User login
Acute-Onset Alopecia
The Diagnosis: Thallium-Induced Alopecia
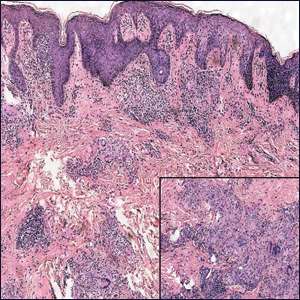
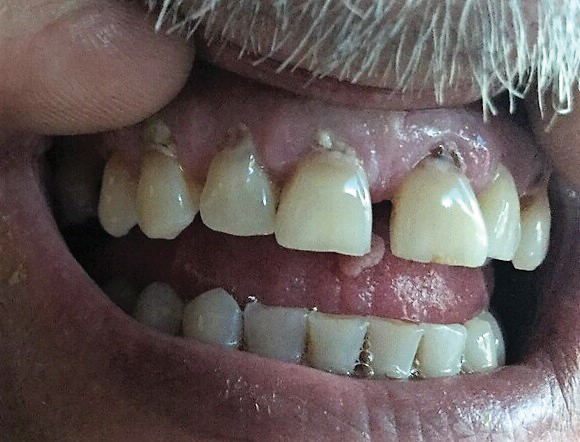
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.

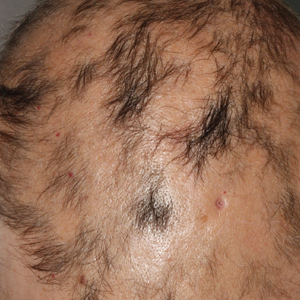
A previously healthy 45-year-old man presented to the dermatology department with abrupt onset of patchy, progressively worsening alopecia of the scalp as well as nausea with emesis and blurry vision of a few weeks' duration. All symptoms were temporally associated with a new demolition job the patient had started at an industrial site. He reported 10 other contractors were similarly affected. The patient denied paresthesia or other skin changes. On physical examination, large patches of smooth alopecia without erythema, scale, scarring, tenderness, or edema that coalesced to involve the majority of the scalp, eyebrows, and eyelashes (inset) were noted.
Acquired Hypertrichosis of the Periorbital Area and Malar Cheek
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
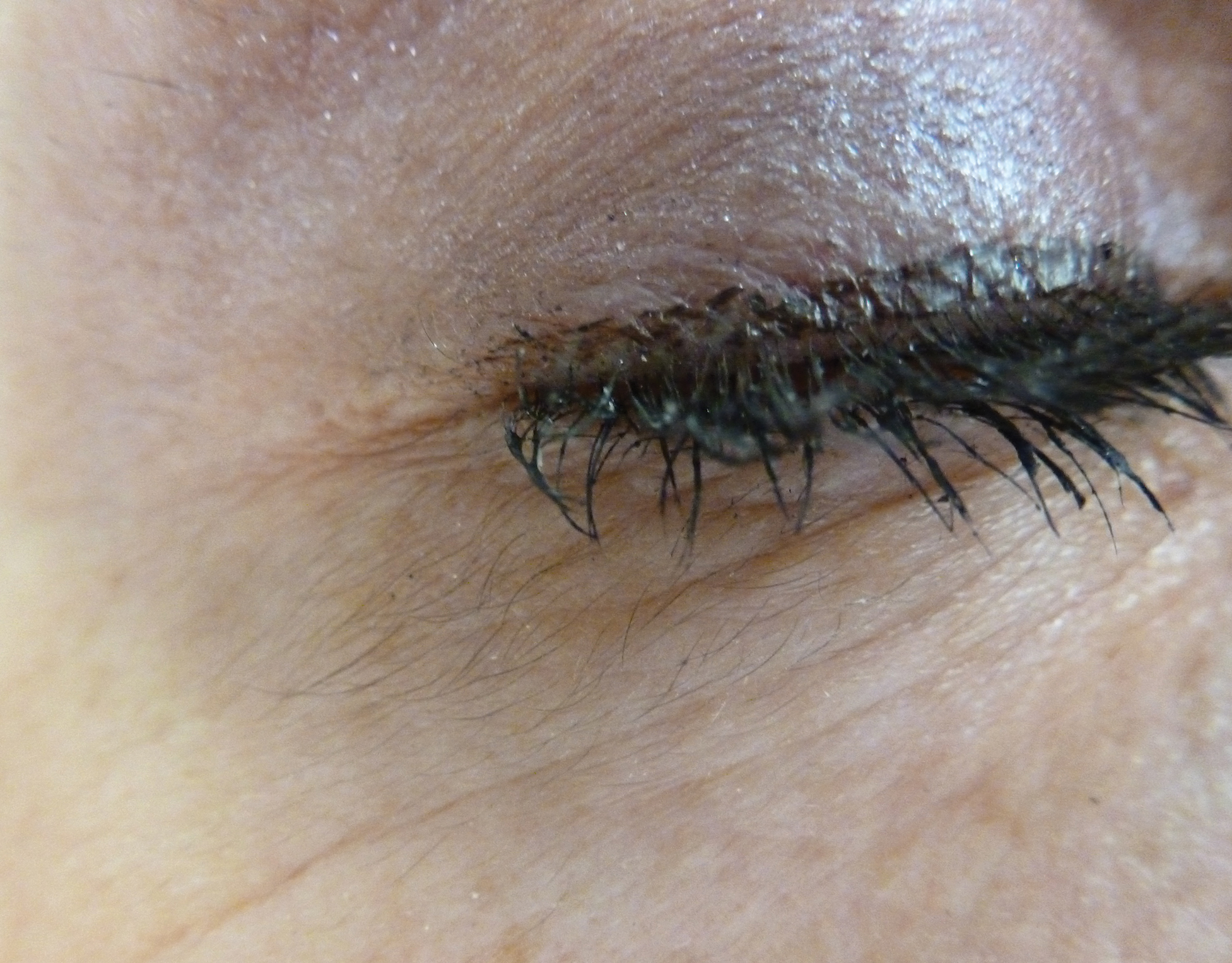
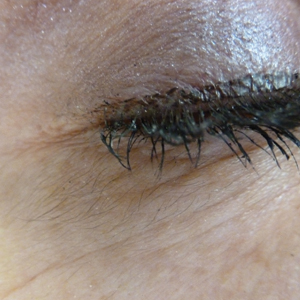
An otherwise healthy woman in her late 50s with Fitzpatrick skin type II presented to the dermatology department for a scheduled cosmetic botulinum toxin injection. Her medical history was notable only for periodic nonsurgical cosmetic procedures including botulinum toxin and dermal fillers, and she was not taking any daily systemic medications. During the preoperative assessment, subtle bilateral and symmetric hypertrichosis with darker terminal hair formation was noted on the periorbital skin and zygomatic cheek. Upon inquiry, the patient admitted to purchasing a “special eye drop” from Mexico and using it regularly. After instillation of 2 to 3 drops per eye, she would laterally wipe the resulting excess drops away from the eyes with her hands and then wash her hands. She denied a change in eye color from their natural brown but did report using blue color contact lenses. She denied an increase in hair growth elsewhere including the upper lip, chin, upper chest, forearms, and hands. She denied deepening of her voice, acne, or hair thinning.
What’s Eating You? Millipede Burns
Clinical Presentation
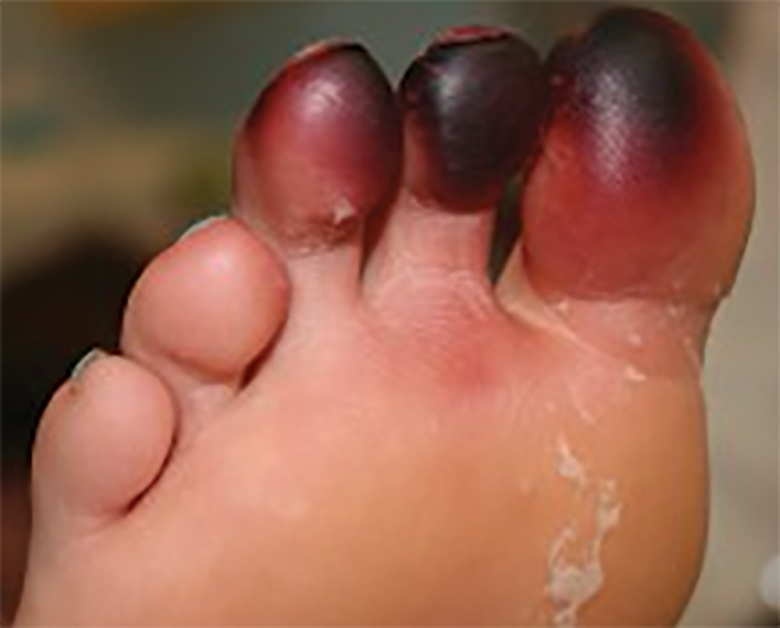
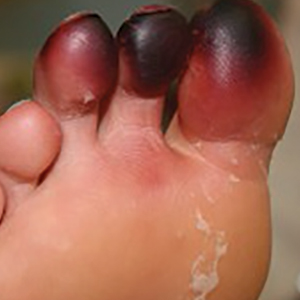
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5

Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
Clinical Presentation
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5
Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
Clinical Presentation
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5
Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
Practice Points
- The most common site of involvement of millipede burns is the foot, followed by other commonly exposed areas such as the arms, face, and eyes. Covered parts of the body are much less commonly affected.
- Millipede burns may resemble child abuse in pediatric patients; therefore, physicians should be aware of this diagnosis when unusual parts of the body are involved.
Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
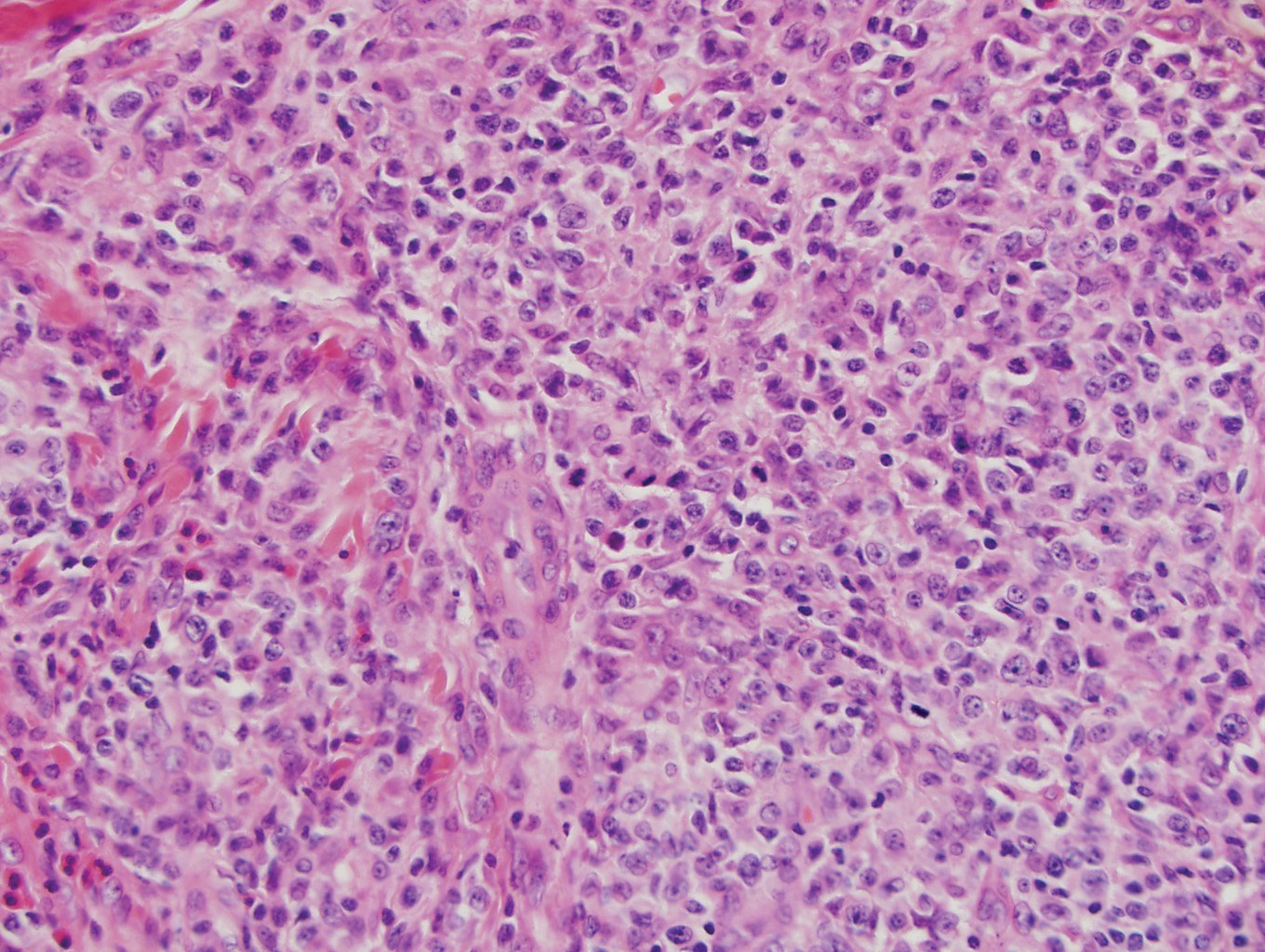
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
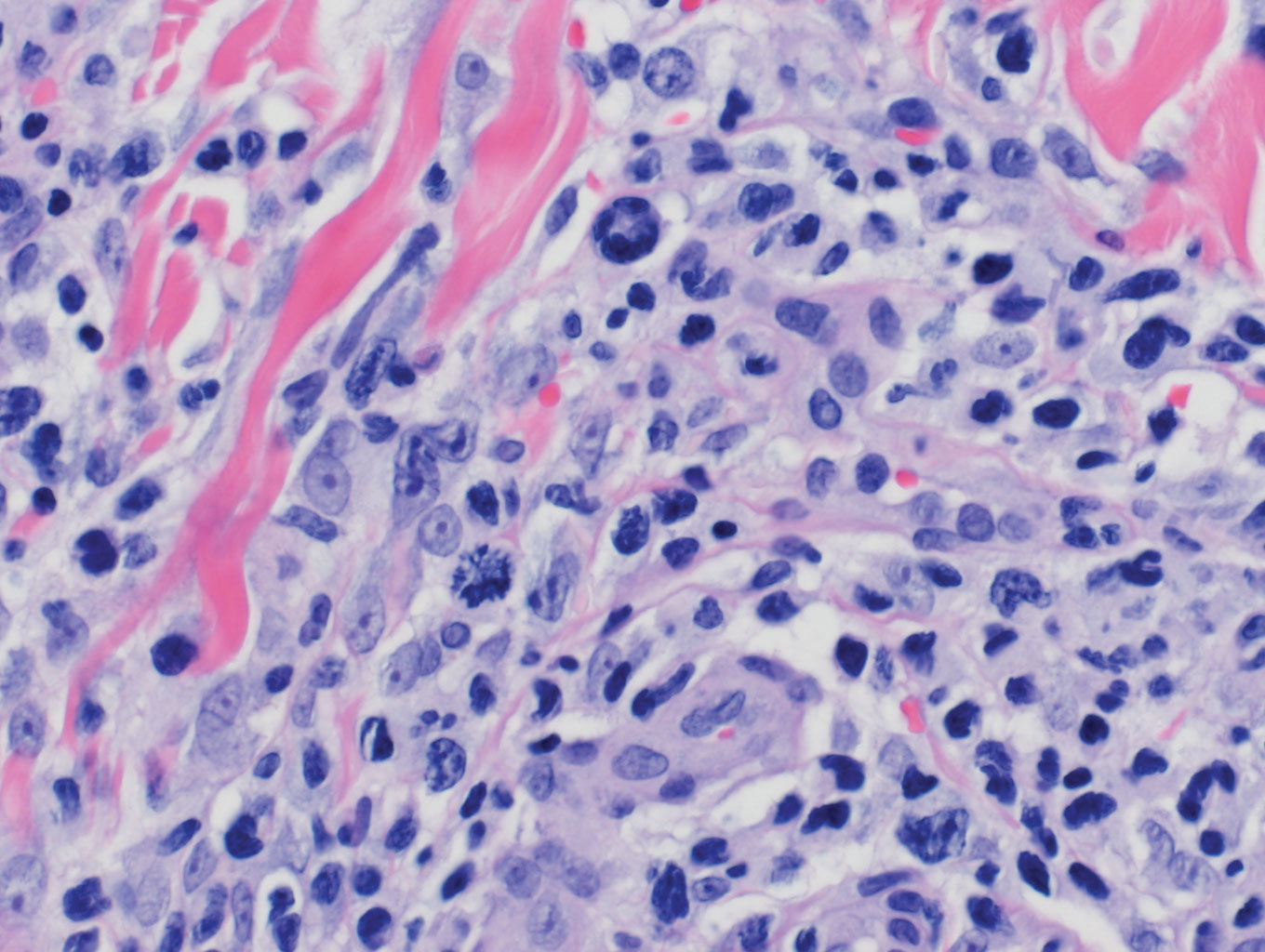
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
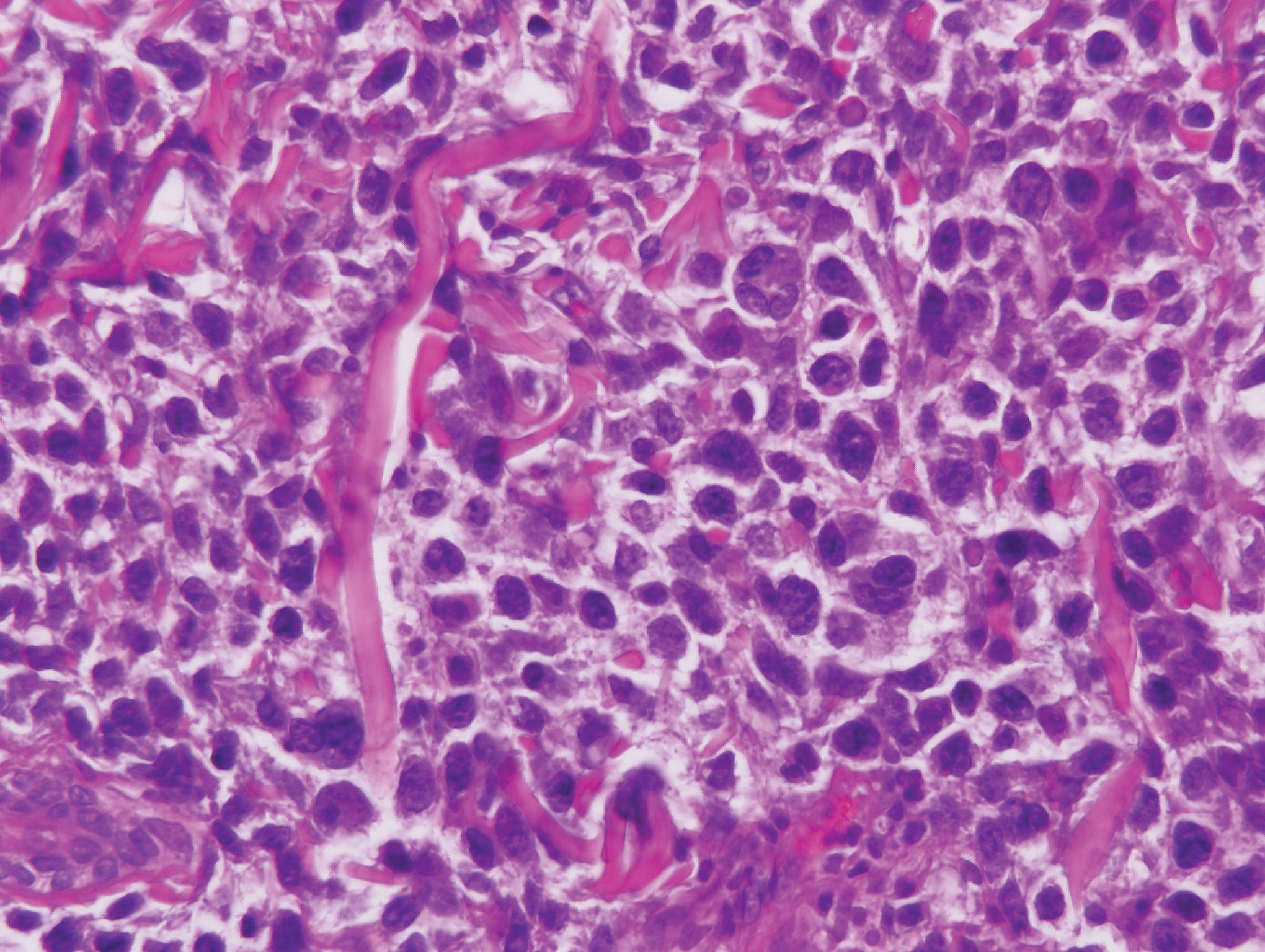
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
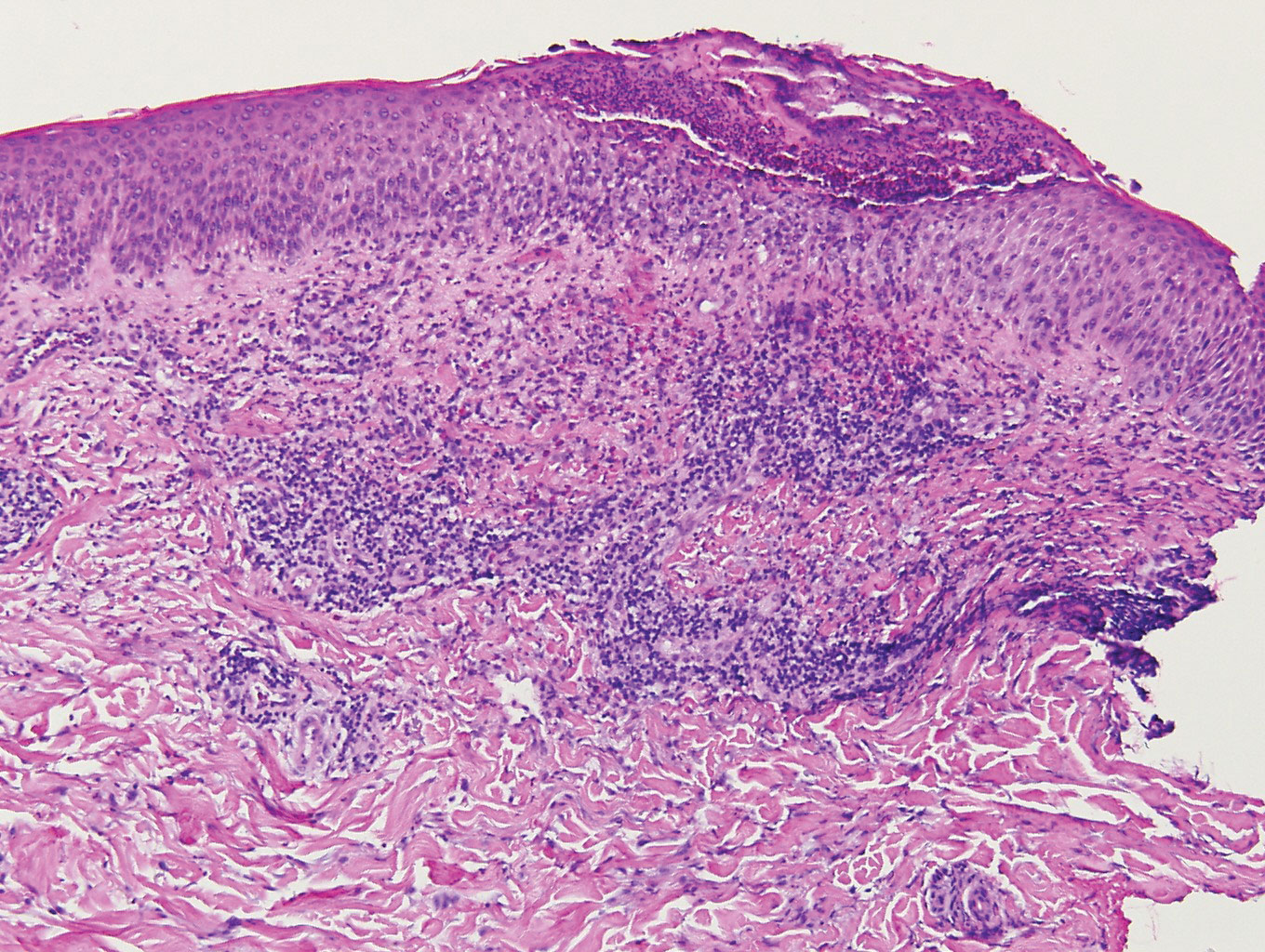
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
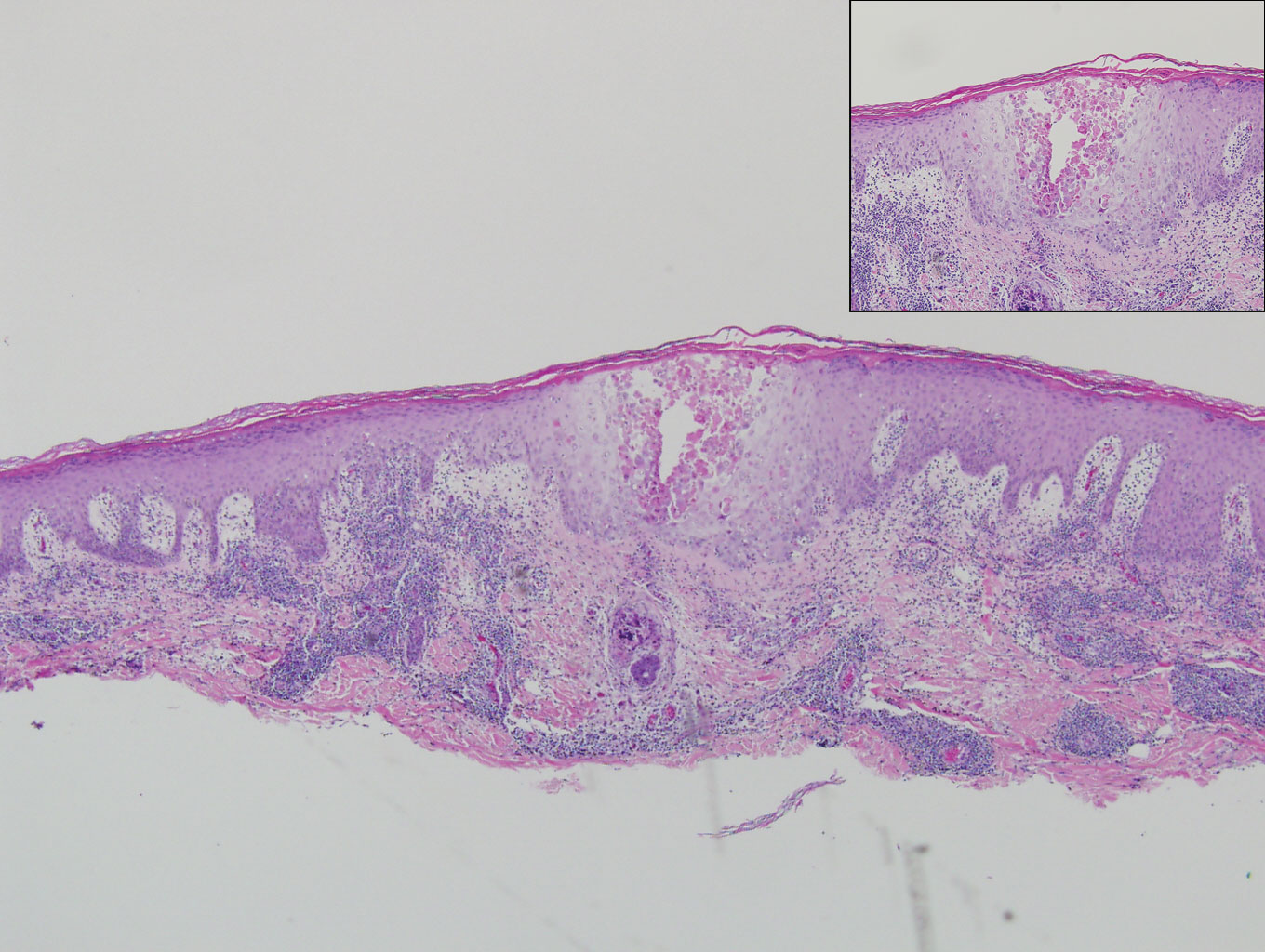
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.
