 plus RIPK3 or RIPK1 plus caspase-8 or FADD—also died soon after birth.
However, mice survived and developed normally when the investigators removed 3 genes—RIPK1, RIPK3, and either caspase-8 or FADD.
“The fact that the mice survived was totally unexpected and made us rethink how these pathways work,” Dr Green said.
The results also demonstrated that other pathways must exist in cells to maintain a balanced response to signals pushing for cell death via apoptosis or necroptosis.
Evidence in this study, for example, suggested one possible new pathway that triggered necroptosis using interferon and other elements of the immune response to infections. ![]()
Method reveals new targets of p53

Credit: A.T. Tikhonenko
A novel sequencing technique has allowed researchers to identify direct targets of p53, providing new insight into this gene’s anticancer activity.
The research, published in eLife, revealed nearly 200 genes that were directly regulated by p53, and many of these had never been identified before.
The study’s authors said this work lays the foundation for investigations into which of these genes are necessary for p53’s cancer-killing effects and how cancer cells evade these genes.
The researchers noted that all cancers must deal with p53’s antitumor effects. Generally, there are 2 ways they do this: by mutating p53 directly or by producing the protein MDM2, which inhibits p53 function. With the current study, the team explored the second strategy.
“MDM2 inhibitors, which are through phase 1 human trials, effectively activate p53 but manage to kill only about 1 in 20 tumors,” said study author Joaquín Espinosa, PhD, of the University of Colorado in Boulder.
“The question is why. What else is happening in these cancer cells that allow them to evade p53?”
According to the researchers, the answer is in the downstream effects of p53. The gene sets in motion a cascade of events that lead to cancer cell destruction. But it has been unclear exactly which other genes are directly activated by p53.
To identify genetic targets of p53, Dr Espinosa and his colleagues used a technique called Global Run-On Sequencing (GRO-Seq). Unlike other methods, GRO-Seq measures new RNA being created, not overall RNA levels.
“Many teams around the world have been getting cancer cells, treating them with MDM2 inhibitors, and waiting hours and hours to see what genes turn on, and then, only imprecisely,” Dr Espinosa said. “GRO-Seq lets us do it in minutes, and the discoveries are massive.”
The technique generates a large quantity of data because it requires counting tens of thousands of RNA molecules before and after p53 activation. So this research required designing algorithms to sort through the data, as well as a computational biologist driving a supercomputer.
But the researchers were able to pinpoint new genes directly regulated by p53. And they believe this could aid the future development of cancer-fighting strategies. ![]()
Credit: A.T. Tikhonenko
A novel sequencing technique has allowed researchers to identify direct targets of p53, providing new insight into this gene’s anticancer activity.
The research, published in eLife, revealed nearly 200 genes that were directly regulated by p53, and many of these had never been identified before.
The study’s authors said this work lays the foundation for investigations into which of these genes are necessary for p53’s cancer-killing effects and how cancer cells evade these genes.
The researchers noted that all cancers must deal with p53’s antitumor effects. Generally, there are 2 ways they do this: by mutating p53 directly or by producing the protein MDM2, which inhibits p53 function. With the current study, the team explored the second strategy.
“MDM2 inhibitors, which are through phase 1 human trials, effectively activate p53 but manage to kill only about 1 in 20 tumors,” said study author Joaquín Espinosa, PhD, of the University of Colorado in Boulder.
“The question is why. What else is happening in these cancer cells that allow them to evade p53?”
According to the researchers, the answer is in the downstream effects of p53. The gene sets in motion a cascade of events that lead to cancer cell destruction. But it has been unclear exactly which other genes are directly activated by p53.
To identify genetic targets of p53, Dr Espinosa and his colleagues used a technique called Global Run-On Sequencing (GRO-Seq). Unlike other methods, GRO-Seq measures new RNA being created, not overall RNA levels.
“Many teams around the world have been getting cancer cells, treating them with MDM2 inhibitors, and waiting hours and hours to see what genes turn on, and then, only imprecisely,” Dr Espinosa said. “GRO-Seq lets us do it in minutes, and the discoveries are massive.”
