User login
Lenalidomide may reduce risk of progression from SMM to MM
Lenalidomide can reduce the risk of progression from smoldering multiple myeloma (SMM) to multiple myeloma (MM), according to a phase 2/3 trial.
At 3 years, the rate of progression-free survival (PFS) was 91% in SMM patients randomized to lenalidomide and 66% in those randomized to observation.
However, more than half of patients randomized to lenalidomide discontinued treatment because of toxicity.
These results are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, discussed the results in a press briefing in advance of the meeting.
A prior trial suggested that lenalidomide plus dexamethasone can improve time to MM development and overall survival in patients with high-risk SMM (Mateos MV et al. NEJM 2013). However, inferior imaging was used in this trial, and the addition of dexamethasone hindered researchers’ ability to isolate the effects of lenalidomide, Dr. Lonial said.
With their trial (NCT01169337), Dr. Lonial and colleagues tested lenalidomide alone and screened patients using magnetic resonance imaging.
The trial enrolled patients with intermediate or high-risk SMM in two phases. In phase 2, all 44 patients received lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle. They also received aspirin at 325 mg on days 1-28.
In the phase 3 portion of the trial, 182 patients were randomized to observation or lenalidomide and aspirin at the aforementioned dose and schedule. Patients were stratified according to time since SMM diagnosis – 1 year or less vs. more than 1 year.
Safety
Dr. Lonial said, in general, lenalidomide was “very well tolerated.” However, 80% of patients in phase 2 and 51% in phase 3 discontinued lenalidomide due to toxicity.
The rates of treatment-related adverse events (AEs) in the phase 2 portion were 34.1% for grade 3 AEs, 11.4% for grade 4, and 4.5% for grade 5. In the phase 3 portion, 35.2% of patients had grade 3 treatment-related AEs, and 5.7% had grade 4 treatment-related AEs.
Common AEs in phase 3 were grade 4 neutrophil count decrease (4.5%) and grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin AEs (5.7%), dyspnea (5.7%), and hypokalemia (3.4%).
Efficacy
“It is worth noting that about 50% of patients had an objective response to lenalidomide in both the phase 2 and the phase 3 trial,” Dr. Lonial said. “I think it’s also important to realize that, in the phase 2 portion of this study, of the 44 patients enrolled, 78% of them did not progress to myeloma with a median follow-up of over 5 years.”
In phase 2, PFS was 98% at 1 year, 87% at 3 years, and 78% at 5 years.
In phase 3, PFS was 98% in the lenalidomide arm and 89% in the observation arm at 1 year. At 2 years, PFS was 93% in the lenalidomide arm and 76% in the observation arm. At 3 years, PFS was 91% in the lenalidomide arm and 66% in the observation arm.
“What’s really quite interesting is that each [risk] group appeared to benefit almost equally from the early intervention of lenalidomide as a single agent,” Dr. Lonial said. “[W]hile the high-risk group may be the target now, this may be a fertile area for investigation in the intermediate-risk group as well.”
Dr. Lonial has relationships with AbbVie, Amgen, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Janssen Oncology, Juno Therapeutics, Merck, Novartis, and Takeda. The trial was funded by the National Institutes of Health.
SOURCE: Lonial S et al. ASCO 2019. Abstract 8001.
Lenalidomide can reduce the risk of progression from smoldering multiple myeloma (SMM) to multiple myeloma (MM), according to a phase 2/3 trial.
At 3 years, the rate of progression-free survival (PFS) was 91% in SMM patients randomized to lenalidomide and 66% in those randomized to observation.
However, more than half of patients randomized to lenalidomide discontinued treatment because of toxicity.
These results are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, discussed the results in a press briefing in advance of the meeting.
A prior trial suggested that lenalidomide plus dexamethasone can improve time to MM development and overall survival in patients with high-risk SMM (Mateos MV et al. NEJM 2013). However, inferior imaging was used in this trial, and the addition of dexamethasone hindered researchers’ ability to isolate the effects of lenalidomide, Dr. Lonial said.
With their trial (NCT01169337), Dr. Lonial and colleagues tested lenalidomide alone and screened patients using magnetic resonance imaging.
The trial enrolled patients with intermediate or high-risk SMM in two phases. In phase 2, all 44 patients received lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle. They also received aspirin at 325 mg on days 1-28.
In the phase 3 portion of the trial, 182 patients were randomized to observation or lenalidomide and aspirin at the aforementioned dose and schedule. Patients were stratified according to time since SMM diagnosis – 1 year or less vs. more than 1 year.
Safety
Dr. Lonial said, in general, lenalidomide was “very well tolerated.” However, 80% of patients in phase 2 and 51% in phase 3 discontinued lenalidomide due to toxicity.
The rates of treatment-related adverse events (AEs) in the phase 2 portion were 34.1% for grade 3 AEs, 11.4% for grade 4, and 4.5% for grade 5. In the phase 3 portion, 35.2% of patients had grade 3 treatment-related AEs, and 5.7% had grade 4 treatment-related AEs.
Common AEs in phase 3 were grade 4 neutrophil count decrease (4.5%) and grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin AEs (5.7%), dyspnea (5.7%), and hypokalemia (3.4%).
Efficacy
“It is worth noting that about 50% of patients had an objective response to lenalidomide in both the phase 2 and the phase 3 trial,” Dr. Lonial said. “I think it’s also important to realize that, in the phase 2 portion of this study, of the 44 patients enrolled, 78% of them did not progress to myeloma with a median follow-up of over 5 years.”
In phase 2, PFS was 98% at 1 year, 87% at 3 years, and 78% at 5 years.
In phase 3, PFS was 98% in the lenalidomide arm and 89% in the observation arm at 1 year. At 2 years, PFS was 93% in the lenalidomide arm and 76% in the observation arm. At 3 years, PFS was 91% in the lenalidomide arm and 66% in the observation arm.
“What’s really quite interesting is that each [risk] group appeared to benefit almost equally from the early intervention of lenalidomide as a single agent,” Dr. Lonial said. “[W]hile the high-risk group may be the target now, this may be a fertile area for investigation in the intermediate-risk group as well.”
Dr. Lonial has relationships with AbbVie, Amgen, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Janssen Oncology, Juno Therapeutics, Merck, Novartis, and Takeda. The trial was funded by the National Institutes of Health.
SOURCE: Lonial S et al. ASCO 2019. Abstract 8001.
Lenalidomide can reduce the risk of progression from smoldering multiple myeloma (SMM) to multiple myeloma (MM), according to a phase 2/3 trial.
At 3 years, the rate of progression-free survival (PFS) was 91% in SMM patients randomized to lenalidomide and 66% in those randomized to observation.
However, more than half of patients randomized to lenalidomide discontinued treatment because of toxicity.
These results are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, discussed the results in a press briefing in advance of the meeting.
A prior trial suggested that lenalidomide plus dexamethasone can improve time to MM development and overall survival in patients with high-risk SMM (Mateos MV et al. NEJM 2013). However, inferior imaging was used in this trial, and the addition of dexamethasone hindered researchers’ ability to isolate the effects of lenalidomide, Dr. Lonial said.
With their trial (NCT01169337), Dr. Lonial and colleagues tested lenalidomide alone and screened patients using magnetic resonance imaging.
