User login
Autoimmune Progesterone Dermatitis
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
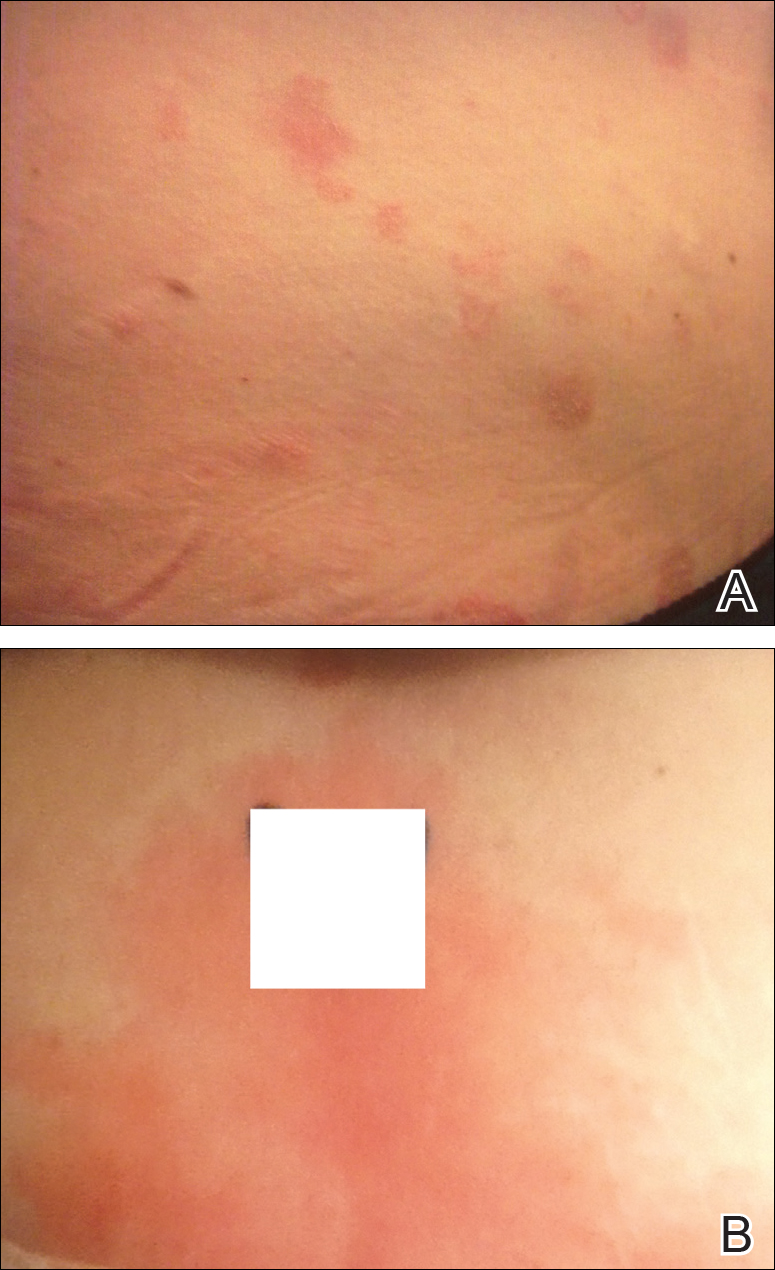
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
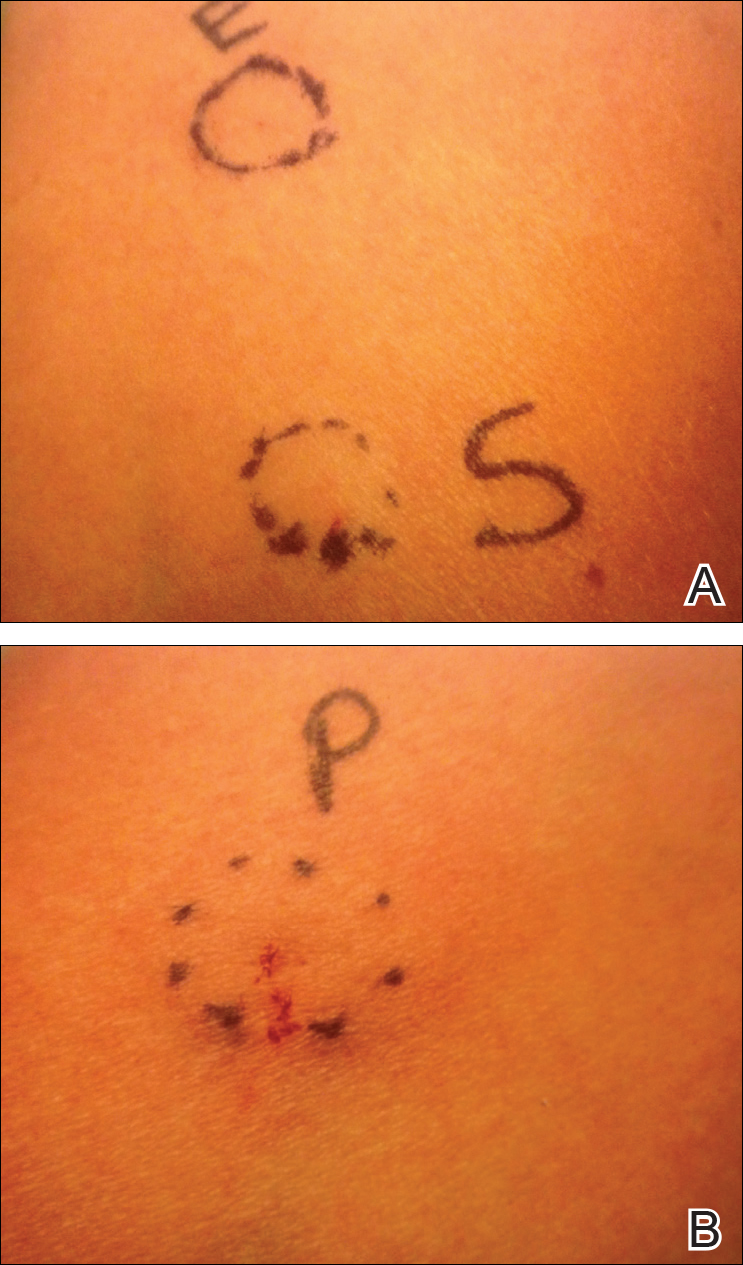
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
Practice Points
- Autoimmune progesterone dermatitis (APD) is a hypersensitivity reaction to the progesterone surge during a woman’s menstrual cycle.