The technique generates a large quantity of data because it requires counting tens of thousands of RNA molecules before and after p53 activation. So this research required designing algorithms to sort through the data, as well as a computational biologist driving a supercomputer.
But the researchers were able to pinpoint new genes directly regulated by p53. And they believe this could aid the future development of cancer-fighting strategies. ![]()
Credit: A.T. Tikhonenko
A novel sequencing technique has allowed researchers to identify direct targets of p53, providing new insight into this gene’s anticancer activity.
The research, published in eLife, revealed nearly 200 genes that were directly regulated by p53, and many of these had never been identified before.
The study’s authors said this work lays the foundation for investigations into which of these genes are necessary for p53’s cancer-killing effects and how cancer cells evade these genes.
The researchers noted that all cancers must deal with p53’s antitumor effects. Generally, there are 2 ways they do this: by mutating p53 directly or by producing the protein MDM2, which inhibits p53 function. With the current study, the team explored the second strategy.
“MDM2 inhibitors, which are through phase 1 human trials, effectively activate p53 but manage to kill only about 1 in 20 tumors,” said study author Joaquín Espinosa, PhD, of the University of Colorado in Boulder.
“The question is why. What else is happening in these cancer cells that allow them to evade p53?”
According to the researchers, the answer is in the downstream effects of p53. The gene sets in motion a cascade of events that lead to cancer cell destruction. But it has been unclear exactly which other genes are directly activated by p53.
To identify genetic targets of p53, Dr Espinosa and his colleagues used a technique called Global Run-On Sequencing (GRO-Seq). Unlike other methods, GRO-Seq measures new RNA being created, not overall RNA levels.
“Many teams around the world have been getting cancer cells, treating them with MDM2 inhibitors, and waiting hours and hours to see what genes turn on, and then, only imprecisely,” Dr Espinosa said. “GRO-Seq lets us do it in minutes, and the discoveries are massive.”
The technique generates a large quantity of data because it requires counting tens of thousands of RNA molecules before and after p53 activation. So this research required designing algorithms to sort through the data, as well as a computational biologist driving a supercomputer.
But the researchers were able to pinpoint new genes directly regulated by p53. And they believe this could aid the future development of cancer-fighting strategies. ![]()
Scientists’ success in job market is predictable, study suggests

Credit: Rhoda Baer
A scientist’s chances of landing a faculty position at an academic institution are predictable based solely on his or her publication record, according to a study published in Current Biology.
Chances depend mostly on the number of publications the scientist has, the impact factor of the journals in which those papers are published, and the number of papers that receive more citations than expected based on the journal in which they were published, the researchers said.
“We’d like to start a discussion on what factors are taken into account when people are selected to become a principal investigator,” said study author David van Dijk, PhD, of the Weizmann Institute of Science in Rehovot, Israel.
“On the one hand, these results are encouraging, because they suggest that people are promoted based on merit. On the other hand, many of the most groundbreaking papers were not published in high-impact-factor journals and did not initially receive a high number of citations. This filtering method will certainly miss some phenomenal and ahead-of-their time scientists.”
Dr Van Dijk said he and his colleagues were motivated by endless conversations with fellow graduate students and post docs, who were dreaming of their first paper in a prestigious journal. There was the sense that those publications were the tickets to success.
So Dr van Dijk and his colleagues wanted to see if they could find evidence to that effect. And, indeed, they could.
The researchers generated publication record data for more than 25,000 scientists and used a machine-learning approach to generate a model of each individual’s chances of moving from the first-author position, typically reserved for trainees, to the last-author position, a place most often held by principal investigators (PIs).
“We find that whether or not a scientist becomes a PI is largely predictable by their publication record, even taking into account only the first few years of publication,” the researchers reported. “Our model is able to predict with relatively high accuracy who becomes a PI and is also able to predict how long this will take.”
To calculate your own likelihood of success using this model, visit: http://www.pipredictor.com.
Dr Van Dijk said the study results suggest the current system is working. Understanding how it works might be useful for those thinking through their careers or for those on hiring committees who might like to allow factors outside of the publication record to factor more significantly in hiring decisions.
The authors don’t recommend that scientists make decisions about their futures based solely on their PI prediction scores, of course. There are surely plenty of other harder-to-quantify factors that can also play a role. And there is some hopeful news for those who are persistent, even if they haven’t landed that stellar paper just yet.