The trial enrolled patients with intermediate or high-risk SMM in two phases. In phase 2, all 44 patients received lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle. They also received aspirin at 325 mg on days 1-28.
In the phase 3 portion of the trial, 182 patients were randomized to observation or lenalidomide and aspirin at the aforementioned dose and schedule. Patients were stratified according to time since SMM diagnosis – 1 year or less vs. more than 1 year.
Safety
Dr. Lonial said, in general, lenalidomide was “very well tolerated.” However, 80% of patients in phase 2 and 51% in phase 3 discontinued lenalidomide due to toxicity.
The rates of treatment-related adverse events (AEs) in the phase 2 portion were 34.1% for grade 3 AEs, 11.4% for grade 4, and 4.5% for grade 5. In the phase 3 portion, 35.2% of patients had grade 3 treatment-related AEs, and 5.7% had grade 4 treatment-related AEs.
Common AEs in phase 3 were grade 4 neutrophil count decrease (4.5%) and grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin AEs (5.7%), dyspnea (5.7%), and hypokalemia (3.4%).
Efficacy
“It is worth noting that about 50% of patients had an objective response to lenalidomide in both the phase 2 and the phase 3 trial,” Dr. Lonial said. “I think it’s also important to realize that, in the phase 2 portion of this study, of the 44 patients enrolled, 78% of them did not progress to myeloma with a median follow-up of over 5 years.”
In phase 2, PFS was 98% at 1 year, 87% at 3 years, and 78% at 5 years.
In phase 3, PFS was 98% in the lenalidomide arm and 89% in the observation arm at 1 year. At 2 years, PFS was 93% in the lenalidomide arm and 76% in the observation arm. At 3 years, PFS was 91% in the lenalidomide arm and 66% in the observation arm.
“What’s really quite interesting is that each [risk] group appeared to benefit almost equally from the early intervention of lenalidomide as a single agent,” Dr. Lonial said. “[W]hile the high-risk group may be the target now, this may be a fertile area for investigation in the intermediate-risk group as well.”
Dr. Lonial has relationships with AbbVie, Amgen, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Janssen Oncology, Juno Therapeutics, Merck, Novartis, and Takeda. The trial was funded by the National Institutes of Health.
SOURCE: Lonial S et al. ASCO 2019. Abstract 8001.
REPORTING FROM ASCO 2019
VA system lags in getting DMARDs to veterans with inflammatory arthritis
MADISON, WISC. – Only half of United States veterans with inflammatory arthritis received disease-modifying medication within 90 days of diagnosis if they received care within the Veterans Health Administration, according to a study presented at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN).
Over the study period, 58.2% of all inflammatory arthritis patients began a disease-modifying antirheumatic drug (DMARD) within 12 months of diagnosis. Rates of DMARD initiation were similar for patients with rheumatoid arthritis (RA, 57.7%) and psoriatic arthritis (PsA, 64.3%), said the first author of the poster presentation, Sogol S. Amjadi, DO, a resident physician at Bingham Memorial Hospital, Blackfoot, Idaho.
However, at 12 months after diagnosis, only 29.6% of ankylosing spondylitis (AS) patients had not been started on a DMARD. “The ankylosing spondylitis group really had the lowest DMARD initiation over time,” said Dr. Amjadi in an interview.
The study used diagnosis codes and natural language processing to look for incident cases of the three inflammatory arthritides (IAs) among patients receiving care within the Veterans Health Administration from 2007 through 2015.
In all, 12,118 patients with incident IA were identified. Of these, 9,711 had RA, 1,472 had PsA, and 935 had AS. Patients were mostly (91.3%) male, with a mean age of 63.7 years.
Over the study period, 41.2% of IA patients were dispensed a DMARD within 30 days of diagnosis, and 50% received a DMARD within 90 days of diagnosis. Patients with PsA or RA had similar rates of DMARD prescription within 30 days of diagnosis (about 42% and 43%, respectively).
The investigators discovered in their analysis that another factor in prompt treatment was access to specialty care.“Timely access to a rheumatology provider is likely important for early DMARD treatment,” wrote Dr. Amjadi and her coauthors in the poster accompanying the presentation. Of patients who did receive a DMARD, 82.7% had received rheumatology specialty care before nonbiologic DMARD dispensing, as had 90.0% of patients receiving biologic DMARDs. Over the entire study period, about 10% of all IA patients had biologic DMARD exposure.
There was a trend over time for increased DMARD dispensing, said the investigators. “The percentage of IA patients with DMARD exposure during the 12-month follow-up period increased from 48.8% in 2008 to 66.4% in 2015.”
For AS patients, early DMARD prescribing rates rose from about 20% in 2007 to nearly 30% in 2015. “DMARD treatment rates during the initial 12 months after diagnosis increased between 2007 and 2015, but nontreatment remained common, particularly in patients with AS,” wrote the investigators. “Delays in treatment for inflammatory arthritis are associated with unfavorable outcomes, including impaired quality of life, irreversible joint damage, and disability.”
The authors reported no conflicts of interest and no outside sources of funding.
MADISON, WISC. – Only half of United States veterans with inflammatory arthritis received disease-modifying medication within 90 days of diagnosis if they received care within the Veterans Health Administration, according to a study presented at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN).
Over the study period, 58.2% of all inflammatory arthritis patients began a disease-modifying antirheumatic drug (DMARD) within 12 months of diagnosis. Rates of DMARD initiation were similar for patients with rheumatoid arthritis (RA, 57.7%) and psoriatic arthritis (PsA, 64.3%), said the first author of the poster presentation, Sogol S. Amjadi, DO, a resident physician at Bingham Memorial Hospital, Blackfoot, Idaho.
However, at 12 months after diagnosis, only 29.6% of ankylosing spondylitis (AS) patients had not been started on a DMARD. “The ankylosing spondylitis group really had the lowest DMARD initiation over time,” said Dr. Amjadi in an interview.
The study used diagnosis codes and natural language processing to look for incident cases of the three inflammatory arthritides (IAs) among patients receiving care within the Veterans Health Administration from 2007 through 2015.
In all, 12,118 patients with incident IA were identified. Of these, 9,711 had RA, 1,472 had PsA, and 935 had AS. Patients were mostly (91.3%) male, with a mean age of 63.7 years.
Over the study period, 41.2% of IA patients were dispensed a DMARD within 30 days of diagnosis, and 50% received a DMARD within 90 days of diagnosis. Patients with PsA or RA had similar rates of DMARD prescription within 30 days of diagnosis (about 42% and 43%, respectively).
The investigators discovered in their analysis that another factor in prompt treatment was access to specialty care.“Timely access to a rheumatology provider is likely important for early DMARD treatment,” wrote Dr. Amjadi and her coauthors in the poster accompanying the presentation. Of patients who did receive a DMARD, 82.7% had received rheumatology specialty care before nonbiologic DMARD dispensing, as had 90.0% of patients receiving biologic DMARDs. Over the entire study period, about 10% of all IA patients had biologic DMARD exposure.
There was a trend over time for increased DMARD dispensing, said the investigators. “The percentage of IA patients with DMARD exposure during the 12-month follow-up period increased from 48.8% in 2008 to 66.4% in 2015.”
