User login
Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
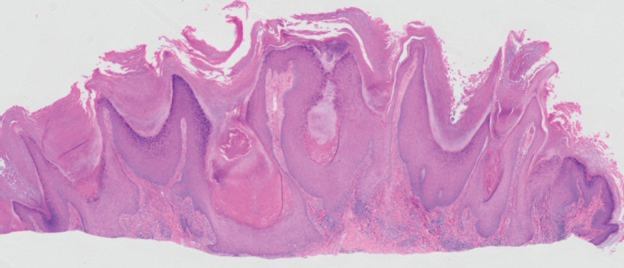
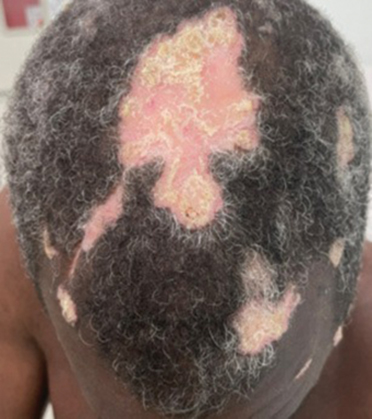
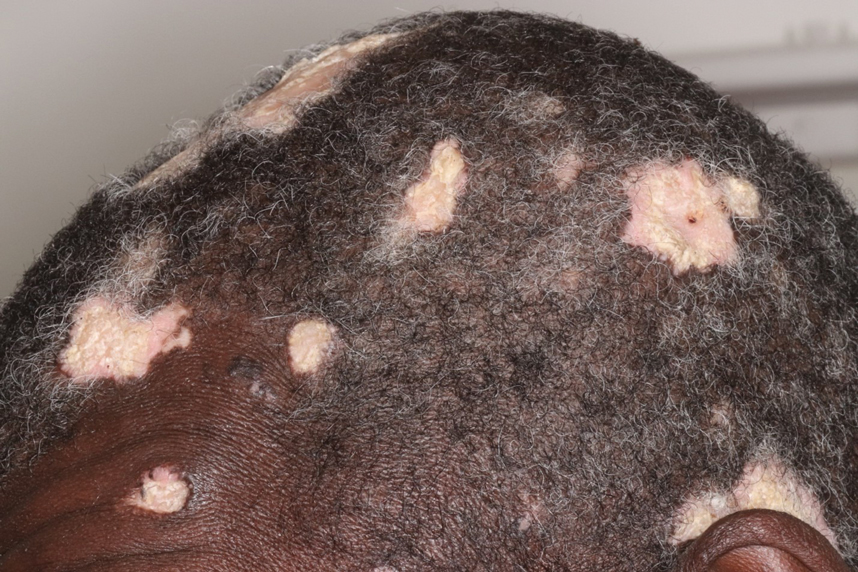
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
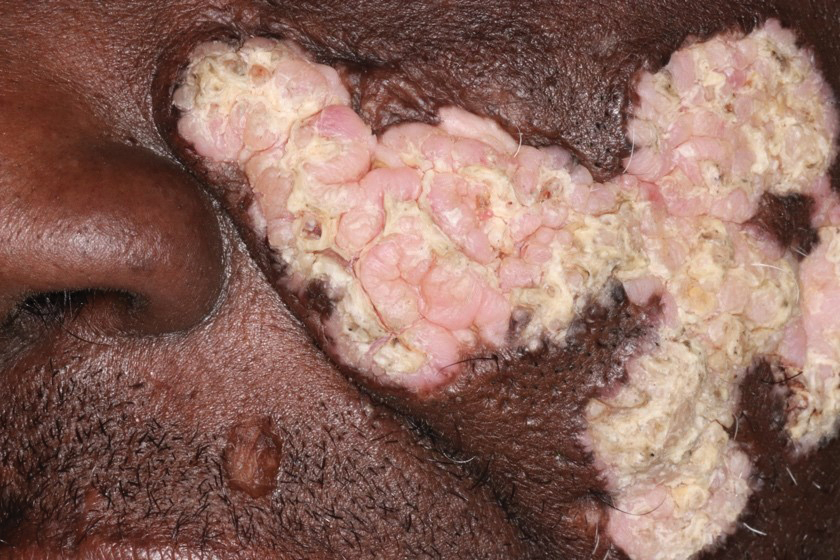
A 54-year-old man with no notable medical history presented to an outpatient dermatology clinic with multiple skin lesions on sun-exposed areas including the face, chest, scalp, and bilateral upper extremities. The patient reported that he had not seen a doctor for 26 years. He noted that the lesions had been present for many years but was unsure of the exact timeframe. Physical examination revealed verrucous plaques with a violaceous rim and central hypopigmentation on the chest, scalp, face, and arms. Scarring alopecia also was noted on the scalp with no associated pain or pruritus. Antinuclear antibody and extractable nuclear antigen tests were negative, and urine analysis was normal. A shave biopsy of the chest was performed for histopathologic evaluation. Rapid plasma reagin tests and HIV antibody tests also were performed.
Biomarker Changes in Systemic Sclerosis–Associated Lung Disease Predict Therapy Response
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Changes in Krebs von den Lungen 6 (KL-6) levels after 12 months of treatment with mycophenolate mofetil (MMF) or cyclophosphamide (CYC) are associated with the development of progressive pulmonary fibrosis (PPF) in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD) in the following year.
METHODOLOGY:
- Despite available treatments, about 25% of patients with SSc-ILD develop PPF, highlighting the need for reliable early treatment response indicators, such as blood biomarkers, which may help predict the risk for PPF.
- Researchers conducted post hoc analyses of a randomized control trial that compared treatment responses to MMF with those to CYC in patients with SSc-ILD. Patients received either oral CYC for 12 months followed by placebo for 12 months or MMF for 24 months.
- A total of 92 patients with complete biomarker measurements at baseline and 12 months were included in the analysis (mean age, 52.2 years; 73.9% women; 68.5% White).
- The analysis included measurement of multiple blood biomarker levels, including C-reactive protein (CRP), interleukin-6, chemokine ligand 4 (CXCL4), CXCL18, and KL-6. Changes in these levels were evaluated from baseline to 12 months.
- The primary outcome was the development of PPF between 12 and 24 months, defined by meeting at least two of these following conditions: Worsening respiratory symptoms, a decline in forced vital capacity ≥ 5% and/or a decline in diffusing capacity for carbon monoxide ≥ 10%, or radiological disease progression.
TAKEAWAY:
- Among 92 patients, 19 developed PPF between 12 and 24 months, with 10 patients in the MMF arm and 9 patients in the CYC arm.
- KL-6 levels increased from baseline to 12 months in patients who developed PPF and decreased in those who did not (mean change, 365.68 vs –207.45 u/mL; P < .001).
- A 0.10-unit increase in KL-6 levels was associated with a 40% increase in the odds of developing PPF in an adjusted analysis (P = .0002).
- In the MMF group, levels of KL-6, CRP, and CXCL4 differed significantly between patients who developed PPF and those who did not (P = .004, P = .04, and P = .038, respectively).
IN PRACTICE:
“Reliable response biomarkers detectable early in the course of SSc-ILD treatment could minimize exposure to toxic therapies that are not conferring benefit and maximize exposure to alternative therapies that do confer benefit,” the authors wrote.
SOURCE:
The study was led by Elizabeth R. Volkmann, MD, MS, University of California, Los Angeles David Geffen School of Medicine. It was published online in Arthritis Care & Research.
LIMITATIONS:
The study population consisted of patients who were treatment-naive to MMF and CYC and had a relatively early disease course, potentially limiting generalizability to patients at later disease stages or with different treatment histories. Additionally, biomarker measurements were conducted at 12 months, when treatment response may be detectable through currently available methods, rather than at earlier timepoints.