- Patients with APD often are misdiagnosed for years due to the variability of each woman’s menstrual cycle, making the correlation difficult.
- It is important to keep APD in mind for any recalcitrant or recurrent rash in females. A thorough history is critical when formulating a diagnosis.
Mohs Micrographic Surgery Overlying a Pacemaker
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.

Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
Practice Points
- Surgical treatment of a cutaneous lesion overlying a cardiac device requires caution, both in avoidance of the device itself as well as electrocautery interference.
- Local anesthesia infiltrates can be used to create a temporary edematous thickening to minimize potential exposure of the device during the procedure.
Intrahepatic Cholestasis of Pregnancy
To the Editor:
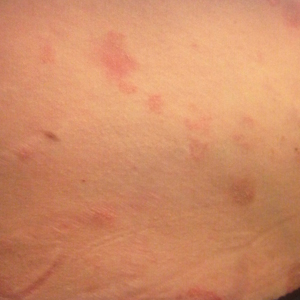
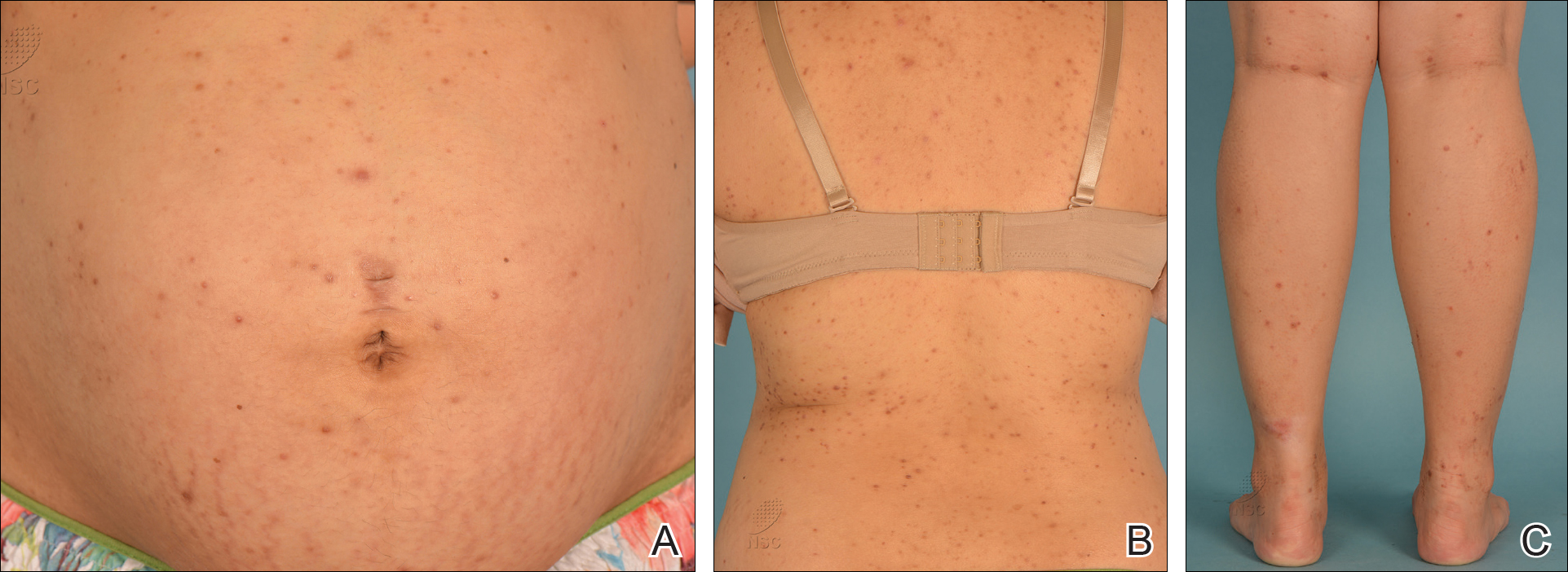
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
To the Editor:
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
To the Editor:
A 28-year-old primigravid woman at 32 weeks’ gestation presented to an outpatient dermatology clinic with a generalized rash and itch of 3 months’ duration. She was distressed with the itch and had tried antihistamines (eg, chlorpheniramine, cetirizine) without relief. She had no notable medical history. Physical examination revealed generalized erythematous papules and nodules on the chest, back, periumbilical region, arms, and legs (Figure). A few pustules were noted on the upper back. No wheals, plaques, vesicles, or bullae were seen.
Laboratory investigations revealed elevated alkaline phosphatase (187 U/L [reference range, 30–120 U/L]), aspartate aminotransferase (45 U/L [reference range, 10–30 U/L]), alanine aminotransferase (120 U/L [reference range, 10–40 U/L]), and γ-glutamyltransferase (48 U/L [reference range, 9–40 U/L]) levels. A fungal scrape of the papules on the upper back demonstrated spores. Subsequent tests included ultrasonography of the liver, which showed fatty changes, as well as rising levels of alkaline phosphatase. Fasting glucose and 2-hour oral glucose tolerance tests showed poorly controlled gestational diabetes mellitus (DM) as well as raised triglycerides.
Based on the patient’s reports of itch, signs of erythematous papules and nodules, and laboratory results of cholestasis, a diagnosis of intrahepatic cholestasis of pregnancy (ICP) was made. The finding of Pityrosporum folliculitis also prompted screening for gestational DM, which was positive.
Treatment with ursodeoxycholic acid (UDCA) 250 mg twice daily was prescribed, which led to some relief of the skin symptoms. Her cutaneous symptoms were discussed with her obstetrician, and a decision was made for emergency cesarean delivery at 37 weeks’ gestation in light of nonreassuring fetal status during her follow-up antenatal ultrasonograph. Her pruritus and poor liver function resolved within 2 weeks after delivery.
Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy. The incidence varies with geographical location and ethnicity.1 It is one of the specific dermatoses of pregnancy and usually presents in the third trimester. It is characterized by pruritus, elevation of serum total bile acids and mild elevations of other liver function tests, and increased rates of adverse fetal outcomes. A positive diagnosis is made by the elevation of the serum total bile acid levels (>11.0 μmol/L [reference range, 0.73–5.63 μmol/L]). It is important for clinicians to recognize ICP because it is associated with fetal prematurity, intrapartal fetal distress, and stillbirths.2
The pathogenesis of ICP is not fully understood. During pregnancy, estrogens interfere with bile acid secretion, and progestins inhibit hepatic glucuronyltransferase. Increased IFN-γ, natural killer cells, and natural killer T cells, as well as decreased T cells in decidua parietalis, also have been reported.3
Mutations in the ATP binding cassette subfamily B member 4 gene, ABCB4, which encodes the multidrug resistance protein 3, a canalicular phosphatidylcholine translocase, have been found in several women with ICP.4 Clinically, patients usually present with pruritus that may precede or follow laboratory abnormalities. The pruritus worsens as the pregnancy advances and can resolve within 48 hours of delivery. Pruritus usually affects the palms and soles but may extend to the legs and abdomen or become generalized.4
Generally, there are no cutaneous signs other than excoriation marks, contrary to primary skin lesions found in other specific dermatoses of pregnancy. Mild jaundice can develop 2 to 4 weeks after the onset of pruritus, which may be associated with subclinical steatorrhea and increased risk of intrapartum and postpartum hemorrhage.5 Of note, ICP may be associated with increased risk for gestational DM, as illustrated in our case.6
Ursodeoxycholic acid currently is the most effective pharmacologic treatment of ICP. It reduces bile acids in cord blood, colostrum, and amniotic fluid.7 A meta-analysis of randomized controlled trials demonstrated that UDCA (450–1200 mg daily) is highly effective in alleviating pruritus and normalizing laboratory abnormalities associated with ICP.8 No severe adverse events have been reported related to UDCA.9,10
Intrahepatic cholestasis of pregnancy has been associated with increased risk for preterm delivery (19%–60%), meconium staining of amniotic fluid (≤27%), fetal bradycardia (≤14%), fetal distress (22%–41%), and fetal loss (0.4%–4.1%).11 The risk for serious fetal complications in ICP makes intensive fetal surveillance mandatory, including weekly nonstress cardiotocography or biophysical assessment from 34 weeks’ gestation. Delivery at 36 weeks or earlier (if lung maturity is achieved and cervix favorable) should be considered for severe cases with jaundice, progressive elevations in serum total bile acids, and suspected fetal distress. At more than 36 weeks’ gestation, amniocentesis and delivery should be considered if cervix is favorable and fetal lung maturity satisfactory.12-14
Our case highlights the importance of diagnosing ICP when a pregnant patient presents with generalized itch associated with elevated liver function tests. Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
- Glantz A, Marschall HU, Mattsson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels and fetal complication rates. Hepatology. 2004;40:467-474.
- Ling B, Yao F, Zhou Y, et al. Cell-mediated immunity imbalance in patients with intrahepatic cholestasis of pregnancy. Cell Mol Immunol. 2007;4:71-75.
- Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genetics. 2000;9:1209-1217.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Martineau M, Raker C, Powrie R, et al. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur J Obstet Gynecol Reprod Biol. 2014;176:80-85.
- Laifer SA, Stiller RJ, Siddiqui DS, et al. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J Matern Fetal Med. 2001;10:131-135.
- Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
- Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129:894-901.
- Tan LK. Obstetric cholestasis: current opinions and management. Ann Acad Med Singapore. 2003;32:294-298.
- Ghosh S, Chaudhuri S. Intra-hepatic cholestasis of pregnancy: a comprehensive review. Indian J Dermatol. 2013;58:327.
- Rioseco AJ, Ivankovic MB, Manzur A, et al. Intrahepatic cholestasis of pregnancy: retrospective case-control study of perinatal outcome. Am J Obstet Gynecol. 1994;170:890-895.
- Saleh MM, Abdo KR. Intrahepatic cholestasis of pregnancy: review of the literature and evaluation of current evidence. J Womens Health (Larchmt). 2007;16:833-841.
- Roncaglia N, Arreghini A, Locatelli A, et al. Obstetric cholestasis: outcome with active management. Eur J Obstet Gynecol Reprod Biol. 2002;100:167-170.
Practice Points
- Intrahepatic cholestasis of pregnancy is a rare form of reversible cholestasis occurring in the second half of pregnancy.
- Interdisciplinary management involving dermatologists, obstetricians, pediatricians, and gastroenterologists is mandatory to acquire a better outcome for the mother and the fetus.