For AS patients, early DMARD prescribing rates rose from about 20% in 2007 to nearly 30% in 2015. “DMARD treatment rates during the initial 12 months after diagnosis increased between 2007 and 2015, but nontreatment remained common, particularly in patients with AS,” wrote the investigators. “Delays in treatment for inflammatory arthritis are associated with unfavorable outcomes, including impaired quality of life, irreversible joint damage, and disability.”
The authors reported no conflicts of interest and no outside sources of funding.
MADISON, WISC. – Only half of United States veterans with inflammatory arthritis received disease-modifying medication within 90 days of diagnosis if they received care within the Veterans Health Administration, according to a study presented at the annual meeting of the Spondyloarthritis Research and Treatment Network (SPARTAN).
Over the study period, 58.2% of all inflammatory arthritis patients began a disease-modifying antirheumatic drug (DMARD) within 12 months of diagnosis. Rates of DMARD initiation were similar for patients with rheumatoid arthritis (RA, 57.7%) and psoriatic arthritis (PsA, 64.3%), said the first author of the poster presentation, Sogol S. Amjadi, DO, a resident physician at Bingham Memorial Hospital, Blackfoot, Idaho.
However, at 12 months after diagnosis, only 29.6% of ankylosing spondylitis (AS) patients had not been started on a DMARD. “The ankylosing spondylitis group really had the lowest DMARD initiation over time,” said Dr. Amjadi in an interview.
The study used diagnosis codes and natural language processing to look for incident cases of the three inflammatory arthritides (IAs) among patients receiving care within the Veterans Health Administration from 2007 through 2015.
In all, 12,118 patients with incident IA were identified. Of these, 9,711 had RA, 1,472 had PsA, and 935 had AS. Patients were mostly (91.3%) male, with a mean age of 63.7 years.
Over the study period, 41.2% of IA patients were dispensed a DMARD within 30 days of diagnosis, and 50% received a DMARD within 90 days of diagnosis. Patients with PsA or RA had similar rates of DMARD prescription within 30 days of diagnosis (about 42% and 43%, respectively).
The investigators discovered in their analysis that another factor in prompt treatment was access to specialty care.“Timely access to a rheumatology provider is likely important for early DMARD treatment,” wrote Dr. Amjadi and her coauthors in the poster accompanying the presentation. Of patients who did receive a DMARD, 82.7% had received rheumatology specialty care before nonbiologic DMARD dispensing, as had 90.0% of patients receiving biologic DMARDs. Over the entire study period, about 10% of all IA patients had biologic DMARD exposure.
There was a trend over time for increased DMARD dispensing, said the investigators. “The percentage of IA patients with DMARD exposure during the 12-month follow-up period increased from 48.8% in 2008 to 66.4% in 2015.”
For AS patients, early DMARD prescribing rates rose from about 20% in 2007 to nearly 30% in 2015. “DMARD treatment rates during the initial 12 months after diagnosis increased between 2007 and 2015, but nontreatment remained common, particularly in patients with AS,” wrote the investigators. “Delays in treatment for inflammatory arthritis are associated with unfavorable outcomes, including impaired quality of life, irreversible joint damage, and disability.”
The authors reported no conflicts of interest and no outside sources of funding.
REPORTING FROM SPARTAN 2019
Key clinical point:
Major finding: Overall, 58.2% of inflammatory arthritis patients received a DMARD within the first year of diagnosis.
Study details: Retrospective review of 12,118 incident cases of inflammatory arthritis in the Veterans Health Administration during the period from 2007 through 2015.
Disclosures: The authors reported no conflicts of interest and no outside sources of funding.
Source: Amjadi SS et al. SPARTAN 2019.
Insomnia meds get boxed warning from FDA
The Food and Drug Administration will now require that certain

Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
The Food and Drug Administration will now require that certain
Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
The Food and Drug Administration will now require that certain
Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
FDA approves Qternmet XR as adjunct therapy for glycemic improvement in type 2 diabetes
The Food and Drug Administration has approved Qternmet XR (dapagliflozin/saxagliptin/metformin) as an oral adjunct therapy to diet and exercise for the improvement of glycemic control in patients with type 2 diabetes, according to AstraZeneca.

FDA approval is based on results from a pair of phase 3 trials that tested different combinations of dapagliflozin and saxagliptin in patients with inadequately controlled type 2 diabetes who were also receiving metformin over a 24-week period. In both trials, treatment with dapagliflozin/saxagliptin/metformin decreased hemoglobin A1c by statistically significant amounts, and increased the number of patients with HbA1c levels below 7%
The safety results of Qternmet XR was consistent with each component medication’s known profile.
The Food and Drug Administration has approved Qternmet XR (dapagliflozin/saxagliptin/metformin) as an oral adjunct therapy to diet and exercise for the improvement of glycemic control in patients with type 2 diabetes, according to AstraZeneca.
FDA approval is based on results from a pair of phase 3 trials that tested different combinations of dapagliflozin and saxagliptin in patients with inadequately controlled type 2 diabetes who were also receiving metformin over a 24-week period. In both trials, treatment with dapagliflozin/saxagliptin/metformin decreased hemoglobin A1c by statistically significant amounts, and increased the number of patients with HbA1c levels below 7%
The safety results of Qternmet XR was consistent with each component medication’s known profile.
The Food and Drug Administration has approved Qternmet XR (dapagliflozin/saxagliptin/metformin) as an oral adjunct therapy to diet and exercise for the improvement of glycemic control in patients with type 2 diabetes, according to AstraZeneca.
FDA approval is based on results from a pair of phase 3 trials that tested different combinations of dapagliflozin and saxagliptin in patients with inadequately controlled type 2 diabetes who were also receiving metformin over a 24-week period. In both trials, treatment with dapagliflozin/saxagliptin/metformin decreased hemoglobin A1c by statistically significant amounts, and increased the number of patients with HbA1c levels below 7%
The safety results of Qternmet XR was consistent with each component medication’s known profile.
Direct pharmacy dispensing of naloxone linked to drop in fatal overdoses
investigators reported.
By contrast, state laws that stopped short of allowing pharmacists to directly dispense the opioid antagonist did not appear to impact mortality, according to the report, which appears in JAMA Internal Medicine (2019 May 6. doi: 10.1001/jamainternmed.2019.0272).
The report, based on state-level trends tracked from 2005 to 2016, indicates that fatal opioid overdoses fell by nearly one-third in states that adopted direct dispensing laws as compared with states that adopted other naloxone laws.
That finding suggests that the policy type determines whether a naloxone law is useful in combating fatal opioid overdoses, said Rahi Abouk, PhD, of William Paterson University, Wayne, N.J. and co-authors of the paper.
“Enabling distribution through various sources, or requiring gatekeepers, will not be as beneficial,” Dr. Abouk and co-authors said in their report.
The current rate of deaths from fentanyl, heroin, and prescription analgesic overdose has outpaced all previous drug epidemics on record, and even surpasses the number of deaths in the peak year of the HIV epidemic of the 1980s, Dr. Abouk and colleagues wrote in their paper.
The number of states with naloxone access laws grew from just 2 in 2005 to 47 by 2016, including 9 states that granted direct authority to pharmacists and 38 that granted indirect authority, according to the researchers.
The analysis of overdose trends from 2005 to 2016 was based on naloxone distribution data from state Medicaid agencies and opioid-related mortality data from a national statistics system. Forty percent of nonelderly adults with an opioid addiction are covered by Medicaid, the researchers said.