DISCLOSURES:
The study was funded by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute and the Department of Defense. MMF was supplied by Hoffmann–La Roche. Some authors reported having financial relationships with pharmaceutical companies, including Hoffmann–La Roche.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Around 5% of US Population Diagnosed With Autoimmune Disease
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In 2022, autoimmune diseases affected over 15 million individuals in the United States, with women nearly twice as likely to be affected as men and more than one third of affected individuals having more than one autoimmune condition.
METHODOLOGY:
- Researchers used electronic health record (EHR) data from six healthcare systems in the United States between 2011 and 2022 to estimate the prevalence of autoimmune diseases according to sex and age.
- They selected 105 autoimmune diseases from the textbook The Autoimmune Diseases and estimated their prevalence in more than 10 million individuals from these healthcare systems; these statistics were subsequently extrapolated to an estimated US population of 333.3 million.
- An individual was considered to have a diagnosis of an autoimmune disease if they had at least two diagnosis codes for the condition, with the codes being at least 30 days apart.
- A software program was developed to compute the prevalence of autoimmune diseases alone and in aggregate, enabling other researchers to replicate or modify the analysis over time.
TAKEAWAY:
- More than 15 million people, accounting for 4.6% of the US population, were diagnosed with at least one autoimmune disease from January 2011 to June 2022; 34% were diagnosed with more than one autoimmune disease.
- Sex-stratified analysis revealed that 63% of patients diagnosed with autoimmune disease were women, and only 37% were men, establishing a female-to-male ratio of 1.7:1; age-stratified analysis revealed increasing prevalence of autoimmune conditions with age, peaking in individuals aged ≥ 65 years.
- Among individuals with autoimmune diseases, 65% of patients had one condition, whereas 24% had two, 8% had three, and 2% had four or more autoimmune diseases (does not add to 100% due to rounding).
- Rheumatoid arthritis emerged as the most prevalent autoimmune disease, followed by psoriasis, type 1 diabetes, Grave’s disease, and autoimmune thyroiditis; 19 of the top 20 most prevalent autoimmune diseases occurred more frequently in women.
IN PRACTICE:
“Accurate data on the prevalence of autoimmune diseases as a category of disease and for individual autoimmune diseases are needed to further clinical and basic research to improve diagnosis, biomarkers, and therapies for these diseases, which significantly impact the US population,” the authors wrote.
SOURCE:
The study was led by Aaron H. Abend, Autoimmune Registry, Guilford, Connecticut, and was published online in The Journal of Clinical Investigation.
LIMITATIONS:
The use of EHR data presented several challenges, including potential inaccuracies in diagnosis codes and the possibility of missing patients with single diagnosis codes because of the two-code requirement. Certain autoimmune diseases evolve over time and involve nonspecific clinical signs and symptoms that can mimic other diseases, potentially resulting in underdiagnosis. Moreover, rare diseases lacking specific diagnosis codes may have been underrepresented.
DISCLOSURES:
The study received support from Autoimmune Registry; the National Institutes of Health National Center for Advancing Translational Sciences; the National Heart, Lung, and Blood Institute; and other sources. Information on potential conflicts of interest was not disclosed.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Valaciclovir Shows Promise in Preventing Herpes Zoster During Anifrolumab Treatment for Lupus
TOPLINE:
The use of valaciclovir as prophylaxis prevents herpes zoster (HZ) in patients with systemic lupus erythematosus (SLE) receiving anifrolumab treatment, with no cases of zoster reported during the follow-up period in patients receiving valaciclovir.
METHODOLOGY:
- Anifrolumab, a human monoclonal antibody binding to type I interferon receptor subunit 1, increases the risk for HZ in patients with SLE; however, specific recommendations to prevent HZ are currently nonexistent for patients with SLE receiving anifrolumab.
- Researchers conducted a multicenter observational study in France from November 2021 to July 2024 to evaluate the prophylactic benefits of valaciclovir in 132 patients with SLE (mean age, 42 years; 92% women) treated with anifrolumab for ≥ 3 months.
- Among these patients, 87 received either 500 mg/d valaciclovir (n = 69) or 1000 mg/d valaciclovir (n = 18) as prophylaxis, whereas 45 did not receive valaciclovir.
- The patients were followed up for a median duration of 234 days under anifrolumab treatment, with monitoring for the development of herpes zoster.
TAKEAWAY:
- The risk for HZ was significantly lower in patients who received valaciclovir than in those who did not (hazard ratio, 0.08; P = .01).
- None of the patients treated with valaciclovir developed HZ during the survey period.
- The frequency of HZ in patients who did not receive valaciclovir increased progressively from 2.2% at 3 months to 6.2% at 6 months, reaching 23% at 12 months.
- None of the reported cases of HZ required hospitalization or led to anifrolumab discontinuation, although one patient developed neuralgia.
IN PRACTICE:
“Prophylactic treatment with valaciclovir is effective for preventing HZ [herpes zoster] infection in SLE patients treated with anifrolumab,” the authors wrote. “This finding is particularly relevant for SLE patients who cannot receive the recombinant HZ vaccine or for whom it is unavailable,” they added.
SOURCE:
The study was led by Ludovic Trefond, MD, PhD, Centre Hospitalier Universitaire de Clermont-Ferrand in France. It was published online on January 4, 2025, in RMD Open.
LIMITATIONS:
The observational design of the study and the low number of herpes zoster events during the follow-up period may have affected the robustness of the findings.
DISCLOSURES:
The authors did not receive any specific grants. Some authors reported having financial relationships with various pharmaceutical companies.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
The use of valaciclovir as prophylaxis prevents herpes zoster (HZ) in patients with systemic lupus erythematosus (SLE) receiving anifrolumab treatment, with no cases of zoster reported during the follow-up period in patients receiving valaciclovir.
METHODOLOGY:
- Anifrolumab, a human monoclonal antibody binding to type I interferon receptor subunit 1, increases the risk for HZ in patients with SLE; however, specific recommendations to prevent HZ are currently nonexistent for patients with SLE receiving anifrolumab.
- Researchers conducted a multicenter observational study in France from November 2021 to July 2024 to evaluate the prophylactic benefits of valaciclovir in 132 patients with SLE (mean age, 42 years; 92% women) treated with anifrolumab for ≥ 3 months.
- Among these patients, 87 received either 500 mg/d valaciclovir (n = 69) or 1000 mg/d valaciclovir (n = 18) as prophylaxis, whereas 45 did not receive valaciclovir.
- The patients were followed up for a median duration of 234 days under anifrolumab treatment, with monitoring for the development of herpes zoster.
TAKEAWAY:
- The risk for HZ was significantly lower in patients who received valaciclovir than in those who did not (hazard ratio, 0.08; P = .01).
- None of the patients treated with valaciclovir developed HZ during the survey period.
- The frequency of HZ in patients who did not receive valaciclovir increased progressively from 2.2% at 3 months to 6.2% at 6 months, reaching 23% at 12 months.
- None of the reported cases of HZ required hospitalization or led to anifrolumab discontinuation, although one patient developed neuralgia.
IN PRACTICE:
“Prophylactic treatment with valaciclovir is effective for preventing HZ [herpes zoster] infection in SLE patients treated with anifrolumab,” the authors wrote. “This finding is particularly relevant for SLE patients who cannot receive the recombinant HZ vaccine or for whom it is unavailable,” they added.
SOURCE:
The study was led by Ludovic Trefond, MD, PhD, Centre Hospitalier Universitaire de Clermont-Ferrand in France. It was published online on January 4, 2025, in RMD Open.
LIMITATIONS:
The observational design of the study and the low number of herpes zoster events during the follow-up period may have affected the robustness of the findings.
DISCLOSURES:
The authors did not receive any specific grants. Some authors reported having financial relationships with various pharmaceutical companies.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
The use of valaciclovir as prophylaxis prevents herpes zoster (HZ) in patients with systemic lupus erythematosus (SLE) receiving anifrolumab treatment, with no cases of zoster reported during the follow-up period in patients receiving valaciclovir.