Sweat Regeneration Following CO2 Fractionated Laser Therapy
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
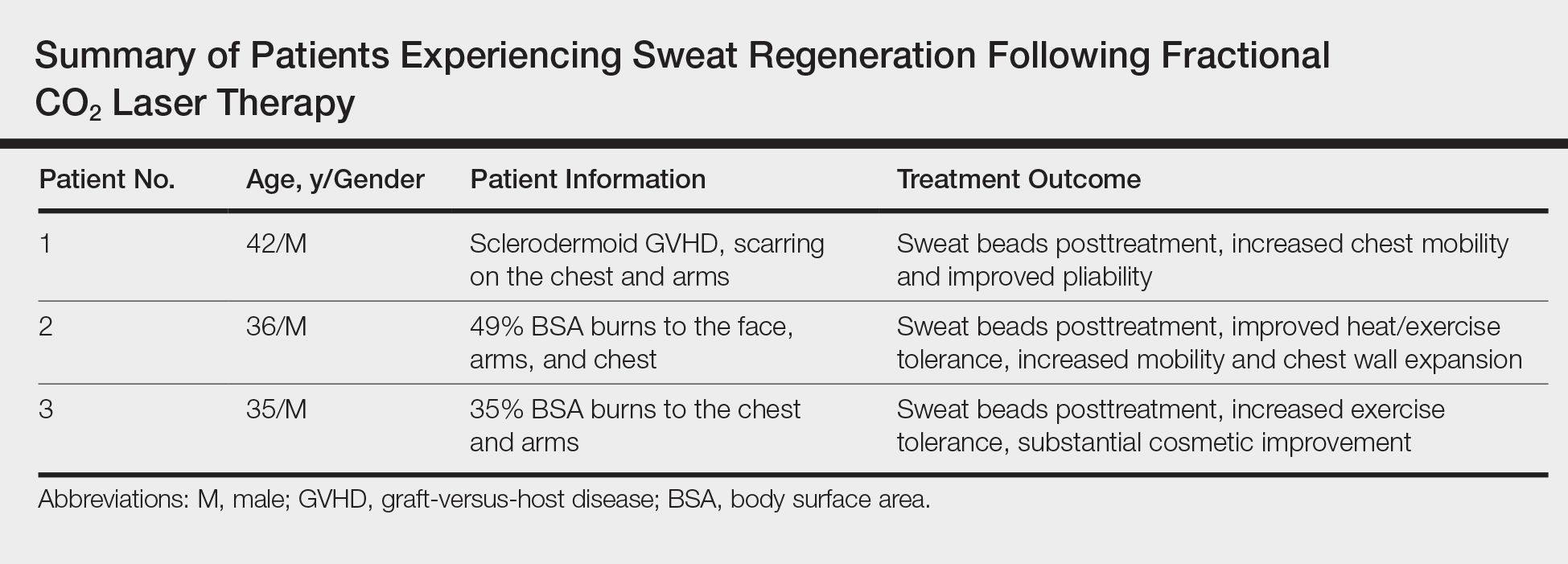
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
To the Editor:
It is not uncommon for patients with extensive dermal scarring to overheat due to the inability to regulate body temperature through evaporative heat loss, as lack of perspiration in areas of prior full-thickness skin injury is well known. One of the authors (C.M.H.) previously reported a case of a patient with considerable hypertrophic scarring after surviving an episode of toxic epidermal necrolysis that was likely precipitated by lamotrigine.1 The patient initially presented to our clinic in consultation for laser therapy to improve the pliability and cosmetic appearance of the scars; however, approximately 3 weeks after initiating treatment with a fractional CO2 laser, the patient noticed perspiration in areas where she once lacked the ability to perspire as well as improved functionality.1 It was speculated that scar remodeling stimulated by the CO2 fractional laser allowed new connections to form between eccrine ducts in the dermis and epidermis.2
These findings are even more notable in light of a study by Rittié et al3 that suggested the primary appendages of the skin involved in human wound healing are the eccrine sweat glands. The investigators were able to demonstrate that eccrine sweat glands are major contributors in reepithelialization and wound healing in humans; therefore, it is possible that stimulating these glands with the CO2 laser may promote enhanced reepithelialization in addition to the reestablishment of perspiration and wound healing.3 Considering inadequate wound repair represents a substantial disturbance to the patient and health care system, this finding offers promise as a potential means to decrease morbidity in patients with dermal scarring from burns and traumatic injuries. We have since evaluated and treated 3 patients who demonstrated sweat regeneration following treatment with the fractional CO2 laser (Table).
A 42-year-old man was our first patient to demonstrate functional scar improvement following bone marrow transplant for acute lymphoblastic leukemia complicated by chronic sclerodermoid graft-versus-host disease and subsequent extensive scarring on the chest and arms. Approximately 2 weeks after the first treatment with the fractional CO2 laser, the patient began to notice the presence of sweat beads in the treated areas. In addition to the reestablishment of perspiration, the patient had perceived increased mobility with improved pliability and “softness” (as described by family members) in treated areas likely related to scar remodeling.
A 36-year-old wounded army veteran presented with burns to the face, arms, and chest affecting 49% of the body surface area. After only 1 treatment, the patient reported that he could subjectively tolerate 10°F more ambient temperature and work all day outside in south Texas when heat intolerance previously would allow him to work only 2 to 3 hours. Additionally, he noted increased mobility and chest wall expansion, which in combination contributed to overall increased exercise tolerance and enhanced quality of life.
A 35-year-old US Marine and firefighter with burns primarily on the chest and arms involving 35% body surface area experienced increased exercise tolerance and sweat regeneration, particularly on the chest after a single treatment with the fractional CO2 laser but continued to experience improvement after a total of 3 treatments. Additionally, the cosmetic improvement was so substantial that the physician (C.M.H) had to review older photographs to verify the location of the scars.
We have now treated 3 patients with various mechanisms of injury and extensive scarring who noticed improved heat tolerance from sweat regeneration following fractional CO2 laser therapy. At this point, we only have anecdotal evidence of subjective functional improvement, and further research is warranted to elucidate the exact mechanism of action to support our findings.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
- Neiner J, Whittemore D, Hivnor C. Buried alive: functional eccrine coils buried under scar tissue? J Am Acad Dermatol. 2011;65:661-663.
- Waibel J, Beer K, Narurkar V, et al. Preliminary observations on fractional ablative resurfacing devices: clinical impressions. J Drugs Dermatol. 2009;8:481-485.
- Rittié L, Sachs D, Orringer J, et al. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am J Pathol. 2013;1:163-171.
Practice Points
- Treatment of dermal scarring with fractional CO2 laser may contribute to eccrine sweat gland regeneration during the remodeling process in addition to increased skin pliability.
- Sweat regeneration has been demonstrated following treatment with fractional CO2 laser in patients with extensive scarring; this case shows sweat regeneration secondary to burns and chronic sclerodermoid graft-versus-host disease.