They found that naloxone laws granting pharmacists direct dispensing authority were linked to a drop in opioid deaths that increased in magnitude over time, according to researchers. The mean number of opioid deaths dropped by 27% in the second year after adoption of direct authority laws, relative to opioid deaths in states with indirect access laws, while in subsequent years, deaths dropped by 34%.
Emergency department visits related to opioids increased by 15% in direct authority states 3 or more years after adoption, as compared to states that did not adopt direct authority laws. According to investigators, that translated into 15 additional opioid-related emergency department visits each month.
That increase suggests that, alongside direct dispensing laws, “useful interventions” and connections to treatment are needed for the emergency department, according to Dr. Abouk and colleagues.
“This is the location where such programs may be the most effective,” they said in their report.
Future research should be done to determine whether removing gatekeepers increases the value of naloxone distribution policies, they concluded in the report.
Dr. Abouk had no disclosures. Co-authors on the study reported funding and conflict of interest disclosures related to the National Institute on Drug Abuse and the Centers for Disease Control and Prevention.
SOURCE: Abouk R, et al. JAMA Intern Med. 2019 May 6. doi:10.1001/jamainternmed.2019.0272.
investigators reported.
By contrast, state laws that stopped short of allowing pharmacists to directly dispense the opioid antagonist did not appear to impact mortality, according to the report, which appears in JAMA Internal Medicine (2019 May 6. doi: 10.1001/jamainternmed.2019.0272).
The report, based on state-level trends tracked from 2005 to 2016, indicates that fatal opioid overdoses fell by nearly one-third in states that adopted direct dispensing laws as compared with states that adopted other naloxone laws.
That finding suggests that the policy type determines whether a naloxone law is useful in combating fatal opioid overdoses, said Rahi Abouk, PhD, of William Paterson University, Wayne, N.J. and co-authors of the paper.
“Enabling distribution through various sources, or requiring gatekeepers, will not be as beneficial,” Dr. Abouk and co-authors said in their report.
The current rate of deaths from fentanyl, heroin, and prescription analgesic overdose has outpaced all previous drug epidemics on record, and even surpasses the number of deaths in the peak year of the HIV epidemic of the 1980s, Dr. Abouk and colleagues wrote in their paper.
The number of states with naloxone access laws grew from just 2 in 2005 to 47 by 2016, including 9 states that granted direct authority to pharmacists and 38 that granted indirect authority, according to the researchers.
The analysis of overdose trends from 2005 to 2016 was based on naloxone distribution data from state Medicaid agencies and opioid-related mortality data from a national statistics system. Forty percent of nonelderly adults with an opioid addiction are covered by Medicaid, the researchers said.
They found that naloxone laws granting pharmacists direct dispensing authority were linked to a drop in opioid deaths that increased in magnitude over time, according to researchers. The mean number of opioid deaths dropped by 27% in the second year after adoption of direct authority laws, relative to opioid deaths in states with indirect access laws, while in subsequent years, deaths dropped by 34%.
Emergency department visits related to opioids increased by 15% in direct authority states 3 or more years after adoption, as compared to states that did not adopt direct authority laws. According to investigators, that translated into 15 additional opioid-related emergency department visits each month.
That increase suggests that, alongside direct dispensing laws, “useful interventions” and connections to treatment are needed for the emergency department, according to Dr. Abouk and colleagues.
“This is the location where such programs may be the most effective,” they said in their report.
Future research should be done to determine whether removing gatekeepers increases the value of naloxone distribution policies, they concluded in the report.
Dr. Abouk had no disclosures. Co-authors on the study reported funding and conflict of interest disclosures related to the National Institute on Drug Abuse and the Centers for Disease Control and Prevention.
SOURCE: Abouk R, et al. JAMA Intern Med. 2019 May 6. doi:10.1001/jamainternmed.2019.0272.
investigators reported.
By contrast, state laws that stopped short of allowing pharmacists to directly dispense the opioid antagonist did not appear to impact mortality, according to the report, which appears in JAMA Internal Medicine (2019 May 6. doi: 10.1001/jamainternmed.2019.0272).
The report, based on state-level trends tracked from 2005 to 2016, indicates that fatal opioid overdoses fell by nearly one-third in states that adopted direct dispensing laws as compared with states that adopted other naloxone laws.
That finding suggests that the policy type determines whether a naloxone law is useful in combating fatal opioid overdoses, said Rahi Abouk, PhD, of William Paterson University, Wayne, N.J. and co-authors of the paper.
“Enabling distribution through various sources, or requiring gatekeepers, will not be as beneficial,” Dr. Abouk and co-authors said in their report.
The current rate of deaths from fentanyl, heroin, and prescription analgesic overdose has outpaced all previous drug epidemics on record, and even surpasses the number of deaths in the peak year of the HIV epidemic of the 1980s, Dr. Abouk and colleagues wrote in their paper.
The number of states with naloxone access laws grew from just 2 in 2005 to 47 by 2016, including 9 states that granted direct authority to pharmacists and 38 that granted indirect authority, according to the researchers.
The analysis of overdose trends from 2005 to 2016 was based on naloxone distribution data from state Medicaid agencies and opioid-related mortality data from a national statistics system. Forty percent of nonelderly adults with an opioid addiction are covered by Medicaid, the researchers said.
They found that naloxone laws granting pharmacists direct dispensing authority were linked to a drop in opioid deaths that increased in magnitude over time, according to researchers. The mean number of opioid deaths dropped by 27% in the second year after adoption of direct authority laws, relative to opioid deaths in states with indirect access laws, while in subsequent years, deaths dropped by 34%.
Emergency department visits related to opioids increased by 15% in direct authority states 3 or more years after adoption, as compared to states that did not adopt direct authority laws. According to investigators, that translated into 15 additional opioid-related emergency department visits each month.
That increase suggests that, alongside direct dispensing laws, “useful interventions” and connections to treatment are needed for the emergency department, according to Dr. Abouk and colleagues.
“This is the location where such programs may be the most effective,” they said in their report.
Future research should be done to determine whether removing gatekeepers increases the value of naloxone distribution policies, they concluded in the report.
Dr. Abouk had no disclosures. Co-authors on the study reported funding and conflict of interest disclosures related to the National Institute on Drug Abuse and the Centers for Disease Control and Prevention.
SOURCE: Abouk R, et al. JAMA Intern Med. 2019 May 6. doi:10.1001/jamainternmed.2019.0272.
FROM JAMA Internal Medicine
Key clinical point: State laws granting pharmacists direct authority to dispense naloxone were linked to significant drops in opioid-related fatal overdoses.
Major finding: The mean number of opioid deaths dropped by 27% in the second year after adoption of direct authority laws relative to opioid deaths in states with indirect access laws, while in subsequent years, deaths dropped by 34%.
Study details: Analysis of naloxone distribution data and opioid-related mortality data from 2005 to 2016 for all 50 states and the District of Columbia.
Disclosures: Study authors reported funding and conflict of interest disclosures related to the National Institute on Drug Abuse and the Centers for Disease Control and Prevention.
Source: Abouk R, et al. JAMA Intern Med. 2019 May 6.