Aquatic Antagonists: Stingray Injury Update
Incidence and Characteristics


Stingrays are dorsoventrally flattened, diamond-shaped fish with light-colored ventral and dark-colored dorsal surfaces. They have strong pectoral wings that allow them to swim forward and backward and even launch off waves.3 Stingrays range in size from the palm of a human hand to 6.5 ft in width. They possess 1 or more spines (2.5 to >30 cm in length) that are disguised by much longer tails.6,7 They often are encountered accidentally because they bury themselves in the sand or mud of shallow coastal waters or rivers with only their eyes and tails exposed to fool prey and avoid predators.
Injury Clinical Presentation
Stingray injuries typically involve the lower legs, ankles, or feet after stepping on a stingray.8 Fishermen can present with injuries of the upper extremities after handling fish with their hands.9 Other rarer injuries occur when individuals are swimming alongside stingrays or when stingrays catapult off waves into moving boats.10,11 Stingrays impale victims by using their tails to direct a retroserrate barb composed of a strong cartilaginous material called vasodentin. The barb releases venom by breaking through the venom-containing integumentary sheath that encapsulates it. Stingray venom contains phosphodiesterase, serotonin, and 5′-nucleotidase. It causes severe pain, vasoconstriction, ischemia, and poor wound healing, along with systemic effects such as disorientation, syncope, seizures, salivation, nausea, vomiting, abdominal pain, diarrhea, muscle cramps or fasciculations, pruritus, allergic reaction, hypotension, cardiac arrhythmias, dyspnea, paralysis, and possibly death.1,8,12,13
Management
Pain Relief
As with many marine envenomations, immersion in hot but not scalding water can inactivate venom and reduce symptoms.8,9 In one retrospective review, 52 of 75 (69%) patients reporting to a California poison center with stingray injuries had improvement in pain within 1 hour of hot water immersion before any analgesics were instituted.8 In another review, 65 of 74 (88%) patients presenting to a California emergency department within 24 hours of sustaining a stingray injury had complete relief of pain within 30 minutes of hot water immersion. Patients who received analgesics in addition to hot water immersion did not require a second dose.9 In concordance with these studies, we suggest immersing areas affected by stingray injuries in hot water (temperature, 43.3°C to 46.1°C [110°F–115°F]; or as close to this range as tolerated) until pain subsides.8,9,14 Ice packs are an alternative to hot water immersion that may be more readily available to patients. If pain does not resolve following hot water immersion or application of an ice pack, additional analgesics and xylocaine without epinephrine may be helpful.9,15
Infection
One major complication of stingray injuries is infection.8,9 Many bacterial species reside in stingray mucus, the marine environment, or on human skin that may be introduced during a single injury. Marine envenomations can involve organisms such as Vibrio, Aeromonas, and Mycobacterium species, which often are resistant to antibiotic prophylaxis covering common causes of soft-tissue infection such as Staphylococcus and Streptococcus species.8,9,16,17 Additionally, physicians should cover for Clostridium species and ensure patients are up-to-date on vaccinations because severe cases of tetanus following stingray injuries have been reported.18 Lastly, fungal infections including fusariosis have been reported following stingray injuries and should be considered if a patient develops an infection.19
Several authors support the use of prophylactic broad-spectrum antibiotics in all but mild stingray injuries.8,9,20,21 Although no standardized definition exists, mild injuries generally represent patients with superficial lacerations or less, while deeper lacerations and puncture wounds require prophylaxis. Several authors agree on the use of fluoroquinolone antibiotics (eg, ciprofloxacin 500 mg twice daily) for 5 to 7 days following severe stingray injuries.1,9,13,22 Other proposed antibiotic regimens include trimethoprim-sulfamethoxazole (160/800 mg twice daily) or tetracycline (500 mg 4 times daily) for 7 days.13 Failure of ciprofloxacin therapy after 7 days has been reported, with resolution of infection after treatment with an intravenous cephalosporin for 7 days.20 Failure of trimethoprim-sulfamethoxazole therapy also has been reported, with one case requiring levofloxacin for a much longer course.21 Clinical follow-up remains essential after prescribing prophylactic antibiotics, as resistance is common.
Foreign Bodies
Stingray injuries also are often complicated by foreign bodies or retained spines.3,8 Although these complications are less severe than infection, all wounds should be explored for material under local anesthesia. Furthermore, there has been support for thorough debridement of necrotic tissue with referral to a hand specialist for deeper injuries to the hands as well as referral to a foot and ankle specialist for deeper injuries of the lower extremities.23,24 More serious injuries with penetration of vital structures, such as through the chest or abdomen, require immediate exploration in an operating room.1,24
Imaging
Routine imaging of stingray injuries remains controversial. In a case series of 119 patients presenting to a California emergency department with stingray injuries, Clark et al9 found that radiographs were not helpful. This finding likely is due in part to an inability to detect hypodense material such as integumentary or glandular tissue via radiography.3 However, radiographs have been used to identify retained stingray barbs in select cases in which retained barbs are suspected.2,25 Lastly, ultrasonography potentially may offer a better first choice when a barb is not readily apparent; magnetic resonance imaging may be indicated for more involved areas and for further visualization of suspected hypodense material, though at a higher expense.2,9
Biopsy
Biopsies of stingray injuries are rarely performed, and the findings are not well characterized. One case biopsied 2 months after injury showed a large zone of paucicellular necrosis with superficial ulceration and granulomatous inflammation. The stingray venom was most likely responsible for the pattern of necrosis noted in the biopsy.21
Avoidance and Prevention
Patients traveling to areas of the world inhabited by stingrays should receive counseling on how to avoid injury. Prior to entry, individuals can throw stones or use a long stick to clear their walking or swimming areas of venomous fish.26 Polarized sunglasses may help spot stingrays in shallow water. Furthermore, wading through water with a shuffling gait can help individuals avoid stepping directly on a stingray and also warns stingrays that someone is in the area. Individuals who spend more time in coastal waters or river systems inhabited by stingrays may invest in protective stingray gear such as leg guards or specialized wading boots.26 Lastly, fishermen should be advised to avoid handling stingrays with their hands and instead cut their fishing line to release the fish.
- Aurbach PS. Envenomations by aquatic vertebrates. In: Auerbach PS. Wilderness Medicine. 5th ed. St. Louis, MO: Mosby; 2007:1730-1749.
- Robins CR, Ray GC. A Field Guide to Atlantic Coast Fishes. New York, NY: Houghton Mifflin Company; 1986.
- Diaz JH. The evaluation, management, and prevention of stingray injuries in travelers. J Travel Med. 2008;15:102-109.
- Haddad V Jr, Neto DG, de Paula Neto JB, et al. Freshwater stingrays: study of epidemiologic, clinical and therapeutic aspects based on 84 envenomings in humans and some enzymatic activities of the venom. Toxicon. 2004;43:287-294.
- Marinkelle CJ. Accidents by venomous animals in Colombia. Ind Med Surg. 1966;35:988-992.
- Last PR, White WT, Caire JN, et al. Sharks and Rays of Borneo. Collingwood VIC, Australia: CSIRO Publishing; 2010.
- Mebs D. Venomous and Poisonous Animals: A Handbook for Biologists, Toxicologists and Toxinologists, Physicians and Pharmacists. Boca Raton, FL: CRC Press; 2002.
- Clark AT, Clark RF, Cantrell FL. A retrospective review of the presentation and treatment of stingray stings reported to a poison control system. Am J Ther. 2017;24:E177-E180.
- Clark RF, Girard RH, Rao D, et al. Stingray envenomation: a retrospective review of clinical presentation and treatment in 119 cases. J Emerg Med. 2007;33:33-37.
- Mahjoubi L, Joyeux A, Delambre JF, et al. Near-death thoracic trauma caused by a stingray in the Indian Ocean. Semin Thorac Cardiovasc Surg. 2017;29:262-263.
- Parra MW, Constantini EN, Rodas EB. Surviving a transfixing cardiac injury caused by a stingray barb. J Thorac Cardiovasc Surg. 2010;139:E115-E116.
- Dos Santos JC, Grund LZ, Seibert CS, et al. Stingray venom activates IL-33 producing cardiomyocytes, but not mast cell, to promote acute neutrophil-mediated injury. Sci Rep. 2017;7:7912.
- Auerbach PS, Norris RL. Marine envenomation. In: Longo DL, Kasper SL, Jameson JL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:144-148.
- Cook MD, Matteucci MJ, Lall R, et al. Stingray envenomation. J Emerg Med. 2006;30:345-347.
- Bowers RC, Mustain MV. Disorders due to physical & environmental agents. In: Humphries RL, Stone C, eds. CURRENT Diagnosis & Treatment Emergency Medicine. 7th ed. New York, NY: McGraw-Hill; 2011:835-861.
- Domingos MO, Franzolin MR, dos Anjos MT, et al. The influence of environmental bacteria in freshwater stingray wound-healing. Toxicon. 2011;58:147-153.
- Auerbach PS, Yajko DM, Nassos PS, et al. Bacteriology of the marine environment: implications for clinical therapy. Ann Emerg Med. 1987;16:643-649.
- Torrez PP, Quiroga MM, Said R, et al. Tetanus after envenomations caused by freshwater stingrays. Toxicon. 2015;97:32-35.
- Hiemenz JW, Kennedy B, Kwon-Chung KJ. Invasive fusariosis associated with an injury by a stingray barb. J Med Vet Mycol. 1990;28:209-213.
- da Silva NJ Jr, Ferreira KR, Pinto RN, et al. A severe accident caused by an ocellate river stingray (Potamotrygon motoro) in central Brazil: how well do we really understand stingray venom chemistry, envenomation, and therapeutics? Toxins (Basel). 2015;7:2272-2288.
- Tartar D, Limova M, North J. Clinical and histopathologic findings in cutaneous sting ray wounds: a case report. Dermatol Online J. 2013;19:19261.
- Jarvis HC, Matheny LM, Clanton TO. Stingray injury to the webspace of the foot. Orthopedics. 2012;35:E762-E765.
- Trickett R, Whitaker IS, Boyce DE. Sting-ray injuries to the hand: case report, literature review and a suggested algorithm for management. J Plast Reconstruct Aesthet Surg. 2009;62:E270-E273.
- Fernandez I, Valladolid G, Varon J, et al. Encounters with venomous sea-life. J Emerg Med. 2011;40:103-112.
- O’Malley GF, O’Malley RN, Pham O, et al. Retained stingray barb and the importance of imaging. Wilderness Environ Med. 2015;26:375-379.
- How to protect yourself from stingrays. Howcast website. https://www.howcast.com/videos/228034-how-to-protect-yourself-from-stingrays/. Accessed July 12, 2018.
Incidence and Characteristics
Stingrays are dorsoventrally flattened, diamond-shaped fish with light-colored ventral and dark-colored dorsal surfaces. They have strong pectoral wings that allow them to swim forward and backward and even launch off waves.3 Stingrays range in size from the palm of a human hand to 6.5 ft in width. They possess 1 or more spines (2.5 to >30 cm in length) that are disguised by much longer tails.6,7 They often are encountered accidentally because they bury themselves in the sand or mud of shallow coastal waters or rivers with only their eyes and tails exposed to fool prey and avoid predators.
Injury Clinical Presentation
Stingray injuries typically involve the lower legs, ankles, or feet after stepping on a stingray.8 Fishermen can present with injuries of the upper extremities after handling fish with their hands.9 Other rarer injuries occur when individuals are swimming alongside stingrays or when stingrays catapult off waves into moving boats.10,11 Stingrays impale victims by using their tails to direct a retroserrate barb composed of a strong cartilaginous material called vasodentin. The barb releases venom by breaking through the venom-containing integumentary sheath that encapsulates it. Stingray venom contains phosphodiesterase, serotonin, and 5′-nucleotidase. It causes severe pain, vasoconstriction, ischemia, and poor wound healing, along with systemic effects such as disorientation, syncope, seizures, salivation, nausea, vomiting, abdominal pain, diarrhea, muscle cramps or fasciculations, pruritus, allergic reaction, hypotension, cardiac arrhythmias, dyspnea, paralysis, and possibly death.1,8,12,13
Management
Pain Relief
As with many marine envenomations, immersion in hot but not scalding water can inactivate venom and reduce symptoms.8,9 In one retrospective review, 52 of 75 (69%) patients reporting to a California poison center with stingray injuries had improvement in pain within 1 hour of hot water immersion before any analgesics were instituted.8 In another review, 65 of 74 (88%) patients presenting to a California emergency department within 24 hours of sustaining a stingray injury had complete relief of pain within 30 minutes of hot water immersion. Patients who received analgesics in addition to hot water immersion did not require a second dose.9 In concordance with these studies, we suggest immersing areas affected by stingray injuries in hot water (temperature, 43.3°C to 46.1°C [110°F–115°F]; or as close to this range as tolerated) until pain subsides.8,9,14 Ice packs are an alternative to hot water immersion that may be more readily available to patients. If pain does not resolve following hot water immersion or application of an ice pack, additional analgesics and xylocaine without epinephrine may be helpful.9,15
Infection
One major complication of stingray injuries is infection.8,9 Many bacterial species reside in stingray mucus, the marine environment, or on human skin that may be introduced during a single injury. Marine envenomations can involve organisms such as Vibrio, Aeromonas, and Mycobacterium species, which often are resistant to antibiotic prophylaxis covering common causes of soft-tissue infection such as Staphylococcus and Streptococcus species.8,9,16,17 Additionally, physicians should cover for Clostridium species and ensure patients are up-to-date on vaccinations because severe cases of tetanus following stingray injuries have been reported.18 Lastly, fungal infections including fusariosis have been reported following stingray injuries and should be considered if a patient develops an infection.19
Several authors support the use of prophylactic broad-spectrum antibiotics in all but mild stingray injuries.8,9,20,21 Although no standardized definition exists, mild injuries generally represent patients with superficial lacerations or less, while deeper lacerations and puncture wounds require prophylaxis. Several authors agree on the use of fluoroquinolone antibiotics (eg, ciprofloxacin 500 mg twice daily) for 5 to 7 days following severe stingray injuries.1,9,13,22 Other proposed antibiotic regimens include trimethoprim-sulfamethoxazole (160/800 mg twice daily) or tetracycline (500 mg 4 times daily) for 7 days.13 Failure of ciprofloxacin therapy after 7 days has been reported, with resolution of infection after treatment with an intravenous cephalosporin for 7 days.20 Failure of trimethoprim-sulfamethoxazole therapy also has been reported, with one case requiring levofloxacin for a much longer course.21 Clinical follow-up remains essential after prescribing prophylactic antibiotics, as resistance is common.
Foreign Bodies
Stingray injuries also are often complicated by foreign bodies or retained spines.3,8 Although these complications are less severe than infection, all wounds should be explored for material under local anesthesia. Furthermore, there has been support for thorough debridement of necrotic tissue with referral to a hand specialist for deeper injuries to the hands as well as referral to a foot and ankle specialist for deeper injuries of the lower extremities.23,24 More serious injuries with penetration of vital structures, such as through the chest or abdomen, require immediate exploration in an operating room.1,24
Imaging
Routine imaging of stingray injuries remains controversial. In a case series of 119 patients presenting to a California emergency department with stingray injuries, Clark et al9 found that radiographs were not helpful. This finding likely is due in part to an inability to detect hypodense material such as integumentary or glandular tissue via radiography.3 However, radiographs have been used to identify retained stingray barbs in select cases in which retained barbs are suspected.2,25 Lastly, ultrasonography potentially may offer a better first choice when a barb is not readily apparent; magnetic resonance imaging may be indicated for more involved areas and for further visualization of suspected hypodense material, though at a higher expense.2,9
Biopsy
Biopsies of stingray injuries are rarely performed, and the findings are not well characterized. One case biopsied 2 months after injury showed a large zone of paucicellular necrosis with superficial ulceration and granulomatous inflammation. The stingray venom was most likely responsible for the pattern of necrosis noted in the biopsy.21
Avoidance and Prevention
Patients traveling to areas of the world inhabited by stingrays should receive counseling on how to avoid injury. Prior to entry, individuals can throw stones or use a long stick to clear their walking or swimming areas of venomous fish.26 Polarized sunglasses may help spot stingrays in shallow water. Furthermore, wading through water with a shuffling gait can help individuals avoid stepping directly on a stingray and also warns stingrays that someone is in the area. Individuals who spend more time in coastal waters or river systems inhabited by stingrays may invest in protective stingray gear such as leg guards or specialized wading boots.26 Lastly, fishermen should be advised to avoid handling stingrays with their hands and instead cut their fishing line to release the fish.