“There is an element of luck in getting a paper in Nature, Cell, or Science, so it can be frustrating if you think you are a good scientist and want to succeed, but that high-impact-factor paper just doesn’t happen,” Dr van Dijk said.
“It’s encouraging that we find that doing good-quality science on a consistent basis—as evidenced by multiple first-author papers of reasonable impact factor—does seem to be rewarded in the end.” ![]()
Credit: Rhoda Baer
A scientist’s chances of landing a faculty position at an academic institution are predictable based solely on his or her publication record, according to a study published in Current Biology.
Chances depend mostly on the number of publications the scientist has, the impact factor of the journals in which those papers are published, and the number of papers that receive more citations than expected based on the journal in which they were published, the researchers said.
“We’d like to start a discussion on what factors are taken into account when people are selected to become a principal investigator,” said study author David van Dijk, PhD, of the Weizmann Institute of Science in Rehovot, Israel.
“On the one hand, these results are encouraging, because they suggest that people are promoted based on merit. On the other hand, many of the most groundbreaking papers were not published in high-impact-factor journals and did not initially receive a high number of citations. This filtering method will certainly miss some phenomenal and ahead-of-their time scientists.”
Dr Van Dijk said he and his colleagues were motivated by endless conversations with fellow graduate students and post docs, who were dreaming of their first paper in a prestigious journal. There was the sense that those publications were the tickets to success.
So Dr van Dijk and his colleagues wanted to see if they could find evidence to that effect. And, indeed, they could.
The researchers generated publication record data for more than 25,000 scientists and used a machine-learning approach to generate a model of each individual’s chances of moving from the first-author position, typically reserved for trainees, to the last-author position, a place most often held by principal investigators (PIs).
“We find that whether or not a scientist becomes a PI is largely predictable by their publication record, even taking into account only the first few years of publication,” the researchers reported. “Our model is able to predict with relatively high accuracy who becomes a PI and is also able to predict how long this will take.”
To calculate your own likelihood of success using this model, visit: http://www.pipredictor.com.
Dr Van Dijk said the study results suggest the current system is working. Understanding how it works might be useful for those thinking through their careers or for those on hiring committees who might like to allow factors outside of the publication record to factor more significantly in hiring decisions.
The authors don’t recommend that scientists make decisions about their futures based solely on their PI prediction scores, of course. There are surely plenty of other harder-to-quantify factors that can also play a role. And there is some hopeful news for those who are persistent, even if they haven’t landed that stellar paper just yet.
“There is an element of luck in getting a paper in Nature, Cell, or Science, so it can be frustrating if you think you are a good scientist and want to succeed, but that high-impact-factor paper just doesn’t happen,” Dr van Dijk said.
“It’s encouraging that we find that doing good-quality science on a consistent basis—as evidenced by multiple first-author papers of reasonable impact factor—does seem to be rewarded in the end.” ![]()
Credit: Rhoda Baer
A scientist’s chances of landing a faculty position at an academic institution are predictable based solely on his or her publication record, according to a study published in Current Biology.
Chances depend mostly on the number of publications the scientist has, the impact factor of the journals in which those papers are published, and the number of papers that receive more citations than expected based on the journal in which they were published, the researchers said.
“We’d like to start a discussion on what factors are taken into account when people are selected to become a principal investigator,” said study author David van Dijk, PhD, of the Weizmann Institute of Science in Rehovot, Israel.
“On the one hand, these results are encouraging, because they suggest that people are promoted based on merit. On the other hand, many of the most groundbreaking papers were not published in high-impact-factor journals and did not initially receive a high number of citations. This filtering method will certainly miss some phenomenal and ahead-of-their time scientists.”
Dr Van Dijk said he and his colleagues were motivated by endless conversations with fellow graduate students and post docs, who were dreaming of their first paper in a prestigious journal. There was the sense that those publications were the tickets to success.
So Dr van Dijk and his colleagues wanted to see if they could find evidence to that effect. And, indeed, they could.