Vemurafenib-Induced Plantar Hyperkeratosis
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
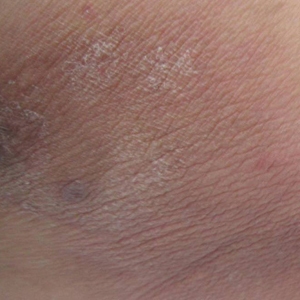
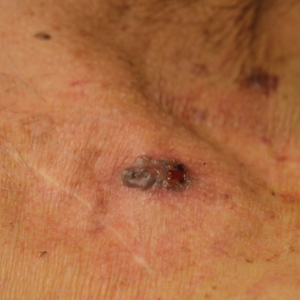
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
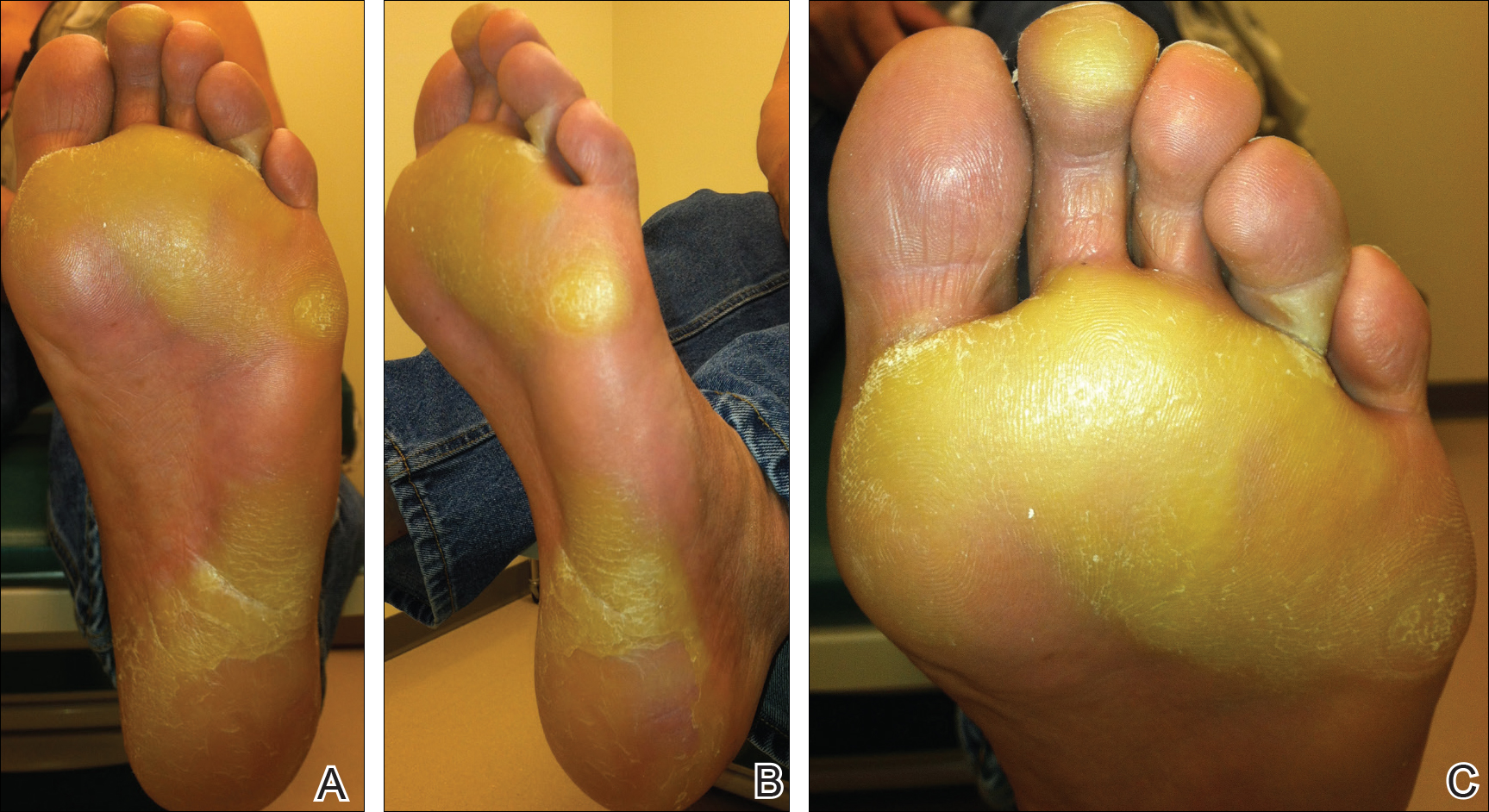
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
To the Editor:
Vemurafenib, a selective BRAF inhibitor, is a chemotherapeutic agent used in the treatment of metastatic melanoma with BRAF mutations. It has been associated with various cutaneous side effects. We report a case of metastatic melanoma with acquired plantar hyperkeratosis secondary to vemurafenib therapy.
A 49-year-old man presented for evaluation of a pigmented plaque on the left pretibial region that had been enlarging over the last 2 months. The lesion had been diagnosed as folliculitis by his primary care physician 1 month prior to the current presentation and was being treated with oral antibiotics. The patient reported occasional bleeding from the lesion but denied other symptoms. Physical examination revealed a 1.4-cm pigmented plaque distributed over the left shin. Excisional biopsy was performed to rule out melanoma. Histopathology revealed well-circumscribed and symmetric proliferation of nested and single atypical melanocytes throughout all layers to the deep reticular dermis, confirming a clinical diagnosis of malignant melanoma. The lesion demonstrated angiolymphatic invasion, mitotic activity, and a Breslow depth of 2.5 mm. The patient underwent wide local excision with 3-cm margins and left inguinal sentinel lymph node biopsy; 2 of 14 lymph nodes were positive for melanoma. Positron emission tomography–computed tomography was negative for further metastatic disease. The patient underwent isolated limb perfusion with ipilimumab, but treatment was discontinued due to regional progression of multiple cutaneous metastases that were positive for the BRAF V600E mutation.
The patient was then started on vemurafenib therapy. Within 2 weeks, the patient reported various cutaneous symptoms, including morbilliform drug eruption covering approximately 70% of the body surface area that resolved with topical steroids and oral antihistamines, as well as the appearance of melanocytic nevi on the posterior neck, back, and abdomen. After 5 months of vemurafenib therapy, the patient began to develop hyperkeratosis of the bilateral soles of the feet (Figure). A diagnosis of acquired plantar hyperkeratosis secondary to vemurafenib therapy was made. Treatment with keratolytics was initiated and vemurafenib was not discontinued. The patient died approximately 1 year after therapy was started.
Metastatic melanoma is challenging to treat and continues to have a high mortality rate; however, newer chemotherapeutic agents targeting specific mutations found in melanoma, including the BRAF V600E mutation, are promising.
