User login
Copresentation of Common Variable Immune Deficiency and Sweet Syndrome
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
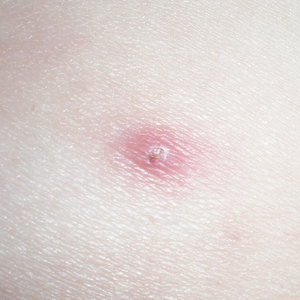
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.


Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
Practice Points
- Suggested workup for Sweet syndrome includes ruling out connective tissue disorders and malignancies.
- Familial common variable immune deficiency is rare and can first manifest in adulthood.
Chilblain Lupus Erythematosus Presenting With Bilateral Hemorrhagic Bullae of Distal Halluces
To the Editor:
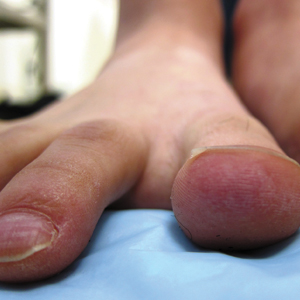
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.

(H&E, original magnification ×400) with deep periadnexal mucin deposition (B)(colloidal iron, original magnification ×40).")
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
Practice Points
- Up to 20% of patients with chilblain lupus erythematosus (CHLE) will develop systemic lupus erythematosus (SLE), necessitating close long-term follow-up.
- Medications such as antihypertensives, antifungals, chemotherapeutic agents, and tumor necrosis factor 11α inhibitors have been reported to trigger CHLE.
- Chilblain lupus erythematosus is less responsive to traditional antimalarial agents commonly used to treat SLE.
- Management of CHLE includes physical protection from cold environments, calcium channel blockers, topical and systemic steroids, and pentoxifylline, among other treatment modalities.
Epidermolysis Bullosa Acquisita in Association With Mantle Cell Lymphoma
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
 and multiple bullae on the sole (B).")
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
.")
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
Practice Points
- Epidermolysis bullosa acquisita (EBA) is an uncommon blistering disorder and few cases have been associated with malignancy.
- Diagnosis of EBA is challenging and requires exclusion of other blistering diseases.
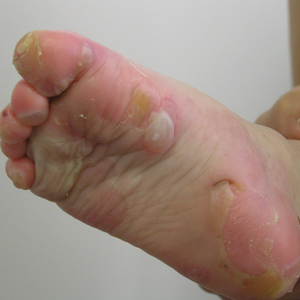
Polypoid Melanoma: An Aggressive Variant of Nodular Melanoma
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.