The researchers generated publication record data for more than 25,000 scientists and used a machine-learning approach to generate a model of each individual’s chances of moving from the first-author position, typically reserved for trainees, to the last-author position, a place most often held by principal investigators (PIs).
“We find that whether or not a scientist becomes a PI is largely predictable by their publication record, even taking into account only the first few years of publication,” the researchers reported. “Our model is able to predict with relatively high accuracy who becomes a PI and is also able to predict how long this will take.”
To calculate your own likelihood of success using this model, visit: http://www.pipredictor.com.
Dr Van Dijk said the study results suggest the current system is working. Understanding how it works might be useful for those thinking through their careers or for those on hiring committees who might like to allow factors outside of the publication record to factor more significantly in hiring decisions.
The authors don’t recommend that scientists make decisions about their futures based solely on their PI prediction scores, of course. There are surely plenty of other harder-to-quantify factors that can also play a role. And there is some hopeful news for those who are persistent, even if they haven’t landed that stellar paper just yet.
“There is an element of luck in getting a paper in Nature, Cell, or Science, so it can be frustrating if you think you are a good scientist and want to succeed, but that high-impact-factor paper just doesn’t happen,” Dr van Dijk said.
“It’s encouraging that we find that doing good-quality science on a consistent basis—as evidenced by multiple first-author papers of reasonable impact factor—does seem to be rewarded in the end.”
Exome sequencing not always accurate, presenter says
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.”
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.”
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.”
Group finds a way to target MDSCs

Researchers say they’ve found a way to target myeloid-derived suppressor cells (MDSCs) while sparing other immune cells.
In preclinical experiments, the team showed they could deplete MDSCs—and shrink tumors—using peptide antibodies.
These “peptibodies” wiped out MDSCs in the blood, spleen, and tumor cells of mice without binding to other white blood cells or dendritic cells.
The researchers described this work in Nature Medicine.
“We’ve known about [MDSCs] blocking immune response for a decade but haven’t been able to shut them down for lack of an identified target,” said the paper’s senior author, Larry Kwak, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“This is the first demonstration of a molecule on these cells that allows us to make an antibody—in this case, a peptide—to bind to them and get rid of them. It’s a brand new immunotherapy target.”
Dr Kwak and his colleagues began this research by applying a peptide phage library to MDSCs, which allowed for a mass screening of candidate peptides that bind to the surface of MDSCs. This revealed 2 peptides, labeled G3 and H6, that bound only to MDSCs.
The researchers fused the 2 peptides to a portion of mouse immune globulin to generate experimental peptibodies called pep-G3 and pep-H6. Both peptibodies bound to both types of MDSCs—monocytic and granulocytic cells.
Dr Kwak and his colleagues then tested the peptibodies in 2 mouse models of thymic tumors, as well as models of melanoma and lymphoma. The team compared pep-G3 and pep-H6 to a control peptibody and an antibody against Gr-1.
Both pep-G3 and pep-H6 depleted monocytic and granulocytic MDSCs in the blood and spleens of all mice. But the Gr-1 antibody only worked against granulocytic MDSCs.
To see whether MDSC depletion would impede tumor growth, the researchers administered the peptibodies to mice with thymic tumors every other day for 2 weeks.
Mice treated with either pep-G3 or pep-H6 had tumors that were about half the size and weight of those in mice treated with the control peptibody or the Gr-1 antibody.
Lastly, the researchers analyzed surface proteins on the MDSCs and found that S100A9 and S100A8 are the likely binding targets for pep-G3 and pep-H6.
Dr Kwak and his colleagues said they are now working to extend these findings to human MDSCs.
Researchers say they’ve found a way to target myeloid-derived suppressor cells (MDSCs) while sparing other immune cells.
In preclinical experiments, the team showed they could deplete MDSCs—and shrink tumors—using peptide antibodies.
These “peptibodies” wiped out MDSCs in the blood, spleen, and tumor cells of mice without binding to other white blood cells or dendritic cells.
The researchers described this work in Nature Medicine.
“We’ve known about [MDSCs] blocking immune response for a decade but haven’t been able to shut them down for lack of an identified target,” said the paper’s senior author, Larry Kwak, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“This is the first demonstration of a molecule on these cells that allows us to make an antibody—in this case, a peptide—to bind to them and get rid of them. It’s a brand new immunotherapy target.”