FDA approves ivosidenib frontline for certain AML patients
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
Mavyret approved for children with any HCV genotype
The Food and Drug Administration has approved glecaprevir/pibrentasvir tablets (Mavyret) for treating any of six identified genotypes of hepatitis C virus in children ages 12-17 years.
The agency noted in its press announcement that, Dosing information now will be provided for patients aged 12 years and older or weighing at least 99 lbs, without cirrhosis or who have compensated cirrhosis. It is not recommended for patients with moderate cirrhosis, and it is contraindicated in patients with severe cirrhosis, as well as patients taking atazanavir and rifampin.
In clinical trials of 47 patients with genotype 1, 2, 3, or 4 HCV without cirrhosis or with only mild cirrhosis, results at 12 weeks after 8 or 16 weeks’ treatment suggested patients’ infections had been cured – 100% had no virus detected in their blood. Adverse reactions observed were consistent with those previously observed in adults during clinical trials.
The most common reactions were headache and fatigue. Hepatitis B virus reactivation has been reported in coinfected adults during or after treatment with direct-acting antivirals, and in those who were not receiving HBV antiviral treatment. Full prescribing information can be found on the FDA website, and more information about this approval can be found in the agency’s announcement.
The Food and Drug Administration has approved glecaprevir/pibrentasvir tablets (Mavyret) for treating any of six identified genotypes of hepatitis C virus in children ages 12-17 years.
The agency noted in its press announcement that, Dosing information now will be provided for patients aged 12 years and older or weighing at least 99 lbs, without cirrhosis or who have compensated cirrhosis. It is not recommended for patients with moderate cirrhosis, and it is contraindicated in patients with severe cirrhosis, as well as patients taking atazanavir and rifampin.
In clinical trials of 47 patients with genotype 1, 2, 3, or 4 HCV without cirrhosis or with only mild cirrhosis, results at 12 weeks after 8 or 16 weeks’ treatment suggested patients’ infections had been cured – 100% had no virus detected in their blood. Adverse reactions observed were consistent with those previously observed in adults during clinical trials.
The most common reactions were headache and fatigue. Hepatitis B virus reactivation has been reported in coinfected adults during or after treatment with direct-acting antivirals, and in those who were not receiving HBV antiviral treatment. Full prescribing information can be found on the FDA website, and more information about this approval can be found in the agency’s announcement.
The Food and Drug Administration has approved glecaprevir/pibrentasvir tablets (Mavyret) for treating any of six identified genotypes of hepatitis C virus in children ages 12-17 years.
The agency noted in its press announcement that, Dosing information now will be provided for patients aged 12 years and older or weighing at least 99 lbs, without cirrhosis or who have compensated cirrhosis. It is not recommended for patients with moderate cirrhosis, and it is contraindicated in patients with severe cirrhosis, as well as patients taking atazanavir and rifampin.
In clinical trials of 47 patients with genotype 1, 2, 3, or 4 HCV without cirrhosis or with only mild cirrhosis, results at 12 weeks after 8 or 16 weeks’ treatment suggested patients’ infections had been cured – 100% had no virus detected in their blood. Adverse reactions observed were consistent with those previously observed in adults during clinical trials.
The most common reactions were headache and fatigue. Hepatitis B virus reactivation has been reported in coinfected adults during or after treatment with direct-acting antivirals, and in those who were not receiving HBV antiviral treatment. Full prescribing information can be found on the FDA website, and more information about this approval can be found in the agency’s announcement.
Long-term antibiotic use may heighten stroke, CHD risk
, according to a study in the European Heart Journal.
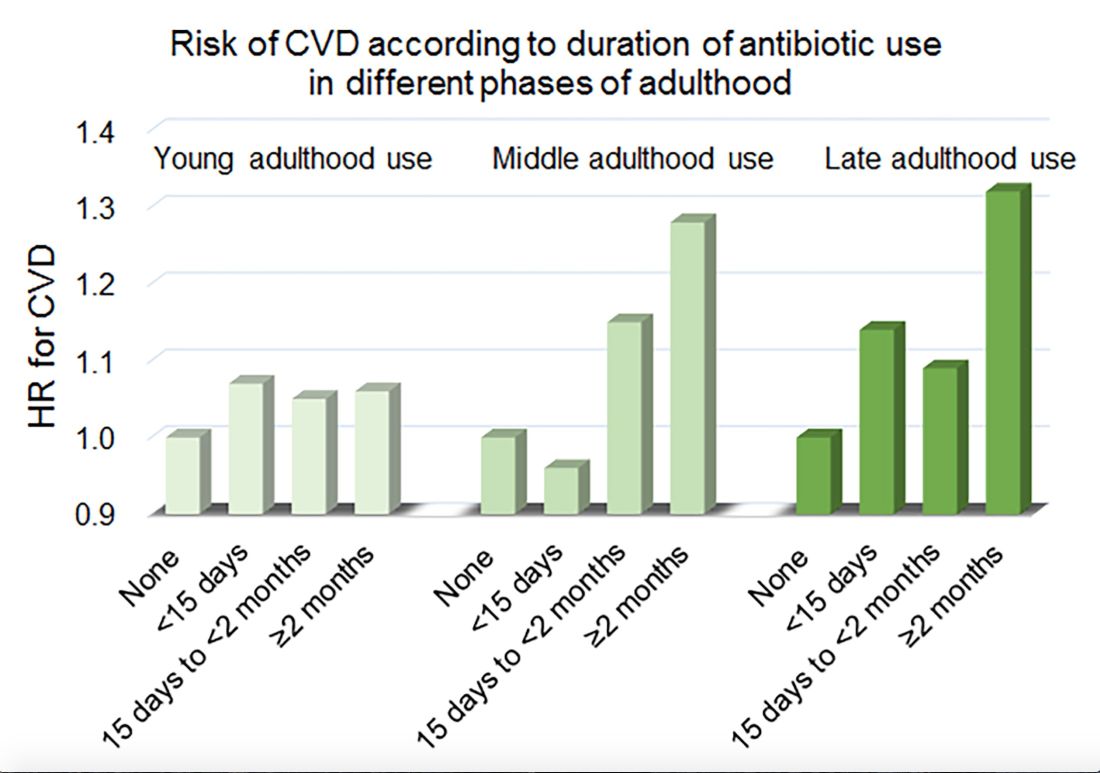
Women in the Nurses’ Health Study who used antibiotics for 2 or more months between ages 40 and 59 years or at age 60 years and older had a significantly increased risk of cardiovascular disease, compared with those who did not use antibiotics. Antibiotic use between 20 and 39 years old was not significantly related to cardiovascular disease.
Prior research has found that antibiotics may have long-lasting effects on gut microbiota and relate to cardiovascular disease risk.
“Antibiotic use is the most critical factor in altering the balance of microorganisms in the gut,” said lead investigator Lu Qi, MD, PhD, in a news release. “Previous studies have shown a link between alterations in the microbiotic environment of the gut and inflammation and narrowing of the blood vessels, stroke, and heart disease,” said Dr. Qi, who is the director of the Tulane University Obesity Research Center in New Orleans and an adjunct professor of nutrition at Harvard T.C. Chan School of Public Health in Boston.