.")
 immunostain (original magnification ×200).")
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
Practice Points
- The differential diagnosis of polypoid melanoma includes pyogenic granuloma and squamous cell carcinoma.
- Polypoid melanoma has a poor prognosis because of its thickness and ulceration at the time of diagnosis and the risk of vascular embolization.
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.
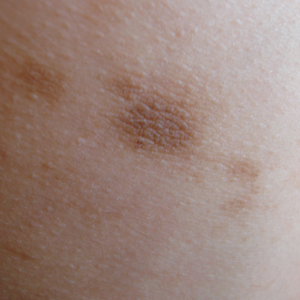
Sweet Syndrome With Aseptic Splenic Abscesses and Multiple Myeloma
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
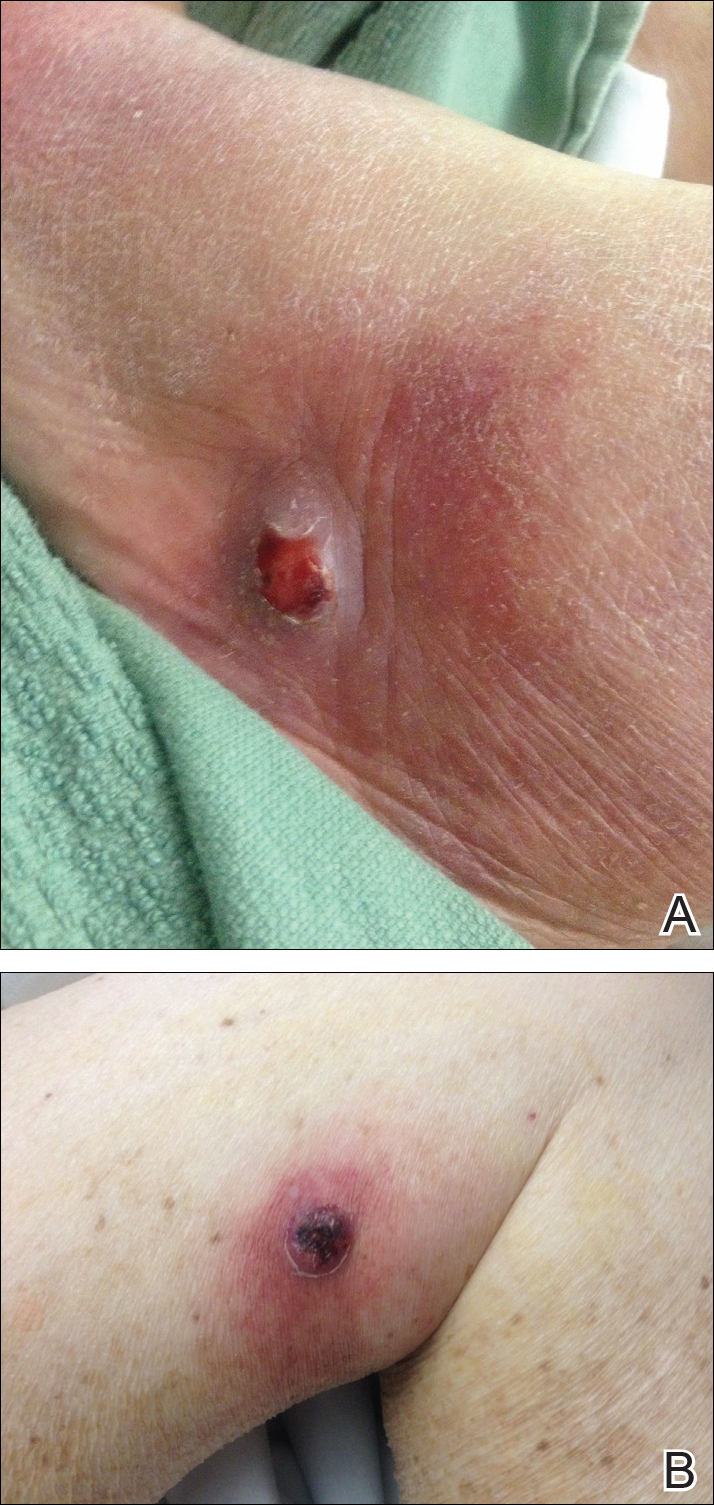