Dr Kwak and his colleagues began this research by applying a peptide phage library to MDSCs, which allowed for a mass screening of candidate peptides that bind to the surface of MDSCs. This revealed 2 peptides, labeled G3 and H6, that bound only to MDSCs.
The researchers fused the 2 peptides to a portion of mouse immune globulin to generate experimental peptibodies called pep-G3 and pep-H6. Both peptibodies bound to both types of MDSCs—monocytic and granulocytic cells.
Dr Kwak and his colleagues then tested the peptibodies in 2 mouse models of thymic tumors, as well as models of melanoma and lymphoma. The team compared pep-G3 and pep-H6 to a control peptibody and an antibody against Gr-1.
Both pep-G3 and pep-H6 depleted monocytic and granulocytic MDSCs in the blood and spleens of all mice. But the Gr-1 antibody only worked against granulocytic MDSCs.
To see whether MDSC depletion would impede tumor growth, the researchers administered the peptibodies to mice with thymic tumors every other day for 2 weeks.
Mice treated with either pep-G3 or pep-H6 had tumors that were about half the size and weight of those in mice treated with the control peptibody or the Gr-1 antibody.
Lastly, the researchers analyzed surface proteins on the MDSCs and found that S100A9 and S100A8 are the likely binding targets for pep-G3 and pep-H6.
Dr Kwak and his colleagues said they are now working to extend these findings to human MDSCs.
Researchers say they’ve found a way to target myeloid-derived suppressor cells (MDSCs) while sparing other immune cells.
In preclinical experiments, the team showed they could deplete MDSCs—and shrink tumors—using peptide antibodies.
These “peptibodies” wiped out MDSCs in the blood, spleen, and tumor cells of mice without binding to other white blood cells or dendritic cells.
The researchers described this work in Nature Medicine.
“We’ve known about [MDSCs] blocking immune response for a decade but haven’t been able to shut them down for lack of an identified target,” said the paper’s senior author, Larry Kwak, MD, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“This is the first demonstration of a molecule on these cells that allows us to make an antibody—in this case, a peptide—to bind to them and get rid of them. It’s a brand new immunotherapy target.”
Dr Kwak and his colleagues began this research by applying a peptide phage library to MDSCs, which allowed for a mass screening of candidate peptides that bind to the surface of MDSCs. This revealed 2 peptides, labeled G3 and H6, that bound only to MDSCs.
The researchers fused the 2 peptides to a portion of mouse immune globulin to generate experimental peptibodies called pep-G3 and pep-H6. Both peptibodies bound to both types of MDSCs—monocytic and granulocytic cells.
Dr Kwak and his colleagues then tested the peptibodies in 2 mouse models of thymic tumors, as well as models of melanoma and lymphoma. The team compared pep-G3 and pep-H6 to a control peptibody and an antibody against Gr-1.
Both pep-G3 and pep-H6 depleted monocytic and granulocytic MDSCs in the blood and spleens of all mice. But the Gr-1 antibody only worked against granulocytic MDSCs.
To see whether MDSC depletion would impede tumor growth, the researchers administered the peptibodies to mice with thymic tumors every other day for 2 weeks.
Mice treated with either pep-G3 or pep-H6 had tumors that were about half the size and weight of those in mice treated with the control peptibody or the Gr-1 antibody.
Lastly, the researchers analyzed surface proteins on the MDSCs and found that S100A9 and S100A8 are the likely binding targets for pep-G3 and pep-H6.
Dr Kwak and his colleagues said they are now working to extend these findings to human MDSCs.
EMA’s transparency plans criticized

to be used in a clinical trial
Credit: Esther Dyson
Many in the research community have lauded the European Medicines Agency’s (EMA’s) plans to make clinical trial data available to the public.
But the agency has also drawn criticism for the way it plans to go about increasing transparency.
The EMA has said it plans to make trial data available online in a “read-only” mode. So readers will not be able to save or transfer the data, making scientific analysis difficult, if not impossible.
The EMA also plans to allow drug manufacturers to omit some trial data from its public posts.