Incidence and Characteristics
Stingrays are dorsoventrally flattened, diamond-shaped fish with light-colored ventral and dark-colored dorsal surfaces. They have strong pectoral wings that allow them to swim forward and backward and even launch off waves.3 Stingrays range in size from the palm of a human hand to 6.5 ft in width. They possess 1 or more spines (2.5 to >30 cm in length) that are disguised by much longer tails.6,7 They often are encountered accidentally because they bury themselves in the sand or mud of shallow coastal waters or rivers with only their eyes and tails exposed to fool prey and avoid predators.
Injury Clinical Presentation
Stingray injuries typically involve the lower legs, ankles, or feet after stepping on a stingray.8 Fishermen can present with injuries of the upper extremities after handling fish with their hands.9 Other rarer injuries occur when individuals are swimming alongside stingrays or when stingrays catapult off waves into moving boats.10,11 Stingrays impale victims by using their tails to direct a retroserrate barb composed of a strong cartilaginous material called vasodentin. The barb releases venom by breaking through the venom-containing integumentary sheath that encapsulates it. Stingray venom contains phosphodiesterase, serotonin, and 5′-nucleotidase. It causes severe pain, vasoconstriction, ischemia, and poor wound healing, along with systemic effects such as disorientation, syncope, seizures, salivation, nausea, vomiting, abdominal pain, diarrhea, muscle cramps or fasciculations, pruritus, allergic reaction, hypotension, cardiac arrhythmias, dyspnea, paralysis, and possibly death.1,8,12,13
Management
Pain Relief
As with many marine envenomations, immersion in hot but not scalding water can inactivate venom and reduce symptoms.8,9 In one retrospective review, 52 of 75 (69%) patients reporting to a California poison center with stingray injuries had improvement in pain within 1 hour of hot water immersion before any analgesics were instituted.8 In another review, 65 of 74 (88%) patients presenting to a California emergency department within 24 hours of sustaining a stingray injury had complete relief of pain within 30 minutes of hot water immersion. Patients who received analgesics in addition to hot water immersion did not require a second dose.9 In concordance with these studies, we suggest immersing areas affected by stingray injuries in hot water (temperature, 43.3°C to 46.1°C [110°F–115°F]; or as close to this range as tolerated) until pain subsides.8,9,14 Ice packs are an alternative to hot water immersion that may be more readily available to patients. If pain does not resolve following hot water immersion or application of an ice pack, additional analgesics and xylocaine without epinephrine may be helpful.9,15
Infection
One major complication of stingray injuries is infection.8,9 Many bacterial species reside in stingray mucus, the marine environment, or on human skin that may be introduced during a single injury. Marine envenomations can involve organisms such as Vibrio, Aeromonas, and Mycobacterium species, which often are resistant to antibiotic prophylaxis covering common causes of soft-tissue infection such as Staphylococcus and Streptococcus species.8,9,16,17 Additionally, physicians should cover for Clostridium species and ensure patients are up-to-date on vaccinations because severe cases of tetanus following stingray injuries have been reported.18 Lastly, fungal infections including fusariosis have been reported following stingray injuries and should be considered if a patient develops an infection.19
Several authors support the use of prophylactic broad-spectrum antibiotics in all but mild stingray injuries.8,9,20,21 Although no standardized definition exists, mild injuries generally represent patients with superficial lacerations or less, while deeper lacerations and puncture wounds require prophylaxis. Several authors agree on the use of fluoroquinolone antibiotics (eg, ciprofloxacin 500 mg twice daily) for 5 to 7 days following severe stingray injuries.1,9,13,22 Other proposed antibiotic regimens include trimethoprim-sulfamethoxazole (160/800 mg twice daily) or tetracycline (500 mg 4 times daily) for 7 days.13 Failure of ciprofloxacin therapy after 7 days has been reported, with resolution of infection after treatment with an intravenous cephalosporin for 7 days.20 Failure of trimethoprim-sulfamethoxazole therapy also has been reported, with one case requiring levofloxacin for a much longer course.21 Clinical follow-up remains essential after prescribing prophylactic antibiotics, as resistance is common.
Foreign Bodies
Stingray injuries also are often complicated by foreign bodies or retained spines.3,8 Although these complications are less severe than infection, all wounds should be explored for material under local anesthesia. Furthermore, there has been support for thorough debridement of necrotic tissue with referral to a hand specialist for deeper injuries to the hands as well as referral to a foot and ankle specialist for deeper injuries of the lower extremities.23,24 More serious injuries with penetration of vital structures, such as through the chest or abdomen, require immediate exploration in an operating room.1,24
Imaging
Routine imaging of stingray injuries remains controversial. In a case series of 119 patients presenting to a California emergency department with stingray injuries, Clark et al9 found that radiographs were not helpful. This finding likely is due in part to an inability to detect hypodense material such as integumentary or glandular tissue via radiography.3 However, radiographs have been used to identify retained stingray barbs in select cases in which retained barbs are suspected.2,25 Lastly, ultrasonography potentially may offer a better first choice when a barb is not readily apparent; magnetic resonance imaging may be indicated for more involved areas and for further visualization of suspected hypodense material, though at a higher expense.2,9
Biopsy
Biopsies of stingray injuries are rarely performed, and the findings are not well characterized. One case biopsied 2 months after injury showed a large zone of paucicellular necrosis with superficial ulceration and granulomatous inflammation. The stingray venom was most likely responsible for the pattern of necrosis noted in the biopsy.21
Avoidance and Prevention
Patients traveling to areas of the world inhabited by stingrays should receive counseling on how to avoid injury. Prior to entry, individuals can throw stones or use a long stick to clear their walking or swimming areas of venomous fish.26 Polarized sunglasses may help spot stingrays in shallow water. Furthermore, wading through water with a shuffling gait can help individuals avoid stepping directly on a stingray and also warns stingrays that someone is in the area. Individuals who spend more time in coastal waters or river systems inhabited by stingrays may invest in protective stingray gear such as leg guards or specialized wading boots.26 Lastly, fishermen should be advised to avoid handling stingrays with their hands and instead cut their fishing line to release the fish.
- Aurbach PS. Envenomations by aquatic vertebrates. In: Auerbach PS. Wilderness Medicine. 5th ed. St. Louis, MO: Mosby; 2007:1730-1749.
- Robins CR, Ray GC. A Field Guide to Atlantic Coast Fishes. New York, NY: Houghton Mifflin Company; 1986.
- Diaz JH. The evaluation, management, and prevention of stingray injuries in travelers. J Travel Med. 2008;15:102-109.
- Haddad V Jr, Neto DG, de Paula Neto JB, et al. Freshwater stingrays: study of epidemiologic, clinical and therapeutic aspects based on 84 envenomings in humans and some enzymatic activities of the venom. Toxicon. 2004;43:287-294.
- Marinkelle CJ. Accidents by venomous animals in Colombia. Ind Med Surg. 1966;35:988-992.
- Last PR, White WT, Caire JN, et al. Sharks and Rays of Borneo. Collingwood VIC, Australia: CSIRO Publishing; 2010.
- Mebs D. Venomous and Poisonous Animals: A Handbook for Biologists, Toxicologists and Toxinologists, Physicians and Pharmacists. Boca Raton, FL: CRC Press; 2002.
- Clark AT, Clark RF, Cantrell FL. A retrospective review of the presentation and treatment of stingray stings reported to a poison control system. Am J Ther. 2017;24:E177-E180.
- Clark RF, Girard RH, Rao D, et al. Stingray envenomation: a retrospective review of clinical presentation and treatment in 119 cases. J Emerg Med. 2007;33:33-37.
- Mahjoubi L, Joyeux A, Delambre JF, et al. Near-death thoracic trauma caused by a stingray in the Indian Ocean. Semin Thorac Cardiovasc Surg. 2017;29:262-263.
- Parra MW, Constantini EN, Rodas EB. Surviving a transfixing cardiac injury caused by a stingray barb. J Thorac Cardiovasc Surg. 2010;139:E115-E116.
- Dos Santos JC, Grund LZ, Seibert CS, et al. Stingray venom activates IL-33 producing cardiomyocytes, but not mast cell, to promote acute neutrophil-mediated injury. Sci Rep. 2017;7:7912.
- Auerbach PS, Norris RL. Marine envenomation. In: Longo DL, Kasper SL, Jameson JL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:144-148.
- Cook MD, Matteucci MJ, Lall R, et al. Stingray envenomation. J Emerg Med. 2006;30:345-347.
- Bowers RC, Mustain MV. Disorders due to physical & environmental agents. In: Humphries RL, Stone C, eds. CURRENT Diagnosis & Treatment Emergency Medicine. 7th ed. New York, NY: McGraw-Hill; 2011:835-861.
- Domingos MO, Franzolin MR, dos Anjos MT, et al. The influence of environmental bacteria in freshwater stingray wound-healing. Toxicon. 2011;58:147-153.
- Auerbach PS, Yajko DM, Nassos PS, et al. Bacteriology of the marine environment: implications for clinical therapy. Ann Emerg Med. 1987;16:643-649.
- Torrez PP, Quiroga MM, Said R, et al. Tetanus after envenomations caused by freshwater stingrays. Toxicon. 2015;97:32-35.
- Hiemenz JW, Kennedy B, Kwon-Chung KJ. Invasive fusariosis associated with an injury by a stingray barb. J Med Vet Mycol. 1990;28:209-213.
- da Silva NJ Jr, Ferreira KR, Pinto RN, et al. A severe accident caused by an ocellate river stingray (Potamotrygon motoro) in central Brazil: how well do we really understand stingray venom chemistry, envenomation, and therapeutics? Toxins (Basel). 2015;7:2272-2288.
- Tartar D, Limova M, North J. Clinical and histopathologic findings in cutaneous sting ray wounds: a case report. Dermatol Online J. 2013;19:19261.
- Jarvis HC, Matheny LM, Clanton TO. Stingray injury to the webspace of the foot. Orthopedics. 2012;35:E762-E765.
- Trickett R, Whitaker IS, Boyce DE. Sting-ray injuries to the hand: case report, literature review and a suggested algorithm for management. J Plast Reconstruct Aesthet Surg. 2009;62:E270-E273.
- Fernandez I, Valladolid G, Varon J, et al. Encounters with venomous sea-life. J Emerg Med. 2011;40:103-112.
- O’Malley GF, O’Malley RN, Pham O, et al. Retained stingray barb and the importance of imaging. Wilderness Environ Med. 2015;26:375-379.
- How to protect yourself from stingrays. Howcast website. https://www.howcast.com/videos/228034-how-to-protect-yourself-from-stingrays/. Accessed July 12, 2018.
- Aurbach PS. Envenomations by aquatic vertebrates. In: Auerbach PS. Wilderness Medicine. 5th ed. St. Louis, MO: Mosby; 2007:1730-1749.
- Robins CR, Ray GC. A Field Guide to Atlantic Coast Fishes. New York, NY: Houghton Mifflin Company; 1986.
- Diaz JH. The evaluation, management, and prevention of stingray injuries in travelers. J Travel Med. 2008;15:102-109.
- Haddad V Jr, Neto DG, de Paula Neto JB, et al. Freshwater stingrays: study of epidemiologic, clinical and therapeutic aspects based on 84 envenomings in humans and some enzymatic activities of the venom. Toxicon. 2004;43:287-294.
- Marinkelle CJ. Accidents by venomous animals in Colombia. Ind Med Surg. 1966;35:988-992.
- Last PR, White WT, Caire JN, et al. Sharks and Rays of Borneo. Collingwood VIC, Australia: CSIRO Publishing; 2010.
- Mebs D. Venomous and Poisonous Animals: A Handbook for Biologists, Toxicologists and Toxinologists, Physicians and Pharmacists. Boca Raton, FL: CRC Press; 2002.
- Clark AT, Clark RF, Cantrell FL. A retrospective review of the presentation and treatment of stingray stings reported to a poison control system. Am J Ther. 2017;24:E177-E180.
- Clark RF, Girard RH, Rao D, et al. Stingray envenomation: a retrospective review of clinical presentation and treatment in 119 cases. J Emerg Med. 2007;33:33-37.
- Mahjoubi L, Joyeux A, Delambre JF, et al. Near-death thoracic trauma caused by a stingray in the Indian Ocean. Semin Thorac Cardiovasc Surg. 2017;29:262-263.
- Parra MW, Constantini EN, Rodas EB. Surviving a transfixing cardiac injury caused by a stingray barb. J Thorac Cardiovasc Surg. 2010;139:E115-E116.
- Dos Santos JC, Grund LZ, Seibert CS, et al. Stingray venom activates IL-33 producing cardiomyocytes, but not mast cell, to promote acute neutrophil-mediated injury. Sci Rep. 2017;7:7912.
- Auerbach PS, Norris RL. Marine envenomation. In: Longo DL, Kasper SL, Jameson JL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:144-148.
- Cook MD, Matteucci MJ, Lall R, et al. Stingray envenomation. J Emerg Med. 2006;30:345-347.
- Bowers RC, Mustain MV. Disorders due to physical & environmental agents. In: Humphries RL, Stone C, eds. CURRENT Diagnosis & Treatment Emergency Medicine. 7th ed. New York, NY: McGraw-Hill; 2011:835-861.
- Domingos MO, Franzolin MR, dos Anjos MT, et al. The influence of environmental bacteria in freshwater stingray wound-healing. Toxicon. 2011;58:147-153.
- Auerbach PS, Yajko DM, Nassos PS, et al. Bacteriology of the marine environment: implications for clinical therapy. Ann Emerg Med. 1987;16:643-649.
- Torrez PP, Quiroga MM, Said R, et al. Tetanus after envenomations caused by freshwater stingrays. Toxicon. 2015;97:32-35.
- Hiemenz JW, Kennedy B, Kwon-Chung KJ. Invasive fusariosis associated with an injury by a stingray barb. J Med Vet Mycol. 1990;28:209-213.
- da Silva NJ Jr, Ferreira KR, Pinto RN, et al. A severe accident caused by an ocellate river stingray (Potamotrygon motoro) in central Brazil: how well do we really understand stingray venom chemistry, envenomation, and therapeutics? Toxins (Basel). 2015;7:2272-2288.
- Tartar D, Limova M, North J. Clinical and histopathologic findings in cutaneous sting ray wounds: a case report. Dermatol Online J. 2013;19:19261.
- Jarvis HC, Matheny LM, Clanton TO. Stingray injury to the webspace of the foot. Orthopedics. 2012;35:E762-E765.
- Trickett R, Whitaker IS, Boyce DE. Sting-ray injuries to the hand: case report, literature review and a suggested algorithm for management. J Plast Reconstruct Aesthet Surg. 2009;62:E270-E273.
- Fernandez I, Valladolid G, Varon J, et al. Encounters with venomous sea-life. J Emerg Med. 2011;40:103-112.
- O’Malley GF, O’Malley RN, Pham O, et al. Retained stingray barb and the importance of imaging. Wilderness Environ Med. 2015;26:375-379.
- How to protect yourself from stingrays. Howcast website. https://www.howcast.com/videos/228034-how-to-protect-yourself-from-stingrays/. Accessed July 12, 2018.
Practice Points
- Acute pain associated with stingray injuries can be treated with hot water immersion.
- Stingray injuries are prone to secondary infection and poor wound healing.

Solitary Nodule on the Thigh
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
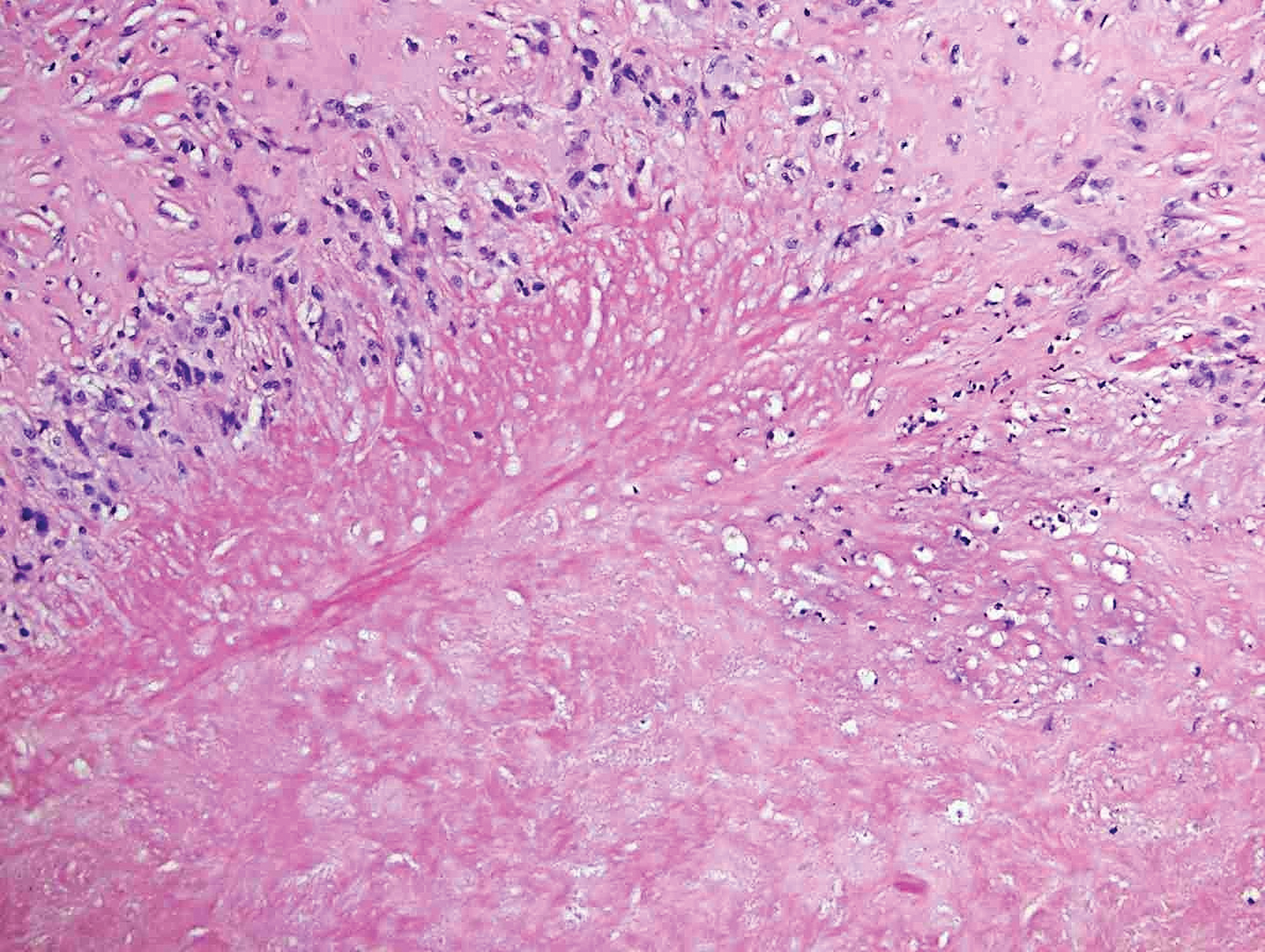
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).

Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
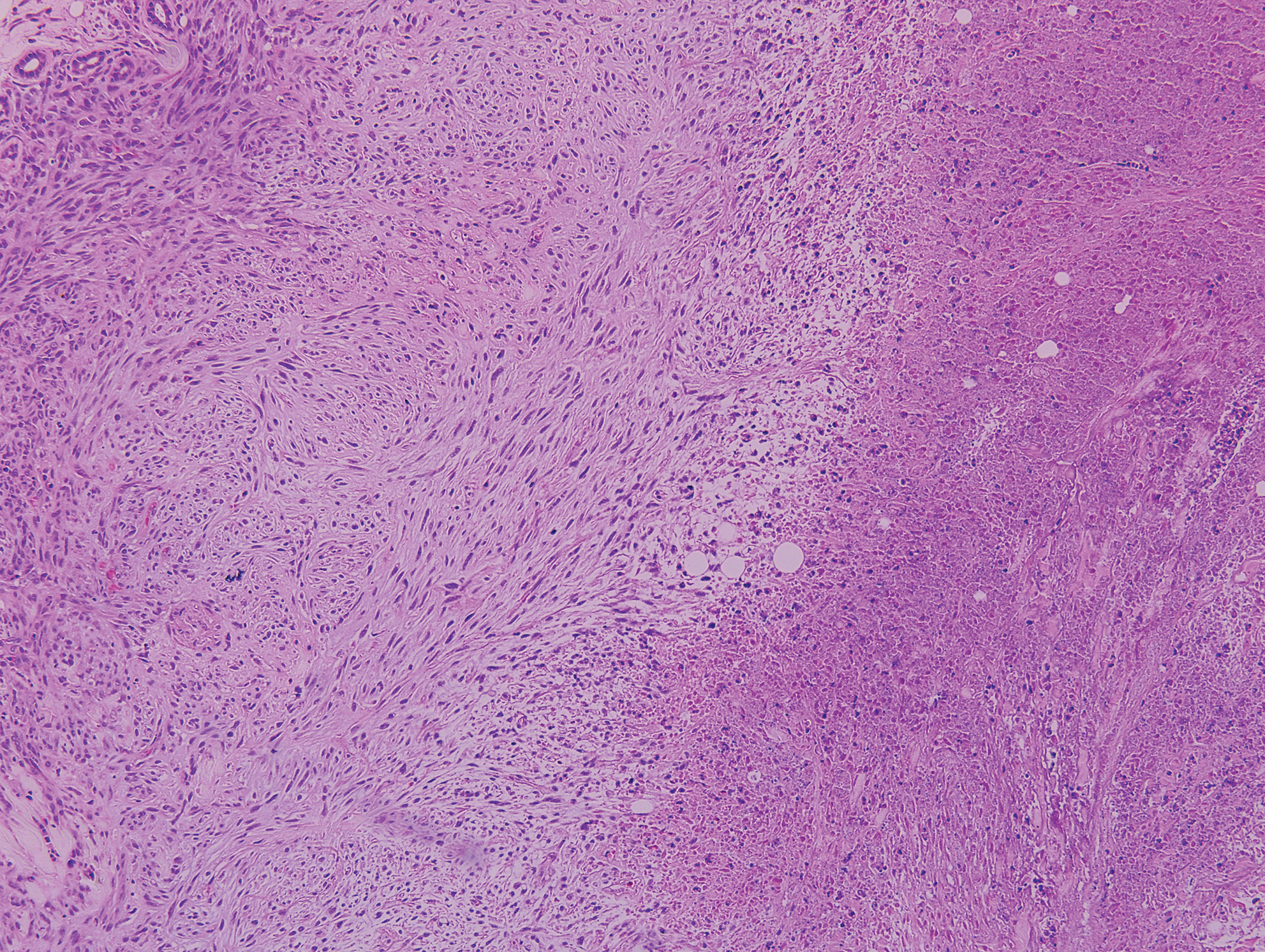
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
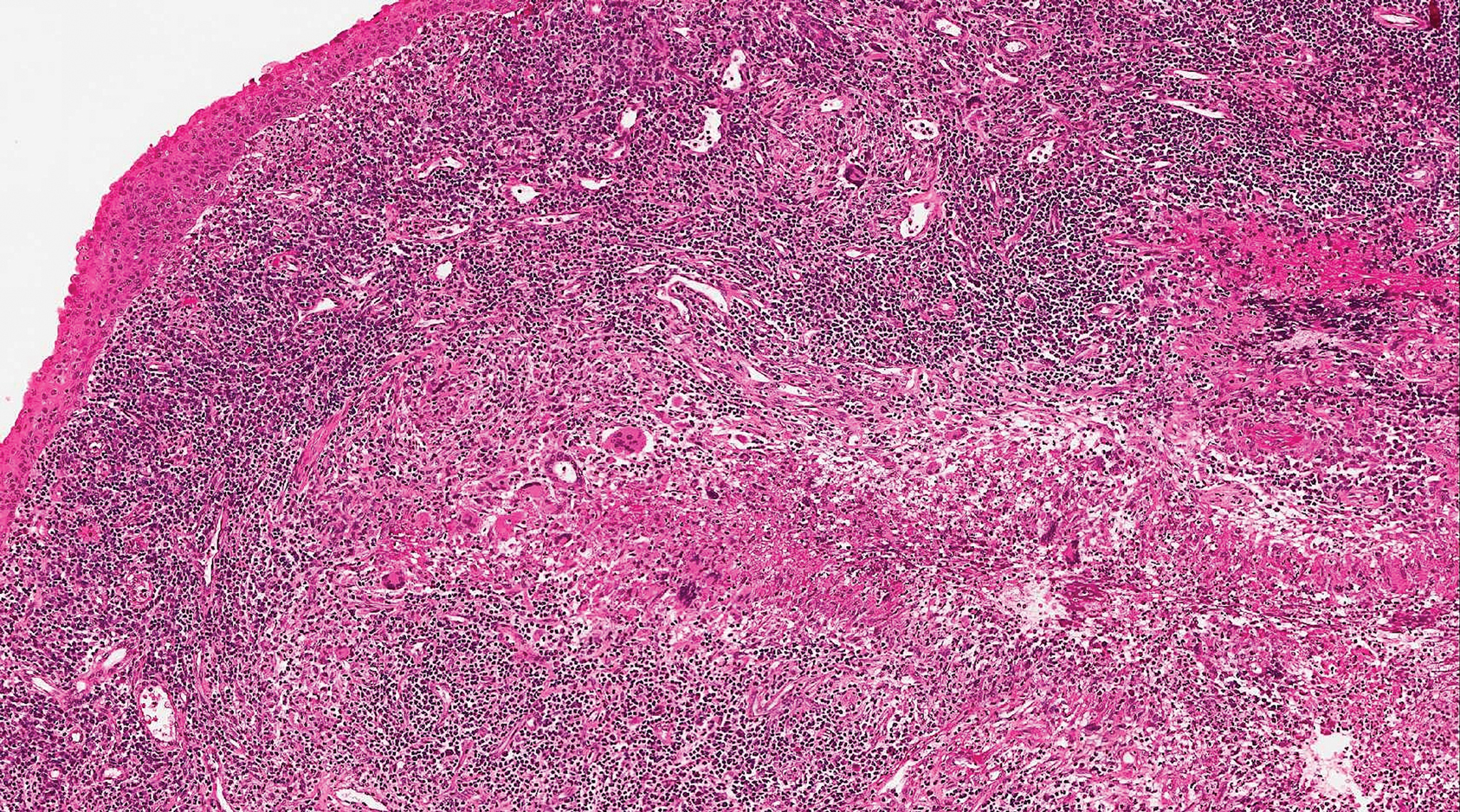
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
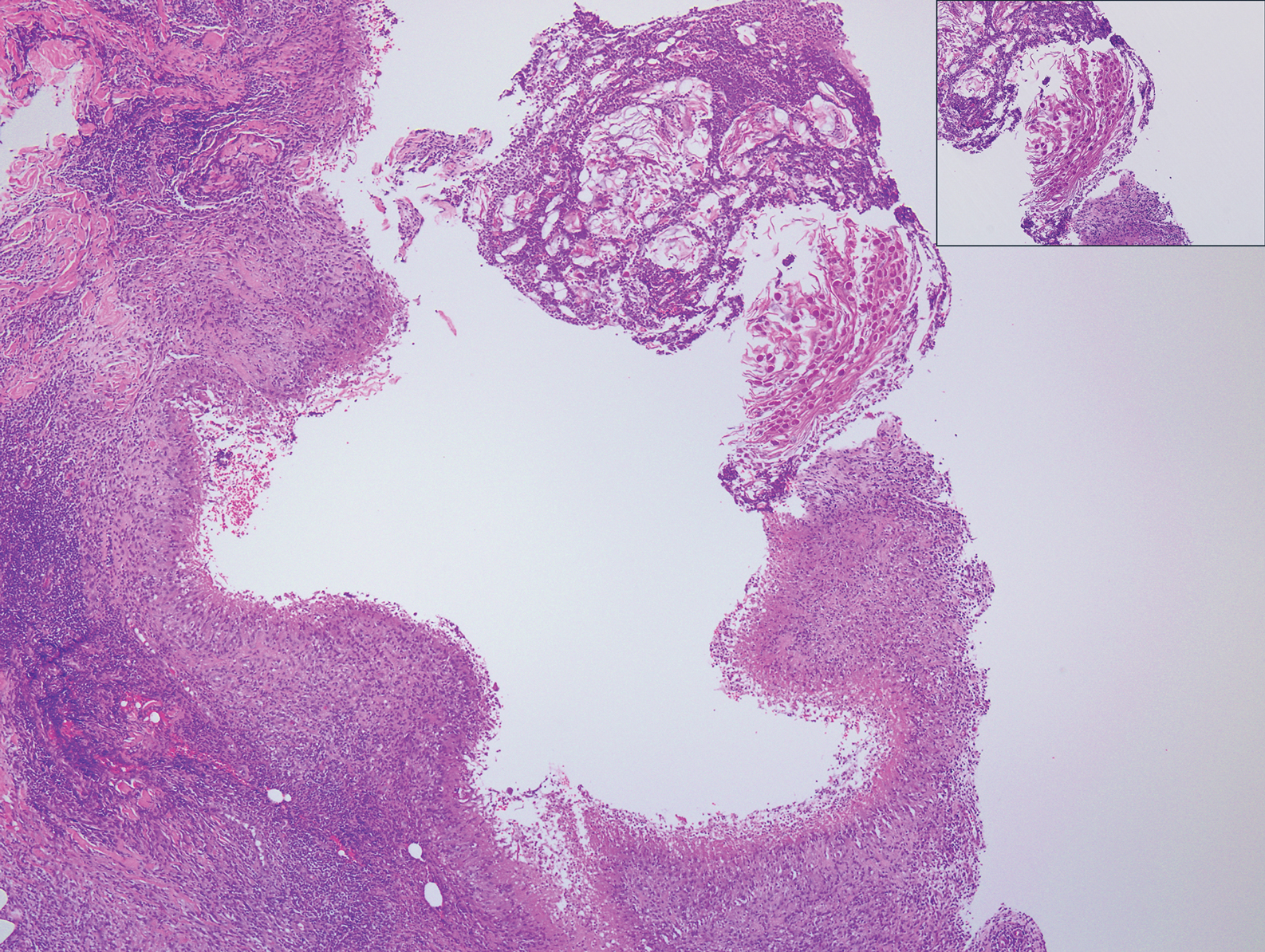
A 17-year-old adolescent girl presented with a discrete nodule on the thigh.
Aquatic Antagonists: Lionfish (Pterois volitans)
The lionfish (Pterois volitans) is a member of the Scorpaenidae family of venomous fish.1-3 Lionfish are an invasive species originally from the Indian and Pacific oceans and the Red Sea that now are widely found throughout tropical and temperate oceans in both hemispheres. They are a popular aquarium fish and were inadvertently introduced in the Atlantic Ocean in South Florida during the late 1980s to early 1990s.2,4 Since then, lionfish have spread into reef systems throughout the Atlantic Ocean, Caribbean Sea, and Gulf of Mexico in rapidly growing numbers, and they are now fo und all along the southeastern coast of the United States.5
Characteristics
Lionfish are brightly colored with red or maroon and white stripes, tentacles above the eyes and mouth, fan-shaped pectoral fins, and spines that deliver an especially painful venomous sting that often results in edema (Figure 1). They have 12 dorsal spines, 2 pelvic spines, and 3 anal spines.

Symptoms of Envenomation
As lionfish continue to spread to popular areas of the southeast Atlantic Ocean and Caribbean Sea, the chances of human contact with lionfish have increased. Lionfish stings are now the second most common marine envenomation injury after those caused by stingrays.4 Lionfish stings usually occur on the hands, fingers, or forearms during handling of the fish in ocean waters or in maintenance of aquariums. The mechanism of the venom apparatus is similar for all venomous fish. The spines have surrounding integumentary sheaths containing venom that rupture and inject venom when they penetrate the skin.6 The venom is a heat-labile neuromuscular toxin that causes edema (Figure 2), plasma extravasation, and thrombotic skin lesions.7
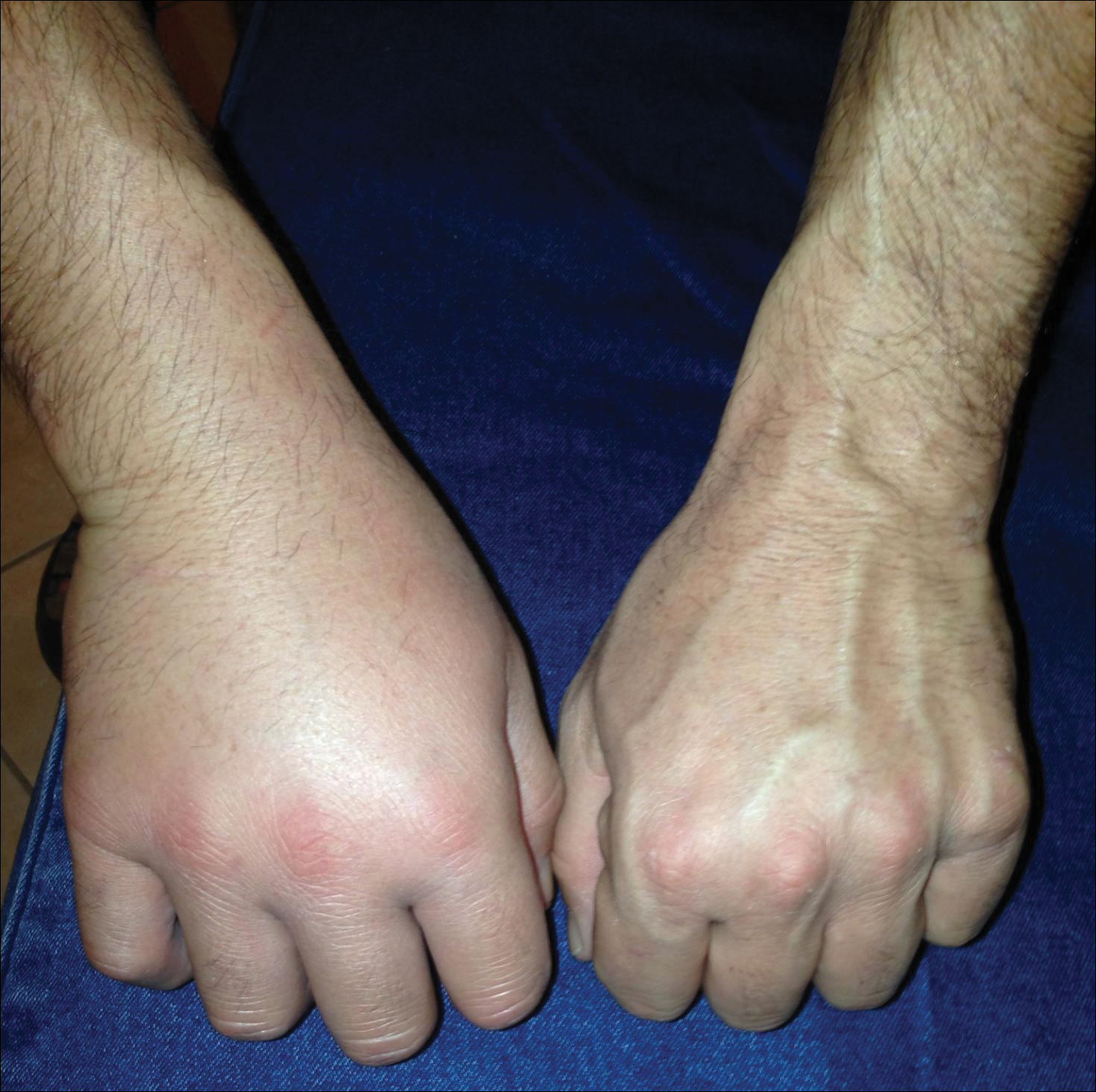
Wounds are classified into 3 categories: grade I consists of local erythema/ecchymosis, grade II involves vesicle or blister formation, and grade III denotes wounds that develop local necrosis.8 The sting causes immediate and severe throbbing pain, often described as excruciating or rated 10/10 on a basic pain scale, typically radiating up the affected limb. Puncture sites may bleed and often have associated redness and swelling. Pain may last up to 24 hours. Occasionally, foreign material may be left in the wound requiring removal. There also is a chance of secondary infection at the wound site, and severe envenomation can lead to local tissue necrosis.8 Systemic effects can occur in some cases, including nausea, vomiting, sweating, headache, dizziness, disorientation, palpitations, and even syncope.9 However, to our knowledge there are no documented cases of human death from a lionfish sting. Anaphylactic reactions are possible and require immediate treatment.6
A study conducted in the French West Indies evaluated 117 patients with lionfish envenomation and found that victims experienced severe pain and local edema (100%), paresthesia (90%), abdominal cramps (62%), extensive edema (53%), tachycardia (34%), skin rash (32%), gastrointestinal tract symptoms (28%), syncope (27%), transient weakness (24%), hypertension (21%), hypotension (18%), and hyperthermia (9%).9 Complications included local infection (18%) such as skin abscess (5%), skin necrosis (3%), and septic arthritis (2%). Twenty-two percent of patients were hospitalized and 8% required surgery. Local infectious complications were more frequent in those with multiple stings (19%). The study concluded that lionfish now represent a major health threat in the West Indies.9 As lionfish numbers have grown, health care providers are seeing increasing numbers of envenomation cases in areas of the coastal southeastern United States and Caribbean associated with considerable morbidity. Providers in nonendemic areas also may see envenomation injuries due to the lionfish popularity in home aquariums.9
Management
Individuals with lionfish stings should immerse the affected area in hot but not scalding water. Those with more serious injuries should seek medical attention. Home remedies that are generally contraindicated include application of topical papain or meat tenderizer.10 Data on ice packs are mixed, but because the toxin is heat labile, the most effective initial step in treatment is immersion of the affected area in water (temperature, 40°C to 45°C) for 30 to 90 minutes.6 The hot water inactivates the heat-labile toxin, leading to near-complete symptomatic relief in 80% of cases and moderate relief in an additional 14%. Immersion time more than 90 minutes considerably increases the risk for burns. Children should always be monitored to prevent burns. If a patient has received a nerve block for analgesia, the wound should not be immersed in hot water to avoid burns to the skin. The wound should be meticulously cleaned with saline irrigation, and radiography or ultrasonography should be performed as deemed necessary to look for any retained foreign bodies.8 Patients may require parenteral or oral analgesia as well as careful follow-up to ensure proper healing.9 Systemic symptoms require supportive care. Venomous fish wounds typically are small and superficial. Empiric antibiotic therapy is not advised for superficial wounds but may be required for clinically infected wounds.8 Tetanus prophylaxis should be given as appropriate to all affected patients. It has been noted that blister fluid contains high concentrations of lionfish venom, and when present, it increases the likelihood of converting the injury from a grade II to grade III wound with tissue necrosis; therefore, blisters should be drained or excised to decrease the chances of subsequent tissue necrosis.11,12 If secondary infection such as cellulitis develops, antibiotics should be chosen to cover likely pathogens including common skin flora such as staphylococci and marine organisms such as Vibrio species. Wounds showing signs of infection should be cultured, with antibiotics adjusted according to sensitivities.5 Deeper wounds should be left open (unsutured) with a proper dressing to heal. Any wounds that involve vascular or joint structures require specialty management. Wounds involving joints may on occasion require surgical exploration and debridement.
Public Health Concerns
In an attempt to slow the growth of their population, human consumption of the fish has been encouraged. The lionfish toxin is inactivated by cooking, and the fish is considered a delicacy; however, a study in the Virgin Islands found that in areas with endemic ciguatera poisoning, 12% of lionfish carried amounts of the toxin above the level considered safe for consumption. This toxin is not inactivated by cooking or freezing and can lead to ciguatera fish poisoning for which there is no antidote and can be associated with prolonged neurotoxicity.13
Conclusion
As lionfish numbers continue to increase, physicians across multiple specialties and regions may see an increase in envenomation injuries. It is important that physicians are aware of how to recognize and treat lionfish stings, as prompt and comprehensive treatment provides benefit to the patient.
- Pterois volitans. Integrated Taxonomic Information System website. https://www.itis.gov/servlet/SingleRpt/SingleRpt?search_topic=TSN&search_value=166883#null. Accessed September 6, 2018.
- Morris JA Jr, Whitfield PE. Biology, Ecology, Control and Management of the Invasive Indopacific Lionfish: An Updated Integrated Assessment. Beaufort, NC: National Oceanic and Atmospheric Administration; 2009. http://aquaticcommons.org/2847/1/NCCOS_TM_99.pdf. Accessed September 6, 2018.
- Pterois volitans/miles. US Geological Survey website. https://nas.er.usgs.gov/queries/FactSheet.aspx?speciesID=963. Revised April 18, 2018. Accessed September 6, 2018.
- Diaz JH. Invasive lionfish (Pterois volitans) pose public health threats [published online August 15, 2015]. J La State Med Soc. 2015;167:166-171.
- Diaz JH. Marine Scorpaenidae envenomation in travelers: epidemiology, management, and prevention. J Travel Med. 2015;22:251-258.
- Hobday D, Chadha P, Din AH, et al. Denaturing the lionfish. Eplasty. 2016;16:ic20.
- Sáenz A, Ortiz N, Lomonte B, et al. Comparison of biochemical and cytotoxic activities of extracts obtained from dorsal spines and caudal fin of adult and juvenile non-native Caribbean lionfish (Pterois volitans/miles). Toxicon. 2017;137:158-167.
- Schult RF, Acquisto NM, Stair CK, et al. A case of lionfish envenomation presenting to an inland emergency department [published online August 13, 2017]. Case Rep Emerg Med. 2017;2017:5893563.
- Resiere D, Cerland L, De Haro L, et al. Envenomation by the invasive Pterois volitans species (lionfish) in the French West Indies—a two-year prospective study in Martinique. Clin Toxicol (Phila). 2016;54:313-318.
- Auerbach PS. Envenomation by aquatic vertebrates. In: Auerback PS. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby Elsevier; 2007:1740-1741.
- Auerbach PS, McKinney HE, Rees RE, et al. Analysis of vesicle fluid following the sting of the lionfish, Pterois volitans. Toxicon. 1987;25:1350-1353.
- Patel MR, Wells S. Lionfish envenomation of the hand. J Hand Surg Am. 1993;18:523-525.
- Robertson A, Garcia AC, Quintana HA, et al. Invasive lionfish (Pterois volitans): a potential human health threat for Ciguatera fish poisoning in tropical waters. Marine Drugs. 2014;12:88-97.
The lionfish (Pterois volitans) is a member of the Scorpaenidae family of venomous fish.1-3 Lionfish are an invasive species originally from the Indian and Pacific oceans and the Red Sea that now are widely found throughout tropical and temperate oceans in both hemispheres. They are a popular aquarium fish and were inadvertently introduced in the Atlantic Ocean in South Florida during the late 1980s to early 1990s.2,4 Since then, lionfish have spread into reef systems throughout the Atlantic Ocean, Caribbean Sea, and Gulf of Mexico in rapidly growing numbers, and they are now fo und all along the southeastern coast of the United States.5
Characteristics
Lionfish are brightly colored with red or maroon and white stripes, tentacles above the eyes and mouth, fan-shaped pectoral fins, and spines that deliver an especially painful venomous sting that often results in edema (Figure 1). They have 12 dorsal spines, 2 pelvic spines, and 3 anal spines.