METHODOLOGY:
- Anifrolumab, a human monoclonal antibody binding to type I interferon receptor subunit 1, increases the risk for HZ in patients with SLE; however, specific recommendations to prevent HZ are currently nonexistent for patients with SLE receiving anifrolumab.
- Researchers conducted a multicenter observational study in France from November 2021 to July 2024 to evaluate the prophylactic benefits of valaciclovir in 132 patients with SLE (mean age, 42 years; 92% women) treated with anifrolumab for ≥ 3 months.
- Among these patients, 87 received either 500 mg/d valaciclovir (n = 69) or 1000 mg/d valaciclovir (n = 18) as prophylaxis, whereas 45 did not receive valaciclovir.
- The patients were followed up for a median duration of 234 days under anifrolumab treatment, with monitoring for the development of herpes zoster.
TAKEAWAY:
- The risk for HZ was significantly lower in patients who received valaciclovir than in those who did not (hazard ratio, 0.08; P = .01).
- None of the patients treated with valaciclovir developed HZ during the survey period.
- The frequency of HZ in patients who did not receive valaciclovir increased progressively from 2.2% at 3 months to 6.2% at 6 months, reaching 23% at 12 months.
- None of the reported cases of HZ required hospitalization or led to anifrolumab discontinuation, although one patient developed neuralgia.
IN PRACTICE:
“Prophylactic treatment with valaciclovir is effective for preventing HZ [herpes zoster] infection in SLE patients treated with anifrolumab,” the authors wrote. “This finding is particularly relevant for SLE patients who cannot receive the recombinant HZ vaccine or for whom it is unavailable,” they added.
SOURCE:
The study was led by Ludovic Trefond, MD, PhD, Centre Hospitalier Universitaire de Clermont-Ferrand in France. It was published online on January 4, 2025, in RMD Open.
LIMITATIONS:
The observational design of the study and the low number of herpes zoster events during the follow-up period may have affected the robustness of the findings.
DISCLOSURES:
The authors did not receive any specific grants. Some authors reported having financial relationships with various pharmaceutical companies.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Patients With Refractory Systemic Sclerosis Have Early Success With CAR T-Cell Therapy
TOPLINE:
CD19-targeting chimeric antigen receptor (CAR) T-cell therapy shows potential to intercept fibrotic organ manifestations and improve disease measures in patients with diffuse cutaneous systemic sclerosis (SSc) who had disease progression despite multiple previous treatments.
METHODOLOGY:
- Researchers conducted a case series to examine the effect of CD19-targeting CAR T-cell therapy on fibrotic and vascular organ manifestations in six patients with diffuse cutaneous SSc (median age, 42 years; four men and two women) who had an insufficient response to at least two previous treatments.
- Participants received CD19-targeting CAR T-cell treatment at a dose of 1 × 106 CAR T cells per kilogram of body weight after lymphodepletion with fludarabine and cyclophosphamide.
- The primary outcome was event-free time or treatment intensification after study entry, with events defined as the progression of interstitial lung disease, onset of congestive heart or renal failure or arterial hypertension, or initiation of new therapy.
- The secondary outcomes included changes in the modified Rodnan skin score (mRSS), imaging and laboratory assessments, patient-reported outcomes, and the modified American College of Rheumatology Composite Response Index in Systemic Sclerosis (ACR-CRISS), assessed at baseline and 3, 6, 9, and 12 months after treatment.
TAKEAWAY:
- No progression of organ manifestations or new lung, cardiac, or renal events occurred within the median follow-up period of 487 days.
- The probability of improvement in the ACR-CRISS score increased to a median value of 100% within 6 and 12 months of CAR T-cell treatment compared with baseline.
- Skin involvement improved in all the patients after CAR T-cell treatment, with a median mRSS decrease of 8 points within 100 days; the improvements were maintained throughout the 1-year follow-up period.
- This treatment also led to a depletion of antinuclear antibodies and SSc-specific autoantibodies.
IN PRACTICE:
“This case series highlights the potential of CAR T-cell therapy to address a crucial unmet need in refractory systemic sclerosis treatment. The study’s most significant contribution is the demonstration that CD19-targeting CAR T-cell therapy can halt or reverse aspects of fibrosis in systemic sclerosis,” Jérôme Avouac, Service de Rhumatologie, Hôpital Cochin, AP-HP Centre-Université Paris Cité, Paris, France, wrote in an accompanying editorial.
SOURCE:
The study was led by Janina Auth, MD, Deutsches Zentrum Immuntherapie, Friedrich-Alexander-Universität Erlangen-Nürnberg and Universitätsklinikum Erlangen in Germany, and was published online on November 11, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study lacked a control group, which limited the ability to draw definitive conclusions about the efficacy of CD19-targeting CAR T-cell therapy compared with standard treatments. The unpredictable nature of SSc, in which periods of stability can occur spontaneously, makes it difficult to attribute the improvements merely to the intervention. Moreover, the effect of CAR T-cell therapy on other disease manifestations, such as pulmonary hypertension, myocardial involvement, and scleroderma renal crisis, remains unclear.
DISCLOSURES:
The study was funded by Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe, ELAN Foundation Erlangen, Interdisziplinäres Zentrum für Klinische Forschung Erlangen, Bundesministerium für Bildung und Forschung, and the European Union. Some authors reported receiving research grants, consulting fees, speaker fees, honoraria, or travel grants from Boehringer Ingelheim, Novartis, Almirall, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
CD19-targeting chimeric antigen receptor (CAR) T-cell therapy shows potential to intercept fibrotic organ manifestations and improve disease measures in patients with diffuse cutaneous systemic sclerosis (SSc) who had disease progression despite multiple previous treatments.
METHODOLOGY:
- Researchers conducted a case series to examine the effect of CD19-targeting CAR T-cell therapy on fibrotic and vascular organ manifestations in six patients with diffuse cutaneous SSc (median age, 42 years; four men and two women) who had an insufficient response to at least two previous treatments.
- Participants received CD19-targeting CAR T-cell treatment at a dose of 1 × 106 CAR T cells per kilogram of body weight after lymphodepletion with fludarabine and cyclophosphamide.
- The primary outcome was event-free time or treatment intensification after study entry, with events defined as the progression of interstitial lung disease, onset of congestive heart or renal failure or arterial hypertension, or initiation of new therapy.
- The secondary outcomes included changes in the modified Rodnan skin score (mRSS), imaging and laboratory assessments, patient-reported outcomes, and the modified American College of Rheumatology Composite Response Index in Systemic Sclerosis (ACR-CRISS), assessed at baseline and 3, 6, 9, and 12 months after treatment.
TAKEAWAY:
- No progression of organ manifestations or new lung, cardiac, or renal events occurred within the median follow-up period of 487 days.
- The probability of improvement in the ACR-CRISS score increased to a median value of 100% within 6 and 12 months of CAR T-cell treatment compared with baseline.
- Skin involvement improved in all the patients after CAR T-cell treatment, with a median mRSS decrease of 8 points within 100 days; the improvements were maintained throughout the 1-year follow-up period.
- This treatment also led to a depletion of antinuclear antibodies and SSc-specific autoantibodies.
IN PRACTICE:
“This case series highlights the potential of CAR T-cell therapy to address a crucial unmet need in refractory systemic sclerosis treatment. The study’s most significant contribution is the demonstration that CD19-targeting CAR T-cell therapy can halt or reverse aspects of fibrosis in systemic sclerosis,” Jérôme Avouac, Service de Rhumatologie, Hôpital Cochin, AP-HP Centre-Université Paris Cité, Paris, France, wrote in an accompanying editorial.