In response to this news, the European Ombudsman, Emily O’Reilly, wrote a letter to the EMA expressing concern about their seemingly significant change in policy.
“We were pleased when EMA announced, in 2012, a new pro-active transparency policy, giving the broadest possible public access to clinical trial data,” O’Reilly said.
“I am now concerned about what appears to be a significant change in EMA’s policy, which could undermine the fundamental right of public access to documents established by EU law. European citizens, doctors, and researchers need maximum information about the medicines they take, prescribe, and analyze.”
Beate Wieseler, PhD, and her colleagues from the German Institute for Quality and Efficiency in Health Care, echoed this sentiment in a “rapid response” published in the British Medical Journal.
The authors said the proposed read-only mode of accessing data makes scientific analyses, such as risk-benefit assessments of drugs, impossible.
For these assessments, an enormous amount of data must be viewed, annotated and saved, pooled from different studies, analyzed statistically, and shared among researchers. But the read-only mode would not allow for that.
Dr Wieseler and other critics also take issue with EMA’s decision to allow drug manufacturers to omit some trial data from its public posts.
The EMA has said that, within the context of market approval, drug manufacturers will be able to submit 2 versions of a clinical study report to the EMA: a complete one, by means of which the agency will decide on approval, and an incomplete one for the public.
The EMA previously decided that individual patient data, which could allow patients to be identified, would be deleted from the study reports. Now, this step has been extended to cover study results.
For example, the EMA believes deleting information is acceptable in cases of results on exploratory outcomes that are not supportive for the approval decision. However, Dr Weiseler and her colleagues noted that such study results often contain analyses of patient-relevant outcomes such as health-related quality of life.
“We are talking about studies in people who participated in a clinical study because they hoped that, with the information gained, better treatments would be developed,” Dr Wieseler said. “This information can only be used to improve patient care if it is publicly available to all.”
About the policy change
In June 2013, the EMA released for public consultation a draft policy on “publication and access to clinical trial data.” In that draft, the agency proposed publishing data submitted in support of marketing-authorization applications (only for centrally approved medicines from 2014 onward).
The idea was that interested parties would no longer need to invoke the European Freedom of Information Regulation (Regulation [EC] N°1049/2001), when exercising their right to access documents held by the EMA.
However, according to documents the EMA shared during a stakeholder consultation earlier this month, the EMA now plans to allow systematic censorship by pharmaceutical companies and impose both strict confidentiality requirements and restrictions on the use of data.
For more information on the policy, see the EMA website.
to be used in a clinical trial
Credit: Esther Dyson
Many in the research community have lauded the European Medicines Agency’s (EMA’s) plans to make clinical trial data available to the public.
But the agency has also drawn criticism for the way it plans to go about increasing transparency.
The EMA has said it plans to make trial data available online in a “read-only” mode. So readers will not be able to save or transfer the data, making scientific analysis difficult, if not impossible.
The EMA also plans to allow drug manufacturers to omit some trial data from its public posts.
In response to this news, the European Ombudsman, Emily O’Reilly, wrote a letter to the EMA expressing concern about their seemingly significant change in policy.
“We were pleased when EMA announced, in 2012, a new pro-active transparency policy, giving the broadest possible public access to clinical trial data,” O’Reilly said.
“I am now concerned about what appears to be a significant change in EMA’s policy, which could undermine the fundamental right of public access to documents established by EU law. European citizens, doctors, and researchers need maximum information about the medicines they take, prescribe, and analyze.”
Beate Wieseler, PhD, and her colleagues from the German Institute for Quality and Efficiency in Health Care, echoed this sentiment in a “rapid response” published in the British Medical Journal.
The authors said the proposed read-only mode of accessing data makes scientific analyses, such as risk-benefit assessments of drugs, impossible.
For these assessments, an enormous amount of data must be viewed, annotated and saved, pooled from different studies, analyzed statistically, and shared among researchers. But the read-only mode would not allow for that.
Dr Wieseler and other critics also take issue with EMA’s decision to allow drug manufacturers to omit some trial data from its public posts.
The EMA has said that, within the context of market approval, drug manufacturers will be able to submit 2 versions of a clinical study report to the EMA: a complete one, by means of which the agency will decide on approval, and an incomplete one for the public.