Symptoms of Envenomation
As lionfish continue to spread to popular areas of the southeast Atlantic Ocean and Caribbean Sea, the chances of human contact with lionfish have increased. Lionfish stings are now the second most common marine envenomation injury after those caused by stingrays.4 Lionfish stings usually occur on the hands, fingers, or forearms during handling of the fish in ocean waters or in maintenance of aquariums. The mechanism of the venom apparatus is similar for all venomous fish. The spines have surrounding integumentary sheaths containing venom that rupture and inject venom when they penetrate the skin.6 The venom is a heat-labile neuromuscular toxin that causes edema (Figure 2), plasma extravasation, and thrombotic skin lesions.7
Wounds are classified into 3 categories: grade I consists of local erythema/ecchymosis, grade II involves vesicle or blister formation, and grade III denotes wounds that develop local necrosis.8 The sting causes immediate and severe throbbing pain, often described as excruciating or rated 10/10 on a basic pain scale, typically radiating up the affected limb. Puncture sites may bleed and often have associated redness and swelling. Pain may last up to 24 hours. Occasionally, foreign material may be left in the wound requiring removal. There also is a chance of secondary infection at the wound site, and severe envenomation can lead to local tissue necrosis.8 Systemic effects can occur in some cases, including nausea, vomiting, sweating, headache, dizziness, disorientation, palpitations, and even syncope.9 However, to our knowledge there are no documented cases of human death from a lionfish sting. Anaphylactic reactions are possible and require immediate treatment.6
A study conducted in the French West Indies evaluated 117 patients with lionfish envenomation and found that victims experienced severe pain and local edema (100%), paresthesia (90%), abdominal cramps (62%), extensive edema (53%), tachycardia (34%), skin rash (32%), gastrointestinal tract symptoms (28%), syncope (27%), transient weakness (24%), hypertension (21%), hypotension (18%), and hyperthermia (9%).9 Complications included local infection (18%) such as skin abscess (5%), skin necrosis (3%), and septic arthritis (2%). Twenty-two percent of patients were hospitalized and 8% required surgery. Local infectious complications were more frequent in those with multiple stings (19%). The study concluded that lionfish now represent a major health threat in the West Indies.9 As lionfish numbers have grown, health care providers are seeing increasing numbers of envenomation cases in areas of the coastal southeastern United States and Caribbean associated with considerable morbidity. Providers in nonendemic areas also may see envenomation injuries due to the lionfish popularity in home aquariums.9
Management
Individuals with lionfish stings should immerse the affected area in hot but not scalding water. Those with more serious injuries should seek medical attention. Home remedies that are generally contraindicated include application of topical papain or meat tenderizer.10 Data on ice packs are mixed, but because the toxin is heat labile, the most effective initial step in treatment is immersion of the affected area in water (temperature, 40°C to 45°C) for 30 to 90 minutes.6 The hot water inactivates the heat-labile toxin, leading to near-complete symptomatic relief in 80% of cases and moderate relief in an additional 14%. Immersion time more than 90 minutes considerably increases the risk for burns. Children should always be monitored to prevent burns. If a patient has received a nerve block for analgesia, the wound should not be immersed in hot water to avoid burns to the skin. The wound should be meticulously cleaned with saline irrigation, and radiography or ultrasonography should be performed as deemed necessary to look for any retained foreign bodies.8 Patients may require parenteral or oral analgesia as well as careful follow-up to ensure proper healing.9 Systemic symptoms require supportive care. Venomous fish wounds typically are small and superficial. Empiric antibiotic therapy is not advised for superficial wounds but may be required for clinically infected wounds.8 Tetanus prophylaxis should be given as appropriate to all affected patients. It has been noted that blister fluid contains high concentrations of lionfish venom, and when present, it increases the likelihood of converting the injury from a grade II to grade III wound with tissue necrosis; therefore, blisters should be drained or excised to decrease the chances of subsequent tissue necrosis.11,12 If secondary infection such as cellulitis develops, antibiotics should be chosen to cover likely pathogens including common skin flora such as staphylococci and marine organisms such as Vibrio species. Wounds showing signs of infection should be cultured, with antibiotics adjusted according to sensitivities.5 Deeper wounds should be left open (unsutured) with a proper dressing to heal. Any wounds that involve vascular or joint structures require specialty management. Wounds involving joints may on occasion require surgical exploration and debridement.
Public Health Concerns
In an attempt to slow the growth of their population, human consumption of the fish has been encouraged. The lionfish toxin is inactivated by cooking, and the fish is considered a delicacy; however, a study in the Virgin Islands found that in areas with endemic ciguatera poisoning, 12% of lionfish carried amounts of the toxin above the level considered safe for consumption. This toxin is not inactivated by cooking or freezing and can lead to ciguatera fish poisoning for which there is no antidote and can be associated with prolonged neurotoxicity.13
Conclusion
As lionfish numbers continue to increase, physicians across multiple specialties and regions may see an increase in envenomation injuries. It is important that physicians are aware of how to recognize and treat lionfish stings, as prompt and comprehensive treatment provides benefit to the patient.
The lionfish (Pterois volitans) is a member of the Scorpaenidae family of venomous fish.1-3 Lionfish are an invasive species originally from the Indian and Pacific oceans and the Red Sea that now are widely found throughout tropical and temperate oceans in both hemispheres. They are a popular aquarium fish and were inadvertently introduced in the Atlantic Ocean in South Florida during the late 1980s to early 1990s.2,4 Since then, lionfish have spread into reef systems throughout the Atlantic Ocean, Caribbean Sea, and Gulf of Mexico in rapidly growing numbers, and they are now fo und all along the southeastern coast of the United States.5
Characteristics
Lionfish are brightly colored with red or maroon and white stripes, tentacles above the eyes and mouth, fan-shaped pectoral fins, and spines that deliver an especially painful venomous sting that often results in edema (Figure 1). They have 12 dorsal spines, 2 pelvic spines, and 3 anal spines.
Symptoms of Envenomation
As lionfish continue to spread to popular areas of the southeast Atlantic Ocean and Caribbean Sea, the chances of human contact with lionfish have increased. Lionfish stings are now the second most common marine envenomation injury after those caused by stingrays.4 Lionfish stings usually occur on the hands, fingers, or forearms during handling of the fish in ocean waters or in maintenance of aquariums. The mechanism of the venom apparatus is similar for all venomous fish. The spines have surrounding integumentary sheaths containing venom that rupture and inject venom when they penetrate the skin.6 The venom is a heat-labile neuromuscular toxin that causes edema (Figure 2), plasma extravasation, and thrombotic skin lesions.7
Wounds are classified into 3 categories: grade I consists of local erythema/ecchymosis, grade II involves vesicle or blister formation, and grade III denotes wounds that develop local necrosis.8 The sting causes immediate and severe throbbing pain, often described as excruciating or rated 10/10 on a basic pain scale, typically radiating up the affected limb. Puncture sites may bleed and often have associated redness and swelling. Pain may last up to 24 hours. Occasionally, foreign material may be left in the wound requiring removal. There also is a chance of secondary infection at the wound site, and severe envenomation can lead to local tissue necrosis.8 Systemic effects can occur in some cases, including nausea, vomiting, sweating, headache, dizziness, disorientation, palpitations, and even syncope.9 However, to our knowledge there are no documented cases of human death from a lionfish sting. Anaphylactic reactions are possible and require immediate treatment.6
A study conducted in the French West Indies evaluated 117 patients with lionfish envenomation and found that victims experienced severe pain and local edema (100%), paresthesia (90%), abdominal cramps (62%), extensive edema (53%), tachycardia (34%), skin rash (32%), gastrointestinal tract symptoms (28%), syncope (27%), transient weakness (24%), hypertension (21%), hypotension (18%), and hyperthermia (9%).9 Complications included local infection (18%) such as skin abscess (5%), skin necrosis (3%), and septic arthritis (2%). Twenty-two percent of patients were hospitalized and 8% required surgery. Local infectious complications were more frequent in those with multiple stings (19%). The study concluded that lionfish now represent a major health threat in the West Indies.9 As lionfish numbers have grown, health care providers are seeing increasing numbers of envenomation cases in areas of the coastal southeastern United States and Caribbean associated with considerable morbidity. Providers in nonendemic areas also may see envenomation injuries due to the lionfish popularity in home aquariums.9
Management
Individuals with lionfish stings should immerse the affected area in hot but not scalding water. Those with more serious injuries should seek medical attention. Home remedies that are generally contraindicated include application of topical papain or meat tenderizer.10 Data on ice packs are mixed, but because the toxin is heat labile, the most effective initial step in treatment is immersion of the affected area in water (temperature, 40°C to 45°C) for 30 to 90 minutes.6 The hot water inactivates the heat-labile toxin, leading to near-complete symptomatic relief in 80% of cases and moderate relief in an additional 14%. Immersion time more than 90 minutes considerably increases the risk for burns. Children should always be monitored to prevent burns. If a patient has received a nerve block for analgesia, the wound should not be immersed in hot water to avoid burns to the skin. The wound should be meticulously cleaned with saline irrigation, and radiography or ultrasonography should be performed as deemed necessary to look for any retained foreign bodies.8 Patients may require parenteral or oral analgesia as well as careful follow-up to ensure proper healing.9 Systemic symptoms require supportive care. Venomous fish wounds typically are small and superficial. Empiric antibiotic therapy is not advised for superficial wounds but may be required for clinically infected wounds.8 Tetanus prophylaxis should be given as appropriate to all affected patients. It has been noted that blister fluid contains high concentrations of lionfish venom, and when present, it increases the likelihood of converting the injury from a grade II to grade III wound with tissue necrosis; therefore, blisters should be drained or excised to decrease the chances of subsequent tissue necrosis.11,12 If secondary infection such as cellulitis develops, antibiotics should be chosen to cover likely pathogens including common skin flora such as staphylococci and marine organisms such as Vibrio species. Wounds showing signs of infection should be cultured, with antibiotics adjusted according to sensitivities.5 Deeper wounds should be left open (unsutured) with a proper dressing to heal. Any wounds that involve vascular or joint structures require specialty management. Wounds involving joints may on occasion require surgical exploration and debridement.
Public Health Concerns
In an attempt to slow the growth of their population, human consumption of the fish has been encouraged. The lionfish toxin is inactivated by cooking, and the fish is considered a delicacy; however, a study in the Virgin Islands found that in areas with endemic ciguatera poisoning, 12% of lionfish carried amounts of the toxin above the level considered safe for consumption. This toxin is not inactivated by cooking or freezing and can lead to ciguatera fish poisoning for which there is no antidote and can be associated with prolonged neurotoxicity.13
Conclusion
As lionfish numbers continue to increase, physicians across multiple specialties and regions may see an increase in envenomation injuries. It is important that physicians are aware of how to recognize and treat lionfish stings, as prompt and comprehensive treatment provides benefit to the patient.
- Pterois volitans. Integrated Taxonomic Information System website. https://www.itis.gov/servlet/SingleRpt/SingleRpt?search_topic=TSN&search_value=166883#null. Accessed September 6, 2018.
- Morris JA Jr, Whitfield PE. Biology, Ecology, Control and Management of the Invasive Indopacific Lionfish: An Updated Integrated Assessment. Beaufort, NC: National Oceanic and Atmospheric Administration; 2009. http://aquaticcommons.org/2847/1/NCCOS_TM_99.pdf. Accessed September 6, 2018.
- Pterois volitans/miles. US Geological Survey website. https://nas.er.usgs.gov/queries/FactSheet.aspx?speciesID=963. Revised April 18, 2018. Accessed September 6, 2018.
- Diaz JH. Invasive lionfish (Pterois volitans) pose public health threats [published online August 15, 2015]. J La State Med Soc. 2015;167:166-171.
- Diaz JH. Marine Scorpaenidae envenomation in travelers: epidemiology, management, and prevention. J Travel Med. 2015;22:251-258.
- Hobday D, Chadha P, Din AH, et al. Denaturing the lionfish. Eplasty. 2016;16:ic20.
- Sáenz A, Ortiz N, Lomonte B, et al. Comparison of biochemical and cytotoxic activities of extracts obtained from dorsal spines and caudal fin of adult and juvenile non-native Caribbean lionfish (Pterois volitans/miles). Toxicon. 2017;137:158-167.
- Schult RF, Acquisto NM, Stair CK, et al. A case of lionfish envenomation presenting to an inland emergency department [published online August 13, 2017]. Case Rep Emerg Med. 2017;2017:5893563.
- Resiere D, Cerland L, De Haro L, et al. Envenomation by the invasive Pterois volitans species (lionfish) in the French West Indies—a two-year prospective study in Martinique. Clin Toxicol (Phila). 2016;54:313-318.
- Auerbach PS. Envenomation by aquatic vertebrates. In: Auerback PS. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby Elsevier; 2007:1740-1741.
- Auerbach PS, McKinney HE, Rees RE, et al. Analysis of vesicle fluid following the sting of the lionfish, Pterois volitans. Toxicon. 1987;25:1350-1353.
- Patel MR, Wells S. Lionfish envenomation of the hand. J Hand Surg Am. 1993;18:523-525.
- Robertson A, Garcia AC, Quintana HA, et al. Invasive lionfish (Pterois volitans): a potential human health threat for Ciguatera fish poisoning in tropical waters. Marine Drugs. 2014;12:88-97.
- Pterois volitans. Integrated Taxonomic Information System website. https://www.itis.gov/servlet/SingleRpt/SingleRpt?search_topic=TSN&search_value=166883#null. Accessed September 6, 2018.
- Morris JA Jr, Whitfield PE. Biology, Ecology, Control and Management of the Invasive Indopacific Lionfish: An Updated Integrated Assessment. Beaufort, NC: National Oceanic and Atmospheric Administration; 2009. http://aquaticcommons.org/2847/1/NCCOS_TM_99.pdf. Accessed September 6, 2018.
- Pterois volitans/miles. US Geological Survey website. https://nas.er.usgs.gov/queries/FactSheet.aspx?speciesID=963. Revised April 18, 2018. Accessed September 6, 2018.
- Diaz JH. Invasive lionfish (Pterois volitans) pose public health threats [published online August 15, 2015]. J La State Med Soc. 2015;167:166-171.
- Diaz JH. Marine Scorpaenidae envenomation in travelers: epidemiology, management, and prevention. J Travel Med. 2015;22:251-258.
- Hobday D, Chadha P, Din AH, et al. Denaturing the lionfish. Eplasty. 2016;16:ic20.
- Sáenz A, Ortiz N, Lomonte B, et al. Comparison of biochemical and cytotoxic activities of extracts obtained from dorsal spines and caudal fin of adult and juvenile non-native Caribbean lionfish (Pterois volitans/miles). Toxicon. 2017;137:158-167.
- Schult RF, Acquisto NM, Stair CK, et al. A case of lionfish envenomation presenting to an inland emergency department [published online August 13, 2017]. Case Rep Emerg Med. 2017;2017:5893563.
- Resiere D, Cerland L, De Haro L, et al. Envenomation by the invasive Pterois volitans species (lionfish) in the French West Indies—a two-year prospective study in Martinique. Clin Toxicol (Phila). 2016;54:313-318.
- Auerbach PS. Envenomation by aquatic vertebrates. In: Auerback PS. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby Elsevier; 2007:1740-1741.
- Auerbach PS, McKinney HE, Rees RE, et al. Analysis of vesicle fluid following the sting of the lionfish, Pterois volitans. Toxicon. 1987;25:1350-1353.
- Patel MR, Wells S. Lionfish envenomation of the hand. J Hand Surg Am. 1993;18:523-525.
- Robertson A, Garcia AC, Quintana HA, et al. Invasive lionfish (Pterois volitans): a potential human health threat for Ciguatera fish poisoning in tropical waters. Marine Drugs. 2014;12:88-97.
Practice Points
- Lionfish are now found all along the southeastern coast of the United States. Physicians may see an increase in envenomation injuries.
- Treat lionfish envenomation with immediate immersion in warm water (temperature, 40°C to 45°C) for 30 to 90 minutes to deactivate heat-labile toxin.
- Infected wounds should be treated with antibiotics for common skin flora and marine organisms such as Vibrio species.
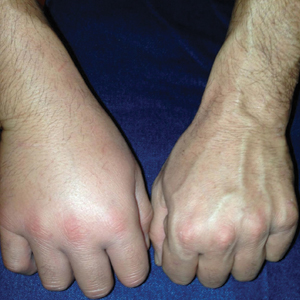
Adult-Onset Still Disease: Persistent Pruritic Papular Rash With Unique Histopathologic Findings
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
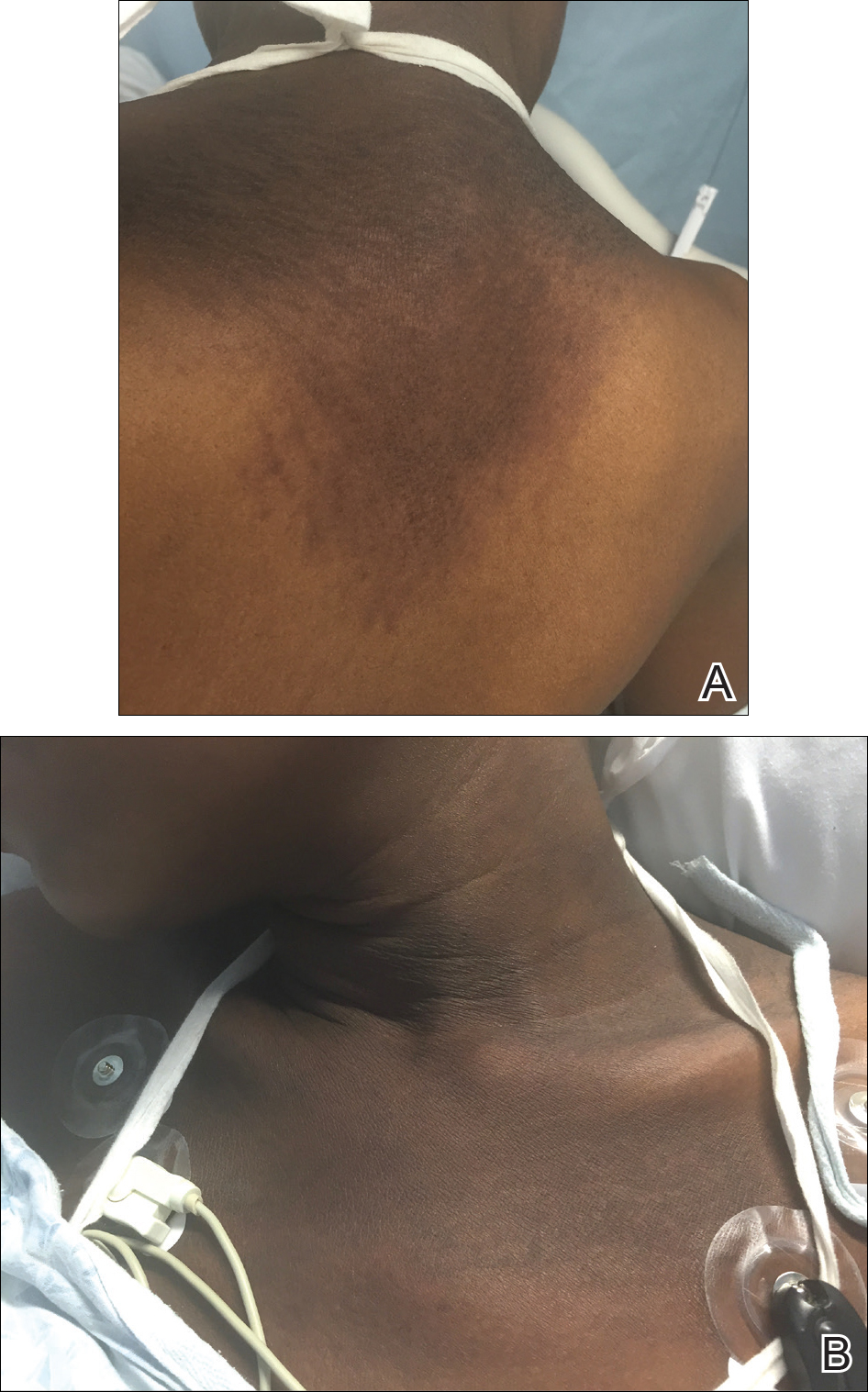
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
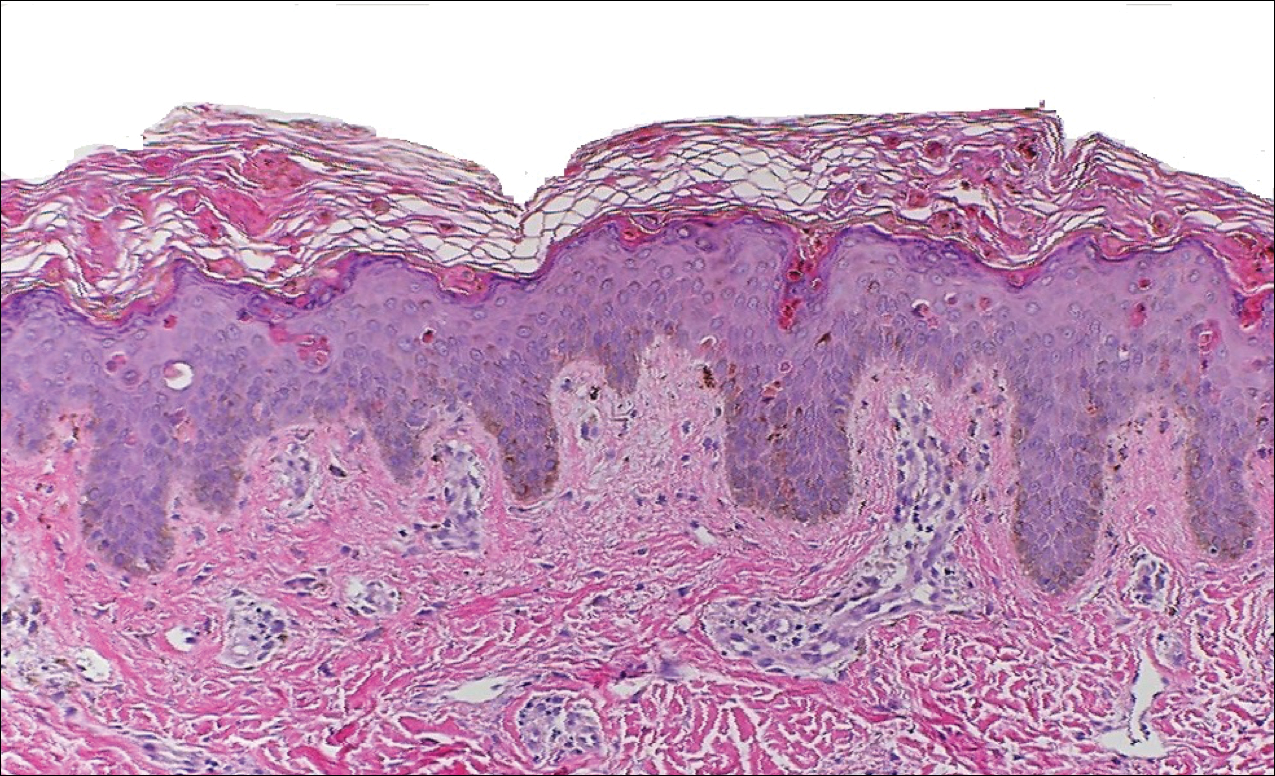
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
Adult-onset Still disease (AOSD) is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, evanescent skin rash, and lymphadenopathy. 1 The most commonly used criteria for diagnosing AOSD are the Yamaguchi criteria. 2 The major criteria include high fever for more than 1 week, arthralgia for more than 2 weeks, leukocytosis, and an evanescent skin rash. The minor criteria consist of sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and negative rheumatoid factor and antinuclear antibodies. Classically, the skin rash is described as an evanescent, salmon-colored erythema involving the extremities. Nevertheless, unusual cutaneous eruptions have been reported in AOSD, including persistent pruritic papules and plaques. 3 Importantly, this atypical rash demonstrates specific histologic findings that are not found on routine histopathology of a typical evanescent rash. We describe 2 patients with this atypical cutaneous eruption along with the unique histopathologic findings of AOSD.