The US Food and Drug Administration first approved vemurafenib, a selective BRAF inhibitor, in 2011 for treatment of metastatic melanoma. Activating BRAF mutations have been detected in up to 60% of cutaneous melanomas.1 In the majority of these mutations, valine (V) is inserted at codon 600 instead of glutamic acid (E); therefore, the mutation is named V600E.2 In a phase 3 trial of 675 metastatic melanoma patients with positive V600E who were randomized to receive either vemurafenib or dacarbazine, the overall survival rate in the vemurafenib group improved by 84% versus 64% in the dacarbazine group at 6 months.3
Vemurafenib and other BRAF inhibitors have been associated with multiple cutaneous side effects, including rash, alopecia, squamous cell carcinoma, photosensitivity, evolution of existing nevi, and less commonly palmoplantar hyperkeratosis.2-5 Constitutional symptoms including arthralgia, nausea, and fatigue also have been commonly reported.2-5 In several large studies comprising 1138 patients, cutaneous side effects were seen in 92% to 95% of patients.3,5 Adverse effects caused interruption or modification of therapy in 38% of patients.3
Palmoplantar keratoderma is a known side effect of vemurafenib therapy, but it is less commonly reported than other cutaneous adverse effects. It is believed that vemurafenib has the ability to paradoxically activate the mitogen-activated protein kinase pathway, leading to keratinocyte proliferation in cells without BRAF mutations.6-8 In the phase 3 trial, approximately 23% to 30% of patients developed some form of hyperkeratosis.5 Comparatively, 64% of patients developed a rash and 23% developed cutaneous squamous cell carcinoma. Incidence of palmoplantar hyperkeratosis was similar in the vemurafenib and dabrafenib groups (6% vs 8%).3,9 Development of keratoderma also has been associated with other multikinase inhibitors (eg, sorafenib, sunitinib).10,11
In our case, the patient displayed multiple side effects while undergoing vemurafenib therapy. Within the first 2 weeks of therapy, he experienced a drug eruption that affected approximately 70% of the body surface area. The eruption resolved with topical steroids and oral antihistamines. The patient also noted the appearance of several new melanocytic nevi on the posterior neck as well as several evolving nevi on the back and abdomen. Five months into the treatment cycle, the patient began to develop hyperkeratosis on the bilateral plantar feet. Treatment consisted of keratolytics. Vemurafenib therapy was not discontinued secondary to any adverse effects.