To evaluate associations between life stage, antibiotic exposure, and subsequent cardiovascular disease, researchers analyzed data from 36,429 participants in the Nurses’ Health Study. The women were at least 60 years old and had no history of cardiovascular disease or cancer when they completed a 2004 questionnaire about antibiotic usage during young, middle, and late adulthood. The questionnaire asked participants to indicate the total time using antibiotics with eight categories ranging from none to 5 or more years.
The researchers defined incident cardiovascular disease as a composite endpoint of coronary heart disease (nonfatal myocardial infarction or fatal coronary heart disease) and stroke (nonfatal or fatal). They calculated person-years of follow-up from the questionnaire return date until date of cardiovascular disease diagnosis, death, or end of follow-up in 2012.
Women with longer duration of antibiotic use were more likely to use other medications and have unfavorable cardiovascular risk profiles, including family history of myocardial infarction and higher body mass index. Antibiotics most often were used to treat respiratory infections. During an average follow-up of 7.6 years, 1,056 participants developed cardiovascular disease.
In a multivariable model that adjusted for demographics, diet, lifestyle, reason for antibiotic use, medications, overweight status, and other factors, long-term antibiotic use – 2 months or more – in late adulthood was associated with significantly increased risk of cardiovascular disease (hazard ratio, 1.32), as was long-term antibiotic use in middle adulthood (HR, 1.28).
Although antibiotic use was self-reported, which could lead to misclassification, the participants were health professionals, which may mitigate this limitation, the authors noted. Whether these findings apply to men and other populations requires further study, they said.
Because of the study’s observational design, the results “cannot show that antibiotics cause heart disease and stroke, only that there is a link between them,” Dr. Qi said. “It’s possible that women who reported more antibiotic use might be sicker in other ways that we were unable to measure, or there may be other factors that could affect the results that we have not been able take account of.”
“Our study suggests that antibiotics should be used only when they are absolutely needed,” he concluded. “Considering the potentially cumulative adverse effects, the shorter time of antibiotic use the better.”
The study was supported by National Institutes of Health grants, the Boston Obesity Nutrition Research Center, and the United States–Israel Binational Science Foundation. One author received support from the Japan Society for the Promotion of Science. The authors had no conflicts of interest.
SOURCE: Heianza Y et al. Eur Heart J. 2019 Apr 24. doi: 10.1093/eurheartj/ehz231.
, according to a study in the European Heart Journal.
Women in the Nurses’ Health Study who used antibiotics for 2 or more months between ages 40 and 59 years or at age 60 years and older had a significantly increased risk of cardiovascular disease, compared with those who did not use antibiotics. Antibiotic use between 20 and 39 years old was not significantly related to cardiovascular disease.
Prior research has found that antibiotics may have long-lasting effects on gut microbiota and relate to cardiovascular disease risk.
“Antibiotic use is the most critical factor in altering the balance of microorganisms in the gut,” said lead investigator Lu Qi, MD, PhD, in a news release. “Previous studies have shown a link between alterations in the microbiotic environment of the gut and inflammation and narrowing of the blood vessels, stroke, and heart disease,” said Dr. Qi, who is the director of the Tulane University Obesity Research Center in New Orleans and an adjunct professor of nutrition at Harvard T.C. Chan School of Public Health in Boston.
To evaluate associations between life stage, antibiotic exposure, and subsequent cardiovascular disease, researchers analyzed data from 36,429 participants in the Nurses’ Health Study. The women were at least 60 years old and had no history of cardiovascular disease or cancer when they completed a 2004 questionnaire about antibiotic usage during young, middle, and late adulthood. The questionnaire asked participants to indicate the total time using antibiotics with eight categories ranging from none to 5 or more years.
The researchers defined incident cardiovascular disease as a composite endpoint of coronary heart disease (nonfatal myocardial infarction or fatal coronary heart disease) and stroke (nonfatal or fatal). They calculated person-years of follow-up from the questionnaire return date until date of cardiovascular disease diagnosis, death, or end of follow-up in 2012.
Women with longer duration of antibiotic use were more likely to use other medications and have unfavorable cardiovascular risk profiles, including family history of myocardial infarction and higher body mass index. Antibiotics most often were used to treat respiratory infections. During an average follow-up of 7.6 years, 1,056 participants developed cardiovascular disease.
In a multivariable model that adjusted for demographics, diet, lifestyle, reason for antibiotic use, medications, overweight status, and other factors, long-term antibiotic use – 2 months or more – in late adulthood was associated with significantly increased risk of cardiovascular disease (hazard ratio, 1.32), as was long-term antibiotic use in middle adulthood (HR, 1.28).
Although antibiotic use was self-reported, which could lead to misclassification, the participants were health professionals, which may mitigate this limitation, the authors noted. Whether these findings apply to men and other populations requires further study, they said.
Because of the study’s observational design, the results “cannot show that antibiotics cause heart disease and stroke, only that there is a link between them,” Dr. Qi said. “It’s possible that women who reported more antibiotic use might be sicker in other ways that we were unable to measure, or there may be other factors that could affect the results that we have not been able take account of.”
“Our study suggests that antibiotics should be used only when they are absolutely needed,” he concluded. “Considering the potentially cumulative adverse effects, the shorter time of antibiotic use the better.”
The study was supported by National Institutes of Health grants, the Boston Obesity Nutrition Research Center, and the United States–Israel Binational Science Foundation. One author received support from the Japan Society for the Promotion of Science. The authors had no conflicts of interest.
SOURCE: Heianza Y et al. Eur Heart J. 2019 Apr 24. doi: 10.1093/eurheartj/ehz231.
, according to a study in the European Heart Journal.
Women in the Nurses’ Health Study who used antibiotics for 2 or more months between ages 40 and 59 years or at age 60 years and older had a significantly increased risk of cardiovascular disease, compared with those who did not use antibiotics. Antibiotic use between 20 and 39 years old was not significantly related to cardiovascular disease.
Prior research has found that antibiotics may have long-lasting effects on gut microbiota and relate to cardiovascular disease risk.
“Antibiotic use is the most critical factor in altering the balance of microorganisms in the gut,” said lead investigator Lu Qi, MD, PhD, in a news release. “Previous studies have shown a link between alterations in the microbiotic environment of the gut and inflammation and narrowing of the blood vessels, stroke, and heart disease,” said Dr. Qi, who is the director of the Tulane University Obesity Research Center in New Orleans and an adjunct professor of nutrition at Harvard T.C. Chan School of Public Health in Boston.
To evaluate associations between life stage, antibiotic exposure, and subsequent cardiovascular disease, researchers analyzed data from 36,429 participants in the Nurses’ Health Study. The women were at least 60 years old and had no history of cardiovascular disease or cancer when they completed a 2004 questionnaire about antibiotic usage during young, middle, and late adulthood. The questionnaire asked participants to indicate the total time using antibiotics with eight categories ranging from none to 5 or more years.
The researchers defined incident cardiovascular disease as a composite endpoint of coronary heart disease (nonfatal myocardial infarction or fatal coronary heart disease) and stroke (nonfatal or fatal). They calculated person-years of follow-up from the questionnaire return date until date of cardiovascular disease diagnosis, death, or end of follow-up in 2012.
Women with longer duration of antibiotic use were more likely to use other medications and have unfavorable cardiovascular risk profiles, including family history of myocardial infarction and higher body mass index. Antibiotics most often were used to treat respiratory infections. During an average follow-up of 7.6 years, 1,056 participants developed cardiovascular disease.