SOURCE:
The study was led by Janina Auth, MD, Deutsches Zentrum Immuntherapie, Friedrich-Alexander-Universität Erlangen-Nürnberg and Universitätsklinikum Erlangen in Germany, and was published online on November 11, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study lacked a control group, which limited the ability to draw definitive conclusions about the efficacy of CD19-targeting CAR T-cell therapy compared with standard treatments. The unpredictable nature of SSc, in which periods of stability can occur spontaneously, makes it difficult to attribute the improvements merely to the intervention. Moreover, the effect of CAR T-cell therapy on other disease manifestations, such as pulmonary hypertension, myocardial involvement, and scleroderma renal crisis, remains unclear.
DISCLOSURES:
The study was funded by Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe, ELAN Foundation Erlangen, Interdisziplinäres Zentrum für Klinische Forschung Erlangen, Bundesministerium für Bildung und Forschung, and the European Union. Some authors reported receiving research grants, consulting fees, speaker fees, honoraria, or travel grants from Boehringer Ingelheim, Novartis, Almirall, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
CD19-targeting chimeric antigen receptor (CAR) T-cell therapy shows potential to intercept fibrotic organ manifestations and improve disease measures in patients with diffuse cutaneous systemic sclerosis (SSc) who had disease progression despite multiple previous treatments.
METHODOLOGY:
- Researchers conducted a case series to examine the effect of CD19-targeting CAR T-cell therapy on fibrotic and vascular organ manifestations in six patients with diffuse cutaneous SSc (median age, 42 years; four men and two women) who had an insufficient response to at least two previous treatments.
- Participants received CD19-targeting CAR T-cell treatment at a dose of 1 × 106 CAR T cells per kilogram of body weight after lymphodepletion with fludarabine and cyclophosphamide.
- The primary outcome was event-free time or treatment intensification after study entry, with events defined as the progression of interstitial lung disease, onset of congestive heart or renal failure or arterial hypertension, or initiation of new therapy.
- The secondary outcomes included changes in the modified Rodnan skin score (mRSS), imaging and laboratory assessments, patient-reported outcomes, and the modified American College of Rheumatology Composite Response Index in Systemic Sclerosis (ACR-CRISS), assessed at baseline and 3, 6, 9, and 12 months after treatment.
TAKEAWAY:
- No progression of organ manifestations or new lung, cardiac, or renal events occurred within the median follow-up period of 487 days.
- The probability of improvement in the ACR-CRISS score increased to a median value of 100% within 6 and 12 months of CAR T-cell treatment compared with baseline.
- Skin involvement improved in all the patients after CAR T-cell treatment, with a median mRSS decrease of 8 points within 100 days; the improvements were maintained throughout the 1-year follow-up period.
- This treatment also led to a depletion of antinuclear antibodies and SSc-specific autoantibodies.
IN PRACTICE:
“This case series highlights the potential of CAR T-cell therapy to address a crucial unmet need in refractory systemic sclerosis treatment. The study’s most significant contribution is the demonstration that CD19-targeting CAR T-cell therapy can halt or reverse aspects of fibrosis in systemic sclerosis,” Jérôme Avouac, Service de Rhumatologie, Hôpital Cochin, AP-HP Centre-Université Paris Cité, Paris, France, wrote in an accompanying editorial.
SOURCE:
The study was led by Janina Auth, MD, Deutsches Zentrum Immuntherapie, Friedrich-Alexander-Universität Erlangen-Nürnberg and Universitätsklinikum Erlangen in Germany, and was published online on November 11, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study lacked a control group, which limited the ability to draw definitive conclusions about the efficacy of CD19-targeting CAR T-cell therapy compared with standard treatments. The unpredictable nature of SSc, in which periods of stability can occur spontaneously, makes it difficult to attribute the improvements merely to the intervention. Moreover, the effect of CAR T-cell therapy on other disease manifestations, such as pulmonary hypertension, myocardial involvement, and scleroderma renal crisis, remains unclear.
DISCLOSURES:
The study was funded by Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe, ELAN Foundation Erlangen, Interdisziplinäres Zentrum für Klinische Forschung Erlangen, Bundesministerium für Bildung und Forschung, and the European Union. Some authors reported receiving research grants, consulting fees, speaker fees, honoraria, or travel grants from Boehringer Ingelheim, Novartis, Almirall, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Painful Oral, Groin, and Scalp Lesions in a Young Man
Painful Oral, Groin, and Scalp Lesions in a Young Man
THE DIAGNOSIS: Pemphigus Vegetans
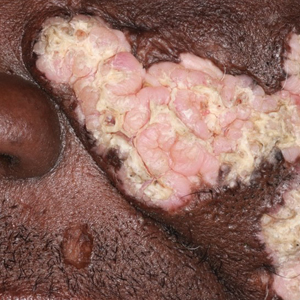