Case Reports
Patient 1
A 23-year-old Chinese woman presented with periodic fevers, persistent rash, and joint pain of 2 years’ duration. Her medical history included splenectomy for hepatosplenomegaly as well as evaluation by hematology for lymphadenopathy; a cervical lymph node biopsy showed lymphoid and follicular hyperplasia.
Twenty days later, the patient was referred to the dermatology department for evaluation of the persistent rash. The patient described a history of flushing of the face, severe joint pain in both arms and legs, aching muscles, and persistent sore throat. The patient did not report any history of drug ingestion. Physical examination revealed a fever (temperature, 39.2°C); swollen nontender lymph nodes in the neck, axillae, and groin; and salmon-colored and hyperpigmented patches and thin plaques over the neck, chest, abdomen, and arms (Figure 1). A splenectomy scar also was noted. Peripheral blood was collected for laboratory analyses, which revealed transaminitis and moderate hyperferritinemia (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. The patient was admitted to the hospital, and a skin biopsy was performed. Histology showed superficial dyskeratotic keratinocytes and sparse perivascular infiltration of neutrophils in the upper dermis (Figure 2).
The patient was diagnosed with AOSD based on fulfillment of the Yamaguchi criteria.2 She was treated with methylprednisolone 60 mg daily and was discharged 14 days later. At 16-month follow-up, the patient demonstrated complete resolution of symptoms with a maintenance dose of prednisolone (7.5 mg daily).
Patient 2
A 23-year-old black woman presented to the emergency department 3 months postpartum with recurrent high fevers, worsening joint pain, and persistent itchy rash of 2 months’ duration. The patient had no history of travel, autoimmune disease, or sick contacts. She occasionally took aspirin for joint pain. Physical examination revealed a fever (temperature, 39.1°C) along with hyperpigmented patches and thin scaly hyperpigmented papules coalescing into a poorly demarcated V-shaped plaque on the upper back and posterior neck, extending to the chest in a shawl-like distribution (Figure 3). Submental lymphadenopathy was present. The spleen was not palpable.
Peripheral blood was collected for laboratory analysis and demonstrated transaminitis and a markedly high ferritin level (Table). An autoimmune panel was negative for rheumatoid factor, antinuclear antibodies, and antineutrophil cytoplasmic antibodies. Skin biopsy was performed and demonstrated many necrotic keratinocytes, singly and in aggregates, distributed from the spinous layer to the stratum corneum. A neutrophilic infiltrate was present in the papillary dermis (Figure 4).
The patient met the Yamaguchi criteria and was subsequently diagnosed with AOSD. She was treated with intravenous methylprednisolone 20 mg every 8 hours and was discharged 1 week later on oral prednisone 60 mg daily to be tapered over a period of months. At 2-week follow-up, the patient continued to experience rash and joint pain; oral methotrexate 10 mg weekly was added to her regimen, as well as vitamin D, calcium, and folic acid supplementation. At the next 2-week follow-up the patient noted improvement in the rash as well as the joint pain, but both still persisted. Prednisone was decreased to 50 mg daily and methotrexate was increased to 15 mg weekly. The patient continued to show improvement over the subsequent 3 months, during which prednisone was tapered to 10 mg daily and methotrexate was increased to 20 mg weekly. The patient showed resolution of symptoms at 3-month follow-up on this regimen, with plans to continue the prednisone taper and maintain methotrexate dosing.
Comment
Adult-onset Still disease is a systemic inflammatory condition that clinically manifests as spiking fevers, arthralgia, salmon-pink evanescent erythema, and lymphadenopathy.2 The condition also can cause liver dysfunction, splenomegaly, pericarditis, pleuritis, renal dysfunction, and a reactive hemophagocytic syndrome.1 Furthermore, one review of the literature described an association with delayed-onset malignancy.4 Early diagnosis is important yet challenging, as AOSD is a diagnosis of exclusion. The Yamaguchi criteria are the most widely used method of diagnosis and demonstrate more than 90% sensitivity.In addition to the Yamaguchi criteria, marked hyperferritinemia is characteristic of AOSD and can act as an indicator of disease activity.5 Interestingly, both of our patients had elevated ferritin levels, with patient 2 showing marked elevation (Table). In both patients, all major criteria were fulfilled, except the typical skin rash.
The skin rash in AOSD, classically consisting of an evanescent, salmon-pink erythema predominantly involving the extremities, has been observed in up to 87% of AOSD patients.5 The histology of the typical evanescent rash is nonspecific, characterized by a relatively sparse, perivascular, mixed inflammatory infiltrate. Notably, other skin manifestations may be found in patients with AOSD.1,2,5-16 Persistent pruritic papules and plaques are the most commonly reported nonclassical rash, presenting as erythematous, slightly scaly papules and plaques with a linear configuration typically on the trunk.2 Both of our patients presented with this atypical eruption. Importantly, the histopathology of this unique rash displays distinctive features, which can aid in early diagnosis. Findings include dyskeratotic keratinocytes in the cornified layers as well as in the epidermis, and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis. These findings were evident in both histopathologic studies of our patients (Figures 2 and 4). Although not present in our patients, dermal mucin deposition has been demonstrated in some reports.1,13,15
A 2015 review of the literature yielded 30 cases of AOSD with pruritic persistent papules and plaques.4 The study confirmed a linear, erythematous or brown rash on the back and neck in the majority of cases. Histologic findings were congruent with those reported in our 2 cases: necrotic keratinocytes in the upper epidermis with a neutrophilic infiltrate in the upper dermis without vasculitis. Most patients showed rapid resolution of the rash and symptoms with the use of prednisone, prednisolone, or intravenous pulsed methylprednisolone. Interestingly, a range of presentations were noted, including prurigo pigmentosalike urticarial papules; lichenoid papules; and dermatographismlike, dermatomyositislike, and lichen amyloidosis–like rashes.4 In our report, patient 2 presented with a rash in a dermat-omyositislike shawl distribution. It has been suggested that patients with dermatomyositislike rashes require more potent immunotherapy as compared to patients with other rash morphologies.4 The need for methotrexate in addition to a prednisone taper in the clinical course of patient 2 lends further support to this observation.
Conclusion
A clinically and pathologically distinct form of cutaneous disease—AOSD with persistent pruritic papules and plaques—was observed in our 2 patients. These histopathologic findings facilitated timely diagnosis in both patients. A range of clinical morphologies may exist in AOSD, an awareness of which is paramount. Adult-onset Still disease should be included in the differential diagnosis of a dermatomyositislike presentation in a shawl distribution. Prompt diagnosis is essential to ensure adequate therapy.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
- Yamamoto T. Cutaneous manifestations associated with adult-onset Still’s disease: important diagnostic values. Rheumatol Int. 2012;32:2233-2237.
- Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424-431.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associates with delayed malignancy [published online June 6, 2015]. J Am Acad Dermatol. 2015;73:294-303.
- Schwarz-Eywill M, Heilig B, Bauer H, et al. Evaluation of serum ferritin as a marker for adult Still’s disease activity. Ann Rheum Dis. 1992;51:683-685.
- Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17:1058-1063.
- Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241-242.
- Lübbe J, Hofer M, Chavaz P, et al. Adult onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710-713.
- Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still’s disease. Dermatology. 2001;202:333-335.
- Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still’s disease. Int J Dermatol. 2003;42:824-825.
- Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still’s disease. Ann Dermatol Venereol. 2005;132:693-696.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still’s disease. Am J Dermatopathol. 2007;49:194-196.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still’s disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still’s disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Azeck AG, Littlewood SM. Adult-onset Still’s disease with atypical cutaneous features. J Eur Acad Dermatol Venereol. 2005;19:360-363.
Practice Points
- Serologic testing and skin biopsy are necessary in the timely and appropriate diagnosis of adult-onset Still disease (AOSD).
- In patients with a persistent pruritic papular rash, consider AOSD if there is a supporting history.
- Skin biopsy is diagnostic of AOSD with the unique histopathologic findings of dyskeratotic keratinocytes in the cornified layers as well as in the epidermis and a sparse neutrophilic and/or lymphocytic infiltrate in the papillary dermis without vasculitis.
What’s Eating You? Ixodes Tick and Related Diseases, Part 2: Diagnosis and Treatment of Regional Tick-borne Diseases
The Ixodes tick is prevalent in temperate climates worldwide. During a blood meal, pathogens may be transmitted from the tick to its host. Borrelia burgdorferi, a spirochete responsible for Lyme disease, is the most prevalent pathogen transmitted by Ixodes ticks.1 Borrelia mayonii recently was identified as an additional cause of Lyme disease in the United States.2
The Ixodes tick also is associated with several less common pathogens, including Babesia microti and the tick-borne encephalitis virus, which have been recognized as Ixodes-associated pathogens for many years.3,4 Other pathogens have been identified, including Anaplasma phagocytophilum, recognized in the 1990s as the cause of human granulocytic anaplasmosis, as well as the Powassan virus and Borrelia miyamotoi.5-7 Additionally, tick paralysis has been associated with toxins in the saliva of various species of several genera of ticks, including some Ixodes species.8 Due to an overlap in geographic distribution (Figure) and disease presentations (eTable), it is important that physicians be familiar with these regional pathogens transmitted by Ixodes ticks.
Human Granulocytic Anaplasmosis
Formerly known as human granulocytic ehrlichiosis, human granulocytic anaplasmosis is caused by A phagocytophilum and is transmitted by Ixodes scapularis, Ixodes pacificus, and Ixodes persulcatus. The incidence of human granulocytic anaplasmosis in the United States increased 12-fold from 2001 to 2011.9
Presenting symptoms generally are nonspecific, including fever, night sweats, headache, myalgias, and arthralgias, often resulting in misdiagnosis as a viral infection. Laboratory abnormalities include mild transaminitis, leukopenia, and thrombocytopenia.9,10 Although most infections resolve spontaneously, 3% of patients develop serious complications. The mortality rate is 0.6%.11
A diagnosis of human granulocytic anaplasmosis should be suspected in patients with a viral-like illness and exposure to ticks in an endemic area. The diagnosis can be confirmed by polymerase chain reaction (PCR), acute- and convalescent-phase serologic testing, or direct fluorescent antibody screening. Characteristic morulae may be present in granulocytes.12 Treatment typically includes doxycycline, which also covers B burgdorferi coinfection. When a diagnosis of human granulocytic anaplasmosis is suspected, treatment should never be delayed to await laboratory confirmation. If no clinical improvement is seen within 48 hours, alternate diagnoses or coinfection with B microti should be considered.10
Babesiosis
The protozoan B microti causes babesiosis in the United States, with Babesia divergens being more common in Europe.13 Reported cases of babesiosis in New York increased as much as 20-fold from 2001 to 2008.14 Transmission primarily is from the Ixodes tick but rarely can occur from blood transfusion.15 Tick attachment for at least 36 hours is required for transmission.13
The clinical presentation of babesiosis ranges from asymptomatic to fatal. Symptoms generally are nonspecific, resembling a viral infection and including headache, nausea, diarrhea, arthralgia, and myalgia. Laboratory evaluation may reveal hemolytic anemia, thrombocytopenia, transaminitis, and elevated blood urea nitrogen and creatinine levels.16 Rash is not typical. Resolution of symptoms generally occurs within 2 weeks of presentation, although anemia may persist for months.13 Severe disease is more common among elderly and immunocompromised patients. Complications include respiratory failure, renal failure, congestive heart failure, and disseminated intravascular coagulation. The mortality rate in the United States is approximately 10%.10,16
A diagnosis of babesiosis is made based on the presence of flulike symptoms, laboratory results, and history of recent travel to an endemic area. A thin blood smear allows identification of the organism in erythrocytes as ring forms or tetrads (a “Maltese cross” appearance).17 Polymerase chain reaction is more sensitive than a blood smear, especially in early disease.18 Indirect fluorescent antibody testing is species-specific but cannot verify active infection.10
Treatment of babesiosis is indicated for symptomatic patients with active infection. Positive serology alone is not an indication for treatment. Asymptomatic patients with positive serology should have diagnostic testing repeated in 3 months with subsequent treatment if parasitemia persists. Mild disease is treated with atovaquone plus azithromycin or clindamycin plus quinine. Severe babesiosis is treated with quinine and intravenous clindamycin and may require exchange transfusion.10 Coinfection with B burgdorferi should be considered in patients with flulike symptoms and erythema migrans or treatment failure. Coinfection is diagnosed by Lyme serology plus PCR for B microti. This is an important consideration because treatment of babesiosis does not eradicate B burgdorferi infection.19
Powassan Virus
Powassan virus is a flavivirus that causes encephalitis. It is transmitted by Ixodes cookei (Powassan virus, lineage I) in the Great Lakes region and by I scapularis (Powassan virus, lineage II, or deer tick virus) in the northeastern United States. Transmission can occur within 15 minutes of tick attachment.6,20,21
Patients typically present with fever, headache, altered mental status, seizures, and focal neurologic deficits. Gastrointestinal symptoms and rash also have been reported.21 The diagnosis is made based on clinical presentation and laboratory testing with PCR or enzyme-linked immunosorbent assay (ELISA). Cross-reactivity on ELISA exists, necessitating confirmation with a neutralizing antibody or PCR. Treatment is supportive. Corticosteroids and intravenous immunoglobulin have been proposed as treatment modalities, but evidence of their efficacy is limited.22
Tick-borne Encephalitis
Tick-borne encephalitis is caused by the flavivirus tick-borne encephalitis virus in Europe and Asia. The tick-borne encephalitis virus is transmitted by Ixodes ricinus in Europe and by Ixodes persulcatus in eastern Russia, China, and Japan. It also has been associated with consumption of unpasteurized milk.23,24
Tick-borne encephalitis presents in a biphasic pattern. The initial viremic phase can persist for as long as 8 days with headache, nausea, myalgia, and fever. One-third of patients then enter an asymptomatic phase, followed by virus penetration into the central nervous system. The neurologic phase produces continued headache and fever with photophobia, focal neurologic deficits, seizures, respiratory depression, or coma. Neurologic sequelae persist in 10% to 20% of patients.25,26
In the viremic stage, diagnosis is made with PCR or culture. During the latent phase or neurologic phase, serologic testing for tick-borne encephalitis virus antibodies is indicated. Neutralizing antibody evaluation may be necessary due to cross-reactivity among flaviviruses.27 Treatment is supportive. An inactivated vaccine is available for high-risk populations.28
Borrelia miyamotoi Disease
Borrelia miyamotoi is a symbiont of the Ixodes tick formerly believed to have no pathogenic significance; however, B miyamotoi was isolated in febrile patients in Russia in 20117 and was identified as a pathogen in both North America29 and Europe in 2013.30 Disease presentation includes nonspecific symptoms of fever, fatigue, headache, arthralgia, myalgia, and nausea. Rash is uncommon. Laboratory abnormalities include leukopenia, thrombocytopenia, and transaminitis.31,32 Meningoencephalitis may occur in immunocompromised patients.29,30
The diagnosis of B miyamotoi disease is confirmed by PCR or serology. An ELISA that is positive for B burgdorferi IgM but negative with confirmatory immunoblot suggests B miyamotoi disease. Seroconversion using a glpQ protein ELISA also can be assessed.31 If ELISA is positive, Lyme disease can be excluded because B burgdorferi does not possess g1pQ. Treatment is with doxycycline.32
Tick Paralysis
Tick paralysis is an intoxication with holocyclotoxin from the saliva of gravid hard ticks. In the United States, intoxication is associated with ticks of various species of Amblyomma, Dermacentor, and Ixodes in the Northwest, Southeast, and Northeast. In Australia, intoxication is associated with Ixodes.33 Patients present with weakness and fatigue, progressing to ascending flaccid paralysis with sensory sparing. The treatment is tick removal.8,33
Conclusion
Arthropods carry many regional pathogens. Physicians outside of those regions should seek a travel history and be alert for imported disease.
- Steere AC, Grodzicki RL, Kornblatt AN, et al. The spirochetal etiology of Lyme disease. N Engl J Med. 1983;308:733-740.
- Dolan MC, Hojgaard A, Hoxmeier JC, et al. Vector competence of the blacklegged tick, Ixodes scapularis, for the recently recognized Lyme borreliosis spirochete Candidatus Borrelia mayonii. Ticks Tick Borne Dis. 2016;7:665-669.
- Rudzinska MA, Spielman A, Riek RF, et al. Intraerythrocytic ‘gametocytes’ of Babesia microti and their maturation in ticks. Can J Zool. 1979;57:424-434.
- Casals J, Olitsky PK. Enduring immunity following vaccination of mice with formalin-inactivated virus of Russian spring-summer (Far Eastern, tick-borne) encephalitis; correlation with serum-neutralizing and complement-fixing antibodies. J Exp Med. 1945;82:431-443.
- Magnarelli LA, Stafford KC III, Mather TN, et al. Hemocytic rickettsia-like organisms in ticks: serologic reactivity with antisera to Ehrlichiae and detection of DNA of agent of human granulocytic ehrlichiosis by PCR. J Clin Microbiol. 1995;33:2710-2714.
- McLean DM, Donohue WL. Powassan virus: isolation of virus from a fatal case of encephalitis. Can Med Assoc J. 1959;80:708-711.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Diaz JH. A 60-year meta-analysis of tick paralysis in the United States: a predictable, preventable, and often misdiagnosed poisoning. J Med Toxicol. 2010;6:15-21.
- Bakken J, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2015;29:341-355.
- Chapman AS, Bakken JS, Folk SM, et al; Tickborne Rickettsial Diseases Working Group; CDC. Diagnosis and management of tickborne rickettsial diseases: Rocky Mountain spotted fever, ehrlichioses, and anaplasmosis—United States: a practical guide for physicians and other health-care and public health professionals. MMWR Recomm Rep. 2006;55(RR-4):1-27.
- Dahlgren FS, Mandel EJ, Krebs JW, et al. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000-2007. Am J Trop Med Hyg. 2011;85:124-130.
- Aguero-Rosenfeld ME. Diagnosis of human granulocytic ehrlichiosis: state of the art. Vector Borne Zoonotic Dis. 2002;2:233-239.
- Vannier EG, Diuk-Wasser MA, Ben Mamoun C, et al. Babesiosis. Infect Dis Clin North Am. 2015;29:357-370.
- Joseph JT, Roy SS, Shams N, et al. Babesiosis in Lower Hudson Valley, New York, USA. Emerg Infect Dis. 2011;17:843-847.
- McQuiston JH, Childs JE, Chamberland ME, et al. Transmission of tickborne agents by blood transfusions: a review of known and potential risks in the United States. Transfusion. 2000;40:274-284.
- Hatcher JC, Greenberg PD, Antique J, et al. Severe babesiosis in Long Island: review of 34 cases and their complications. Clin Infect Dis. 2001;32:1117-1125.
- Healy GR, Ruebush TK. Morphology of Babesia microti in human blood smears. Am J Clin Pathol. 1980;73:107-109.
- Kowalski TJ, Jobe DA, Dolan EC, et al. The emergence of clinically relevant babesiosis in southwestern Wisconsin. WMJ. 2015;114:152-157.
- Krause PJ, Telford SR III, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Centers for Disease Control and Prevention. Statistics & maps. http://www.cdc.gov/powassan/statistics.html. Updated February 14, 2017. Accessed December 11, 2017.
- Piantadosi A, Rubin DB, McQuillen DP, et al. Emerging cases of Powassan virus encephalitis in New England: clinical presentation, imaging, and review of the literature. Clin Infect Dis. 2016;62:707-713.
- El Khoury MY, Camargo JF, White JL, et al. Potential role of deer tick virus in Powassan encephalitis cases in Lyme disease-endemic areas of New York, U.S.A. Emerg Infect Dis. 2013;19:1926-1933.
- World Health Organization (WHO). Vaccines against tick-borne encephalitis: WHO position paper. Wkly Epidemiol Rec. 2011;86:241-256.
- Centers for Disease Control and Prevention (CDC). Tick-borne encephalitis among U.S. travelers to Europe and Asia—2000-2009. JAMA. 2010;303:2132-2135.
- Valarcher JF, Hägglund S, Juremalm M, et al. Tick-borne encephalitits. Rev Sci Tech. 2015;34:453-466.
- Schultze D, Dollenmaier G, Rohner A, et al. Benefit of detecting tick-borne encephalitis viremia in the first phase of illness. J Clin Virol. 2007;38:172-175.
- Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine. 2003;21(suppl 1):S36-S40.
- Zavadska D, Anca I, André F, et al. Recommendations for tick-borne encephalitis vaccination from the Central European Vaccination Awareness Group. Hum Vaccin Immunother. 2013;9:362-374.
- Gugliotta JL, Goethert HK, Berardi VP, et al. Meningoencephalitis from Borrelia miyamotoi in an immunocompromised patient. N Engl J Med. 2013;368:240-245.
- Hovius JW, de Wever B, Sohne M, et al. A case of meningoencephalitis by the relapsing fever spirochaete Borrelia miyamotoi in Europe. Lancet. 2013;382:658.