In a multivariable model that adjusted for demographics, diet, lifestyle, reason for antibiotic use, medications, overweight status, and other factors, long-term antibiotic use – 2 months or more – in late adulthood was associated with significantly increased risk of cardiovascular disease (hazard ratio, 1.32), as was long-term antibiotic use in middle adulthood (HR, 1.28).
Although antibiotic use was self-reported, which could lead to misclassification, the participants were health professionals, which may mitigate this limitation, the authors noted. Whether these findings apply to men and other populations requires further study, they said.
Because of the study’s observational design, the results “cannot show that antibiotics cause heart disease and stroke, only that there is a link between them,” Dr. Qi said. “It’s possible that women who reported more antibiotic use might be sicker in other ways that we were unable to measure, or there may be other factors that could affect the results that we have not been able take account of.”
“Our study suggests that antibiotics should be used only when they are absolutely needed,” he concluded. “Considering the potentially cumulative adverse effects, the shorter time of antibiotic use the better.”
The study was supported by National Institutes of Health grants, the Boston Obesity Nutrition Research Center, and the United States–Israel Binational Science Foundation. One author received support from the Japan Society for the Promotion of Science. The authors had no conflicts of interest.
SOURCE: Heianza Y et al. Eur Heart J. 2019 Apr 24. doi: 10.1093/eurheartj/ehz231.
FROM THE EUROPEAN HEART JOURNAL
Key clinical point: Among middle-aged and older women, 2 or more months’ exposure to antibiotics is associated with an increased risk of coronary heart disease or stroke.
Major finding: Long-term antibiotic use in late adulthood was associated with significantly increased risk of cardiovascular disease (hazard ratio, 1.32), as was long-term antibiotic use in middle adulthood (HR, 1.28).
Study details: An analysis of data from nearly 36,500 women in the Nurses’ Health Study.
Disclosures: The study was supported by National Institutes of Health grants, the Boston Obesity Nutrition Research Center, and the United States–Israel Binational Science Foundation. One author received support from the Japan Society for the Promotion of Science. The authors had no conflicts of interest.
Source: Heianza Y et al. Eur Heart J. 2019 Apr 24. doi: 10.1093/eurheartj/ehz231.
Medical cannabis relieved pain, decreased opioid use in elderly
results of a recent retrospective chart review suggest. Treatment with medical cannabis improved pain, sleep, anxiety, and neuropathy in patients aged 75 years of age and older, and was associated with reduced use of opioids in about one-third of cases, according to authors of the study, which will be presented at the annual meeting of the American Academy of Neurology.

“Our findings are promising and can help fuel further research into medical marijuana as an additional option for this group of people who often have chronic conditions,” said lead investigator Laszlo Mechtler, MD, of Dent Neurologic Institute in Buffalo, N.Y., in a news release. However, additional randomized, placebo-controlled studies are needed to confirm results of this study, Dr. Mechtler added.
The chart review focused on 204 elderly patients who participated in New York State’s medical marijuana program and were followed in a neurologic outpatient setting. The cohort included 129 female and 75 male patients, ranging in age from 75 to 102 years, with a mean age of 81 years. The medical marijuana was taken by mouth as a liquid extract tincture, capsule, or in an electronic vaporizer.
With an average exposure time of 16.8 weeks, 69% of patients experienced symptomatic benefit, according to patient self-report. The most commonly reported benefit was relief of chronic pain in 49%, while improvements in sleep, neuropathy, and anxiety were reported in 18%, 15%, and 10%, respectively. Reductions in opioid pain medication were noted in about one-third of cases, they found.
While 34% of patients had adverse effects on medical marijuana, only 21% reported adverse effects after cannabinoid doses were adjusted, investigators said. Adverse effects led to discontinuation of medical cannabis in seven patients, or 3.4% of the overall cohort. Somnolence, disequilibrium, and gastrointestinal disturbance were the most common adverse effects, occurring in 13%, 7%, and 7% of patients, respectively. Euphoria was reported in 3% of patients.
Among patients who had no reported adverse effects, the most commonly used formulation was a balanced 1:1 tincture of tetrahydrocannabinol to cannabidiol, investigators said.
Further trials could explore optimal dosing of medical cannabis in elderly patients and shed more light on adverse effects such as somnolence and disequilibrium, according to Dr. Mechtler and colleagues.
The study was supported by the Dent Family Foundation.
SOURCE: Bargnes V et al. AAN 2019, Abstract P4.1-014.
results of a recent retrospective chart review suggest. Treatment with medical cannabis improved pain, sleep, anxiety, and neuropathy in patients aged 75 years of age and older, and was associated with reduced use of opioids in about one-third of cases, according to authors of the study, which will be presented at the annual meeting of the American Academy of Neurology.
“Our findings are promising and can help fuel further research into medical marijuana as an additional option for this group of people who often have chronic conditions,” said lead investigator Laszlo Mechtler, MD, of Dent Neurologic Institute in Buffalo, N.Y., in a news release. However, additional randomized, placebo-controlled studies are needed to confirm results of this study, Dr. Mechtler added.
The chart review focused on 204 elderly patients who participated in New York State’s medical marijuana program and were followed in a neurologic outpatient setting. The cohort included 129 female and 75 male patients, ranging in age from 75 to 102 years, with a mean age of 81 years. The medical marijuana was taken by mouth as a liquid extract tincture, capsule, or in an electronic vaporizer.
With an average exposure time of 16.8 weeks, 69% of patients experienced symptomatic benefit, according to patient self-report. The most commonly reported benefit was relief of chronic pain in 49%, while improvements in sleep, neuropathy, and anxiety were reported in 18%, 15%, and 10%, respectively. Reductions in opioid pain medication were noted in about one-third of cases, they found.
While 34% of patients had adverse effects on medical marijuana, only 21% reported adverse effects after cannabinoid doses were adjusted, investigators said. Adverse effects led to discontinuation of medical cannabis in seven patients, or 3.4% of the overall cohort. Somnolence, disequilibrium, and gastrointestinal disturbance were the most common adverse effects, occurring in 13%, 7%, and 7% of patients, respectively. Euphoria was reported in 3% of patients.
Among patients who had no reported adverse effects, the most commonly used formulation was a balanced 1:1 tincture of tetrahydrocannabinol to cannabidiol, investigators said.
Further trials could explore optimal dosing of medical cannabis in elderly patients and shed more light on adverse effects such as somnolence and disequilibrium, according to Dr. Mechtler and colleagues.
The study was supported by the Dent Family Foundation.
SOURCE: Bargnes V et al. AAN 2019, Abstract P4.1-014.
results of a recent retrospective chart review suggest. Treatment with medical cannabis improved pain, sleep, anxiety, and neuropathy in patients aged 75 years of age and older, and was associated with reduced use of opioids in about one-third of cases, according to authors of the study, which will be presented at the annual meeting of the American Academy of Neurology.
“Our findings are promising and can help fuel further research into medical marijuana as an additional option for this group of people who often have chronic conditions,” said lead investigator Laszlo Mechtler, MD, of Dent Neurologic Institute in Buffalo, N.Y., in a news release. However, additional randomized, placebo-controlled studies are needed to confirm results of this study, Dr. Mechtler added.