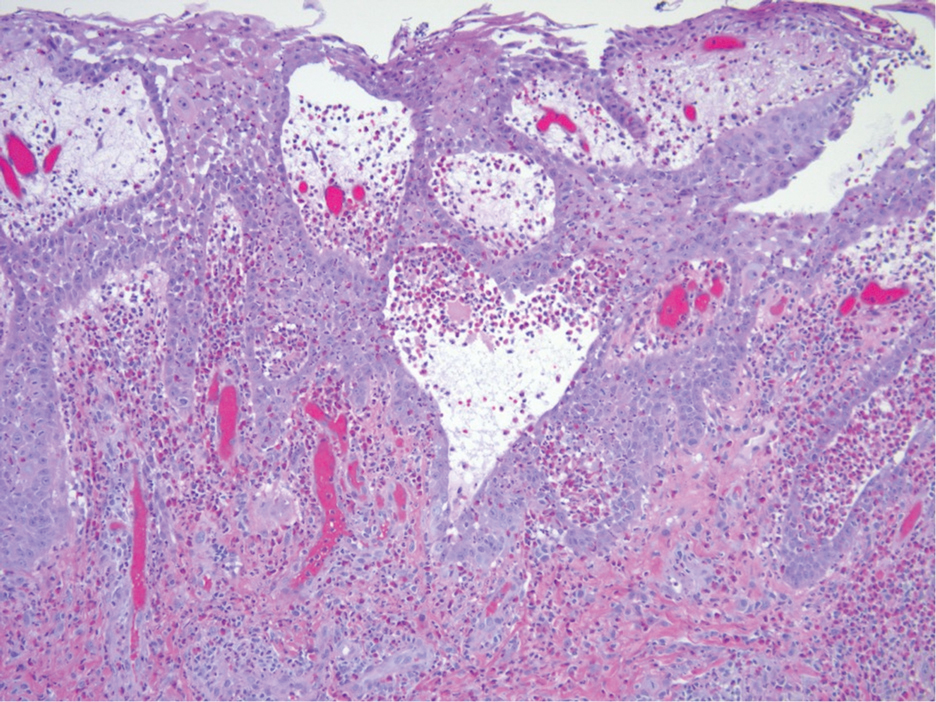
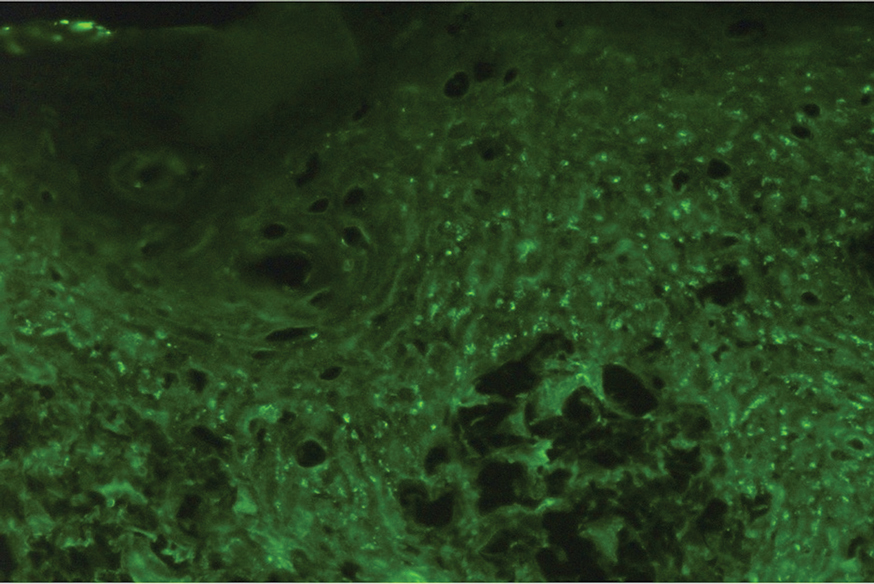
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
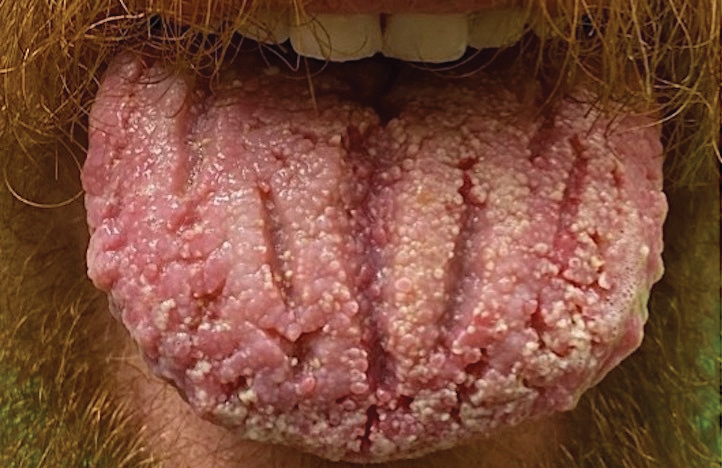
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
THE DIAGNOSIS: Pemphigus Vegetans
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
THE DIAGNOSIS: Pemphigus Vegetans
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
Painful Oral, Groin, and Scalp Lesions in a Young Man
Painful Oral, Groin, and Scalp Lesions in a Young Man
A 27-year-old man presented to the dermatology department with painful oral and groin lesions of 2 years’ duration as well as lip ulceration that had been present for 1 month. The patient also reported moderately tender scalp and face lesions that had been present for several weeks. The lip ulceration was previously treated by his primary care provider with valacyclovir (1 g daily for 2 weeks) without improvement. Six months prior to the current presentation, we treated the groin lesions as condyloma involving the perineum and genital region at our clinic with no response to cryotherapy, topical imiquimod, or extensive surgical excision with skin grafting. Pathology at the time showed condyloma but was negative for human papillomavirus. Physical examination at the current presentation revealed superficial erosions along the vermilion border. The oral mucosa exhibited cobblestoning, and fissures were present on the tongue. Eroded pink plaques studded with vesicles were present on the vertex scalp and left chin. The bilateral inguinal regions extending to anterior-lateral upper thighs and posterior buttocks revealed erythematous, arcuate, and annular erosive plaques with verrucous hyperkeratotic borders and fissuring on the leading edge. Pink erosive and verrucous erythematous plaques were noted on the penile shaft, scrotum, and perineum. Punch biopsies of the scalp and left anterior thigh as well as direct immunofluorescence were performed.
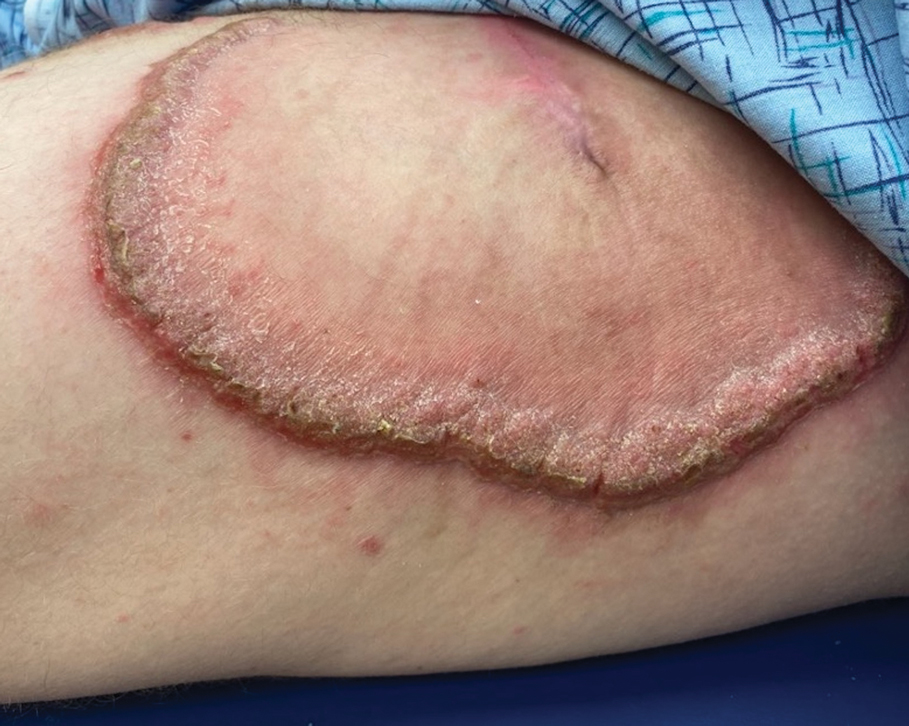

Cutaneous Lupus Associated with Greater Risk for Atherosclerotic Cardiovascular Disease
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
than with psoriasis.
METHODOLOGY:
- A retrospective matched longitudinal study compared the incidence and prevalence of ASCVD of 8138 individuals with CLE; 24,675 with SLE; 192,577 with psoriasis; and 81,380 control individuals.
- The disease-free control population was matched in a 10:1 ratio to the CLE population on the basis of age, sex, insurance type, and enrollment duration.
- Prevalent ASCVD was defined as coronary artery disease, prior myocardial infarction, or cerebrovascular accident, with ASCVD incidence assessed by number of hospitalizations over 3 years.
TAKEAWAY:
- Persons with CLE had higher ASCVD risk than control individuals (odds ratio [OR], 1.72; P < .001), similar to those with SLE (OR, 2.41; P < .001) but unlike those with psoriasis (OR, 1.03; P = .48).
- ASCVD incidence at 3 years was 24.8 per 1000 person-years for SLE, 15.2 per 1000 person-years for CLE, 14.0 per 1000 person-years for psoriasis, and 10.3 per 1000 person-years for controls.
- Multivariable Cox proportional regression modeling showed ASCVD risk was highest in those with SLE (hazard ratio [HR], 2.23; P < .001) vs CLE (HR, 1.32; P < .001) and psoriasis (HR, 1.06; P = .09).
- ASCVD prevalence was higher in individuals with CLE receiving systemic therapy (2.7%) than in those receiving no therapy (1.6%), suggesting a potential link between disease severity and CVD risk.
IN PRACTICE:
“Persons with CLE are at higher risk for ASCVD, and guidelines for the evaluation and management of ASCVD may improve their quality of care,” the authors wrote.
SOURCE:
The study was led by Henry W. Chen, MD, Department of Dermatology, University of Texas Southwestern Medical Center, Dallas. It was published online on December 4, 2024, in JAMA Dermatology.
LIMITATIONS:
The study was limited by its relatively young population (median age, 49 years) and the exclusion of adults aged > 65 years on Medicare insurance plans. The database lacked race and ethnicity data, and the analysis was restricted to a shorter 3-year period. The study could not fully evaluate detailed risk factors such as blood pressure levels, cholesterol measurements, or glycemic control, nor could it accurately assess smoking status.
DISCLOSURES:
The research was supported by the Department of Dermatology at the University of Texas Southwestern Medical Center and a grant from the National Institutes of Health. Several authors reported receiving grants or personal fees from various pharmaceutical companies. One author reported being a deputy editor for diversity, equity, and inclusion at JAMA Cardiology. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Study Addresses Lichen Planus Prevalence, Treatment
TOPLINE:
METHODOLOGY:
- To evaluate the prevalence of LP, researchers analyzed 566,851 eligible patients from the Explorys database, comprising electronic medical records from over 40 healthcare networks and 53 million patients across the United States.