The EMA previously decided that individual patient data, which could allow patients to be identified, would be deleted from the study reports. Now, this step has been extended to cover study results.
For example, the EMA believes deleting information is acceptable in cases of results on exploratory outcomes that are not supportive for the approval decision. However, Dr Weiseler and her colleagues noted that such study results often contain analyses of patient-relevant outcomes such as health-related quality of life.
“We are talking about studies in people who participated in a clinical study because they hoped that, with the information gained, better treatments would be developed,” Dr Wieseler said. “This information can only be used to improve patient care if it is publicly available to all.”
About the policy change
In June 2013, the EMA released for public consultation a draft policy on “publication and access to clinical trial data.” In that draft, the agency proposed publishing data submitted in support of marketing-authorization applications (only for centrally approved medicines from 2014 onward).
The idea was that interested parties would no longer need to invoke the European Freedom of Information Regulation (Regulation [EC] N°1049/2001), when exercising their right to access documents held by the EMA.
However, according to documents the EMA shared during a stakeholder consultation earlier this month, the EMA now plans to allow systematic censorship by pharmaceutical companies and impose both strict confidentiality requirements and restrictions on the use of data.
For more information on the policy, see the EMA website.
to be used in a clinical trial
Credit: Esther Dyson
Many in the research community have lauded the European Medicines Agency’s (EMA’s) plans to make clinical trial data available to the public.
But the agency has also drawn criticism for the way it plans to go about increasing transparency.
The EMA has said it plans to make trial data available online in a “read-only” mode. So readers will not be able to save or transfer the data, making scientific analysis difficult, if not impossible.
The EMA also plans to allow drug manufacturers to omit some trial data from its public posts.
In response to this news, the European Ombudsman, Emily O’Reilly, wrote a letter to the EMA expressing concern about their seemingly significant change in policy.
“We were pleased when EMA announced, in 2012, a new pro-active transparency policy, giving the broadest possible public access to clinical trial data,” O’Reilly said.
“I am now concerned about what appears to be a significant change in EMA’s policy, which could undermine the fundamental right of public access to documents established by EU law. European citizens, doctors, and researchers need maximum information about the medicines they take, prescribe, and analyze.”
Beate Wieseler, PhD, and her colleagues from the German Institute for Quality and Efficiency in Health Care, echoed this sentiment in a “rapid response” published in the British Medical Journal.
The authors said the proposed read-only mode of accessing data makes scientific analyses, such as risk-benefit assessments of drugs, impossible.
For these assessments, an enormous amount of data must be viewed, annotated and saved, pooled from different studies, analyzed statistically, and shared among researchers. But the read-only mode would not allow for that.
Dr Wieseler and other critics also take issue with EMA’s decision to allow drug manufacturers to omit some trial data from its public posts.
The EMA has said that, within the context of market approval, drug manufacturers will be able to submit 2 versions of a clinical study report to the EMA: a complete one, by means of which the agency will decide on approval, and an incomplete one for the public.
The EMA previously decided that individual patient data, which could allow patients to be identified, would be deleted from the study reports. Now, this step has been extended to cover study results.
For example, the EMA believes deleting information is acceptable in cases of results on exploratory outcomes that are not supportive for the approval decision. However, Dr Weiseler and her colleagues noted that such study results often contain analyses of patient-relevant outcomes such as health-related quality of life.
“We are talking about studies in people who participated in a clinical study because they hoped that, with the information gained, better treatments would be developed,” Dr Wieseler said. “This information can only be used to improve patient care if it is publicly available to all.”
About the policy change
In June 2013, the EMA released for public consultation a draft policy on “publication and access to clinical trial data.” In that draft, the agency proposed publishing data submitted in support of marketing-authorization applications (only for centrally approved medicines from 2014 onward).
The idea was that interested parties would no longer need to invoke the European Freedom of Information Regulation (Regulation [EC] N°1049/2001), when exercising their right to access documents held by the EMA.
However, according to documents the EMA shared during a stakeholder consultation earlier this month, the EMA now plans to allow systematic censorship by pharmaceutical companies and impose both strict confidentiality requirements and restrictions on the use of data.
For more information on the policy, see the EMA website.