- Molloy PJ, Telford SR III, Chowdri HR, et al. Borrelia miyamotoi disease in the northeastern United States: a case series. Ann Intern Med. 2015;163:91-98.
- Telford SR 3rd, Goethert HK, Molloy PJ, et al. Borrelia miyamotoi disease: neither Lyme disease nor relapsing fever. Clin Lab Med. 2015;35:867-882.
- Diaz JH. A comparative meta-analysis of tick paralysis in the United States and Australia. Clin Toxicol (Phila). 2015;53:874-883.
The Ixodes tick is prevalent in temperate climates worldwide. During a blood meal, pathogens may be transmitted from the tick to its host. Borrelia burgdorferi, a spirochete responsible for Lyme disease, is the most prevalent pathogen transmitted by Ixodes ticks.1 Borrelia mayonii recently was identified as an additional cause of Lyme disease in the United States.2
The Ixodes tick also is associated with several less common pathogens, including Babesia microti and the tick-borne encephalitis virus, which have been recognized as Ixodes-associated pathogens for many years.3,4 Other pathogens have been identified, including Anaplasma phagocytophilum, recognized in the 1990s as the cause of human granulocytic anaplasmosis, as well as the Powassan virus and Borrelia miyamotoi.5-7 Additionally, tick paralysis has been associated with toxins in the saliva of various species of several genera of ticks, including some Ixodes species.8 Due to an overlap in geographic distribution (Figure) and disease presentations (eTable), it is important that physicians be familiar with these regional pathogens transmitted by Ixodes ticks.
Human Granulocytic Anaplasmosis
Formerly known as human granulocytic ehrlichiosis, human granulocytic anaplasmosis is caused by A phagocytophilum and is transmitted by Ixodes scapularis, Ixodes pacificus, and Ixodes persulcatus. The incidence of human granulocytic anaplasmosis in the United States increased 12-fold from 2001 to 2011.9
Presenting symptoms generally are nonspecific, including fever, night sweats, headache, myalgias, and arthralgias, often resulting in misdiagnosis as a viral infection. Laboratory abnormalities include mild transaminitis, leukopenia, and thrombocytopenia.9,10 Although most infections resolve spontaneously, 3% of patients develop serious complications. The mortality rate is 0.6%.11
A diagnosis of human granulocytic anaplasmosis should be suspected in patients with a viral-like illness and exposure to ticks in an endemic area. The diagnosis can be confirmed by polymerase chain reaction (PCR), acute- and convalescent-phase serologic testing, or direct fluorescent antibody screening. Characteristic morulae may be present in granulocytes.12 Treatment typically includes doxycycline, which also covers B burgdorferi coinfection. When a diagnosis of human granulocytic anaplasmosis is suspected, treatment should never be delayed to await laboratory confirmation. If no clinical improvement is seen within 48 hours, alternate diagnoses or coinfection with B microti should be considered.10
Babesiosis
The protozoan B microti causes babesiosis in the United States, with Babesia divergens being more common in Europe.13 Reported cases of babesiosis in New York increased as much as 20-fold from 2001 to 2008.14 Transmission primarily is from the Ixodes tick but rarely can occur from blood transfusion.15 Tick attachment for at least 36 hours is required for transmission.13
The clinical presentation of babesiosis ranges from asymptomatic to fatal. Symptoms generally are nonspecific, resembling a viral infection and including headache, nausea, diarrhea, arthralgia, and myalgia. Laboratory evaluation may reveal hemolytic anemia, thrombocytopenia, transaminitis, and elevated blood urea nitrogen and creatinine levels.16 Rash is not typical. Resolution of symptoms generally occurs within 2 weeks of presentation, although anemia may persist for months.13 Severe disease is more common among elderly and immunocompromised patients. Complications include respiratory failure, renal failure, congestive heart failure, and disseminated intravascular coagulation. The mortality rate in the United States is approximately 10%.10,16
A diagnosis of babesiosis is made based on the presence of flulike symptoms, laboratory results, and history of recent travel to an endemic area. A thin blood smear allows identification of the organism in erythrocytes as ring forms or tetrads (a “Maltese cross” appearance).17 Polymerase chain reaction is more sensitive than a blood smear, especially in early disease.18 Indirect fluorescent antibody testing is species-specific but cannot verify active infection.10
Treatment of babesiosis is indicated for symptomatic patients with active infection. Positive serology alone is not an indication for treatment. Asymptomatic patients with positive serology should have diagnostic testing repeated in 3 months with subsequent treatment if parasitemia persists. Mild disease is treated with atovaquone plus azithromycin or clindamycin plus quinine. Severe babesiosis is treated with quinine and intravenous clindamycin and may require exchange transfusion.10 Coinfection with B burgdorferi should be considered in patients with flulike symptoms and erythema migrans or treatment failure. Coinfection is diagnosed by Lyme serology plus PCR for B microti. This is an important consideration because treatment of babesiosis does not eradicate B burgdorferi infection.19
Powassan Virus
Powassan virus is a flavivirus that causes encephalitis. It is transmitted by Ixodes cookei (Powassan virus, lineage I) in the Great Lakes region and by I scapularis (Powassan virus, lineage II, or deer tick virus) in the northeastern United States. Transmission can occur within 15 minutes of tick attachment.6,20,21
Patients typically present with fever, headache, altered mental status, seizures, and focal neurologic deficits. Gastrointestinal symptoms and rash also have been reported.21 The diagnosis is made based on clinical presentation and laboratory testing with PCR or enzyme-linked immunosorbent assay (ELISA). Cross-reactivity on ELISA exists, necessitating confirmation with a neutralizing antibody or PCR. Treatment is supportive. Corticosteroids and intravenous immunoglobulin have been proposed as treatment modalities, but evidence of their efficacy is limited.22
Tick-borne Encephalitis
Tick-borne encephalitis is caused by the flavivirus tick-borne encephalitis virus in Europe and Asia. The tick-borne encephalitis virus is transmitted by Ixodes ricinus in Europe and by Ixodes persulcatus in eastern Russia, China, and Japan. It also has been associated with consumption of unpasteurized milk.23,24
Tick-borne encephalitis presents in a biphasic pattern. The initial viremic phase can persist for as long as 8 days with headache, nausea, myalgia, and fever. One-third of patients then enter an asymptomatic phase, followed by virus penetration into the central nervous system. The neurologic phase produces continued headache and fever with photophobia, focal neurologic deficits, seizures, respiratory depression, or coma. Neurologic sequelae persist in 10% to 20% of patients.25,26
In the viremic stage, diagnosis is made with PCR or culture. During the latent phase or neurologic phase, serologic testing for tick-borne encephalitis virus antibodies is indicated. Neutralizing antibody evaluation may be necessary due to cross-reactivity among flaviviruses.27 Treatment is supportive. An inactivated vaccine is available for high-risk populations.28
Borrelia miyamotoi Disease
Borrelia miyamotoi is a symbiont of the Ixodes tick formerly believed to have no pathogenic significance; however, B miyamotoi was isolated in febrile patients in Russia in 20117 and was identified as a pathogen in both North America29 and Europe in 2013.30 Disease presentation includes nonspecific symptoms of fever, fatigue, headache, arthralgia, myalgia, and nausea. Rash is uncommon. Laboratory abnormalities include leukopenia, thrombocytopenia, and transaminitis.31,32 Meningoencephalitis may occur in immunocompromised patients.29,30
The diagnosis of B miyamotoi disease is confirmed by PCR or serology. An ELISA that is positive for B burgdorferi IgM but negative with confirmatory immunoblot suggests B miyamotoi disease. Seroconversion using a glpQ protein ELISA also can be assessed.31 If ELISA is positive, Lyme disease can be excluded because B burgdorferi does not possess g1pQ. Treatment is with doxycycline.32
Tick Paralysis
Tick paralysis is an intoxication with holocyclotoxin from the saliva of gravid hard ticks. In the United States, intoxication is associated with ticks of various species of Amblyomma, Dermacentor, and Ixodes in the Northwest, Southeast, and Northeast. In Australia, intoxication is associated with Ixodes.33 Patients present with weakness and fatigue, progressing to ascending flaccid paralysis with sensory sparing. The treatment is tick removal.8,33
Conclusion
Arthropods carry many regional pathogens. Physicians outside of those regions should seek a travel history and be alert for imported disease.
The Ixodes tick is prevalent in temperate climates worldwide. During a blood meal, pathogens may be transmitted from the tick to its host. Borrelia burgdorferi, a spirochete responsible for Lyme disease, is the most prevalent pathogen transmitted by Ixodes ticks.1 Borrelia mayonii recently was identified as an additional cause of Lyme disease in the United States.2
The Ixodes tick also is associated with several less common pathogens, including Babesia microti and the tick-borne encephalitis virus, which have been recognized as Ixodes-associated pathogens for many years.3,4 Other pathogens have been identified, including Anaplasma phagocytophilum, recognized in the 1990s as the cause of human granulocytic anaplasmosis, as well as the Powassan virus and Borrelia miyamotoi.5-7 Additionally, tick paralysis has been associated with toxins in the saliva of various species of several genera of ticks, including some Ixodes species.8 Due to an overlap in geographic distribution (Figure) and disease presentations (eTable), it is important that physicians be familiar with these regional pathogens transmitted by Ixodes ticks.
Human Granulocytic Anaplasmosis
Formerly known as human granulocytic ehrlichiosis, human granulocytic anaplasmosis is caused by A phagocytophilum and is transmitted by Ixodes scapularis, Ixodes pacificus, and Ixodes persulcatus. The incidence of human granulocytic anaplasmosis in the United States increased 12-fold from 2001 to 2011.9
Presenting symptoms generally are nonspecific, including fever, night sweats, headache, myalgias, and arthralgias, often resulting in misdiagnosis as a viral infection. Laboratory abnormalities include mild transaminitis, leukopenia, and thrombocytopenia.9,10 Although most infections resolve spontaneously, 3% of patients develop serious complications. The mortality rate is 0.6%.11
A diagnosis of human granulocytic anaplasmosis should be suspected in patients with a viral-like illness and exposure to ticks in an endemic area. The diagnosis can be confirmed by polymerase chain reaction (PCR), acute- and convalescent-phase serologic testing, or direct fluorescent antibody screening. Characteristic morulae may be present in granulocytes.12 Treatment typically includes doxycycline, which also covers B burgdorferi coinfection. When a diagnosis of human granulocytic anaplasmosis is suspected, treatment should never be delayed to await laboratory confirmation. If no clinical improvement is seen within 48 hours, alternate diagnoses or coinfection with B microti should be considered.10
Babesiosis
The protozoan B microti causes babesiosis in the United States, with Babesia divergens being more common in Europe.13 Reported cases of babesiosis in New York increased as much as 20-fold from 2001 to 2008.14 Transmission primarily is from the Ixodes tick but rarely can occur from blood transfusion.15 Tick attachment for at least 36 hours is required for transmission.13
The clinical presentation of babesiosis ranges from asymptomatic to fatal. Symptoms generally are nonspecific, resembling a viral infection and including headache, nausea, diarrhea, arthralgia, and myalgia. Laboratory evaluation may reveal hemolytic anemia, thrombocytopenia, transaminitis, and elevated blood urea nitrogen and creatinine levels.16 Rash is not typical. Resolution of symptoms generally occurs within 2 weeks of presentation, although anemia may persist for months.13 Severe disease is more common among elderly and immunocompromised patients. Complications include respiratory failure, renal failure, congestive heart failure, and disseminated intravascular coagulation. The mortality rate in the United States is approximately 10%.10,16
A diagnosis of babesiosis is made based on the presence of flulike symptoms, laboratory results, and history of recent travel to an endemic area. A thin blood smear allows identification of the organism in erythrocytes as ring forms or tetrads (a “Maltese cross” appearance).17 Polymerase chain reaction is more sensitive than a blood smear, especially in early disease.18 Indirect fluorescent antibody testing is species-specific but cannot verify active infection.10
Treatment of babesiosis is indicated for symptomatic patients with active infection. Positive serology alone is not an indication for treatment. Asymptomatic patients with positive serology should have diagnostic testing repeated in 3 months with subsequent treatment if parasitemia persists. Mild disease is treated with atovaquone plus azithromycin or clindamycin plus quinine. Severe babesiosis is treated with quinine and intravenous clindamycin and may require exchange transfusion.10 Coinfection with B burgdorferi should be considered in patients with flulike symptoms and erythema migrans or treatment failure. Coinfection is diagnosed by Lyme serology plus PCR for B microti. This is an important consideration because treatment of babesiosis does not eradicate B burgdorferi infection.19
Powassan Virus
Powassan virus is a flavivirus that causes encephalitis. It is transmitted by Ixodes cookei (Powassan virus, lineage I) in the Great Lakes region and by I scapularis (Powassan virus, lineage II, or deer tick virus) in the northeastern United States. Transmission can occur within 15 minutes of tick attachment.6,20,21
Patients typically present with fever, headache, altered mental status, seizures, and focal neurologic deficits. Gastrointestinal symptoms and rash also have been reported.21 The diagnosis is made based on clinical presentation and laboratory testing with PCR or enzyme-linked immunosorbent assay (ELISA). Cross-reactivity on ELISA exists, necessitating confirmation with a neutralizing antibody or PCR. Treatment is supportive. Corticosteroids and intravenous immunoglobulin have been proposed as treatment modalities, but evidence of their efficacy is limited.22
Tick-borne Encephalitis
Tick-borne encephalitis is caused by the flavivirus tick-borne encephalitis virus in Europe and Asia. The tick-borne encephalitis virus is transmitted by Ixodes ricinus in Europe and by Ixodes persulcatus in eastern Russia, China, and Japan. It also has been associated with consumption of unpasteurized milk.23,24
Tick-borne encephalitis presents in a biphasic pattern. The initial viremic phase can persist for as long as 8 days with headache, nausea, myalgia, and fever. One-third of patients then enter an asymptomatic phase, followed by virus penetration into the central nervous system. The neurologic phase produces continued headache and fever with photophobia, focal neurologic deficits, seizures, respiratory depression, or coma. Neurologic sequelae persist in 10% to 20% of patients.25,26
In the viremic stage, diagnosis is made with PCR or culture. During the latent phase or neurologic phase, serologic testing for tick-borne encephalitis virus antibodies is indicated. Neutralizing antibody evaluation may be necessary due to cross-reactivity among flaviviruses.27 Treatment is supportive. An inactivated vaccine is available for high-risk populations.28
Borrelia miyamotoi Disease
Borrelia miyamotoi is a symbiont of the Ixodes tick formerly believed to have no pathogenic significance; however, B miyamotoi was isolated in febrile patients in Russia in 20117 and was identified as a pathogen in both North America29 and Europe in 2013.30 Disease presentation includes nonspecific symptoms of fever, fatigue, headache, arthralgia, myalgia, and nausea. Rash is uncommon. Laboratory abnormalities include leukopenia, thrombocytopenia, and transaminitis.31,32 Meningoencephalitis may occur in immunocompromised patients.29,30
The diagnosis of B miyamotoi disease is confirmed by PCR or serology. An ELISA that is positive for B burgdorferi IgM but negative with confirmatory immunoblot suggests B miyamotoi disease. Seroconversion using a glpQ protein ELISA also can be assessed.31 If ELISA is positive, Lyme disease can be excluded because B burgdorferi does not possess g1pQ. Treatment is with doxycycline.32
Tick Paralysis
Tick paralysis is an intoxication with holocyclotoxin from the saliva of gravid hard ticks. In the United States, intoxication is associated with ticks of various species of Amblyomma, Dermacentor, and Ixodes in the Northwest, Southeast, and Northeast. In Australia, intoxication is associated with Ixodes.33 Patients present with weakness and fatigue, progressing to ascending flaccid paralysis with sensory sparing. The treatment is tick removal.8,33
Conclusion
Arthropods carry many regional pathogens. Physicians outside of those regions should seek a travel history and be alert for imported disease.
- Steere AC, Grodzicki RL, Kornblatt AN, et al. The spirochetal etiology of Lyme disease. N Engl J Med. 1983;308:733-740.
- Dolan MC, Hojgaard A, Hoxmeier JC, et al. Vector competence of the blacklegged tick, Ixodes scapularis, for the recently recognized Lyme borreliosis spirochete Candidatus Borrelia mayonii. Ticks Tick Borne Dis. 2016;7:665-669.
- Rudzinska MA, Spielman A, Riek RF, et al. Intraerythrocytic ‘gametocytes’ of Babesia microti and their maturation in ticks. Can J Zool. 1979;57:424-434.
- Casals J, Olitsky PK. Enduring immunity following vaccination of mice with formalin-inactivated virus of Russian spring-summer (Far Eastern, tick-borne) encephalitis; correlation with serum-neutralizing and complement-fixing antibodies. J Exp Med. 1945;82:431-443.
- Magnarelli LA, Stafford KC III, Mather TN, et al. Hemocytic rickettsia-like organisms in ticks: serologic reactivity with antisera to Ehrlichiae and detection of DNA of agent of human granulocytic ehrlichiosis by PCR. J Clin Microbiol. 1995;33:2710-2714.
- McLean DM, Donohue WL. Powassan virus: isolation of virus from a fatal case of encephalitis. Can Med Assoc J. 1959;80:708-711.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Diaz JH. A 60-year meta-analysis of tick paralysis in the United States: a predictable, preventable, and often misdiagnosed poisoning. J Med Toxicol. 2010;6:15-21.
- Bakken J, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2015;29:341-355.
- Chapman AS, Bakken JS, Folk SM, et al; Tickborne Rickettsial Diseases Working Group; CDC. Diagnosis and management of tickborne rickettsial diseases: Rocky Mountain spotted fever, ehrlichioses, and anaplasmosis—United States: a practical guide for physicians and other health-care and public health professionals. MMWR Recomm Rep. 2006;55(RR-4):1-27.
- Dahlgren FS, Mandel EJ, Krebs JW, et al. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000-2007. Am J Trop Med Hyg. 2011;85:124-130.
- Aguero-Rosenfeld ME. Diagnosis of human granulocytic ehrlichiosis: state of the art. Vector Borne Zoonotic Dis. 2002;2:233-239.
- Vannier EG, Diuk-Wasser MA, Ben Mamoun C, et al. Babesiosis. Infect Dis Clin North Am. 2015;29:357-370.
- Joseph JT, Roy SS, Shams N, et al. Babesiosis in Lower Hudson Valley, New York, USA. Emerg Infect Dis. 2011;17:843-847.
- McQuiston JH, Childs JE, Chamberland ME, et al. Transmission of tickborne agents by blood transfusions: a review of known and potential risks in the United States. Transfusion. 2000;40:274-284.
- Hatcher JC, Greenberg PD, Antique J, et al. Severe babesiosis in Long Island: review of 34 cases and their complications. Clin Infect Dis. 2001;32:1117-1125.
- Healy GR, Ruebush TK. Morphology of Babesia microti in human blood smears. Am J Clin Pathol. 1980;73:107-109.
- Kowalski TJ, Jobe DA, Dolan EC, et al. The emergence of clinically relevant babesiosis in southwestern Wisconsin. WMJ. 2015;114:152-157.
- Krause PJ, Telford SR III, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Centers for Disease Control and Prevention. Statistics & maps. http://www.cdc.gov/powassan/statistics.html. Updated February 14, 2017. Accessed December 11, 2017.
- Piantadosi A, Rubin DB, McQuillen DP, et al. Emerging cases of Powassan virus encephalitis in New England: clinical presentation, imaging, and review of the literature. Clin Infect Dis. 2016;62:707-713.
- El Khoury MY, Camargo JF, White JL, et al. Potential role of deer tick virus in Powassan encephalitis cases in Lyme disease-endemic areas of New York, U.S.A. Emerg Infect Dis. 2013;19:1926-1933.
- World Health Organization (WHO). Vaccines against tick-borne encephalitis: WHO position paper. Wkly Epidemiol Rec. 2011;86:241-256.
- Centers for Disease Control and Prevention (CDC). Tick-borne encephalitis among U.S. travelers to Europe and Asia—2000-2009. JAMA. 2010;303:2132-2135.
- Valarcher JF, Hägglund S, Juremalm M, et al. Tick-borne encephalitits. Rev Sci Tech. 2015;34:453-466.
- Schultze D, Dollenmaier G, Rohner A, et al. Benefit of detecting tick-borne encephalitis viremia in the first phase of illness. J Clin Virol. 2007;38:172-175.
- Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine. 2003;21(suppl 1):S36-S40.
- Zavadska D, Anca I, André F, et al. Recommendations for tick-borne encephalitis vaccination from the Central European Vaccination Awareness Group. Hum Vaccin Immunother. 2013;9:362-374.
- Gugliotta JL, Goethert HK, Berardi VP, et al. Meningoencephalitis from Borrelia miyamotoi in an immunocompromised patient. N Engl J Med. 2013;368:240-245.
- Hovius JW, de Wever B, Sohne M, et al. A case of meningoencephalitis by the relapsing fever spirochaete Borrelia miyamotoi in Europe. Lancet. 2013;382:658.
- Molloy PJ, Telford SR III, Chowdri HR, et al. Borrelia miyamotoi disease in the northeastern United States: a case series. Ann Intern Med. 2015;163:91-98.
- Telford SR 3rd, Goethert HK, Molloy PJ, et al. Borrelia miyamotoi disease: neither Lyme disease nor relapsing fever. Clin Lab Med. 2015;35:867-882.
- Diaz JH. A comparative meta-analysis of tick paralysis in the United States and Australia. Clin Toxicol (Phila). 2015;53:874-883.
- Steere AC, Grodzicki RL, Kornblatt AN, et al. The spirochetal etiology of Lyme disease. N Engl J Med. 1983;308:733-740.
- Dolan MC, Hojgaard A, Hoxmeier JC, et al. Vector competence of the blacklegged tick, Ixodes scapularis, for the recently recognized Lyme borreliosis spirochete Candidatus Borrelia mayonii. Ticks Tick Borne Dis. 2016;7:665-669.
- Rudzinska MA, Spielman A, Riek RF, et al. Intraerythrocytic ‘gametocytes’ of Babesia microti and their maturation in ticks. Can J Zool. 1979;57:424-434.