Vemurafenib and other BRAF inhibitors are efficacious in the treatment of metastatic melanoma with V600E mutations. The use of these therapies is likely to continue and increase in the future. BRAF inhibitors have been associated with a variety of side effects, including palmoplantar hyperkeratosis. Awareness of and appropriate response to adverse reactions is essential to proper patient care and continuation of potentially life-extending therapies.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
- Davies H, Bignell GR, Cox C, et al. Mutations in the BRAF gene in human cancer. Nature. 2002;417:949-954.
- Cohen PR, Bedikian AY, Kim KB. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Rinderknecht JD, Goldinger SM, Rozati S, et al. RASopathic skin eruptions during vemurafenib therapy [published online March 13, 2014]. PLoS One. 2013;8:e58721.
- Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18:314-322.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969-978.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Autier J, Escudier B, Wechsler J, et al. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886-892.
- Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy—classification and management. J Dtsch Dermatol Ges. 2010;8:652-661.
Practice Points
- BRAF inhibitors such as vemurafenib are associated with a high incidence of cutaneous side effects, including rash, hyperkeratosis, and cutaneous squamous cell carcinoma.
- Practitioners should be aware of these side effects and their management to avoid discontinuation or interruption of therapy.
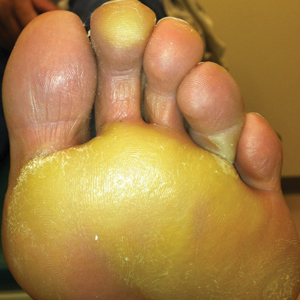
Terra Firma-Forme Dermatosis Mimicking Livedo Racemosa
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
Practice Points
- Clinicians should include terra firma-forme dermatosis in the differential diagnosis of any hyperpigmented condition, regardless of pattern of presentation.
- Clean the skin with an alcohol wipe to rule out a diagnosis of terra firma-forme dermatosis.
Microcystic Adnexal Carcinoma of the External Auditory Canal
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.

Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
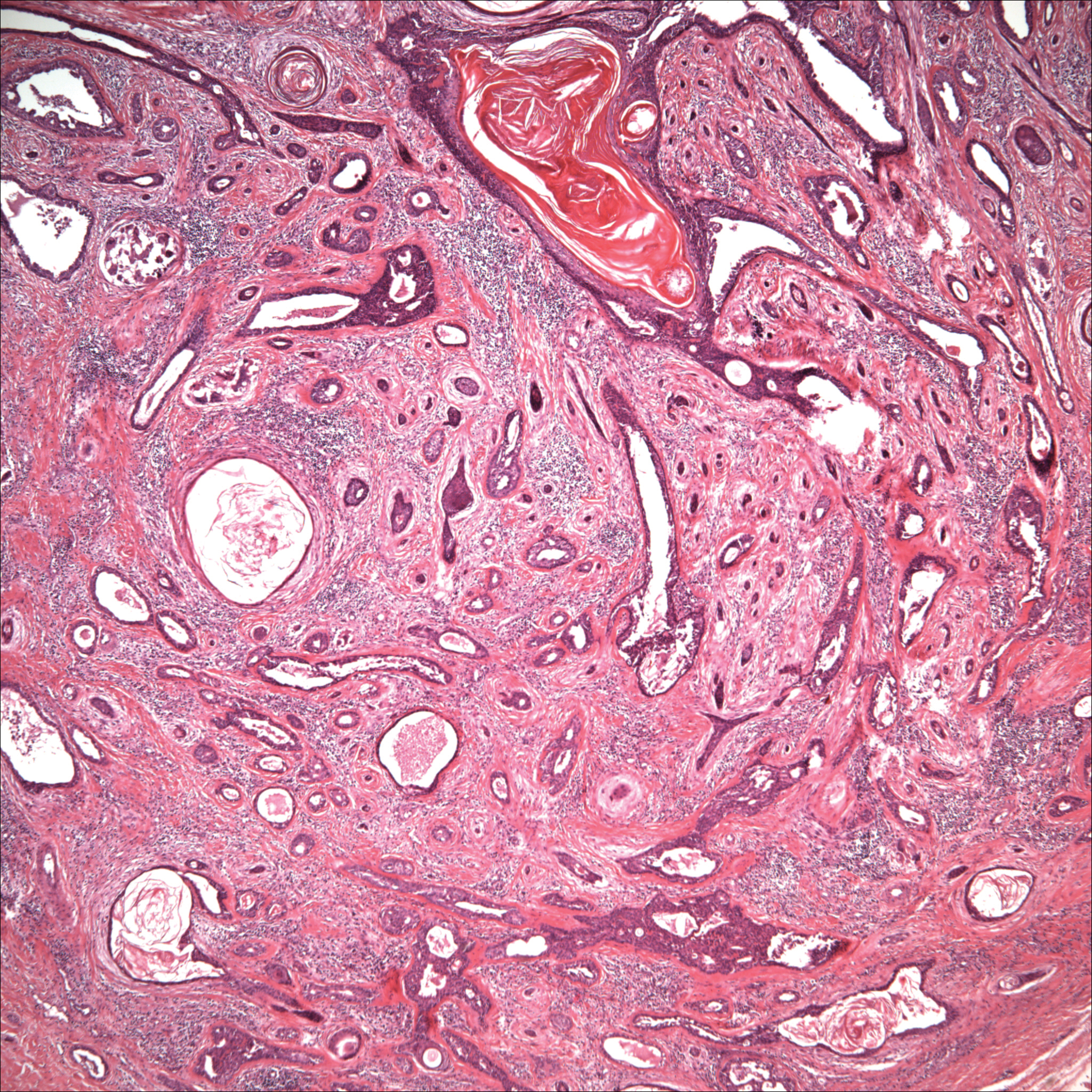
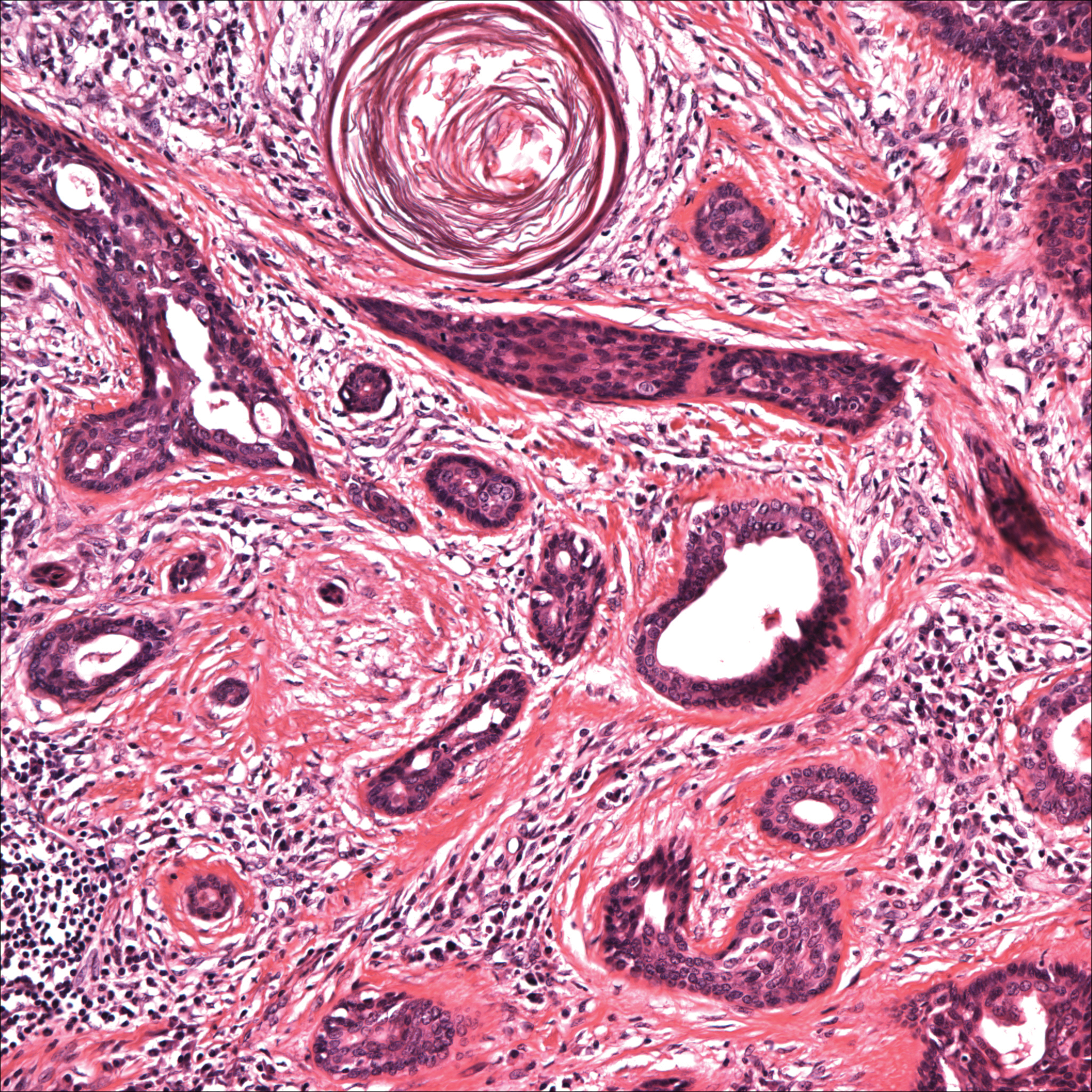
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
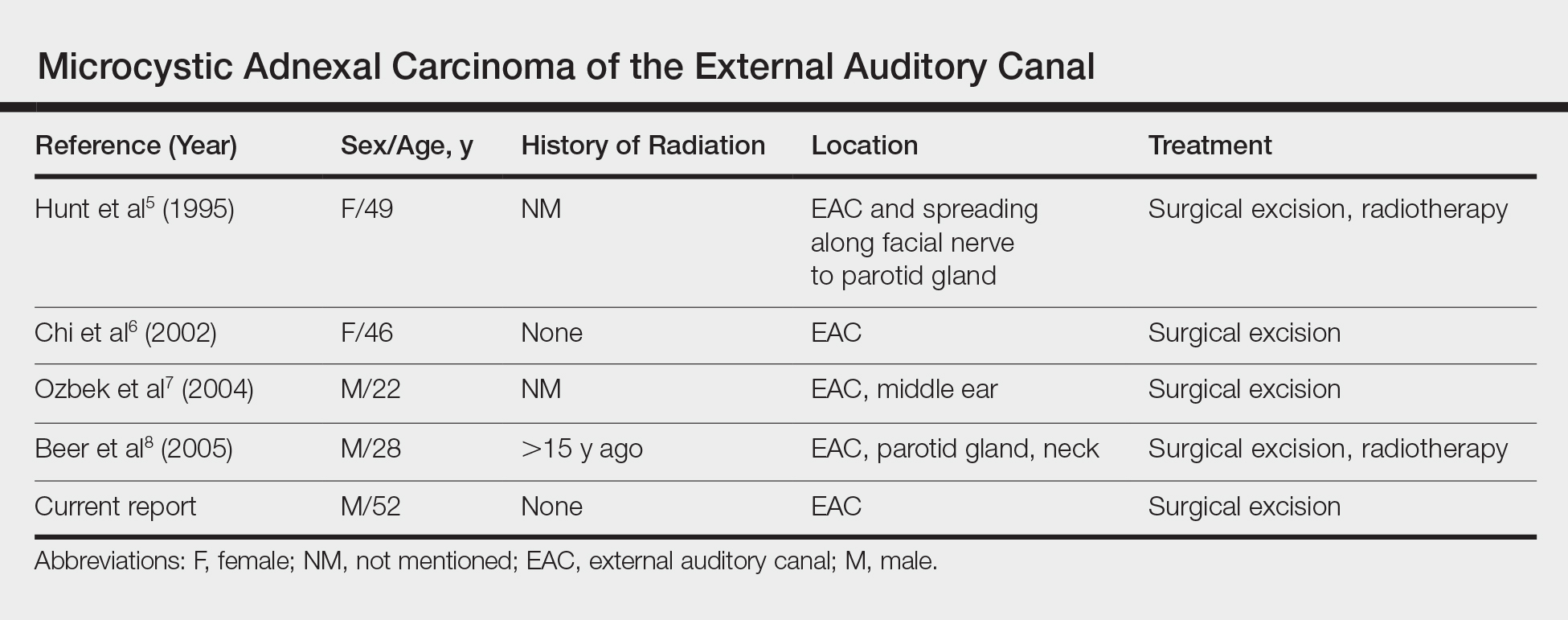
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
Practice Points
- Microcystic adnexal carcinoma is a locally aggressive malignant adnexal tumor with a high potential for local recurrence.