- They also assessed treatment plans separately among 1998 newly diagnosed patients with LP between October 2015 and January 2020, who required at least one dermatology encounter within the first year following diagnosis.
- The primary outcome was overall prevalence of LP in the United States, including prevalence across specific age, sex, and racial subgroups. Additionally, dermatologist-prescribed treatments for non-oral LP were also reported.
TAKEAWAY:
- Overall, there were 1098 cases of LP (median age, 66 years; 74% women); the crude prevalence of LP was 0.19% and the age- and sex-standardized overall prevalence was 0.15%. Prevalence in women was 1.77 times higher than in men.
- Asian patients showed the highest standardized prevalence (0.2%), followed by Black patients (0.16). Prevalence increased with age, ranging from 0.04% among those aged 18-29 years to 0.26% among those aged 60-69 years and 0.33% among those aged 70-79 years.
IN PRACTICE:
“LP is a fairly common disease, which disproportionately affects women and individuals older than 60 years of age,” the authors wrote. “Future research to help identify patients who may need systemic treatment and determine appropriate treatments for patients with LP to limit sequelae is important as no medication is currently FDA approved for LP.”
SOURCE:
The study was led by Natalia Pelet Del Toro, MD, Department of Dermatology, Northwell Health, New Hyde Park, New York, and was published online in The Journal of the American Academy of Dermatology.
LIMITATIONS:
The absence of a precise diagnosis code for non-oral LP introduces potential misclassification risks. Additionally, the study design did not allow for the establishment of disease severity levels, limiting the ability to correlate treatment choices with disease severity.
DISCLOSURES:
The study did not receive any funding. Two authors reported to have received advisory fees, grants, and/or honoraria from several pharmaceutical companies. Pelet Del Toro and another author did not declare any conflict of interests.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- To evaluate the prevalence of LP, researchers analyzed 566,851 eligible patients from the Explorys database, comprising electronic medical records from over 40 healthcare networks and 53 million patients across the United States.
- They also assessed treatment plans separately among 1998 newly diagnosed patients with LP between October 2015 and January 2020, who required at least one dermatology encounter within the first year following diagnosis.
- The primary outcome was overall prevalence of LP in the United States, including prevalence across specific age, sex, and racial subgroups. Additionally, dermatologist-prescribed treatments for non-oral LP were also reported.
TAKEAWAY:
- Overall, there were 1098 cases of LP (median age, 66 years; 74% women); the crude prevalence of LP was 0.19% and the age- and sex-standardized overall prevalence was 0.15%. Prevalence in women was 1.77 times higher than in men.
- Asian patients showed the highest standardized prevalence (0.2%), followed by Black patients (0.16). Prevalence increased with age, ranging from 0.04% among those aged 18-29 years to 0.26% among those aged 60-69 years and 0.33% among those aged 70-79 years.
IN PRACTICE:
“LP is a fairly common disease, which disproportionately affects women and individuals older than 60 years of age,” the authors wrote. “Future research to help identify patients who may need systemic treatment and determine appropriate treatments for patients with LP to limit sequelae is important as no medication is currently FDA approved for LP.”
SOURCE:
The study was led by Natalia Pelet Del Toro, MD, Department of Dermatology, Northwell Health, New Hyde Park, New York, and was published online in The Journal of the American Academy of Dermatology.
LIMITATIONS:
The absence of a precise diagnosis code for non-oral LP introduces potential misclassification risks. Additionally, the study design did not allow for the establishment of disease severity levels, limiting the ability to correlate treatment choices with disease severity.
DISCLOSURES:
The study did not receive any funding. Two authors reported to have received advisory fees, grants, and/or honoraria from several pharmaceutical companies. Pelet Del Toro and another author did not declare any conflict of interests.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- To evaluate the prevalence of LP, researchers analyzed 566,851 eligible patients from the Explorys database, comprising electronic medical records from over 40 healthcare networks and 53 million patients across the United States.
- They also assessed treatment plans separately among 1998 newly diagnosed patients with LP between October 2015 and January 2020, who required at least one dermatology encounter within the first year following diagnosis.
- The primary outcome was overall prevalence of LP in the United States, including prevalence across specific age, sex, and racial subgroups. Additionally, dermatologist-prescribed treatments for non-oral LP were also reported.
TAKEAWAY:
- Overall, there were 1098 cases of LP (median age, 66 years; 74% women); the crude prevalence of LP was 0.19% and the age- and sex-standardized overall prevalence was 0.15%. Prevalence in women was 1.77 times higher than in men.
- Asian patients showed the highest standardized prevalence (0.2%), followed by Black patients (0.16). Prevalence increased with age, ranging from 0.04% among those aged 18-29 years to 0.26% among those aged 60-69 years and 0.33% among those aged 70-79 years.
IN PRACTICE:
“LP is a fairly common disease, which disproportionately affects women and individuals older than 60 years of age,” the authors wrote. “Future research to help identify patients who may need systemic treatment and determine appropriate treatments for patients with LP to limit sequelae is important as no medication is currently FDA approved for LP.”
SOURCE:
The study was led by Natalia Pelet Del Toro, MD, Department of Dermatology, Northwell Health, New Hyde Park, New York, and was published online in The Journal of the American Academy of Dermatology.
LIMITATIONS:
The absence of a precise diagnosis code for non-oral LP introduces potential misclassification risks. Additionally, the study design did not allow for the establishment of disease severity levels, limiting the ability to correlate treatment choices with disease severity.
DISCLOSURES:
The study did not receive any funding. Two authors reported to have received advisory fees, grants, and/or honoraria from several pharmaceutical companies. Pelet Del Toro and another author did not declare any conflict of interests.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Phase 3 Lupus Trial Shows Promising Results for Dapirolizumab Pegol
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — The investigational anti-CD40 ligand agent dapirolizumab pegol (DZP) outperformed placebo in improving disease activity and reducing high-dose corticosteroid use in patients with systemic lupus erythematosus (SLE) in the phase 3 PHOENYCS GO trial.
“We really think that dapirolizumab pegol may represent a novel treatment for lupus, particularly given its broad immune modulatory effects,” said Megan Clowse, MD, MPH, associate professor of medicine and chief of the Division of Rheumatology and Immunology at Duke University School of Medicine in Durham, North Carolina. She presented the study in a late-breaking poster session at the American College of Rheumatology (ACR) 2024 Annual Meeting.
There is a “huge unmet need” for drugs for lupus, Clowse told this news organization. Patients with SLE continue to have high disease burden, including ongoing symptoms often driven by inflammation. Corticosteroids are often the best medications to control disease activity, she said, but they can result in long-term toxicity.
What Makes DZP Unique?
Through CD40 ligand signaling, DZP has been shown to reduce B- and T-cell activation and to downregulate interferon pathways. Previous antibodies targeting the CD40 ligand have been associated with an increased risk for thromboembolic events. However, DZP lacks the Fc portion of the antibody, which can bind to platelets and cause clotting. Data from phase 1, 2, and 3 trials thus far do not show an elevated risk for these events, Clowse explained. In fact, safety signals were strong enough that patients with antiphospholipid antibodies — a key driver for blood clots in patients with SLE — were included in the trial.
In PHOENYCS GO, investigators enrolled 321 patients with moderate to severe SLE with persistently active or frequently flaring/relapsing-remitting disease activity despite stable standard of care (SOC) medications such as antimalarials, corticosteroids, and immunosuppressants.
Patients were randomized 2:1 to receive intravenous DZP (24 mg/kg) plus SOC or intravenous placebo plus SOC every 4 weeks, with patients and investigators blinded to treatment assignments.