- Casals J, Olitsky PK. Enduring immunity following vaccination of mice with formalin-inactivated virus of Russian spring-summer (Far Eastern, tick-borne) encephalitis; correlation with serum-neutralizing and complement-fixing antibodies. J Exp Med. 1945;82:431-443.
- Magnarelli LA, Stafford KC III, Mather TN, et al. Hemocytic rickettsia-like organisms in ticks: serologic reactivity with antisera to Ehrlichiae and detection of DNA of agent of human granulocytic ehrlichiosis by PCR. J Clin Microbiol. 1995;33:2710-2714.
- McLean DM, Donohue WL. Powassan virus: isolation of virus from a fatal case of encephalitis. Can Med Assoc J. 1959;80:708-711.
- Platonov AE, Karan LS, Kolyasnikova NM, et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg Infect Dis. 2011;17:1816-1823.
- Diaz JH. A 60-year meta-analysis of tick paralysis in the United States: a predictable, preventable, and often misdiagnosed poisoning. J Med Toxicol. 2010;6:15-21.
- Bakken J, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2015;29:341-355.
- Chapman AS, Bakken JS, Folk SM, et al; Tickborne Rickettsial Diseases Working Group; CDC. Diagnosis and management of tickborne rickettsial diseases: Rocky Mountain spotted fever, ehrlichioses, and anaplasmosis—United States: a practical guide for physicians and other health-care and public health professionals. MMWR Recomm Rep. 2006;55(RR-4):1-27.
- Dahlgren FS, Mandel EJ, Krebs JW, et al. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000-2007. Am J Trop Med Hyg. 2011;85:124-130.
- Aguero-Rosenfeld ME. Diagnosis of human granulocytic ehrlichiosis: state of the art. Vector Borne Zoonotic Dis. 2002;2:233-239.
- Vannier EG, Diuk-Wasser MA, Ben Mamoun C, et al. Babesiosis. Infect Dis Clin North Am. 2015;29:357-370.
- Joseph JT, Roy SS, Shams N, et al. Babesiosis in Lower Hudson Valley, New York, USA. Emerg Infect Dis. 2011;17:843-847.
- McQuiston JH, Childs JE, Chamberland ME, et al. Transmission of tickborne agents by blood transfusions: a review of known and potential risks in the United States. Transfusion. 2000;40:274-284.
- Hatcher JC, Greenberg PD, Antique J, et al. Severe babesiosis in Long Island: review of 34 cases and their complications. Clin Infect Dis. 2001;32:1117-1125.
- Healy GR, Ruebush TK. Morphology of Babesia microti in human blood smears. Am J Clin Pathol. 1980;73:107-109.
- Kowalski TJ, Jobe DA, Dolan EC, et al. The emergence of clinically relevant babesiosis in southwestern Wisconsin. WMJ. 2015;114:152-157.
- Krause PJ, Telford SR III, Spielman A, et al. Concurrent Lyme disease and babesiosis. evidence for increased severity and duration of illness. JAMA. 1996;275:1657-1660.
- Centers for Disease Control and Prevention. Statistics & maps. http://www.cdc.gov/powassan/statistics.html. Updated February 14, 2017. Accessed December 11, 2017.
- Piantadosi A, Rubin DB, McQuillen DP, et al. Emerging cases of Powassan virus encephalitis in New England: clinical presentation, imaging, and review of the literature. Clin Infect Dis. 2016;62:707-713.
- El Khoury MY, Camargo JF, White JL, et al. Potential role of deer tick virus in Powassan encephalitis cases in Lyme disease-endemic areas of New York, U.S.A. Emerg Infect Dis. 2013;19:1926-1933.
- World Health Organization (WHO). Vaccines against tick-borne encephalitis: WHO position paper. Wkly Epidemiol Rec. 2011;86:241-256.
- Centers for Disease Control and Prevention (CDC). Tick-borne encephalitis among U.S. travelers to Europe and Asia—2000-2009. JAMA. 2010;303:2132-2135.
- Valarcher JF, Hägglund S, Juremalm M, et al. Tick-borne encephalitits. Rev Sci Tech. 2015;34:453-466.
- Schultze D, Dollenmaier G, Rohner A, et al. Benefit of detecting tick-borne encephalitis viremia in the first phase of illness. J Clin Virol. 2007;38:172-175.
- Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine. 2003;21(suppl 1):S36-S40.
- Zavadska D, Anca I, André F, et al. Recommendations for tick-borne encephalitis vaccination from the Central European Vaccination Awareness Group. Hum Vaccin Immunother. 2013;9:362-374.
- Gugliotta JL, Goethert HK, Berardi VP, et al. Meningoencephalitis from Borrelia miyamotoi in an immunocompromised patient. N Engl J Med. 2013;368:240-245.
- Hovius JW, de Wever B, Sohne M, et al. A case of meningoencephalitis by the relapsing fever spirochaete Borrelia miyamotoi in Europe. Lancet. 2013;382:658.
- Molloy PJ, Telford SR III, Chowdri HR, et al. Borrelia miyamotoi disease in the northeastern United States: a case series. Ann Intern Med. 2015;163:91-98.
- Telford SR 3rd, Goethert HK, Molloy PJ, et al. Borrelia miyamotoi disease: neither Lyme disease nor relapsing fever. Clin Lab Med. 2015;35:867-882.
- Diaz JH. A comparative meta-analysis of tick paralysis in the United States and Australia. Clin Toxicol (Phila). 2015;53:874-883.
Practice Points
- Apart from the more familiar Borrelia burgdorferi, several less common pathogens associated with diseases transmitted by Ixodes ticks include Anaplasma phagocytophilum, Babesia microti, Borrelia miyamotoi, the Powassan virus, and the tick-borne encephalitis virus.
- Overlap in both the geographic distribution and the clinical presentations of these uncommon pathogens underscores the importance of being familiar with their capacity for causing illness and effective treatment.
- Intoxication with the saliva of some Ixodes species can cause an ascending flaccid tick paralysis.
Perianal Condyloma Acuminatum-like Plaque
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
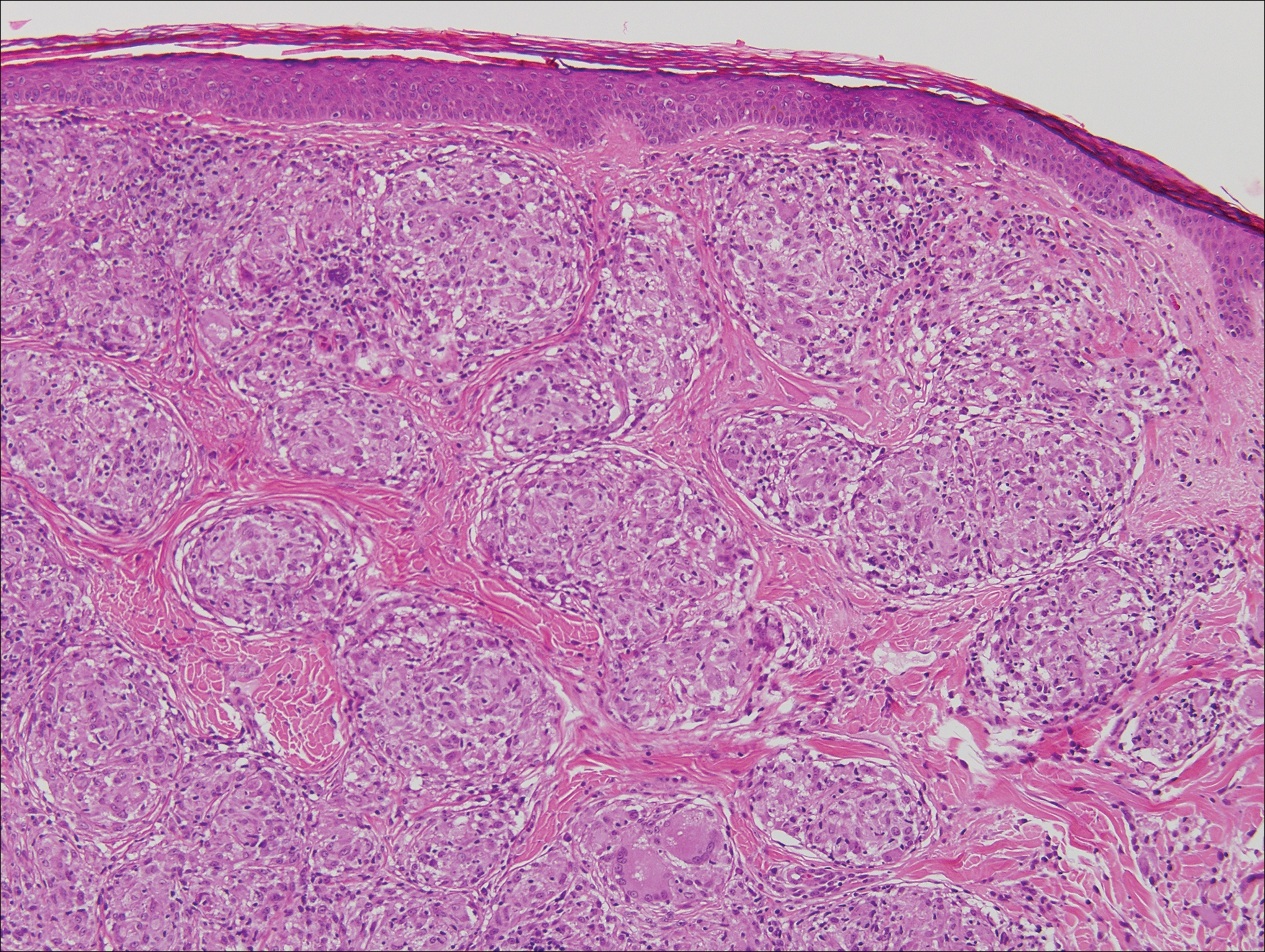
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
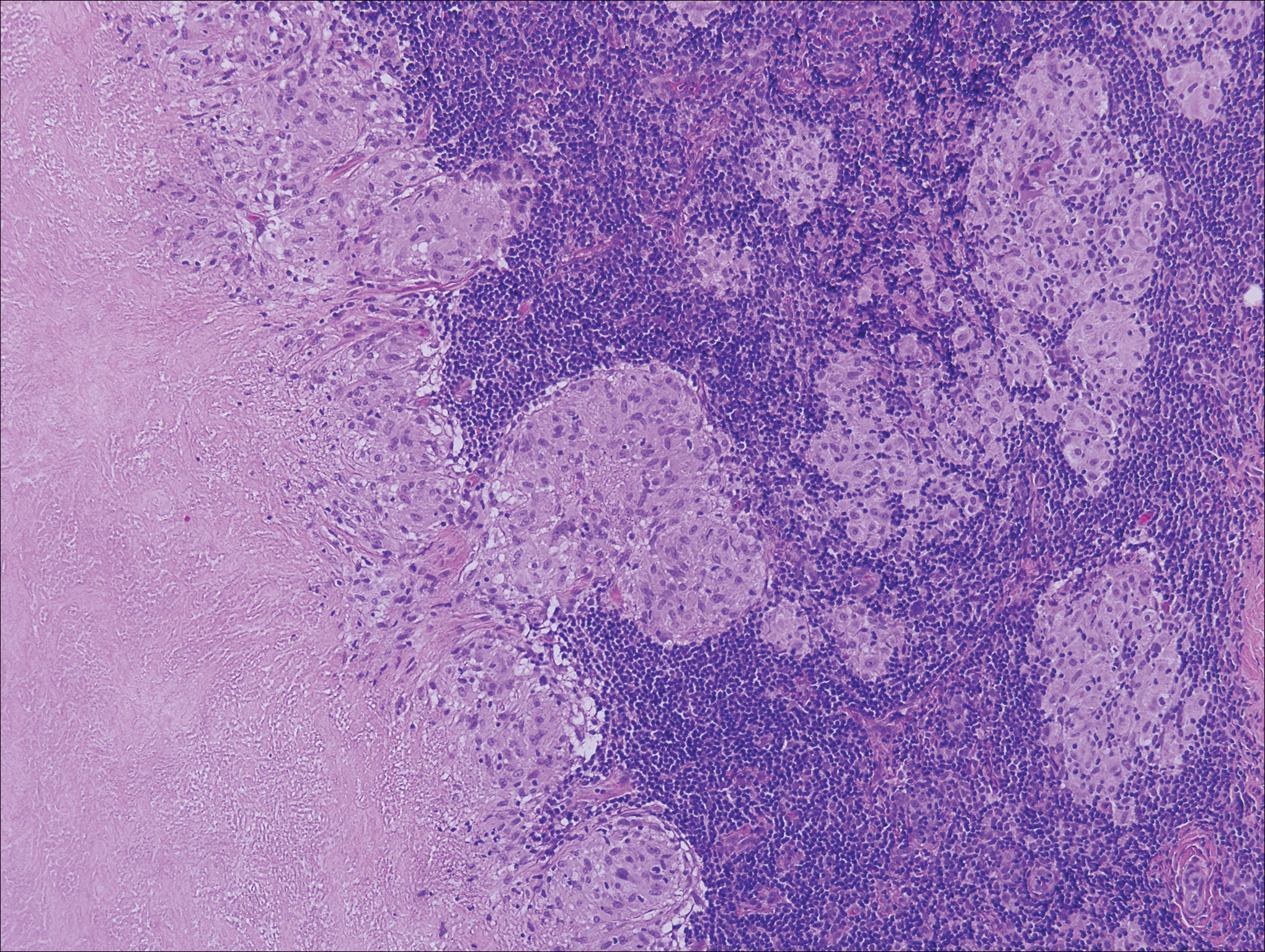
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
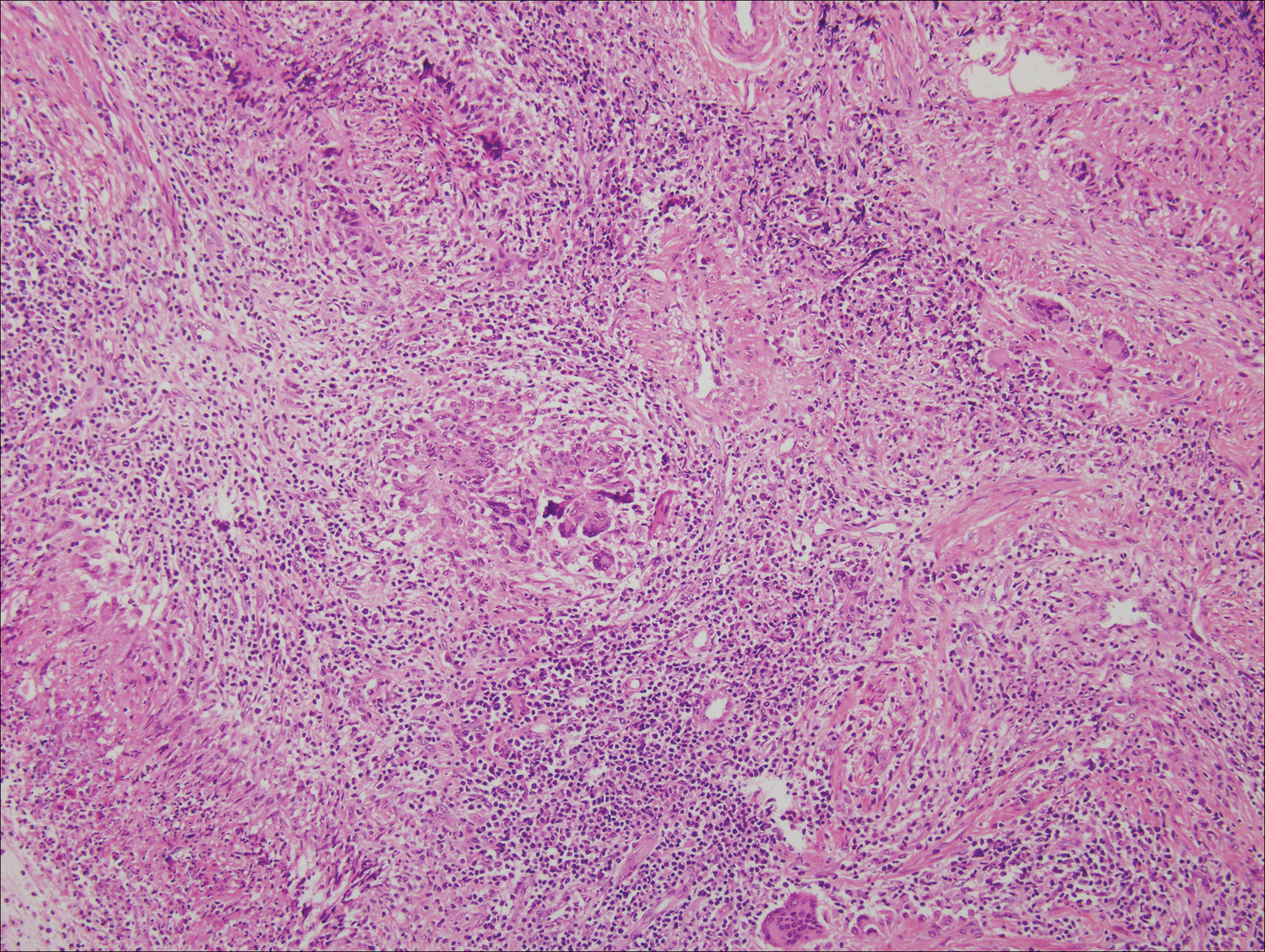
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
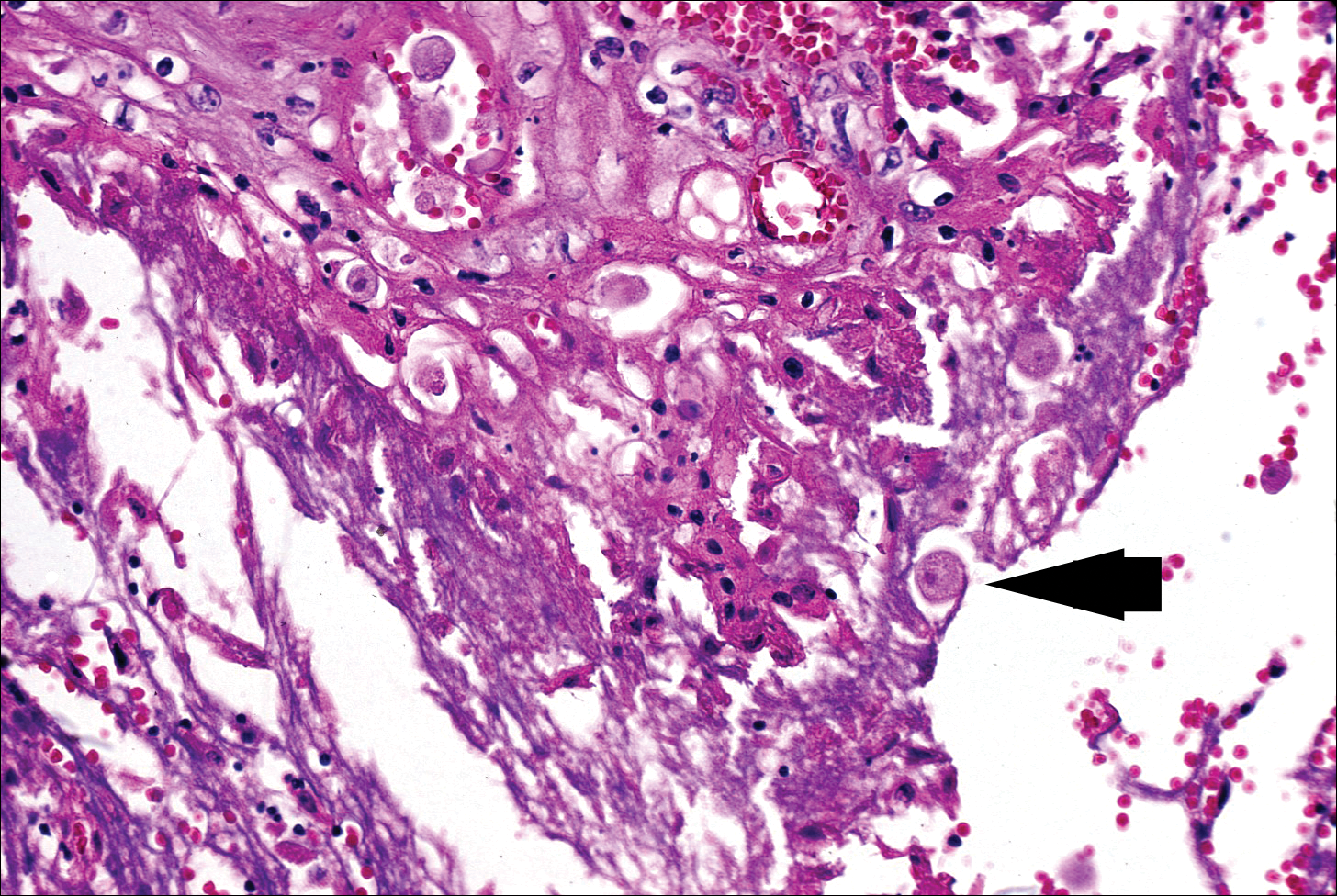
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
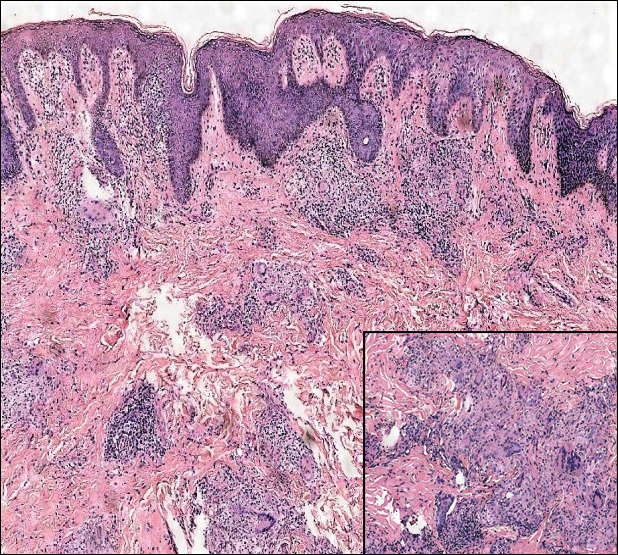
A 19-year-old man presented with a perianal condyloma acuminatum-like plaque of 2 years' duration and a 6-month history of diarrhea.