The chart review focused on 204 elderly patients who participated in New York State’s medical marijuana program and were followed in a neurologic outpatient setting. The cohort included 129 female and 75 male patients, ranging in age from 75 to 102 years, with a mean age of 81 years. The medical marijuana was taken by mouth as a liquid extract tincture, capsule, or in an electronic vaporizer.
With an average exposure time of 16.8 weeks, 69% of patients experienced symptomatic benefit, according to patient self-report. The most commonly reported benefit was relief of chronic pain in 49%, while improvements in sleep, neuropathy, and anxiety were reported in 18%, 15%, and 10%, respectively. Reductions in opioid pain medication were noted in about one-third of cases, they found.
While 34% of patients had adverse effects on medical marijuana, only 21% reported adverse effects after cannabinoid doses were adjusted, investigators said. Adverse effects led to discontinuation of medical cannabis in seven patients, or 3.4% of the overall cohort. Somnolence, disequilibrium, and gastrointestinal disturbance were the most common adverse effects, occurring in 13%, 7%, and 7% of patients, respectively. Euphoria was reported in 3% of patients.
Among patients who had no reported adverse effects, the most commonly used formulation was a balanced 1:1 tincture of tetrahydrocannabinol to cannabidiol, investigators said.
Further trials could explore optimal dosing of medical cannabis in elderly patients and shed more light on adverse effects such as somnolence and disequilibrium, according to Dr. Mechtler and colleagues.
The study was supported by the Dent Family Foundation.
SOURCE: Bargnes V et al. AAN 2019, Abstract P4.1-014.
FROM AAN 2019
FDA approves IL-23 inhibitor risankizumab for treating plaque psoriasis
Risankizumab, an interleukin-23 inhibitor, has been approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the manufacturer announced on April 23.
Risankizumab selectively inhibits interleukin-23 (IL-23), a key inflammatory protein, by binding to its p19 subunit. The drug is administered at a dose of 150 mg, in two subcutaneous injections, every 12 weeks, after starting doses at weeks 0 and 4. It will be available in early May, according to an AbbVie press release announcing the approval.
The approval was based in part on data from two phase 3, 2-year studies, In UltIMMA-1 and UltIMMA-2, at 16 weeks, 75% of risankizumab patients in both studies achieved a Psoriasis Area and Severity Index (PASI 90), compared with 5% and 2% of those on placebo, respectively. These results were published in 2018 (Lancet. 2018 Aug 25;392[10148]:650-61).
At 1 year, 82% and 81% of those treated with risankizumab in the two studies achieved a PASI 90, and 56% and 60% achieved a PASI 100, respectively, according to the company.
Approval was also based on additional phase 3 studies, IMMhance and IMMvent.
Upper respiratory infections were among the most common adverse events associated with risankizumab in trials, reported in 13%, according to the company. Other adverse events associated with treatment included headache (3.5 %), fatigue (2.5 %), injection site reactions (1.5%) and tinea infections (1.1%). The AbbVie release states that candidates for treatment should be evaluated for tuberculosis before starting therapy, and patients should be instructed to report signs and symptoms of infection.
Risankizumab, which will be marketed as Skyrizi, was recently approved in Canada for the same indication, and in Japan, for plaque psoriasis, generalized pustular psoriasis, erythrodermic psoriasis and psoriatic arthritis in adults. It currently is under review in Europe.
AbbVie and Boehringer Ingelheim are collaborating on the development of risankizumab, according to an AbbVie press release. Studies of risankizumab for treatment of psoriatic arthritis and Crohn’s disease are underway.
Risankizumab, an interleukin-23 inhibitor, has been approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the manufacturer announced on April 23.
Risankizumab selectively inhibits interleukin-23 (IL-23), a key inflammatory protein, by binding to its p19 subunit. The drug is administered at a dose of 150 mg, in two subcutaneous injections, every 12 weeks, after starting doses at weeks 0 and 4. It will be available in early May, according to an AbbVie press release announcing the approval.
The approval was based in part on data from two phase 3, 2-year studies, In UltIMMA-1 and UltIMMA-2, at 16 weeks, 75% of risankizumab patients in both studies achieved a Psoriasis Area and Severity Index (PASI 90), compared with 5% and 2% of those on placebo, respectively. These results were published in 2018 (Lancet. 2018 Aug 25;392[10148]:650-61).
At 1 year, 82% and 81% of those treated with risankizumab in the two studies achieved a PASI 90, and 56% and 60% achieved a PASI 100, respectively, according to the company.
Approval was also based on additional phase 3 studies, IMMhance and IMMvent.
Upper respiratory infections were among the most common adverse events associated with risankizumab in trials, reported in 13%, according to the company. Other adverse events associated with treatment included headache (3.5 %), fatigue (2.5 %), injection site reactions (1.5%) and tinea infections (1.1%). The AbbVie release states that candidates for treatment should be evaluated for tuberculosis before starting therapy, and patients should be instructed to report signs and symptoms of infection.
Risankizumab, which will be marketed as Skyrizi, was recently approved in Canada for the same indication, and in Japan, for plaque psoriasis, generalized pustular psoriasis, erythrodermic psoriasis and psoriatic arthritis in adults. It currently is under review in Europe.
AbbVie and Boehringer Ingelheim are collaborating on the development of risankizumab, according to an AbbVie press release. Studies of risankizumab for treatment of psoriatic arthritis and Crohn’s disease are underway.
Risankizumab, an interleukin-23 inhibitor, has been approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the manufacturer announced on April 23.
Risankizumab selectively inhibits interleukin-23 (IL-23), a key inflammatory protein, by binding to its p19 subunit. The drug is administered at a dose of 150 mg, in two subcutaneous injections, every 12 weeks, after starting doses at weeks 0 and 4. It will be available in early May, according to an AbbVie press release announcing the approval.
The approval was based in part on data from two phase 3, 2-year studies, In UltIMMA-1 and UltIMMA-2, at 16 weeks, 75% of risankizumab patients in both studies achieved a Psoriasis Area and Severity Index (PASI 90), compared with 5% and 2% of those on placebo, respectively. These results were published in 2018 (Lancet. 2018 Aug 25;392[10148]:650-61).
At 1 year, 82% and 81% of those treated with risankizumab in the two studies achieved a PASI 90, and 56% and 60% achieved a PASI 100, respectively, according to the company.
Approval was also based on additional phase 3 studies, IMMhance and IMMvent.
Upper respiratory infections were among the most common adverse events associated with risankizumab in trials, reported in 13%, according to the company. Other adverse events associated with treatment included headache (3.5 %), fatigue (2.5 %), injection site reactions (1.5%) and tinea infections (1.1%). The AbbVie release states that candidates for treatment should be evaluated for tuberculosis before starting therapy, and patients should be instructed to report signs and symptoms of infection.
Risankizumab, which will be marketed as Skyrizi, was recently approved in Canada for the same indication, and in Japan, for plaque psoriasis, generalized pustular psoriasis, erythrodermic psoriasis and psoriatic arthritis in adults. It currently is under review in Europe.
AbbVie and Boehringer Ingelheim are collaborating on the development of risankizumab, according to an AbbVie press release. Studies of risankizumab for treatment of psoriatic arthritis and Crohn’s disease are underway.