Patients taking a corticosteroid dose > 7.5 mg/day began a mandatory steroid taper by week 8 of the trial, with the goal of reducing that to < 7.5 mg/day. The tapering regimen was at the discretion of providers and was adapted to each patient’s individual disease activity.
The primary endpoint was British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response at week 48.
Patients in the DZP and placebo groups were on average 43.5 and 41.5 years old, respectively. More than 90% of patients were women, all on concomitant SLE medications. About half of the participants took a daily corticosteroid dose > 7.5 mg.
At 48 weeks, half of the DZP group (49.5%) achieved BICLA response compared with 34.6% in the placebo group (P = .0110). A higher proportion of patients taking DZP achieved SLE Responder Index-4 response than those taking placebo (60.1% vs 41.1%, respectively; P = .0014), and the rate of severe British Isles Lupus Assessment Group flares in the DZP group was half that of the placebo group (11.6% vs 23.4%; P = .0257). In the subgroup of patients who underwent corticosteroid tapering, 72.4% receiving DZP and 52.9% taking placebo reduced their dose to < 7.5 mg/day by 48 weeks (P = .0404).
DZP was generally well tolerated. Over 48 weeks, 82.6% of the DZP group and 75% of the placebo group reported treatment-emergent adverse events, but serious occurrences were more common in the placebo group (14.8%) than in the DZP group (9.9%). Herpes viral infections were higher in the placebo group, although there were three ophthalmic herpes cases in the DZP group. There was one case of acute myocardial infarction and one death linked to gangrene-related sepsis in patients receiving DZP.
A ‘Mild to Moderate’ Response
Although these are definitely positive results, they show a “mild to moderate response” to DZP, commented Gregory Gardner, MD, an emeritus professor in the Division of Rheumatology at the University of Washington, Seattle, and chair of the American College of Rheumatology’s annual meeting planning committee. He moderated the session where the research was presented. Although DZP showed efficacy among some patients, he noted, “there were still 51% patients that it didn’t work for.”
The drug uses an alternative pathway to current lupus drugs, Gardner added, and more research is needed to understand how best to use this medication in practice.
Clowse noted that DZP could be particularly beneficial for patients with SLE who want to get pregnant. Many drugs used to treat the disease are teratogenic; however, “because of the lack of Fc portion on this drug, it very likely does not cross the placenta in any kind of significant amount,” she said. Although there are not yet any reproductive safety data on DZP, she added, “that is a great potential niche.”
Biogen and UCB, which are jointly developing DZP, aim to start a second phase 3 trial of DZP in patients with SLE, called PHOENYCS FLY, in 2024.
The trial was sponsored by UCB. Clowse is a consultant and has received grant/research support from GSK and UCB. Gardner had no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2024
Cancer Mortality Not Higher for Patients With Autoimmune Disease on Checkpoint Inhibitors
WASHINGTON — Immune checkpoint inhibitor (ICI) therapy does not increase mortality in people with preexisting autoimmune diseases, new research has found.
Results from a large database analysis of patients with and without autoimmune diseases suggest it is safe to treat them with ICI if they develop a cancer for which it is indicated, Greg Challener, MD, a postdoctoral fellow at the Rheumatology and Allergy Clinical Epidemiology Research Center, Massachusetts General Hospital, Boston, said at the American College of Rheumatology 2024 Annual Meeting.
“One message is that, when rheumatologists are asked by oncologists about patients with rheumatoid arthritis or vasculitis or other autoimmune diseases and whether it’s safe to treat them with immune checkpoint inhibitors, this result provides some evidence that it probably is safe…. Checkpoint inhibitors are really incredible drugs, and they’ve improved mortality for a lot of cancers, particularly melanoma, and so I think there should be a pretty high threshold for us to say a patient shouldn’t receive them because of an autoimmune condition,” he told this news organization.
Another implication, Challener said, is that people with autoimmune diseases shouldn’t routinely be excluded from clinical trials of ICIs. Currently they are excluded because of concerns about exacerbation of underlying autoimmunity, possible interference between the ICI and the immunosuppressive drugs used to treat the autoimmune condition, and a theoretical risk for serious adverse events.
“Clinical trials are continuing to exclude these patients, and they paint with a very broad brush anyone with underlying autoimmunity ... I’m hoping that that changes. I don’t think there’s a great evidence base to support that practice, and it’s unfortunate that patients with underlying autoimmune diseases are excluded from important studies,” Challener said.
Asked to comment, session moderator Matlock Jeffries, MD, director of the Arthritis Research Unit at the Oklahoma Medical Research Foundation, Oklahoma City, told this news organization that he agrees the data are generally reassuring. “If one of our patients gets cancer and their oncologist wants to use a checkpoint inhibitor, we’d obviously still monitor them for complications, but we wouldn’t automatically assume the combination of a checkpoint inhibitor and autoimmune disease would increase their mortality.”
No Difference in Mortality for Those With and Without Autoimmune Disease
Challener and colleagues used administrative health data from the TriNetX Diamond network of 92 US healthcare sites with 212 million patients. All patients included in the study were receiving anti-programmed death protein 1/programmed death ligand 1 to treat malignancies involving the skin, lung/bronchus, digestive organs, or urinary tract. The study population also had at least one rheumatologic, gastrointestinal, neurologic, dermatologic, or endocrine autoimmune disease.
Propensity score matching between those with and without autoimmune disease was performed for about 100 covariates. Prior to the matching, the autoimmune disease group had significantly higher rates of cardiovascular and other comorbidities. The matching yielded 23,714 individuals with autoimmune disease and the same number without who had similar demographics and comorbidity rates, as well as malignancy type, alcohol/tobacco use, and medication use.
At a median follow-up of 250 days, the risk for mortality prior to propensity matching was 40.0% in the autoimmune disease group and 38.1% for those without, a significant difference with hazard ratio 1.07 (95% CI, 1.05-1.10). But after the matching, the difference was no longer significant: 39.8% vs 40.2%, respectively (0.97, 0.94-1.00).
The Kaplan-Meier curves for survival probability for those with or without autoimmune disease were nearly superimposed, showing no difference up to 1600 days. An analysis of just the patients with rheumatic diseases yielded similar results, Challener said.
Some Caveats About the Data
Jeffries, who is also an associate professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, and the Oklahoma VA, said he would like to see additional data on outcomes, both for the autoimmune conditions and the cancers. Challener said there are plans to look at other hard endpoints such as myocardial infarction and end-stage renal disease, but that the database is limited.
Both Challener and Jeffries also cautioned that the reassurance may not apply to patients with active disease.
“One thing this research doesn’t address is whether active autoimmune disease might have a different outcome compared to more kind of quiet disease…. If you have a patient who has extremely active rheumatoid arthritis or extremely active giant cell arthritis, for instance, I think that could be more challenging. I would be frightened to put a patient with really active GCA on pembrolizumab or say that it’s safe without their disease being controlled. But for someone who has well-controlled disease or minimally active disease, this is very reassuring,” Challener told this news organization.
“I think this may also be important in that it’s a good argument to tell the drug companies to include autoimmune patients in these trials so we can get better data,” Jeffries said.
Challener and Jeffries had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — Immune checkpoint inhibitor (ICI) therapy does not increase mortality in people with preexisting autoimmune diseases, new research has found.
Results from a large database analysis of patients with and without autoimmune diseases suggest it is safe to treat them with ICI if they develop a cancer for which it is indicated, Greg Challener, MD, a postdoctoral fellow at the Rheumatology and Allergy Clinical Epidemiology Research Center, Massachusetts General Hospital, Boston, said at the American College of Rheumatology 2024 Annual Meeting.
“One message is that, when rheumatologists are asked by oncologists about patients with rheumatoid arthritis or vasculitis or other autoimmune diseases and whether it’s safe to treat them with immune checkpoint inhibitors, this result provides some evidence that it probably is safe…. Checkpoint inhibitors are really incredible drugs, and they’ve improved mortality for a lot of cancers, particularly melanoma, and so I think there should be a pretty high threshold for us to say a patient shouldn’t receive them because of an autoimmune condition,” he told this news organization.