- The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.
- Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence is essential.
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.
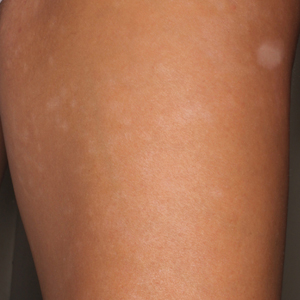
Gottron Papules Mimicking Dermatomyositis: An Unusual Manifestation of Systemic Lupus Erythematosus
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
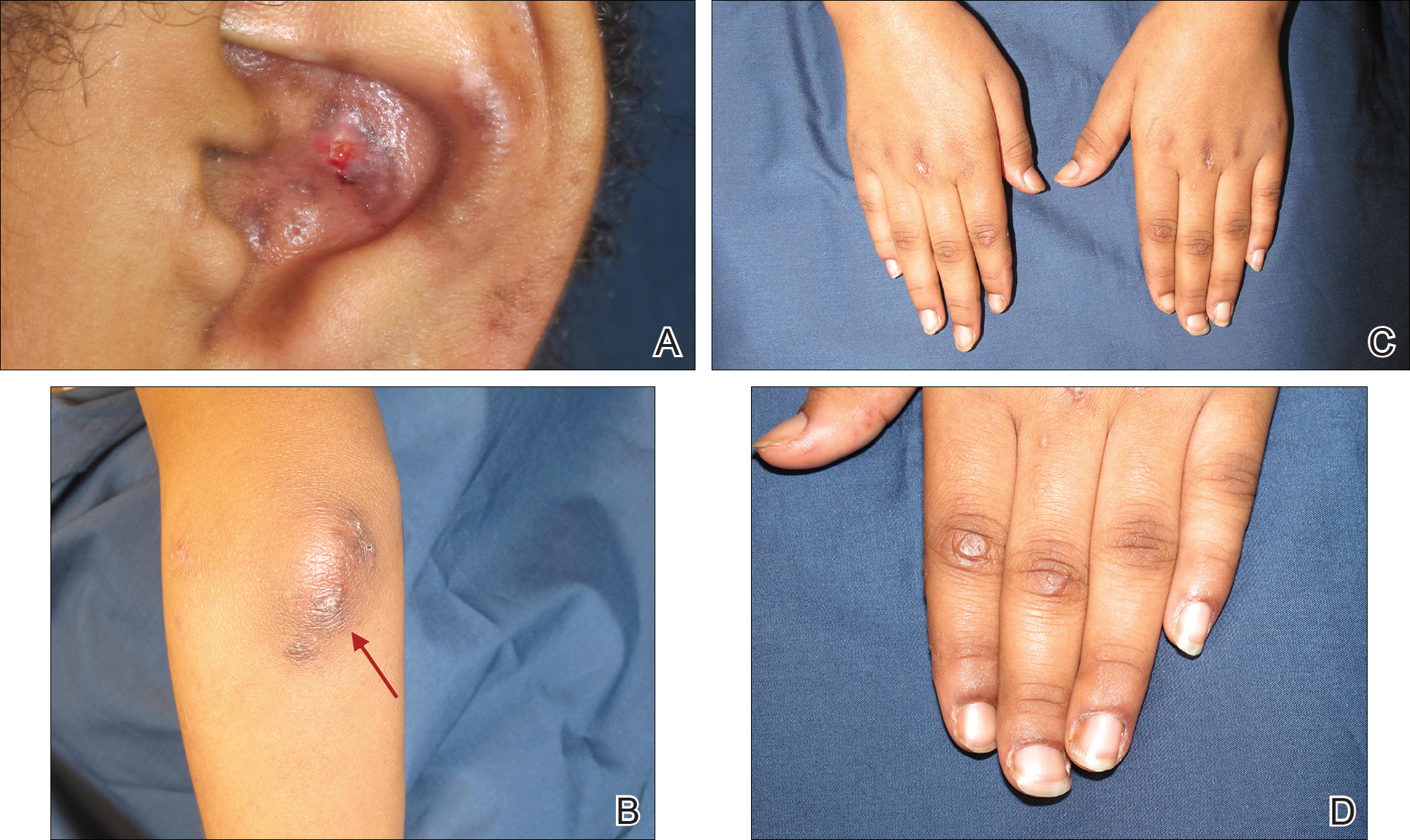
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
Practice Points
- Gottron-like papules can be a dermatologic presentation of lupus erythematosus.
- When present along with other findings of lupus erythematosus without any clinical manifestations of dermatomyositis, Gottron-like papules can be thought of as a manifestation of lupus erythematosus rather than dermatomyositis.

Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.