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
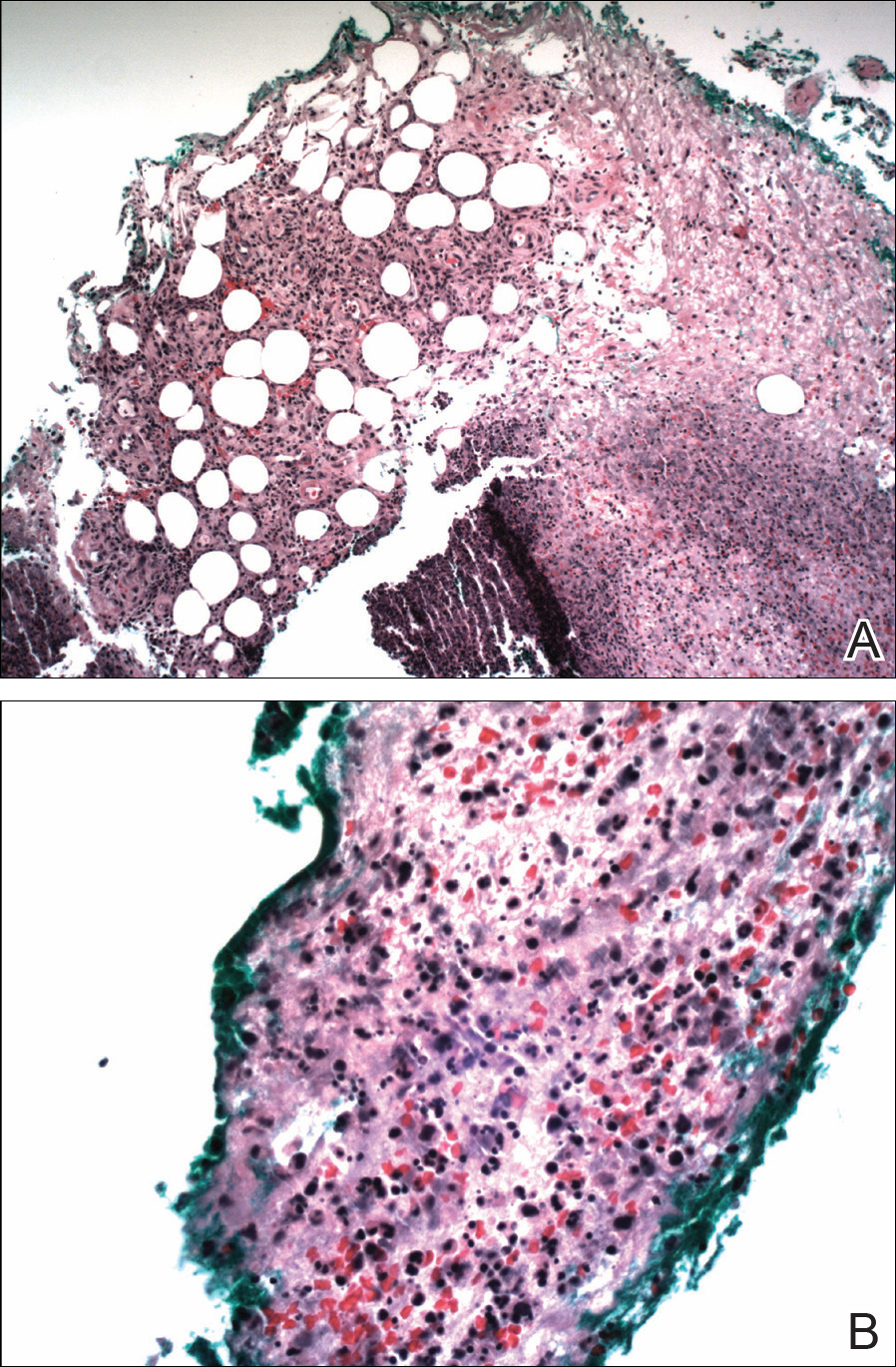
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
Practice Points
- Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an inflammatory process characterized by a diffuse dermal neutrophilic infiltrate in the absence of vasculitis.
- A diagnosis of SS warrants further investigation due to its association with malignancy, especially hematologic malignancy.
- Other organs in SS also may have aseptic involvement.
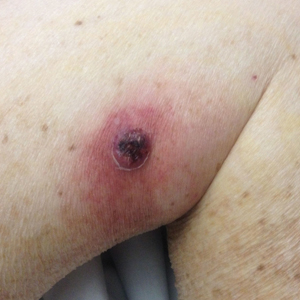
Beltlike Lichen Planus Pigmentosus Complicated With Focal Amyloidosis
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.

Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.

.")
.")
.")
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
Practice Points
- Lichen planus pigmentosus can present in a unique beltlike distribution pattern.
- Focal amyloidosis due to chronic friction and scratching cannot be excluded from the differential diagnosis.
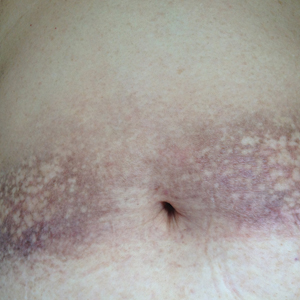
Acute Generalized Exanthematous Pustulosis Caused by Pantoprazole
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
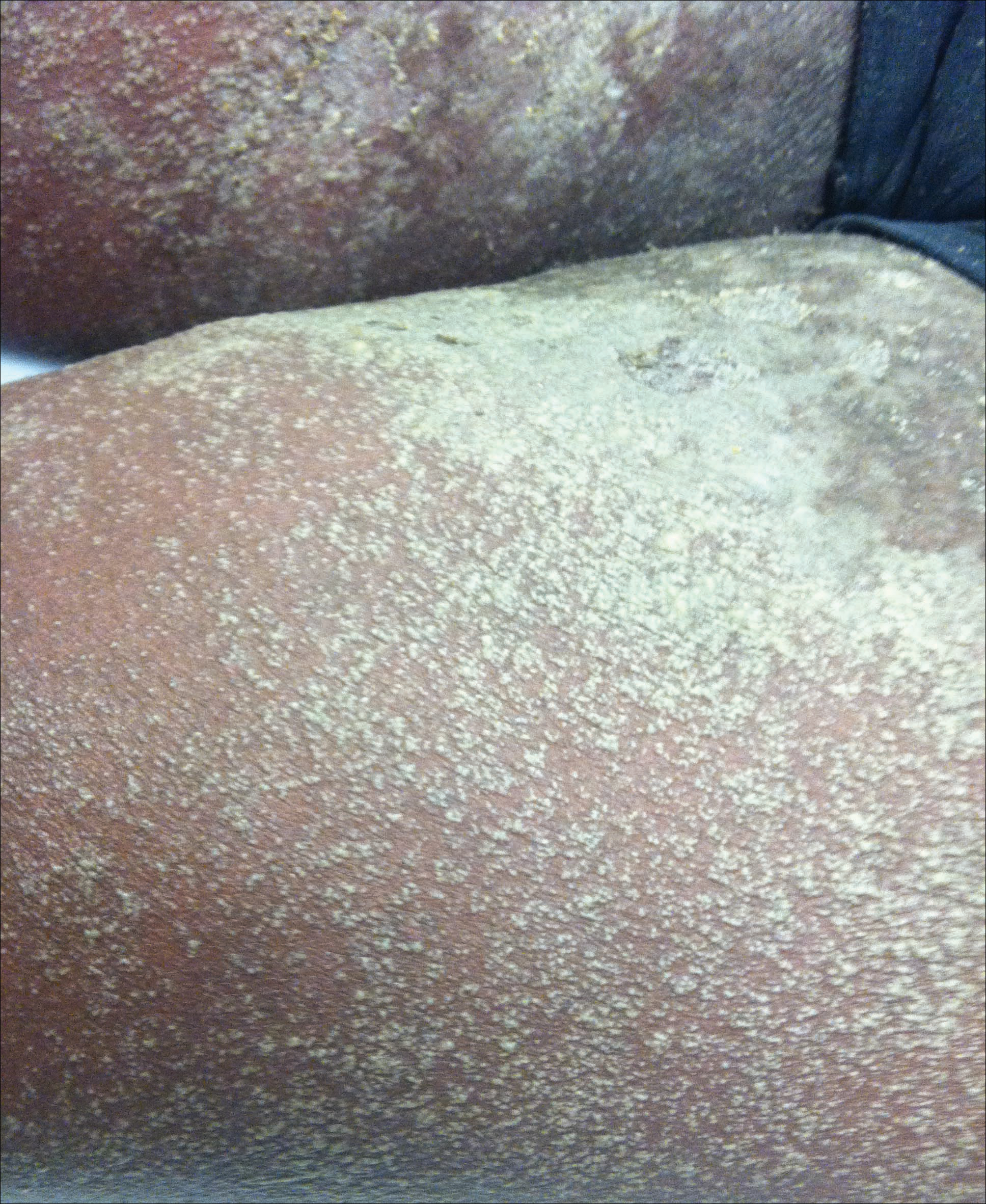
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.

Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
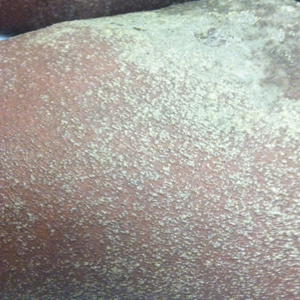
Interstitial Granulomatous Dermatitis and Palisaded Neutrophilic Granulomatous Dermatitis
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.
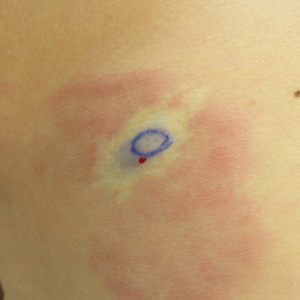
Tinea Incognito in a Tattoo
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.

Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
Practice Points
- Health care providers should have a low threshold to perform a potassium hydroxide preparation when the possibility of a superficial fungal infection is considered.
- Tinea incognito occurs when a superficial fungal infection has unusual clinical features in the setting of local immune suppression.