Another implication, Challener said, is that people with autoimmune diseases shouldn’t routinely be excluded from clinical trials of ICIs. Currently they are excluded because of concerns about exacerbation of underlying autoimmunity, possible interference between the ICI and the immunosuppressive drugs used to treat the autoimmune condition, and a theoretical risk for serious adverse events.
“Clinical trials are continuing to exclude these patients, and they paint with a very broad brush anyone with underlying autoimmunity ... I’m hoping that that changes. I don’t think there’s a great evidence base to support that practice, and it’s unfortunate that patients with underlying autoimmune diseases are excluded from important studies,” Challener said.
Asked to comment, session moderator Matlock Jeffries, MD, director of the Arthritis Research Unit at the Oklahoma Medical Research Foundation, Oklahoma City, told this news organization that he agrees the data are generally reassuring. “If one of our patients gets cancer and their oncologist wants to use a checkpoint inhibitor, we’d obviously still monitor them for complications, but we wouldn’t automatically assume the combination of a checkpoint inhibitor and autoimmune disease would increase their mortality.”
No Difference in Mortality for Those With and Without Autoimmune Disease
Challener and colleagues used administrative health data from the TriNetX Diamond network of 92 US healthcare sites with 212 million patients. All patients included in the study were receiving anti-programmed death protein 1/programmed death ligand 1 to treat malignancies involving the skin, lung/bronchus, digestive organs, or urinary tract. The study population also had at least one rheumatologic, gastrointestinal, neurologic, dermatologic, or endocrine autoimmune disease.
Propensity score matching between those with and without autoimmune disease was performed for about 100 covariates. Prior to the matching, the autoimmune disease group had significantly higher rates of cardiovascular and other comorbidities. The matching yielded 23,714 individuals with autoimmune disease and the same number without who had similar demographics and comorbidity rates, as well as malignancy type, alcohol/tobacco use, and medication use.
At a median follow-up of 250 days, the risk for mortality prior to propensity matching was 40.0% in the autoimmune disease group and 38.1% for those without, a significant difference with hazard ratio 1.07 (95% CI, 1.05-1.10). But after the matching, the difference was no longer significant: 39.8% vs 40.2%, respectively (0.97, 0.94-1.00).
The Kaplan-Meier curves for survival probability for those with or without autoimmune disease were nearly superimposed, showing no difference up to 1600 days. An analysis of just the patients with rheumatic diseases yielded similar results, Challener said.
Some Caveats About the Data
Jeffries, who is also an associate professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, and the Oklahoma VA, said he would like to see additional data on outcomes, both for the autoimmune conditions and the cancers. Challener said there are plans to look at other hard endpoints such as myocardial infarction and end-stage renal disease, but that the database is limited.
Both Challener and Jeffries also cautioned that the reassurance may not apply to patients with active disease.
“One thing this research doesn’t address is whether active autoimmune disease might have a different outcome compared to more kind of quiet disease…. If you have a patient who has extremely active rheumatoid arthritis or extremely active giant cell arthritis, for instance, I think that could be more challenging. I would be frightened to put a patient with really active GCA on pembrolizumab or say that it’s safe without their disease being controlled. But for someone who has well-controlled disease or minimally active disease, this is very reassuring,” Challener told this news organization.
“I think this may also be important in that it’s a good argument to tell the drug companies to include autoimmune patients in these trials so we can get better data,” Jeffries said.
Challener and Jeffries had no relevant disclosures.
A version of this article appeared on Medscape.com.
WASHINGTON — Immune checkpoint inhibitor (ICI) therapy does not increase mortality in people with preexisting autoimmune diseases, new research has found.
Results from a large database analysis of patients with and without autoimmune diseases suggest it is safe to treat them with ICI if they develop a cancer for which it is indicated, Greg Challener, MD, a postdoctoral fellow at the Rheumatology and Allergy Clinical Epidemiology Research Center, Massachusetts General Hospital, Boston, said at the American College of Rheumatology 2024 Annual Meeting.
“One message is that, when rheumatologists are asked by oncologists about patients with rheumatoid arthritis or vasculitis or other autoimmune diseases and whether it’s safe to treat them with immune checkpoint inhibitors, this result provides some evidence that it probably is safe…. Checkpoint inhibitors are really incredible drugs, and they’ve improved mortality for a lot of cancers, particularly melanoma, and so I think there should be a pretty high threshold for us to say a patient shouldn’t receive them because of an autoimmune condition,” he told this news organization.
Another implication, Challener said, is that people with autoimmune diseases shouldn’t routinely be excluded from clinical trials of ICIs. Currently they are excluded because of concerns about exacerbation of underlying autoimmunity, possible interference between the ICI and the immunosuppressive drugs used to treat the autoimmune condition, and a theoretical risk for serious adverse events.
“Clinical trials are continuing to exclude these patients, and they paint with a very broad brush anyone with underlying autoimmunity ... I’m hoping that that changes. I don’t think there’s a great evidence base to support that practice, and it’s unfortunate that patients with underlying autoimmune diseases are excluded from important studies,” Challener said.
Asked to comment, session moderator Matlock Jeffries, MD, director of the Arthritis Research Unit at the Oklahoma Medical Research Foundation, Oklahoma City, told this news organization that he agrees the data are generally reassuring. “If one of our patients gets cancer and their oncologist wants to use a checkpoint inhibitor, we’d obviously still monitor them for complications, but we wouldn’t automatically assume the combination of a checkpoint inhibitor and autoimmune disease would increase their mortality.”
No Difference in Mortality for Those With and Without Autoimmune Disease
Challener and colleagues used administrative health data from the TriNetX Diamond network of 92 US healthcare sites with 212 million patients. All patients included in the study were receiving anti-programmed death protein 1/programmed death ligand 1 to treat malignancies involving the skin, lung/bronchus, digestive organs, or urinary tract. The study population also had at least one rheumatologic, gastrointestinal, neurologic, dermatologic, or endocrine autoimmune disease.
Propensity score matching between those with and without autoimmune disease was performed for about 100 covariates. Prior to the matching, the autoimmune disease group had significantly higher rates of cardiovascular and other comorbidities. The matching yielded 23,714 individuals with autoimmune disease and the same number without who had similar demographics and comorbidity rates, as well as malignancy type, alcohol/tobacco use, and medication use.
At a median follow-up of 250 days, the risk for mortality prior to propensity matching was 40.0% in the autoimmune disease group and 38.1% for those without, a significant difference with hazard ratio 1.07 (95% CI, 1.05-1.10). But after the matching, the difference was no longer significant: 39.8% vs 40.2%, respectively (0.97, 0.94-1.00).
The Kaplan-Meier curves for survival probability for those with or without autoimmune disease were nearly superimposed, showing no difference up to 1600 days. An analysis of just the patients with rheumatic diseases yielded similar results, Challener said.
Some Caveats About the Data
Jeffries, who is also an associate professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, and the Oklahoma VA, said he would like to see additional data on outcomes, both for the autoimmune conditions and the cancers. Challener said there are plans to look at other hard endpoints such as myocardial infarction and end-stage renal disease, but that the database is limited.
Both Challener and Jeffries also cautioned that the reassurance may not apply to patients with active disease.
“One thing this research doesn’t address is whether active autoimmune disease might have a different outcome compared to more kind of quiet disease…. If you have a patient who has extremely active rheumatoid arthritis or extremely active giant cell arthritis, for instance, I think that could be more challenging. I would be frightened to put a patient with really active GCA on pembrolizumab or say that it’s safe without their disease being controlled. But for someone who has well-controlled disease or minimally active disease, this is very reassuring,” Challener told this news organization.
“I think this may also be important in that it’s a good argument to tell the drug companies to include autoimmune patients in these trials so we can get better data,” Jeffries said.
Challener and Jeffries had no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2024